

Abstract
The homeostatic link between oxidative stress and autophagy plays an important role in cellular responses to a wide variety of physiological and pathological conditions. However, the regulatory pathway and outcomes remain incompletely understood. Here, we show that reactive oxygen species (ROS) function as signaling molecules that regulate autophagy through ataxia‐telangiectasia mutated (ATM) and cell cycle checkpoint kinase 2 (CHK2), a DNA damage response (DDR) pathway activated during metabolic and hypoxic stress. We report that CHK2 binds to and phosphorylates Beclin 1 at Ser90/Ser93, thereby impairing Beclin 1‐Bcl‐2 autophagy‐regulatory complex formation in a ROS‐dependent fashion. We further demonstrate that CHK2‐mediated autophagy has an unexpected role in reducing ROS levels via the removal of damaged mitochondria, which is required for cell survival under stress conditions. Finally, CHK2−/− mice display aggravated infarct phenotypes and reduced Beclin 1 p‐Ser90/Ser93 in a cerebral stroke model, suggesting an in vivo role of CHK2‐induced autophagy in cell survival. Taken together, these results indicate that the ROS‐ATM‐CHK2‐Beclin 1‐autophagy axis serves as a physiological adaptation pathway that protects cells exposed to pathological conditions from stress‐induced tissue damage.
Keywords: autophagy, Beclin 1, CHK2, oxidative stress, ROS
Subject Categories: Autophagy & Cell Death, Metabolism
ROS accumulation activates protective autophagy to prevent stress‐induced tissue damage.

Introduction
Nutrient deprivation and metabolic fluctuations cause oxidative stress due to increased reactive oxygen species (ROS) production coupled with an impaired antioxidant system, leading to redox state imbalance and cell death (Finkel & Holbrook, 2000; D'Autreaux & Toledano, 2007; Holmstrom & Finkel, 2014). Therefore, mechanisms of suppressing excessive ROS and maintaining cell and tissue homeostasis that are independent of the classic antioxidant systems are evolutionarily advantageous. We previously showed that cells deficient in essential autophagy genes have increased basal levels of ROS (Lee et al, 2012). Indeed, autophagy is an essential cellular process that plays a crucial role in recycling cellular components and damaged organelles to eliminate sources of ROS in response to diverse stress conditions (Kroemer et al, 2010; Scherz‐Shouval & Elazar, 2011; Filomeni et al, 2015).
Growing evidence indicates that ROS is one of the main intracellular signal transducers crucial for sustaining autophagy progression. For instance, the essential autophagy Cys protease ATG4 appears to be a direct target of oxidants generated during starvation. The redox‐dependent inactivation of ATG4 leads in turn to increased autophagosome formation (Scherz‐Shouval et al, 2007). Starvation‐induced ROS has also been shown to oxidize high mobility group box 1, triggering its translocation from the nucleus to the cytoplasm where it interacts with Beclin 1, causing disassociation of Beclin 1 from Bcl‐2 and induction of autophagy (Tang et al, 2010). ROS can also activate the lysosomal Ca2+ channel, mucolipin‐1 (MCOLN1), triggering the release of Ca2+ into cytosol that results in calcineurin‐dependent translocation of transcription factor EB and autophagy induction (Zhang et al, 2016b). However, other pathways presumably exist to couple redox status with autophagic flux and thereby help cells maintain homeostasis during nutrient deprivation or metabolic fluctuations.
The ATM/CHK2 protein kinases have a intriguing connection to autophagy. The ATM kinases are activated by DNA double‐strand breaks through the Mre11‐Rad50‐Nbs1 DNA repair complex. ATM kinases orchestrate signaling cascades that initiate the DDR and coordinate many processes including DNA repair, regulation of cell cycle checkpoints, and ultimately the induction of programmed cell death (Matsuoka et al, 1998; Lee et al, 2000; Yang et al, 2002; Bakkenist & Kastan, 2003; Lukas et al, 2003; Roos & Kaina, 2006). However, growing evidence indicates that ATM/CHK2 activation induced by H2O2 can also occur in the absence of DNA damage (Guo et al, 2010), indicating that ATM is an important sensor of ROS in human cells. Other studies suggest that ATM activity is required for ROS‐induced autophagy. For instance, in response to elevated ROS, ATM activates the TSC2 tumor suppressor via the LKB1/AMPK metabolic pathway in the cytoplasm to repress mTORC1 and induce autophagy (Alexander et al, 2010). A recent study showed that ATM signaling activates ULK1 and inhibits mTORC1 to induce autophagy of peroxisomes (pexophagy) in response to ROS (Zhang et al, 2015). However, the direct crosstalk between the DDR signaling and the autophagic machinery remains unclear.
A key factor in autophagy induction is the Beclin 1 protein. Beclin 1 was the first autophagy‐related protein identified in mammals (Liang et al, 1998), and BECN is an essential gene required during normal embryonic development (Yue et al, 2003). Beclin 1 also protects against the development of degenerative diseases (Shibata et al, 2006; Pickford et al, 2008; Lipinski et al, 2010; Ashkenazi et al, 2017) and functions as a tumor suppressor (Liang et al, 1999). Beclin 1 regulates both the initiation and the maturation of the autophagosome, by forming distinct PI3K complexes together with the core lipid kinase VPS34 and the regulatory component VPS15. Binding of either ATG14L or UVRAG to the complex promotes the initiation of autophagy and the formation of autophagosomes (Itakura et al, 2008; Liang et al, 2008). Following binding of RUBICON or Bcl‐2 to the complex, VPS34 kinase activity is inhibited and autophagic flux is reduced (Liang et al, 1998; Matsunaga et al, 2009; Zhong et al, 2009). Post‐transcriptional modifications of Beclin 1, in particular phosphorylation, play an important role in the regulation of autophagy by affecting VPS34 kinase activity and its interaction with various autophagy‐regulatory proteins (Hill et al, 2019). For instance, ULK1, AMPK, MAPKAPK2/3, DAPK, CaMKII, and PGK1 can phosphorylate Beclin 1 to promote autophagy (Zalckvar et al, 2009; Kim et al, 2013; Russell et al, 2013; Wei et al, 2015; Fujiwara et al, 2016; Zhang et al, 2016a; Li et al, 2017; Qian et al, 2017). In contrast, AKT1, EGFR, FAK, HER2, and MST1 phosphorylate Beclin 1 to inhibit autophagy (Wang et al, 2012; Maejima et al, 2013; Wei et al, 2013; Cheng et al, 2017; Vega‐Rubin‐de‐Celis et al, 2018). Beclin 1 phosphorylation is also precisely regulated by diverse signals including glucose starvation, serum starvation, amino acid deprivation, glutamine deprivation, ionomycin treatment, and hypoxia (Kim et al, 2013; Russell et al, 2013; Wei et al, 2013; Zhang et al, 2016a; Li et al, 2017; Qian et al, 2017). However, the redox‐dependent regulation of the Beclin 1‐interacting complex has not been characterized.
In this study, we report an unexpected function of CHK2‐mediated autophagy in limiting ROS levels during nutrient deprivation and metabolic fluctuations to maintain cell and tissue homeostasis. In response to ROS stimulation, CHK2 binds to and phosphorylates Beclin 1 at Ser90/Ser93, promoting autophagy via Beclin 1 release from Bcl‐2 sequestration. Our findings thereby establish a novel and critical role for the ATM/CHK2/Beclin 1 axis in protecting cells from oxidative stress and delineate a pathway whereby ROS functions as a signaling molecule to modulate autophagic flux.
Results
CHK2 is involved in oxidative stress‐induced autophagy
To investigate the link between oxidative stress and autophagy, we explored candidate pathways that are involved in the ROS‐mediated genotoxic stress under autophagic conditions. We found a potential correlation between CHK2 activation and autophagy biomarkers under starvation conditions with or without addition of N‐acetyl cysteine (NAC), a scavenger of free radicals. Starvation caused the expected increase in autophagy, as demonstrated by the decrease in the autophagy substrate p62 and increase in the conversion of the nonlipidated form (LC3‐I) to the phosphatidyl ethanolamine‐conjugated form (LC3‐II) of LC3. This nutrient stress was also accompanied by increased phosphorylation of CHK2 at Thr68, indicating the activation of CHK2. Interestingly, the ROS scavenger NAC could largely inhibit the activation of CHK2, as well as the upregulation of autophagy (Figs 1A and EV1A). These results demonstrate that starvation‐induced ROS production is upstream of both endogenous CHK2 activation and autophagy induction.
Figure 1. CHK2 is involved in oxidative stress‐induced autophagy.
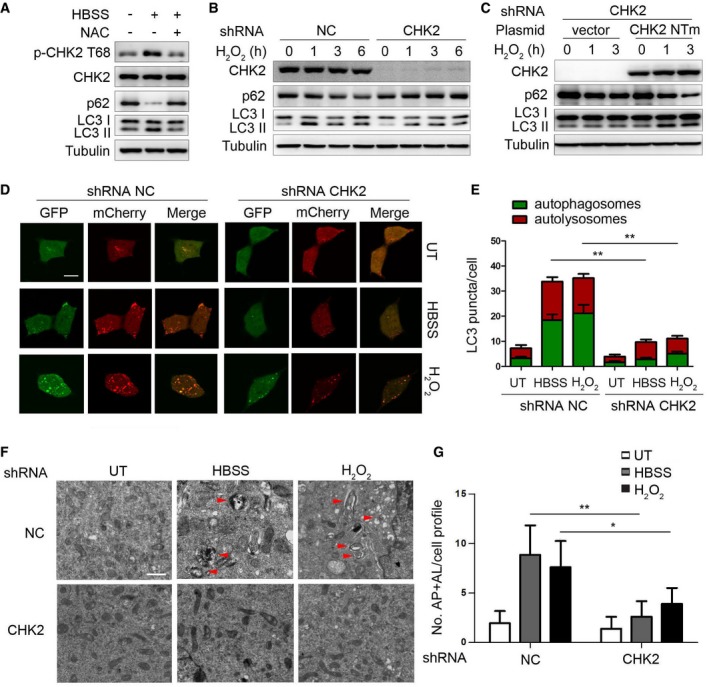
- Western blot detection of p‐CHK2 Thr68, CHK2, p62, and LC3 in H1299 cells in normal medium (HBSS “−”) or after 1 h HBSS starvation (HBSS “+”) pretreated with or without NAC (3 mM).
- Western blot detection of p62 and LC3 in H1299 cells transfected with the indicated shRNA in normal medium or after H2O2 (500 μM) treatment.
- Western blot detection of p62 and LC3 in H1299 cells cotransfected with the indicated shRNA and the indicated plasmid and cultured in normal medium or after H2O2 (500 μM) treatment. CHK2 NTm, a shRNA nontargetable mutant CHK2 rescue plasmid.
- Autophagic flux is shown by representative confocal microscopic images of 293 cells stably expressing GFP‐mCherry‐LC3 transfected with the indicated shRNA following HBSS starvation and H2O2 (500 μM) treatment for 3 h. Scale bar, 10 μm.
- Quantitation of autophagosomal (yellow) and autolysosomal (red) LC3 puncta following HBSS starvation and H2O2 (500 μM) treatment for 1 h (n = 30). Data are presented as mean ± s.e.m. from three independent experiments; **P < 0.01 (Student's t‐test).
- Representative electron microscopic image of autophagic vesicles or autophagosomes in H1299 cells transfected with the indicated shRNA in normal medium or after 3 h HBSS starvation or treated with H2O2 (500 μM). Scale bars, 500 nm. The red arrows indicate double‐membraned autophagic structures.
- Electron microscopic quantification of autophagy vacuole in H1299 cells transfected with the indicated shRNA in normal medium or after 3 h HBSS starvation or treated with H2O2 (500 μM). Data are presented as mean ± s.e.m. from three independent experiments; *P < 0.05; **P < 0.01 (Student's t‐test).
Source data are available online for this figure.
Figure EV1. CHK2 is involved in oxidative stress‐induced autophagy.
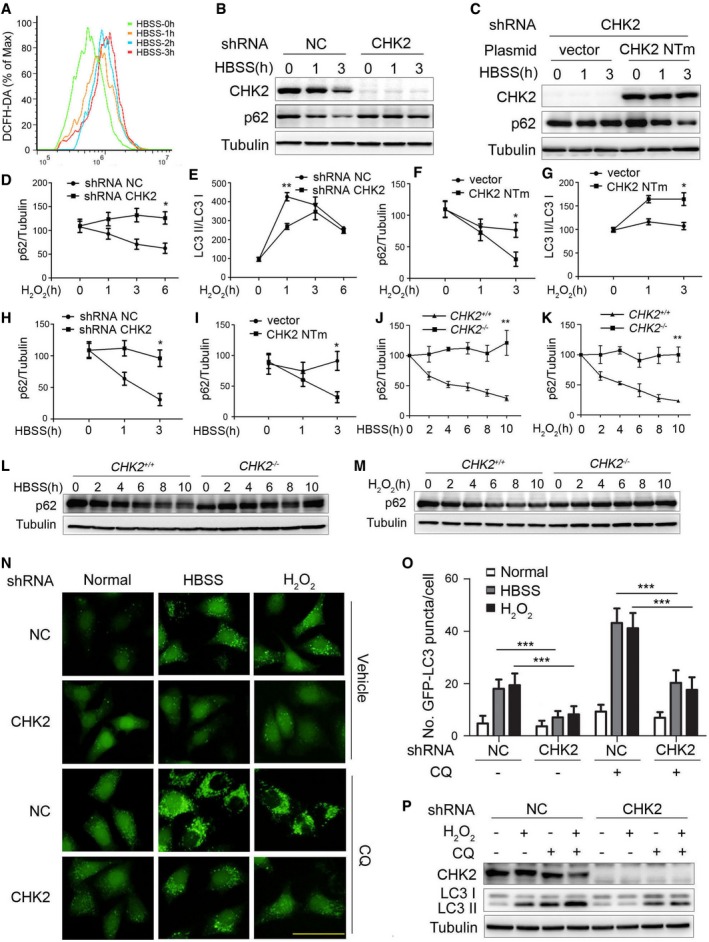
-
AIntracellular ROS levels detected in H1299 cells treated with HBSS starvation.
-
BWestern blot detection of p62 in H1299 cells transfected with the indicated shRNA in normal medium or after HBSS starvation.
-
CWestern blot detection of p62 in H1299 cells cotransfected with the indicated shRNA and plasmid and cultured in normal or HBSS starvation medium. CHK2 NTm, shRNA nontargetable mutant CHK2 rescue plasmid.
-
D, EQuantification of p62 and LC3‐II/I protein levels in H1299 cells transfected with the indicated shRNA in normal medium or after H2O2 (500 μM) treatment. Results are presented as mean ± s.e.m. from three independent experiments, *P < 0.05, **P < 0.01 (Student's t‐test).
-
F, GQuantification of p62 and LC3‐II/I protein levels in H1299 cells cotransfected with the indicated shRNA and plasmid and cultured in normal medium or after H2O2 (500 μM) treatment. Results are presented as mean ± s.e.m. from three independent experiments, *P < 0.05 (Student's t‐test).
-
HQuantification of p62 protein levels in H1299 cells transfected with the indicated shRNA in normal medium or after HBSS starvation. Results are presented as mean ± s.e.m. from three independent experiments, *P < 0.05 (Student's t‐test).
-
IQuantification of p62 protein levels in H1299 cells cotransfected with the indicated shRNA and plasmid and cultured in normal medium or after HBSS starvation. Results are presented as mean ± s.e.m. from three independent experiments, *P < 0.05 (Student's t‐test).
-
JQuantification of p62 protein levels in MEFs of indicated genotype during growth in normal medium or after HBSS starvation. Results are presented as mean ± s.e.m. from three independent experiments, **P < 0.01 (Student's t‐test).
-
KQuantification of p62 protein levels in MEFs of the indicated genotype during growth in normal medium or after H2O2 (500 μM) treatment. Results are presented as mean ± s.e.m. from three independent experiments, **P < 0.01 (Student's t‐test).
-
LWestern blot detection of p62 in MEFs of the indicated genotype during growth in normal medium or after HBSS starvation.
-
MWestern blot detection of p62 in MEFs of the indicated genotype during growth in normal medium or after H2O2 (500 μM) treatment.
-
NRepresentative images of GFP‐LC3 puncta in HeLa/GFP‐LC3 cells transfected with the indicated shRNA, pretreated with 60 nM CQ or vehicle (DMSO), and cultured for 1 h in normal, HBSS starvation medium or treated with H2O2 (500 μM). Scale bar, 50 μm.
-
OQuantification of GFP‐LC3 puncta in HeLa/GFP‐LC3 cells transfected with the indicated shRNA, pretreated with 60 nM CQ or vehicle (DMSO), and cultured for 1 h in normal, HBSS starvation medium or treated with H2O2 (500 μM; n = 30). Results are presented as mean ± s.e.m. from three independent experiments, ***P < 0.001 (Student's t‐test).
-
PWestern blot detection of LC3 in H1299 cells transfected with the indicated shRNA, pretreated with 60 nM CQ or vehicle (DMSO), and cultured for 3 h in normal or treated with H2O2 (500 μM).
Source data are available online for this figure.
To further probe the requirement for CHK2 in autophagy, we evaluated the consequence of shRNA‐mediated knockdown of CHK2 in H1299 cells. Silencing of CHK2 in cells under metabolic and oxidative stresses resulted in increased p62 levels and reduced conversion of the LC3‐I to LC3‐II (Figs 1B and EV1B). The potent inhibitory effect of CHK2 silencing on stress‐induced autophagy was not observed when a nontargetable CHK2 construct was expressed in cells (Figs 1C and EV1C–I). Consistent with these observations, stress‐induced autophagy was more pronounced in MEFs derived from CHK2 +/+ mice than from CHK2 −/− mice (Fig EV1J–M).
Next, we monitored autophagic flux using a tandem‐labeled GFP‐mCherry‐LC3B constructed in HEK293 cells. GFP fluorescence is quenched in autolysosomes by the low‐pH environment, whereas mCherry fluorescence is detectable in both autophagosomes and autolysosomes. The fusion of autophagosomes with autolysosomes resulted in the loss of yellow puncta and the appearance of red‐only puncta due to GFP quenching (Kimura et al, 2007). Autophagic flux in response to metabolic and oxidative stresses was significantly decreased following CHK2 knockdown (Fig 1D and E). Similarly, HeLa cells expressing the GFP‐LC3 reporter in the presence or absence of metabolic or oxidative stress showed a significant decrease in autophagosome formation as determined by persistence of GFP puncta. This analysis revealed autophagosome numbers per cell were significantly decreased following CHK2 knockdown. The decrease in autophagosomes during metabolic and oxidative stresses was not due to an acceleration of autophagolysosomal maturation, as the number of autophagosomes and conversion of LC3‐I to LC3‐II were also decreased in CHK2 shRNA‐treated cells in the presence of the lysosomal inhibitor chloroquine (CQ; Fig EV1N–P).
Finally, quantitative electron microscopic analysis showed that CHK2 knockdown resulted in fewer stress‐induced autophagic structures (autophagosomes and autolysosomes) than the nonspecific RNAi‐treated cells (Fig 1F and G). Taken together, these data indicate that endogenous CHK2 plays an important role in the induction of stress‐induced autophagy.
Oxidative stress enhances CHK2‐Beclin 1 interaction
To elucidate how CHK2 regulates autophagy, we investigated whether CHK2 directly interacts with a set of autophagy‐associated proteins including Ulk1, Atg5, Beclin 1, Atg7, or LC3. Immunoprecipitation analysis revealed that endogenous Beclin 1 could interact with CHK2. By contrast, none of the other autophagy‐associated proteins interacted with CHK2, with the possible exception Ulk1 that showed a weak interaction (Fig 2A). To further identify the regions of CHK2 responsible for the Beclin 1 interaction, we constructed full‐length (FL), as well as various truncated versions of CHK2. The forkhead‐associated (FHA) domain (residues 115–175) of CHK2 was sufficient to bind Beclin 1 (Fig EV2A). In a parallel study, we mapped the Beclin 1 regions required for CHK2 binding. We found that both of the coiled‐coil domain (CCD; residues 141–270) and the evolutionarily conserved domain (ECD; residues 271–450) of Beclin 1 were required for the interaction with CHK2 (Fig EV2B).
Figure 2. Oxidative stress enhances CHK2‐Beclin 1 interaction.
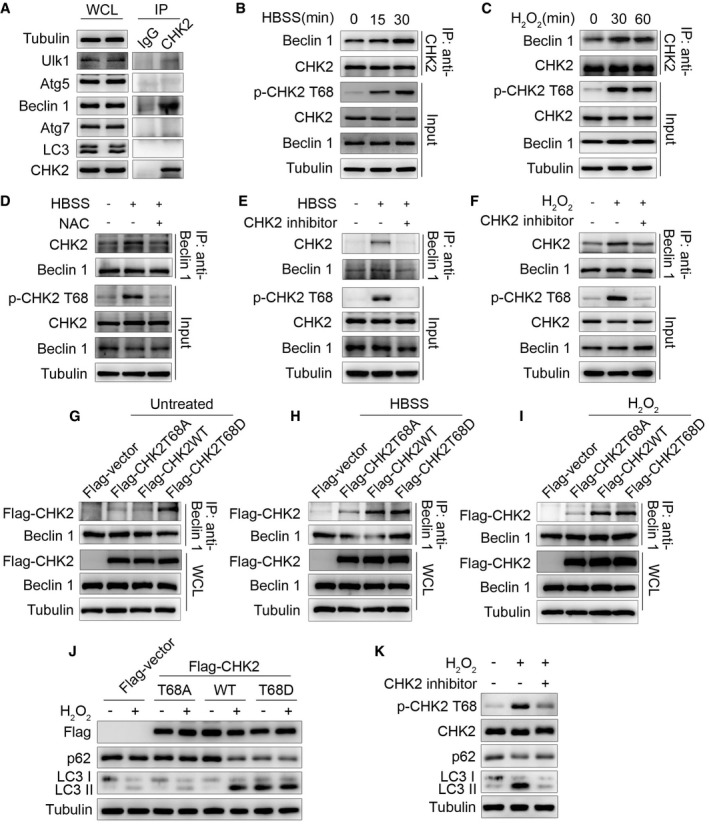
-
AImmunoprecipitation assays testing the physical interaction between CHK2 and proteins encoded by autophagy‐related genes in HCT116 cells. Lysates were extracted for immunoprecipitation with CHK2‐specific antibody or control IgG, followed by probing with antibodies specific for Ulk1, Atg5, Beclin 1, Atg7, or LC3.
-
BInteraction between endogenous CHK2 and Beclin 1 in HCT116 cells under fed conditions (t = 0) and after HBSS starvation in the cytoplasmic fractions.
-
CInteraction between endogenous CHK2 and Beclin 1 in HCT116 cells under fed conditions (t = 0) and after H2O2 (500 μM) stimulation in the cytoplasmic fractions.
-
DHCT116 cells were pretreated with NAC in RPMI 1640 complete medium for 4 h and then cultured for 1 h in HBSS starvation. The cytoplasmic lysates were subjected to immunoprecipitation with anti‐Beclin 1 antibody followed by immunoblotting with anti‐CHK2 and anti‐Beclin 1 antibodies. The expression of p‐CHK2 Thr68, CHK2, and Beclin 1 was monitored by immunoblotting in the cytoplasmic fractions. Tubulin was used as a loading control.
-
E, FHCT116 cells were pretreated with CHK2 inhibitor II in RPMI 1640 complete medium for 4 h and then cultured for 1 h in HBSS starvation (E) or H2O2 (500 μM) stimulation (F). The cytoplasmic lysates were subjected to immunoprecipitation with anti‐Beclin 1 antibody followed by immunoblotting with anti‐CHK2 and anti‐Beclin 1 antibodies. The expression of p‐CHK2 Thr68, CHK2, and Beclin 1 was monitored by immunoblotting in the cytoplasmic fractions. Tubulin was used as a loading control.
-
G–IHCT116 cells were transiently transfected with the expression plasmids as indicated. After 36 h post‐transfection, cells were treated or untreated (G) with HBSS starvation (H) or H2O2 (500 μM) stimulation (I) for 1 h and then collected for immunoprecipitation and Western blotting analysis. Immunoprecipitation was performed using anti‐Beclin 1 antibody followed by immunoblotting with anti‐Flag or anti‐Beclin 1 antibody.
-
JWestern blot detection of p62 and LC3 in H1299 cells transfected with indicated plasmid in normal medium or after H2O2 (500 μM) cultured for 3 h.
-
KH1299 cells were pretreated with CHK2 inhibitor II in complete medium for 4 h and then cultured for 3 h in H2O2 (500 μM) stimulation. Western blot detection of p‐CHK2 Thr68, CHK2, p62, and LC3.
Source data are available online for this figure.
Figure EV2. The interaction domain between CHK2 and Beclin 1 and characterization of Beclin 1 Ser90 phosphorylation antibody.
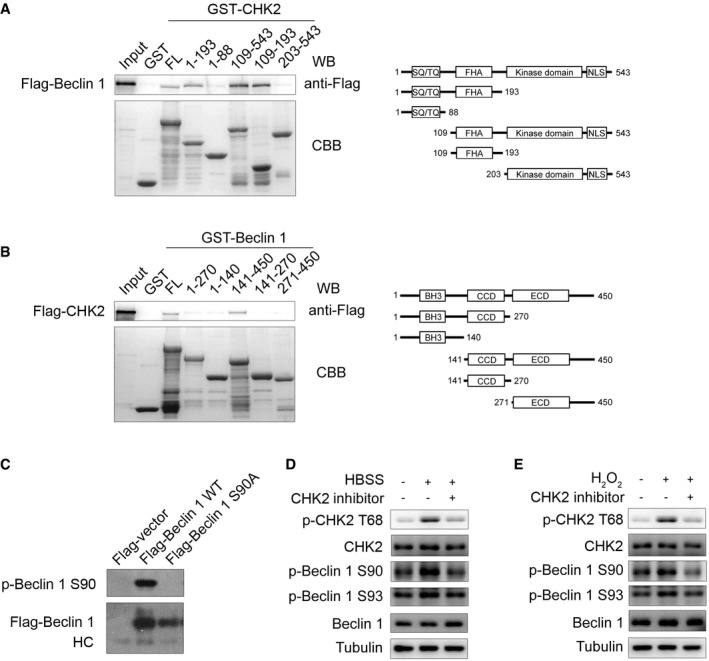
-
AGST‐CHK2 full‐length (FL) and fragments fusion proteins were expressed in bacteria and purified; these proteins were then incubated with in vitro‐translated Flag‐Beclin 1 protein. Western blotting was performed to detect the interaction of Beclin 1 with CHK2.
-
BGST‐Beclin 1 FL and fragments fusion proteins were expressed in bacteria and purified; these proteins were then incubated with in vitro‐translated Flag‐CHK2 protein. Western blotting was performed to detect the interaction of CHK2 with Beclin 1.
-
CCell lysates from 293 cells transfected with WT or mutant forms of Flag‐Beclin 1 were immunoprecipitated with Flag‐M2 beads followed by Western blot using antibody against Beclin 1 Ser90 or Flag.
-
D, EH1299 cells were pretreated with CHK2 inhibitor II in complete medium for 4 h and then cultured for 1 h in HBSS starvation (D) or H2O2 (500 μM) stimulation (E). Western blot detection of p‐CHK2 Thr68, CHK2, p‐Beclin 1 Ser90, p‐Beclin 1 Ser93, and Beclin 1.
Source data are available online for this figure.
We next tested whether ROS affects the interaction between CHK2 and Beclin 1. Indeed, we observed enhanced interaction in cells exposed to metabolic (Fig 2B) and oxidative stresses (Fig 2C). The interaction was markedly reduced by NAC treatment (Fig 2D), suggesting a role for ROS in promoting the interaction between these two proteins. CHK2 phosphorylation at Thr68 was also observed under conditions that promote the CHK2‐Beclin 1 interaction, indicating CHK2 activation might facilitate the interaction with Beclin 1. Consistent with this hypothesis, a pharmacological CHK2 inhibitor dramatically diminished Thr68 phosphorylation, as well as the interaction between CHK2 and Beclin 1 under both metabolic (Fig 2E) and oxidative stresses (Fig 2F). Furthermore, although wild‐type (WT) CHK2 coimmunoprecipitated with Beclin 1 in the setting of metabolic and oxidative stresses, a mutant CHK2 (T68A) did not. Mimicking Thr68 phosphorylation with a negatively charged amino acid Asp (T68D) promoted CHK2 interaction with Beclin 1 even under basal, nonstressed conditions (Fig 2G–I). Furthermore, we found that CHK2‐WT increased H2O2‐induced autophagy, whereas the CHK2 T68A mutant did not. Notably, the CHK2 T68D mutant was sufficient to markedly increase basal autophagy, even in cells lacking H2O2 treatment (Fig 2J). Consistently, CHK2 T68 site‐specific inhibitors blocked phosphorylation of CHK2 at T68 and autophagy induced by H2O2 (Fig 2K). Together, these results demonstrate that Thr68 phosphorylation facilitates the interaction between Beclin 1 and CHK2 in response to metabolic and oxidative stresses.
CHK2 phosphorylates Beclin 1 at Ser90 and Ser93
To determine whether CHK2 can phosphorylate Beclin 1, we performed in vitro kinase assays using various lengths of Beclin 1 as a substrate. We found that CHK2 could phosphorylate FL Beclin 1 (GST‐Beclin 1 WT) and a truncated Beclin 1 protein spanning amino acids 88–185, but not other domains of Beclin 1 (Fig 3A and B). Ser90 of Beclin 1 conforms to the consensus CHK2 motif that includes both the RXXS and RXXT sequences (Seo et al, 2003). Ser90 is considered to be the initial event that catalyzes subsequent phosphorylation at Ser93 (Fogel et al, 2013). We found that site‐directed mutation of Ser90 or Ser93 to alanine reduced the phosphorylation of Beclin 1 by CHK2, whereas the combined Ser90 and Ser93 mutants (AA) abrogated nearly all of the Beclin 1 phosphorylation signal (Fig 3C).
Figure 3. CHK2 phosphorylates Beclin 1 at Ser90 and Ser93.
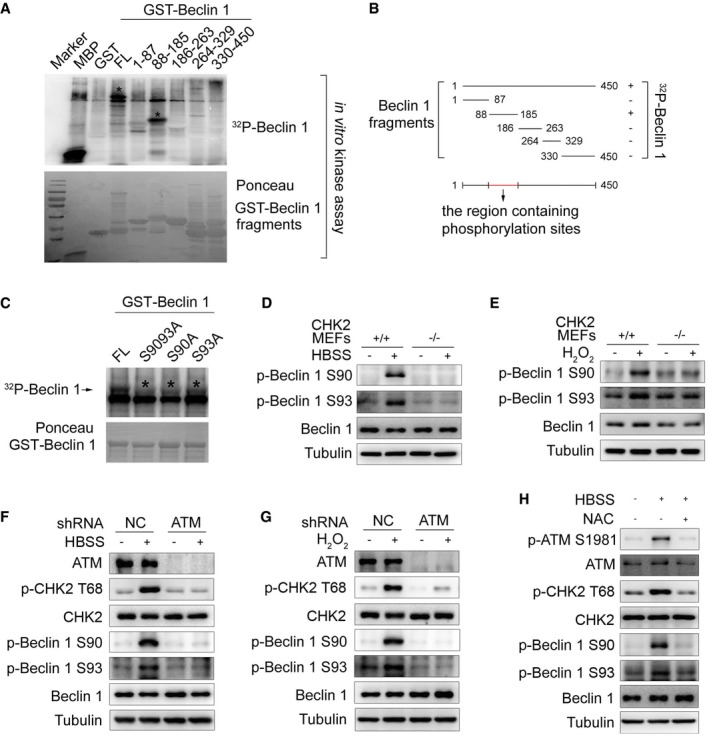
-
AThe indicated recombinant fragments of GST‐Beclin 1 were incubated with recombinant CHK2 in the presence of 32P‐labeled ATP for in vitro kinase assays. MBP was used as a positive control. Ponceau staining indicates the expression of GST‐Beclin 1 fragments for in vitro kinase assays. The asterisks in the blot indicate the domain of phosphorylated Beclin 1.
-
BSchematic representation of recombinant fragments of Beclin 1.
-
CIn vitro kinase assays to test the ability of recombinant CHK2 to phosphorylate recombinant GST‐Beclin 1 WT, S9093A (AA), S90A, and S93A protein. Reactions were analyzed by SDS–PAGE followed by autoradiography. The asterisks in the blot indicate the phosphorylation of Beclin 1 mutants.
-
D, EWestern blot detection of p‐Beclin 1 Ser90, p‐Beclin 1 Ser93, and Beclin 1 in MEFs indicated genotype in normal medium or after 1 h HBSS starvation (D) or H2O2 (500 μM) stimulation (E).
-
F, GWestern blot detection of ATM, p‐CHK2 Thr68, CHK2, p‐Beclin 1 Ser90, p‐Beclin 1 Ser93, and Beclin 1 in H1299 cells transfected with indicated shRNA in normal medium or after HBSS starvation (F) or H2O2 (500 μM) stimulation (G).
-
HWestern blot detection of p‐ATM Ser1981, ATM, p‐CHK2 Thr68, CHK2, p‐Beclin 1 Ser90, p‐Beclin 1 Ser93, and Beclin 1 in H1299 cells pretreated with NAC for 4 h and then cultured for 1 h in HBSS starvation.
Source data are available online for this figure.
To further establish phosphorylation of Beclin 1 in vivo, we generated Ser90 and Ser93 phospho‐specific antibodies for Beclin 1 (Fig EV2C). Using these phospho‐specific antibodies, we observed that phosphorylation of endogenous Beclin 1 Ser90/Ser93 was enhanced under metabolic (Fig 3D, Appendix Fig S1A and B) and oxidative stress (Fig 3E, Appendix Fig S1C and D) conditions. This increase in phosphorylation was not observed in CHK2 KO MEFs, demonstrating that CHK2 is required for Beclin 1 phosphorylation. Similarly, pharmacological CHK2 inhibition also blocked Beclin 1 phosphorylation in cells under metabolic (Fig EV2D, Appendix Fig S1E and F) or oxidative stress (Fig EV2E, Appendix Fig S1G and H). Moreover, CHK2 phosphorylation and Beclin 1 phosphorylation in response to both metabolic (Fig 3F, Appendix Fig S1I and J) and oxidative stresses (Fig 3G, Appendix Fig S1K and L) were reduced in ATM shRNA‐treated cells, demonstrating that ATM is upstream of CHK2 and Beclin 1 activation. Whereas the ATM/CHK2/Beclin 1 pathway was activated in cells under metabolic stress, treatment with the antioxidant NAC blocked this activation (Fig 3H, Appendix Fig S1M and N). These data establish an important redox‐dependent role for CHK2 in the phosphorylation of Beclin 1 Ser90/Ser93 in response to metabolic or oxidative stress.
CHK2‐mediated Beclin 1 phosphorylation regulates autophagy
Previous studies have found that Bcl‐2 binds to the BH3 domain of Beclin 1 to inhibit autophagy. Disruption of the Bcl‐2–Beclin 1 complex is crucial for stimulus‐induced autophagy in mammalian cells (He & Levine, 2010). We therefore tested whether the interaction between Beclin 1 and Bcl‐2 can be disrupted by CHK2. Indeed, in vitro binding assays using recombinant proteins showed a reduced interaction between GST‐Beclin 1 WT and Bcl‐2 in the presence of CHK2. GST‐Beclin 1 containing the two nonphosphorylatable mutations S90/93A (AA) interacted with Bcl‐2, and treatment of GST‐Beclin 1 AA with CHK2 had no effect on its ability to interact with Bcl‐2. Importantly, the GST‐Beclin 1 S90/93D (DD) phosphomimetic mutant showed minimal interaction with Bcl‐2 and was unaffected by CHK2 (Fig 4A), supporting a role for CHK2‐mediated phosphorylation in regulating the interaction.
Figure 4. CHK2‐mediated Beclin 1 phosphorylation regulates autophagy.
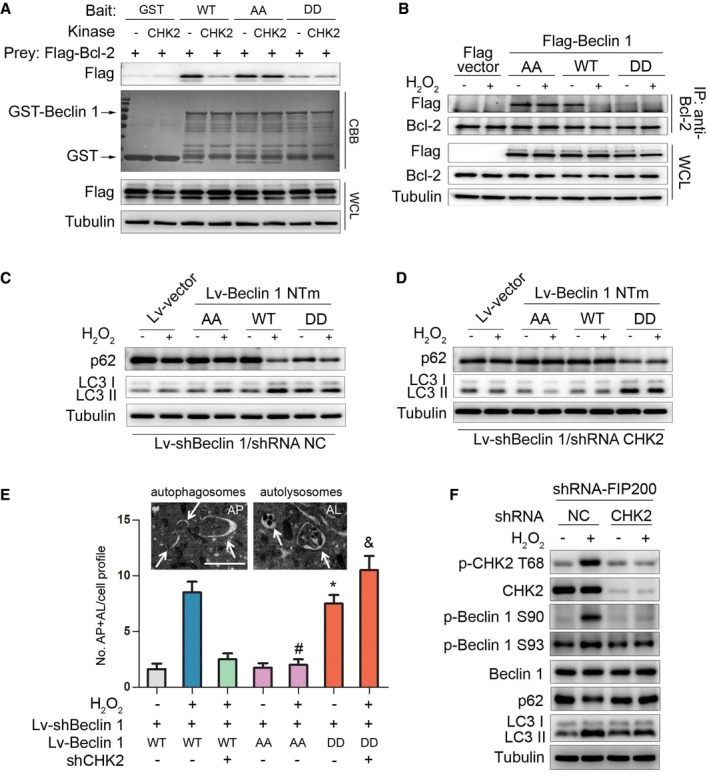
-
AThe effect of Beclin 1 Ser90/93 phosphorylation on the interaction between Beclin 1 and Bcl‐2. GST‐Beclin 1 WT, AA mutant, and DD mutant (bait) were incubated with or without recombinant CHK2 in the presence of ATP and then incubated with Flag‐Bcl‐2 (preys).
-
BImmunoprecipitation of Bcl‐2 with Flag‐Beclin 1 WT, AA mutant, and DD mutant in HCT116 cells in normal medium or after H2O2 (500 μM) treatment for 1 h.
-
C, DAutophagy levels were determined in Beclin 1‐depleted H1299 cells with reconstituted expression of Beclin 1 WT, AA mutant, or DD mutant in normal medium or after H2O2 (500 μM) treatment for 3 h in the presence (C) or absence (D) of CHK2.
-
ERepresentative electron microscopic image of an autophagosome (arrow, left panel, AP) and an autolysosome (arrow, right panel, AL) in H1299 Beclin 1 WT cells treated with H2O2 (500 μM) for 6 h. Scale bars, 500 nm. Electron microscopic quantification of autophagosomes (AP) and autolysosomes (AL) in H1299 cells with the indicated plasmids cultured for 6 h in normal or H2O2 (500 μM) treatment. All quantitative data are presented as mean ± s.e.m. from three independent experiments; *P < 0.001 compared to Beclin 1 WT; # P < 0.001 compared to Beclin 1 WT treated with H2O2; & P < 0.001 compared to Beclin 1 WT treated with H2O2 in the absence of CHK2.
-
FWestern blot detection of p‐CHK2 Thr68, CHK2, p‐Beclin 1 Ser90, p‐Beclin 1 Ser93, Beclin 1, p62, and LC3 in H1299 cells transfected with the indicated shRNA in normal medium or after H2O2 (500 μM) treatment for 3 h.
Source data are available online for this figure.
We next examined the interaction between Beclin 1 and Bcl‐2 under stress conditions. Notably, the interaction between Beclin 1 WT and Bcl‐2 decreased in the setting of metabolic and oxidative stresses. In contrast, the Beclin 1 AA mutant remained similarly bound to Bcl‐2 under basal and stress conditions, whereas the Beclin 1 DD mutant had limited interaction with Bcl‐2 during basal and stress conditions (Figs 4B and EV3A).
Figure EV3. CHK2‐mediated Beclin 1 phosphorylation promotes autophagy.
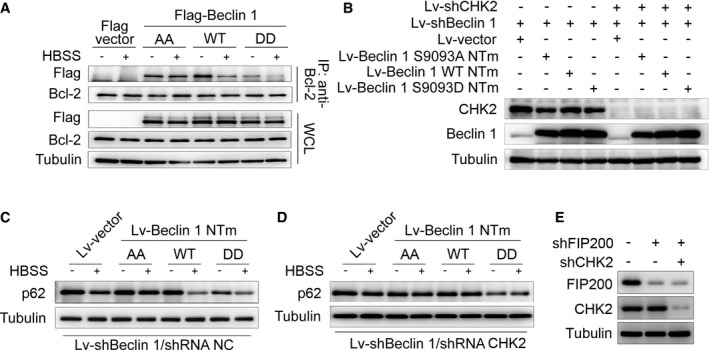
-
AImmunoprecipitation of Bcl‐2 with Flag‐Beclin 1 WT, AA mutant, and DD mutant in HCT116 cells in normal medium or after HBSS starvation.
-
BBeclin 1‐depleted H1299 cells with reconstituted expression of the indicated proteins.
-
C, DAutophagy level was determined in Beclin 1‐depleted H1299 cells with reconstituted expression of Beclin 1 WT, AA mutant, or DD mutant in normal medium or after HBSS starvation in the presence (C) or absence (D) of CHK2.
-
EFIP200‐ and CHK2‐depleted H1299 cells with reconstituted expression of the indicated proteins.
Source data are available online for this figure.
To determine whether CHK2 positively regulates autophagy through a mechanism that involves Beclin 1 Ser90/Ser93 phosphorylation, we generated Beclin 1‐depleted H1299 cells that expressed Beclin 1 WT, AA, or DD mutant in the presence or absence of CHK2 (Fig EV3B). Expression of Beclin 1 WT resulted in increased starvation and oxidative stress‐induced autophagy, but had no effect in CHK2 knockdown cells. The Beclin 1 AA mutant did not enhance starvation and oxidative stress‐induced autophagy, whereas the Beclin 1 DD mutant was sufficient to markedly increase basal autophagy, even in cells lacking CHK2 (Figs 4C and D, and EV3C and D, Appendix Fig S2A–F). This is consistent with phosphorylation of Beclin 1 playing an important role in autophagy induction.
Quantitative electron microscopic analysis led to a similar conclusion. The number of autophagic structures (autophagosomes and autolysosomes) in H1299 cells increased in the presence of the Beclin 1 DD mutant compared to the Beclin 1 WT and Beclin 1 AA mutant under baseline conditions. During oxidative stress, the Beclin 1 AA mutant abolished the CHK2‐induced increase in the number of autophagic structures. Conversely, Beclin 1 DD mutant increased the number of autophagic structures under oxidative stress, even in the absence of CHK2. These results suggest that phosphorylation of the Ser90/Ser93 residues of Beclin 1 by CHK2 positively regulates autophagy (Fig 4E).
In addition, we conducted FIP200‐depleted H1299 cells with or without CHK2. We found that CHK2 phosphorylation and Beclin 1 phosphorylation as well as autophagy were increased in FIP200 shRNA‐treated cells in response to H2O2 stimulation, demonstrating that CHK2‐Beclin 1‐mediated autophagy occurs even in FIP200‐deficient cells. Beclin 1 phosphorylation and autophagy were reduced in FIP200 and CHK2 shRNA‐treated cells. These data establish an important role for the CHK2/Beclin 1 pathway in regulating autophagy in response to oxidative stress in a FIP200‐independent manner (Figs 4F and EV3E).
ROS acts as a signaling molecule to activate the CHK2‐Beclin 1 axis under glucose starvation and hypoxic stress
We next asked whether the ATM/CHK2/Beclin 1 axis acts as a regulator of autophagy in response to ROS perturbations. We found that under glucose deprivation and hypoxia stimulation, either CHK2 (Fig 5A and B, Appendix Fig S3A–F) or ATM (Fig 5C and D, Appendix Fig S3G–J) shRNA knockdown decreased the phosphorylation of Beclin 1 Ser90/Ser93, indicating that Beclin 1 Ser90/Ser93 phosphorylation is dependent on the ATM/CHK2 pathway. We also found that silencing CHK2 caused inhibition of autophagy induced by glucose withdrawal and hypoxia (Fig EV4A–C). It is interesting to note that glucose deprivation induced early AMPK Thr172 phosphorylation (3 h), whereas CHK2 Thr68 phosphorylation was elevated nearly 12 h after glucose withdrawal (Fig 5A). Similarly, hypoxia‐induced CHK2 Thr68 phosphorylation was enhanced 36 h after being placed in low oxygen conditions (Fig 5B). Taken together, these findings suggest that glucose deprivation and hypoxia might be not the proximal signals activating the ATM/CHK2/Beclin 1 axis.
Figure 5. ROS acts as a signaling molecule to activate the CHK2‐Beclin 1 axis under glucose starvation and hypoxic stress.
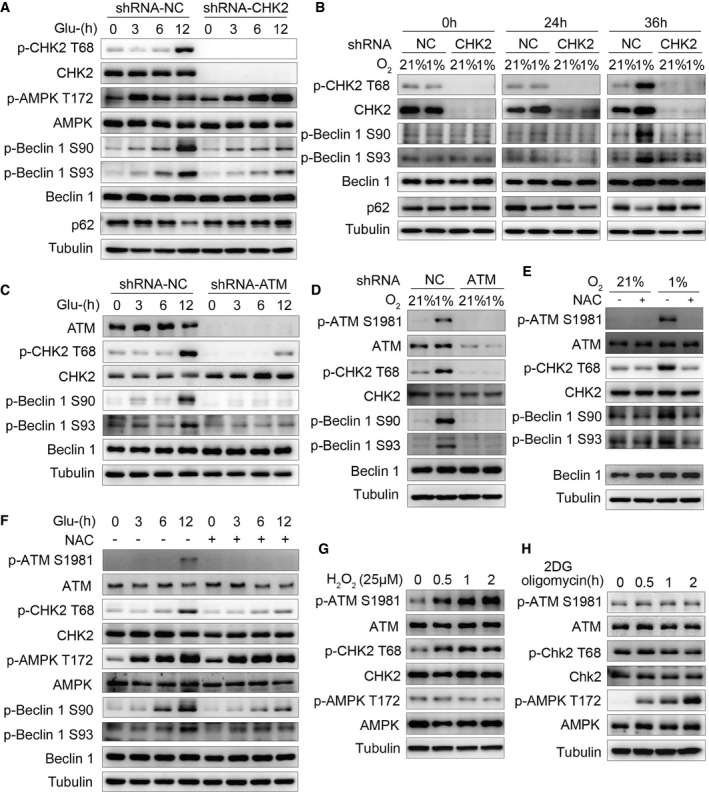
- Western blot detection of p‐CHK2 Thr68, CHK2, p‐AMPKα Thr172, AMPKα, p‐Beclin 1 Ser90, p‐Beclin 1 Ser93, and Beclin 1 in H1299 cells transfected with the indicated shRNA in normal medium or after glucose starvation.
- Western blot detection of p‐CHK2 Thr68, CHK2, p‐Beclin 1 Ser90, p‐Beclin 1 Ser93, and Beclin 1 in H1299 cells transfected with the indicated shRNA in normal medium or after hypoxia.
- Western blot detection of ATM, p‐CHK2 Thr68, CHK2, p‐Beclin 1 Ser90, p‐Beclin 1 Ser93, and Beclin 1 in H1299 cells transfected with the indicated shRNA in normal medium or after glucose starvation.
- Western blot detection of p‐ATM Ser1981, ATM, p‐CHK2 Thr68, CHK2, p‐Beclin 1 Ser90, p‐Beclin 1 Ser93, and Beclin 1 in H1299 cells transfected with the indicated shRNA in normal medium or after hypoxia.
- P‐ATM Ser1981, ATM, p‐CHK2 Thr68, CHK2, p‐Beclin 1 Ser90, p‐Beclin 1 Ser93, and Beclin 1 were analyzed in H1299 cells, and the cells were subjected to hypoxia with or without pretreatment with NAC.
- P‐ATM Ser1981, ATM, p‐CHK2 Thr68, CHK2, p‐AMPKα Thr172, AMPKα, p‐Beclin 1 Ser90, p‐Beclin 1 Ser93, and Beclin 1 were analyzed in H1299 cells, and the cells were subjected to glucose starvation with or without pretreatment with NAC.
- Western blot detection of p‐ATM Ser1981, ATM, p‐CHK2 Thr68, CHK2, p‐AMPKα Thr172, and AMPKα in H1299 cells in normal medium or after H2O2 (25 μM) stimulation.
- Western blot detection of p‐ATM Ser1981, ATM, p‐CHK2 Thr68, CHK2, p‐AMPKα Thr172, and AMPKα in H1299 cells in normal medium or after 2DG (5 mM) and oligomycin (2.5 μM) treatment.
Source data are available online for this figure.
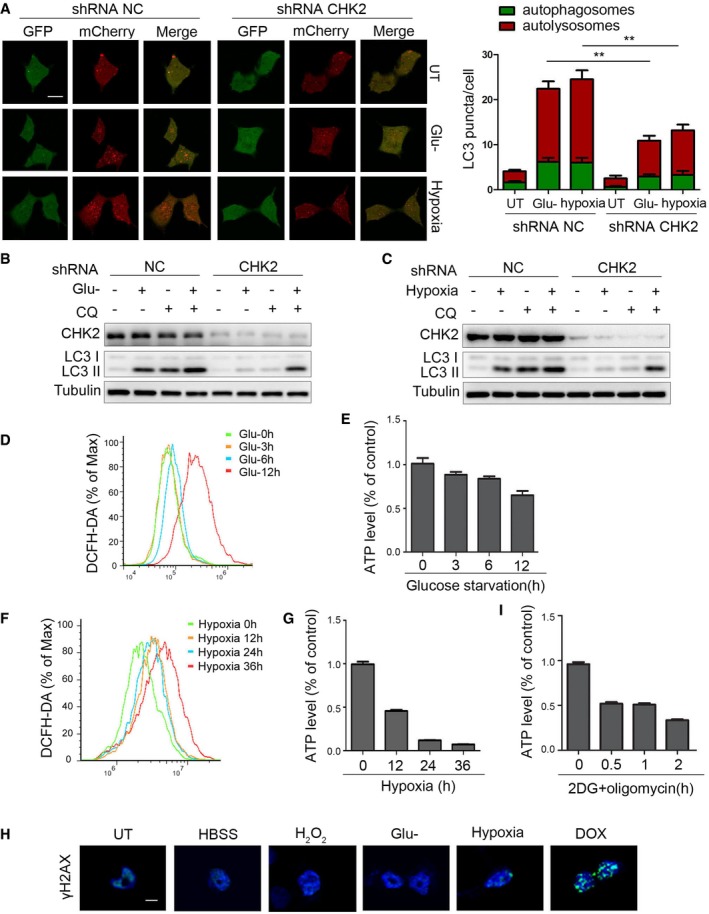
Figure EV4. ROS acts as a signaling molecule to activate the ATM/CHK2/Beclin 1 axis under glucose starvation and hypoxic stress.
-
AAutophagic flux is shown by representative confocal microscopic images for 293 cells stably expressing GFP‐mCherry‐LC3 transfected with the indicated shRNA. Scale bar, 10 μm. Quantitation of autophagosomal (green) and autolysosomal (red) LC3 puncta following 12‐h glucose starvation and 36‐h hypoxia (n = 30). Data are presented as mean ± s.e.m. from three independent experiments; **P < 0.01 (Student's t‐test).
-
BWestern blot detection of LC3 in H1299 cells transfected with the indicated shRNA, pretreated with 30 nM CQ or vehicle (DMSO), and cultured for 12 h in normal or glucose starvation.
-
CWestern blot detection of LC3 in H1299 cells transfected with the indicated shRNA, pretreated with 30 nM CQ or vehicle (DMSO), and cultured for 36 h in normal or hypoxia.
-
D, EIntracellular ROS (D) or ATP (E) levels detected in H1299 cells treated with glucose starvation. Data are presented as mean ± s.e.m. from three independent experiments.
-
F, GIntracellular ROS (F) or ATP (G) levels detected in H1299 cells treated with hypoxia. Data are presented as mean ± s.e.m. from three independent experiments.
-
HH1299 cells detected with γH2AX staining followed by HBSS starvation (1 h), H2O2 treatment (200 μM, 3 h), glucose starvation (10 h), hypoxia (36 h), and DOX (0.5 μM, 3 h). Scale bar, 10 μm.
-
IIntracellular ATP levels detected in H1299 cells treated with 2DG (5 mM) and oligomycin (2.5 μM) treatment. Data are presented as mean ± s.e.m. from three independent experiments.
Source data are available online for this figure.
During our search for the signal inducing ATM/CHK2/Beclin 1 axis activation, we noted that glucose starvation and hypoxia caused an increase in intracellular ROS levels and a decrease in ATP levels, but without DNA damage (Fig EV4D–H). Furthermore, treating cells with NAC decreased glucose deprivation‐ and hypoxia‐induced phosphorylation of ATM Ser1981, CHK2 Thr68, and Beclin 1 Ser90/Ser93 (Fig 5E and F, Appendix Fig S3K–N). In addition, we found that CHK2 was activated with low concentrations of hydrogen peroxide, but AMPKα1/2 was not (Fig 5G). On the other hand, treating cells with 2DG and oligomycin to induce energy depletion (Fig EV4I) activated AMPKα1/2 without affecting ATM/CHK2 (Fig 5H). Taken together, these findings suggest that ATM/CHK2/Beclin 1 axis is a sensitive and specific sensing mechanism that uses intracellular ROS to stimulate autophagic flux.
CHK2‐mediated autophagy limits ROS levels by clearing damaged mitochondria
Reactive oxygen species and oxidative stress can activate autophagy, which generally serves as a negative feedback cytoprotective mechanism to selectively eliminate sources of ROS such as damaged mitochondria. To further explore the role of CHK2 in sensing intracellular ROS changes and initiating autophagy, we measured the level of intracellular ROS, mitophagy, and mitochondrial health in CHK2 knockdown cells in comparison with cells treated with the control shRNA. Much higher ROS production was detected in CHK2‐silenced cells under conditions of glucose starvation and hypoxia (Fig 6A and B). To investigate the possible link between CHK2‐deficiency and these higher levels of ROS, we evaluated the role of CHK2 in mitophagy. Immunoblot analysis showed that the mitochondrial proteins Tom20 (outer membrane), TIM23 (inner membrane), and HSP60 (mitochondrial matrix) were much less abundant in the cells cultured under glucose starvation and hypoxic conditions than in the cells cultured in complete growth medium under normoxic conditions, demonstrating the likely induction of mitophagy in the stressed cells. Significantly, under the same conditions, the decrease in mitochondrial proteins was inhibited in CHK2 knockdown cells (shRNA‐CHK2; Fig 6C).
Figure 6. CHK2‐mediated autophagy limits ROS levels by clearing damaged mitochondria.
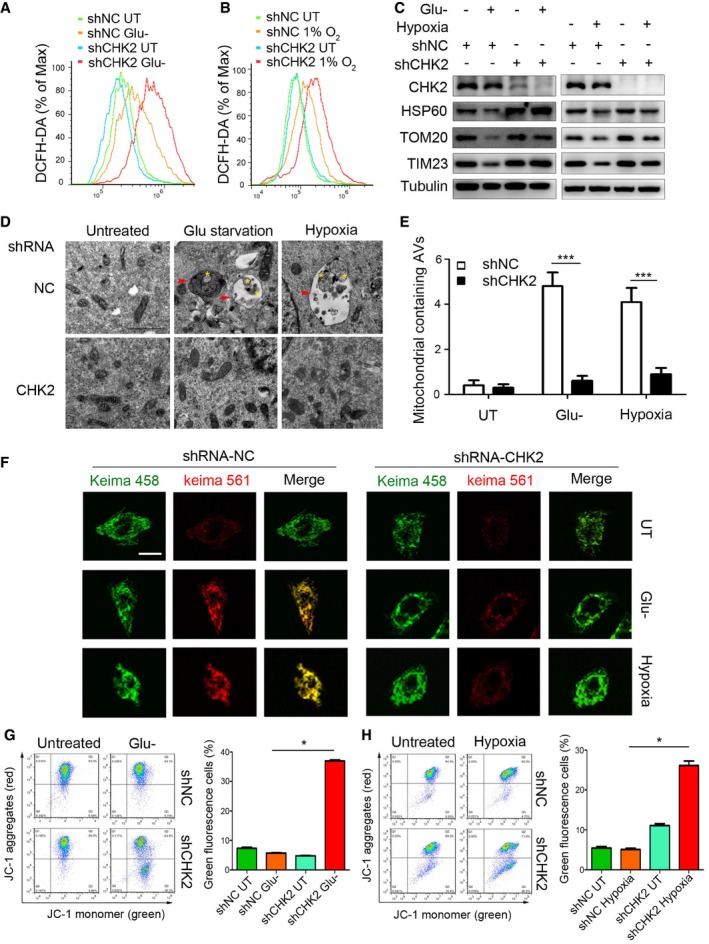
-
A, BIntracellular ROS levels detected in H1299 cells transfected with the indicated shRNA in normal medium or after 12‐h glucose starvation (Glu‐) (A) or 36‐h hypoxia (B).
-
CWestern blot detection of CHK2, HSP60, TOM20, and TIM23 in H1299 cells transfected with the indicated shRNA in normal medium or after 12‐h glucose starvation (Glu‐) or 36‐h hypoxia.
-
D, ETransmission electron microscopy of indicated H1299 cells cultured in glucose starvation (Glu‐) medium or hypoxia. The yellow asterisks mark mitochondria. The red arrows indicate autophagic structures. All quantitative data are presented as mean ± s.e.m. from three independent experiments; ***P < 0.001 compared to shNC treated with H2O2 or hypoxia (Mann–Whitney test); Scale bar, 1 μm.
-
FRepresentative confocal images of the shift in the fluorescence emission of Keima from 458 to 561 nm. Scale bar, 10 μm.
-
G, HThe mitochondrial membrane potential analysis of 293 cells transfected with the indicated shRNA in normal medium or after 18‐h glucose starvation (G) or 48‐h hypoxia (H). All results are from three independent experiments. All quantitative data are presented as mean ± s.e.m. from three independent experiments; *P < 0.05 (Mann–Whitney test).
Source data are available online for this figure.
Our transmission electron microscopy analysis revealed that in the setting of glucose withdrawal and hypoxic conditions, mitophagy occurred in CHK2‐WT cells but not in CHK2 knockdown cells, demonstrating the important role of CHK2 in promoting mitophagy and the removal of damaged mitochondria (Fig 6D and E). Moreover, we used Keima, a pH‐sensitive fluorescent protein targeted to the mitochondria, to measure mitophagic flux in cells (Katayama et al, 2011). Compared with the control, silencing of CHK2 inhibited the shifting of Keima from 458 nm to 561 nm, indicating a decrease in mitochondria entering the acidic environment of the lysosome in response to glucose starvation and hypoxia (Fig 6F). We also found that the fraction of puncta with green fluorescent protein (GFP)‐tagged LC3 that localized with mitochondria (TOM20) was significantly lower in the cells expressing CHK2 T68A than in the cells expressing CHK2‐WT or CHK2 T68D in response to glucose starvation and hypoxia (Fig EV5A). Moreover, the fraction of puncta with GFP‐tagged LC3 that localized with mitochondria (TOM20) was significantly lower in the cells expressing Beclin 1 S9093A than in the cells expressing Beclin 1 WT or Beclin 1 S9093D in response to glucose starvation and hypoxia (Fig EV5B).
Figure EV5. CHK2‐mediated autophagy limits ROS levels by clearing damaged mitochondria.
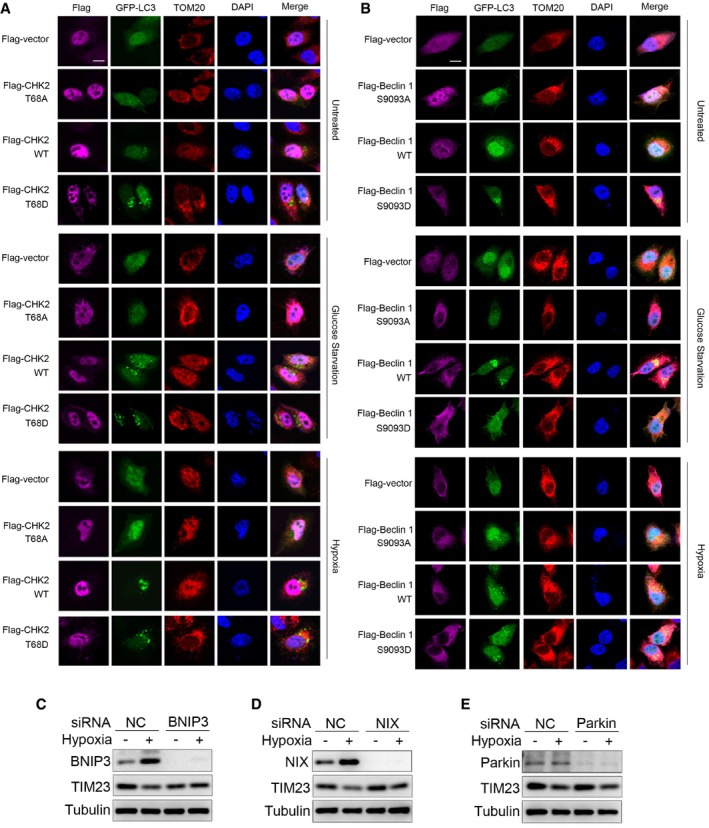
-
A, BFluorescence microscopy of the colocalization of GFP‐LC3 with mitochondria (identified with the mitochondrial stain TOM20) in H1299 cells transfected with the indicated plasmid and cultured in untreated (UT), glucose starvation for 12 h, or subjected to hypoxia for 36 h. DAPI, DNA‐binding dye. Scale bar, 10 μm.
-
CWestern blot detection of BNIP3 and TIM23 in H1299 cells transfected with the indicated siRNA and untreated or subjected to hypoxia for 36 h.
-
DWestern blot detection of NIX and TIM23 in H1299 cells transfected with the indicated siRNA and untreated or subjected to hypoxia for 36 h.
-
EWestern blot detection of parkin and TIM23 in H1299 cells transfected with the indicated siRNA and untreated or subjected to hypoxia for 36 h.
Source data are available online for this figure.
We also investigated how CHK2‐Beclin 1 mediated autophagy that targets damaged mitochondria. We found that in parallel with the initiation of autophagy through CHK2‐Beclin 1 under hypoxia, selective autophagy receptors NIX and BNIP3 were induced under hypoxia. Furthermore, we found that silencing NIX and BNIP3 inhibited mitophagy; however, silencing parkin had no effect on mitophagy in this setting (Fig EV5C–E). These data suggest that CHK2‐Beclin 1‐mediated autophagy might target damaged mitochondria through the induced NIX and BNIP3 under hypoxia.
Mitophagy can be induced by stresses to remove damaged mitochondria with lower membrane potential than that of healthy mitochondria. We used JC‐1 staining assays to determine mitochondrial health under stress in the presence or absence of CHK2. Compared with the control, CHK2 silencing caused an increase in the abundance of damaged mitochondria with low potential (JC‐1 monomer with green fluorescence) in response to glucose starvation and hypoxia (Fig 6G and H). Taken together, these data suggest that CHK2‐mediated autophagy is needed to maintain healthy mitochondria in response to glucose starvation and hypoxia.
CHK2‐mediated autophagy protects against cell death and tissue damage following cerebral ischemia
To further explore the role of CHK2 in maintaining ROS homeostasis, we examined cell apoptosis using both in vitro and in vivo experiments. For our in vitro studies, we performed cell apoptosis assays after prolonged glucose starvation and hypoxia. The results revealed that silencing of CHK2 exacerbated the apoptotic cell death of H1299 cells induced by glucose deprivation and hypoxia stimulation (Fig 7A and B). In addition, to test the involvement of CHK2‐mediated Beclin 1 phosphorylations in this phenomenon, we determined the effect of Beclin 1 phosphomimetic (DD) and nonphosphorylatable (AA) mutants on apoptosis under glucose deprivation and hypoxia. We found that the number of cells undergoing apoptosis decreased in the presence of the Beclin 1 WT and S9093D mutant compared to the vector control and the Beclin 1 AA mutant under glucose deprivation and hypoxia (Fig 7C and D).
Figure 7. CHK2‐mediated autophagy protects against cell death and tissue damage following cerebral ischemia.
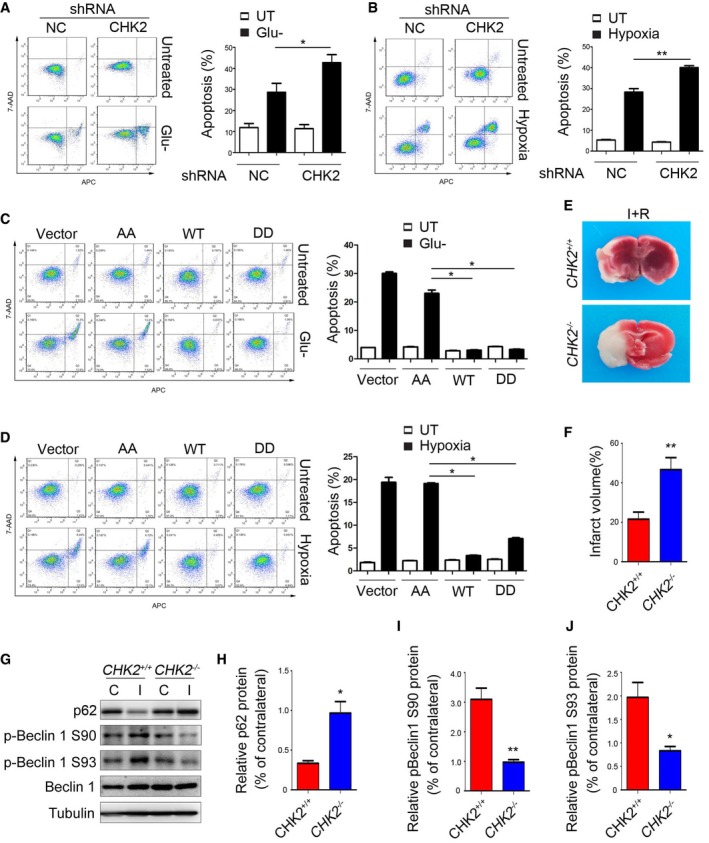
-
A, BH1299 cells transfected with the indicated shRNA in normal medium or after 12‐h glucose starvation (A) or 48‐h hypoxia stimulation (B). Representative FACS analysis of apoptosis. Data are presented as mean ± s.e.m. from three independent experiments; *P < 0.05 compared to NC glucose starvation (A); **P < 0.01 compared to NC hypoxia (B) (Student's t‐test).
-
C, DH1299 cells transfected with the indicated plasmids in normal medium or after 12‐h glucose starvation (C) or 48‐h hypoxia stimulation (D). Representative FACS analysis of apoptosis. Results from three independent experiments are presented as a histogram. Data are presented as mean ± s.e.m. from three independent experiments; *P < 0.05 compared to Beclin 1 S9093A (AA) treated with H2O2 or hypoxia (Mann–Whitney test).
-
ECHK2 +/+ and CHK2 −/− mice were subjected to MCAO for 1 h and reperfusion for 12 h. Contralateral (C) and ipsilateral (I) tissues of the mouse brain were coronally sectioned and stained with 2% TTC.
-
FThe infarct volume was determined by measuring infarct size relative to normal in the slice. (n = 5 mice). Data are presented as mean ± s.e.m.; **P < 0.01 (Student's t‐test).
-
G–JImmunoblots for p62, p‐Beclin 1 Ser90, p‐Beclin 1 Ser93, and Beclin 1 in the cortical extracts from ischemia‐ and reperfusion‐treated CHK2 +/+ and CHK2 −/− mice. Quantification of p62, p‐Beclin 1 Ser90, and p‐Beclin 1 Ser93 protein levels (n = 3 mice). Data are presented as mean ± s.e.m.; *P < 0.05, **P < 0.01 (Student's t‐test).
Source data are available online for this figure.
Because brain ischemia can initiate a complex cascade of metabolic events resulting in ROS‐induced tissue damage, we investigated the in vivo role of CHK2 in autophagy activation using the in vivo mouse stroke model. We employed a middle cerebral artery occlusion (MCAO) assay in CHK2 knockout (CHK2 −/−) mice. Twelve hours after MCAO, the measured infarct areas were much larger in the brains of CHK2 −/− mice than in the brains of WT mice (Fig 7E and F). In addition, Western blotting analysis showed that the level of p62 decreased and the phosphorylation of Beclin 1 increased in the ipsilateral region of WT, but not in CHK2 −/− mice (Fig 7G–J). These data suggest that the CHK2‐Beclin 1 autophagy pathway might contribute to cell survival in ischemic stroke.
Discussion
Our study establishes a mechanism by which cells sense oxidative stress signals to increase autophagy and thereby maintain redox homeostasis in the setting of nutrient deprivation or metabolic fluctuations. We have shown that in response to glucose deprivation and hypoxia, the ATM/CHK2/Beclin 1 axis is activated to positively regulate autophagy, which maintains cell and tissue homeostasis by suppressing ROS and clearing damaged mitochondria.
In addition to its role in DNA damage repair, growing evidence indicates that ATM/CHK2 activation induced by H2O2 can occur in the absence of DNA damage (Alexander et al, 2010; Guo et al, 2010). Consistent with previous studies, our results show that in the early stages of glucose starvation or hypoxia, although energetic stress exists as evidenced by AMPK activation, the ATM/CHK2 axis is not activated. However, with prolonged glucose starvation and hypoxia, ROS levels increase, and the ATM/CHK2/Beclin 1 pathway is activated. Moreover, this activation can be suppressed by the antioxidant NAC. Previous studies have shown that AMPK is not only an energy sensor, but also subject to regulation by ROS (Choi et al, 2001). However, researchers have found that ROS‐activated AMPK is dependent on its AMP‐sensing mechanism. When using AMPKγ2 R531G mutant cell lines, which generate an AMP insensitive AMPK complex, AMPK cannot be activated by H2O2. These findings suggest that the target for exogenous H2O2 may not be AMPK itself but rather the components of the respiratory chain, leading to a secondary effect on AMPK through increased AMP/ATP ratio (Hawley et al, 2010). In this study, we found that AMPK can be activated following ATP deprivation, but it was not activated by low concentration of H2O2 (without ATP deprivation). The activation of the ATM/CHK2 pathway under these conditions suggests this pathway is a more sensitive signaling pathway for sensing redox perturbations induced by changes in metabolic pathways.
Reactive oxygen species is a product of various stress conditions, and excessive ROS is also the main cause of cell death (Ren & Shen, 2019). Therefore, how to control ROS at a moderate level is important for cell fate. Our study found that with glucose starvation or with hypoxic conditions, the ATM/CHK2/Beclin 1 axis promotes autophagy by sensing ROS, controls excessive ROS accumulation, clears damaged mitochondria, and inhibits apoptosis. This ATM/CHK2/Beclin 1 axis might be an important supplement to classic antioxidant responses. This suggests that the DDR pathway acts as an important component of the oxidative stress response in the cytoplasm, as well as functioning in nuclear DNA repair.
Our results show that CHK2 phosphorylates Beclin 1 at Ser90/93 within the Bcl‐2 binding domain of Beclin 1. This phosphorylation disrupts the interaction between Beclin 1 and Bcl‐2, thereby promoting autophagy under oxidative stress. These results are consistent with the previous observations that the phosphorylation of Beclin 1 at Ser90/93 (Fogel et al, 2013) or monophosphorylation of Beclin 1 at Ser90 (Wei et al, 2015; Fujiwara et al, 2016; Li et al, 2017) positively regulates autophagy. It should be noted that some studies found an autophagy‐independent role for Beclin 1 phosphorylation at Ser90/93/96 by AMPK, which enables Beclin 1 to interact with SLC7A11 and initiate ferroptosis, a type of programmed cell death (Song et al, 2018). The basis for whether Beclin 1 phosphorylation triggers autophagy or ferroptosis remains unknown, but could relate to the intensity or duration of the perturbation.
Accumulating results indicate that autophagy is indeed involved in the pathophysiological changes in ischemic stroke (Scherz‐Shouval & Elazar, 2011). However, whether autophagy is a friend or a foe is still controversial. Our study found that in the model of MCAO, CHK2 WT mice showed increased p‐Beclin 1 S90/93 activation, higher levels of autophagy, and reduced brain infarct size than CHK2 −/− mice. This indicates that the CHK2‐Beclin 1 autophagy pathway might contribute to cell survival in ischemic stroke. Due to the lethality of BECN 1 knockout mice, we currently have no way to prove it with transgenic mice (Yue et al, 2003). However, there remains the possibility that CHK2 protects cells from I/R‐induced cell death via other mechanisms as well.
Given that nutrient deprivation and metabolic fluctuations can cause redox imbalance, it is important to evolve homeostatic mechanisms to cope with acute ROS accumulation. Our data suggest that the ATM/CHK2/Beclin 1 signaling pathway represents one such mechanism.
Materials and Methods
Regents and antibodies
Antibodies against Atg5 (#2630, 1:1,000 dilution), Beclin 1 (#3495, 1:1,000 dilution), Phospho‐Beclin‐1 (Ser93; #14717, 1:1,000 dilution), LC3A/B (#4108, 1:1,000 dilution), Phospho‐CHK2 (Thr68; #2661, 1:1,000 dilution), ATM (#2873, 1:1,000 dilution), Phospho‐ATM (Ser1981; #13050, 1:1,000 dilution), AMPKα (#2532, 1:1,000 dilution), Phospho‐AMPKα (Thr172; #2531, 1:1,000 dilution), BNIP3 (#44060, 1:1,000 dilution), NIX (#12396, 1:1,000 dilution), Parkin (#4211, 1:500 dilution), TOM20 (#42406, 1:1,000 dilution), Phospho‐Histone H2AX (Ser139; #2577, 1:1,000 dilution), and α‐Tubulin (#2144, 1:2,000 dilution) were from Cell Signaling Technology. Antibodies against p62/SQSTM1 (P0067, 1:2,000 dilution) and ATG7 (SAB1407006, 1:2,000 dilution) were from Sigma. CHK2 antibody (05‐649, 1:1,000 dilution) was from Merch Millipore. Bcl‐2 antibody (sc‐7382, 1:500 dilution) was from Santa Cruz Biotechnology. TIM23 antibody (NBP2‐13432, 1:1,000 dilution) was from NOVUS Biologicals. FIP200 antibody (WL03276, 1:500 dilution) was from Wanlei Biotechnology. Flag‐tag antibody (SG4110‐16, 1:1,000 dilution) was from Shanghai Genomics Technology. A rabbit antiserum against Beclin 1 phosphorylated at Ser90 was raised against peptide PPARMMS(P)TESANS, of which the serine is phosphorylated and indicated as S (P). The antiserum was precleaned using the corresponding nonphosphorylated peptide coupled to SulfoLink Coupling Resin (Thermo Scientific) and purified by affinity chromatography.
2, 3, 5‐triphenyltetrazolium chloride (TTC), N‐acetyl cysteine (NAC), ATP, puromycin, and CHK2 inhibitor II were from Sigma (St. Louis, MO). [γ‐32P]ATP was purchased from China Isotope & Radiation Corporation. Glutathione Sepharose™ 4B was from GE Healthcare (Uppsala, Sweden). CHK2 recombinant human protein and chloroquine were from Thermo Fisher Scientific. HBSS was from GIBCO. Protein A/G agarose was from Santa Cruz.
Cell culture and mice
H1299, HEK293T, HEK293, HeLa with mt‐Keima expression (a generous gift from Xin Pan), and HeLa with GFP‐LC3 expression (a generous gift from Weiguo Zhu) cells were cultured at 37°C in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (heat inactivated at 56°C for 45 min) and 100 U/ml penicillin, and 100 μg/ml streptomycin, in a 37°C incubator with a humidified, 5% CO2 atmosphere. HCT116 cells were cultured in RPMI1640 medium supplemented with 10% fetal bovine serum. Primary mouse embryonic fibroblasts (MEFs) were prepared in standard fashion and cultured in DMEM supplemented with 15% fetal bovine serum. For hypoxia treatment, cells were cultured under hypoxic (1% oxygen) condition.
CHK2 +/− mice were a kind gift from Takai H (Takai et al, 2002). CHK2 +/+ and CHK2 −/− mice were generated by mating CHK2 +/− males and females. The genotyping was performed by standard PCR. Mice were maintained in a specific pathogen‐free conditions, and all animal experiments were approved and performed in accordance with the guidelines of the Institutional Animal Care and Use Committee (IACUC) of China Medical University.
Plasmid constructs, mutagenesis and viral infection
CHK2 and Bcl‐2 expression plasmids were purchased from ADDGENE and GENECHEM. Beclin 1 wild type was provided by Feng Li. We used the pGEX‐5X‐1 vector to generate GST‐CHK2‐WT, GST‐CHK2 (amino acids 1–88), GST‐CHK2 (amino acids 1–193), GST‐CHK2 (amino acids 109–193), GST‐CHK2 (amino acids 109–543), GST‐CHK2 (amino acids 203–543), GST‐Beclin 1 WT, GST‐Beclin 1 (amino acids 1–87), GST‐Beclin 1 (amino acids 88–185), GST‐Beclin 1 (amino acids 186–263), GST‐Beclin 1 (amino acids 264–329), GST‐Beclin 1 (amino acids 330–450), GST‐Beclin 1 (amino acids 1–140), GST‐Beclin 1 (amino acids 1–270), GST‐Beclin 1 (amino acids 141–270), GST‐Beclin 1 (amino acids 141–450), and GST‐Beclin 1 (amino acids 271–450) plasmids. Mutagenesis (CHK2 T68A, CHK2 T68D, Beclin 1 S9093A, and Beclin 1 S9093D) was performed based on Quick Change Site‐Directed Mutagenesis Kit (Stratagene, La Jolla, CA). The CHK2 and Beclin 1 RNAi nontargetable mutant plasmids were generated by mutating three nucleotide base pairs to alternative nucleotides within the shRNA target region without altering the amino acid sequences. The sequences of all constructs were confirmed by DNA sequencing.
For lentiviral production and infection, control shRNA (shCtrl) lentivirus, shRNA against CHK2 (shCHK2), shRNA against Beclin 1 (shBeclin 1), and shRNA against FIP200 (shFIP200) lentivirus were purchased from Shanghai GeneChem Company. The CHK2 sequence was 5′‐ACAGATAAATACCGAACAT‐3′; the Beclin 1 sequence was 5′‐GGAGCCATTTATTGAAACT‐3′; and the FIP200 sequence was 5′‐AAAGAAATTAGGGAATCTT‐3′. GFP‐mCherry‐LC3 lentivirus was purchased from Beijing Syngentech Co., Ltd., China. Stably silencing and control cell lines were selected with puromycin (2 g/ml) after lentivirus infection. BNIP3, NIX, and parkin siRNA were purchased from RiboBio Co., Ltd., Guangzhou, China. The BNIP3 targeting sequence was 5′‐GAACTGCACTTCAGCAATA‐3′; the NIX targeting sequence was 5′‐CAGACACCCTAAACGTTCT‐3′; and the parkin targeting sequence was 5′‐AGTCGGAACATCACTTGCA‐3′.
Western blot and immunoprecipitation
Cells were lysed with IP lysis buffer (50 mM Tris–Cl at pH 7.4, 1% Triton X‐100, 1% NP40, 150 mM NaCl, 1 mM EDTA, 0.25% sodium deoxycholate and protease inhibitor cocktail), and lysates were centrifuged at 18,300 g for 20 min at 4°C. Then, lysates were used for Western blot analyses. For immunoprecipitation, cells were lysed with IP lysis buffer. Primary antibody was coupled with protein A/G beads (Santa Cruz), and then, immune complex was added to the cell lysates and incubated at 4°C overnight. After immunoprecipitation, the samples were washed with IP lysis buffer for three times. Proteins were eluted with 2× SDS sample buffer. The eluates were subjected to SDS–PAGE, and proteins were detected by immunoblot.
In vitro GST pull‐down
Proteins fused to glutathione S‐transferase (GST) were expressed in Escherichia coli BL21 cultured in LB medium containing 100 μg/ml ampicillin. Cells were induced to protein overexpression by addition of 1 mM IPTG (isopropyl β‐D‐1‐thiogalactopyranoside) at 30°C for 3 h, and then, they were resuspended in bacterial lysis buffer (20% glucose, 10% glycerol, 2 mM MgCl2, 50 mM Tris–Cl at pH 8.0), followed by ultrasonication. The proteins were purified with glutathione sepharose 4B according to the manufacturer's protocol, in which a Flag‐tagged protein synthesized by transcription and translation kit in vitro (Promega, P2221) was added, further incubation for 4 h at 4°C in binding buffer (20 mM Tris–Cl at pH 7.5, 50 mM NaCl, 10% glycerol, 1% NP40). After washing three times with binding buffer, proteins were eluted with 2× SDS sample buffer. The eluates were subjected to SDS–PAGE, and proteins were detected by immunoblot.
GST‐Beclin 1 (wild type, S9093A, or S9093D) was purified as indicated. The proteins were treated with or without recombinant CHK2 (200 ng) in kinase buffer at 30°C for 45 min, and then, they were incubated with Flag‐tagged Bcl‐2 that was overexpressed in HCT116 cells at 4°C overnight, followed by IP lysis buffer washing three times. The eluates were subjected to SDS–PAGE, and proteins were detected by immunoblot.
In vitro CHK2 kinase assay
The kinase reactions were performed as described previously (Guo et al, 2014). In brief, recombinant CHK2 (200 ng) was incubated with purified GST‐tagged FL or fragmented Beclin 1 in a kinase buffer (50 mM HEPES [pH 7.4], 10 mM MgCl2, 10 mM MnCl2, DTT 0.2 mM containing 100 μM ATP or 5 μM ATP, and 10 μCi [γ‐32P] ATP per reaction) at 30°C for 45 min. Phosphorylated proteins were separated by SDS–PAGE and analyzed by autoradiography.
Fluorescence microscopy
Fluorescence of mt‐Keima was imaged in two channels via two sequential excitations (458 nm, green; 561 nm, red) and using a 609‐ to 735‐nm emission range. For immunofluorescence analysis, the cells were grown on coverslips and were fixed in 4% paraformaldehyde for 15 min and then permeabilized with 0.1% Triton X‐100. Five percent BSA in PBS was used for blocking. Fixed cells were incubated overnight at 4°C with the corresponding primary antibodies: rabbit anti‐TOM20 (Cell Signaling Technology, #42406, 1:200 dilution), goat anti‐Flag (Abcam, ab1257; 1:400 dilution), and rabbit anti‐Phospho‐Histone H2A.X (Ser139; Cell Signaling Technology, #9718, 1:200 dilution). Coverslips were then washed in phosphate‐buffered saline (PBS) and stained for 60 min with Alexa Fluor 594‐, Alexa Fluor 630‐, and Alexa Fluor 488‐conjugated secondary antibodies (Invitrogen; 1:400). Coverslips were washed again with PBS and mounted with DAPI (Sigma, 32670). Confocal images were obtained using a 60× oil lens objective on an inverted fluorescence microscope (Nikon, Ti‐E, DS‐Ri2, NY USA). HEK293 stably expressing GFP‐mCherry‐LC3 and HeLa stably expressing GFP‐LC3 were plated onto 6‐wells plate. The following day, the medium was replaced with the complete culture medium for 4 h before experiment. Autophagy was induced by glucose starvation for 12 h, hypoxia for 36 h, and HBSS starvation or H2O2 (500 μM) for 1 h with or without CQ treatment. Cells were mounted and visualized under a microscope (Nikon, Ti‐E, DS‐Ri2).
Measurement of cellular ROS production and ATP concentration
Cells were treated with glucose starvation or hypoxia, followed by incubation with 5 μM DCFDA (Sigma) at 37°C for 15–20 min, and were analyzed by flow cytometry. The same number of cells was lysed to measure the ATP concentration following the manufacturer's protocol of the ATP Determination Kit (Beyotime).
Flow cytometric analysis
To determine cell apoptosis, 1 × 106 cells were treated with glucose starvation for 12 h or hypoxia 36 h, followed by incubation with APC and 7‐AAD. The frequency of apoptotic cells was measured following the manufacturer's protocol for Annexin V‐APC/7‐AAD kit (KeyGEN BioTECH, KGA1026). For the mitochondrial membrane potential analysis, cells were treated with glucose starvation for 18 h or hypoxia for 60 h. The Mitochondrial Membrane Potential Assay Kit with JC‐1 (Beyotime Institute of Biotechnology, China) was used according to the manufacturer's protocol. In cells with high potential, J‐aggregates formed red fluorescence, while in cells with low potential, the JC‐1 monomer formed green fluorescence.
Transmission electron microscopy
Cells grown in 10‐cm dishes were fixed in 2.5% glutaraldehyde for 12 h at 4°C. After fixation, cell monolayers were washed three times in PBS and then post‐fixed with 1% osmium tetroxide in 0.1 M cacodylate buffer for 1 h. After PBS washed three times, the samples were dehydrated through a graded series of ethanol and embedded in Poly/bed 812 epoxy resin (Polysciences, 02597‐50). Ultrathin (60 nm) sections were collected on copper grids and stained with 2% uranyl acetate in 50% methanol for 10 min, followed by 1% lead citrate for 7 min. Images were photographed with a JEOL JEM 1210 transmission electron microscope (JEOL).
MCAO
The experimental protocols were approved by the Institutional Animal Care and Use Committee of China Medical University, Shenyang, China. CHK2‐WT or KO male mice (5‐ to 6‐month old) were anesthetized with isoflurane, and transient MCAO was conducted. Briefly, the left common carotid artery was exposed through a midline incision in the neck and unilateral MCAO was conducted by inserting a silicone rubber‐coated monofilament (Jialing, Guangzhou, China). The MCAO was occluded for 1 h, and then, the suture was withdrawn to allow 12 h of reperfusion. After that, the animals were killed and brains were rapidly removed, coronally sectioned at 2 mm, and stained with 2% 2,3,5‐triphenyltetrazolium chloride (TTC, in 0.9% saline) for 15 minutes at 37°C. The infarct volume for each brain was calculated as I% = (volume of contralateral − normal volume of ipsilateral)/volume of contralateral.
Statistical analysis
Statistical comparisons between only two groups were carried out using unpaired Student's t‐test or the Mann–Whiney U‐test when a normal distribution could not be assumed. Data are presented as mean ± s.e.m. We tested data for normality and variance, and considered a P value of < 0.05 as significant. Statistical analyses were done using sas 9.4.
Author contributions
Q‐QG, SH, X‐YS, and LC designed the experiments, and Q‐QG carried out most of the experiments; Q‐QG, S‐SW, and S‐SZ performed the Western blot, immunoprecipitation, and GST pull‐down experiments. Q‐QG performed plasmids constructs, mutagenesis, viral infection, and the kinase assay in vitro. S‐SW and T‐TZ performed measurement of cellular ROS production, ATP concentration, and cell apoptosis. Q‐QG, S‐SW, S‐SZ, and G‐FZ performed in vivo experiments. H‐DX, X‐ML, Y‐LF, W‐DG, M‐TM, YG, FY, BJ, NB, X‐YS, G‐JF, C‐GW, ZW, XW, and S‐PZ provided reagents; Q‐QG and LC wrote the paper, and L‐YC, BPO, S‐HL, and P‐YW contributed to editing the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Acknowledgements
We thank Xin Pan for providing expressing mt‐Keima HeLa cells. We thank Weiguo Zhu for providing expressing GFP‐LC3 HeLa cells. We also thank Du Feng, Chen Liu, and Jiabin Li for assistance with electron microscopy assay and identification of autophagy structures. This work was supported by National Key R&D Program of China (2016YFC1302400); Natural Science Foundation of China (81502400, 81702738, 31300963); Ministry of Education Innovation Team Development Plan (IRT13101/17R107); and Natural Science Foundation of Liaoning Province of China (L201571).
The EMBO Journal (2020) 39: e103111
Contributor Information
Shuai Han, Email: hanshuai537197@sina.com.
Xiao‐Yu Song, Email: xysong@cmu.edu.cn.
Liu Cao, Email: lcao@cmu.edu.cn.
References
- Alexander A, Cai SL, Kim J, Nanez A, Sahin M, MacLean KH, Inoki K, Guan KL, Shen J, Person MD et al (2010) ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS. Proc Natl Acad Sci USA 107: 4153–4158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashkenazi A, Bento CF, Ricketts T, Vicinanza M, Siddiqi F, Pavel M, Squitieri F, Hardenberg MC, Imarisio S, Menzies FM et al (2017) Polyglutamine tracts regulate beclin 1‐dependent autophagy. Nature 545: 108–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakkenist CJ, Kastan MB (2003) DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 421: 499–506 [DOI] [PubMed] [Google Scholar]
- Cheng Z, Zhu Q, Dee R, Opheim Z, Mack CP, Cyr DM, Taylor JM (2017) Focal adhesion kinase‐mediated phosphorylation of Beclin1 protein suppresses cardiomyocyte autophagy and initiates hypertrophic growth. J Biol Chem 292: 2065–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SL, Kim SJ, Lee KT, Kim J, Mu J, Birnbaum MJ, Soo Kim S, Ha J (2001) The regulation of AMP‐activated protein kinase by H(2)O(2). Biochem Biophys Res Commun 287: 92–97 [DOI] [PubMed] [Google Scholar]
- D'Autreaux B, Toledano MB (2007) ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol 8: 813–824 [DOI] [PubMed] [Google Scholar]
- Filomeni G, De Zio D, Cecconi F (2015) Oxidative stress and autophagy: the clash between damage and metabolic needs. Cell Death Differ 22: 377–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkel T, Holbrook NJ (2000) Oxidants, oxidative stress and the biology of ageing. Nature 408: 239–247 [DOI] [PubMed] [Google Scholar]
- Fogel AI, Dlouhy BJ, Wang C, Ryu SW, Neutzner A, Hasson SA, Sideris DP, Abeliovich H, Youle RJ (2013) Role of membrane association and Atg14‐dependent phosphorylation in beclin‐1‐mediated autophagy. Mol Cell Biol 33: 3675–3688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara N, Usui T, Ohama T, Sato K (2016) Regulation of Beclin 1 protein phosphorylation and autophagy by protein phosphatase 2A (PP2A) and death‐associated protein kinase 3 (DAPK3). J Biol Chem 291: 10858–10866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Z, Kozlov S, Lavin MF, Person MD, Paull TT (2010) ATM activation by oxidative stress. Science 330: 517–521 [DOI] [PubMed] [Google Scholar]
- Guo Q, Su N, Zhang J, Li X, Miao Z, Wang G, Cheng M, Xu H, Cao L, Li F (2014) PAK4 kinase‐mediated SCG10 phosphorylation involved in gastric cancer metastasis. Oncogene 33: 3277–3287 [DOI] [PubMed] [Google Scholar]
- Hawley SA, Ross FA, Chevtzoff C, Green KA, Evans A, Fogarty S, Towler MC, Brown LJ, Ogunbayo OA, Evans AM et al (2010) Use of cells expressing gamma subunit variants to identify diverse mechanisms of AMPK activation. Cell Metab 11: 554–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He C, Levine B (2010) The Beclin 1 interactome. Curr Opin Cell Biol 22: 140–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill SM, Wrobel L, Rubinsztein DC (2019) Post‐translational modifications of Beclin 1 provide multiple strategies for autophagy regulation. Cell Death Differ 26: 617–629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmstrom KM, Finkel T (2014) Cellular mechanisms and physiological consequences of redox‐dependent signalling. Nat Rev Mol Cell Biol 15: 411–421 [DOI] [PubMed] [Google Scholar]
- Itakura E, Kishi C, Inoue K, Mizushima N (2008) Beclin 1 forms two distinct phosphatidylinositol 3‐kinase complexes with mammalian Atg14 and UVRAG. Mol Biol Cell 19: 5360–5372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katayama H, Kogure T, Mizushima N, Yoshimori T, Miyawaki A (2011) A sensitive and quantitative technique for detecting autophagic events based on lysosomal delivery. Chem Biol 18: 1042–1052 [DOI] [PubMed] [Google Scholar]
- Kim J, Kim YC, Fang C, Russell RC, Kim JH, Fan W, Liu R, Zhong Q, Guan KL (2013) Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell 152: 290–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura S, Noda T, Yoshimori T (2007) Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent‐tagged LC3. Autophagy 3: 452–460 [DOI] [PubMed] [Google Scholar]
- Kroemer G, Marino G, Levine B (2010) Autophagy and the integrated stress response. Mol Cell 40: 280–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JS, Collins KM, Brown AL, Lee CH, Chung JH (2000) hCds1‐mediated phosphorylation of BRCA1 regulates the DNA damage response. Nature 404: 201–204 [DOI] [PubMed] [Google Scholar]
- Lee IH, Kawai Y, Fergusson MM, Rovira II, Bishop AJ, Motoyama N, Cao L, Finkel T (2012) Atg7 modulates p53 activity to regulate cell cycle and survival during metabolic stress. Science 336: 225–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Wu XQ, Deng R, Li DD, Tang J, Chen WD, Chen JH, Ji J, Jiao L, Jiang S et al (2017) CaMKII‐mediated Beclin 1 phosphorylation regulates autophagy that promotes degradation of Id and neuroblastoma cell differentiation. Nat Commun 8: 1159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang XH, Kleeman LK, Jiang HH, Gordon G, Goldman JE, Berry G, Herman B, Levine B (1998) Protection against fatal Sindbis virus encephalitis by beclin, a novel Bcl‐2‐interacting protein. J Virol 72: 8586–8596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, Levine B (1999) Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 402: 672–676 [DOI] [PubMed] [Google Scholar]
- Liang C, Lee JS, Inn KS, Gack MU, Li Q, Roberts EA, Vergne I, Deretic V, Feng P, Akazawa C et al (2008) Beclin1‐binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nat Cell Biol 10: 776–787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski MM, Zheng B, Lu T, Yan Z, Py BF, Ng A, Xavier RJ, Li C, Yankner BA, Scherzer CR et al (2010) Genome‐wide analysis reveals mechanisms modulating autophagy in normal brain aging and in Alzheimer's disease. Proc Natl Acad Sci USA 107: 14164–14169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukas C, Falck J, Bartkova J, Bartek J, Lukas J (2003) Distinct spatiotemporal dynamics of mammalian checkpoint regulators induced by DNA damage. Nat Cell Biol 5: 255–260 [DOI] [PubMed] [Google Scholar]
- Maejima Y, Kyoi S, Zhai P, Liu T, Li H, Ivessa A, Sciarretta S, Del Re DP, Zablocki DK, Hsu CP et al (2013) Mst1 inhibits autophagy by promoting the interaction between Beclin1 and Bcl‐2. Nat Med 19: 1478–1488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsunaga K, Saitoh T, Tabata K, Omori H, Satoh T, Kurotori N, Maejima I, Shirahama‐Noda K, Ichimura T, Isobe T et al (2009) Two Beclin 1‐binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat Cell Biol 11: 385–396 [DOI] [PubMed] [Google Scholar]
- Matsuoka S, Huang M, Elledge SJ (1998) Linkage of ATM to cell cycle regulation by the Chk2 protein kinase. Science 282: 1893–1897 [DOI] [PubMed] [Google Scholar]
- Pickford F, Masliah E, Britschgi M, Lucin K, Narasimhan R, Jaeger PA, Small S, Spencer B, Rockenstein E, Levine B et al (2008) The autophagy‐related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J Clin Invest 118: 2190–2199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian X, Li X, Cai Q, Zhang C, Yu Q, Jiang Y, Lee JH, Hawke D, Wang Y, Xia Y et al (2017) Phosphoglycerate kinase 1 phosphorylates beclin1 to induce autophagy. Mol Cell 65: 917–931.e916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Y, Shen HM (2019) Critical role of AMPK in redox regulation under glucose starvation. Redox Biol 101154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roos WP, Kaina B (2006) DNA damage‐induced cell death by apoptosis. Trends Mol Med 12: 440–450 [DOI] [PubMed] [Google Scholar]
- Russell RC, Tian Y, Yuan H, Park HW, Chang YY, Kim J, Kim H, Neufeld TP, Dillin A, Guan KL (2013) ULK1 induces autophagy by phosphorylating Beclin‐1 and activating VPS34 lipid kinase. Nat Cell Biol 15: 741–750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherz‐Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z (2007) Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J 26: 1749–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherz‐Shouval R, Elazar Z (2011) Regulation of autophagy by ROS: physiology and pathology. Trends Biochem Sci 36: 30–38 [DOI] [PubMed] [Google Scholar]
- Seo GJ, Kim SE, Lee YM, Lee JW, Lee JR, Hahn MJ, Kim ST (2003) Determination of substrate specificity and putative substrates of Chk2 kinase. Biochem Biophys Res Commun 304: 339–343 [DOI] [PubMed] [Google Scholar]
- Shibata M, Lu T, Furuya T, Degterev A, Mizushima N, Yoshimori T, MacDonald M, Yankner B, Yuan J (2006) Regulation of intracellular accumulation of mutant Huntingtin by Beclin 1. J Biol Chem 281: 14474–14485 [DOI] [PubMed] [Google Scholar]
- Song X, Zhu S, Chen P, Hou W, Wen Q, Liu J, Xie Y, Klionsky DJ, Kroemer G, Lotze MT et al (2018) AMPK‐mediated BECN1 phosphorylation promotes ferroptosis by directly blocking system Xc(‐) activity. Curr Biol 28: 2388–2399.e2385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takai H, Naka K, Okada Y, Watanabe M, Harada N, Saito S, Anderson CW, Appella E, Nakanishi M, Suzuki H et al (2002) Chk2‐deficient mice exhibit radioresistance and defective p53‐mediated transcription. EMBO J 21: 5195–5205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang D, Kang R, Livesey KM, Cheh CW, Farkas A, Loughran P, Hoppe G, Bianchi ME, Tracey KJ, Zeh HJ 3rd et al (2010) Endogenous HMGB1 regulates autophagy. J Cell Biol 190: 881–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vega‐Rubin‐de‐Celis S, Zou Z, Fernandez AF, Ci B, Kim M, Xiao G, Xie Y, Levine B (2018) Increased autophagy blocks HER2‐mediated breast tumorigenesis. Proc Natl Acad Sci USA 115: 4176–4181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang RC, Wei Y, An Z, Zou Z, Xiao G, Bhagat G, White M, Reichelt J, Levine B (2012) Akt‐mediated regulation of autophagy and tumorigenesis through Beclin 1 phosphorylation. Science 338: 956–959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y, Zou Z, Becker N, Anderson M, Sumpter R, Xiao G, Kinch L, Koduru P, Christudass CS, Veltri RW et al (2013) EGFR‐mediated Beclin 1 phosphorylation in autophagy suppression, tumor progression, and tumor chemoresistance. Cell 154: 1269–1284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y, An Z, Zou Z, Sumpter R, Su M, Zang X, Sinha S, Gaestel M, Levine B (2015) The stress‐responsive kinases MAPKAPK2/MAPKAPK3 activate starvation‐induced autophagy through Beclin 1 phosphorylation. Elife 4: e05289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Kuo C, Bisi JE, Kim MK (2002) PML‐dependent apoptosis after DNA damage is regulated by the checkpoint kinase hCds1/Chk2. Nat Cell Biol 4: 865–870 [DOI] [PubMed] [Google Scholar]
- Yue Z, Jin S, Yang C, Levine AJ, Heintz N (2003) Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci USA 100: 15077–15082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zalckvar E, Berissi H, Mizrachy L, Idelchuk Y, Koren I, Eisenstein M, Sabanay H, Pinkas‐Kramarski R, Kimchi A (2009) DAP‐kinase‐mediated phosphorylation on the BH3 domain of beclin 1 promotes dissociation of beclin 1 from Bcl‐XL and induction of autophagy. EMBO Rep 10: 285–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Tripathi DN, Jing J, Alexander A, Kim J, Powell RT, Dere R, Tait‐Mulder J, Lee JH, Paull TT et al (2015) ATM functions at the peroxisome to induce pexophagy in response to ROS. Nat Cell Biol 17: 1259–1269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Wang W, Sun X, Xu D, Wang C, Zhang Q, Wang H, Luo W, Chen Y, Chen H et al (2016a) AMPK regulates autophagy by phosphorylating BECN1 at threonine 388. Autophagy 12: 1447–1459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Cheng X, Yu L, Yang J, Calvo R, Patnaik S, Hu X, Gao Q, Yang M, Lawas M et al (2016b) MCOLN1 is a ROS sensor in lysosomes that regulates autophagy. Nat Commun 7: 12109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Y, Wang QJ, Li X, Yan Y, Backer JM, Chait BT, Heintz N, Yue Z (2009) Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1‐phosphatidylinositol‐3‐kinase complex. Nat Cell Biol 11: 468–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7