

Abstract
An assessment of the C−H activation catalyst [(COD)Ir(IMes)(PPh3)]PF6 (COD=1,5‐cyclooctadiene, IMes=1,3‐bis(2,4,6‐trimethylphenyl)imidazol‐2‐ylidene) in the deuteration of phenyl rings containing different functional directing groups is divulged. Competition experiments have revealed a clear order of the directing groups in the hydrogen isotope exchange (HIE) with an iridium (I) catalyst. Through DFT calculations the iridium–substrate coordination complex has been identified to be the main trigger for reactivity and selectivity in the competition situation with two or more directing groups. We postulate that the competition concept found in this HIE reaction can be used to explain regioselectivities in other transition‐metal‐catalyzed functionalization reactions of complex drug‐type molecules as long as a C−H activation mechanism is involved.
Keywords: C−H functionalization, deuterium, directing groups, hydrogen isotope exchange, iridium catalysis
Assessment of an iridium(I) catalyst for C−H activation in the deuteration of phenyl compounds substituted with different functional directing groups is disclosed. Competition experiments and DFT calculations have revealed a clear order of the directing groups in hydrogen isotope exchange reactions.
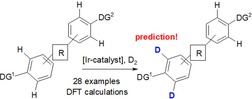
In recent years C−H functionalization of complex molecules has become a strong tool in lead optimization of bioactive molecules in the life‐science industry.1 To increase speed and decrease costs and resources in drug‐discovery research C−H functionalization reactions enable the modification of complex structures at the latest possible linear step. In this way novel chemical space can be explored and new intellectual property (IP) of compounds with already optimized pharmaceutical value can be generated. This concept is also followed in the hydrogen isotope exchange (HIE) reaction, the most fundamental of all C−H functionalization reactions (Scheme 1). The HIE reaction has become a broadly utilized and elegant method for the incorporation of deuterium or tritium into organic molecules, circumventing the need for a tedious multi‐step synthesis.2 In drug discovery, radioactive tritium substances are used as research tools3, 4 for example, for understanding tissue distribution,5 in covalent binding assays,6 or for ADME (absorption distribution metabolism excretion) profiling of new drug candidates.7 Numerous HIE protocols utilizing homogeneous or heterogeneous catalysts have already been described.2, 8 Even though a new generation of bidentate iridium catalyst systems such as those from Pfaltz,9 Burgess,10 and Tamm11 have extended the scope of the aromatic ortho‐directing HIE reactions, (Scheme 1) the commercially available Crabtree's12 and Kerr's catalyst13, 14 1 remain the most regularly applied iridium catalysts in industry today. Interestingly new HIE applications have recently been published with great success utilizing other metals, such as iron,15 cobalt,16 or ruthenium,17, 18, 19 or photoredox reactions20 broadening the scope also to C(sp3)‐carbon atoms. Nevertheless, although there are numerous publications on the use of different directing groups such as ketones, esters, amides, carboxylic acids, nitro groups, sulfones, sulfonamides, and others, there is still a lack of understanding and prediction for how the different directing groups enable HIE if more than one directing group is present in the molecule. This understanding is especially important because the introduction of a radioactive label in a metabolically stable position is essential for the application of tritiated compounds in in vivo experiments.3 Furthermore, these predictions could also help to plan late‐stage functionalization reactions and result in more complex molecules in general.
Scheme 1.

Iridium‐catalyzed ortho‐directed aromatic HIE reactions. (DG=directing group).
We identified several functional groups, for example, imidazole (N heterocycle), ketones, esters, amides, carbamates, acids, nitro groups, sulfones, sulfonamides, and phenols, among others, that are present repeatedly in aromatic drug molecules (based on an overview of 200 prescribed drugs in 2016 reported by Njarðarson and co‐workers21). Based on these findings we studied the order of the aforementioned functional groups as directing groups in a standard HIE reaction with deuterium gas as the isotope source.22
Initial experiments were performed under standard HIE conditions in the presence of the commercial Kerr catalyst 1 [1 atm D2, rt, dichloromethane (DCM), 2 h] to make our results as comparable with literature results23 as possible and consistent across the set. We conducted a series of competition experiments,24 employing two simple mono‐substituted aromatic substrates in one reaction vessel (Scheme 2 A), along with catalyst 1 to show which of the two directing groups enables greater deuteration in the HIE experiment. The analysis of deuterium incorporation was performed by either mass spectrometry or by 1H NMR spectroscopy. Applying this methodology to aromatic compounds 2–10 (Table 1), we were able to determine experimentally the general relative directing‐group strength in the order: imidazole 2 (heterocycle25)>acetylanilide 3≈ketone 4>nitro 5>tertiary amide 6>aldehyde 7>ester 8>primary sulfonamide 9>carbamate 10 (Scheme 2 B).
Scheme 2.

Competition HIE experiment of aromatic compounds with catalysts 1 at room temperature in DCM. (Ar=Phenyl; except for 4, Ar*=4‐biphenyl).
Table 1.
Deuterium incorporation results of the competition HIE reactions with catalyst 1 and substrates 2–10.[a,b]
|
|
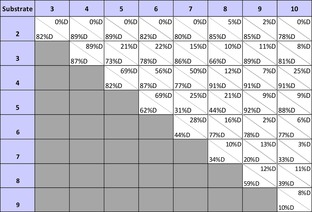
[a] Conditions: each substrate (1 equiv.), catalyst 1 (5 mol %, 0.05 equiv.), DCM (6 mL), D2 (1 atm), rt, 2 h. [b] The analysis was done either by mass spectrometry (if substrate was ionizable and there was no overlapping between the two substrates) or by 1H NMR spectroscopy.
With these results in hand a prediction of the labelling positions with catalyst 1 in more complex molecules bearing these directing groups should be possible. However, to understand the theoretical background of our proposal we applied density functional theory (DFT) calculations (M06 functional),26 following the approach successfully applied by Kerr and co‐workers,14c, 27, 28 based on the mechanism suggested by Heys and co‐workers.29 The constructed free energy profiles (ΔG rel) with catalyst 1 are shown in Figure 1 for the first step of the HIE reaction. It is reported that the rate‐determining step is the most significant step in HIE reactions and the free activation energy of Ir–C−H insertion (ΔΔG rel, 2–10 a→2–10 b) is considered to be the main parameter to explain the formation of the deuterated products.27, 28 However, the order of directing‐group significance in compounds 2–10 should have differed compared with the order observed experimentally. This result is especially significant for compounds 3 and 6 with their relative free activation energies (ΔΔG rel, 2–10 a→2–10 b) calculated to be higher than those of compounds 5, 7, and 8 (Table 2, Figure 1). However, in these competition experiments, which take place readily at room temperature, the relative populations of the coordination complex 2–10 a strongly influence the outcome of the overall reaction. Once the coordination complex 2–10 a has been formed, the reaction will proceed, via transition state 2–10 b and insertion product 2–10 c, to completion. This influence is reflected in the order of the calculated relative free energies ΔG rel(CC) of the coordination complexes 2–10 a, which matches well the order observed experimentally. For similarly coordinating directing groups (same ΔG rel(CC) values), in which the coordination complexes are populated almost equally (e.g. 7–9), relative free activation energies (ΔΔG rel, 2–10 a→2–10 b) become dominant again. This can be seen clearly for compound pairs 4 a versus 6 a (−0.3 vs. 4.6 kcal mol−1) and 7 a versus 8 a (0.4 vs. 3.6 kcal mol−1). This contribution is significant to understanding the processes of the C−H activation mechanism with complex substrates. It is not the rate‐limiting14b, 27, 28 transition state 2–10 b that is responsible for the HIE reaction outcome in competition cases but the lower free energy ΔG rel(CC) of the initially formed coordination complexes 2–10 a. After the success and observations of the competition experiments, we sought to obtain more insight into the role of competing directing groups in the same molecule. We studied aromatic compounds 11–20 with at least two different substituents. Owing to the higher complexity of 1H NMR analysis in 1,2‐ or 1,3‐disubstituted benzenes, we focused our study on 1,4‐disubstituted systems (Scheme 3). Furthermore, we have calculated the ΔΔG rel(CC) values of the two directing groups from Table 2 to demonstrate the predicted energy difference in the coordination complex combined with the experimental result. All predicted results for the deuteration reaction strengthen the reported order obtained in the competition experiments (Table 2 and Figure 1).
Figure 1.

Relative free energies ΔG rel of strong, moderate, and low substrate–iridium catalyst coordination complexes 2–10 a, transition states 2–10 b, and C−H insertion products 2–10 c.
Table 2.
For different directing groups the relative free energies to form the coordination complexes 2–10 a (sorted by ΔG rel) and the free activation energies ΔΔG rel to generate 2–10 c via transition states 2–10 b are shown.
|
|
DG |
ΔG rel (CC) 2–10 a |
ΔΔG rel (2–10 a→2–10 b) |
|---|---|---|---|
|
Aryl (Ph) |
−2‐pyridine |
−71.8 |
22.9 |
|
−N‐oxide |
−32.9 |
27.2 |
|
|
−NH2 |
−29.6 |
33.3 |
|
|
−CO‐NH2 |
−28.6 |
27.5 |
|
|
−2‐imidazole 2 |
−27.5 |
23.9 |
|
|
−PO(NMe2)2 |
−25.4 |
26.5 |
|
|
−CO‐NMe2 |
−25.1 |
26.0 |
|
|
−N‐CO‐Me 3 |
−23.7 |
27.4 |
|
|
−CH2COOMe |
−23.1 |
28.5 |
|
|
−CO‐Me |
−23.0 |
22.7 |
|
|
−CO‐NEt2 6 |
−23.0 |
27.6 |
|
|
−COOH |
−22.0 |
22.3 |
|
|
−SO2NH2 9 |
−21.5 |
23.8 |
|
|
−COOMe 8 |
−20.9 |
24.4 |
|
|
−OH |
−20.9 |
35.8 |
|
|
−CO‐H 7 |
−20.3 |
20.6 |
|
|
−SO2‐Me |
−20.2 |
23.4 |
|
|
−CH2COMe |
−19.8 |
37.5 |
|
|
−NSO2Me |
−17.7 |
28.1 |
|
|
−OMe |
−16.8 |
29.2 |
|
|
−NO2 5 |
−16.6 |
18.5 |
|
|
−SO3H |
−16.6 |
22.9 |
|
|
−SO2NHCONH‐Me |
−15.4 |
24.5 |
|
|
−OCOMe 10 |
−14.3 |
23.0 |
Scheme 3.
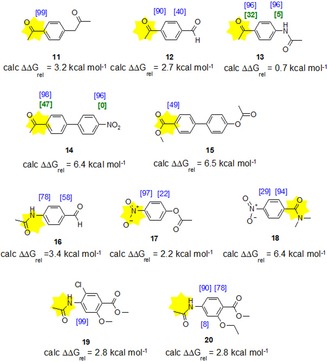
HIE experiment of aromatic substrates 11–20 with deuterium gas and catalyst 1 (blue: 25 °C, %D; green: 0 °C, %D); the DG, as predicted by ΔΔG rel(CC), is labelled in yellow.
However, by this method we are able to predict the main product, but not the final deuteration result in %D. With compounds 13 and 14 it was shown that by lowering the reaction temperature to 0 °C the selectivity of deuteration was shifted in favor of the ketone as the directing group (green numbers, Scheme 3), indicating that the calculated transition activation energy ΔΔG rel(CC) plays a much bigger role at temperatures below 0 °C. To our delight we found a general agreement in spite of the selectivity of the main deuterated product according to our prediction based on the DFT calculation. To rule out an empirical finding we performed DFT calculations with additional less common directing groups that feature in drug molecules, including sulfone, carboxylate, phenol, and phenylacetic ester groups. The directing groups were ordered according to their ΔG rel energy for the formation of their coordination complex 2–10 a (Table 2).
For comparison we also added the free activation energy (ΔΔG rel, 2–10 a→2–10 b) for the C−H activation step showing that there are significant differences between these two parameters. In groups including N‐oxide, phenol, carboxylic and sulfonic acid there is a strong pH dependency present as the negative charge adds strongly to the negative ΔG rel(CC) in the coordination complex 2–10 a. A positive charge on the potentially coordinating heteroatom (N, O, or S) inhibits the formation of complex 2–10 a completely, explaining why drug salts from strong acids (HCl, TFA, etc.) generally fail in this HIE reaction. Furthermore, we observed a strong steric effect in the formation of complex 2–10 a demonstrated in the case of the benzamides: primary amide −28.6 kcal mol−1, tertiary amides CONMe2 −25.1 kcal mol−1, and CONEt2 −23.0 kcal mol−1. We conclude that Table 2 summarizes many of the common directing groups in drug discovery and therefore should be a great help for synthesis planning and predicting outcomes in HIE reactions. However, we identified some directing groups, such as NO2, where there is not a perfect match between the theoretical and experimental result.
Finally, we have performed HIE reactions with catalyst 1 on complex molecules 21–29 where at least two different directing groups are present (Scheme 4). The red arrow shows the predicted main deuteration or tritiation position based on the order shown in Table 2, the blue numbers in brackets show the %D at the relevant position from the performed deuteration experiment, the red numbers show the corresponding %T from the tritium experiment. In nearly all cases where the HIE occurs with catalyst 1 we were able to predict the correct labelling position in a competition situation independently from the hydrogen isotope source.
Scheme 4.

HIE experiment of drugs 21–29 with deuterium gas and catalyst 1; red arrow indicates the predicted major deuteration/tritiation position based on the directing‐group strength shown in Table 2.
We consider that this finding will be a useful and general principle to understand the outcome of competition reactions in which different directing groups are present in one molecule and this knowledge can likely be transferred to further transition‐metal‐catalyzed C−H functionalization reactions. Other research groups have found similar regioselectivities in directed C−H functionalization competition reactions for C−C or C−X bond formations in complex molecules,30 by applying catalysts such as iridium,31 rhodium,32 palladium,33 cobalt,34 or ruthenium35 metals. Establishing convenient prediction models will be extremely beneficial.36 We consider that with the help of improved computer‐aided tools the prediction of late‐stage functionalization of complex molecules can be significantly improved by taking this competition principle into account. Together with the understanding of directing‐group‐enhancing transformations recently described by Dai, Yu, and co‐workers37 this research could lead to a much broader use of C−H functionalization in lead optimization in the future.
In summary we have provided a clear order of the influence of the directing group in HIE reactions with catalyst 1. These results not only allow predictions in regioselective HIE reactions of complex molecules, but also give a perspective to the concept of directing‐group‐induced late‐stage functionalization in general. Understanding the influence of the directing groups could allow for a much better planning of syntheses and an increase in the successful prediction of late‐stage functionalization pathways. Therefore, the success rate of these reactions could be improved, and strategic synthetic management positively influenced. With this concept we aim to improve the outcome of computer‐aided predictions of HIE reactions and potentially increase the effectiveness of late‐stage functionalization reactions in research.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
M.V. is participant in the EU ITN Isotopics consortium. The ISOTOPICS project has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Sklodowska‐Curie grant agreement No. 675071.
M. Valero, T. Kruissink, J. Blass, R. Weck, S. Güssregen, A. T. Plowright, V. Derdau, Angew. Chem. Int. Ed. 2020, 59, 5626.
References
- 1. Cernak T., Dykstra K. D., Tyagarajan S., Vachal P., Krska S. W., Chem. Soc. Rev. 2016, 45, 546–576. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Atzrodt J., Derdau V., Kerr W. J., Reid M., Angew. Chem. Int. Ed. 2018, 57, 3022–3047; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 3074–3101; [Google Scholar]
- 2b. Atzrodt J., Derdau V., Fey T., Zimmermann J., Angew. Chem. Int. Ed. 2007, 46, 7744–7765; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 7890–7911; [Google Scholar]
- 2c. Junk T., Catallo W. J., Chem. Soc. Rev. 1997, 26, 401–406; [Google Scholar]
- 2d. Di Giuseppe A., Castarlenas R., Oro L. A., C. R. Chim. 2015, 18, 713–741; [Google Scholar]
- 2e. Voges R., Heys J. R., Moenius T., Preparation of Compounds Labeled with Tritium and Carbon-14, Wiley, New York, 2009; [Google Scholar]
- 2f. Valero M., Derdau V., J. Labelled Compd. Radiopharm. 2019, 10.1002/jlcr.3783. [DOI] [PubMed] [Google Scholar]
- 3. Atzrodt J., Derdau V., Kerr W. J., Reid M., Angew. Chem. Int. Ed. 2018, 57, 1758–1784; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 1774–1802. [Google Scholar]
- 4.For selected reviews, see:
- 4a. Elmore C. S., Bragg R. A., Bioorg. Med. Chem. Lett. 2015, 25, 167–171; [DOI] [PubMed] [Google Scholar]
- 4b. Krauser A., J. Labelled Compd. Radiopharm. 2013, 56, 441–446; [DOI] [PubMed] [Google Scholar]
- 4c. Lockley W. J. S., McEwen A., Cooke R., J. Labelled Compd. Radiopharm. 2012, 55, 235–257; [Google Scholar]
- 4d. Mutlib A. E., Chem. Res. Toxicol. 2008, 21, 1672–1689. [DOI] [PubMed] [Google Scholar]
- 5.For selected reviews, see:
- 5a. Solon E. G., Chem. Res. Toxicol. 2012, 25, 543–555; [DOI] [PubMed] [Google Scholar]
- 5b. Harrell A. W., Sychterz C., Ho M. Y., Weber A., Valko K., Negash K., Pharm. Res. Per. 2015, 3, e00173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.For selected reviews, see:
- 6a. Maguire J. J., Kuc R. E., Davenport A. P., Methods Mol. Biol. 2012, 897, 31–77; [DOI] [PubMed] [Google Scholar]
- 6b. Hulme E. C., Trevethick M. A., Br. J. Pharmacol. 2010, 161, 1219–1237; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6c. Harder D., Fotiadis D., Nat. Protoc. 2012, 7, 1569–1578; [DOI] [PubMed] [Google Scholar]
- 6d. Berry J., Price-Jones M., Killian B., Methods Mol. Biol. 2012, 897, 79–94; [DOI] [PubMed] [Google Scholar]
- 6e. Buscher B., Laakso S., Mascher H., Pusecker K., Doig M., Dillen L., Wagner-Redeker W., Pfeifer T., Delrat P., Timmerman P., Bioanalysis 2014, 6, 673–682. [DOI] [PubMed] [Google Scholar]
- 7.For selected reviews, see:
- 7a. Penner N., Xu L., Prakash C., Chem. Res. Toxicol. 2012, 25, 513–531; [DOI] [PubMed] [Google Scholar]
- 7b. Isin E. M., Elmore C. S., Nilsson G. N., Thompson R. A., Weidolf L., Chem. Res. Toxicol. 2012, 25, 532–542. [DOI] [PubMed] [Google Scholar]
- 8.For selected reviews, see:
- 8a. Heys J. R., J. Labelled Compd. Radiopharm. 2007, 50, 770–778; [Google Scholar]
- 8b. Nilsson G. N., Kerr W. J., J. Labelled Compd. Radiopharm. 2010, 53, 662–667; [Google Scholar]
- 8c. Salter R., J. Labelled Compd. Radiopharm. 2010, 53, 645–657; [Google Scholar]
- 8d. Allen P. H., Hickey M. J., Kingston L. P., Wilkinson D. J., J. Labelled Compd. Radiopharm. 2010, 53, 731–738; [Google Scholar]
- 8e. Atzrodt J., Derdau V., J. Labelled Compd. Radiopharm. 2010, 53, 674–685. [Google Scholar]
- 9. Parmentier M., Hartung T., Pfaltz A., Muri D., Chem. Eur. J. 2014, 20, 11496–11504. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Perry M. C., Cui X., Powell M. T., Hou D. R., Reibenspies J. H., Burgess K., J. Am. Chem. Soc. 2003, 125, 113—123; [DOI] [PubMed] [Google Scholar]
- 10b. Burhop A., Weck R., Atzrodt J., Derdau V., Eur. J. Org. Chem. 2017, 1418–1424; [Google Scholar]
- 10c. Burhop A., Prohaska R., Weck R., Atzrodt J., Derdau V., J. Labelled Compd. Radiopharm. 2017, 60, 343–348; [DOI] [PubMed] [Google Scholar]
- 10d. Valero M., Mishra A., Blass J., Weck R., Derdau V., ChemistryOpen 2019, 8, 1183–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.
- 11a. Jess K., Derdau V., Weck R., Atzrodt J., Freytag M., Jones P. G., Tamm M., Adv. Synth. Catal. 2017, 359, 629–638; [Google Scholar]
- 11b. Valero M., Burhop A., Jess K., Weck R., Tamm M., Atzrodt J., Derdau V., J. Labelled Compd. Radiopharm. 2018, 61, 380–385; [DOI] [PubMed] [Google Scholar]
- 11c. Valero M., Becker D., Jess K., Weck R., Bannenberg T., Atzrodt J., Derdau V., Tamm M., Chem. Eur. J. 2019, 25, 6517–6522. [DOI] [PubMed] [Google Scholar]
- 12. Crabtree R., Acc. Chem. Res. 1979, 12, 331–337. [Google Scholar]
- 13.
- 13a. Brown J. A., Irvine S., Kennedy A. R., Kerr W. J., Andersson S., Nilsson G. N., Chem. Commun. 2008, 1115–1117. [DOI] [PubMed] [Google Scholar]
- 14.
- 14a. Kerr W. J., Lindsay D. M., Reid M., Atzrodt J., Derdau V., Rojahn P., Weck R., Chem. Commun. 2016, 52, 6669–6672; [DOI] [PubMed] [Google Scholar]
- 14b. Brown J. A., Cochrane A. R., Irvine S., Kerr W. J., Mondal B., Parkinson J. A., Paterson L. C., Reid M., Tuttle T., Andersson S., Nilson G. N., Adv. Synth. Catal. 2014, 356, 3551–3562; [Google Scholar]
- 14c. Cochrane A. R., Idziak C., Kerr W. J., Mondal B., Paterson L. C., Tuttle T., Andersson S., Nilsson G. N., Org. Biomol. Chem. 2014, 12, 3598–3603. [DOI] [PubMed] [Google Scholar]
- 15. Yu R. P., Hesk D., Rivera N., Pelczer I., Chirik P. J., Nature 2016, 529, 195–199. [DOI] [PubMed] [Google Scholar]
- 16. Palmer W. N., Chirik P. J., ACS Catal. 2017, 7, 5674–5678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Neubert L., Michalik D., Bähn S., Imm S., Neumann H., Atzrodt J., Derdau V., Holla W., Beller M., J. Am. Chem. Soc. 2012, 134, 12239–12244. [DOI] [PubMed] [Google Scholar]
- 18. Taglang C., Martinez-Prieto L. M., del Rosal I., Maron L., Poteau R., Philippot K., Chaudret B., Perato S., Sam Lone A., Puente C., Dugave C., Rousseau B., Pieters G., Angew. Chem. Int. Ed. 2015, 54, 10474–10477; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 10620–10623. [Google Scholar]
- 19. Gao L., Perato S., Garcia-Argote S., Taglang C., Martinez-Prieto L. M., Chollet C., Buisson D.-A., Dauvois V., Lesot P., Chaudret B., Rousseau B., Feuillastrea S., Pieters G., Chem. Commun. 2018, 54, 2986–2989. [DOI] [PubMed] [Google Scholar]
- 20. Loh Y. Y., Nagao K., Hoover A. J., Hesk D., Rivera N. R., Coletti S. L., Davies I. W., MacMillan D. W. C., Science 2017, 358, 1182–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.https://njardarson.lab.arizona.edu/content/top-pharmaceuticals-poster;; Smith D. T., Delost M. D., Qureshi H., Njarðarson J. T., J. Chem. Educ. 2010, 87, 1348; Poster information gathered by the Njarðarson group with data from DrugTopics & Pharmacompass. [Google Scholar]
- 22.Please note that there is no standard protocol to apply phenols or carboxylic acids in iridium-catalyzed HIE reactions. Therefore, these directing groups have not been included.
- 23.
- 23a. Kennedy A. R., Kerr W. J., Moir R., Reid M., Org. Biomol. Chem. 2014, 12, 7927–7931; [DOI] [PubMed] [Google Scholar]
- 23b. Kerr W. J., Mudd R. J., Paterson L. C., Brown J. A., Chem. Eur. J. 2014, 20, 14604–14607; [DOI] [PubMed] [Google Scholar]
- 23c. Kerr W. J., Mudd R. J., Owens P. K., Reid M., Brown J. A., Campos S., J. Labelled Compd. Radiopharm. 2016, 59, 601–603. [DOI] [PubMed] [Google Scholar]
- 24.For an account on the synthetic relevance of competition experiments, see: Collins K. D., Glorius F., Nat. Chem. 2013, 5, 597.23787750 [Google Scholar]
- 25. Atzrodt J., Derdau V., Kerr W. J., Reid M., Rojahn P., Weck R., Tetrahedron 2015, 71, 1924–1924. [Google Scholar]
- 26.Details of the calculation method along with further calculation results can be found in the Supporting Information.
- 27. Kerr W. J., Reid M., Tuttle T., Angew. Chem. Int. Ed. 2017, 56, 7808–7812; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 7916–7920. [Google Scholar]
- 28.
- 28a. Kerr W. J., Reid M., Tuttle T., ACS Catal. 2015, 5, 402–410; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28b. Kerr W. J., Lindsay D. M., Owens P. K., Reid M., Tuttle T., Campos S., ACS Catal. 2017, 7, 7182–7186. [Google Scholar]
- 29. Shu A. Y. L., Chen W., Heys J. R., J. Organomet. Chem. 1996, 524, 87–93. [Google Scholar]
- 30. Abrams D. J., Provencher P. A., Sorensen E. J., Chem. Soc. Rev. 2018, 47, 8925–8967. [DOI] [PubMed] [Google Scholar]
- 31. Wu G., Ouyang W., Chen Q., Huo Y., Li X., Org. Chem. Front. 2019, 6, 284–289. [Google Scholar]
- 32. He J., Hamann L. G., Davies H. M. L., Beckwith R. E. J., Nat. Commun. 2015, 6, 5943. [DOI] [PubMed] [Google Scholar]
- 33.
- 33a. Rosen B. R., Simke L. R., Thuy-Boun P. S., Dixon D. D., Yu J. Q., Baran P. S., Angew. Chem. Int. Ed. 2013, 52, 7317–7320; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 7458–7461; [Google Scholar]
- 33b. Oh Y., Jang Y. J., Jeon M., Kim H. S., Kwak J. H., Chung K. H., Pyo S., Jung Y. H., Kim I. S., J. Org. Chem. 2017, 82, 11566–11572; [DOI] [PubMed] [Google Scholar]
- 33c. Orito K., Horibata A., Nakamura T., Ushito H., Nagasaki H., Yuguchi M., Yamashita S., Tokuda M., J. Am. Chem. Soc. 2004, 126, 14342–14343. [DOI] [PubMed] [Google Scholar]
- 34. Qu S., Cramer C. J., J. Org. Chem. 2017, 82, 1195–1204. [DOI] [PubMed] [Google Scholar]
- 35.
- 35a. Shi P., Li S., Hu L.-M., Wang C., Loh T.-P., Hu X.-H., Chem. Commun. 2019, 55, 11115–11118; [DOI] [PubMed] [Google Scholar]
- 35b. Leitch J. A., Frost C. G., Chem. Soc. Rev. 2017, 46, 7145–7153; [DOI] [PubMed] [Google Scholar]
- 35c. Mei R., Zhu C., Ackermann L., Chem. Commun. 2016, 52, 13171–13174. [DOI] [PubMed] [Google Scholar]
- 36.
- 36a.T. J. Struble, C. W. Coley, K. F. Jensen, ChemRxiv 2019 DOI 10.26434/chemrxiv.9735599.v1; [DOI]
- 36b. Margrey K. A., McManus J. B., Bonazzi S., Zecri F., Nicewicz D. A., J. Am. Chem. Soc. 2017, 139, 11288–11299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shang M., Wang M.-M., Saint-Denis T. G., Li M.-H., Dai H.-X., Yu J.-Q., Angew. Chem. Int. Ed. 2017, 56, 5317–5321; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 5401–5405. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary