

Abstract
Toll-like receptor 3 (TLR3)-mediated signaling is important for host defense against RNA virus. Upon viral RNA stimulation, toll and interleukin-1 receptor domain-containing adaptor inducing IFN-β (TRIF) is recruited to TLR3 and then undergoes oligomerization, which is required for the recruitment of downstream molecules to transmit signals. Here, we identified zinc finger CCHC-type containing 3 (ZCCHC3) as a positive regulator of TLR3-mediated signaling. Overexpression of ZCCHC3 promoted transcription of downstream antiviral genes stimulated by the synthetic TLR3 ligand poly(I:C). ZCCHC3-deficiency markedly inhibited TLR3- but not TLR4-mediated induction of type I interferons (IFNs) and proinflammatory cytokines. Zcchc3−/− mice were more resistant to poly(I:C)- but not lipopolysaccharide-induced inflammatory death. Mechanistically, ZCCHC3 promoted recruitment of TRIF to TLR3 after poly(I:C) stimulation. Our findings reveal that ZCCHC3 plays an important role in TLR3-mediated innate immune response by promoting the recruitment of TRIF to TLR3 after ligand stimulation.
Keywords: TLR3, TRIF, ZCCHC3, innate immune response, signaling
Graphical Abstract
Graphical Abstract.

Introduction
Toll-like receptors (TLRs) are evolutionarily conserved pattern recognition receptors (PRRs), which play important roles in host defense against a wide variety of pathogens (Kawai and Akira, 2010, 2011). TLRs contain an extracellular domain consisting of leucine rich repeats, a transmembrane domain, and a toll and interleukin-1 receptor (TIR) domain that is responsible for mediating protein–protein interaction (Jin and Lee, 2008). Most TLRs signal through the adaptor MyD88, but TLR3 signals through TIR domain-containing adaptor inducing IFN-β (TRIF), while TLR4 utilizes both MyD88 and TRIF for signaling. TLR3 is expressed mostly in immune cells and is responsible for recognition of extracellular viral dsRNA as well as its synthetic analog poly(I:C) (Alexopoulou et al., 2001; Yamamoto et al., 2003). Upon binding to its ligand, TLR3 is phosphorylated by protein tyrosine kinases and then recruit the critical downstream adaptor protein TRIF (Johnsen et al., 2006; Garcia-Cattaneo et al., 2012; Yamashita et al., 2012; Toscano et al., 2013). The activated TRIF dissociates from TLR3 and forms a speckle-like structure, which acts as a platform for the recruitment of TRAF3-TBK1/IKK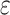 and TRAF6-IKKα/β/γ complexes, leading to activation of transcriptional factors including IRF3 and NF-κB. These transcription factors collaborate to induce transcription of downstream antiviral genes, such as type I interferons (IFNs) and inflammatory cytokines, to induce innate immune and inflammatory responses as well as facilitate adaptive immunity (Sharma et al., 2003; Yamamoto, et al., 2003; Takeuchi and Akira, 2010; Qu et al., 2015; Huang et al., 2019).
and TRAF6-IKKα/β/γ complexes, leading to activation of transcriptional factors including IRF3 and NF-κB. These transcription factors collaborate to induce transcription of downstream antiviral genes, such as type I interferons (IFNs) and inflammatory cytokines, to induce innate immune and inflammatory responses as well as facilitate adaptive immunity (Sharma et al., 2003; Yamamoto, et al., 2003; Takeuchi and Akira, 2010; Qu et al., 2015; Huang et al., 2019).
Zinc finger CCHC-type containing 3 (ZCCHC3) is critical for regulation of innate immune responses to cytosolic dsRNA and dsDNA (Lian et al., 2018a, b). In this study, we found that ZCCHC3 positively regulates TLR3-mediated signaling pathway. ZCCHC3-deficiency inhibits TLR3- but not TLR4-mediated innate immune and inflammatory responses. Biochemical analysis indicates that ZCCHC3 promotes the recruitment of TRIF to TLR3 and facilitates TRIF oligomerization. Our findings reveal that ZCCHC3 is a critical component in TLR3-mediated innate immune response.
Results
ZCCHC3 positively regulates poly(I:C)-triggered signaling
Our previous studies have revealed that ZCCHC3 is important for efficient innate immune responses to RNA and DNA viruses by acting as co-receptors of the cytosolic RIG-I, MDA5, and cGAS sensors (Lian et al., 2018a, b). Since the endosomal TLR3 also recognizes viral RNA, we wondered whether ZCCHC3 is involved in TLR3-mediated innate immune and inflammatory responses. Reporter assays indicated that overexpression of ZCCHC3 and its truncations potentiated poly(I:C)-induced activation of the IFN-β promoter and ISRE in a dose-dependent manner in HEK293 cells stably expressing TLR3 (293-TLR3 cells) (Figure 1A; Supplementary Figure S1A). Quantitative PCR (qPCR) experiments indicated that overexpression of ZCCHC3 potentiated poly(I:C)-induced transcription of IFNB1, ISG56, and IL6 genes in HT1080 cells, which express endogenous TLR3 (Yamashita et al., 2012) (Supplementary Figure S1B). These results suggest that overexpression of ZCCHC3 potentiates TLR3-mediated signaling.
Figure 1.

ZCCHC3 positively regulates poly(I:C)-triggered signaling. (A) ZCCHC3 promotes poly(I:C)-induced activation of the IFN-β promoter and ISRE in a dose-dependent manner. 293-TLR3 cells (1 × 105) were transfected with the IFN-β promoter or ISRE reporter plasmids (0.1 μg) and increased amounts of ZCCHC3 plasmids (5, 10, 20 ng) for 18 h, and then left untreated or treated with poly(I:C) (20 μg/ml) for 6 h before luciferase assays. (B) Efficiencies of ZCCHC3-RNAi plasmids on ZCCHC3 levels. 293 cells (4 × 105) were transfected with ZCCHC3-RNAi and control RNAi plasmids for 24 h before immunoblotting. (C) Effects of ZCCHC3 knockdown on poly(I:C)-induced activation of the IFN-β promoter and ISRE. 293-TLR3 (1 × 105) cells were transfected with the indicated luciferase reporter and RNAi plasmids. Aftre 20 h, cells were treated with poly(I:C) (20 μg/ml) or left untreated for 6 h before luciferase assays (**P < 0.01, ***P < 0.001). (D) Effects of ZCCHC3 knockdown on poly(I:C)-induced transcription of downstream genes in 293-TLR3 cells. The cells (4 × 105) were transduced with control or ZCCHC3-RNAi by retroviral-mediated gene transfer, and then either untreated or treated with poly(I:C) (20 μg/ml) for the indicated times before qPCR experiments (*P < 0.05, **P < 0.01, ***P < 0.001). (E) Effects of ZCCHC3 knockdown on LPS-induced transcription of downstream genes in 293-TLR4 cells. The cells (4 × 105) were transduced with control or ZCCHC3-RNAi by retroviral-mediated gene transfer, and then either untreated or treated with LPS (200 ng/ml) for the indicated times before qPCR experiments. (F) Effects of ZCCHC3 knockdown on poly(I:C)-induced phosphorylation of TBK1, p65, and IRF3. ZCCHC3-RNAi and control 293-TLR3 cells (4 × 105) were left untreated or treated with poly(I:C) (50 μg/ml) for the indicated times before immunoblotting. The line graphs show the relative intensities of the indicated phosphorylated proteins, which were quantitated by densitometry using ImageJ and normalized by their respective total protein levels. (G) Effects of ZCCHC3 knockdown on LPS-induced phosphorylation of TBK1 and p65. ZCCHC3-RNAi and control 293-TLR4 cells (4 × 105) were left untreated or treated with LPS (200 ng/ml) for the indicated times before immunoblotting.
We next determined the roles of endogenous ZCCHC3 in TLR3-mediated signaling by RNAi-mediated knockdown experiments. We constructed three RNAi plasmids for ZCCHC3. Both #1 and #3 RNAi plasmids could markedly inhibit the expression of endogenous ZCCHC3 in HEK293 cells (Figure 1B). Knockdown of ZCCHC3 inhibited poly(I:C)-induced activation of the IFN-β promoter and ISRE in 293-TLR3 cells (Figure 1C). qPCR experiments indicated that knockdown of ZCCHC3 inhibited poly(I:C)-induced transcription of IFNB1, ISG56, and CXCL10 genes in 293-TLR3 cells (Figure 1D) and HT1080 cells (Supplementary Figure S1C). In similar experiments, knockdown of ZCCHC3 had no marked effects on lipopolysaccharide (LPS, a ligand for TLR4)-induced transcription of IFNB1, TNFα, and CXCL10 genes in HEK293 cells stably expressing TLR4 (293-TLR4 cells) (Figure 1E). Consistently, phosphorylation of TBK1, IRF3, and p65 induced by poly(I:C), which are hallmarks of TBK1, IRF3, and NF-κB activation, was impaired in ZCCHC3-RNAi 293-TLR3 or HT1080 cells compared with their respective control cells (Figure 1F; Supplementary Figure S1D). In contrast, phosphorylation of TBK1 and p65 induced by LPS was comparable between ZCCHC3-knockdown and control 293-TLR4 cells (Figure 1G). These results suggest that ZCCHC3 positively regulates TLR3-mediated signaling pathway.
Zcchc3-deficiency impairs TLR3-mediated response in mice
To further investigate the roles of ZCCHC3 in vivo, we utilized Zcchc3-deficient mice. qPCR experiments indicated that transcription of downstream genes including Ifnb1, Isg56, and Il6 following treatment with poly(I:C) but not LPS was significantly inhibited in Zcchc3−/− mouse bone-marrow-derived macrophages (BMDMs) (Figure 2A), mouse bone-marrow-derived dendrite cells (BMDCs) (Figure 2B), and mouse lung fibroblasts (MLFs) (Supplementary Figure S2A). However, the transcription of downstream genes triggered by monophosphoryl lipid A (MPLA), a derivate of LPS that mainly induces TRIF- but not MyD88-dependent signaling, peptidoglycan (PGN; a ligand for TLR2), or R848 (a ligand for TLR7/8) was comparable between Zcchc3+/+ and Zcchc3−/− BMDCs (Figure 2C). Consistently, Zcchc3-deficiency inhibited phosphorylation of Tbk1 and p65 induced by poly(I:C) but not LPS in BMDCs (Figure 2D), BMDMs (Supplementary Figure S2B), and MLFs (Supplementary Figure S2C). Collectively, these data suggest that Zcchc3 specifically potentiates TLR3-mediated signaling in mouse primary immune cells and fibroblasts.
Figure 2.

Zcchc3-deficiency impairs TLR3-mediated signaling in primary mouse cells. (A and B) Effects of Zcchc3-deficiency on poly(I:C)- or LPS-induced transcription of Ifnb1, Isg56, and Il6 in BMDMs (A) and BMDCs (B). Zcchc3+/+ and Zcchc3−/− cells (3 × 105) were treated with poly(I:C) (20 μg/ml) or LPS (50 ng/ml) for the indicated times before qPCR was performed (*P < 0.05, **P < 0.01). (C) Effects of Zcchc3-deficiency on MPLA-, PGN-, or R848-induced transcription of downstream genes in BMDCs. Zcchc3+/+ and Zcchc3−/− cells (3 × 105) were treated with MPLA (200 ng/ml), PGN (20 μg/ml), or R848 (40 μM) for the indicated times before qPCR was performed. (D) Effects of Zcchc3-deficiency on poly(I:C)- or LPS-induced phosphorylation of Tbk1 and p65 in BMDCs. Zcchc3+/+ and Zcchc3−/− BMDCs (5 × 105) were treated with poly(I:C) (50 μg/ml) or LPS (100 ng/ml) for the indicated times before immunoblotting. The line graphs show the relative intensities of the indicated phosphorylated proteins, which were quantitated by densitometry using ImageJ and normalized by their respective total protein levels.
To further explore the roles of ZCCHC3 in regulation of TLR3-mediated innate immune and inflammatory responses in vivo, age- and sex-matched Zcchc3+/+ and Zcchc3−/− mice were injected with poly(I:C) plus D-galactosamine or LPS through intraperitoneal (i.p.) route. Poly(I:C)- but not LPS-induced production of IFN-β, IL-6, and TNFα was significantly decreased in the sera of Zcchc3−/− compared to Zcchc3+/+ mice (Figure 3A). Hematoxylin–eosin (H&E) staining analysis indicated that poly(I:C)- but not LPS-induced inflammatory damage of lungs was relieved in Zcchc3−/− mice (Figure 3B). Consistently, inflammatory death of Zcchc3−/− mice induced by poly(I:C) but not LPS was delayed and reduced in comparison with Zcchc3+/+ mice (Figure 3C). These results indicate that Zcchc3 plays an important role in regulating TLR3-mediated production of IFN-β and proinflammatory cytokines, as well as inflammatory death in mice.
Figure 3.

Zcchc3-deficiency impairs TLR3-mediated immune responses in vivo. (A) Serum cytokine concentrations in Zcchc3+/+ and Zcchc3−/− mice. Sex- and age-matched Zcchc3+/+ and Zcchc3−/− mice (n = 6 or 7) were infected with poly(I:C) (2 μg/g) plus D-galactosamine (1 mg/g) or LPS (10 μg/g) for 4 h, and the concentrations of IFN-β, IL-6, and TNFα in the serum were determined by ELISA (***P < 0.001; N.S., not significant). (B) Effects of Zcchc3-deficiency on poly(I:C)- or LPS-induced inflammation in the lungs of mice. Sex- and age-matched Zcchc3+/+ and Zcchc3−/− mice (n = 8) were injected i.p. with poly(I:C) (2 μg/g) plus D-galactosamine (1 mg/g) or LPS (10 μg/g) for 6 h and lung sections were used for histological analysis (H&E staining). The integrated optical densities of injured areas of the whole lungs (8 mice for each group) were quantified and analyzed by Image-Pro software (***P < 0.001; N.S., not significant). (C) Effects of Zcchc3-deficiency on poly(I:C)- or LPS-induced inflammatory death of mice. Zcchc3+/+ and Zcchc3−/− mice (n = 7 or 8) were injected i.p. with poly(I:C) (1 μg/g) plus D-galactosamine (0.5 mg/g) or LPS (5 μg/g) per mouse, and the survival rates of mice were monitored for 40 h.
ZCCHC3 mediates the recruitment of TRIF to TLR3
We next investigated the molecular mechanisms responsible for the roles of ZCCHC3 in TLR3-mediated signaling. We determined whether ZCCHC3 is associated with signaling components of TLR3-mediated signaling pathway. Co-immunoprecipitation experiments indicated that ZCCHC3 interacted with TLR3 and TRIF but no other components including TLR4, MyD88, TRAM, TRAF6, or TRAF3 in mammalian overexpression system (Figure 4A). Domain mapping experiments indicated that ZCCHC3 interacted with the TIR domain (728–904) of TLR3. ZCCHC3 could interact with either the N-terminal (1–394) or the middle TIR domain of TRIF (Figure 4B). Similar experiments indicated that the C-terminal zinc finger (ZF) domain (300–404) and the N-terminal domain (1–300) of ZCCHC3 were required for its interaction with TLR3 and TRIF, respectively (Figure 4C). These results suggest that ZCCHC3 is associated with TLR3 and TRIF via distinct domains. Endogenous co-immunoprecipitation experiments indicated that ZCCHC3 did not interact with TLR3 or TRIF under physiological condition, but the association of ZCCHC3 with TLR3 and TRIF was induced at 0.5 h after poly(I:C) stimulation (Figure 4D). Very interestingly, the interaction of ZCCHC3 with TLR3 was decreased at later phase after poly(I:C) stimulation, while ZCCHC3–TRIF interaction was increased at 2 h after poly(I:C) stimulation (Figure 4D). It is possible that the TRIF–ZCCHC3 complex is released from TLR3 after ligand stimulation, which results in the decrease of ZCCHC3–TLR3 interaction at later phases after poly(I:C) stimulation. The increase of ZCCHC3–TRIF interaction at 2 h after poly(I:C) stimulation may be explained by the possibility that ZCCHC3 recruits more TRIF at the later phase to promote the downstream response.
Figure 4.

ZCCHC3 is associated with TLR3 and TRIF. (A–C) HEK293 cells (2 × 106) were transfected with the indicated plasmids before co-immunoprecipitation and immunoblotting were performed with anti-HA-peroxidase and anti-FLAG-M2-peroxidase. (A) ZCCHC3 is associated with TLR3 and TRIF. (B) Interaction of ZCCHC3 with TLR3 mutants and TRIF mutants. (C) Interaction of ZCCHC3 mutants with TLR3 and TRIF. (D) Endogenous ZCCHC3 is associated with TLR3 and TRIF in HT1080 cells. HT1080 cells (1 × 107) were left untreated or treated with poly(I:C) (50 μg/ml) for the indicated times before co-immunoprecipitation and immunoblotting.
Previous experiments have shown that ZCCHC3 acts as a co-receptor of RIG-I-like receptors or cGAS for dsRNA or dsDNA recognition in innate immune responses (Lian et al., 2018a, b). We next investigated whether ZCCHC3 regulates TLR3-mediated signaling in similar manner. Pull-down experiments indicated that ZCCHC3 had no effects on the binding of TLR3 to poly(I:C) (Figure 5A and B). We also investigated whether ZCCHC3 regulates TLR3-mediated signaling is dependent of its function on RIG-I-like receptors- or cGAS-mediated signaling. As shown in Figure 1, knockdown of ZCCHC3 inhibited TLR3-mediated innate immune response in 293-TLR3 (a cell line naturally lacking of cGAS protein), which suggests that the effects of ZCCHC3 on TLR3-mediated signaling are independent of its function on cGAS-mediated signaling. We also made MDA5 and RIG-I double deficient 293-TLR3 cells by the CRISPR-Cas9 method. (Supplementary Figure S3A). We found that knockdown of ZCCHC3 inhibited poly(I:C)-induced activation of ISRE (Supplementary Figure S3B) or transcription of IFNB1 and CXCL10 genes (Supplementary Figure S3C) in MDA5/RIG-I-deficient 293-TLR3 cells to similar degrees as in control cells. These results suggest that ZCCHC3 regulates TLR3-mediated signaling is independent of its function on MDA5/RIG-I-mediated signaling.
Figure 5.

ZCCHC3 promotes the recruitment of TRIF to TLR3. (A) Overexpression of ZCCHC3 has no effects on the binding of TLR3 to poly(I:C). HEK293 cells (2 × 106) were transfected with the indicated plasmids. After 20 h, the cell lysates were incubated with the indicated biotinylated-poly(I:C) for 1 h before pull-down assays were performed with streptavidin-sepharose beads. Precipitates and cell lysates were analyzed by immunoblotting with the indicated antibodies. (B) Endogenous ZCCHC3 has no effects on the binding of TLR3 to poly(I:C). Lysate of 293-TLR3 cells (1 × 107) was incubated with the indicated biotinylated-poly(I:C) for 1 h before pull-down assays and immunoblotting. (C) ZCCHC3 localizes in the cytosolic but not the lumen of endosomes. 293-TLR3 cells (4 × 108) were collected and washed twice with ice-cold PBS. Endosomal and lysosomal fractions were isolated and left untreated or treated with proteinase K (2 μg/ml) or proteinase K plus 0.2% Triton X-100 for 15 min at room temperature. The samples were then analyzed by immunoblotting with the indicates antibodies. (D) ZCCHC3 promotes TLR3–TRIF but not TLR4–TRIF interaction in mammalian overexpression system. HEK293 cells (2 × 106) were transfected with the indicated plasmids. After 20 h, the cells were lysed for co-immunoprecipitation and immunoblotting with the indicated antibodies. (E) Knockdown of ZCCHC3 disrupts TLR3–TRIF but not TLR4–TRIF interaction. ZCCHC3-knockdown or control 293-TLR3 cells (2 × 106) were transfected with the indicated plasmids. After 20 h, the cell lysates were harvested and lysed for co-immunoprecipitation before immunoblotting with the indicated antibodies. (F) Knockdown of ZCCHC3 disrupts the recruitment of TRIF to TLR3. ZCCHC3-knockdown or control 293-TLR3 cells (1 × 107) were treated with poly(I:C) (50 μg/ml) for the indicated times. The cell lysates were immunoprecipitated with anti-TLR3 and analyzed by immunoblotting with the indicated antibodies. (G) Stimulation of poly(I:C) but not LPS induces the interaction of ZCCHC3 with TRIF. The indicated cells (1 × 107) were left untreated or treated with poly(I:C) (50 μg/ml) or LPS (200 ng/ml) for the indicated times before co-immunoprecipitation and immunoblotting with the indicated antibodies.
We further determined whether ZCCHC3 localizes to the cytosolic or luminal sides of endosomes/lysosomes by proteinase K protection assays with purified endosomes/lysosomes. The results showed that ZCCHC3 was diminished upon proteinase K treatment (Figure 5C), suggesting that ZCCHC3 is localized at the cytosolic but not the luminal sides of endosomes/lysosomes. Next, we explored whether ZCCHC3 acts as a bridge protein for TLR3–TRIF association. Co-immunoprecipitation experiments indicated that overexpression of ZCCHC3 enhanced the interaction of TLR3–TRIF but not TLR4–TRIF (Figure 5D). Conversely, knockdown of ZCCHC3 inhibited the interaction of TLR3–TRIF but not TLR4–TRIF (Figure 5E). Consistently, knockdown of ZCCHC3 markedly inhibited the recruitment of TRIF to TLR3 upon poly(I:C) stimulation but had no effects on the tyrosine phosphorylation of TLR3 (Figure 5F). These results suggest that ZCCHC3 is responsible for recruitment of TRIF to TLR3 after poly(I:C) stimulation. Interestingly, poly(I:C) but not LPS stimulation induced endogenous association between ZCCHC3 and TRIF in their respective response cells (Figure 5G).
ZCCHC3 promotes TRIF activity
Since ZCCHC3 facilitates recruitment of TRIF to TLR3, we next determined whether ZCCHC3 promotes TRIF activity. ZCCHC3(1–300), which interacts with TRIF but not TLR3, enhanced TRIF-mediated activation of the IFN-β promoter, whereas ZCCHC3(300–404), which interacts with TLR3 but not TRIF, had no effects on TRIF-mediated activation of the IFN-β promoter (Figure 6A). The oligomerization of TRIF is required for recruitment of downstream adaptors to transmit signals (Funami et al., 2007, 2008). Co-immunoprecipitation experiments indicated that overexpression of ZCCHC3 enhanced self-association of TRIF (Figure 6B), whereas knockdown of ZCCHC3 had opposite effects (Figure 6C). Once TLR3/4 binds to ligands, TRIF is recruited to interact with TLR3/4 and undergoes oligomerization. We further performed semi-denaturing detergent agarose gel electrophoresis (SDD–AGE) to detect whether ZCCHC3 regulates TRIF oligomerization (Lang et al., 2017). The results showed that overexpression of ZCCHC3 enhanced TRIF oligomerization (Figure 6D). The oligomerization of TRIF induced by poly(I:C) but not LPS was decreased in ZCCHC3-knockdown cells (Figure 6E). Consistently, knockdown of ZCCHC3 markedly inhibited the recruitment of TRAF6 and TBK1 to TRIF (Figure 6F). Collectively, these results suggest that ZCCHC3 promotes poly(I:C)-induced oligomerization and activation of TRIF.
Figure 6.

ZCCHC3 promotes TRIF activity. (A) ZCCHC3 and ZCCHC3(1–300) potentiate TRIF-mediated activation of the IFN-β promoter. HEK293 cells (1 × 105) were transfected with the IFN-β reporter (50 ng) and the indicated plasmids for 18 h before luciferase assays (**P < 0.01). (B) ZCCHC3 potentiates TRIF–TRIF interaction in mammalian overexpression system. HEK293 cells (2 × 106) were transfected with the indicated plasmids. After 20 h, the cells were lysed for co-immunoprecipitation and immunoblotting with the indicated antibodies. (C) Knockdown of ZCCHC3 disrupts TRIF–TRIF interaction. ZCCHC3-knockdown or control 293-TLR3 cells (2 × 106) were transfected with the indicated plasmids. After 20 h, the cells were lysed for coimmunoprecipitation and immunoblotting. (D) ZCCHC3 potentiates the oligomerization of TRIF in mammalian overexpression system. HEK293 cells (2 × 106) were transfected with the indicated plasmids. After 20 h, cell lysates were fractionated by SDD–AGE and SDS–PAGE and then analyzed by immunoblotting with the indicated antibodies. (E) Effects of ZCCHC3-RNAi on poly(I:C)- or LPS-induced oligomerization of TRIF. ZCCHC3-knockdown or control 293-TLR3 or 293-TLR4 cells (1 × 107) were left untreated or treated with poly(I:C) (50 μg/ml) or LPS (200 ng/ml) for the indicated times. Cell lysates were fractionated by SDD–AGE and SDS–PAGE and then analyzed by immunoblotting with the indicated antibodies. (F) Effects of ZCCHC3-RNAi on poly(I:C)-induced recruitment of TRAF6 and TBK1 to TRIF. ZCCHC3-knockdown or control 293-TLR3 cells (1 × 107) were left untreated or treated with poly(I:C) (50 μg/ml) for the indicated times. The cell lysates were immunoprecipitated with anti-TRIF and analyzed by immunoblotting with the indicated antibodies. (G) A model on the roles of ZCCHC3 in regulation of TLR3-mediated signaling.
Discussion
In this study, we identified ZCCHC3 as a positive regulator of TLR3-mediated signaling, which is important for dsRNA-induced innate immune and inflammatory responses.
There are several lines of evidence support a critical role of ZCCHC3 in TLR3-mediated signaling. Overexpression of ZCCHC3 and its truncations markedly potentiated poly(I:C)-induced activation of the IFN-β promoter and ISRE, whereas knockdown of ZCCHC3 had opposite effects. ZCCHC3-deficiency inhibited poly(I:C)- but not LPS-induced transcription of Ifnb1, Isg56, and Il6 genes in BMDMs, BMDCs, and MLFs. In addition, Zcchc3−/− mice produced lower levels of serum IFN-β, IL-6, and TNFα after poly(I:C) but not LPS administration and were more resistant to poly(I:C)- but not LPS-triggered inflammatory death compared with Zcchc3+/+ mice. However, ZCCHC3-deficiency did not affect the transcription of Ifnb1 and downstream genes induced by TLR2 and TLR7/8 ligands in mouse primary immune cells. These findings suggest that ZCCHC3 positively regulates TLR3-mediated innate immune and inflammatory responses.
It has been shown that ZCCHC3 acts as a co-sensor of RIG-I/MDA5 and cGAS to regulate both RNA and DNA virus-triggered induction of type I IFNs (Lian et al., 2018a, b). In this study, we found that ZCCHC3 acts in TLR3-mediated signaling by a distinct mechanism. ZCCHC3 was found to localize at the cytosolic but not the luminal sides of endosomes/lysosomes, and pull-down experiments indicated that ZCCHC3 was not required for the binding of TLR3 to poly(I:C). These results suggest that ZCCHC3 does not act to promote ligand binding of TLR3 in the lumen of endosomes/lysosomes. Domain-mapping experiments indicated that the C-terminal domain (300–404) and N-terminal domain (1–300) of ZCCHC3 were required for its interaction with the cytosolic side TIR domain of TLR3 and the N-terminal/TIR domains of TRIF, respectively. ZCCHC3 promoted the interaction of TLR3 and TRIF, whereas knockdown of ZCCHC3 inhibited the interaction of TLR3 and TRIF. Most importantly, endogenous ZCCHC3 was associated with TLR3 and TRIF after poly(I:C) but not LPS stimulation. Knockdown of ZCCHC3 inhibited the recruitment of TRIF to TLR3 after poly(I:C) stimulation. Taken together, our findings suggest that ZCCHC3 promotes recruitment of TRIF to TLR3 after ligand stimulation, and then dissociates from TLR3 in complex with TRIF. Consistently, ZCCHC3 is important for TRIF-mediated signaling, including TRIF oligomerization and its recruitment of downstream components, including TRAF6 and TBK1. In conclusion, our findings suggest that ZCCHC3 is an important regulator of TLR3-mediated innate immune and inflammatory response by linking TRIF to TLR3.
Materials and methods
Mice
Zcchc3−/− mice on the C57BL/6 background were generated by the CRISPR/Cas9 method (Lian et al., 2018a, b). The protocols and procedures for mice experiments in this study were approved by the Wuhan University College of Life Sciences Animal Care and Use Committee (approval number WDSKY0200902-2).
Reagents, antibodies, and cells
poly(I:C), R848, PGN, and Lipofectamine 2000 (InvivoGen); MPLA and LPS (Sigma); polybrene (Millipore); SYBR (Bio-Rad); FuGene and Dual-Specific Luciferase Assay Kit (Promega); puromycin and EZ-link Psoralen–PEG3–Biotin (Thermo); streptavidin agarose (Solulink); ELISA kits for murine IFN-β (PBL), IL-6, and TNFα (BioLegend).
Mouse monoclonal antibodies against HA (Origene), FLAG, and β-actin, anti-HA-Peroxidase, and anti-FLAG-M2-Peroxidase (Sigma) and rabbit monoclonal antibodies against TLR3, phosphor-IRF3, p65 (Cell Signaling Technology), phosphor-TBK1, TBK1, TRIF (Abcam), and IRF3 (Santa Cruz Biotechnology) were purchased from the indicated manufacturers. Antisera against ZCCHC3, RIG-I, and MDA5 were generated by immunizing rabbits or mice with purified recombinant ZCCHC3 (133–404), RIG-I(231–925), and MDA5(239–1020). HEK293 and HT1080 cells were obtained from ATCC.
Preparation of primary mouse cells
The preparations of BMDMs, BMDCs, and MLFs were previously described (Luo et al., 2016a, 2017; Yang et al., 2017). Briefly, for preparation of BMDMs, mouse bone marrow cells (5 × 106) were cultured in 100-mm dishes in 5 ml of 10% M-CSF-containing conditional medium from L929 cells for 3–5 days. For preparation of BMDCs, mouse bone marrow cells (5 × 106) were cultured in medium containing murine GM-CSF (50 ng/ml) for 6–8 days. For preparation of lung fibroblasts, lungs were minced and digested in calcium and magnesium-free HBSS containing 10 μg/ml type II collagenase and 20 μg/ml DNase I for 1 h at 37°C with shaking. Cell suspensions were centrifuged at 1500 rpm for 5 min. The cells were then plated in culture medium (1:1 [v/v] DMEM/Ham’s F-12 containing 10% FBS, 50 U/ml penicillin, 50 μg/ml streptomycin, 15 mM HEPES, and 2 mM L-glutamine).
Transfection and reporter assays
Transfection and reporter assays were performed as previously described (Xu et al., 2005; Lei et al., 2010; He et al., 2013; Luo et al., 2016b; Fu et al., 2017). HEK293 cells were transfected by standard calcium phosphate precipitation method. Empty control plasmid was added to ensure that each transfection receives the same amount of total DNA. The pRL-TK (Renilla luciferase) reporter plasmid (0.02 μg) was added to each transfection for normalization of transfection efficiency. Luciferase assays were performed using a Dual-Specific Luciferase Assay Kit (Promega). Firefly luciferase activities were normalized on the basis of Renilla luciferase activities.
qPCR
Total RNA was isolated using the Trizol reagent (Invitrogen) and then reverse-transcription using HiScript II qRT SuperMix (Vazyme). Aliquots of products were subjected to qPCR analysis to measure mRNA abundance of the indicated genes. Data shown are the relative abundance of the indicated mRNA normalized to that of GAPDH. Gene-specific primer sequences were as followed:GAPDH: GACAAGCTTCCCGTTCTCAG (forward) and GAGTCAACGGATTTGGTGGT (reverse); IFNB1: TTGTTGAGAACCTCCTGGCT (forward) and TGACTATGGTCCAGGCACAG (reverse); CXCL10: GGTGAGAAGAGATGTCTGAATCC (forward) and GTCCATCCTTGGAAGCACTGCA (reverse); ISG56: TCATCAGGTCAAGGATAGTC (forward) and CCACACTGTATTTGGTGTCTACG (reverse); IL6: GCCGCATCGCCGTCTCCTAC (forward) and CCTCAGCCCCCTCTGGGGTC (reverse); TNFα: GCCGCATCGCCGTCTCCTAC (forward) and CCTCAGCCCCCTCTGGGGTC (reverse); Gapdh: ACGGCCGCATCTTCTTGTGCA (forward) and ACGGCCAAATCCGTTCACACC (reverse); Ifnb1: TCCTGCTGTGCTTCTCCACCACA (forward) and AAGTCCGCCCTGTAGGTGAGGTT (reverse); Il6: TCTGCAAGAGACTTCCATCCAGTTGC (forward) and AGCCTCCGACTTGTGAAGTGGT (reverse); Isg56: ACAGCAACCATGGGAGAGAATGCTG (forward) and ACGTAGGCCAGGAGGTTGTGCAT (reverse); Tnfα: GGTGATCGGTCCCCAAAGGGATGA (forward) and TGGTTTGCTACGACGTGGGCT (reverse).
ELISA
Eight-week-old Zcchc3+/+ and Zcchc3−/− mice were injected i.p. with poly(I:C) (2 μg/g body weight) plus D-galactosamine (1 mg/g body weight) (n = 6) or with LPS (10 μg/g body weight) (n = 7) for 4 h, and then the sera of mice were collected for measurement of IFN-β, IL-6, and TNFα.
Co-immunoprecipitation and immunoblotting
Cells were lysed in pre-lysis buffer (20 mM Tris-HCl pH 7.4, 150 mM NaCl, 1 mM EDTA, and 1% Triton X-100). Co-immunoprecipitation and immunoblotting were performed as previously described (Zhong et al., 2009, 2010; Yan et al., 2014; Xu et al., 2016; Wei et al., 2017).
Proteinase K protection assays
Proteinase K protection assays were performed as previously described (Yang et al., 2016). The 293-TLR3 cells (4 × 108) were collected and washed twice with ice-cold PBS. The endosome/lysosome fractions were isolated from cells by calcium precipitation, and left untreated, treated with 2 μg/ml proteinase K or treated with proteinase K plus 0.2% Triton X-100 for 15 min at room temperature. The samples were then mixed with 2× SDS loading buffer and analyzed by immunoblotting.
In vitro pull-down assays
In vitro pull-down assays were previously described (Li et al., 2012; Yang, et al., 2016). Briefly, HEK293 cells transfected with the indicated plasmids were lysed in pre-lysis buffer. Lysates were incubated with biotinylated-poly(I:C) for 1 h at room temperature, and then incubated with streptavidin beads for another 2 h at room temperature. The beads were washed four times with lysis buffer and analyzed by immunoblotting with the indicated antibodies.
Immunohistochemistry analysis
Immunohistochemistry analysis was previously described (Yang et al., 2017). Lungs from mice were fixed in formalin and embedded into paraffin blocks for H&E staining (Servicebio).
Semi-denaturing detergent agarose gel electrophoresis
SDD–AGE was performed as previously described (Hou et al., 2011; Liu et al., 2013). HEK293, 293-TLR3, or 293-TLR4 cells were lysed in pre-lysis buffer, and the cell lysates were mixed in 1× sample buffer (0.5× TBE, 10% glycerol, 2% SDS, and 0.0025% bromophenol blue) and loaded onto a vertical 2% agarose gel (Bio-Rad). After electrophoresis in the running buffer (1× TBE and 0.1% SDS) for about 2 h with a constant voltage of 100 V at 4°C, the proteins were transferred to immobilon membrane (Millipore) for immunoblotting.
Statistical analysis
Unpaired Student’s t-test was used for statistical analysis with GraphPad Prism Software. For the mouse survival study, Kaplan–Meier survival curves were generated and analyzed by log-rank test. Densitometry quantification was made with ImageJ Software. The integrated optical density of injured areas of lung was analyzed by Image-Pro software.
Supplementary Material
Acknowledgements
H.-B.S., H.L., and R.Z. conceived and designed the study; R.Z. and X.Z. performed the experiments; H.-B.S., R.Z., H.L., and Q.Y. analyzed the data; and H.-B.S., R.Z., and Q.Y. wrote the manuscript.
Funding
This work was supported by grants from the State Key R&D Program of China (2017YFA0505800 and 2016YFA0502102) and the National Natural Science Foundation of China (31830024, 31630045, and 31800728).
Conflict of interest: none declared.
References
- Alexopoulou L., Holt A.C., Medzhitov R., et al. (2001). Recognition of double-stranded RNA and activation of NF-κB by toll-like receptor 3. Nature 413, 732–738. [DOI] [PubMed] [Google Scholar]
- Fu Y.Z., Su S., Gao Y.Q., et al. (2017). Human cytomegalovirus tegument protein UL82 inhibits STING-mediated signaling to evade antiviral immunity. Cell Host Microbe 21, 231–243. [DOI] [PubMed] [Google Scholar]
- Funami K., Sasai M., Ohba Y., et al. (2007). Spatiotemporal mobilization of toll/IL-1 receptor domain-containing adaptor molecule-1 in response to dsRNA. J. Immunol. 179, 6867–6872. [DOI] [PubMed] [Google Scholar]
- Funami K., Sasai M., Oshiumi H., et al. (2008). Homo-oligomerization is essential for toll/interleukin-1 receptor domain-containing adaptor molecule-1-mediated NF-κB and interferon regulatory factor-3 activation. J. Biol. Chem. 283, 18283–18291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Cattaneo A., Gobert F.X., Muller M., et al. (2012). Cleavage of toll-like receptor 3 by cathepsins B and H is essential for signaling. Proc. Natl Acad. Sci. USA 109, 9053–9058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X., Li Y., Li C., et al. (2013). USP2a negatively regulates IL-1β- and virus-induced NF-κB activation by deubiquitinating TRAF6. J. Mol. Cell Biol. 5, 39–47. [DOI] [PubMed] [Google Scholar]
- Hou F., Sun L., Zheng H., et al. (2011). MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell 146, 448–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X., Li Y., Li X., et al. (2019). TRIM14 promotes endothelial activation via activating NF-κB signaling pathway. J. Mol. Cell Biol. 12, 176–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin M.S., and Lee J.O. (2008). Structures of the toll-like receptor family and its ligand complexes. Immunity 29, 182–191. [DOI] [PubMed] [Google Scholar]
- Johnsen I.B., Nguyen T.T., Ringdal M., et al. (2006). Toll-like receptor 3 associates with c-Src tyrosine kinase on endosomes to initiate antiviral signaling. EMBO J. 25, 3335–3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai T., and Akira S. (2010). The role of pattern-recognition receptors in innate immunity: update on toll-like receptors. Nat. Immunol. 11, 373–384. [DOI] [PubMed] [Google Scholar]
- Kawai T., and Akira S. (2011). Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 34, 637–650. [DOI] [PubMed] [Google Scholar]
- Lang X., Tang T., Jin T., et al. (2017). TRIM65-catalized ubiquitination is essential for MDA5-mediated antiviral innate immunity. J. Exp. Med. 214, 459–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei C.Q., Zhong B., Zhang Y., et al. (2010). Glycogen synthase kinase 3β regulates IRF3 transcription factor-mediated antiviral response via activation of the kinase TBK1. Immunity 33, 878–889. [DOI] [PubMed] [Google Scholar]
- Li Y., Chen R., Zhou Q., et al. (2012). LSm14A is a processing body-associated sensor of viral nucleic acids that initiates cellular antiviral response in the early phase of viral infection. Proc. Natl Acad. Sci. USA 109, 11770–11775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian H., Wei J., Zang R., et al. (2018a). ZCCHC3 is a co-sensor of cGAS for dsDNA recognition in innate immune response. Nat. Commun. 9, 3349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian H., Zang R., Wei J., et al. (2018b). The zinc-finger protein ZCCHC3 binds RNA and facilitates viral RNA sensing and activation of the RIG-I-like receptors. Immunity 49, 438–448e435. [DOI] [PubMed] [Google Scholar]
- Liu Z., Wu S.W., Lei C.Q., et al. (2013). Heat shock cognate 71 (HSC71) regulates cellular antiviral response by impairing formation of VISA aggregates. Protein Cell 4, 373–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo W.W., Li S., Li C., et al. (2016a). iRhom2 is essential for innate immunity to DNA viruses by mediating trafficking and stability of the adaptor STING. Nat. Immunol. 17, 1057–1066. [DOI] [PubMed] [Google Scholar]
- Luo W.W., Li S., Li C., et al. (2017). iRhom2 is essential for innate immunity to RNA virus by antagonizing ER- and mitochondria-associated degradation of VISA. PLoS Pathog. 13, e1006693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo W.W., Lian H., Zhong B., et al. (2016b). Kruppel-like factor 4 negatively regulates cellular antiviral immune response. Cell. Mol. Immunol. 13, 65–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu L., Ji Y., Zhu X., et al. (2015). hCINAP negatively regulates NF-κB signaling by recruiting the phosphatase PP1 to deactivate IKK complex. J. Mol. Cell Biol. 7, 529–542. [DOI] [PubMed] [Google Scholar]
- Sharma S., tenOever B.R., Grandvaux N., et al. (2003). Triggering the interferon antiviral response through an IKK-related pathway. Science 300, 1148–1151. [DOI] [PubMed] [Google Scholar]
- Takeuchi O., and Akira S. (2010). Pattern recognition receptors and inflammation. Cell 140, 805–820. [DOI] [PubMed] [Google Scholar]
- Toscano F., Estornes Y., Virard F., et al. (2013). Cleaved/associated TLR3 represents the primary form of the signaling receptor. J. Immunol. 190, 764–773. [DOI] [PubMed] [Google Scholar]
- Wei J., Guo W., Lian H., et al. (2017). SNX8 mediates IFNγ-triggered noncanonical signaling pathway and host defense against listeria monocytogenes. Proc. Natl Acad. Sci. USA 114, 13000–13005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L.G., Wang Y.Y., Han K.J., et al. (2005). VISA is an adapter protein required for virus-triggered IFN-β signaling. Mol. Cell 19, 727–740. [DOI] [PubMed] [Google Scholar]
- Xu Z.S., Zhang H.X., Zhang Y.L., et al. (2016). PASD1 promotes STAT3 activity and tumor growth by inhibiting TC45-mediated dephosphorylation of STAT3 in the nucleus. J. Mol. Cell Biol. 8, 221–231. [DOI] [PubMed] [Google Scholar]
- Yamamoto M., Sato S., Hemmi H., et al. (2003). Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science 301, 640–643. [DOI] [PubMed] [Google Scholar]
- Yamashita M., Chattopadhyay S., Fensterl V., et al. (2012). Epidermal growth factor receptor is essential for toll-like receptor 3 signaling. Sci. Signal. 5, ra50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J., Li Q., Mao A.P., et al. (2014). TRIM4 modulates type I interferon induction and cellular antiviral response by targeting RIG-I for K63-linked ubiquitination. J. Mol. Cell Biol. 6, 154–163. [DOI] [PubMed] [Google Scholar]
- Yang Q., Liu T.T., Lin H., et al. (2017). TRIM32-TAX1BP1-dependent selective autophagic degradation of TRIF negatively regulates TLR3/4-mediated innate immune responses. PLoS Pathog. 13, e1006600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y., Wang S.Y., Huang Z.F., et al. (2016). The RNA-binding protein Mex3B is a coreceptor of toll-like receptor 3 in innate antiviral response. Cell Res. 26, 288–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong B., Zhang L., Lei C., et al. (2009). The ubiquitin ligase RNF5 regulates antiviral responses by mediating degradation of the adaptor protein MITA. Immunity 30, 397–407. [DOI] [PubMed] [Google Scholar]
- Zhong B., Zhang Y., Tan B., et al. (2010). The E3 ubiquitin ligase RNF5 targets virus-induced signaling adaptor for ubiquitination and degradation. J. Immunol. 184, 6249–6255. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.