

Abstract
Aims/Introduction
Neonatal diabetes mellitus is created by alterations in the genes responsible for beta‐cell mass and/or function. The present study aimed to evaluate the genetic variants in the insulin gene (INS) in four Chinese infants aged <12 months with diabetes onset, and to explore the clinical and genetic characteristics of permanent neonatal diabetes mellitus caused by INS mutations.
Materials and Methods
The complete coding sequences of KCNJ11, ABCC8 and INS were detected using Sanger sequencing. The pathogenicity of the mutations was determined based on the American College of Medical Genetics and Genomics, and the structure of wild‐type and mutant proteins was predicted using the web‐based tool, Phyre2.
Results
One novel mutation (p.I99_C100insSI) and three previously reported variants (p.G32S, p.R89C and p.C96R) in INS were identified in four infants with early‐onset diabetes. All the mutations in the four patients were de novo. Except for mutation R89C, which causes permanent neonatal diabetes mellitus through the addition of an additional cysteine residue at the cleavage site of the A chain and C‐peptide, the other three mutations affected disulfide bonds. The patients had diabetes with marked hyperglycemia or diabetic ketoacidosis, and were then treated with exogenous insulin. Mutations in crucial regions of the INS might give rise to diabetes with varying severity.
Conclusions
This study enriches our awareness of the mutant spectrum in INS, and suggests the important role of INS in the development of permanent neonatal diabetes mellitus.
Keywords: Chinese patients, INS mutation, Neonatal diabetes mellitus
In this study, we discovered four mutations in the insulin gene (INS), one of which was novel and was considered to cause permanent neonatal diabetes mellitus. We explored the clinical and genetic characteristics of Chinese permanent neonatal diabetes mellitus caused by INS mutations. This study enriches our awareness of the mutant spectrum in INS, and suggests the important role of INS in the development of permanent neonatal diabetes mellitus.

Introduction
Neonatal diabetes mellitus is a subtype of diabetes with onset within the first 6 months of life. It occurs in approximately one in 90,000–160,000 live births, is an uncommon monogenic disease owing to genetic defects of beta‐cell function and/or mass, and 80% of cases possess a known genetic dysfunction in Europe1. In China, Li et al. 2 reported that the genetic etiology could be determined in 84% (19/23) of Chinese patients with neonatal diabetes mellitus, which was higher than the outcomes from Cao et al. 3 (60%; 15/25). According to the clinical consequences, neonatal diabetes mellitus can be divided into two forms, transient neonatal diabetes mellitus and permanent neonatal diabetes mellitus4. Transient neonatal diabetes mellitus accounts for 50–60% of neonatal diabetes mellitus, which is 28–56.5% in Chinese patients2, 3. Transient neonatal diabetes mellitus is generally diagnosed within 1 month after birth (median 6 days), and is characterized by intrauterine growth retardation with a low original dose of insulin for replacement and yields a “Diabetes–Remission–Relapse” natural history, which enters remission after 12 weeks, whereas it might relapse in young adults5. Permanent neonatal diabetes mellitus shows a lifelong disorder without remission.
To date, >20 genes have been recognized as key elements that cause permanent neonatal diabetes mellitus6, of which mutations in KCNJ11 and ABCC8 accounted for 40–60% of all occasions, which was 53.3–68.4% in China2, 3. They are separately responsible for Kir6.2 and SUR1 of the adenosine triphosphate‐sensitive potassium channel6. Mutations in the two genes can result in consistent opening of the adenosine triphosphate‐sensitive potassium channel, which induces membrane hyperpolarization and insulin secretion disorder7. Mutations in the insulin gene (INS) are the secondary cause (23%)8. Other mutations include GATA6, EIF2AK3, GCK, PTF1A, FOXP3, ZFP57, GLIS3, PDX1, SLC2A2, SLC19A2, GATA4, NEUROD1, NEUROG3, NKX2‐2, RFX6, IER3IP1, MNX1, HNF1B and STAT3 1. The overexpression of genes at chromosome 6q24 is the most common cause of transient neonatal diabetes mellitus (OMIM 601410), which encompasses 70% of all cases9. Basic and clinical studies suggest that sulfonylurea can help with the closing of the adenosine triphosphate‐sensitive potassium channel, and improve glycemic control and neurological outcomes10, which was also confirmed in Chinese patients2, 3. Thus, molecular diagnosis is vital for accurate diagnosis and prognosis assessment for neonatal diabetes mellitus. Here, we carried out genetic sequencing in four Chinese infants with diabetes, and identified four mutations in the INS gene.
Methods
Participants
Four infants were recruited from the outpatient clinic of Beijing Peking Union Medical College Hospital, Beijing, China, with a diagnosis of diabetes within 1 year‐of‐age and without remission. Our research team has focused on neonatal diabetes mellitus from 2007 to the present; during this time period, 25 patients were diagnosed with neonatal diabetes mellitus (with diabetes onset <6 months), and all patients underwent genetic testing. Among them, 21 patients carried mutations causing neonatal diabetes mellitus, and three patients harbored mutations in INS. Additionally, one person with diabetes onset at 8 months (>6 months) also carried mutations in INS. Thus, four patients were included in the present study. The families were all unrelated and of Chinese descent. Written consent was obtained from responsible family members. All four cases had no family history of diabetes and were islet autoantibody negative, and no neurological abnormalities (such as developmental delay) were detected in their clinical history.
Ethical consideration
All procedures carried out in studies involving human participants were in accordance with the ethical standards of the Peking Union Medical College Hospital Ethics Committee, and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Genetic analysis
Genomic DNA of the four pedigrees together with the parents of the four probands was isolated from peripheral blood samples and used to screen the mutations in KCNJ11, ABCC8 and INS by polymerase chain reaction direct sequencing. All exons of the aforementioned three genes were screened by primer sequences devised by Premier 5 software (Premier company, Canada) (Table S1). The entire coding sequence of these genes was screened in the four infants by direct sequencing. Polymerase chain reaction products corresponding to abnormal electrophoretic patterns were directly sequenced to characterize nucleotide variants on the ABI 377 automated sequencer (Perkin‐Elmer Corp., Foster City, CA, USA). The NCBI BLAST database was then used to identify variants by aligning with reference sequences ABCC8 (NM_000352), KCNJ11 (NM_000525) and INS (NM_000207). Databases including Exome Variant Server (http://evs.gs.washington.edu/EVS), dbSNP database in UCSC genome bank and NCBI (http://www.ncbi.nlm.nih.gov/snp/) were used to exclude single nucleotide polymorphisms. Furthermore, the determined variants should not have been present in 100 non‐diabetic healthy controls (blood donors associated with the clinic). Mutations in the parents were detected by targeting sequencing of the affected exons. The functional effects of variants were predicted by PolyPhen2 (http://genetics.bwh.harvard.edu/pph2/), SIFT (http://sift.jcvi.org) and MutationTaster (http://www.mutationtaster.org). The pathogenicity of the mutations was assessed according to the American College of Medical Genetics and Genomics11.
The key variables included sex, age, diabetic onset age and family history of diabetes. Clinical indicators included fasting glucose, fasting C‐peptide and glycated hemoglobin A1c (HbA1c). HbA1c (before and after treatment) was measured using dedicated high‐performance liquid chromatography. Islet cell antibodies were measured using indirect immunofluorescence; glutamic acid decarboxylase and insulinoma‐associated antigen 2 were determined by enzyme‐linked immunosorbent assay (SIEMENS ADVIA Centaur XP, Erlangen, Germany).
Results
Four infants were diagnosed with permanent neonatal diabetes mellitus. Among them, the diabetic onset age varied from 12 weeks to 8 months; the birthweight ranged from 2,600 g to 3,300 g. All patients showed diabetic ketoacidosis or marked hyperglycemia, with fasting C‐peptide <0.5 ng/mL, and were treated with insulin from diabetes diagnosis. After treatment, HbA1c was controlled from 7.3% to 8.4% (Table 1).
Table 1.
Clinical manifestations of the four patients
| Mutations | Location and function | Sex | Birthweight (g) | Clinical manifestations | Onset age (week) | FBG at onset (mmol/L) | C‐peptide (ng/mL) | Treatment | HbA1c before/after treatment (%) |
|---|---|---|---|---|---|---|---|---|---|
| C96R | A7, affecting disulfide bonds | Male | 3,300 (25–50th) | DKA | 24 | NA | 0.03 | Insulin | 8.1%/7.5% |
| I99_C100insSI | A10‐A11, affecting disulfide bonds | Female | 2,600 (3–10th) | DKA | 16 | NA | 0.05 | Insulin | 9.7%/7.8% |
| G32S | B8, affecting disulfide bonds | Male | 2,900 (10–25th) | Hyperglycemia | 12 | 33 | 0.42 | Insulin | 9.1%/8.4% |
| R89C | Cleavage site between A chain and C‐peptide | Female | 2,950 (10–25th) | Hyperglycemia | 32 | 30.32 | 0.37 | Insulin | 8.6%/7.3% |
DKA, diabetic ketoacidosis; NA, not available.
Case 1 was a 1‐year‐old girl with a birthweight of 2,600 g (3–10th). She was diagnosed with diabetes at 4 months after birth and was referred to the hospital because of diabetic ketoacidosis. Biochemical examination showed that the fasting C‐peptide level was very low (0.05 ng/mL). Subsequently, she was treated with basal and preprandial insulin. Case 2 was a 6‐month‐old boy whose birthweight was 2,900 g (10–25th). The diabetes manifested at 3 months after birth. He was sent to the hospital due to hyperglycemia, and was treated with basal and preprandial insulin. He also showed a low fasting C‐peptide level (0.42 ng/mL). Case 3 was a 2‐year‐old boy who had suffered from diabetes since he was aged 6 months, and he had received insulin injections until the study. His birthweight was 3,300 g (25–50th), and his fasting C‐peptide was 0.03 ng/mL. Case 4 had the oldest diabetic onset age (8 months after her birth) of the four patients. Her birthweight was 2,950 g (10–25th), and the fasting C‐peptide was 0.37 ng/mL. Among them, an attempt by case 2 to switch from insulin to oral sulfonylureas failed.
Genetic testing showed four mutations, including one novel (p.I99_C100insSI [case 1]) and three previously reported mutations (p.G32S [case 2], p.C96R [case 3] and p.R89C [case 4]; Figure 1), and no mutations were found in KCNJ11 and ABCC8. The positions of these mutations in proinsulin are shown in Figure 2. The novel mutation, p. I99_C100insSI, caused the insertion of two amino acids (Ser and Ile) into the key position of the disulfide bond in the A chain (A10 and A11). Mutation p.C96R (c.286 T>C) induced an amino acid change (T>C) at c.286, which is also located at the site of the disulfide bond. Mutation p. R89C affects the cleavage site between the A chain and C‐peptide. Mutation p.G32S plays a role in affecting proinsulin folding in the endoplasmic reticulum and disulfide bonds12. The parents of the four cases had no mutations and no clinical manifestations of diabetes (Figure 3).
Figure 1.
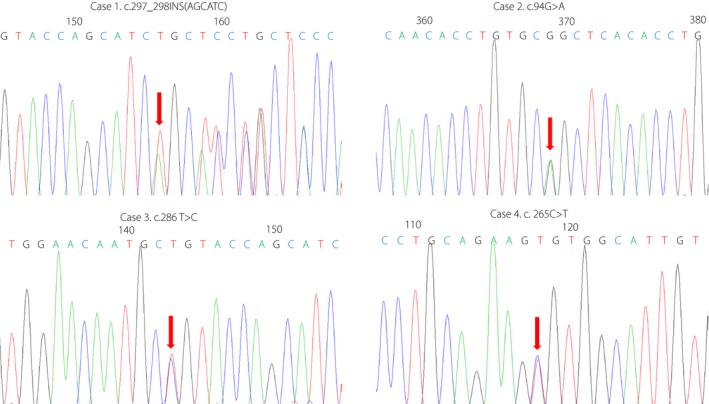
Sequencing chromatogram of the mutations in the INS gene. Arrows indicate the changed nucleotide base.
Figure 2.

Diagram representing the human proinsulin molecule showing the location of mutations causing neonatal diabetes. Blue, insulin A chain; dark yellow, insulin B chain; yellow, C‐peptide; blank, basic residues that are the cleavage site for conversion from proinsulin to insulin; green, mutations in INS; boxes, mutations found in the present study, in which the red box was the novel mutation found in the study.
Figure 3.

Pedigree of families of the four infants in the present study. Squares represent male family members, whereas circles represent female family members. Black symbols represent individuals with diabetes, and blank symbols represent normal individuals. Arrows indicate probands in the families. Variant carriers presented as N, normal allele; M, mutation.
Of the four variants, three were predicted to be deleterious by three bioinformatics tools (p. C96R, p. R89C and p. G32S; Table 2), and all of them were considered pathogenic in a previous study13, 14. Furthermore, the novel mutation, p.I99_C100insSI, was considered as “likely pathogenic” based on the American College of Medical Genetics and Genomics 11: PM2 (absent from controls in ESP, 1,000 Genomes or ExAC), PM4 (variant affecting protein length), PP1 (cosegregation with permanent neonatal diabetes mellitus in family members) and PP4 (the clinical phenotype is gene‐specific). Phyre2 (http://www.sbg.bio.ic.ac.uk/~phyre2/html/page.cgi?xml:id=index) was used to predict the structure of the proteins with missense mutations. We found no differences in the structure of wild‐type and mutant‐type mutations p. R89C and p.C96R; however, structural damage was yielded by mutation p. G32S (Figure 4).
Table 2.
Prediction of the missense mutations in INS found in the present study
| Mutations | SIFT | PolyPhen‐2 | MutationTaster |
|---|---|---|---|
| p.C96R | Deleterious (0) | Probably damaging with a score of 0.993 | Disease‐causing |
| p.R89C | Deleterious (0) | Probably damaging with a score of 0.987 | Disease‐causing |
| p.G32S | Deleterious (0) | Probably damaging with a score of 1.000 | Disease‐causing |
Figure 4.

Structure modeling of wild‐type and mutant proinsulin (p. G32S). The red circles indicate the amino acid residue of wild‐type and mutant proinsulin.
Discussion
In the present study, four patients were diagnosed with hyperglycemia during their first year of life and have been treated with insulin since diagnosis. There was no history of familial diabetes. Based on the age of onset of diabetes, negative autoantibodies and persistent hyperglycemia, they were diagnosed with permanent neonatal diabetes mellitus. Because up to 90–95% of patients with neonatal diabetes mellitus caused by KCNJ11 and ABCC8 can be successfully transitioned from insulin therapy to sulfonylureas1, one of the patients was treated with oral sulfonylureas, but it was not successful. Thus, we carried out genetic testing focusing on the most common genes (ABCC8, KCNJ11 and INS) causing permanent neonatal diabetes mellitus, and discovered four mutations in INS, one of which was novel and was considered to cause permanent neonatal diabetes mellitus.
INS encodes preproinsulin, which includes 110 amino acids, and then preproinsulin forms proinsulin through cleavage of the signal peptide in the endoplasmic reticulum in beta‐cells1. Furthermore, the folding action of proinsulin is managed, and three native disulfide bonds (A6‐A11, B7‐A7 and B19‐A20) are formed before the cleaving of C‐peptide12, 15. The mutation in INS causing hyperinsulinemia was first reported in 1983 by Tager et al. 16, and then, many researchers proceeded to identify mutations in INS by genome technology (e.g., microarray and sequencing). To date, >50 INS gene mutations (grch37.ensembl.org/) have been identified to cause neonatal diabetes mellitus. A previous study showed that cysteine‐correlated mutations, which introduce or remove a cysteine, lead to pancreatic beta‐cell dysfunction and diabetes, whereas non‐cysteine‐associated mutations might impair the efficiency of chain combination15.
The novel mutation found in the present study, p. I99_C100insSI, is considered “likely pathogenic,” which inserted amino acids Ser and Ile between p. I99 and p.C100 in the molecular structure of insulin. The mutation is located at the disulfide bonds (A11); thus, we supposed that the insertion mutation damaged the disulfide bond of the A chain, resulting in permanent neonatal diabetes mellitus. However, molecular biology testing is required to elucidate the function of the mutation. The patient carrying this mutation showed the lowest birthweight and significantly reduced fasting C‐peptide levels in addition to other typical clinical performance of neonatal diabetes mellitus, indicating that this mutation might lead to more severe destruction of beta‐cell function.
B8 glycine has a positive dihedral angle, which prevents L‐amino acids. The L‐amino acids could lead to the rotation of B1–B8, thus turning B7 away from A713. That is, the substitution of the amino acid (G32S) presumably destabilizes the thermodynamic stability of the molecule and further affects the disulfide bond, which ultimately leads to beta‐cell apoptosis by impairing proinsulin folding and severe endoplasmic reticulum stress12, 13, 17. Additionally, the glycine residue is conserved within insulin‐like growth factors and is unchanged in INS sequences13, suggesting that this mutation was likely pathogenic. In line with the results, the mutation found in the present study was considered pathogenic and cosegregated with clinical phenotypes of permanent neonatal diabetes mellitus within the family. Furthermore, Phyre2 predicts structural damage from this mutation.
Mutation p.C96R caused the changeable amino acid T>C at c.286 in the INS gene. This mutation is believed to cause permanent neonatal diabetes mellitus, because of its disruption in normal disulfide bonds14, 18. Its pathogenicity was supported by the three functional prediction software programs, SIFT (0), PolyPhen‐2 (0.993) and MutationTaster (disease‐causing), and American College of Medical Genetics and Genomics standards: PM1 (the position locates in the hot spots for mutations in INS gene), PM2 (absent from controls in ESP, 1,000 Genomes or ExAC), PM5 (missense mutation leads to amino acid substitution, and the mutation at the same locus has been confirmed as pathogenic) and PP3 (pathogenicity was confirmed by three bioinformatics tools).
R89C was another mutation in INS found in the present study. The mutation caused the changeable amino acid C>T at c.265 in INS. The mutation influences the proteolytic processing of proinsulin and attracts an additional exposed cysteine residue at the site in proinsulin sequences, and is considered as a transition at CpG dinucleotides19. The pathogenicity of the mutation causing permanent neonatal diabetes mellitus has been reported13 and was confirmed by three different software programs: SIFT (0), PolyPhen‐2 (0.987) and MutationTaster (disease‐causing).
All four mutations were de novo in the families, which was in line with the previous study from Edghill et al. 8, who speculated that the majority (80%) of patients with INS gene mutations were sporadic cases that resulted from de novo mutations.
Previous studies have suggested that permanent neonatal diabetes mellitus caused by INS mutations within 6 months after birth accounts for 12–20% of permanent neonatal diabetes mellitus, but recent studies showed that there were also reports about neonatal diabetes mellitus with diabetes occurring up to 6 months‐of‐age, albeit at a reduced frequency20. In the present study, one of the patients diagnosed with diabetes in her 8 months of life carried the mutation p. R89C in INS. The antibody related to type 1 diabetes was negative, and the C‐peptide was very low. This is the first case report of permanent neonatal diabetes mellitus caused by the R89C mutation in INS in China. The results suggested that genetic screening was required, even in patients with older diabetic onset age (>6 months), especially with clinical manifestations of permanent neonatal diabetes mellitus.
Patients with permanent neonatal diabetes mellitus usually presented with diabetic ketoacidosis or marked hyperglycemia, and are not associated with beta‐cell autoantibodies. The C‐peptide levels were low, and lifelong insulin therapy in replacement dose was required. Usually, patients require insulin 0.6–0.8 U/kg/day, which is similar to type 1 diabetes (initial dosage: 0.4–0.5 U/kg/day, prepuberty: 0.7–1.0 U/kg/day). All of these features suggest that most patients are insulin‐dependent. Most of the patients had poor glucose control after insulin treatment, and the possible reasons were as follows: (i) insulin secretion was deficient due to INS gene mutation; and (ii) in order to meet growth and development milestones, and avoid hypoglycemia, a loose target was set. Among them, patients with mutations at p.G32 had worse glycemic control (HbA1c 8.0~9.0%) than the other positions (median HbA1c was 7.9%)13. Structural damage might help explain the phenomenon, and close monitoring during treatment is vital in patients carrying mutations located at B8. There might be other reasons to explain the heterogeneous clinical manifestations. Additionally, participants with permanent neonatal diabetes mellitus showed low birthweight (2.8 kg)13, which is consistent with the present study, where patients also showed low birthweight (2.9 kg, 10–25th).
In conclusion, we discovered one novel mutation in INS, p.I99_C100insSI, which was likely to cause permanent neonatal diabetes mellitus. It is predicted that the substitution mutation, p.G32, might lead to more severe hyperglycemia because of structural destruction. Additionally, because INS mutations are another common cause of neonatal diabetes mellitus, patients with ineffective treatment with sulfonylureas should be particularly concerned with the detection of this gene, especially for diabetes patients who were diagnosed before the age of 12 months.
Disclosure
The authors declare no conflict of interest.
Supporting information
Table S1 | Primer sequence of INS.
Acknowledgments
This work was supported by grants from National Key R&D Program of China (2017YFC1309603), National Key Research and Development Program of China (2016YFA0101002), National Natural Science Foundation of China (No. 81170736, 81570715, 81870579), and Medical Epigenetics Research Center, Chinese Academy of Medical Sciences (2017PT31036, 2018PT31021).
J Diabetes Investig 2020; 11: 578–584
References
- 1. Lemelman MB, Letourneau L, Greeley SAW. Neonatal diabetes mellitus: an update on diagnosis and management. Clin Perinatol 2018; 45: 41–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Li X, Xu A, Sheng H, et al. Early transition from insulin to sulfonylureas in neonatal diabetes and follow‐up: Experience from China. Pediatr Diabetes 2018; 19: 251–258. [DOI] [PubMed] [Google Scholar]
- 3. Cao B, Gong C, Wu D, et al. Genetic analysis and follow‐up of 25 neonatal diabetes mellitus patients in China. J Diabetes Res 2016; 2016: 6314368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kylat RI, Senguttuvan R, Bader MY. Personalized precision medicine in extreme preterm infants with transient neonatal diabetes mellitus. J Pediatr Endocrinol Metab 2017; 30: 593–596. [DOI] [PubMed] [Google Scholar]
- 5. Yorifuji T, Higuchi S, Hosokawa Y, et al. Chromosome 6q24‐related diabetes mellitus. Clin Pediatr Endocrinol 2018; 27: 59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. De Franco E, Flanagan SE, Houghton JA, et al. The effect of early, comprehensive genomic testing on clinical care in neonatal diabetes: an international cohort study. Lancet (London, England) 2015; 386: 957–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gloyn AL, Pearson ER, Antcliff JF, et al. Activating mutations in the gene encoding the ATP‐sensitive potassium‐channel subunit Kir6.2 and permanent neonatal diabetes. N Engl J Med 2004; 350: 1838–1849. [DOI] [PubMed] [Google Scholar]
- 8. Edghill EL, Flanagan SE, Patch AM, et al. Insulin mutation screening in 1,044 patients with diabetes: mutations in the INS gene are a common cause of neonatal diabetes but a rare cause of diabetes diagnosed in childhood or adulthood. Diabetes 2008; 57: 1034–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Greeley SA, Tucker SE, Naylor RN, et al. Neonatal diabetes mellitus: a model for personalized medicine. Trends Endocrinol Metab 2010; 21: 464–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shah RP, Spruyt K, Kragie BC, et al. Visuomotor performance in KCNJ11‐related neonatal diabetes is impaired in children with DEND‐associated mutations and may be improved by early treatment with sulfonylureas. Diabetes Care 2012; 35: 2086–2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015; 17: 405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liu M, Sun J, Cui J, et al. INS‐gene mutations: from genetics and beta cell biology to clinical disease. Mol Aspects Med 2015; 42: 3–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Stoy J, Edghill EL, Flanagan SE, et al. Insulin gene mutations as a cause of permanent neonatal diabetes. Proc Natl Acad Sci USA 2007; 104: 15040–15044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Moritani M, Yokota I, Tsubouchi K, et al. Identification of INS and KCNJ11 gene mutations in type 1B diabetes in Japanese children with onset of diabetes before 5 years of age. Pediat Diabetes 2013; 14: 112–120. [DOI] [PubMed] [Google Scholar]
- 15. Liu M, Weiss MA Biosynthesis, structure, and folding of the insulin precursor protein. Diabetes Obes Metab 2018; 20(Suppl 2): 28–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shoelson S, Fickova M, Haneda M, et al. Identification of a mutant human insulin predicted to contain a serine‐for‐phenylalanine substitution. Proc Natl Acad Sci USA 1983; 80: 7390–7394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nakagawa SH, Zhao M, Hua QX, et al. Chiral mutagenesis of insulin. Foldability and function are inversely regulated by a stereospecific switch in the B chain. Biochemistry 2005; 44: 4984–4999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Busiah K, Drunat S, Vaivre‐Douret L, et al. Neuropsychological dysfunction and developmental defects associated with genetic changes in infants with neonatal diabetes mellitus: a prospective cohort study [corrected]. Lancet Diabetes Endocrinol 2013; 1: 199–207. [DOI] [PubMed] [Google Scholar]
- 19. Ron D. Proteotoxicity in the endoplasmic reticulum: lessons from the Akita diabetic mouse. J Clin Investig 2002; 109: 443–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Letourneau LR, Carmody D, Wroblewski K, et al. Diabetes presentation in infancy: high risk of diabetic ketoacidosis. Diabetes Care 2017; 40: e147–e148. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 | Primer sequence of INS.