

Abstract
We previously identified that hepatitis B virus (HBV) encodes a microRNA (HBV-miR-3) that restrains HBV replication by targeting the HBV transcript. However, whether HBV-miR-3 affects host innate immunity to modulate HBV replication remains unclear. Here, we examined the vital functions of HBV-miR-3 in the innate immune response after HBV infection. We found that HBV-miR-3 expression gradually increased in a dose- and time-dependent manner in HBV-infected HepG2-NTCP cells. HBV-miR-3 activated the JAK/STAT signaling pathway by downregulating SOCS5 in hepatocytes, thereby enhancing the IFN-induced anti-HBV effect. In addition, HBV-miR-3 in exosomes facilitated the M1 polarization of macrophages. Furthermore, exosomes containing HBV-miR-3 enhanced the secretion of IL-6 via inhibiting the SOCS5-mediated ubiquitination of EGFR. In short, these results demonstrate that HBV-miR-3 activates the innate immune response to restrain HBV replication by multiple pathways, which may suppress HBV-induced acute liver cell injury and affect the progression of persistent HBV infection.
Keywords: hepatitis B virus, HBV-miR-3, IFN, M1 polarization, IL-6
Introduction
Hepatitis B virus (HBV) is a small hepatotropic, noncytopathic DNA virus infecting humans (Guidotti and Chisari, 2006). HBV infection causes acute or chronic liver diseases characterized by different levels of liver inflammation and viral replication, which are determined by a complex interplay of host and viral factors, including the host genetic background, viral load, viral genotype, and age of acquisition (Mcmahon, 2009). Unlike many other viral infections, HBV infection is characterized by a long period of noncytolytic chronic infection with indirect liver cell injury. This trait is attributed to the complex interactions between HBV and the immune system, including adaptive and innate immunity.
Type I interferons (IFNs) are the most important innate immune response to viral infection. Recently, IFN-I and its derivatives have become the first choice to treat some viral infectious diseases, including hepatitis B and hepatitis C (Thio, 2010). IFN-I interacts with IFN-I receptor (IFNAR1) and activates the Janus kinase (JAK)/STAT pathway. Phosphorylated STAT1/2 then translocates into the nucleus and binds to the IFN-stimulated response element (ISRE), thus initiating the transcription of hundreds of IFN-stimulated genes (Pardo et al., 2007). This antiviral mechanism of infected cells is negatively regulated by the suppressor of cytokine signaling (SOCS) family (Nicholson and Hilton, 1998). Linossi et al. (2013) found that SOCS5, an E3 ubiquitin ligase, interacts with JAKs via its JAK interacting region (JIR) and inhibits the autophosphorylation of JAK1 and JAK2, leading to inhibition of JAK kinase activity. Although SOCS5 can regulate the replication of Japanese encephalitis virus and infectious bursal disease virus (Sharma et al., 2016; Fu et al., 2018), the role of SOCS5 in HBV infection has not been well studied.
Macrophages are pivotal cells in innate immunity, as they detect and respond to pathogenic and tissue-derived signals to clear pathogens, repair injured tissue, and restore tissue homeostasis (Murray and Wynn, 2011). The extent of macrophage heterogeneity and the polarizing stimuli that exist in vivo are far from clear, but two main functional phenotypes have been well described experimentally (Gordon and Taylor, 2005; Mosser and Edwards, 2008). Classical or M1-activated macrophages mainly induced by microbial products with or without IFN-γ are associated with pathogen infection, tumors, and proinflammation, while alternative or M2 macrophages induced by IL-4 or IL-13 are involved in anti-inflammatory effects. These diverse but polarized functional phenotypes, driven by microenvironmental cues or multiple factors, allow macrophages to adapt readily to changing conditions within tissues (Martinez et al., 2008). The type of macrophage differentiation in the process of HBV infection affects the development of hepatitis B, and there is growing interest in how macrophage polarization can be manipulated in vivo to alter disease outcomes (Gordon and Taylor, 2005; Murray, 2015).
Immune cells can control HBV replication in a noncytolytic fashion via the secretion of cytokines and other immune mediators (Xu et al., 2014). IL-6 is a typical pleiotropic cytokine associated with a variety of biological processes (Rincon and Irvin, 2012) and plays important roles in balancing the differentiation of pro- and anti-inflammatory cells in the progression of HBV infection (Lan et al., 2015). Compared with chronic active HBV patients, patients with severe, acute HBV infections have significantly higher serum IL-6 levels (Heinz et al., 2010), and the increased IL-6 expression may inhibit HBV entry through the downregulation of an HBV-specific receptor (Bouezzedine et al., 2015) and induce an inhibitory effect on HBV replication (Kuo et al., 2009). In addition, the secretion of IL-6 is regulated by many factors. For example, HBV-encoded proteins such as HBx and HBc could stimulate/inhibit the secretion of IL-6 (Xia et al., 2015). Enhanced epidermal growth factor receptor (EGFR) could increase IL-6 expression in Kupffer cells under infection and inflammatory conditions (Lanaya et al., 2014). However, it remains unclear whether HBV-encoded microRNAs (miRNAs) regulate IL-6 secretion.
MicroRNA (miRNA) is a post-transcriptional regulator that generally binds to the 3′ untranslated regions (UTRs) of target mRNAs to silence their expression and participate in physiological and pathogenic processes (Bartel, 2004). Recent studies have demonstrated that many miRNAs, such as miR-146a, miR-155, and miR-223, play important roles in the innate immune response (Gottwein and Cullen, 2008). To eliminate virus immediately after infection, host-encoded miRNAs can directly interfere with virus replication (Mahajan et al., 2009). Furthermore, virus-encoded miRNAs can evolve to regulate viral gene expression to accommodate the virus life cycle and maintain latency, and they affect cellular gene expression by directly participating in host gene expression or by mimicking cellular miRNAs to hijack unclear cellular regulatory networks (Sullivan et al., 2005). Regarding HBV infection, we previously reported that cellular miR-210 and miR-199a can target transcripts of HBV to attenuate its replication (Zhang et al., 2010). More interestingly, we first revealed that HBV encodes an miRNA, HBV-miR-3, that is not only released into the circulation by exosomes and HBV virions but also inhibit HBV replication by targeting its own transcript (Yang et al., 2017).
In this study, we investigated the functional and regulatory roles of HBV-miR-3 in innate immunity during HBV infection. Our data showed that HBV-miR-3 expression was increased during HBV infection. By stimulating the expression of IFN-stimulated genes and the secretion of IL-6, HBV-miR-3 affected the polarization of macrophages and resulted in the restriction of HBV replication, thus potentially contributing to the elimination of HBV from infected host cells and alleviation of cell injury. Our findings provide a new understanding of the mechanism underlying HBV–host cell interactions, which may yield new insights into the potential applications of viral miRNAs.
Results
HBV-miR-3 is highly expressed in HepG2-NTCP cells during HBV infection
Recent studies have reported that HepG2-NTCP cells (HepG2 cells stably expressing sodium-taurocholate cotransporting polypeptide) are permissive for HBV infection in vitro (Yan et al., 2012). Accordingly, we first established the HepG2-NTCP cell line by transfection with the pcDNA3/NTCP vector and subsequent selection by G418 for 3 months. Western blotting and immunofluorescence (IF) staining validated NTCP expression and its cell membrane localization (Figure 1A and B). Next, HepG2-NTCP cells were inoculated with HBV at different loads (MOI, genome equivalents per cell), and the cells were infected for different amounts of time. At the indicated time points (MOI of 300; 3, 5, and 7 days post-infection) or at a fixed time point with different MOIs (MOI of 50, 200, and 1000; 7 days post-infection), total RNAs were extracted, and HBV-miR-3 expression was determined by reverse transcription quantitative polymerase chain reaction (RT-qPCR). Compared with that in the mock-infected (MI) cells, the expression of HBV-miR-3 in infected cells was gradually increased in a dose- and time-dependent manner (Figure 1C and D). We further obtained similar results in Huh7 cells after transfection with pHBV1.3 (Figure 1E). To determine whether HBV-miR-3 exists in exosomes from HBV-infected cells, we isolated exosomes from the supernatant of HBV-infected HepG2-NTCP cells, and RT-qPCR analysis verified that the exosomes contained HBV-miR-3 (Figure 1F). Taken together, our results suggest that HBV-miR-3 is highly expressed in HBV-infected HepG2-NTCP cells and is secreted extracellularly through exosomes.
Figure 1.
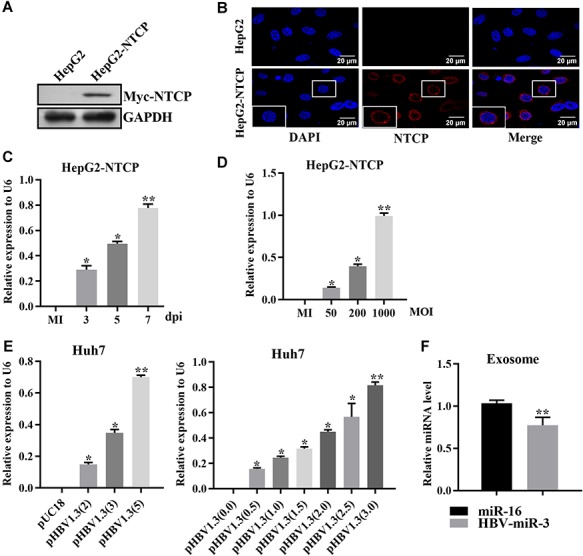
HBV-miR-3 is highly expressed in HepG2-NTCP cells during HBV infection. (A and B) Verification of the established stable HepG2-NTCP cells as detected by western blot (A) and IF analyses (B). A Myc tag antibody was used to detect the myc-tagged NTCP. The parental HepG2 cells served as a negative control. Scale bar, 20 μm. (C and D) Relative HBV-miR-3 expression in HepG2-NTCP cells infected with HBV for different times at a fixed MOI of 300 (genome equivalents per cell) (C) or with HBV at different MOIs for 7 days (D) was determined by RT-qPCR and normalized with U6 RNA. The uninfected HepG2-NTCP cells served as a control. MI, mock-infected. (E) HBV-miR-3 levels in Huh7 cells transfected with pHBV1.3 or the empty vector pUC18 were measured by RT-qPCR and normalized with U6 RNA. Numbers in brackets represent the days post-transfection (dpi, left panel) or the amount of plasmid (μg/ml, right panel). (F) RT-qPCR was performed to detect the expression of HBV-miR-3 in HBV-infected HepG2-NTCP cell-derived exosomes, and miR-16 was used as an internal control for exosome miRNAs due to its stable expression. Data are presented as mean ± SD from three independent experiments. *P < 0.05, **P < 0.01.
HBV-miR-3 enhances the IFN-induced anti-HBV effect
IFN-α plays an important role in antiviral infection by inducing antiviral effectors, such as OAS-1, MX1, IFIT2, and IFIT3. To explore whether HBV-miR-3 is involved in the antiviral response of IFN during HBV infection, we first collected 20 serum samples from patients infected with HBV and investigated the correlation between HBV-miR-3 and other hepatitis-related parameters, including alanine aminotransferase (ALT), aspartate transaminase (AST), and type I IFNs (IFN-α and IFN-β). As shown in Figure 2A and B, the expression of HBV-miR-3 was significantly positively correlated with ALT, AST, IFN-α, and IFN-β expression. These data indicated that HBV-miR-3 levels might reflect liver injury and the antiviral effect, suggesting that HBV-miR-3 induces the inhibition of HBV replication. Next, HepG2.2.15 and HepG2 cells were treated with an HBV-miR-3 inhibitor (ASO-HBV-miR-3) or an HBV-miR-3 expression vector (pHBV-miR-3), followed by IFN-α stimulation. As shown in Figure 2C and D, inhibition of HBV-miR-3 decreased the typical IFN-α-induced antiviral effectors OAS-1, MX1, IFIT2, and IFIT3 at the mRNA and protein levels in HepG2.2.15 cells. In contrast, HBV-miR-3 overexpression increased production of the antiviral effectors OAS-1, MX1, IFIT2, and IFIT3 in HepG2 cells (Figure 2E and F). These results suggest that HBV-miR-3 positively regulates IFN-α-induced signal transduction. As HBV-miR-3 augments IFN-α signaling, we further assessed the effect of HBV-miR-3 on HBV replication in HepG2.2.15 cells, followed by IFN-α stimulation. The HBV replication capacity was assessed by ELISA and RT-qPCR analysis. Inhibition of HBV-miR-3 increased the production of HBsAg, HBeAg, pregenomic RNA (pgRNA), total RNA, and viral DNAs (Figure 2G), suggesting that HBV-miR-3 markedly enhanced the IFN-α-induced anti-HBV effects.
Figure 2.
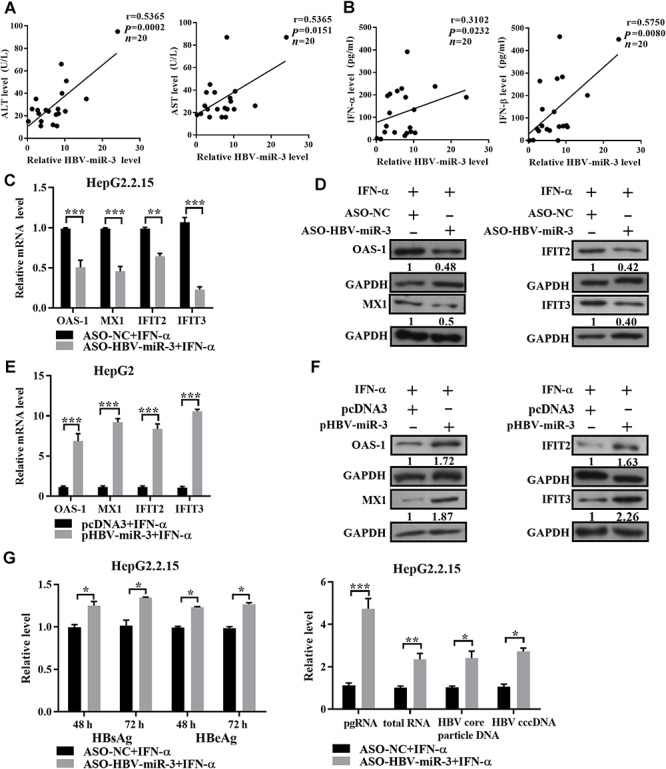
HBV-miR-3 enhances the IFN-induced anti-HBV effect. (A and B) Pearson’s correlation analysis indicated positive correlations between HBV-miR-3 expression and the ALT, AST, IFN-α, and IFN-β levels in the sera of HBV-infected patients (n = 20). (C–F) HepG2.2.15 and HepG2 cells were transfected with ASO-HBV-miR-3/ASO-NC or HBV-miR-3/pcDNA3 for 24 h and then treated with IFN-α (50 U/ml). (C and E) After 12 h, the mRNA levels of OAS-1, MX1, IFIT2, and IFIT3 were measured by RT-qPCR, and the results are expressed as the fold change compared to the control (ASO-NC/pcDNA3). (D and F) After 24 h, the protein levels of OAS-1, MX1, IFIT2, and IFIT3 were measured by western blotting. (G) ASO-HBV-miR-3/ASO-NC was transfected into HepG2.2.15 cells. After 24 h, these cells were treated with IFN-α (100 U/ml) for further 12, 48, and 72 h. The levels of HBsAg and HBeAg in the cell culture supernatants were detected by ELISA. Total cellular RNA, pregenomic RNA, HBV DNA, and cccDNA levels were analyzed by RT-qPCR, and the results are expressed as the fold change compared to the control (ASO-NC). Data are presented as mean ± SD from three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001.
HBV-miR-3 targets and downregulates SOCS5 expression during HBV infection
To gain insights into the mechanism underlying the functions of HBV-miR-3 and its potential association with innate immunity, we predicted the potential targets of HBV-miR-3 by TargetScan5.2 Custom (http://www.targetscan.org/vert_50/) and selected SOCS5 for further validation. We constructed EGFP reporter vectors containing the wild-type (WT) or mutant (Mut) form of the HBV-miR-3-binding sequence on the SOCS5 3′UTR (Figure 3A) downstream of the EGFP reporter gene. As shown in Figure 3B, HBV-miR-3 decreased the fluorescence intensity of the WT EGFP reporter but not that of the Mut form. Furthermore, overexpression of HBV-miR-3 significantly decreased SOCS5 protein expression, but no significant mRNA changes were observed in Huh7 cells (Figure 3C and D). In addition, the expression of SOCS5 was gradually reduced in HepG2-NTCP cells during HBV infection as the HBV load increased and the infection period extended and dramatically declined at 7 days post-infection (Figure 3E), which was opposite to the expression patterns of HBV-miR-3, as indicated in Figure 1C and D. These data suggest an inverse correlation between HBV-miR-3 and SOCS5 expression.
Figure 3.

HBV-miR-3 targets and downregulates SOCS5 expression during HBV infection. (A) Predicted HBV-miR-3 binding sites in the 3′UTR of SOCS5 mRNA. The WT 3′UTR contains an HBV-miR-3-binding site, whereas the Mut 3′UTR contains mutations in the seed region matched with HBV-miR-3. (B) Huh7 cells were co-transfected with either WT or Mut pEGFP-SOCS5-3′UTR construct and pHBV-miR-3 or pcDNA3 for 48 h, and the cells were harvested to analyze the EGFP fluorescence intensities. pDsRed2-N1 was used for normalization. Data are presented as mean ± SD from three independent experiments. (C and D) Huh7 cells were transfected with HBV-miR-3 or pcDNA3. (C) After 36 h, SOCS5 mRNA expression was detected by RT-qPCR, and the results are expressed as the fold change compared to the control (pcDNA3). (D) After 48 h, SOCS5 protein expression was detected by western blotting. (E) HepG2-NTCP cells were either infected with HBV at an MOI of 300 for different times or infected with HBV at different MOIs for 5 days. SOCS5 expression was detected by western blotting. MI, mock-infected. (F) HepG2-NTCP cells were co-transfected with either ASO-HBV-miR-3 or ASO-NC together with WT or Mut pEGFP-SOCS5-3′UTR constructs, followed by HBV infection for 3 days, and the fluorescence intensities were measured. Data are presented as mean ± SD of three individual experiments. (G) HepG2-NTCP cells were transfected with ASO-HBV-miR-3/ASO-NC and either infected with HBV at an MOI of 300 genomes/cell for different times or infected with HBV at different MOIs for 5 days. The expression of SOCS5 was detected by western blotting. MI, mock-infected. *P < 0.05, **P < 0.01, ns = not significant.
To further investigate whether HBV infection alters the regulatory effect of HBV-miR-3 on SOCS5 expression, we co-transfected HepG2-NTCP cells with ASO-HBV-miR-3/ASO-NC and EGFP-SOCS5 3′UTR reporter vectors (WT or Mut form) and subsequently infected the cells with HBV for 3 days. The EGFP fluorescence intensities were significantly increased in the cells co-transfected with the WT (but not the mutated) SOCS5 3′UTR reporter and ASO-HBV-miR-3 compared with those in the respective control group (Figure 3F). The SOCS5 protein levels in HepG2-NTCP cells transfected with ASO-HBV-miR-3 combined with HBV infection for different amounts of time or at different viral loads were further analyzed by western blotting. As shown in Figure 3G, blocking HBV-miR-3 counteracted the SOCS5 reduction induced by HBV infection compared to that in the control group in a time- and viral load-dependent manner. These data indicate that HBV-miR-3 binds to the 3′UTR of SOCS5 and suppresses its protein expression during HBV infection.
HBV-miR-3 activates the JAK/STAT signaling pathway in liver cancer cells
As a main feedback regulatory protein of the JAK/STAT signaling pathway, SOCS5 conducts negative feedback inhibition (Smith et al., 2005; Linossi et al., 2013). Our abovementioned data showed that HBV-miR-3 enhanced the IFN-induced anti-HBV effect and suppressed SOCS5 expression during HBV infection. Therefore, we speculated that SOCS5 might be a mediator of HBV-miR-3 to exert anti-HBV effects. To address this hypothesis, HepG2 cells were transfected with pshR-SOCS5 to knock down SOCS5 expression, and the phosphorylation of STAT1 and the downstream target genes of IFN were examined after treatment with IFN-α. As shown in Figure 4A and B, phosphorylation of STAT1 on the Tyr701 residue was markedly increased, and the selected IFN-stimulated genes OAS-1, MX1, IFTT2, and IFTT3 exhibited significantly increased expression at the mRNA and protein levels. In addition, knockdown of SOCS5 reduced the transcription of HBV RNA, replication of DNA (including cccDNA and virion DNA), and generation of HBsAg and HBeAg in HepG2.2.15 cells (Figure 4C). Conversely, overexpression of SOCS5 significantly decreased the phosphorylation of STAT1 and the expression of OAS-1, MX1, IFTT2, and IFTT3 in HepG2 cells (Figure 4D and E). Furthermore, overexpression of SOCS5 enhanced the transcription of HBV RNA, replication of DNA (including cccDNA and virion DNA), and HBsAg and HBeAg production in HepG2.2.15 cells (Figure 4F). Due to the involvement of SOCS5 in the JAK/STAT–IFN pathway in the context of viral infection, we further investigated the effects of HBV-miR-3 on the JAK/STAT pathway. Expression of HBV-miR-3 in HepG2 cells enhanced the phosphorylation of STAT1 after IFN-α treatment (Figure 4G), while overexpression of SOCS5 attenuated the effect of HBV-miR-3 on STAT1 phosphorylation (Figure 4H). In summary, these results indicate that HBV-miR-3 may suppress viral replication mainly by suppressing SOCS5 expression, resulting in STAT1 activation and the subsequent promotion of IFN-responsive effector activation.
Figure 4.
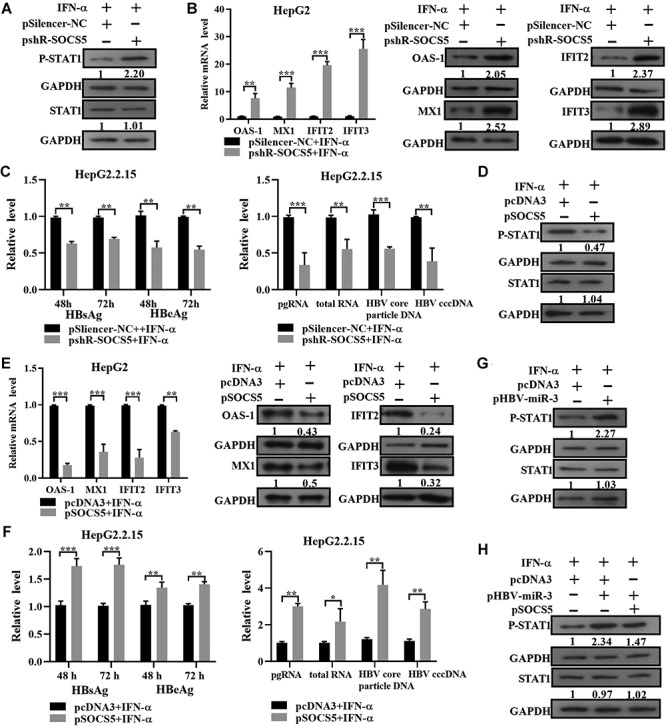
HBV-miR-3 activates the JAK/STAT signaling pathway in liver cancer cells. (A–F) pshR-SOCS5/pSilencer-NC and SOCS5/pcDNA3 were transfected into HepG2 or HepG2.2.15 cells for 24 h, followed by IFN-α treatment (50 U/ml for HepG2 cells and 100 U/ml for HepG2.2.15 cells). (A and D) After 24 h, phosphorylated STAT1 (p-STAT1) and total STAT1 in HepG2 cells were detected by western blotting. (B and E) After 12 /24 h, the mRNA/protein levels of OAS-1, MX1, IFIT2, and IFIT3 were measured by RT-qPCR/western blotting, and the results are expressed as the fold change compared to the control (pSilencer-NC/pcDNA3). (C and F) The levels of HBsAg and HBeAg in the cell culture supernatants were detected by ELISA. Total cellular RNA, pregenomic RNA, HBV DNA, and cccDNA levels in HepG2.2.15 cells were analyzed by RT-qPCR. (G) Enhanced phosphorylation of STAT1 upon HBV-miR-3 overexpression in HepG2 cells treated with IFN-α (50 U/ml) for 24 h was determined by western blotting. (H) Co-transfection of HBV-miR-3 and SOCS5 in HepG2 cells. p-STAT1 and total STAT1 were detected by western blotting. Quantitative data are presented as mean ± SD from three independent experiments, and representative western blot images are shown. *P < 0.05, **P < 0.01, ***P < 0.001, ns = not significant.
HBV-miR-3 in exosomes affects macrophage polarization
Macrophages are highly heterogeneous cells that can rapidly alter their phenotype and function in response to microenvironmental signals, and studies have documented the flexibility of macrophage activation (Jiang, 2005; Mylonas et al., 2009; E et al., 2011). Here, we demonstrated that exosomes released from HBV-infected hepatocytes contained HBV-miR-3. To determine whether HBV-miR-3 has the ability to promote polarization of the M1 macrophage phenotype, THP-1 cells were treated with HBV-miR-3 exosomes for 48 h, and some classical M1 and M2 markers were then detected by RT-qPCR. Compared with those in the control group, elevated expression levels of MCP-1, NOS2, and CXCL10 (classical M1 markers) and decreased expression levels of CD206, Arg-1, and KLF4 (classical M2 markers) were observed in HBV-miR-3 exosome-treated THP-1 cells (Figure 5A). Western blotting also showed similar expression patterns of CXCL10 and KLF4 (Figure 5B). Consistent with the changes in the abovementioned surface markers, the macrophages treated with HBV-miR-3 exosomes secreted more IL-6 and less IL-10 than the control, as evaluated by ELISA (Figure 5C). To eliminate the effects of other regulatory molecules in the exosomes, we first transfected ASO-HBV-miR-3 into THP-1 cells. Twenty-four hours later, these cells were incubated with HBV-miR-3 exosomes for 72 h. The protein levels of CXCL10 and KLF4 (Figure 5D) and the secretion of IL-6 and IL-10 (Figure 5E) were not altered after blocking HBV-miR-3. Taken together, our results suggest that HBV-miR-3 stimulates macrophage differentiation into the M1 type.
Figure 5.

HBV-miR-3 in exosomes affects macrophage polarization. THP-1 cells were incubated with Huh7-derived HBV-miR-3 exosomes (100 μg/ml) or the control (100 μg/ml, Huh7-derived pcDNA3 exosomes) for further analyses. (A) After 48 h, RT-qPCR analyses were performed to determine the indicated macrophage differentiation-associated molecules, and the results are expressed as the fold change compared to the control. (B and C) After 96 h incubation, protein levels of CXCL10 and KLF4 were determined by western blotting, and levels of IL-6 and IL-10 secretion were evaluated by ELISA, respectively. (D and E) ASO-HBV-miR-3/ASO-NC were transfected into THP-1 cells. After 24 h, the THP-1 cells were incubated with HBV-miR-3 exosomes or the control for 72 h. Western blot and ELISA analyses were performed to measure CXCL10 and KLF4 expression and IL-6 and IL-10 secretion, respectively. (F) p-STAT1, total STAT1, and CXCL10 were detected by western blotting. (G) The expression levels of SOCS5 were detected by RT-qPCR and western blotting. (H–K) Macrophages were transfected with pshR-SOCS5/pSilencer-NC. Activation of STAT1 was assessed by western blotting (H), the polarization of macrophages was determined by RT-qPCR and western blotting by analyzing the M1 and M2 surface markers at 24 h (I, RT-qPCR) and 48 h post-transfection (J, western blot), and IL-6 and IL-10 secretion was quantified by ELISA at 48 h post-transfection (K). (L and M) p-STAT1 and total STAT1 were detected by western blotting (L), and the levels of IL-6 and IL-10 secretion were quantification by ELISA (M) at 48 h after co-transfection with HBV-miR-3 and SOCS5 in macrophages. Data are shown as mean ± SEM, n = 3; *P < 0.05, **P < 0.01, and ***P < 0.001.
To further investigate the molecular mechanism of HBV-miR-3 on the polarization of macrophages, we examined the expression and phosphorylation of STAT1 in macrophages treated with HBV-miR-3 exosomes (100 μg/ml). As shown in Figure 5F (left panel), HBV-miR-3 exosomes increased the phosphorylated STAT1 levels, when HBV-miR-3 was blocked, the phosphorylation of STAT1 was not altered (Figure 5F, middle panel). In addition, we treated THP-1 cells with 15 mM fludarabine (STAT1 activation inhibitor) 2 h prior to HBV-miR-3 exosome treatment. The stimulation of phosphorylated STAT1 and the enhancement of the macrophage M1 marker CXCL10 were both lost, implying that phosphorylation of STAT1 played an important role in the M1 polarization induced by HBV-miR-3 exosomes (Figure 5F, right panel). The SOCS family was recently reported to regulate the STAT-mediated activation of macrophages (Wang et al., 2014). Here, we showed reduced expression of SOCS5 in macrophages treated with HBV-miR-3 exosomes, whereas the expression of SOCS5 was not altered when HBV-miR-3 was blocked (Figure 5G). We silenced SOCS5 expression in macrophages using pshR-SOCS5 and observed that the expression of p-STAT1 and M1-related markers was increased and that the expression of M2-related markers was suppressed (Figure 5H–K). In addition, when THP-1 cells were co-transfected with HBV-miR-3 and SOCS5, overexpression of SOCS5 attenuated the effect of HBV-miR-3 on STAT1 phosphorylation and the secretion of IL-6 and augmented the secretion of IL-10 (Figure 5L and M). Altogether, these data indicate that HBV-miR-3 activates the STAT1 signaling pathway by downregulating SOCS5 and promoting the M1 macrophage polarization.
HBV-miR-3 induces IL-6 secretion by SOCS5-mediated EGFR ubiquitination in M1 macrophages
IL-6 is a key mediator of inflammation in the acute phase response of HBV infection and a cytokine secreted by M1 macrophages (Hösel et al., 2009). To assess the regulation of IL-6 production by HBV-miR-3, THP-1 cells were treated with 10 ng/ml LPS and 20 ng/ml IFN-γ to induce macrophage polarization into proinflammatory M1 phenotypes. Next, M1 macrophages were treated with HBV-miR-3 exosomes, which resulted in the increased secretion of IL-6 (Figure 6A), and ELISA analysis showed increased IL-6 expression upon overexpression of EGFR in M1 macrophages (Figure 6B). In addition, treatment with HBV-miR-3 exosomes significantly increased the EGFR levels in M1 macrophages as determined by RT-qPCR and western blotting (Figure 6C and D). SOCS5 has been reported to inhibit EGFR signaling in CHO cells (Kario et al., 2005), and our data showed that HBV-miR-3 could negatively regulate SOCS5; thus, we next investigated the interactions between SOCS5 and EGFR. Coimmunoprecipitation (Co-IP) and IF analyses showed that SOCS5 and EGFR were directly bound together and colocalized in the cytoplasm of M1 macrophages (Figure 6E and F). We further showed that the protein expression of EGFR was significantly decreased by overexpression of SOCS5, while the EGFR protein expression was increased upon silencing of SOCS5 in M1 macrophages (Figure 6G). We also found that ectopic expression of SOCS5 significantly promoted the ubiquitination and degradation of EGFR via the proteasome pathway in macrophages (Figure 6H), as well as decreased the expression of IL-6 and vice versa (Figure 6I). In addition, HBV-miR-3 exosomes could rescue the effect of pshR-EGFR on EGFR expression (Figure 6J) and IL-6 production (Figure 6K). It has been reported that IL-6 can inhibit HBV transcription by regulating HBV cccDNA (Palumbo et al., 2015). Our results above also indicated the effect of SOCS5 and HBV-miR-3 on HBV cccDNA (Figures 2G, 4C and F). Taken together, these findings demonstrate that in M1 macrophages, HBV-miR-3 promotes EGFR expression by suppressing SOCS5-mediated EGFR degradation via the ubiquitination–proteasome pathway and subsequently stimulates the secretion of IL-6 to regulate HBV replication.
Figure 6.

HBV-miR-3 induces IL-6 secretion by SOCS5-mediated EGFR ubiquitination in M1 macrophages. (A) M1 macrophages were incubated with Huh7-derived HBV-miR-3 exosomes (100 μg/ml) or the control for further analyses. After 72 h, the levels of IL-6 were determined by ELISA. (B) ELISA analysis of IL-6 secreted by M1 macrophages after transfection with either pcDNA3 or EGFR. (C and D) M1 macrophages were treated with HBV-miR-3 exosomes (100 μg/ml) or the control. The levels of EGFR were determined by RT-qPCR/western blotting. (E) Co-IP experiments exploring the interactions between EGFR and SOCS5 in M1 macrophages. (F) Colocalization of EGFR and SOCS5 in M1 macrophages. Scale bar, 20 μm. (G) Western blot showing the impact of SOCS5 on EGFR expression at 48 h after transfection in M1 macrophages. (H) Western blot detecting the ubiquitination of EGFR in M1 macrophages after transfection with Myc-SOCS5 and the controls. (I) pSOCS5 and pshR-SOCS5 were transfected into M1 macrophages. After 72 h, the levels of IL-6 were determined by ELISA. (J and K) pshR-EGFR/pSilencer-NC were transfected into M1 macrophages. After 24 h, M1 macrophages were incubated with Huh7-derived HBV-miR-3 exosomes (100 μg/ml) or the control. The expression levels of EGFR were determined by western blotting (J). The levels of IL-6 were determined by ELISA (K). Data are presented as mean ± SD; *P < 0.05, **P < 0.01.
Discussion
The virus–host immune system interaction is a key process determining the status of viral infection. Viruses not only evade cellular immune responses by various strategies but also restrict the number of virions in infected cells to maintain a persistent replication status. Many viruses and viral proteins reportedly adopt the host miRNA-mediated regulation of antiviral pathways to alleviate their pathogenesis (Sharma et al., 2016). Virus-encoded miRNAs can also regulate the gene expression profiles of cells or of the virus itself (Grundhoff and Sullivan, 2011). Our laboratory first reported an HBV-encoded miRNA (called HBV-miR-3), which is located at nucleotides (nt) 373 to 393 of the HBV genome and generated from 3.5-kb, 2.4-kb, and 2.1-kb HBV (Yang et al., 2017). Currently, hepatitis B patients can be treated well in the early stage regardless of whether the virus is in its active phase, and liver enzymes are maintained at normal levels in most patients. Since the expression of HBV-miR-3 is highly correlated with the activity of HBV, we speculate that HBV-miR-3 expression maintains a certain correlation with liver enzymes even if liver enzyme changes are not obvious. In addition, the collection of only 20 samples limited our results. In the current study, we aimed to elucidate the interactions between HBV-miR-3 and innate immunity, and we made three key observations: (i) HBV-miR-3 enhances the IFN-induced anti-HBV effect, (ii) HBV-miR-3 in exosomes affects macrophage polarization, and (iii) HBV-miR-3 induces IL-6 production in M1 macrophages.
In previous clinical studies on IFN treatment in HBV-infected patients, the genotype and viral load of HBV were considered the key indicators for IFN therapeutic efficacy (Kao et al., 2000; Simon et al., 2000). Although many studies have been focused on HBV, studies on HBV-encoded microRNAs that may alter IFN antiviral effects have not been performed. Moreover, only a few studies on the correlation between HBV and SOCS proteins have been performed. Here, we showed that HBV-miR-3 suppressed SOCS5 expression by binding to its 3′UTR, which led to increased expression of IFN-stimulated genes and restrained HBV replication. Based on this study, we hypothesize that the regulation of HBV-miR-3 can be used to explain the characteristics of HBV chronicity, latency, and low immunogenicity. On the other hand, our findings also provide a new direction for the treatment of hepatitis B patients.
Extracellular vesicles, including exosomes, have important functions in intercellular communication (Schorey and Harding, 2016). Here, we demonstrated that exosomes released from HBV-infected hepatocytes contained HBV-miR-3, and HBV-miR-3 exosomes could stimulate macrophage differentiation into the M1 type. Notably, SOCS5 was recently shown to contribute to M1 polarization by binding IL-4Rα and blocking STAT6 phosphorylation (Mccormick and Heller, 2015). Here, we demonstrate that HBV-miR-3 exosomes can inhibit the expression of SOCS5 in macrophages and that the loss of SOCS5 expression might direct the proinflammatory M1 effect of macrophages by activating the JAK/STAT pathway. To date, miR-125, miR-155, miR-378, miR-9, miR-21, miR-146, miR-147, and miR-187 have been reported to be involved in controlling macrophage activation (Squadrito et al., 2013). Macrophage polarization is involved in various signaling pathways and in the actions of transcription factors, and polarized macrophages are associated with the occurrence and development of many diseases (Murray et al., 2014; Zhou et al., 2014). Therefore, research on the plasticity mechanism of macrophage polarization is of great significance.
IL-6 is a pleiotropic cytokine-mediated inflammatory mediator that plays a role in the acute phase response of the liver (Tacke et al., 2009). A recent study showed that IL-6 production in Kupffer cells is strictly dependent on EGFR expression (Lanaya et al., 2014). Our results also revealed increased IL-6 expression upon overexpression of EGFR in M1 macrophages. Here, we provided solid evidence that HBV-miR-3 promotes EGFR expression by suppressing SOCS5-mediated ubiquitination and subsequently stimulates the secretion of IL-6 to inhibit HBV replication. Human macrophages or THP cells were reported to respond to HBV with a rapid induction of TNF-α and IL-6 (Cheng et al., 2017). At this point, the author did not explain which viral component(s) was responsible for the activation of macrophages. Based on our findings, we speculated that HBV-miR-3 might play an important role. First, HBV-miR-3 promoted the differentiation of THP-1 cells into M1 macrophages (one of the main cell types that secrete IL-6). Second, HBV-miR-3 enhanced the expression of EGFR by downregulating the expression of SOCS5 in M1-type macrophages, thus promoting the secretion of IL-6.
In the present study, we showed that the expression of HBV-encoded HBV-miR-3 increased in an infection time- and viral load-dependent manner during HBV infection. Functionally, HBV-miR-3 enhanced the IFN-induced anti-HBV effect, stimulated the differentiation of macrophages to the M1 phenotype and promoted the secretion of IL-6 in M1 macrophages. Mechanically, we identified SOCS5 as a direct target of HBV-miR-3. Downregulation of SOCS5 activated STAT1, resulting in the enhanced transcription of IFN-stimulated genes in HBV-infected liver cancer cells. On the other hand, exosomes containing HBV-miR-3 promoted the M1 polarization of macrophages via the SOCS5/STAT1 pathway. In addition, HBV-miR-3 induced IL-6 secretion by increasing EGFR expression via inhibiting the SOCS5-mediated ubiquitination-proteasome degradation of EGFR (Figure 7). To the best of our knowledge, we are the first to report that HBV-miR-3 restricts HBV replication by modulating the innate immune response and helps to reduce liver cell injury, which may contribute to the development of persistent infections in HBV patients. In addition, SOCS5, a direct target gene of HBV-miR-3, not only regulates the antiviral effect of IFNs in hepatocytes but also participates in the polarization of macrophages and the secretion of cytokines. Thus, we reveal a new mechanism by which HBV-encoded miRNAs control the process of HBV replication during infection by activating innate immune responses, which might provide a novel target for the treatment of hepatitis B. Although our in vitro data cannot completely reflect the natural occurrences in vivo, they can still indicate the roles of miRNAs in HBV immunopathogenesis and suggest potential antiviral strategies based on targeting inhibitory immune miRNAs.
Figure 7.
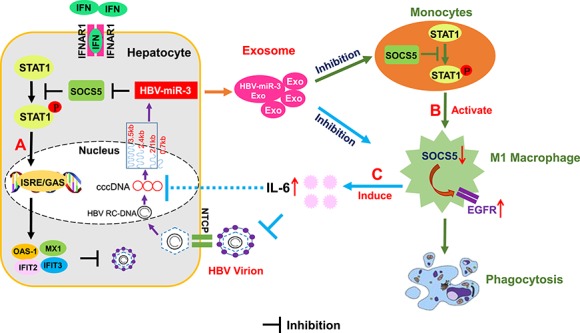
Diagram of the HBV-miR-3 activation of innate immunity to restrain HBV replication. (A) An HBV-encoded miRNA (HBV-miR-3) decreases the expression of SOCS5 in hepatocytes, leading to activation of the JAK/STAT pathway, thus initiating the transcription of hundreds of IFN-stimulated response element (ISRE) and enhancing the IFN-induced anti-HBV effect. (B) HBV-miR-3 activates the STAT1 signaling pathway by downregulating SOCS5 and thus promotes macrophage differentiation into the M1 type, which allows the elimination of physiologically aging, infected, or cancerous cells. (C) In M1 macrophages, HBV-miR-3 promotes EGFR expression by inhibiting SOCS5, thereby regulating IL-6 secretion and inhibiting HBV replication.
Materials and methods
Human serum samples
Serum samples from 20 hepatitis B patients were obtained from Tianjin Medical University General Hospital. All participants were adults. Written informed consent was obtained from each patient, and ethical approval for this work was issued by the Ethics Committee of Tianjin Medical University. Detailed information was listed in Supplementary Table S2.
Cell cultures and transfections
The human hepatoma cell lines Huh7 and HepG2.2.15 and the monocytic cell line THP-1 were purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). HepG2, HepG2.2.15, and Huh-7 cells were cultured and transfected with plasmids as described previously (Yang et al., 2017). For the experiments shown in Figures 2 and 4, human IFN-α was added to the cells after transfection. Recombinant human IFN-α was produced by Changsheng Gene Pharm Ltd. THP-1 cells were maintained in RPMI 1640 medium (Invitrogen) containing 10% FBS. Cultures were incubated in a humidified atmosphere at 37°C with 5% CO2. THP-1 cells were transfected with GenePORTER® 2 Transfection Reagent (Genlantis) according to the manufacturer’s recommendations. To induce differentiation into macrophages, THP-1 cells (1 × 106/well) were incubated with 100 ng/ml phorbol 12-myristate 13-acetate (PMA, Sigma-Aldrich) for 48 h. To mimic a proinflammatory environment and consequently promote M1 polarization, THP-1 cells were stimulated with 100 ng/ml lipopolysaccharide (LPS) and 20 ng/ml IFN-γ (Sigma-Aldrich).
Establishment of the HepG2-NTCP cell line and infection of HepG2-NTCP cells with HBV
To establish an HBV infectious stable line, HepG2 cells were transfected with pcDNA3-NTCP and selected with G418 according to a previously described procedure (Yan et al., 2012). Screened single HepG2-NTCP clones were used to verify NTCP expression by western blotting and IF microscopy (Zeiss). HepG2-NTCP cells (6 × 104/well) were seeded into 48-well plates in 200 μl of DMEM with G418. Two hundred microliters of PMM medium was exchanged after 3 h. For HBV infection, cells were incubated with 300 HBV genomes/cell in medium containing 5% PEG 8000 (Sigma-Aldrich) for 16 h. The viral mixture was discarded, and the cells were washed twice with 200 μl of DMEM each and then transferred into PMM medium for further incubation. Supernatants were collected at 3, 5, and 7 days post-infection.
RT-qPCR
Total RNA was extracted using TRIzol reagent (Invitrogen) according to the manufacturer’s instructions. cDNA was synthesized using Moloney murine leukemia virus reverse transcriptase (Promega). qPCR was performed using SYBR Mixture (CWBio). β-actin and U6 snRNA were used as the endogenous controls for mRNA and miRNA, respectively. HBV core particle DNA and cccDNA were extracted and detected by qPCR as previously described (Palumbo et al., 2015). Primers and oligonucleotides used were detailed in Supplementary Table S1.
Core particle-associated HBV DNA purification and quantitation
To purify HBV DNA from intracellular core particles, transfected cells were washed once with ice-cold phosphate-buffered saline (PBS) and lysed in lysis buffer A (10 mM Tris–HCl, pH 7.4, 1 mM EDTA, 50 mM NaCl, and 1% NP-40) for 30 min. Nuclei were pelleted by centrifugation for 5 min at 10000 g. The supernatant was treated with 0.1 mg/ml DNase I and 0.1 mM MgCl2 for 30 min at 37°C, followed by inactivation of the enzyme with 1 mM EDTA. Viral core particles were precipitated in a 0.8 M NaCl/8% polyethylene glycol solution at 4°C for 1 h, centrifuged (10 min at 10000 g), resuspended in a mixture containing 10 mM Tris–HCl, 100 mM NaCl, 1 mM EDTA, and 1% SDS with 0.5 mg/ml proteinase K, and incubated at 56°C for 2 h. Viral DNA released from the lysed core particles was extracted with phenol–chloroform (1:1), precipitated with isopropanol and quantified by qPCR.
HBV cccDNA quantification
HepG2 or Huh7 cells were collected at the indicated time points after transfection and resuspended in lysis buffer A at 4°C for 10 min. The whole lysates were centrifuged for 5 min at 10000 g; pelleted nuclei were resuspended in lysis buffer B (10 mM Tris–HCl, 10 mM EDTA, 150 mM NaCl, 0.5% SDS, and 0.5 mg/ml proteinase K), sonicated by one or two pulses at 80% power, and incubated overnight at 37°C. Nucleic acids were then extracted by phenol–chloroform (1:1) and ethanol precipitation. Five hundred nanogram aliquots of each extracted DNA sample were treated with 10 U of Plasmid-Safe DNase I (Epicentre, Inc.) at 37°C for 45 min, and the enzyme was inactivated by heating to 70°C for 30 min.
ELISA
Secretions of HBsAg, HBeAg, IL-6, IL-10, IFN-α, and IFN-β in the culture supernatants were detected by ELISA kits according to the manufacturers’ protocols. The ELISA detection kits used in this study were purchased from InTec Products (for HBsAg and HBeAg), Ray Biotech (for IL-6 and IL-10), and JYM (for IFN-α and IFN-β). The absorbance was determined using a microtiter plate reader (BioTek Instruments).
Plasmid constructions
The coding sequences (CDSs) of SOCS5 and EGFR were amplified from the cDNA of gastric cancer (GC) cells by PCR and cloned into the Flag/pcDNA3 or myc/pcDNA3 vectors between the BamHI/Xhol sites or KpnI/Xhol sites, respectively. The shRNA vector targeting SOCS5 (pshR-SOCS5) was constructed by annealing synthesized oligos into pSilencer 2.1-U6 neo between the HindIII and BamHI sites. The 3′UTR fragments of the SOCS5 gene containing the putative HBV-miR-3 binding site and its respective mutated form were inserted into pcDNA3-EGFP vectors between the BamHI and EcoRI sites by oligo annealing and ligation. Primers and oligonucleotides used were detailed in Supplementary Table S1.
Fluorescent reporter assay
Huh7 and HepG2-NTCP cells were cotransfected with HBV-miR-3 (400 ng) or ASO-HBV-miR-3 (50 nM) together with pcDNA3/EGFP-SOCS5 3′UTR WT or mutated reporter plasmids (0.2 μg). pDsRed2-N1 (Clontech) was used for normalization. After 48 h, the cells were lysed, and the intensity of EGFP and RFP fluorescence was determined using an F-4500 fluorescence spectrophotometer (Hitachi).
Western blot analysis
Transfected cells were lysed in RIPA buffer. Proteins were separated by 10% SDS-PAGE, transferred to nitrocellulose membranes, and then detected using the appropriate antibodies. The rabbit anti-GAPDH, anti-SOCS5, anti-OAS-1, anti-MX1, anti-IFIT2, anti-IFIT3, anti-STAT1, anti-KLF4, and anti-iNOS antibodies were purchased from Saierbio. The p-STAT1 (Tyr701) and Flag antibodies were purchased from Abcam.
Exosome isolation
To prepare exosomes, the culture supernatants of HepG2-NTCP and Huh7 cells stably expressing HBV-miR-3 and pcDNA3 were harvested and extracted as previously described (Yang et al., 2017).
Co-IP and ubiquitination analysis
M1 macrophages were transfected with SOCS5 and EGFR. At 48 h post-transfection, M1 macrophages cells were harvested and analyzed according to a previously described protocol (Zhu et al., 2014).
IF analysis
Cells were fixed with 4% paraformaldehyde for 10 min, permeabilized with 0.05% Triton X-100, and blocked in 10% donkey serum at room temperature for 2 h. After overnight incubation with the primary antibody at 4°C, the fluorescence-labeled secondary antibody was added, and the mixture was incubated for 2 h at room temperature in the dark. The cells were mounted with DAPI Fluoromount-G (SouthernBiotech), and images were captured with an LSM 700 confocal microscope (Zeiss).
Statistical analysis
All statistical analyses were performed using GraphPad Prism 6 (GraphPad Software Inc.). Comparisons between the treatment and control were made using the paired Student’s t-test. The correlations between HBV-miR-3 and the indicated hepatitis-related factors were analyzed by Pearson correlation. All analyses were two-tailed, and P ≤ 0.05 was considered statistically significant.
Supplementary Material
Funding
This work was supported in part by the National Natural Science Foundation of China (91629302, 81830094, 81572790, 31270818, and 81773002) and the Natural Science Foundation of Tianjin (19JCZDJC35900 and 16JCYBJC42400).
Conflicts of interest: none declared.
References
- Bartel D.P. (2004). MicroRNAs: genomics, biogenesis, mechanism and function. Cell 116, 281–297. [DOI] [PubMed] [Google Scholar]
- Bouezzedine F., Fardel O., and Gripon P. (2015). Interleukin 6 inhibits HBV entry through NTCP down regulation. Virology 481, 34–42. [DOI] [PubMed] [Google Scholar]
- Cheng X., Xia Y., Serti E., et al. (2017). Hepatitis B virus evades innate immunity of hepatocytes but activates macrophages during infection. Hepatology 66, 1779–1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- E S., A P., FJ B., et al. (2011). Activin A skews macrophage polarization by promoting a proinflammatory phenotype and inhibiting the acquisition of anti-inflammatory macrophage markers. Blood 117, 5092–5101. [DOI] [PubMed] [Google Scholar]
- Fu M., Wang B., Chen X., et al. (2018). MicroRNA gga-miR-130b suppresses infectious bursal disease virus replication via targeting of the viral genome and cellular suppressors of cytokine signaling 5. J. Virol. 92, e01646-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon S., and Taylor P.R. (2005). Monocyte and macrophage heterogeneity. Nat. Rev. Immunol. 5, 953–964. [DOI] [PubMed] [Google Scholar]
- Gottwein E., and Cullen B.R. (2008). Viral and cellular microRNAs as determinants of viral pathogenesis and immunity. Cell Host Microbe 3, 375–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundhoff A., and Sullivan C.S. (2011). Virus-encoded microRNAs. Virology 411, 325–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidotti L.G., and Chisari F.V. (2006). Immunobiology and pathogenesis of viral hepatitis. Annu. Rev. Pathol. 1, 23–61. [DOI] [PubMed] [Google Scholar]
- Heinz D., Peters M., Prange R., et al. (2010). Possible role of human interleukin-6 and soluble interleukin-6 receptor in hepatitis B virus infection. J. Viral Hepat. 8, 186–193. [DOI] [PubMed] [Google Scholar]
- Hösel M., Quasdorff M., Wiegmann K., et al. (2009). Not interferon, but interleukin-6 controls early gene expression in hepatitis B virus infection. Hepatology 50, 1773–1782. [DOI] [PubMed] [Google Scholar]
- Jiang C.X. (2005). Macrophages sequentially change their functional phenotype in response to changes in microenvironmental influences. J. Immunol. 175, 342–349. [DOI] [PubMed] [Google Scholar]
- Kao J.H., Chen P.J., Lai M.Y., et al. (2000). Hepatitis B genotypes correlate with clinical outcomes in patients with chronic hepatitis B. Gastroenterology 118, 554–559. [DOI] [PubMed] [Google Scholar]
- Kario E., Marmor M.D., Adamsky K., et al. (2005). Suppressors of cytokine signaling 4 and 5 regulate epidermal growth factor receptor signaling. J. Biol. Chem. 280, 7038. [DOI] [PubMed] [Google Scholar]
- Kuo T.M., Hu C.P., Chen Y.L., et al. (2009). HBV replication is significantly reduced by IL-6. J. Biomed. Sci. 16, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan T., Chang L., Wu L., et al. (2015). IL-6 plays a crucial role in HBV infection. J. Clin. Transl. Hepatol. 3, 271–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanaya H., Natarajan A., Komposch K., et al. (2014). EGFR has a tumour-promoting role in liver macrophages during hepatocellular carcinoma formation. Nat. Cell Biol. 16, 972–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linossi E.M., Chandrashekaran I.R., Kolesnik T.B., et al. (2013). Suppressor of cytokine signaling (SOCS) 5 utilises distinct domains for regulation of JAK1 and interaction with the adaptor protein Shc-1. PLoS One 8, e70536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahajan V.S., Adam D., and Jianzhu C. (2009). Virus-specific host miRNAs: antiviral defenses or promoters of persistent infection? Trends Immunol. 30, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez F.O., Sica A., Mantovani A., et al. (2008). Macrophage activation and polarization. Front. Biosci. 13, 453–461. [DOI] [PubMed] [Google Scholar]
- Mccormick S.M., and Heller N.M. (2015). Regulation of macrophage, dendritic cell, and microglial phenotype and function by the SOCS proteins. Front. Immunol. 6, 549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mcmahon B.J. (2009). The natural history of chronic hepatitis B virus infection. Hepatology 49, S45–S55. [DOI] [PubMed] [Google Scholar]
- Mosser D.M., and Edwards J.P. (2008). Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 8, 958–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray P.J. (2015). Macrophage activation and polarization. Semin. Immunol. 27, 235–236. [DOI] [PubMed] [Google Scholar]
- Murray P.J., Allen J.E., Biswas S.K., et al. (2014). Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity 41, 14–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray P.J., and Wynn T.A. (2011). Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 11, 723–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mylonas K.J., Nair M.G., Prietolafuente L., et al. (2009). Alternatively activated macrophages elicited by helminth infection can be reprogrammed to enable microbial killing. J. Immunol. 182, 3084–3094. [DOI] [PubMed] [Google Scholar]
- Nicholson S.E., and Hilton D.J. (1998). The SOCS proteins: a new family of negative regulators of signal transduction. J. Leukoc. Biol. 63, 665–668. [DOI] [PubMed] [Google Scholar]
- Palumbo G.A., Scisciani C., Pediconi N., et al. (2015). Correction: IL6 inhibits HBV transcription by targeting the epigenetic control of the nuclear cccDNA minichromosome. PLoS One 10, e0142599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardo M., Bartolomé J., and Carreño V. (2007). Current therapy of chronic hepatitis B. Arch. Med. Res. 38, 661–677. [DOI] [PubMed] [Google Scholar]
- Rincon M., and Irvin C.G. (2012). Role of IL-6 in asthma and other inflammatory pulmonary diseases. Int. J. Biol. Sci. 8, 1281–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schorey J.S., and Harding C.V. (2016). Extracellular vesicles and infectious diseases: new complexity to an old story. J. Clin. Invest. 126, 1181–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma N., Kumawat K.L., Rastogi M., et al. (2016). Japanese encephalitis virus exploits the microRNA-432 to regulate the expression of suppressor of cytokine signaling (SOCS) 5. Sci. Rep. 6, 27685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon K., Rotter K., Zalewska M., et al. (2000). HBV-DNA level in blood serum as a predictor of good response to therapy with interferon-α-2b of patients with chronic hepatitis B. Med. Sci. Monit. 6, 971–975. [PubMed] [Google Scholar]
- Smith P.L., Lombardi G., and Foster G.R. (2005). Type I interferons and the innate immune response--more than just antiviral cytokines. Mol. Immunol. 42, 869–877. [DOI] [PubMed] [Google Scholar]
- Squadrito M.L., Etzrodt M., De P.M., et al. (2013). MicroRNA-mediated control of macrophages and its implications for cancer. Trends Immunol. 34, 350–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan C.S., Grundhoff A.T., Satvir T., et al. (2005). SV40-encoded microRNAs regulate viral gene expression and reduce susceptibility to cytotoxic T cells. Nature 435, 682–686. [DOI] [PubMed] [Google Scholar]
- Tacke F., Luedde T., and Trautwein C. (2009). Inflammatory pathways in liver homeostasis and liver injury. Clin. Rev. Allergy Immunol. 36, 4–12. [DOI] [PubMed] [Google Scholar]
- Thio C.L. (2010). Review of hepatitis B therapeutics. Clin. Infect. Dis. 51, 1201–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang N., Liang H., and Zen K. (2014). Molecular mechanisms that influence the macrophage M1–M2 polarization balance. Front. Immunol. 5, 614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia C., Liu Y., Chen Z., et al. (2015). Involvement of interleukin 6 in hepatitis B viral infection. Cell. Physiol. Biochem. 37, 677–686. [DOI] [PubMed] [Google Scholar]
- Xu L., Yin W., Sun R., et al. (2014). Kupffer cell-derived IL-10 plays a key role in maintaining humoral immune tolerance in hepatitis B virus-persistent mice. Hepatology 59, 443–452. [DOI] [PubMed] [Google Scholar]
- Yan G.Z.H., Xu G.W., He W.H., et al. (2012). Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. eLife 1, e00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X., Li H., Sun H., et al. (2017). Hepatitis B virus-encoded miRNA controls viral replication. J. Virol. 91, pii: e01919-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G.L., Li Y.X., Zheng S.Q., et al. (2010). Suppression of hepatitis B virus replication by microRNA-199a-3p and microRNA-210. Antiviral Res. 88, 169–175. [DOI] [PubMed] [Google Scholar]
- Zhou D., Huang C., Lin Z., et al. (2014). Macrophage polarization and function with emphasis on the evolving roles of coordinated regulation of cellular signaling pathways. Cell. Signal. 26, 192–197. [DOI] [PubMed] [Google Scholar]
- Zhu Y., Zhang Y., Sui Z., et al. (2014). USP14 de-ubiquitinates vimentin and miR-320a modulates USP14 and vimentin to contribute to malignancy in gastric cancer cells. Oncotarget 8, 48725–48736. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.