

Summary
Kingdonia uniflora, an alpine herb, has an extremely narrow distribution and represents a model for studying evolutionary mechanisms of species that have adapted to undisturbed environments for evolutionarily long periods of time. We assembled a 1,004.7-Mb draft genome (encoding 43,301 genes) of K. uniflora and found significant overrepresentation in gene families associated with DNA repair, underrepresentation in gene families associated with stress response, and loss of most plastid ndh genes. During the evolutionary process, the overrepresentation of gene families involved in DNA repair could help asexual K. uniflora reduce the accumulation of deleterious mutations, while reducing genetic diversity, which is important in responding to environment fluctuations. The underrepresentation of gene families related to stress response and functional loss of ndh genes could be due to lack or loss of ability to respond to environmental changes caused by long-term adaptation to a relatively stable ecological environment.
Subject Areas: Biological Sciences, Plant Genetics, Plant Ecology, Plant Evolution
Graphical Abstract
Highlights
-
•
Provides a high-quality assembly for K. uniflora genome
-
•
K. uniflora shows overrepresentation in gene families associated with DNA repair
-
•
K. uniflora shows underrepresentation in gene families associated with stress response
-
•
The current endangered status of K. uniflora is related with adaptive degeneration
Biological Sciences; Plant Genetics; Plant Ecology; Plant Evolution
Introduction
Habitat destruction caused by changing climate and human activities has driven numerous plant species to endangered status (Yang et al., 2018). Current management of endangered species includes in situ and ex situ measurements (Nuijten et al., 2016). Owing to the rapidly growing number of threatened species, optimizing conservation practices based on evolutionary history and endangered mechanisms is imperative. In recent years, the development of genomic techniques has opened up new opportunities for species conservation. The main improvement is the enormous increase in loci allowing for a more detailed study of previous research questions (Nuijten et al., 2016). Specifically, genomic approaches provide the possibility to detect adaptive genetic variation across the entire genome (Kohn et al., 2006, Ouborg et al., 2010, Angeloni et al., 2012, Nuijten et al., 2016), which may help to address long-standing questions in conservation biology not fully resolved with traditional methods (Primmer, 2009, Avise, 2010, Frankham, 2010, Ouborg et al., 2010, Allendorf et al., 2010, Angeloni et al., 2012, Funk et al., 2012, Steiner et al., 2013, McMahon et al., 2014, Shafer et al., 2015; Garner et al., 2016, Benestan et al., 2016, Fuentes-Pardo and Ruzzante, 2017).
Plant lineages vary widely in their geographic distributions owing to numerous factors, e.g., ecological niche range, dispersal capacity, reproductive investments, and time since origin (Debussche and Thompson, 2003, Lavergne et al., 2004, Becker, 2010, Youssef et al., 2011), among which one crucial factor is adaptive capacity to respond to environmental changes. Lineages maintaining a small distribution are likely to possess low adaptive capacity to respond to geological and climatic changes at large scales, as well as habitat changes at small scales. For example, some asexual lineages, generating genetically and phenotypically identical individuals, are limited in their capacity to respond quickly to environmental fluctuation owing to low genetic diversity, which can lead to a status of endangerment or even extinction (Bell and Collins, 2008). In addition, species living in an equable environment for long periods might lack or lose the ability to defend against rapidly fluctuating environmental stress. Once the habitat is altered or destroyed, these species are incapable of colonizing new habitats. Shrinking habitats and low adaptive ability to new environments together lead some plants to endangered status. A recent study has shown that endangered species have more vulnerable genomes than that of congeneric non-endangered species (Hamabata et al., 2019); in fact, the endangered species exhibited significantly lower genetic diversity and proportion of duplicated genes and accumulated more deleterious variations than non-endangered species. Owing to limited genomic studies, genomic changes of endangered plants are largely unknown. To clarify which genetic factors contribute to vulnerability in endangered species, genome-wide genetic changes in functionality should be assessed (Hamabata et al., 2019).
Kingdonia uniflora Balf. f. and W.W. Sm. (Circaeasteraceae, Ranunculales), an alpine herb (diploid, 2n = 18), has a very narrow distribution (Figure S1). The habitat of K. uniflora represents an ecological environment of primeval forest with few disturbances. Specifically, K. uniflora is restricted to growing in high altitudes (∼2,800–4,000 m); cold, damp climates with deep humus; and usually under species of Abies. K. uniflora and Circaeaster agrestis Maxim. together constitute the early-diverging eudicot family Circaeasteraceae (Ranunculales) (Angiosperm Phylogeny Group, 2016). Previous estimates showed K. uniflora diverged from C. agrestis around 52 mya (Ruiz-Sanchez et al., 2012). Although no fossil record is known for K. uniflora, fossil fruits similar to those of C. agrestis have been reported from the mid-Albian of Virginia, USA (Crane et al., 1994, Drinnan et al., 1994, Sun et al., 2017). Remarkably, different from all other angiosperms, K. uniflora and C. agrestis possess an unusual dichotomous venation (Figure S2) similar to that found in ferns and Ginkgo (Sun et al., 2017). Old age for the lineage and dichotomous veins indicate an ancient relictual character for K. uniflora. Additionally, K. uniflora typically reproduces asexually, relying on rhizome systems to produce new individuals. These indications raise multiple questions, including: As an ancient relictual lineage, what history has K. uniflora experienced? As an endangered and habitat-dependent species, what genomic traits does K. uniflora possess? K. uniflora provides an ideal model to study the evolutionary mechanisms of ancient plant lineages that have an extremely narrow distribution and rely on a highly specialized habitat.
The ndh genes encode subunits of the thylakoid NADPH complex that mediate cyclic electron flow around Photosystem I and facilitates chlororespiration (Martín and Sabater, 2010). A series of studies suggests that the ndh genes can be dispensable under mild non-stressing environments (e.g., Casano et al., 2001, Martin et al., 2004, Rumeau et al., 2007, Martín and Sabater, 2010). A previous study (Sun et al., 2017) found rampant loss and pseudogenization of ndh genes in the K. uniflora plastome, which may indicate an association between the vulnerability of K. uniflora and genomic changes. In the present study, we provide a de novo genome sequence of K. uniflora using both Illumina and Pacbio sequencing technologies. We aim to investigate the evolutionary history of K. uniflora and reveal the potential mechanisms of its evolutionary specialization.
Results
Genome Assembly and Annotation
Genome size estimation using flow cytometry suggested a haploid genome size of 1,150 Mb for K. uniflora (Figure S3), whereas k-mer statistics indicated a similar genome size of 1,170 Mb, with very low heterozygosity (Figure S4 and Table S1). In the present study, we generated 236 Gb of Illumina reads and 106 Gb Pacbio reads with an N50 length of 12.876 kb (Table S2). A total assembly of 1,004.7 Mb (representing ∼86% of the estimated genome size), consisting of 2,932 scaffolds (scaffold N50 of 2.09 Mb; longest scaffold of 11.5 Mb) was achieved (Table 1). A total of 43,301 protein-coding genes were predicted (Table 1), among which 35,953 genes (83.03%) were functionally annotated (Table S3). In addition to protein-coding genes, various noncoding RNA sequences were identified and annotated (Table S4), including 1,124 transfer RNAs, 715 ribosomal RNAs, 125 microRNAs, and 1,751 small nuclear RNAs. The completeness of gene regions assessed by BUSCO (Benchmarking Universal Single Copy Orthologs) showed that 90.6% of the green plant single-copy orthologs were complete (Table S5).
Table 1.
Genome Assembly of Kingdonia uniflora
| Genome Features | Contigs/Scaffolds |
|---|---|
| Total length, bp | 1,004,656,313 |
| Total number of contigs | 2,932 |
| Longest length, bp | 11,531,354 |
| Length of N50, bp | 2,099,369 |
| Length of N90, bp | 292,588 |
| GC content, % | 38.04% |
| No. of genes | 43,301 |
We compared the draft genome of K. uniflora with the well-annotated genomes of the model plant Arabidopsis thaliana (Brassicaceae) and the Ranunculales species Aquilegia coerulea (Ranunculaceae). The genome size of K. uniflora is much larger than that of both references. The K. uniflora genome showed strong synteny with the genome of Aq. coerulea (Figure 1), but weak synteny with that of A. thaliana (Figure S5), which is not surprising given their placement in the angiosperm Tree of Life. The gene density in K. uniflora is lower than that in Aq. coerulea and A. thaliana, whereas the density of TEs (transposable elements) in K. uniflora was higher than that in Aq. coerulea and A. thaliana (Figures 1 and S5).
Figure 1.

Comparative Analyses of Genomic Features between Kingdonia uniflora and Aquilegia coerulea
Tracks from inside to outside are collinearity between both genomes, number of chromosomes/contigs, gene density, GC content, and TE density.
We also compared our draft genome sequence with five other draft genomes of Ranunculales taxa, representing three of the seven Ranunculales families (Table S6); the quality of our assembly is comparable with that of all the five species, generating the longest N50 length and relatively fewer scaffolds. Comparatively, the genome of K. uniflora is larger than other Ranunculales species with sequenced genomes, such as Aq. coerulea (293.08 Mb), Eschscholzia californica (489.065 Mb), and Macleaya cordata (377.83 Mb) (Table S6).
Repeat Elements
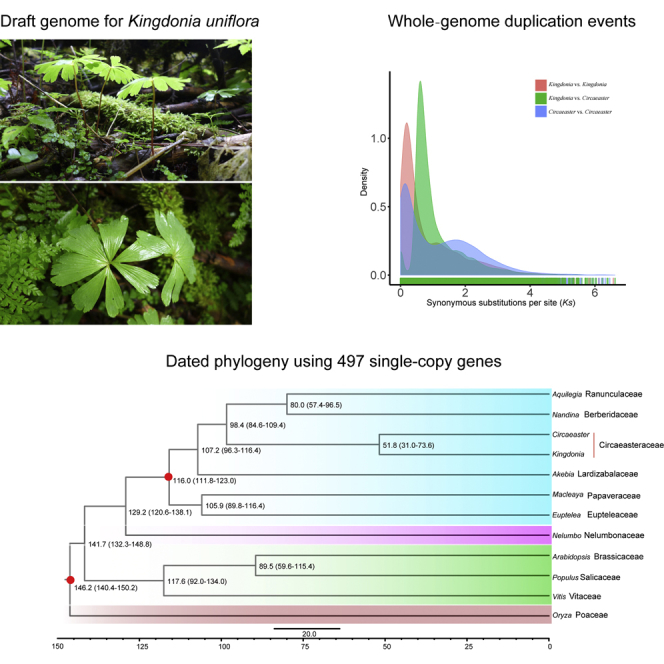
Through a combination of approaches, we annotated 66.83% of the assembly as repetitive elements, among which long terminal repeats (LTRs) were the most abundant, occupying 40.62% of the genome assembly length; DNA elements and long interspersed nuclear elements occupied 5.0% and 3.0% of the genome, respectively (Table S7). The proliferation of LTR retrotransposons in K. uniflora was estimated to peak around 2.7 mya (Figure 2A). Analyses of age distributions built from synonymous substitutions per synonymous site (Ks) indicated that K. uniflora has undergone one recent whole-genome duplication (WGD) event, which occurred after its divergence from C. agrestis (Figure 2B). The inferred WGD event in the K. uniflora genome was further supported by dot-plot analysis of representative scaffolds, in which numerous paralogs derived from this event were identified (Figure 2C).
Figure 2.

Evolutionary Analyses of the K. uniflora Genome
(A) Insertion time distribution of LTR retrotransposons. (B) Distribution of synonymous substitution rates (Ks) for pairs of paralogs/orthologs in/between K. uniflora and C. agrestis. (C) Dot plots of paralogs identified across contigs in the K. uniflora genome. (D) Venn diagram showing unique and shared gene families between genomes of K. uniflora and four other Ranunculales species.
Phylogenetic Tree Construction and Estimation of Divergence Times
Applying OrthoFinder (Emms and Kelly, 2015) to eight whole-genome and four transcriptome sequences including monocots, basal eudicots, and core eudicots, we identified a total of 18,742 orthogroups, among which 6,883 were shared by Kingdonia and four other Ranunculales species (Figure 2D). Among these orthogroups, 497 were identified as putative single-copy gene families. To further investigate the phylogenetic relationships within Ranunculales, we conducted both concatenated and coalescent analyses using the sequences of 497 single-copy genes in 12 species. The topologies from two analyses were identical, confirming the sister relationship between K. uniflora and C. agrestis and resolving Circaeasteraceae as sister to the clade formed by Ranunculaceae and Berberidaceae (Figure 3); Papaveraceae + Eupteleaceae was placed as the earliest-diverging clade (Figure 3). K. uniflora and C. agrestis were estimated to have diverged ∼51.8 mya in our analyses using MCMCtree with two calibration points (Figure 3). In addition, the phylogenetic analysis with an expanded group of taxa, which correspond to a larger taxonomic sampling but fewer loci indicated a similar phylogeny of Ranunculales, except the placement of Eupteleaceae (Figure S6).
Figure 3.

Dated Phylogeny for 12 Plant Species with Oryza as an Outgroup
A timescale is shown at the bottom, and red points in some nodes indicate fossil calibration points.
Gene Family Overrepresentation and Underrepresentation
Comparisons of the genomes among 12 species identified a total of 111 gene families that are significantly (p < 0.01) overrepresented in K. uniflora and 22 gene families that are significantly underrepresented (Table S8). The results from Kyoto Encyclopedia of Genes and Genomes (KEGG) and Gene Ontology (GO) annotations showed that overrepresented gene families were considerably enriched in DNA repair pathways, such as homologous recombination, mismatch repair, DNA replication and nucleotide excision repair, whereas gene families showing significant underrepresentation in the K. uniflora genome were found to be involved in pathways related to stress or pest responses, such as the phenylpropanoid biosynthesis and secondary metabolites biosynthesis (Table 2).
Table 2.
Functional Annotation of the Significantly Overrepresented and Underrepresented Gene Families in Kingdonia uniflora
| Gene Families | KEGG Terms | Input No. | Background No. | p Value | Corrected p Value |
|---|---|---|---|---|---|
| Overrepresented gene families | Glycosphingolipid biosynthesis - globo series | 9 | 9 | 5.28 × 10−7 | 1.59 × 10−5 |
| Homologous recombination | 17 | 56 | 2.87 × 10−6 | 6.74 × 10−5 | |
| Mismatch repair | 12 | 39 | 7.31 × 10−5 | 0.000968079 | |
| Sphingolipid metabolism | 9 | 26 | 0.000282866 | 0.003082136 | |
| DNA replication | 12 | 50 | 0.000516123 | 0.005446878 | |
| Nucleotide excision repair | 14 | 69 | 0.000789672 | 0.007779277 | |
| Peroxisome | 16 | 87 | 0.000878589 | 0.008505414 | |
| Galactose metabolism | 12 | 55 | 0.001064483 | 0.010034842 | |
| Plant hormone signal transduction | 33 | 271 | 0.002344688 | 0.020456082 | |
| Underrepresented gene families | Cyanoamino acid metabolism | 5 | 60 | 2.06 × 10−9 | 1.57 × 10−7 |
| Phenylpropanoid biosynthesis | 5 | 157 | 2.06 × 10−7 | 6.24 × 10−6 | |
| Starch and sucrose metabolism | 5 | 202 | 6.95 × 10−7 | 1.96 × 10−5 | |
| Biosynthesis of secondary metabolites | 6 | 1,076 | 0.00020813 | 0.001073645 | |
| Metabolic pathways | 6 | 1,910 | 0.004094269 | 0.015173186 |
Dispensability of Plastid ndh Genes
To detect whether the ndh genes/segments lost from K. uniflora plastome were transferred to the nuclear genome, we conducted a BLASTN search using 11 intact ndh sequences extracted from C. agrestis as the query, using the assembled K. uniflora genome sequences as target. The result showed that no intact sequences for ndh plastid genes were discovered with the exception of ndhE and ndhJ, indicating functional copies of these genes likely have been lost (Table S9 and Figure 4).
Figure 4.

Length Comparison of ndh Genes between K. uniflora and C. agrestis
Discussion
Species that live in stable habitats face less stress, which can cause lack or loss of ability to respond to environmental changes. In the present study, we de novo assembled the genome of K. uniflora, an ancient relictual species exclusively found in China. Given that K. uniflora has a larger genome than many of its close relatives, we hypothesize that the proliferation of LTR retrotransposons and the WGD event together are likely responsible for the increased genome size of K. uniflora (Michael, 2014). Several studies (Tenaillon et al., 2010, Michael, 2014) have suggested that the proliferation of TEs and specifically long terminal repeat retrotransposons (LTRs) in genomes is the primary driver of genome size differences in plants. This is because LTRs are expressed as RNA and reverse-transcribed into a new DNA element that can be inserted in every replication cycle (Wicker et al., 2007). A comparative study using high-quality genomes detected a correlation between intact LTR retrotransposons and genome size (El Baidouri et al., 2013). Abundant LTR retrotransposons were detected in the genome of K. uniflora, whereas considerably fewer LTR retrotransposons were identified in other members of the order with sequenced genomes (Figure 1). To investigate the evolutionary dynamics of the LTR retrotransposons, we estimated their insertion dates. The results indicate that the proliferation of LTR retrotransposons in K. uniflora was likely triggered around one rapid uplift of the Hengduan Mountain region, occurring between the late Miocene and late Pliocene (Kirby et al., 2002, Clark et al., 2005, Sun et al., 2011, Wang et al., 2012, Wang et al., 2014, Meng et al., 2016, Xing and Ree, 2017). WGDs have been shown to pervade the evolutionary history of angiosperms (Landis et al., 2018), and K. uniflora is no different (Figures 2B and 2C). Therefore, the relatively larger genome size of K. uniflora compared with close relatives might be promoted by both LTRs proliferation and WGD events.
Based on phylogenetic inference and estimated divergence times, we speculate that the speciation of K. uniflora was promoted by the Himalayan orogeny. Previous studies based on DNA commonly recognize K. uniflora being closely related to C. agrestis (Kim et al., 2004, Wang et al., 2009, Sun et al., 2017). Our phylogenetic analyses confirmed the sister relationship between K. uniflora and C. agrestis. These species were resolved as sister to the clade formed by Ranunculaceae and Berberidaceae, a result incongruent with previous placements of Circaeasteraceae and Lardizabalaceae as sister pairs (Kim et al., 2004, Wang et al., 2009, Sun et al., 2017). The divergence estimation between K. uniflora and C. agrestis (∼51.8 mya) is consistent with a previous estimate of ∼52 mya (Ruiz-Sanchez et al., 2012) and also coincides with the timing of the first stage of Himalayan orogeny (Rowley, 1996, Huang et al., 2015). Similar to K. uniflora, C. agrestis has a narrow distribution confined to China and the Himalayas. However, the distribution range of Circaeaster is relatively larger; in places where Kingdonia occur Circaeaster can always be found, whereas in most regions that Circaeaster is distributed Kingdonia is absent. Previous studies have shown that orogeny could create conditions favoring speciation of resident lineages (e.g., Hoorn et al., 2013, Wen et al., 2014, Favre et al., 2015). Hence, we hypothesize that the divergence between K. uniflora and C. agrestis was likely driven by the rapid uplift of contemporary Himalayan orogeny.
An asexual reproductive system and overrepresentation of DNA repair genes together reduce genetic diversity of K. uniflora. Levels of genetic diversity are often associated with reproductive strategies of species (Otálora et al., 2013). Colonization, population persistence, and extinction probabilities are all influenced by the reproductive systems of species (Stephenson et al., 2000, Barbará et al., 2009, Wornik and Grube, 2010, Beatty and Provan, 2011, Otálora et al., 2013). K. uniflora primarily reproduces asexually via rhizomes. This reproductive mode could produce identical individuals rapidly but lacks recombination and the possibility to create genetic variation in offspring, reducing the opportunities for adaptive evolution (Eckert, 2001, Castonguay and Angers, 2012). In addition, without segregation and recombination, obligate asexual multiplication may push a species into extinction owing to the steady accumulation of deleterious mutations (Thomas et al., 2016). During the long-term evolutionary history of K. uniflora, deleterious mutation accumulation cannot be ruled out; if something goes awry, such as the occurrence of a fatal mutation, whole clusters of clones can be wiped out. The integrated DNA-repair mechanism overrepresented in K. uniflora would allow for a reduction in the accumulation of deleterious mutations. This DNA-repair system might also reduce genetic diversity produced by mutations. Having high genetic diversity is important for plants to respond to environmental changes. We thus speculate that the extremely narrow distribution range of K. uniflora is associated with low genetic diversity, which restricted suitable environments to simplex, equable habitats rather than multiple, divergent habitats. Specifically, K. uniflora can only live in high elevations with minor human disturbances, while being characterized by perennial cold temperatures of below 0°C. The extreme temperature is likely to cause DNA, RNA, and protein damage. The overrepresentation of gene families for DNA repair might also be one kind of protection from extremely low temperature.
Underrepresentation of genes associated with stress response in K. uniflora leads to degeneration of adaptive ability to environmental changes. Phenylpropanoids are believed to contribute to all aspects of plant responses toward biotic and abiotic stimuli (Vogt, 2010). As concluded by La Camera et al. (2004), when plants suffer environmental stress, pests, or diseases, phenylpropanoids could evoke relevant response mechanisms to protect the plant from damage. Similarly, secondary metabolites also play a role in plant defense against environmental stresses, pests, and diseases (Bennett and Wallsgrove, 1994). Given the relatively undisturbed ecological environment of K. uniflora and lack of habitat stress during growth, adaptation to such conditions resulted in the functional degeneration of stress response systems. In addition, phenylpropanoids can also promote invasion of new habitats (Bais et al., 2003, Vogt, 2010). Our results indicate the underrepresentation of gene families involved in phenylpropanoid biosynthesis, which might be one reason causing low ability of K. uniflora to invade new habitats.
We conclude long-term living in highly equable habitats lead to the underrepresentation of stress response genes, which finally resulted in loss of ability to adapt to changing environments. The asexual reproductive strategy promoted overrepresentation of DNA repair genes, which reduced genetic diversity associated with adaptive capacity to environmental changes. Hence, both the underrepresentation of stress response genes and overrepresentation of DNA repair genes are responsible for the low adaptive ability of K. uniflora.
Equable habitats likely promoted the dispensability of most ndh genes in K. uniflora. Most angiosperms contain 11 plastid ndh genes, whereas all ndh genes, except for ndhE and ndhJ, were found to be either pseudogenized (ΨndhA, ΨndhB, ΨndhD, ΨndhH, and ΨndhK) or absent (ndhC, ndhF, ndhI, and ndhG) in the K. uniflora plastome (Sun et al., 2017). All 11 plastid ndh genes are intact in C. agrestis (Sun et al., 2017), indicating that the loss of ndh genes from K. uniflora occurred after the split between K. uniflora and C. agrestis, suggesting that within 52 million years most of the plastid ndh genes were lost from K. uniflora not only in the plastome but also in the nuclear genome. Among land plants, the plastid ndh loci have also been found absent in non-photosynthetic plants, epiphytes, Gnetales, conifers, and Erodium (Geraniaceae) (Kim et al., 2015, Lin et al., 2017, Nie et al., 2017). Evidence suggests that the thylakoid NADPH complex could optimize photosynthesis for plants under environmental stresses, while being found dispensable for plant growth under optimal growth conditions (Martín and Sabater, 2010). K. uniflora is extremely selective in habitat preference (Li et al., 2001) and is known as the indicator for natural ecological environment without disturbance. We hence speculate that the current habitats of K. uniflora might have promoted the dispensability of the plastid ndh genes. Additionally, within plants, NADPH supplies hydrogen for many anabolic processes (Antal et al., 2015). The underrepresentation of gene families related to metabolic pathways, as detected from our CAFÉ-based analyses, is likely related with the nonfunction state of plastid ndh genes.
Changing climate, shrinking habitats, and low adaptive ability to environmental changes together contributed to the extremely narrow distribution of K. uniflora. The overrepresentation of gene families involved in DNA repair could help reduce the accumulation of deleterious mutations during asexual reproduction, which is the dominant mode of reproduction in K. uniflora, while reducing genetic diversity, which is important in responding to environment fluctuations. The underrepresentation of gene families in charge of stress response and nonfunction of plastid ndh genes could be due to the adaptive degeneration caused by long-term adaptation to living in relatively stress-free environments. Considering the long evolutionary history of K. uniflora, and the fossil records from C. agrestis in the mid-Albian of Virginia, USA (Crane et al., 1994, Drinnan et al., 1994), we speculate it should have been widespread around the world. Changing climate, shrinking habitats, asexual reproduction, and adaptive degeneration caused by relying on easeful environment together led it to a current status of endangerment.
Limitations of the Study
We reported the draft genome of the ancient and currently endangered K. uniflora. Although we revealed the evolutionary history and potential endangered mechanism of K. uniflora through primary analyses, population genomic analyses are required to estimate genetic diversity, reveal genetic structure and its causes, and identify loci associated with environmental adaption.
Resource Availability
Lead Contact
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, Yanxia Sun (sunyanxia@wbgcas.cn).
Materials Availability
This study did not generate new reagents.
Data and Code Availability
The accession number for the genome assembly and raw reads reported in this paper is GenBank: PRJNA587615 and PRJNA611722, respectively.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
This work was supported by the Strategic Priority Research Program of Chinese Academy of Sciences (XDA20050203), the Program Foundation for the Backbone of Scientific Research by Wuhan Botanical Garden, Chinese Academy of Sciences (Y855241G01), the Major Program of National Natural Science Foundation of China (31590823), and the National Key R and D Program of China (2017YFC0505200).
Author Contributions
H.W., H.S., and Xi. Zhang conceived the study Y.S., H.Z., and N.L. collected the materials. Y.S. and T.D. prepared DNA and RNA for sequencing. Y.S., A.Z., and Xu Zhang performed the genome assembly and genome annotation. Y.S. and A.Z. designed genomics analyses. Y.S., T.D., M.J.M., J.B.L., and J.H. wrote the manuscript with the input of all co-authors.
Declaration of Interests
The authors declare no competing interests.
Published: May 22, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101124.
Contributor Information
Xiujun Zhang, Email: zhangxj@wbgcas.cn.
Hang Sun, Email: sunhang@mail.kib.ac.cn.
Hengchang Wang, Email: hcwang@wbgcas.cn.
Supplemental Information
References
- Allendorf F.W., Hohenlohe P.A., Luikart G. Genomics and the future of conservation genetics. Nat. Rev. Genet. 2010;11:697–709. doi: 10.1038/nrg2844. [DOI] [PubMed] [Google Scholar]
- Angiosperm Phylogeny Group An update of the angiosperm phylogeny group classification for the orders and families of flowering plants: APG IV. Bot. J. Linn. Soc. 2016;181:1–20. [Google Scholar]
- Antal T.K., Krendeleva T.E., Tyystjärvi E. Multiple regulatory mechanisms in the chloroplast of green algae: relation to hydrogen production. Photosynth. Res. 2015;125:357–381. doi: 10.1007/s11120-015-0157-2. [DOI] [PubMed] [Google Scholar]
- Angeloni F., Wagemaker N., Vergeer P., Ouborg J. Genomic toolboxes for conservation biologists. Evol. Appl. 2012;5:130–143. doi: 10.1111/j.1752-4571.2011.00217.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avise J.C. Perspective: conservation genetics enters the genomics era. Conserv. Genet. 2010;11:665–669. [Google Scholar]
- Becker T. Explaining rarity of the dry grassland perennial Astragalus exscapus. Folia Geobot. 2010;45:303–321. [Google Scholar]
- Bais H.P., Vepachedu R., Gilroy S., Callaway R.M., Vivanco J.M. Allelopathy and exotic plant invasion: from molecules and genes to species interactions. Science. 2003;301:1377–1380. doi: 10.1126/science.1083245. [DOI] [PubMed] [Google Scholar]
- Barbará T., Martinelli G., Palma-Silva C., Fay M.F., Mayo S., Lexer C. Genetic relationships and variation in reproductive strategies in four closely related bromeliads adapted to neotropical ‘inselbergs’: Alcantarea glaziouana, A. regina, A. geniculata and A. imperialis (Bromeliaceae) Ann. Bot. 2009;103:65–77. doi: 10.1093/aob/mcn226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beatty G.E., Provan J. High clonal diversity in threatened peripheral populations of the yellow bird’s nest (Hypopitys monotropa; syn. Monotropa hypopitys) Ann. Bot. 2011;107:663–670. doi: 10.1093/aob/mcr003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell G., Collins S. Adaptation, extinction and global change. Evol. Appl. 2008;1:3–16. doi: 10.1111/j.1752-4571.2007.00011.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benestan L.M., Ferchaud A.-L., Hohenlohe P.A., Garner B.A., Naylor G.J.P., Baums I.B., Schwartz M.K., Kelley J.L., Luikart G. Conservation genomics of natural and managed populations: building a conceptual and practical framework. Mol. Ecol. 2016;25:2967–2977. doi: 10.1111/mec.13647. [DOI] [PubMed] [Google Scholar]
- Bennett R.N., Wallsgrove R.M. Secondary metabolites in plant defense mechanisms. New Phytol. 1994;127:617–633. doi: 10.1111/j.1469-8137.1994.tb02968.x. [DOI] [PubMed] [Google Scholar]
- Casano L.M., Martín M., Sabater B. Hydrogen peroxide mediates the induction of chloroplast Ndh complex under photooxidative stress in barley. Plant Physiol. 2001;125:1450e1458. doi: 10.1104/pp.125.3.1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castonguay E., Angers B. The key role of epigenetics in the persistence of asexual lineages. Genet. Res. Int. 2012;2012:1–9. doi: 10.1155/2012/534289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark M.K., House M.A., Royden L.H., Whipple K., Burchfiel B.C., Zhang X., Tang W. Late cenozoic uplift of southeastern tibet. Geology. 2005;33:525–528. [Google Scholar]
- Crane P.R., Friis E.M., Pedersen K.R. Paleobotanical evidence on the early radiation of magnoliid angiosperms. Plant Syst. Evol. 1994;8:51–72. [Google Scholar]
- Debussche M., Thompson J.D. Habitat differentiation between two closely related Mediterranean plant species, the endemic Cyclamen balearicum and the widespread C. repandum. Acta Oecol. 2003;24:35–45. [Google Scholar]
- Drinnan A.N., Crane P.R., Hoot S.B. Patterns of floral evolution in the early diversification of non-magnoliid dicotyledons (eudicots) Plant Syst. Evol. 1994;8:93–122. [Google Scholar]
- Eckert C.G. The loss of sex in clonal plants. Evol. Ecol. 2001;15:501–520. [Google Scholar]
- El Baidouri M., Panaud O. Comparative genomic paleontology across plant kingdom reveals the dynamics of TE-driven genome evolution. Genome Biol. Evol. 2013;5:954–965. doi: 10.1093/gbe/evt025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emms D.M., Kelly S. OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 2015;16:157. doi: 10.1186/s13059-015-0721-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favre A., Päckert M., Pauls S.U., Jähnig S.C., Uhl D., Michalak I., Muellner-Riehl A.N. The role of the uplift of the Qinghai-Tibetan Plateau for the evolution of Tibetan biotas. Biol. Rev. 2015;90:236–253. doi: 10.1111/brv.12107. [DOI] [PubMed] [Google Scholar]
- Frankham R. Challenges and opportunities of genetic approaches to biological conservation. Biol. Conserv. 2010;143:1919–1927. [Google Scholar]
- Fuentes-Pardo A.P., Ruzzante D.E. Whole-genome sequencing approaches for conservation biology: advantages, limitations and practical recommendations. Mol. Ecol. 2017;26:5369–5406. doi: 10.1111/mec.14264. [DOI] [PubMed] [Google Scholar]
- Funk W.C., McKay J.K., Hohenlohe P.A., Allendorf F.W. Harnessing genomics for delineating conservation units. Trends Ecol. Evol. 2012;27:489–496. doi: 10.1016/j.tree.2012.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garner B.A., Hand B.K., Amish S.J., Bernatchez L., Foster J.T., Miller K.M., Morin P.A., Narum S.R., O’Brien S.J., Roffler G. Genomics in Conservation: case studies and bridging the gap between data and application. Trends Ecol. Evol. 2016;31:81–83. doi: 10.1016/j.tree.2015.10.009. [DOI] [PubMed] [Google Scholar]
- Hamabata T., Kinoshita G., Kurita K., Cao P.-L., Ito M., Murata J., Komaki Y., Isagi Y., Makino T. Endangered island endemic plants have vulnerable genomes. Commun. Biol. 2019;2:244. doi: 10.1038/s42003-019-0490-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoorn C., Mosbrugger V., Mulch A., Antonelli A. Biodiversity from mountain building. Nat. Geosci. 2013;6:154. [Google Scholar]
- Huang W., van Hinsbergen D.J.J., Lippert P.C., Guo Z., Dupont-Nivet G. Paleomagnetic tests of tectonic reconstructions of the India-Asia collision zone. Geophys. Res. Lett. 2015;42:2642–2649. [Google Scholar]
- Kim H.T., Kim J.S., Moore M.J., Neubig K.M., Williams N.H., Whitten W.M., Kim J.-H. Seven new complete plastome sequences reveal rampant independent loss of the ndh gene family across Orchids and associated instability of the inverted repeat/small single-copy region boundaries. PLoS One. 2015;10:e0142215. doi: 10.1371/journal.pone.0142215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S., Soltis D.E., Soltis P.S., Zanis M.J., Suh Y. Phylogenetic relationships among early-diverging eudicots based on four genes: were the eudicots ancestrally woody? Mol. Phylogenet. Evol. 2004;31:16–30. doi: 10.1016/j.ympev.2003.07.017. [DOI] [PubMed] [Google Scholar]
- Kirby E., Reiners P.W., Krol M.A., Whipple K.X., Hodges K.V., Farley K.A., Tang W.Q., Chen Z. Late Cenozoic evolution of the eastern margin of the Tibetan Plateau: inferences from 40ar/39ar and (U-Th)/He thermochronology. Tectonics. 2002;21:1–20. [Google Scholar]
- Kohn M.H., Murphy W.J., Ostrander E.A., Wayne R.K. Genomics and conservation genetics. Trends Ecol. Evol. 2006;21:629–637. doi: 10.1016/j.tree.2006.08.001. [DOI] [PubMed] [Google Scholar]
- La Camera S., Gouzerh G., Dhondt S., Hoffmann L., Fritig B., Legrand M., Heitz T. Metabolic reprogramming in plant innate immunity: the contributions of phenylpropanoid and oxylipin pathways. Immunol. Rev. 2004;198:267–284. doi: 10.1111/j.0105-2896.2004.0129.x. [DOI] [PubMed] [Google Scholar]
- Landis J.B., Soltis D.E., Li Z., Marx H.E., Barker M.S., Tank D.C., Soltis P.S. Impact of whole-genome duplication events on diversification rates in angiosperms. Am. J. Bot. 2018;105:348–363. doi: 10.1002/ajb2.1060. [DOI] [PubMed] [Google Scholar]
- Lavergne S., Thompson J.D., Garnier E., Debussche M. The biology and ecology of narrow endemic and widespread plants: a comparative study of trait variation in 20 congeneric pairs. Oikos. 2004;107:505–508. [Google Scholar]
- Li J.X., Zhang W.H., Li H. A study on geological distribution and ecological characteristics of Kingdonia uniflora. J. Northwest For. Univ. 2001;16:1–4. [Google Scholar]
- Martín M., Sabater B. Plastid ndh genes in plant evolution. Plant Physiol. Biochem. 2010;48:636e645. doi: 10.1016/j.plaphy.2010.04.009. [DOI] [PubMed] [Google Scholar]
- Lin C.-S., Chen J.J.W., Chiu C.-C., Hsiao H.C.W., Yang C.-J., Jin X.-H., Leebens-Mack J., de Pamphilis C.W., Huang Y.-T., Yang L.-H. Concomitant loss of NDH complex-related genes within chloroplast and nuclear genomes in some orchids. Plant J. 2017;90:994–1006. doi: 10.1111/tpj.13525. [DOI] [PubMed] [Google Scholar]
- Martin M., Casano L.M., Zapata J.M., Guera A., del Campo E.M., Schmitz-Linneweber C., Maier R.M., Sabater B. Role of thylakoid Ndh complex and peroxidase in the protection against photo-oxidative stress: fluorescence and enzyme activities in wild-type and ndhF-deficient tobacco. Physiol. Plant. 2004;122:443e452. [Google Scholar]
- McMahon B.J., Teeling E.C., Höglund J. How and why should we implement genomics into conservation? Evol. Appl. 2014;7:999–1007. doi: 10.1111/eva.12193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng K., Wang E., Wang G. Uplift of the Emei Shan, western Sichuan basin: implication for eastward propagation of the Tibetan plateau in early Miocene. J. Asian Earth. Sci. 2016;115:29–39. [Google Scholar]
- Michael T.P. Plant genome size variation: bloating and purging DNA. Brief Funct. Genomics. 2014;13:308–317. doi: 10.1093/bfgp/elu005. [DOI] [PubMed] [Google Scholar]
- Nie Z.X., Ye Y.J., Bai T.D., Xu M., Xu L.-A. Complete chloroplast genome of Pinus massoniana (Pinaceae): gene rearrangements, loss of ndh Genes, and short inverted repeats contraction, expansion. Molecules. 2017;22:1528. doi: 10.3390/molecules22091528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuijten R.J.M., Bosse M., Crooijmans R.P.M.A., Madsen O., Schaftenaar W., Ryder O.A., Groenen M.A.M., Megens H.-J. The Use of genomics in conservation management of the endangered Visayan warty pig (Sus cebifrons) Int. J. Genomics. 2016;2016:1–9. doi: 10.1155/2016/5613862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouborg N.J., Pertoldi C., Loeschcke V., Bijlsma R. (Kuke), Hedrick P.W. Conservation genetics in transition to conservation genomics. Trends Genet. 2010;26:177–187. doi: 10.1016/j.tig.2010.01.001. [DOI] [PubMed] [Google Scholar]
- Otálora M.A.G., Salvador C., Martínez I., Aragón G. Does the reproductive strategy affect the transmission and genetic diversity of bionts in cyanolichens? A case study using two closely related species. Microb. Ecol. 2013;65:517–530. doi: 10.1007/s00248-012-0136-5. [DOI] [PubMed] [Google Scholar]
- Primmer C.R. From conservation genetics to conservation genomics. Ann. NY Acad. Sci. 2009;1162:357–368. doi: 10.1111/j.1749-6632.2009.04444.x. [DOI] [PubMed] [Google Scholar]
- Rowley D.B. Age of initiation of collision between India and Asia: a review of stratigraphic data. Earth Planet. Sc. Lett. 1996;145:1–13. [Google Scholar]
- Ruiz-Sanchez E., Rodriguez-Gomez F., Sosa V. Refugia and geographic barriers of populations of the desert poppy, Hunnemannia fumariifolia (Papaveraceae) Org. Divers. Evol. 2012;12:133–143. [Google Scholar]
- Rumeau D., Peltier G., Cornac L. Chlororespiration and cyclic electron flow around PSI during photosynthesis and plant stress response. Plant Cell Environ. 2007;30:1041e1051. doi: 10.1111/j.1365-3040.2007.01675.x. [DOI] [PubMed] [Google Scholar]
- Shafer A.B.A., Wolf J.B.W., Alves P.C., Bergström L., Bruford M.W., Brännström I., Colling G., Dalén L., De Meester L., Ekblom R. Genomics and the challenging translation into conservation practice. Trends Ecol. Evol. 2015;30:78–87. doi: 10.1016/j.tree.2014.11.009. [DOI] [PubMed] [Google Scholar]
- Steiner C.C., Putnam A.S., Hoeck P.E.A., Ryder O.A. Conservation genomics of threatened animal species. Annu. Rev. Anim. Biosci. 2013;1:261–281. doi: 10.1146/annurev-animal-031412-103636. [DOI] [PubMed] [Google Scholar]
- Stephenson A.G., Good S.V., Vogler D.W. Interrelationships among inbreeding depression, plasticity in the self-incompatibility system, and the breeding system of Campanula rapunculoides L. (Campanulaceae) Ann. Bot. 2000;85:211–219. [Google Scholar]
- Sun B.-N., Wu J.-Y., Liu Y.-S.(Christopher)., Ding S.-T., Li X.-C., Xie S.-P., Yan D.-F., Lin Z.-C. Reconstructing Neogene vegetation and climates to infer tectonic uplift in western Yunnan, China. Palaeogeogr. Palaeoclimatol. Palaeoecol. 2011;304:328–336. [Google Scholar]
- Sun Y., Moore M.J., Lin N., Adelalu K.F., Meng A., Jian S., Yang L.S., Li J.Q., Wang H. Complete plastome sequencing of both living species of Circaeasteraceae (Ranunculales) reveals unusual rearrangements and the loss of the ndh gene family. BMC Genomics. 2017;18:592. doi: 10.1186/s12864-017-3956-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenaillon M.I., Hollister J.D., Gaut B.S. A triptych of the evolution of plant transposable elements. Trends Plant Sci. 2010;15:471–478. doi: 10.1016/j.tplants.2010.05.003. [DOI] [PubMed] [Google Scholar]
- Thomas G.E., Geetha K.A., Augustine L., Mamiyil S., Thomas G. Analyses between reproductive behavior, genetic diversity and pythium responsiveness in Zingiber spp. reveal an adaptive significance for hemiclonality. Front. Plant Sci. 2016;7:1913. doi: 10.3389/fpls.2016.01913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt T. Phenylpropanoid biosynthesis. Mol. Plant. 2010;3:2–20. doi: 10.1093/mp/ssp106. [DOI] [PubMed] [Google Scholar]
- Wang P., Scherler D., Liu-Zeng J., Mey J., Avouac J.-P., Zhang Y., Shi D. Tectonic control of yarlung tsangpo gorge revealed by a buried canyon in Southern Tibet. Science. 2014;346:978–981. doi: 10.1126/science.1259041. [DOI] [PubMed] [Google Scholar]
- Wang W., Lu A.-M., Ren Y., Endress M.E., Chen Z.-D. Phylogeny and classification of Ranunculales: evidence from four molecular loci and morphological data. Perspect. Plant Ecol. 2009;11:81–110. [Google Scholar]
- Wang Y., Tang H., DeBarry J.D., Tan X., Li J., Wang X., Lee T., Jin H., Marler B., Guo H. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012;40:e49. doi: 10.1093/nar/gkr1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen J., Zhang J.-Q., Nie Z.-L., Zhong Y., Sun H. Evolutionary diversifications of plants on the Qinghai-Tibetan plateau. Front. Genet. 2014;5:1–16. doi: 10.3389/fgene.2014.00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicker T., Sabot F., Hua-Van A., Bennetzen J.L., Capy P., Chalhoub B., Flavell A., Leroy P., Morgante M., Panaud O. A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 2007;8:973–982. doi: 10.1038/nrg2165. [DOI] [PubMed] [Google Scholar]
- Wornik S., Grube M. Joint dispersal does not imply maintenance of partnerships in lichen symbioses. Microb. Ecol. 2010;59:150–157. doi: 10.1007/s00248-009-9584-y. [DOI] [PubMed] [Google Scholar]
- Xing Y., Ree R.H. Uplift-driven diversification in the Hengduan Mountains, a temperate biodiversity hotspot. Proc. Natl. Acad. Sci. 2017;114:E3444–E3451. doi: 10.1073/pnas.1616063114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y., Ma T., Wang Z., Lu Z., Li Y., Fu C., Chen X., Zhao M., Olson M.S., Liu J. Genomic effects of population collapse in a critically endangered ironwood tree Ostrya rehderiana. Nat. Commun. 2018;9:5449. doi: 10.1038/s41467-018-07913-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youssef S., Baumel A., Véla E., Juin M., Dumas E., Affre L., Tatoni T. Factors underlying the narrow distribution of the mediterranean annual plant Arenaria provincialis (Caryophyllaceae) Folia Geobot. 2011;46:327–350. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the genome assembly and raw reads reported in this paper is GenBank: PRJNA587615 and PRJNA611722, respectively.