

Abstract
Aims/Introduction
We aimed to investigate the nationwide incidence, treatment details and outcomes of patients with endogenous hyperinsulinemic hypoglycemia (EHH), including those with transient/persistent congenital hyperinsulinism (CHI), insulinoma, non‐insulinoma pancreatogenous hypoglycemia syndrome and insulin autoimmune syndrome (Hirata’s disease) in Japan.
Materials and Methods
A nationwide, questionnaire‐based survey was carried out to determine the number of patients with EHH who were treated for hypoglycemia or hypoglycemia‐related complications in 2017−2018. The questionnaires were sent to all hospitals in Japan with >300 beds, and with pediatric and/or adult clinics likely managing EHH patients. The secondary questionnaires were sent to obtain the patients’ date of birth, sex, age at onset, treatment details and post‐treatment outcomes.
Results
A total of 447 patients with CHI (197 transient CHI, 225 persistent CHI and 25, unknown histology), 205 with insulinoma (118 benign, 18 malignant and 69 unknown subtype), 111 with non‐insulinoma pancreatogenous hypoglycemia syndrome (33 post‐gastric surgery HH, 57 postprandial HH, 10 nesidioblastosis and 11 unknown subtype) and 22 with insulin autoimmune syndrome were identified. Novel findings included: (i) marked improvement in the prognosis of persistent CHI over the past 10 years; (ii) male dominance in the incidence of transient CHI; (iii) non‐insulinoma pancreatogenous hypoglycemia syndrome emerging as the second most common form of EHH in adults; (iv) frequent association of diabetes mellitus with insulin autoimmune syndrome; and (v) frequent post‐treatment residual hypoglycemia and impaired quality of life.
Conclusions
The first nationwide, all age group survey of EHH showed the current status of each type of EHH disorder and the unmet needs of the patients.
Keywords: Hyperinsulinism, Hypoglycemia, Surveys
The first nationwide, all age‐group survey of endogenous hyperinsulinemic hypoglycemia showed the current status of each type of endogenous hyperinsulinemic hypoglycemia disorder and the unmet needs of the patients.

Introduction
Endogenous hyperinsulinemic hypoglycemia (EHH) is defined as a group of disorders causing hyperinsulinemic hypoglycemia without exogenous administration of insulin or its secretagogues, with various etiologies. The effector arm, however, is common across these disorders – persistent endogenous hyperinsulinemia causing neuroglycopenic and neurogenic symptoms1. These disorders have the same differential diagnosis and common treatment strategies. Compared with hypoglycemia associated with diabetes mellitus, few standard clinical guidelines exist for the treatment of EHH, and few formal clinical trials have been carried out to treat patients with this condition, causing difficulty for patients and medical caregivers. In the present study, we aimed to carry out a nationwide survey to explore the incidence of EHH and the treatment details, post‐treatment outcomes, and quality of life of patients with this condition.
Methods
Definition of EHH
Before the study, EHH was defined as a group of disorders causing hyperinsulinemic hypoglycemia without exogenous administration of insulin or its secretagogues. It included congenital hyperinsulinism (CHI), insulinoma, non‐insulinoma pancreatogenous hypoglycemia syndrome (NIPHS)2 and insulin autoimmune syndrome (Hirata’s disease)3. CHI was further categorized into transient or persistent CHI, depending on its resolution before 3 months of life4. Similarly, NIPHS was further divided into three categories, post‐gastric surgery HH, postprandial HH and adult‐onset nesidioblastosis (β‐cell hyperplasia)5, 6, 7.
Survey procedure
The primary questionnaires were administered to determine the number of patients with each of the EHH disorders who were treated for hypoglycemia or hypoglycemia‐related complications in 2017–2018. The questionnaires were sent to all hospitals in Japan with >300 beds (https://hospia.jp/hoslist, accessed 1/12/2018), and with pediatric and/or adult clinics that are likely managing EHH patients. Briefly, when departments of endocrinology, metabolism or diabetology were present, these were prioritized over general internal medicine or pediatrics as addressees. For pediatric clinics, all neonatology clinics were also included as addressees.
Secondary questionnaires were sent to the responding clinics with at least one EHH patient and contained disorder‐specific questions. For CHI, the questionnaire contained questions on the patients’ sociodemographic information (date of birth, sex, age at onset), treatment details (nutritional treatment including tube‐feeding, gastrostomy, uncooked cornstarch; medications including diazoxide, somatostatin analogs, glucagon, glucocorticoids, alpha‐glucosidase inhibitors, calcium channel blockers, and mammalian target of rapamycin [mTOR] inhibitors; or pancreatectomy with its extent) and post‐treatment outcomes (residual hypoglycemia, diabetes mellitus, neurological abnormalities and quality of life) of the patients. Similarly, for insulinoma, the questionnaire included questions related to the patients’ date of birth, sex, age at onset, histological diagnosis, treatment details (nutritional treatment, medications, pancreatectomy with its extent) and post‐treatment outcomes. For NIPHS, the questionnaire contained questions related to the patients’ date of birth, sex, age at onset, disease category, treatment details and post‐treatment outcomes. For insulin autoimmune syndrome, the questionnaire contained questions related to the patients’ date of birth, sex, age at onset, comorbidities, treatment details and post‐treatment outcomes.
The study protocol was approved by the institutional review board of Osaka City General Hospital (approval no. 1812106).
Results
Nationwide survey
We sent primary questionnaires to 1,717 clinics (939 adult clinics and 778 pediatric clinics) of 947 hospitals, and received responses from 359 (38.2%) adult and 530 (68.1%) pediatric clinics. Of these, 312 clinics (144 adult clinics and 168 pediatric clinics) experienced EHH patients during the study period. Secondary questionnaires were sent to these clinics, and responses were obtained from 255 clinics (108 adult clinics and 147 pediatric clinics).
After excluding overlapping patients with the date of birth, sex and age at onset, 785 with EHH were finally identified. Of them, 447 had CHI (197 transient, 225 persistent and 25 unknown subtype), 205 had insulinoma (118 benign, 18 malignant and 69 unknown histology), 111 had NIPHS (33 post‐gastric surgery, 57 postprandial, 10 adult‐onset nesidioblastosis and 11 unknown etiology) and 22 had insulin autoimmune syndrome.
Congenital hyperinsulinism
Table 1 summarizes the survey results for CHI. The subtypes could not be specified in 25 newborns at the time of the survey.
Table 1.
Summary of the survey results for congenital hyperinsulinism
| Transient CHI | Persistent CHI | Unknown | Total | |
|---|---|---|---|---|
| No. patients (%) | ||||
| Total | 197 | 225 | 25 | 447 |
| Male | 125 (63.5) | 120 (53.3) | 14 (56.0) | 259 (57.9) |
| Female | 72 (36.5) | 105 (46.7) | 11 (44.0) | 188 (42.1) |
| Age at onset | ||||
| Median | 0 day | 0 day | 0 day | 0 day |
| Range | 0 day–1 month | 0 day–2 years 4 months | 0 day–2 months | 0 day–2 years 4 months |
| Treatment (%) | ||||
| Nutritional treatment | 80 (40.6) | 124 (55.1) | 10 (40.0) | 214 (47.9) |
| Diazoxide | 99 (50.3) | 213 (94.7) | 14 (56.0) | 326 (72.9) |
| Somatostatin analogs | 1 (0.5) | 58 (25.8) | 0 (0) | 59 (13.2) |
| Glucagon | 9 (4.6) | 29 (12.9) | 3 (12.0) | 41 (9.2) |
| Glucocorticoids | 20 (10.2) | 31 (13.8) | 3 (12.0) | 54 (12.1) |
| Alpha‐glucosidase inhibitors | 0 (0) | 3 (1.3) | 0 (0) | 3 (0.7) |
| Calcium channel blockers | 0 (0) | 2 (0.9) | 0 (0) | 2 (0.4) |
| mTOR inhibitors | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Pancreatectomy | 0 (0) | 25 (11.1) | 0 (0) | 25 (5.6) |
| Posttreatment complications (%) | ||||
| Residual hypoglycaemia | 1 (0.5) | 80 (35.6) | 2 (8.0) | 83 (18.6) |
| Diabetes mellitus | 2 (1.0) | 14 (6.2) | 0 (0) | 16 (3.6) |
| Developmental delay | 23 (11.7) | 63 (28.0) | 4 (16.0) | 90 (20.1) |
| Epilepsy | 4 (2.0) | 32 (14.2) | 1 (4.0) | 37 (8.3) |
Abbreviations: CHI, congenital hyperinsulinism; mTOR, mammalian target of rapamycin.
Although most of these CHI patients developed hypoglycemia soon after birth, some patients, especially in the persistent CHI group, developed hypoglycemia later in life (Figure 1).
Figure 1.
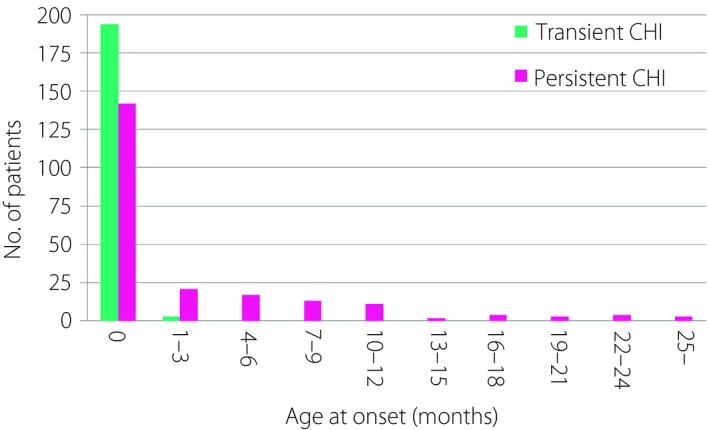
Age at onset of patients with transient and persistent congenital hyperinsulinism. CHI, congenital hyperinsulinism.
In patients with transient CHI, other than intravenous dextrose infusion, treatment modalities mostly included nutritional support and/or diazoxide therapy. Although glucocorticoids were used in 10.2% of the patients, other medications including somatostatin analogs and glucagon were used only for a limited number of patients, and none of them received alpha‐glucosidase inhibitors, calcium channel blockers, mTOR inhibitors or underwent pancreatectomy. In patients with persistent CHI, other treatment modalities were used more often: somatostatin analogs (25.8%), glucagon (12.9%), glucocorticoid (13.8%), alpha‐glucosidase inhibitors (1.3%), calcium channel blockers (0.9%) and pancreatectomy (11.1%).
As expected, even with the intensified treatment, residual hypoglycemia was prevalent (35.6%) in patients with persistent HI. Neurological complications, such as developmental delay and epilepsy, were also commonly observed in 28.0% and 14.2% of the patients, respectively.
Some patients, even those with transient CHI, developed developmental delay (12%), epilepsy (2.0%), ketotic hypoglycemia (0.5%) and diabetes mellitus (1.0%). Two patients with transient CHI who developed diabetes mellitus as a late complication had mutations in the ABCC8 gene.
CHI patients born in 2017–2018
As many transient CHI patients without complications were expected to be lost to follow up and not represented in Table 1, we then focused on CHI patients who were born during the survey period (2017–2018; Table 2).
Table 2.
Treatment modalities and outcomes of patients with transient or persistent congenital hyperinsulinism born in 2017–2018
| Transient CHI | Persistent CHI | |
|---|---|---|
| No. patients (%) | ||
| Total | 137 | 59 |
| Male | 83 (60.6) | 35 (59.3) |
| Female | 54 (39.4) | 24 (40.7) |
| Treatment (%) | ||
| Nutritional treatment | 59 (43.1) | 32 (54.2) |
| Diazoxide | 68 (49.6) | 57 (96.6) |
| Somatostatin analogs | 0 (0) | 8 (13.6) |
| Glucagon | 5 (3.6) | 4 (6.8) |
| Glucocorticoids | 12 (8.8) | 8 (13.6) |
| mTOR inhibitors | 0 (0) | 0 (0) |
| Pancreatectomy | 0 (0) | 1 (1.7) |
| Posttreatment complications (%) | ||
| Residual hypoglycemia | 0 (0) | 22 (37.3) |
| Diabetes mellitus | 0 (0) | 1 (1.7) |
| Developmental delay (%) | ||
| Total | 11 (8.0) | 11 (18.6) |
| Mild | 7 (5.1) | 2 (3.4) |
| Moderate | 2 (1.5) | 3 (5.1) |
| Severe | 2 (1.5) | 6 (10.2) |
| epilepsy | 2 (1.5) | 6 (10.2) |
Abbreviations: Mild, moderate and severe developmental delay were defined as developmental or intelligence quotient of 50–70, 30–49 and <30, respectively. mTOR, mammalian target of rapamycin.
Of the 197 patients with transient CHI, 137 were born in 2017–2018, translating to the annual incidence of transient CHI of at least one in 13,600 births. Transient CHI was more prevalent in males than in females (P = 0.0355 by the χ2‐test). Similarly, of the 225 patients with persistent CHI, 59 were born in 2017–2018, translating to the annual incidence of persistent CHI of at least one in 31,600 births. Contrary to transient CHI, there was no significant sex difference in the incidence of persistent CHI (P = 0.266).
When the treatment modalities and outcomes of transient and persistent CHI were compared, residual hypoglycemia and post‐treatment diabetes mellitus were found only in patients with persistent CHI. Notably, neurological complications, including developmental delay or epilepsy, were more common and more severe in patients with persistent CHI than in those with transient CHI.
Secular changes in pancreatectomy and outcomes of persistent CHI
Next, we compared the treatment modalities and the outcomes of patients with persistent CHI diagnosed before and after 2009 (Table 3).
Table 3.
Secular changes in the surgical treatment and outcomes of patients with persistent congenital hyperinsulinism
| Year at diagnosis | Before 2009 | 2009–2018 |
|---|---|---|
| No. (%) | ||
| Total | 62 | 162 |
| Male | 29 (46.8) | 91 (56.2) |
| Female | 33 (53.2) | 71 (43.8) |
| Treatment (%) | ||
| Nutritional treatment | 33 (53.2) | 92 (56.8) |
| Diazoxide | 57 (91.9) | 155 (95.7) |
| Somatostatin analogs | 13 (21.0) | 45 (27.8) |
| Glucagon | 7 (11.3) | 22 (13.6) |
| Glucocorticoids | 8 (12.9) | 23 (14.2) |
| Alpha‐glucosidase inhibitors | 2 (3.2) | 1 (0.5) |
| Calcium channel blockers | 1 (1.6) | 1 (0.5) |
| mTOR inhibitors | 0 (0) | 0 (0) |
| Pancreatectomy (%) | ||
| Total | 11 (17.7) | 14 (8.6) |
| Near/subtotal | 10 (16.1) | 4 (2.5) |
| Partial | 1 (1.6) | 9 (5.6) |
| Unknown | 0 (0) | 1 (0.5) |
| Posttreatment complications (%) | ||
| Residual hypoglycemia | 18 (29.0) | 62 (38.3) |
| Diabetes mellitus (%) | ||
| Total | 13 (21.0) | 1 (6.2) |
| Post‐pancreatectomy | 10 (16.1) | 0 (0) |
| Developmental delay | 25 (40.3) | 38 (23.5) |
| Epilepsy | 15 (24.4) | 17 (10.5) |
Patients diagnosed before and after 2009 were compared. mTOR, mammalian target of rapamycin.
In terms of treatment, the most significant change was the clear shift toward partial pancreatectomy from near/subtotal pancreatectomy (Table 3). Before 2009, 91.0% of the pancreatectomies for CHI were near/subtotal; after 2009, partial pancreatectomy represented 64.3%, whereas just four underwent near/subtotal pancreatectomy. Because of the shift toward partial pancreatectomy, there had been a dramatic decrease in the number of patients with post‐treatment diabetes mellitus over the years. In total, 14 patients with post‐treatment diabetes mellitus were identified in the study. Of them, 13 were treated before 2009, 10 with a history of near/subtotal pancreatectomy. In contrast, there was only one patient with diabetes who was treated after 2009.
Insulinoma
Table 4 shows the survey results for insulinoma. The estimated prevalence was 0.16 per 100,000 population. As previously described8, insulinoma was more prevalent among female patients than among male patients (140/65), although the sex difference was smaller in those with malignant cases (10/8). The median ages of onset for benign and malignant insulinoma were 53 and 49 years, respectively (Figure 2). Reflecting the nature of the study, pediatric patients were represented better in the present study compared with those in previous studies, which mostly focused on adult Japanese patients with neuroendocrine tumors8, 9, 10. In total, 11 patients developed insulinoma before the age of 20 years: one at the age of 5 years, one at the age of 6 years, one at the age of 7 years and the remaining eight had onset between the ages of 12 and 17 years.
Table 4.
Summary of the survey results for insulinoma
| Benign | Malignant | Unknown | Total | |
|---|---|---|---|---|
| No. patients (%) | ||||
| Total | 118 | 18 | 69 | 205 |
| Male | 38 (32.2) | 8 (44.4) | 19 (27.5) | 65 (31.7) |
| Female | 80 (67.8) | 10 (55.6) | 50 (72.5) | 140 (68.3) |
| Age at onset | ||||
| Median (years) | 53 | 49 | 58 | 55 |
| Range | 5–88 | 24–86 | 6–98 | 5–98 |
| Treatment (%) | ||||
| Nutritional treatment | 32 (27.1) | 3 (16.7) | 14 (20.3) | 49 (23.9) |
| Diazoxide | 31 (26.3) | 12 (66.7) | 24 (34.8) | 67 (32.7) |
| Somatostatin analogs | 8 (6.8) | 13 (72.2) | 12 (17.4) | 33 (16.1) |
| Glucagon | 0 (0) | 0 (0) | 2 (2.9) | 2 (1.0) |
| Glucocorticoids | 2 (1.7) | 0 (0) | 4 (5.8) | 6 (3.0) |
| Alpha‐glucosidase inhibitors | 4 (3.4) | 5 (27.8) | 12 (17.4) | 21 (10.2) |
| Calcium channel blockers | 4 (3.4) | 0 (0) | 2 (2.9) | 6 (2.9) |
| mTOR inhibitors | 0 (0) | 11 (61.1) | 2 (2.9) | 13 (6.3) |
| Pancreatectomy (%) | ||||
| Total | 118 (100) | 11 (61.1) | 30 (43.5) | 159 (77.6) |
| Tumor enucleation | 43 (36.4) | 0 (0) | 9 (13.0) | 52 (25.4) |
| Body and/or tail resection | 58 (49.2) | 9 (50.0) | 13 (18.8) | 80 (39.0) |
| Pancreatoduodenectomy | 13 (11.0) | 2 (11.1) | 4 (5.8) | 19 (9.3) |
| Subtotal resection | 0 (0) | 0 (0) | 1 (1.4) | 1 (0.5) |
| Unknown | 4 (3.4) | 0 (0) | 3 (4.3) | 7 (3.4) |
| Posttreatment complications (%) | ||||
| Residual hypoglycemia | 1 (0.8) | 9 (50.0) | 19 (27.5) | 29 (14.1) |
| Diabetes mellitus | 16 (13.6) | 4 (22.2) | 7 (10.1) | 27 (13.2) |
| Dementia | 0 (0) | 0 (0) | 7 (10.1) | 7 (3.4) |
| Epilepsy | 6 (5.1) | 0 (0) | 1 (1.4) | 7 (3.4) |
| Other neurological deficits | 4 (3.4) | 1 (5.6) | 3 (4.3) | 8 (3.9) |
Abbreviation: mTOR, mammalian target of rapamycin.
Figure 2.
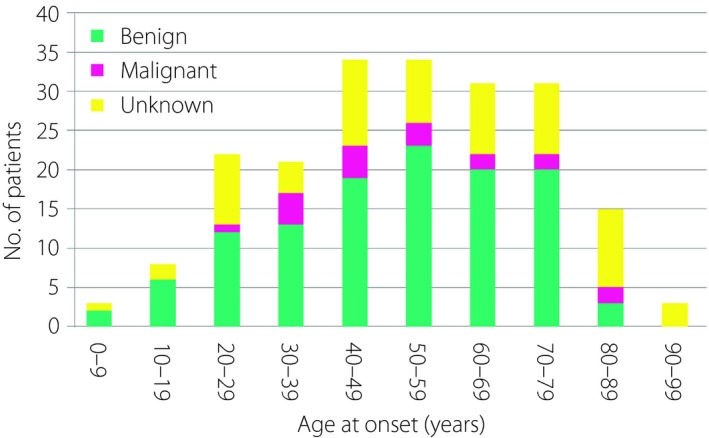
Age at onset of patients with insulinoma.
Medical treatments were more aggressive as compared with those for other forms of EHH; diazoxide was used in 32.7% of patients with insulinoma, somatostatin analog in 16.1%, glucagon in 1.0%, glucocorticoid in 3.0%, alpha‐glucosidase inhibitors in 10.2%, calcium channel blockers in 2.9% and mTOR inhibitors in 6.3%. Pancreatectomy was carried out in 159 patients (77.6%): 52 underwent tumor enucleation, 80 underwent body and/or tail resection, 19 underwent pancreatoduodenectomy, and one underwent subtotal resection (Tables 4,5). In seven patients, the extent of surgery was unknown. Post‐treatment residual hypoglycemia was found in 29 patients (14.1%; Table 5), and was common among those who did not undergo pancreatectomy (21 patients). Post‐surgery residual hypoglycemia was found in eight patients: one had benign, five had malignant, and two had unknown histology. By contrast, post‐treatment diabetes mellitus was more prevalent and found in 25 (15.8%) patients who underwent pancreatectomy: 3.8% of those who had tumor enucleation, 21.3% of body and/or tail resection, 26.3% of pancreatoduodenectomy, and 100% of subtotal resection. Post‐treatment diabetes mellitus was less frequent among patients who did not undergo surgery; just two (4.3%) developed diabetes.
Table 5.
Post‐treatment complications for insulinoma
| Patients with pancreatectomy | Patients without pancreatectomy | ||||||
|---|---|---|---|---|---|---|---|
| Total | Tumor enucleation | Body and/or tail resection | Pancreato‐ duodenectomy | Subtotal resection | Unknown | ||
| No. patients | 159 | 52 | 80 | 19 | 1 | 7 | 46 |
| Residual hypoglycemia | 8 (5.1%) | 0 (0%) | 6 (7.5%) | 1 (5.3%) | 1 (100%) | 0 (0%) | 21 (44.7%) |
| Diabetes mellitus | 25 (15.8%) | 2 (3.8%) | 17 (21.3%) | 5 (26.3%) | 1 (100%) | 0 (0%) | 2 (4.3%) |
| Dementia | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 7 (14.9%) |
| Epilepsy | 6 (3.8%) | 1 (1.9%) | 4 (5.0%) | 1 (5.3%) | 0 (0%) | 0 (0%) | 1 (2.1%) |
| Other neurological deficits | 5 (3.2%) | 1 (1.9%) | 4 (5.0%) | 0 (0%) | 0 (0%) | 0 (0%) | 3 (6.4%) |
Even after undergoing these extensive treatments, neurological impairment still occurred in 22 (10.7%) patients with insulinoma: seven had dementia, seven had epilepsy and eight had other neurological deficits. In addition, impaired quality of life was often reported in patients with residual hypoglycemia (9/29), which included persistent hypoglycemia requiring frequent snacks causing obesity or frequent hypoglycemic loss of consciousness.
Non‐insulinoma pancreatogenous hypoglycemia syndrome
Table 6 summarizes the survey results for NIPHS. The estimated prevalence was 0.09 per 100,000 population. Of the 33 patients with post‐gastric surgery HH (23 males and 10 females), the median age of onset was 69 years (range 1–89 years), although the age distribution was bimodal: the first peak was observed at ages 0–9 years, whereas the second peak occurred after 30 years (Figure 3). Although the treatment modalities were mostly nutritional support and/or administration of alpha‐glucosidase inhibitors, one patient underwent pancreatectomy. After a median follow up of 3.2 years (range 0.4–34.2 years), 13 patients (39.4%) had residual hypoglycemia. The associated comorbidities included cerebral infarction and dementia after an episode of atrial fibrillation (onset 69 years), cerebral infarction associated with asplenia and single ventricle (onset 1 year), and severe multiple handicaps (onset 4 years). One adult‐onset patient (onset 75 years) reported frequent episodes of morning hypoglycemia associated with muscle weakness.
Table 6.
Summary of the survey results for non‐insulinoma pancreatogenous hypoglycemia syndrome and insulin autoimmune syndrome (Hirata’s disease)
| Non‐insulinoma pancreatogenous insulin autoimmune syndrome | Insulin autoimmune syndrome (Hirata’s disease) | ||||
|---|---|---|---|---|---|
| Post‐gastric surgery HH | No surgery | ||||
| Postprandial HH | Adult nesidioblastosis | Unknown | |||
| No. patients (%) | |||||
| Total | 33 | 57 | 10 | 11 | 22 |
| Male | 23 (69.7) | 21 (36.8) | 5 (50.0) | 4 (36.4) | 10 (45.5) |
| Female | 10 (30.3) | 36 (63.2) | 5 (50.0) | 7 (63.6) | 12 (54.4) |
| Age at onset | |||||
| Median (years) | 69 | 41 | 49 | 43 | 63 |
| Range | 1–89 | 0.7–91 | 19–79 | 12–82 | 3–88 |
| Treatment (%) | |||||
| Nutritional Treatment | 18 (54.5) | 26 (45.6) | 1 (10.0) | 4 (36.4) | 8 (36.4) |
| Diazoxide | 2 (6.1) | 0 (0) | 7 (70.0) | 3 (27.3) | 0 (0) |
| Somatostatin analogs | 0 (0) | 0 (0) | 2 (20.0) | 0 (0) | 0 (0) |
| Glucagon | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (4.5) |
| Glucocorticoids | 2 (6.1) | 4 (7.0) | 1 (10.0) | 1 (9.1) | 3 (13.6) |
| Alpha‐glucosidase inhibitors | 18 (54.5) | 19 (33.3) | 3 (30.0) | 1 (9.1) | 11 (50.0) |
| Calcium channel blockers | 0 (0) | 1 (1.8) | 0 (0) | 0 (0) | 1 (4.5) |
| mTOR inhibitors | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Pancreatectomy | 1 (3.0) | 0 (0) | 6 (60.0) | 0 (0) | 0 (0) |
| Posttreatment complications (%) | |||||
| Residual hypoglycemia | 13 (39.4) | 17 (29.8) | 4 (40.0) | 5 (45.5) | 4 (18.2) |
| Diabetes mellitus | 4 (12.1) | 6 (10.5) | 1 (10.0) | 1 (9.1) | 8 (36.4) |
| Developmental delay | 1 (3.0) | 7 (12.3) | 2 (20.0) | 1 (9.1) | 0 (0) |
| Epilepsy | 2 (6.1) | 4 (7.0) | 1 (10.0) | 0 (0) | 0 (0) |
Abbreviations: HH, hyperinsulinemic hypoglycaemia; mTOR, mammalian target of rapamycin.
Figure 3.
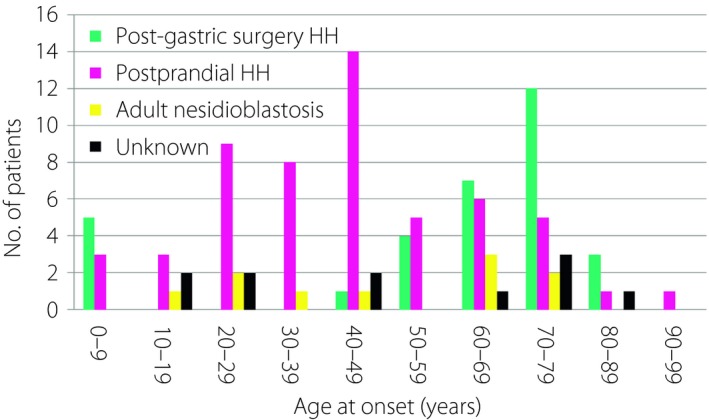
Age at onset of non‐insulinoma pancreatogenous hypoglycemia syndrome. HH, hyperinsulinemic hypoglycemia.
Of the 57 patients with postprandial HH (21 males and 36 females), the median age at onset was 41 years (range 8 months–91 years; Figure 3). Management consisted mostly of dietary treatment and/or administration of alpha‐glucosidase inhibitors. After a median follow up of 2.5 years (range 0.5–19.4 years), 17 patients (29.3%) still had residual hypoglycemia. Six patients (10.5%) had intellectual disabilities, whereas four (7.0%) had epilepsy. Five patients with severe developmental delay had congenital abnormalities: cerebral palsy in two, Fukuyama‐type congenital muscular dystrophy in one, congenital cytomegalovirus infection in one and de Lange syndrome in one. Two adult‐onset patients reported difficulties in driving a car, and one reported an episode of hypoglycemic coma.
Of the 10 patients with adult‐onset nesidioblastosis (five men and five women), the median age of onset was 49 years (range 19–79 years; Figure 3). Medical management included dietary treatment in one patient, diazoxide in seven, somatostatin analog in two, glucocorticoid in one, calcium channel blockers in one and alpha‐glucosidase inhibitors in three. Six patients underwent pancreatic resection resulting in the histological diagnosis of nesidioblastosis. The surgical procedures included body and/or tail resection in four patients, and pancreatoduodenectomy in one. In one patient, the extent of pancreatic resection was unknown.
After a median follow up of 5.65 years (range 3.9–16.2 years), four patients (40%) still presented with residual hypoglycemia. Post‐treatment diabetes mellitus was reported in one patient, and neurological abnormalities in two patients.
Of the 11 patients (four males and seven females) with NIPHS of unknown etiologies, the median age of onset was 43 years (range 12–82 years). Medical management included dietary treatment in four patients, diazoxide in three, glucocorticoid in one and alpha‐glucosidase inhibitor in one. None of the patients underwent pancreatectomy.
After a median follow up of 1.8 years (range 0.6–49 years), five patients (45.4%) still presented with residual hypoglycemia, one with diabetes mellitus and one with intellectual disabilities. Two patients reported psychological problems: one had impaired cognitive function, while the other had schizophrenia.
Insulin autoimmune syndrome
Table 6 summarizes the survey results for insulin autoimmune syndrome (Hirata’s disease). The estimated prevalence was 0.017 per 100,000 population. The median age of onset was 63 years (range 3–88 years; Figure 4). The associated comorbidities were diabetes mellitus (12), Graves’ disease (2), rheumatoid arthritis (2) and vasculitis (1). Management mostly included dietary treatment and/or by the administration of alpha‐glucosidase inhibitors. After a median follow up of 4.54 years (range 0.55–21.9 years), four patients (18.2%) still presented with hypoglycemia, and eight (36.4%) with diabetes mellitus. Two patients had deteriorated cognitive function.
Figure 4.
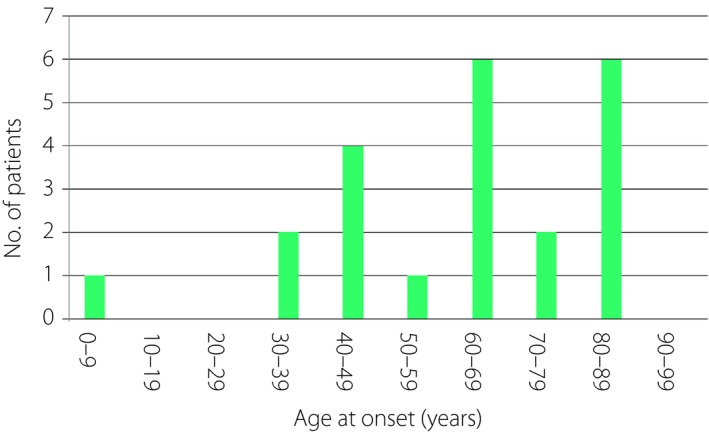
Age at onset of patients with insulin autoimmune syndrome (Hirata’s disease).
Discussion
This is the first nationwide survey to report the different etiologies of EHH in all ages: neonates, childhood and adulthood. As we used a more systematic approach, we were able to identify more patients compared with previous studies, which focused on a limited number of specialized centers11.
The estimated incidence of transient CHI (1/13,600 births) and persistent CHI (1/31,600 births) in 2017–2018 was higher than those reported by Kawakita et al. 12, 13 for patients born in 2007–2009: 1/17,000 for transient CHI and 1/35,400 for persistent CHI. It remains unclear whether this increase reflects the actual increasing trend or is caused by other factors, such as increased capture rate or increased awareness of these disorders.
Interestingly, transient CHI was significantly more common in males (male/female = 1.74), but there was no sex difference in the incidence of persistent CHI. This finding was also reported in a previous study carried out in 2007–2009 in Japan (male/female = 2.20)12. This difference cannot be explained by the increased male/female ratio (approximately 1.05) of live births or by the increased incidence of male neonates with lower birth weights (Vital Statistics by the Ministry of Health, Labor and Welfare of Japan, https://www.mhlw.go.jp/english/database/db-hw/vs01.html).
As previously reported, neurological sequelae were frequently observed in patients with CHI14, 15, 16. In patients born in 2017–2018, developmental delay and epilepsy were more commonly observed in patients with persistent CHI (developmental delay 18.6%, epilepsy 10.2%) than in those with transient CHI (developmental delay 8.0%, epilepsy 1.5%; Table 3). The developmental delay was more severe in patients with persistent CHI. This finding is in contrast with the results of a previous study, which reported a similar incidence, and the severity of neurological complications in patients with transient and persistent CHI17. Although the reason for this difference is unclear, this might be due to the smaller number of patients in the previous study (33 transient, 34 persistent)17. In fact, in the previous study, although there was no statistical significance, the proportion of patients with neurological abnormality was larger in persistent CHI (47%), as compared with transient (30%)17.
As the severity of initial hypoglycemia does not significantly differ between patients with transient CHI and those with persistent CHI12, the present findings might support the encouraging notion that the neurological outcome can be improved by better medical care after the initial episodes to avoid further hypoglycemic episodes.
With regard to the post‐treatment diabetes mellitus, there was a significant decline in its incidence over the past 10 years. In Japan, the pre‐emptive identification of the focal form of adenosine triphosphate‐sensitive potassium channel CHI by fluorine‐18‐l‐dihydroxyphenylalanine positron emission tomography followed by local surgical resection was first reported in 2009, and has gradually become the standard management13, 18. With the progress in the diagnosis of focal CHI, more aggressive medical treatments of diazoxide‐unresponsive CHI using continuous octreotide infusion and/or enteral nutrition using gastrostomy or nasogastric feeding have also become the standard care19, 20 (management flowchart available in Yorifuji et al.13). These factors led to the remarkable decrease in the number of patients treated with near/subtotal pancreatectomy (only one since 2009), thus causing a decline in the incidence of post‐treatment diabetes.
The number of insulinoma patients, the age distribution, female predominance and the proportion of patients with malignant pathology (8.8%) were similar to those reported in previous studies8, 9, 10.
Reflecting the more severe nature of the disorder, medical treatments other than diet and alpha‐glucosidase inhibitors were more often used in these patients. Off‐label use of somatostatin analogs or mTOR inhibitors was common in patients with insulinoma.
However, post‐surgical diabetes mellitus and residual hypoglycemia remain the major endocrinological problems of these patients. The incidence of residual hypoglycemia was lower in patients who underwent pancreatectomy than in those who received medical treatment alone. However, the incidence of post‐surgery diabetes mellitus is clearly elevated in patients who underwent body/tail resection, pancreatoduonectomy or subtotal pancreatectomy compared with those who had tumor enucleation.
Non‐insulinoma pancreatogenous hypoglycemia syndrome was first described as a syndrome characterized by endogenous hyperinsulinemic hypoglycemia in adults not caused by insulinoma2. Histologically, these patients show features of nesidioblastosis, which is similar to that observed in patients with CHI, and the clinical features are postprandial hypoglycemia without fasting hypoglycemia5. Post‐gastric surgery hypoglycemia, which has similar clinical and histological features, is regarded as a different entity, but is often described as a form of NIPHS5.
The current survey included post‐gastric surgery HH, postprandial HH and adult‐onset nesidioblastosis as forms of NIPHS. Although post‐gastric surgery HH can be clearly distinguished by history, the distinction between postprandial HH and adult‐onset nesidioblastosis is not entirely clear. Nesidioblastosis is a histological term for islet hyperplasia associated with the continuous budding of islet cells from the ductal epithelium6, 7, 21; therefore, the diagnosis of adult‐onset nesidioblastosis should not be made without the results of patients’ pancreatic histology. In real life, however, the diagnosis is often used to describe patients with features of NIPHS without histological confirmation7.
Although few systematic epidemiological studies were carried out to explore this condition, NIPHS is believed to be a very rare condition affecting <100 patients described in the literature during 1975–20147. The current nationwide survey identified 67 patients, excluding 33 with post‐gastric surgery HH. The incidence, therefore, is approximately one‐third of that for insulinoma: rare, but not extremely rare.
As previously reported, postprandial HH and adult‐onset nesidioblastosis appeared to be more common in females, whereas post‐gastric surgery HH was more common in males (Table 6), which is one of the features that distinguishes post‐gastric surgery HH from other forms of NIPHS5.
Although the median ages at onset for post‐gastric surgery HH, postprandial HH and adult‐onset nesidioblastosis were 69, 41 and 49 years, respectively, the age distribution of post‐gastric surgery HH was bimodal; its first peak occurred at the age of <10 years, whereas the second peak occurred after 40 years. Each of these findings reflects the specific age when surgical intervention can be carried out in children with gastroesophageal reflux (fundoplication) and in adults with obesity (gastric bypass) or gastric cancer (gastrectomy)22, 23.
Nutritional management and alpha‐glucosidase inhibitors were more commonly used in patients with NIPHS compared with those with persistent CHI or insulinoma. However, hypoglycemia was not well controlled in a significant fraction of these patients (29.8–40.0%). As all forms of EHH have similar pathophysiological backgrounds, intensified treatment strategies used for persistent CHI or insulinoma might be applicable in these patients24, 25, 26.
Insulin autoimmune syndrome (Hirata’s disease) was first described in 19703. It is characterized by the production of low‐affinity, high‐binding capacity autoantibodies against insulin, which causes spontaneous HH in patients without exogenous insulins27. The disorder appears to be more common in Asian countries including Japan where the majority of cases are drug induced (e.g., methimazole and alpha‐lipoic acids) and transient28, 29, 30. In other parts of the world, however, the syndrome is more often associated with autoimmune or hematological disorders30. In this survey, 22 patients with insulin autoimmune syndrome were registered. As previously reported30, the median age at onset was 63 years, and males and females were equally affected.
In contrast to previous reports, 50% of these patients had comorbid diabetes mellitus, and one patient developed this disease at the age of 3 years. In addition, 18.2% of the patients still presented with hypoglycemia 2.1–21.9 years after the disease onset.
Whether these patients with diabetes mellitus were on insulin therapy or not was not specifically asked in the survey and remained unclear. Generally, when administered exogenous insulins, many patients develop antibodies against insulin; normally, these antibodies have high affinity and do not cause hypoglycemia27. However, there have been reports of insulin‐treated patients who developed low‐affinity, high‐capacity insulin autoantibodies similar to those found in patients with insulin autoimmune syndrome, resulting in unstable blood glucose control31, 32, 33. These patients with diabetes as comorbidity might have such conditions.
Hirata’s disease has also been reported as the cause of transient hypoglycemia in children34, 35, 36, who could have lower levels of blood insulin than in older patients, making this disorder as an important pitfall in the differential diagnosis of childhood HH.
There were limitations of the present study. First, compared with the pediatric clinics, the response rate from the adult clinics was lower (68.1% vs 38.2%), which might have affected the estimated prevalence of each EHH disorder. However, the response rate was similar to the average response rate for academic surveys in non‐Western countries (37.8%)37, and was higher than that of the previous Japanese national survey for the estimation of neuroendocrine tumors (10.8%)8. Second, to place more emphasis on the overall landscape, we did not go into the details of the clinical course enough to analyze the contribution of different factors affecting the clinical outcome. This needs to be addressed in future studies.
The differences from previous Japanese reports on insulin autoimmune syndrome might suggest that the disease structure in Japanese is changing.
Disclosure
The authors declare no conflict of interest.
Acknowledgments
We thank Mitsuyasu Takahashi of the Patients Support Group for his help in the study, and all our colleagues in Japan who responded to the survey. This work was supported by a grant‐in‐aid for scientific research from the Ministry of Health, Labor and Welfare of Japan (Research on Measures for Intractable Diseases 19FC1008).
J Diabetes Investig 2020; 11: 554–563
References
- 1. Cryer PE. Symptoms of hypoglycemia, thresholds for their occurrence, and hypoglycemia unawareness. Endocrinol Metab Clin North Am 1999; 28: 495–500. [DOI] [PubMed] [Google Scholar]
- 2. Service FJ, Natt N, Thompson GB, et al Noninsulinoma pancreatogenous hypoglycemia: a novel syndrome of hyperinsulinemic hypoglycemia in adults independent of mutations in Kir6.2 and SUR1 genes. J Clin Endocrinol Metab 1999; 84: 1582–1589. [DOI] [PubMed] [Google Scholar]
- 3. Hirata Y, Ishizu H, Ouchi N, et al Insulin autoimmunity in a case of spontaneous hypoglycemia. J Jpn Diabetes Soc 1970; 13: 312–320. (Japanese). [Google Scholar]
- 4. Yorifuji T. Congenital hyperinsulinism: current status and future perspectives. Ann Pediatr Endocrinol Metab 2014; 19: 57–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Service FJ, Vella A Noninsulinoma pancreatogenous hypoglycemia syndrome. In UpToDate® Available from: http://www.uptodate.com. Accessed August 11, 2019.
- 6. Klöppel G, Anlauf M, Raffel A, et al Adult diffuse nesidioblastosis: genetically or environmentally induced? Hum Pathol 2008; 39: 3–8. [DOI] [PubMed] [Google Scholar]
- 7. Dravecka I, Lazurova I. Nesidioblastosis in adults. Neoplasma 2014; 61: 252–256. [DOI] [PubMed] [Google Scholar]
- 8. Ito T, Sasano H, Tanaka M, et al Epidemiological study of gastroenteropancreatic neuroendocrine tumors in Japan. J Gastroenterol 2010; 45: 234–243. [DOI] [PubMed] [Google Scholar]
- 9. Ito T, Igarashi H, Nakamura K, et al Epidemiological trends of pancreatic and gastrointestinal neuroendocrine tumors in Japan: a nationwide survey analysis. J Gastroenterol 2015; 50: 58–64. [DOI] [PubMed] [Google Scholar]
- 10. Ito T, Lee L, Hijioka M, et al The up‐to‐date review of epidemiological pancreatic neuroendocrine tumors in Japan. J Hepatobiliary Pancreat Sci 2015; 22: 574–577. [DOI] [PubMed] [Google Scholar]
- 11. Woo CY, Jeong JY, Jang JE, et al Clinical features and causes of endogenous hyperinsulinemic hypoglycemia in Korea. Diabetes Metab J 2015; 39: 126–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kawakita R, Sugimine H, Nagai S, et al Clinical characteristics of congenital hyperinsulinemic hypoglycemia in infant: a nationwide epidemiological survey in Japan. J Jpn Pediatr Soc 2011; 115: 563–569. (Japanese). [Google Scholar]
- 13. Yorifuji T, Horikawa R, Hasegawa T, et al Clinical practice guidelines for congenital hyperinsulinism. Clin Pediatr Endocrinol 2017; 26: 127–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Menni F, de Lonlay P, Sevin C, et al Neurologic outcomes of 90 neonates and infants with persistent hyperinsulinemic hypoglycemia. Pediatrics 2001; 107: 476–495. [DOI] [PubMed] [Google Scholar]
- 15. Ludwig A, Ziegenhorn K, Empting S, et al Glucose metabolism and neurological outcome in congenital hyperinsulinism. Semin Pediatr Surg 2011; 20: 45–49. [DOI] [PubMed] [Google Scholar]
- 16. Ludwig A, Enke S, Heindorf J, et al Formal neurocognitive testing in 60 patients with congenital hyperinsulinism. Horm Res Paediatr 2018; 89: 1–6. [DOI] [PubMed] [Google Scholar]
- 17. Avatapalle HB, Banerjee I, Shah S, et al Abnormal neurodevelopmental outcomes are common in children with transient congenital hyperinsulinism. Front Endocrinol (Lausanne) 2013; 4: 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kanamori Y, Watanabe T, Yorifuji T, et al Congenital hyperinsulinism treated by surgical resection of the hyperplastic lesion which had been preoperatively diagnosed by 18F‐DOPA PET examination in Japan: a nationwide survey. Pediatr Surg Int 2018; 34: 1093–1098. [DOI] [PubMed] [Google Scholar]
- 19. Yorifuji T, Kawakita R, Hosokawa Y, et al Efficacy and safety of long‐term, continuous subcutaneous octreotide infusion for patients with different subtypes of K(ATP) ‐channel hyperinsulinism. Clin Endocrinol (Oxf) 2013; 78: 891–897. [DOI] [PubMed] [Google Scholar]
- 20. Hosokawa Y, Kawakita R, Yokoya S, et al Efficacy and safety of octreotide for the treatment of congenital hyperinsulinism: a prospective, open‐label clinical trial and an observational study in Japan using a nationwide registry. Endocr J 2017; 64: 867–880. [DOI] [PubMed] [Google Scholar]
- 21. Rumilla KM, Erickson LA, Service FJ, et al Hyperinsulinemic hypoglycemia with nesidioblastosis: histologic features and growth factor expression. Mod Pathol 2009; 22: 239–245. [DOI] [PubMed] [Google Scholar]
- 22. Calabria AC, Charles L, Givler S, et al Postprandial hypoglycemia in children after gastric surgery: clinical characterization and pathophysiology. Horm Res Paediatr 2016; 85: 140–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cardoso JB, Sousa D, Pereira C, et al Dumping syndrome: a rare complication following nissen fundoplication. Eur J Case Rep Intern Med 2019; 6: 001177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Davi MV, Pia A, Guarnotta V, et al The treatment of hyperinsulinemic hypoglycaemia in adults: an update. J Endocrinol Invest 2017; 40: 9–20. [DOI] [PubMed] [Google Scholar]
- 25. Schwetz V, Horvath K, Kump P, et al Successful medical treatment of adult nesidioblastosis with pasireotide over 3 years: a case report. Medicine (Baltimore) 2016; 95: e3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Guseva N, Phillips D, Mordes JP. Successful treatment of persistent hyperinsulinemic hypoglycemia with nifedipine in an adult patient. Endocr Pract 2010; 16: 107–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Eguchi Y, Uchigata Y, Yao K, et al Longitudinal changes of serum insulin concentration and insulin antibody features in persistent insulin autoimmune syndrome (Hirata's disease). Autoimmunity 1994; 19: 279–284. [DOI] [PubMed] [Google Scholar]
- 28. Uchigata Y, Eguchi Y, Takayama‐Hasumi S, et al Insulin autoimmune syndrome (Hirata disease): clinical features and epidemiology in Japan. Diabetes Res Clin Pract 1994; 22: 89–94. [DOI] [PubMed] [Google Scholar]
- 29. Uchigata Y, Hirata Y, Iwamoto Y. Drug‐induced insulin autoimmune syndrome. Diabetes Res Clin Pract 2009; 83: e19–e20. [DOI] [PubMed] [Google Scholar]
- 30. Uchigata Y, Hirata Y, Iwamoto Y. Insulin autoimmune syndrome (Hirata disease): epidemiology in Asia, including Japan. Diabetol Int 2010; 1: 21–25. [Google Scholar]
- 31. Ishizuka T, Ogawa S, Mori T, et al Characteristics of the antibodies of two patients who developed daytime hyperglycemia and morning hypoglycemia because of insulin antibodies. Diabetes Res Clin Pract 2009; 84: e21–23. [DOI] [PubMed] [Google Scholar]
- 32. Iizuka K, Tomita R, Horikawa Y, et al A case of glycemic instability and insulin allergy due to anti‐insulin antibodies in a patient with type 2 diabetes. Diabetol Int 2012; 3: 233–238. [Google Scholar]
- 33. Tamura Y, Kimbara Y, Funatsuki S, et al A case of insulin antibody‐induced glucose instability in an elderly woman with type 2 diabetes on hemodialysis, successfully ameliorated with liraglutide. Diabetol Int 2013; 4: 71–75. [Google Scholar]
- 34. Alves C, Constança J, De León DD, et al A novel atypical presentation of insulin autoimmune syndrome (Hirata's disease) in a child. J Pediatr Endocrinol Metab 2013; 26: 1163–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dos Santos TJ, Passone CGB, Ybarra M, et al Pitfalls in the diagnosis of insulin autoimmune syndrome (Hirata’s disease) in a hypoglycemic child: a case report and review of the literature. J Pediatr Endocrinol Metab 2019; 32: 421–428. [DOI] [PubMed] [Google Scholar]
- 36. Censi S, Mian C, Betterle C. Insulin autoimmune syndrome: from diagnosis to clinical management. Ann Transl Med. 2018; 6: 335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Baruch Y. Response Rate in Academic Studies-A Comparative Analysis. Human Relations. 2016; 52(4): 421–438. [Google Scholar]