

Abstract
Aims/Introduction
Insulin resistance syndrome (IRS) of type A or B is triggered by gene abnormalities of or autoantibodies to the insulin receptor, respectively. Rabson–Mendenhall/Donohue syndrome is also caused by defects of the insulin receptor gene (INSR), but is more serious than type A IRS. Here, we carried out a nationwide survey of these syndromes in Japan.
Materials and Methods
We sent questionnaires to a total of 1,957 academic councilors or responsible individuals at certified facilities of the Japan Diabetes Society, as well as at the department pediatrics or neonatology in medical centers with >300 beds.
Results
We received 904 responses with information on 23, 30 and 10 cases of type A or B IRS and Rabson–Mendenhall/Donohue syndrome, respectively. Eight cases with type A IRS‐like clinical features, but without an abnormality of INSR, were tentatively designated type X IRS, with five of these cases testing positive for PIK3R1 mutations. Fasting serum insulin levels at diagnosis (mean ± standard deviation) were 132.0 ± 112.4, 1122.1 ± 3292.5, 2895.5 ± 3181.5 and 145.0 ± 141.4 μU/mL for type A IRS, type B IRS, Rabson–Mendenhall/Donohue syndrome and type X IRS, respectively. Type A and type X IRS, as well as Rabson–Mendenhall/Donohue syndrome were associated with low birthweight. Type B IRS was diagnosed most frequently in older individuals, and was often associated with concurrent autoimmune conditions and hypoglycemia.
Conclusions
Information yielded by this first nationwide survey should provide epidemiological insight into these rare conditions and inform better healthcare for affected patients.
Keywords: Insulin resistance syndrome, Nationwide survey
(a) Rabson–Mendenhall/Donohue syndrome and type B insulin resistance syndrome (IRS) showed the highest and second highest levels of hyperinsulinemia, respectively, and those with type A and type X IRS were similar. Fasting serum insulin levels for 26 of the 27 (96.3%) type A and type X IRS patients for whom such data were available were >30 μU/mL. (b) Type A and type X IRS, as well as Rabson–Mendenhall/Donohue syndrome were associated with low birthweight.

Introduction
Insulin resistance syndromes (IRSs), formerly known as insulin receptor abnormalities, are characterized by insulin resistance as a result of dysfunction of the insulin receptor1. These syndromes have classically been categorized as type A or type B, in which insulin receptor function is impaired as a result of mutations in the receptor gene (INSR) or the presence of autoantibodies to the receptor, respectively2, 3. Rabson–Mendenhall syndrome and Donohue syndrome are also caused by abnormalities of INSR, but these syndromes are characterized by more serious symptoms resulting from profound defects in receptor function4.
In type B IRS, autoantibodies block insulin binding to its receptor and thereby cause insulin resistance, whereas some patients with type B IRS paradoxically manifest episodic hypoglycemia5. This syndrome is often accompanied by a variety of autoimmune conditions5, 6, 7. There is no established therapy for type B IRS, although immunological interventions – such as the administration of immunosuppressive drugs8 or immunoglobulin9 or the performance of plasmapheresis10 – have been found to be effective in some, but not all,2, 11 cases. In addition, a case of type B IRS accompanied by idiopathic thrombocytopenic purpura (ITP) was reported in which eradication of Helicobacter pylori for treatment of ITP cured not only ITP, but also the IRS12.
Whereas a number of case reports and some case series have been published2, 3, 13, information relating to epidemiological surveillance of IRSs has not been available. Furthermore, clinical features similar to those of type A IRS (such as early disease onset, as well as persistent and severe insulin resistance without apparent humoral or metabolic causes) have also been reported for individuals who do not harbor INSR defects. Whereas some of these conditions are likely attributable to genetic abnormalities of postreceptor signaling14, 15, data for such patients are limited. Information, such as disease prevalence, sex differences, peak age at onset, proportions of patients with hypoglycemia or autoimmune disease and the effectiveness of therapy (in particular, eradication of H. pylori), is lacking for type B IRS.
To provide insight into the characteristics of IRSs, we carried out a nationwide survey for type A IRS, type B IRS, Rabson–Mendenhall syndrome and Donohue syndrome in Japan. We also collected information on patients with type A IRS‐like features, but without a mutation in INSR, a condition that we here term type X IRS.
Methods
Collection of information
This study was approved by the ethics committees of Iwate Medical University (approval no. H28‐40) and Kobe University Graduate School of Medicine (approval no. 160116), and carried out in accordance with the Declaration of Helsinki and its amendments. In November 2014, we sent a questionnaire to a total of 1,063 academic councilors or responsible individuals at certified facilities of Board Certified Diabetologists of the Japan Diabetes Society. The questionnaire asked whether they had undertaken care of patients with type A IRS, type B IRS, Rabson–Mendenhall syndrome or Donohue syndrome between October 2008 and December 2014. We then sent a second questionnaire in January 2016 to the facilities that had undertaken such patient care during the specified period to collect detailed information. We also sent a questionnaire in October 2016 to a total of 894 responsible individuals at the department of pediatrics or neonatology in medical centers with >300 beds in Japan. In this latter questionnaire, we asked whether the pediatricians had experienced care of patients with the syndromes between October 2008 and December 2014, and, if they had, to answer questions relating to the characteristics of the patients.
Definition of syndromes
A confirmed case of type A IRS was defined by the presence of insulin resistance without an apparent humoral or metabolic cause in an individual harboring a mutation of INSR. A suspected case of type A IRS was defined by suspicion based on clinical and laboratory findings in an individual who did not undergo genetic testing. Confirmed cases of Rabson–Mendenhall syndrome or Donohue syndrome were defined by the presence of insulin resistance without an apparent humoral or metabolic cause in early infancy in an individual harboring a mutation of INSR. Suspected cases of these two syndromes were defined by suspicion based on clinical and laboratory findings in an individual who did not undergo genetic testing. Given that the difference between Rabson–Mendenhall syndrome and Donohue syndrome is unclear16, 17, we combined information on these syndromes and considered them together as Rabson–Mendenhall/Donohue syndrome in our analysis. We defined type B IRS as the presence of hyperglycemia or hypoglycemia (or both) in individuals positive for autoantibodies to the insulin receptor.
We also collected information on patients who were suspected of having type A IRS or Rabson–Mendenhall/Donohue syndrome on the basis of clinical and laboratory findings, but who had undergone genetic testing that showed no abnormality of INSR. This condition was here designated type X IRS. Lipodystrophy or lipodystrophic diabetes, diagnosed either by genetic testing or on the basis of clinical manifestations, was excluded from the survey.
Genetic testing
We sequenced all 22 exons of INSR for suspected cases of type A IRS and of Rabson–Mendenhall/Donohue syndrome and all 16 exons of PIK3R1 for patients categorized as having type X IRS if they and their attending physicians desired it.
Results
Number of participants with each type of IRS
We sent a total of 1,957 questionnaires and received 904 responses. We obtained information on 17 and nine confirmed and suspected cases of type A IRS, respectively, eight and two confirmed and suspected cases of Rabson–Mendenhall/Donohue syndrome, respectively, five cases of type X IRS, and 30 cases of type B IRS (Figure 1).
Figure 1.
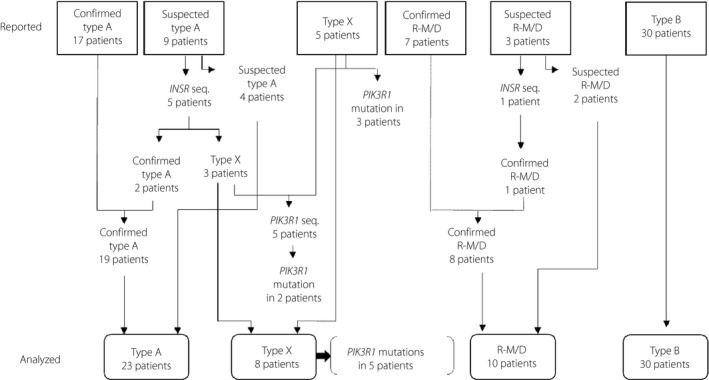
Numbers of patients with type A, type X or type B insulin resistance syndrome and of Rabson–Mendenhall/Donohue (R‐M/D) syndrome. The numbers of patients reported in the questionnaires (upper) and those after re‐categorization on the basis of gene sequencing (lower) are shown.
We sequenced INSR for one and five suspected cases of Rabson–Mendenhall/Donohue syndrome and type A IRS, respectively, and found mutations in one and two cases, respectively, which were then re‐categorized as confirmed cases. The three suspected cases of type A IRS that tested negative for a mutation of INSR were re‐categorized as type X IRS (Figure 1). After INSR sequencing, the numbers of confirmed and suspected cases of type A IRS were thus 19 and four, respectively, and those of confirmed and suspected cases of Rabson–Mendenhall/Donohue syndrome were nine and one, respectively (Figure 1).
After we received the questionnaire responses, three patients with type X IRS were shown to harbor mutations in PIK3R1, which encodes a regulatory subunit of phosphoinositide 3‐kinase (PI 3‐kinase)18, 19, 20, as a result of analysis carried out independently of the present study21, 22. We sequenced PIK3R1 in the remaining five patients with type X IRS, and detected PIK3R1 mutations in two patients. Among the eight patients with type X IRS, five patients were thus found to be positive for mutations in PIK3R1 (Figure 1).
Characteristics of type A IRS
Information for 23 patients (4 males, 19 females) with type A IRS, including 19 confirmed and four suspected cases, is presented in Table 1. Age and hemoglobin A1c (HbA1c) level at the time of clinical diagnosis (mean ± standard deviation) were 17.6 ± 13.8 years (range 0–66 years) and 8.0 ± 2.6% (range 5.2–14.9%), respectively. The fasting serum insulin concentration at the time of clinical diagnosis and birthweight for these patients are shown in Figure 2. Treatment for diabetes included insulin (8 patients, 35%), recombinant human insulin‐like growth factor‐1 (2 patients, 9%), metformin (13 patients, 57%), a sodium–glucose cotransporter (SGLT)‐2 inhibitor (4 patients, 17%), a dipeptidyl peptidase‐4 inhibitor (3 patients, 13%), an α‐glucosidase inhibitor (2 patients, 9%), a sulfonylurea (1 patient, 4%) and diet alone (2 patients, 9%), and the HbA1c level at the time of the survey was 8.1 ± 2.9% (range 4.8–14.9%; Table 1).
Table 1.
Clinical information for patients with type A insulin resistance syndrome
| Patient | Age at diagnosis (years)/sex | Fasting serum insulin at diagnosis (μU/mL) | HbA1c at diagnosis (%) | Birthweight (g) | Age at survey (years) | BMI at survey (kg/m2) | HbA1c at survey (%) | Therapy for diabetes at survey | INSR mutation |
|---|---|---|---|---|---|---|---|---|---|
| 143 | 11/M | 71.9 | 6.1 | 2,392 | 16 | 27.8 | 5.6 | Metformin | Gly1146Arg heterozygous |
| 244 | 9/F | 44.1 | 6.6 | 1,894 | 12 | 21.8 | 5.1 | Metformin | Gln205Ter heterozygous |
| 3 | 13/F | 59.9 | 5.3 | NA | 16 | 18.4 | 4.8 | Metformin | Gly1035Val heterozygous |
| 4 | 11/F | NA | 9.2 | 1,926 | 11 | 17.4 | 5.8 | Metformin | Arg1201Trp heterozygous |
| 545 | 11/F | 65.7 | 6.9 | 2,495 | 21 | NA | 6.9 | Metformin | Arg331Ter heterozygous |
| 6 | 12/F | 279.3 | 8.8 | 2,090 | 16 | 19.3 | 9.3 | Insulin (CSII, 58 U), metformin, α‐GI, DPP‐4i, SGLT‐2i | Asn489Asp heterozygous, Val1054Met heterozygous |
| 746 | 10/F | 389 | 8.1 | 1,511 | 13 | 19.8 | 12.2 | Metformin, rhIGF‐1 | Ser835Ile heterozygous, Ala842Val heterozygous |
| 847 | 13/F | 234 | NA | NA | 43 | 20.5 | 8.4 | Bolus insulin | Arg252His homozygous |
| 9 | 35/F | 88.6 | 7.1 | NA | 42 | 22.9 | 8.0 | Basal insulin | Arg120Gln heterozygous |
| 10 | 35/F | 169.1 | 9.9 | NA | 46 | 21.1 | 10.0 | SGLT‐2i | Arg120Gln heterozygous |
| 1148 | 0/F | 199 | 5.6 | NA | 34 | 19.1 | 6.2 | Metformin | Asn462Ser heterozygous |
| 1245 | 66/F | 30.2 | 7.8 | NA | 77 | 19.7 | 7.7 | Metformin, SU, DPP‐4i | Arg331Ter heterozygous |
| 13 | 20/F | 168 | 13.8 | NA | 35 | 21.5 | 13.8 | Basal insulin (18 U) | Leu1199Phe heterozygous |
| 14 | 17/F | 105 | 7.2 | NA | 25 | 21.9 | 7.2 | Metformin, bolus insulin (15 U) | Pro1205Leu heterozygous |
| 1549 | 32/F | 68.8 | 7.5 | NA | 40 | 22.7 | 7.5 | Metformin, SGLT‐2i, α‐GI, basal (30 U) and bolus (78 U) insulin | Leu999del heterozygous |
| 1649 | 16/F | 61 | 5.8 | NA | 49 | 18.7 | 5.8 | Diet | Leu999del heterozygous |
| 1750 | 14/F | 55 | 8.3 | NA | 47 | 19.1 | 8.3 | Bolus insulin (26 U) | Ala145Val heterozygous |
| 18 | 12/F | 61.1 | 5.2 | NA | 31 | 22.5 | 5.2 | Metformin | Arg1201Gln heterozygous |
| 1949 | 27/M | NA | 10.5 | NA | 51 | 24.8 | 10.5 | NA | Leu999del heterozygous |
| 20 | 9/M | 404 | NA | NA | 16 | 18.8 | 4.8 | Diet | ND |
| 21 | 7/M | 150 | 14.9 | 1,950 | 43 | 15.9 | 14.9 | Basal (40 U) and bolus (120 U) insulin | ND |
| 22 | 14/F | 35 | 8.1 | NA | 25 | 32.3 | 11.3 | DPP‐4i, SGLT‐2i | ND |
| 2351 | 10/F | 21.1 | 5.7 | NA | 37 | 16.5 | 7.2 | Metformin, rhIGF‐1 | ND |
α‐GI, α‐glucosidase inhibitor; BMI, body mass index; F, female; HbA1c, hemoglobin A1c; DPP‐4i, dipeptidyl peptidase‐4 inhibitor; M, male; NA, information not available; ND, not determined; SGLT‐2i, sodium–glucose cotransporter‐2 inhibitor; rhIGF‐1, recombinant human insulin‐like growth factor–1; SU, sulfonylurea.
Figure 2.
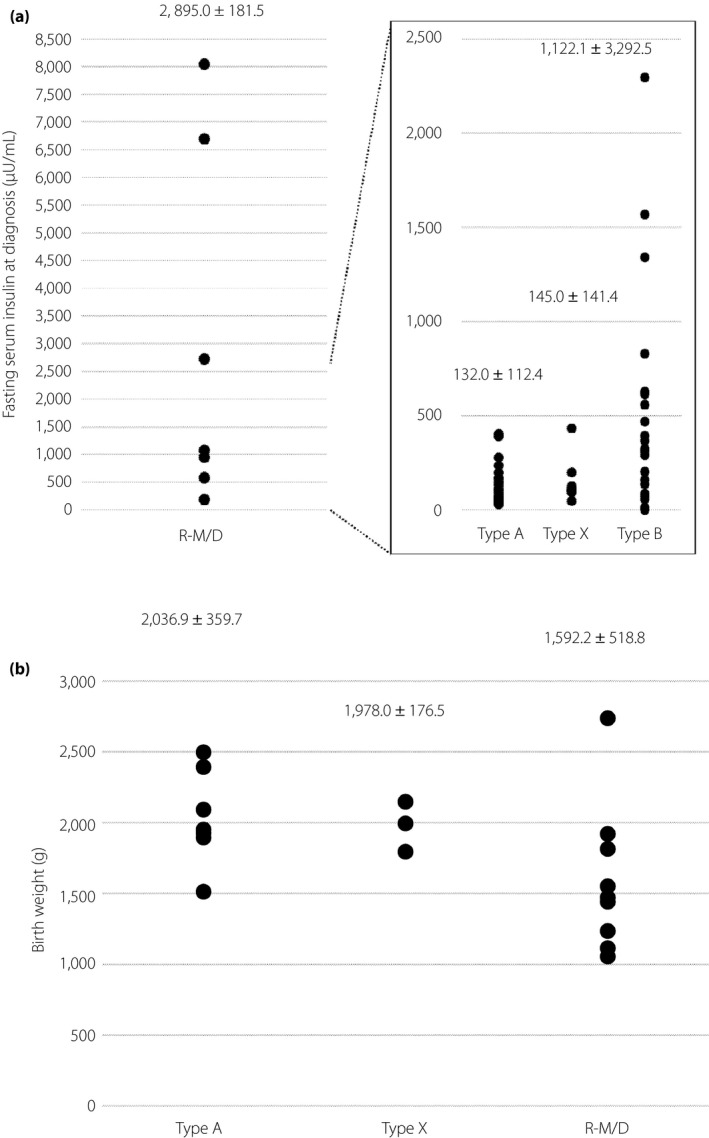
Fasting serum insulin level at (a) clinical diagnosis and (b) birth weight for cases of type A, type X, or type B insulin resistance syndrome and of Rabson–Mendenhall/Donohue (R‐M/D) syndrome. Mean ± standard deviation values are shown.
Among the 19 confirmed cases, 16, one and two cases harbored a single heterozygous, a single homozygous and multiple heterozygous mutations, respectively, in INSR (Table 1). The INSR mutations identified in two patients (patients 14 and 18) were previously detected in patients with type A IRS23, 24.
Characteristics of Rabson–Mendenhall/Donohue syndrome
Information for 10 patients (5 males, 5 females) with Rabson–Mendenhall/Donohue syndrome, including eight confirmed and two suspected cases, is shown in Table 2. All the patients were clinically diagnosed before 1 year‐of‐age. The fasting serum insulin concentration at the time of clinical diagnosis and birthweight are shown in Figure 2. Four patients had died by the time of the survey, with their age at death being 3 months for two patients, 17 months for one patient and 3 years for one patient. Treatment for diabetes included insulin (10 patients, 100%), recombinant human insulin‐like growth factor‐1 (9 patients, 90%), metformin (6 patients, 60%), an SGLT‐2 inhibitor (1 patient, 10%), an α‐glucosidase inhibitor (3 patients, 30%) and a dipeptidyl peptidase‐4 inhibitor (2 patients, 20%), and the HbA1c level at the time of the survey was 8.7 ± 3.0% (3.6–12.5%; Table 2).
Table 2.
Clinical information for patients with Rabson–Mendenhall/Donohue syndrome
| Patient | Age at diagnosis (years)/sex | Fasting serum insulin at diagnosis (μU/mL) | HbA1c at diagnosis (%) | Birthweight (g) | Age at survey (years) | BMI at survey (kg/m2) | HbA1c at survey (%) | Therapy for diabetes at survey | INSR mutation |
|---|---|---|---|---|---|---|---|---|---|
| 152 | 0/M | 8,038 | 2.8 | 1,440 | 7 | 14.3 | 10.3 | Metformin, basal insulin, rhIGF‐1 | Thr937Met heterozygous,Ala1204Thr heterozygous |
| 243 | 0/F | 1,070 | 7.1 | 1,234 | Died at 17 months | NA | 3.6 | Insulin, rhIGF‐1 | Ser98Arg homozygous |
| 3 | 0/M | NA | NA | 1,470 | Died at 3 years | NA | NA | Insulin, rhIGF‐1 | Val657Phe heterozygous, deletion including exon 2 † heterozygous |
| 453 | 0/F | NA | 6.5 | 2,736 | 26 | 20.3 | 8.1 | Metformin, α‐GI, DPP‐4i, insulin, rhIGF‐1 | Met910Thr heterozygous,1‐bp deletion in exon 19 † heterozygous |
| 5 | 0/M | 586.8 | NA | 1,814 | 18 | 21.2 | NA | Metformin, α‐GI, DPP‐4i SGLT‐2i, basal insulin (70 U), rhIGF‐1 | Cys981Asp heterozygous, Cys1126Asp heterozygous, Phe1144Ser heterozygous |
| 654 | 0/M | 6,702 | NA | 1,920 | 6 | 17.5 | 10.0 | Insulin, metformin, rhIGF‐1 | Thr910Met heterozygous,Glu1047Lys heterozygous |
| 755 | 0/F | 185 | 6.6 | 1,550 | 27 | 15.3 | 12.5 | Metformin, α‐GI, basal (240 U) and bolus (240 U) insulin | Arg1201Trp heterozygous, deletion including exons 4–6 † heterozygous |
| 8 | 0/F | 2,730 | 5.8 | NA | 10 | 14.9 | 7.6 | Insulin, metformin, rhIGF‐1 | Ile925Ser heterozygous |
| 956 | 0/F | 956.4 | 23 | 1,054 | Died at 3 months | 10.3 | NA | Insulin, rhIGF‐1 | ND |
| 10 | 0/M | NA | NA | 1,112 | Died at 3 months | NA | NA | Insulin, rhIGF‐1 | ND |
α‐GI, α‐glucosidase inhibitor; BMI, body mass index; DPP‐4i, dipeptidyl peptidase‐4 inhibitor; F, female; HbA1c, hemoglobin A1c; M, male; NA, information not available; ND, not determined; rhIGF‐1, recombinant human insulin‐like growth factor‐1; SGLT‐2i, sodium–glucose cotransporter‐2 inhibitor.
Detailed information not available.
Among the eight confirmed patients, one, one and six patients harbored a single heterozygous, a single homozygous and multiple heterozygous mutations, respectively, in INSR (Table 2). The INSR mutation identified in one patient in the present study (patient 8) had not previously been described in a patient with either type A IRS or Rabson–Mendenhall/Donohue syndrome.
Characteristics of type X IRS
Information for eight patients (3 males, 5 females) with type X IRS is shown in Table 3. Age and HbA1c level at the time of clinical diagnosis were 13.4 ± 1.7 years and 7.8 ± 0.8%, respectively. The fasting serum insulin concentration at the time of clinical diagnosis and birthweight are shown in Figure 2. Treatment for diabetes included insulin (1 patient, 13%), metformin (5 patients, 63%), an SGLT‐2 inhibitor (1 patients, 13%), an α‐glucosidase inhibitor (2 patients, 25%), a thiazolidinedione (1 patient, 13%) and diet alone (3 patients, 38%), and the HbA1c level at the time of the survey was 6.8 ± 0.7% (range 5.7–7.6%; Table 3).
Table 3.
Clinical information for patients with type X insulin resistance syndrome
| Patient | Age at diagnosis (years)/sex | Fasting serum insulin at diagnosis (μU/mL) | HbA1c at diagnosis (%) | Birthweight (g) | Age at survey (years) | BMI at survey (kg/m2) | HbA1c at survey (%) | Therapy for diabetes at survey | Mutation |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 13/F | 199 | 7.4 | NA | 15 | 25.0 | 7.4 | Diet | ND |
| 2 | 15/M | 46.1 | 6.8 | 1,994 | 16 | 13.0 | 7.3 | Metformin | PIK3R1 Arg649Trp heterozygous |
| 3 | 12/F | 104.4 | 7.4 | NA | 17 | 19.6 | 6.2 | Metformin, α‐GI | ND |
| 4 | 14/M | 92.5 | 7.2 | 2,146 | 20 | 16.7 | 6.7 | Metformin, α‐GI | PIK3R1 † |
| 521 | NA/M | NA | NA | 1,794 | 2 | 12.5 | NA | None | PIK3R1 Arg649Trp heterozygous |
| 6 | 11/F | NA | 8.9 | NA | 19 | 21.1 | 5.7 | Diet | ND |
| 7 | 13/F | 433.8 | 8.8 | NA | 20 | 23.9 | 6.9 | Metformin, thiazolidinedione | PIK3R1 Arg649Trp heterozygous |
| 821 | 16/F | 128 | 8.0 | NA | 38 | 19.3 | 7.6 | Metformin, SGLT‐2i, insulin (80U) | PIK3R1 Arg649Trp heterozygous |
α‐GI, α‐glucosidase inhibitor; BMI, body mass index; F, female; HbA1c, hemoglobin A1c; M, male; NA, information not available; ND, not determined; SGLT‐2i, sodium–glucose cotransporter‐2 inhibitor.
Detailed information not available.
Five patients were found to harbor mutations in PIK3R1. Mutations of this gene are responsible for SHORT (Short stature, Hyperextensibility of joints and/or inguinal hernia, Ocular depression, Rieger anomaly, and Teething delay) syndrome18, 19, 20, 25, 26, and four of the five patients who where type X IRS‐positive for such mutations harbored Arg649Trp (Table 3), one of the most frequent mutations previously described in SHORT syndrome27. These four patients, including a mother and son (patients 5 and 8), manifested a recognizable facial gestalt (including a triangular face, prominent forehead, small chin and ocular depression) that is characteristic of SHORT syndrome. Other bodily characteristics of SHORT syndrome, including hyperextensibility of joints, inguinal hernia and Rieger anomaly27, 28, were not apparent in these patients. The height of the four adult patients with PIK3R1 mutations was 158.5 ± 13.3 cm.
Characteristics of type B IRS
Information for 30 patients (19 males, 11 females) with type B IRS is shown in Table 4. The age and HbA1c level at the time of clinical diagnosis were 59.6 ± 16.5 years (range 13–85 years) and 8.2 ± 2.5% (range 4.3–14.1%), respectively. The age at diagnosis showed a peak in the 60s (Figure 3a). The fasting serum insulin concentration at the time of clinical diagnosis is shown in Figure 2a. A total of 18 patients had experienced episodic hypoglycemia (Figure 3b), and 17 patients were accompanied by confirmed autoimmune conditions, including SLE, Sjögren’s syndrome, Hashimoto’s disease, mixed connective tissue disease, Graves’ disease, ITP, scleroderma and rheumatoid arthritis (Table 5).
Table 4.
Clinical information for patients with type B insulin resistance syndrome
| Patient | Age at diagnosis (years)/ sex | Fasting serum insulin at diagnosis (μU/mL) | BMI at diagnosis (kg/m2) | HbA1c at diagnosis (%) | Concurrent autoimmune disease | Immunomodulating therapy | Therapy for diabetes | Hypoglycemia |
|---|---|---|---|---|---|---|---|---|
| 1 | 13/F | 395 | 19.5 | 14.1 | SLE | mPSL, azathioprine | Insulin (180 U) | – |
| 257 | 45/F | 73.6 | 18.0 | 10.8 | MCTD | Insulin (525 U), liraglutide | + | |
| 358 | 69/F | 324.6 | 19.1 | 9.8 | NI | Eradication of Helicobacter pylori | SU, DPP‐4i, α‐GI, metformin, SGLT‐2i | – |
| 4 | 68/M | ND | 20.0 | ND | MCTD | ND | + | |
| 559 | 56/M | ND | 22.7 | ND | NI | ND | + | |
| 660 | 65/M | 310 | 21.4 | 7.9 | NI | PSL (12.5 mg/day) | α‐GI, metformin | + |
| 7 | 62/M | 627.4 | 18.5 | 4.9 | NI | Insulin (18 U), liraglutide | – | |
| 861 | 60/M | 620.4 | 20.0 | 13.0 | SLE Hashimoto’s disease | Cyclosporine (25 mg), PSL (10 mg) | Insulin (100 U), metformin | – |
| 9 | 52/F | 617.7 | 14.6 | 8.0 | RA | Thiazolidinedione, metformin, bolus insulin (65 U) | + | |
| 10 | 17/F | 12.1 | 20.8 | 5.1 | SLE | PSL (17.5 mg) | DPP‐4i, glinide | + |
| 11 | 69/M | 292.4 | 23.2 | 8.3 | NI | Insulin (64 U) | + | |
| 12 | 61/F | 620 | 27.6 | 8.7 | SLE, SJS, Hashimoto’s disease | Insulin (34 U) | – | |
| 13 | 72/F | 615 | 21.2 | 11.1 | SJS | rhIGF‐1, α‐GI | + | |
| 1462 | 32/F | 0.5 | 22.4 | 4.3 | ITP | Hydrocortisone (15 mg/day), human immunoglobulin (27.5 g/day) | + | |
| 15 | 69/M | 493 | 23.5 | 5.5 | NI | PSL | – | |
| 1612 | 84/M | 138 | 37.4 | 9.0 | ITP | Eradication of H. pylori | – | |
| 17 | 61/F | 560 | 19.8 | 12.1 | SJS, Hashimoto’s disease | PSL | Insulin, rhIGF‐1 | – |
| 18 | 67/M | 88 | 16.4 | 5.7 | Graves’ disease | Eradication of H. pylori, PSL (50 mg) | + | |
| 19 | 85/M | 1,568 | 26.4 | 6.4 | NI | PSL (25 mg) | + | |
| 20 | 72/M | 2,294 | 18.0 | 8.3 | SJS | Insulin (8 U), liraglutide, thiazolidinedione, DPP‐4i, α‐GI | + | |
| 21 | 42/M | 13.3 | 27.8 | 5.8 | NI | SU, DPP‐4i, metformin | – | |
| 2263 | 59/M | 316 | 15.6 | 9.5 | SLE | PSL (30 mg) | Insulin (610 U), rhIGF‐1 | + |
| 2363 | 50/F | 830 | 23.3 | 8.0 | SLE | Cyclosporine (250 mg), PSL (30 mg) | Insulin (68 U), rhIGF‐1, metformin, thiazolidinedione | + |
| 24 | 75/F | 202 | 24.0 | 8.4 | Graves’ disease | Insulin (60 U), thiazolidinedione, SU | – | |
| 25 | 65/M | 1,7390 | ND | 6.5 | NI | Insulin (20 U), DPP‐4i, metformin, | + | |
| 26 | 67/M | 471.1 | 27.7 | 9.1 | NI | Insulin (30 U), DPP‐4i, metformin | + | |
| 2764 | 70/M | 366 | 21.1 | 8.6 | NI | α‐GI | + | |
| 28 | 68/M | 1,340 | 29.0 | 7.2 | NI | PSL (20 mg/day) | Glinide, α‐GI | – |
| 29 | 41/M | 10 | 37.4 | 4.7 | NI | α‐GI | – | |
| 30 | 54/M | 158 | 17.0 | 9.6 | Scleroderma | Liraglutide, α‐GI | + |
ssssα‐GI, α‐glucosidase inhibitor; BMI, body mass index; DPP‐4i, dipeptidyl peptidase‐4 inhibitor; F, female; HbA1c, hemoglobin A1c; ITP, idiopathic thrombocytopenic purpura; M, male; MCTD, mixed connective tissue disease; mPSL, methylprednisolone; NI, not identified; PSL, prednisolone; RA, rheumatoid arthritis; rhIGF‐1, recombinant human insulin‐like growth factor‐1; SLE, systemic lupus erythematosus; SGLT‐2i, sodium‐glucose cotransporter‐2 inhibitor; SJS, Sjögren’s syndrome; SU, sulfonylurea.
Figure 3.
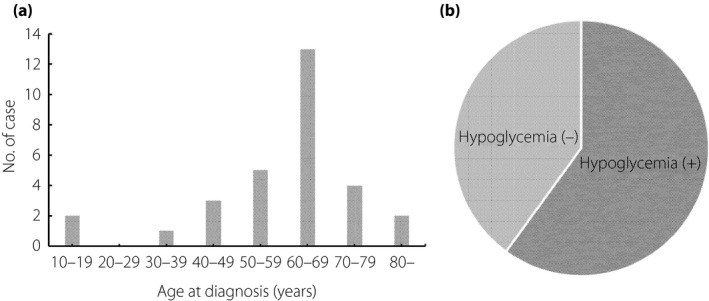
Prevalence of (a) age at diagnosis and (b) hypoglycemia for cases of type B insulin resistance syndrome.
Table 5.
Concomitant autoimmune diseases for patients with type B insulin resistance syndrome
| Autoimmune disease | No. patients |
|---|---|
| Systemic lupus erythematosus | 6 (20.0%) |
| Mixed connective tissue disease | 2 (6.7%) |
| Sjögren’s syndrome | 4 (13.3%) |
| Hashimoto’s or Graves’ disease | 5 (16.7%) |
| Idiopathic thrombocytopenic purpura | 2 (6.7%) |
| Scleroderma | 1 (3.3%) |
| Rheumatoid arthritis | 1 (3.3%) |
| Not identified | 13 (43.3%) |
A total of 14 patients received immunomodulation therapy, including glucocorticoid, immune suppressant or immunoglobulin administration, as well as eradication of H. pylori (Table 6). Treatment for diabetes included insulin (14 patients, 47%), metformin (8 patients, 27%), recombinant human insulin‐like growth factor‐1 (4 patients, 13%), a glucagon‐like peptide‐1 receptor agonist (4 patients, 13%), a dipeptidyl peptidase‐4 inhibitor (6 patients, 20%), an α‐glucosidase inhibitor (8 patients, 27%), a thiazolidinedione (4 patients, 13%), a sulfonylurea (3 patients, 10%), a glinide (2 patients, 7%) and an SGLT‐2 inhibitor (1 patient, 3%), and the HbA1c level at the time of the survey was 6.5 ± 1.4% (range 4.8–10.6%).
Table 6.
Immunomodulation therapy for patients with type B insulin resistance syndrome
| Immunomodulation therapy | No. patients |
|---|---|
| Steroid | 12 (40.0%) |
| Helicobacter pylori eradication therapy | 3 (10.0%) |
| Cyclosporine | 2 (6.7%) |
| Azathioprine | 1 (3.3%) |
| Human immunoglobulin | 1 (3.3%) |
| None | 16 (53.3%) |
Physical findings
Acanthosis nigricans and hirsutism were commonly observed in patients with type A IRS or Rabson–Mendenhall/Donohue syndrome, but were relatively uncommon with type B IRS (Table 7). Scarce fat tissue was apparent more frequently in Rabson–Mendenhall/Donohue syndrome than in the other syndromes. Characteristic facial appearance was also common in Rabson–Mendenhall/Donohue syndrome. All patients with type X IRS who manifested the characteristic facial appearance harbored mutations in PIK3R1.
Table 7.
Physical findings for patients with insulin resistance syndromes
| Physical findings | Type A | R‐M/D | Type X | Type B |
|---|---|---|---|---|
| Characteristic facial appearance | 4 (17%) | 9 (90%) | 5 (63%) | 0 (0%) |
| Acanthosis nigricans | 17 (74%) | 7 (70%) | 6 (75%) | 5 (17%) |
| Hirsutism | 9 (39%) | 9 (90%) | 5 (63%) | 1 (3%) |
| Tooth hypoplasia | 4 (17%) | 4 (40%) | 1 (13%) | 0 (0%) |
| Scarce fat tissue | 1 (4%) | 8 (80%) | 2 (25%) | 0 (0%) |
| Fatty liver | 0 (0%) | 1 (10%) | 3 (38%) | 2 (7%) |
| Androgen excess | 5 (22%) | 5 (50%) | 5 (63%) | 0 (0%) |
R–M/D, Rabson–Mendenhall/Donohue syndrome.
Triggers for diagnosis
Low birthweight and urinalysis abnormalities at school were the most frequent triggers for diagnosis of Rabson–Mendenhall/Donohue syndrome or of type A or type X IRS, respectively (Table 8). Whereas type B IRS was diagnosed most frequently by recognition of hyperglycemia, hypoglycemia triggered the diagnosis of this syndrome in some patients.
Table 8.
Triggers for diagnosis of insulin resistance syndromes
| Trigger for diagnosis | Type A | R‐M/D | Type X | Type B |
|---|---|---|---|---|
| Low birthweight | 0 (0%) | 5 (50%) | 0 (0%) | 0 (0%) |
| Physical findings | 4 (17%) | 2 (20%) | 1 (13%) | 1 (3%) † |
| Hyperglycemia | 5 (22%) | 3 (30%) | 0 (0%) | 20 (67%) |
| Hypoglycemia | 0 (0%) | 0 (0%) | 0 (0%) | 7 (23%) |
| Urinalysis at school | 9 (39%) | 0 (0%) | 6 (75%) | 0 (0%) |
| Diagnosis of relatives | 3 (13%) | 0 (0%) | 1 (13%) | 0 (0%) |
| Amenorrhea | 1 (4%) | 0 (0%) | 0 (0%) | 0 (0%) |
R‐M/D, Rabson–Mendenhall/Donohue syndrome.
One patient with mixed connective tissue disease was diagnosed on the basis of hand swelling.
Discussion
We carried out a nationwide survey of type A and type B IRS, as well as Rabson–Mendenhall/Donohue syndrome in Japan. As far as we are aware, this is the first such nationwide epidemiological survey for these syndromes in any country.
We sent questionnaires to healthcare professionals who treat mostly adult or non‐adult patients, thereby likely covering most of the institutions that provide care for these rare diseases in Japan. In Japan, only one clinical testing company undertakes testing for autoantibodies to the insulin receptor. An enquiry to the company revealed that they had tested samples from 1,796 individuals, and that 88 of these samples had tested positive between April 2009 and March 2013. Whereas these individuals included those who had already been diagnosed with type B IRS before April 2009, it is likely that approximately 20 cases of this syndrome are diagnosed per year in Japan. Given that we collected information on 30 patients with this syndrome who had received care during an approximately 6‐year period, our survey likely covered about one‐quarter of such patients nationwide.
Among patients for whom information on INSR mutations was available, 84.2% (16/19) of type A IRS patients harbored a single heterozygous mutation, whereas 87.5% (7/8) of Rabson–Mendenhall/Donohue syndrome patients harbored either a homozygous or multiple heterozygous mutations, consistent with the notion that the more serious nature of Rabson–Mendenhall/Donohue syndrome is due to defects in both INSR alleles29, 30. Given that 10 patients were found to harbor INSR mutations in both alleles (3 with type A IRS and 7 with Rabson–Mendenhall/Donohue syndrome) and that the population of Japan is approximately 126 million, at least 0.05% of the population might harbor a pathological INSR mutation in one allele according to a calculation based on the Hardy–Weinberg principle: 2 × (10 / 126,000,000)1/2 × 100 ≈ 0.05. Given that the coverage of our survey was not 100%, it is likely that the proportion of the population harboring such mutations is actually >0.05%. It is likely that most individuals who harbor a mutation of INSR in one allele manifest minimal metabolic disturbance and remain unidentified. A recent study found that, among individuals who underwent a health checkup, 0.4% (33/8630) showed hyperinsulinemia (>15 μU/mL during fasting) without obesity (body mass index of <25 kg/m2), and that two of the 11 such individuals tested harbored a single heterozygous INSR mutation31.
Rabson–Mendenhall/Donohue syndrome and type B IRS showed the highest and second highest levels of hyperinsulinemia, respectively, and those in type A and type X IRS were similar. Fasting serum insulin levels for 26 of the 27 (96.3%) type A and type X IRS patients for whom such data were available were >30 μU/mL. By contrast, we measured the fasting insulin levels of 86 Japanese type 2 diabetes patients with a body mass index of <25 kg/m2 and without insulin administration (age 61.3 ± 12.7 years, BMI 21.8 ± 3.1 kg/m2), and found that they were all <30 μU/mL (1–26, 5.1 ± 4.0). A diagnosis of type A or type X IRS should thus be suspected in an individual with a fasting insulin level of >30 μU/mL and no other apparent cause of insulin resistance including obesity.
Type A IRS‐like features in the absence of an INSR mutation are potentially attributable to defects in signaling downstream of the insulin receptor. Mutations in the gene for Akt2, a protein kinase that plays a key role in insulin action, and in the gene for TBC1D4, a substrate of Akt2 that contributes to the regulation of glucose transport, have been found to trigger the development of severe insulin resistance in humans14, 15. PI 3‐kinase mediates regulation of various actions of insulin32. Mutations in PIK3R1, a gene that encodes a regulatory subunit of PI 3‐kinase, were recently identified in individuals with SHORT syndrome27. Together with its characteristic body features, this syndrome is associated with the development of diabetes mellitus33. Furthermore, some individuals with PIK3R1 mutations have been followed up as cases of insulin‐resistant diabetes, not of SHORT syndrome18, as were individuals with type X IRS in the present study. Given that five of the eight patients with type X IRS in our survey harbored PIK3R1 mutations, PIK3R1 is likely the most common responsible gene for severe insulin resistance triggered by a genetic defect in postreceptor insulin signaling. Four of the five patients with type X IRS with mutations in PIK3R1 harbored the Arg649Trp mutation. Arg649 of the regulatory subunit of PI 3‐kinase resides in a Scr‐homology‐2 domain, a domain that is essential for the binding of the enzyme to insulin receptor substrates, and the Arg649Trp mutant protein inhibits the binding in a dominant negative manner34. Furthermore, forced expression of a dominant negative mutant of the regulatory subunit of PI 3‐kinase has been shown to trigger glucose intolerance in mice35.
We found that 60% of patients with type B IRS experienced hypoglycemia. Of note, the death of a patient with type B IRS from hypoglycemia has been reported36, 37, 38, 39. The trigger for diagnosis of type B IRS was hypoglycemia in 23% of the present patients. Hypoglycemia is thus important both as a trigger of diagnosis and as a serious event that warrants careful attention in this syndrome. Although the precise mechanisms underlying occasional hypoglycemia remain unclear, one possible explanation is the presence of both inhibitory and stimulatory antibodies to the insulin receptor8. Autoantibodies associated with type B IRS are usually polyclonal1, 39, and such antibodies with different characteristics might exist simultaneously. Alternatively, dissociation of inhibitory antibodies from the insulin receptor by an unknown mechanism might result in a sudden burst of insulin signaling that gives rise to hypoglycemia.
In our survey, patients with type B IRS were defined as individuals who manifest hyperglycemia or hypoglycemia (or both) and possess autoantibodies to the insulin receptor. However, two such patients (patients 4 and 14) showed hypoglycemia without hyperinsulinemia. Given that such cases are not technically “insulin resistant,” it might be inappropriate to classify them as type B IRS, with a new pathological categorization possibly being required. Regardless, healthcare providers should be aware that autoantibodies to the insulin receptor might induce hypoglycemia in the absence of insulin resistance or hyperinsulinemia.
The age at clinical diagnosis of type B IRS peaked in the 60s, and the male/female ratio was 19/11, suggesting that this syndrome occurs in mature individuals and that females are not at greater risk, in contrast to a previous study describing female predominance13. A total of 57% of patients were associated with concurrent autoimmune diseases, and 47% had been treated with immunomodulation therapy. Three patients with type B IRS had undergone eradication therapy for H. pylori, but only one of these patients, who also had ITP, responded to the treatment. Such therapy is thus unlikely to be effective for type B IRS patients in general. However, a patient with type B IRS and scleroderma was recently reported for whom H. pylori eradication eliminated the autoantibodies to the insulin receptor and markedly improved the effects of combined immune suppressants40. Given that ITP41 and scleroderma42 have been linked to H. pylori infection, type B IRS might be ameliorated by therapies effective for accompanying autoimmune diseases. Indeed, among the patients in our survey, the onset of type B IRS triggered the diagnosis of accompanying autoimmune diseases, and the treatment of these diseases also alleviated glycemic fluctuations. It seems reasonable to screen patients with type B IRS for other autoimmune diseases and to treat these accompanying disorders.
With regard to limitations, questionnaire‐based surveys are associated with a certain level of inaccuracy in the collection of information. Of note, the effectiveness of therapies might not have been accurately estimated. In addition, we sequenced only the exons of INSR and PIK3R1, and therefore cannot exclude the possibility that some genetic abnormalities, including long deletions of the genes, were not identified. Finally, the cases of type A IRS and Rabson–Mendenhall/Donohue syndrome analyzed included suspected cases, given that genetic testing is not always carried out in the clinical setting.
Disclosure
YI has received research support from MSD and Ono, as well as lecture fees from Bayer, Kowa, MSD, Novartis, Novo Nordisk, Ono, Sanofi and Takeda Pharmaceutical. Y Hirota has received lecture fees from Eli Lilly, Sanofi and Takeda Pharmaceutical. TA has received research support from Astellas, Cmic, Cosmic, Daiichi Sankyo, Eli Lilly, Kowa, MSD, Novartis, Ono, Pfizer and Takeda Pharmaceutical, as well as lecture fees from Ono. WO has received research support from Abbot Diabetes Care UK, Abbot Japan, Astellas, AstraZeneca, Boehringer Ingelheim, Dainippon‐Sumitomo Pharma, Daiichi Sankyo, Eli Lilly, Kowa, Kyorin Pharmaceutical, Mitsubishi Tanabe Pharma, MSD, Novartis, Novo Nordisk, Sanofi, Taisho Toyama Pharmaceutical, Takeda Pharmaceutical and Teijin Pharma, as well as lecture fees from Abbot Japan, Astellas, Boehringer Ingelheim, Dainippon‐Sumitomo Pharma, Mitsubishi Tanabe Pharma, MSD, Novartis, Sanofi and Takeda Pharmaceutical. H Katagiri has received research support from Otsuka Pharmaceutical and Astellas; endowments from MSD, Eli Lilly, Novo Nordisk, Takeda Pharmaceutical, Daiichi Sankyo, Astellas, Sanofi, Novartis, Mitsubishi Tanabe Pharma, Ono and Taisho Pharma; and lecture fees from Takeda Pharmaceutical, MSD, Daiichi Sankyo, Astellas, Taisho Pharma, Novartis, Mitsubishi Tanabe Pharma and Boehringer Ingelheim. All other authors declare no conflict of interest.
Acknowledgments
We thank the Japan Diabetes Society, the Japanese Society of Pediatric Endocrinology and healthcare professionals who responded to the questionnaires for their cooperation. This study was supported by the Research Program for Intractable Disease of the Ministry of Health, Labor and Welfare of Japan (to TA, WO and HK), the Practical Research Project for Rare/Intractable Diseases of the Japan Agency for Medical Research and Development (AMED; to TA, WO and HK), and a Grant‐in‐Aid for Scientific Research (C) from the Ministry of Education, Culture, Sports, Science and Technology of Japan (to Y Hirota).
J Diabetes Investig 2020; 11: 603–616
Contributor Information
Wataru Ogawa, Email: ogawa@med.kobe-u.ac.jp.
Hideki Katagiri, Email: katagiri@med.tohoku.ac.jp.
References
- 1. Kahn CR, Flier JS, Bar RS, et al The syndromes of insulin resistance and acanthosis nigricans. Insulin‐receptor disorders in man. N Engl J Med 1976; 294: 739–745. [DOI] [PubMed] [Google Scholar]
- 2. Arioglu E, Andewelt A, Diabo C, et al Clinical course of the syndrome of autoantibodies to the insulin receptor (type B insulin resistance): a 28‐year perspective. Medicine 2002; 81: 87–100. [DOI] [PubMed] [Google Scholar]
- 3. Musso C, Cochran E, Moran SA, et al Clinical course of genetic diseases of the insulin receptor (type A and Rabson‐Mendenhall syndromes): a 30‐year prospective. Medicine 2004; 83: 209–222. [DOI] [PubMed] [Google Scholar]
- 4. Semple RK, Savage DB, Cochran EK, et al Genetic syndromes of severe insulin resistance. Endocr Rev 2011; 32: 498–514. [DOI] [PubMed] [Google Scholar]
- 5. Flier JS, Bar RS, Muggeo M, et al The evolving clinical course of patients with insulin receptor autoantibodies: spontaneous remission or receptor proliferation with hypoglycemia. J Clin Endocrinol Metab 1978; 47: 985–995. [DOI] [PubMed] [Google Scholar]
- 6. Kawanishi K, Kawamura K, Nishina Y, et al Successful immunosuppressive therapy in insulin resistant diabetes caused by anti‐insulin receptor autoantibodies. J Clin Endocrinol Metab 1977; 44: 15–21. [DOI] [PubMed] [Google Scholar]
- 7. Hirano T, Adachi M. Insulin‐like growth factor 1 therapy for type B insulin resistance. Ann Intern Med 1997; 127: 245–246. [DOI] [PubMed] [Google Scholar]
- 8. Taylor SI, Grunberger G, Marcus‐Samuels B, et al Hypoglycemia associated with antibodies to the insulin receptor. N Engl J Med 1982; 307: 1422–1426. [DOI] [PubMed] [Google Scholar]
- 9. Tran HA, Reeves GE. Treatment of type B insulin resistance with immunoglobulin: novel use of an old therapy. Med J Aust 2009; 190: 168. [DOI] [PubMed] [Google Scholar]
- 10. Muggeo M, Flier JS, Abrams RA, et al Treatment by plasma exchange of a patient with autoantibodies to the insulin receptor. N Engl J Med 1979; 300: 477–480. [DOI] [PubMed] [Google Scholar]
- 11. Yamamoto T, Sato T, Mori T, et al Clinical efficacy of insulin‐like growth factor‐1 in a patient with autoantibodies to insulin receptors: a case report. Diabetes Res Clin Pract 2000; 49: 65–69. [DOI] [PubMed] [Google Scholar]
- 12. Imai J, Yamada T, Saito T, et al Eradication of insulin resistance. Lancet 2009; 374: 264. [DOI] [PubMed] [Google Scholar]
- 13. Kasuga M, Kadowaki T. Insulin receptor disorders in Japan. Diabetes Res Clin Pract. 1994; 24: S145–151. [DOI] [PubMed] [Google Scholar]
- 14. George S, Rochford JJ, Wolfrum C, et al A family with severe insulin resistance and diabetes due to a mutation in AKT2 . Science 2004; 304: 1325–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dash S, Sano H, Rochford JJ, et al A truncation mutation in TBC1D4 in a family with acanthosis nigricans and postprandial hyperinsulinemia. Proc Natl Acad Sci USA. 2009; 106: 9350–9355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rabson S, Mendenhall E. Familial hypertrophy of pineal body, hyperplasia of adrenal cortex and diabetes mellitus; report of 3 cases. Am J Clin Pathol 1956; 26: 283–290. [DOI] [PubMed] [Google Scholar]
- 17. Donohue WL, Uchida I. Leprechaunism: a euphemism for a rare familial disorder. J Pediatr 1954; 45: 505–519. [DOI] [PubMed] [Google Scholar]
- 18. Thauvin‐Robinet C, Auclair M, Duplomb L, et al PIK3R1 mutations cause syndromic insulin resistance with lipoatrophy. Am J Hum Genet 2013; 93: 141–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chudasama KK, Winnay J, Johansson S, et al SHORT syndrome with partial lipodystrophy due to impaired phosphatidylinositol 3 kinase signaling. Am J Hum Genet 2013; 93: 150–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dyment DA, Smith AC, Alcantara D, et al Mutations in PIK3R1 cause SHORT syndrome. Am J Hum Genet 2013; 93: 158–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hamaguchi T, Hirota Y, Takeuchi T, et al Treatment of a case of severe insulin resistance as a result of a PIK3R1 mutation with a sodium‐glucose cotransporter 2 inhibitor. J Diabetes Investig 2018; 9: 1224–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ohkubo K, Ishiyama K, Oyama N, et al PIK3R1 heni ni yori SHORT shoukougun to shindan shita shibouishukusei tounyoubyou no ichi rei (A case of lipoatrophic diabetes diagnosed as SHORT syndrome based on PIK3R1 mutation). Nihon Naibunpi Gakkai Zasshi 2017; 93: 536. (Japanese). [Google Scholar]
- 23. Kim H, Kadowaki H, Sakura H, et al Detection of mutations in the insulin receptor gene in patients with insulin resistance by analysis of single‐stranded conformational polymorphisms. Diabetologia 1992; 35: 261–266. [DOI] [PubMed] [Google Scholar]
- 24. Moritz W, Froesch ER, Böni‐Schnetzler M. Functional properties of a heterozygous mutation (Arg1174–>Gln) in the tyrosine kinase domain of the insulin receptor from a type A insulin resistant patient. FEBS Lett 1994; 351: 276–280. [DOI] [PubMed] [Google Scholar]
- 25. Chudasama KK, Winnay J, Johansson S, et al SHORT syndrome with partial lipodystrophy due to impaired phosphatidylinositol 3 kinase signaling. Am J Hum Gene. 2013; 93: 150–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schroeder C, Riess A, Bonin M, et al PIK3R1 mutations in SHORT syndrome. Clin Genet 2014; 86: 292–294. [DOI] [PubMed] [Google Scholar]
- 27. Avila M, Dyment DA, Sagen JV, et al Clinical reappraisal of SHORT syndrome with PIK3R1 mutations: toward recommendation for molecular testing and management. Clin Genet 2016; 89: 501–506. [DOI] [PubMed] [Google Scholar]
- 28. Aarskog D, Ose L, Pande H, et al Autosomal dominant partial lipodystrophy associated with Rieger anomaly, short stature, and insulinopenic diabetes. Am J Med Genet 1983; 15: 29–38. [DOI] [PubMed] [Google Scholar]
- 29. Longo N, Wang Y, Smith SA, et al Genotype‐phenotype correlation in inherited severe insulin resistance. Hum Mol Genet 2002; 11: 1465–1475. [DOI] [PubMed] [Google Scholar]
- 30. Kadowaki T, Bevins CL, Cama A, et al Two mutant alleles of the insulin receptor gene in a patient with extreme insulin resistance. Science 1988; 240: 787–790. [DOI] [PubMed] [Google Scholar]
- 31. Fujita S, Kuroda Y, Fukui K, et al Hyperinsulinemia and insulin receptor gene mutation in nonobese healthy subjects in Japan. J Endocr Soc 2017; 1: 1351–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Taniguchi CM, Emanuelli B, Kahn CR. Critical nodes in signalling pathways: insights into insulin action. Nat Rev Mol Cell Biol 2006; 7: 85–96. [DOI] [PubMed] [Google Scholar]
- 33. Schwingshandl J, Mache CJ, Rath K, et al SHORT syndrome and insulin resistance. Am J Med Genet 1993; 47: 907–909. [DOI] [PubMed] [Google Scholar]
- 34. Winnay JN, Solheim MH, Dirice E, et al PI3‐kinase mutation linked to insulin and growth factor resistance in vivo. J Clin Invest 2016; 126: 1401–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Miyake K, Ogawa W, Matsumoto M, et al Hyperinsulinemia, glucose intolerance, and dyslipidemia induced by acute inhibition of phosphoinositide 3‐kinase signaling in the liver. J Clin Invest. 2002; 110: 1483–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tsokos GC, Gorden P, Antonovych T, et al Lupus nephritis and other autoimmune features in patients with diabetes mellitus due to autoantibody to insulin receptors. Ann Intern Med 1985; 102: 176–181. [DOI] [PubMed] [Google Scholar]
- 37. Bloise W, Wajchenberg BL, Moncada VY, et al Atypical antiinsulin receptor antibodies in a patient with type B insulin resistance and scleroderma. J Clin Endocrinol Metab 1989; 68: 227–231. [DOI] [PubMed] [Google Scholar]
- 38. O’Brien TD, Rizza RA, Carney JA, et al Islet amyloidosis in a patient with chronic massive insulin resistance due to antiinsulin receptor antibodies. J Clin Endocrinol Metab 1994; 79: 290–292. [DOI] [PubMed] [Google Scholar]
- 39. Flier JS, Kahn CR, Roth J, et al Antibodies that impair insulin receptor binding in an unusual diabetic syndrome with severe insulin resistance. Science 1975; 190: 63–65. [DOI] [PubMed] [Google Scholar]
- 40. Yang Guo‐Qing, Li Yi‐Jun, Dou Jing‐Tao, et al Type B insulin resistance syndrome with Scleroderma successfully treated with multiple immune suppressants after eradication of Helicobacter pylori infection: a case report. BMC Endocr Disord 2016; 16: 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Stasi R, Sarpatwari A, Segal JB, et al Effects of eradication of Helicobacter pylori infection in patients with immune thrombocytopenic purpura: a systematic review. Blood 2009; 113: 1231–1240. [DOI] [PubMed] [Google Scholar]
- 42. Radić M, Martinović Kaliterna D, Bonacin D, et al Correlation between Helicobacter pylori infection and systemic sclerosis activity. Rheumatology 2010; 49: 1784–1785. [DOI] [PubMed] [Google Scholar]
- 43. Takasawa K, Tsuji‐Hosokawa A, Takishima S, et al Clinical characteristics of adolescent cases with Type A insulin resistance syndrome caused by heterozygous mutations in the β‐subunit of the insulin receptor (INSR) gene. J Diabetes 2019; 11: 46–54. [DOI] [PubMed] [Google Scholar]
- 44. Saito‐Hakoda A, Nishii A, Uchida T, et al A follow‐up during puberty in a Japanese girl with type A insulin resistance due to a novel mutation in INSR . Clin Pediatr Endocrinol 2018; 27: 53–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Takahashi I, Yamada Y, Kadowaki H, et al Phenotypical variety of insulin resistance in a family with a novel mutation of the insulin receptor gene. Endocr J 2010; 57: 509–516. [DOI] [PubMed] [Google Scholar]
- 46. Hosoe J, Kadowaki H, Miya F, et al Structural Basis and Genotype‐Phenotype Correlations of INSR Mutations Causing Severe Insulin Resistance. Diabetes 2017; 66: 2713–2723. [DOI] [PubMed] [Google Scholar]
- 47. Nakashima N, Miyamura T, Yamashita T, et al Type A‐insulin resistance with lipopexia on extremities: a case report. Endocrinol Jpn 1992; 39: 347–53. [DOI] [PubMed] [Google Scholar]
- 48. Hashimoto N, Yokoi N, Komada H, et al A case of type A insulin resistance associated with heterozygous Asn462Ser mutation of the insulin receptor gene. Diabetol Int 2012; 3: 239–243. [Google Scholar]
- 49. Ando A, Yatagai T, Rokkaku K, et al Obesity is a critical risk factor for worsening of glucose tolerance in a family with the mutant insulin receptor. Diabetes Care 2002; 25: 1484–1485. [DOI] [PubMed] [Google Scholar]
- 50. Taira M, Taira M, Hashimoto N, et al Human diabetes associated with a deletion of the tyrosine kinase domain of the insulin receptor. Science 1989; 245: 63–66. [DOI] [PubMed] [Google Scholar]
- 51. Harada M, Uchigata Y, Kataoka N, et al A case of diabetes mellitus caused by a type A insulin receptor abnormality who has been treated with insulin‐like growth factor 1 and oral glycemic agents for 19 years. J Jpn Diabet Soc 2010; 53: 626–630 (Japanese). [Google Scholar]
- 52. Abe Y, Sato T, Takagi M, et al A case of Rabson‐Mendenhall syndrome with a novel mutation in the tyrosine kinase domain of the insulin receptor gene complicated by medullary sponge kidney. J Pediatr Endocrinol Metab. 2012; 25: 587–590. [DOI] [PubMed] [Google Scholar]
- 53. Fukunaga T, Murakami T, Tanaka H, et al Dental and craniofacial characteristics in a patient with leprechaunism treated with insulin‐like growth factor‐I. Angle Orthod. 2008; 78: 745–751. [DOI] [PubMed] [Google Scholar]
- 54. Kawashima Y, Nishimura R, Utsunomiya A, et al Leprechaunism (Donohue syndrome): a case bearing novel compound heterozygous mutations in the insulin receptor gene. Endocr J. 2013; 60: 107–112. [DOI] [PubMed] [Google Scholar]
- 55. Nakae J, Kato M, Murashita M, et al Long‐term effect of recombinant human insulin‐like growth factor I on metabolic and growth control in a patient with leprechaunism. J Clin Endocrinol Metab. 1998; 83: 542–549. [DOI] [PubMed] [Google Scholar]
- 56. Satomi T, Kanetsugu Y, Noro A, et al rh IGF‐1 ni yoru chiryou wo okonatta youseishou no ichirei (A case of leprecbaunism treated by rh IGF‐1). J Sapporo City Gen Hosp 2013; 73: 53–59. [Google Scholar]
- 57. Takesue K, Obata T, Isse N, et al Report; A case of type B insulin‐resistance syndrome ameliorated with immune‐suppression therapies. Nihon Naika Gakkai Zasshi 2016; 105: 710–714. (Japanese). [DOI] [PubMed] [Google Scholar]
- 58. Kawasaki F, Anno T, Takai M, et al Saibokuto as a possible therapy for type B insulin resistance syndrome: the disappearance of anti‐insulin receptor antibody and a marked amelioration of glycemic control by Saibokuto treatment. Intern Med 2018; 57: 2359–2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sato N, Ohsawa I, Takagi M, et al Type B insulin resistance syndrome with systemic lupus erythematosus. Clin Nephrol 2010; 73: 157–162. [DOI] [PubMed] [Google Scholar]
- 60. Yamada H, Asano T, Kusaka I, et al Type B insulin resistance syndrome with fasting hypoglycemia and postprandial hyperglycemia. Diabetol Int 2015; 6: 144–148. [Google Scholar]
- 61. Takei M, Ishii H, Kawai Y, et al Efficacy of oral glucocorticoid and cyclosporine in a case of rituximab‐refractory type B insulin resistance syndrome. J Diabetes Investig 2015; 6: 734–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Toshihiro M, Katagiri H, Kataoka K, et al Recurrent hypoglycemia during pregnancies in a woman with multiple autoantibodies including anti‐insulin receptor antibody and anti‐platelet antibody, whose serum lowered murine blood glucose levels and phosphorylated insulin receptor of CHO‐IR cells. Endocr J 2011; 58: 1037–1043. [DOI] [PubMed] [Google Scholar]
- 63. Horie I, Yamasaki H, Kawashiri S, et al Marked Different Responses of rhIGF‐1 and Steroid Therapy in Two Patients with Type B Insulin Resistance Complicated by Systemic Lupus Erythematosus. J Jpn Diabet Soc 2009; 52: 957–963 (Japanese). [Google Scholar]
- 64. Uchida S, Yamaguchi M, Hasegawa H, et al A Case of Type B Insulin Resistance Syndrome With Postprandial Hyperglycemia Successfully Treated With Liraglutide. J Jpn Diabet Soc 2017; 60: 244–252 (Japanese). [Google Scholar]