

Abstract
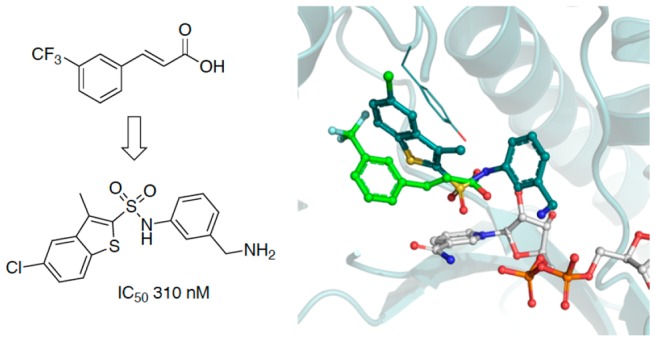
Tuberculosis (TB) remains a leading cause of mortality among infectious diseases worldwide. InhA has been the focus of numerous drug discovery efforts as this is the target of the first line pro-drug isoniazid. However, with resistance to this drug becoming more common, the aim has been to find new clinical candidates that directly inhibit this enzyme and that do not require activation by the catalase peroxidase KatG, thus circumventing the majority of the resistance mechanisms. In this work, the screening and validation of a fragment library are described, and the development of the fragment hits using a fragment growing strategy was employed, which led to the development of InhA inhibitors with affinities of up to 250 nM.
Introduction
Mycobacterium tuberculosis (Mtb), the causative agent of tuberculosis (TB), remains the deadliest infectious disease worldwide with approximately 10.4 million affected cases and 1.7 million deaths per year.1 The emergence of a number of multidrug-resistant TB (MDR-TB) and extensively drug resistant (XDR) strains as well as a deadly synergy with HIV1 present many challenges. Despite recent successes in identifying new chemical entities to combat TB and the prospect of new regimens that will shorten TB treatment from the current six months, there remains an urgent need to find new drugs to fight this disease.
Mycolic acids, synthesized by the fatty acid synthase complex II (FAS II), are essential components of the unique cell envelope of mycobacteria. The essentiality of this pathway is explored clinically by isoniazid (INH), a first-line drug, and ethionamide (ETH), a second-line drug, used to treat MDR-TB. Both of these drugs target the FAS II enzyme InhA, an NADH dependent enzyme involved in the reduction of long-chain fatty acids. Both INH and ETH are pro-drugs that need to be activated within the mycobacteria to form a covalent adduct with the nicotinamide adenine dinucleotide (INH-NAD adduct), which binds strongly into the catalytic site of InhA.2 Resistance to these two pro-drugs can be predominantly explained by mutations in the activating enzymes (KatG for INH and EthA for ETH) or in the upstream promoter region of InhA and less commonly in InhA itself due to the essentiality of the enzyme.2 Many recent drug discovery efforts have focused on several of the enzymes of this pathway such as KasA,3 Pks13,4,5 Ag85C,6,7 and FadD328 with InhA receiving most of the attention.9−13
InhA has been a major target for TB drug discovery with many novel and diverse potent inhibitors, which include triclosan derivatives, pyridomycin, 4-hydroxy-2-pyridones, thiadiazoles, proline series, and pyrrolidine carboxamide, that have been reported in the literature. These inhibitors show direct inhibition of InhA without the need for activation, thus circumventing the most common resistance mechanism to the current drugs targeting this enzyme.13
In this work, fragment-based drug discovery (FBDD) was used as an alternative approach to previously reported drug discovery methods for InhA, where the aim is to find novel, potent inhibitors of the enzyme. FBDD is now established as a powerful tool to discover small-molecule ligands or fragments as hits, which are further elaborated into lead compounds for drug development, and this approach has been widely used in the TB field.14,15 Researchers at the University of Dundee recently reported the screening of a fragment library against InhA; however, further development of the fragment hits has not yet been reported.16 Herein, a three-stage biophysical screening cascade, thermal shift, ligand-based NMR, and X-ray crystallography, led to the identification of several fragment hits. Using a fragment growing strategy supported by enzymatic assays, X-ray crystallography, and molecular docking, one fragment, which displays a novel binding mode in the X-ray crystal structure, was further elaborated into a new class of nanomolar inhibitors of InhA.
Results and Discussion
Fragment Screening Cascade
Differential scanning fluorimetry (DSF) was used to screen an in-house library of 800 rule-of-three-compliant fragments. A saturating concentration of the cofactor in oxidized form (1 mM NAD+) was used in all the experiments to ensure that the cofactor binding site would be occupied by NAD+ and that fragments would not bind there. InhA was shown to have a melting temperature (Tm) of 53 °C in the presence of 1 mM NAD+. Triclosan, a known inhibitor of InhA,17 was used as a positive control and presented a thermal shift (ΔTm) of +3.0 °C with a melting temperature of 56 °C at a concentration of 0.5 mM in the presence of NAD+. Upon screening of the fragment library, a hit was defined where the melting point was increased by at least +3 °C at a concentration of 5 mM (Figure 1a). A total of 42 fragment hits were identified using DSF, representing a hit rate of 5.2% (Table S1).
Figure 1.

(a) Histogram of fragments (5 mM) found to increase the melting point (ΔTm) of InhA (10 μM) in the presence of NAD+ (1 mM) by a fluorescence-based thermal-shift assay. (b) Chemical structures of the X-ray hits obtained. (c) X-ray structure of fragment 1 (teal) bound in an InhA–NAD+ complex (gray), showing the cavity surface. The substrate binding regions of InhA are divided into three sites: site I, site II, and site III. Fragment 1 occupies the catalytic site I and a part of the hydrophobic pocket site II, and Y158 adopts an “in” conformation. (d) Overlaid X-ray crystal structures of fragments 1 (green) and 3 (magenta) binding to InhA in the presence of NAD+ with Y158 in an open conformation for fragment 1 (green, PDB Code 6SQ5) and in a new, previously unseen, conformation for fragment 3 (magenta, PDB Code 6SQ9).
In order to validate the hits obtained in the thermal shift assay, ligand-based NMR techniques, CPMG (Carr–Purcell–Meiboom–Gill, which examines the T2 relaxation in the presence of the protein), WaterLOGSY (which examines the transfer of magnetization from the bulk water molecules in the protein to the ligand), and STD (saturated transfer difference, which involved the selective saturation of protein resonances and subsequent transfer of magnetization), were used.18−20 The fragments were screened at a concentration of 1 mM in the presence of NAD+ (0.5 mM) with 20 μM InhA using three techniques. Eighteen fragments showed interactions with InhA in at least two ligand-based NMR techniques, and these were considered as confirmed hits. The validated hit rate was 42% in which 83% (15 fragments) contained carboxylic acids (Table S1).
The 18 fragment hits validated by NMR were then soaked into InhA crystals, and X-ray diffraction data was collected. Of these fragments, five showed sufficiently clear electron density in the X-ray crystal structures that allowed them to be modeled with confidence (Figures 1b and S1).
The binding pocket of the InhA inhibitors that are substrate competitive can be divided into three distinct sites that have different properties, site I being the catalytic site, site II being a hydrophobic region that binds fatty acid chains, and site III being a solvent exposed site that is known as the size-limiting region (Figure 1c).21 All of the obtained X-ray crystal structures of the fragments contain NAD+, and the compounds occupy the same area as the InhA substrate covering regions of sites I and II. The X-ray data collection and refinement statistics are reported in Table S2. In the X-ray crystal structures of all the initial fragments, with the exception of compound 3, Y158 adopts a previously described conformation known as “Y158-in” (Figure S2). The “Y158-in” conformation has previously been observed with InhA inhibitors (triclosan, 4-hydroxypyridones, and pyrrolidine carboxamides) where the hydroxy group of Y158 is oriented toward the inhibitor in site I. The aromatic group of the fragments occupies the hydrophobic site II of the InhA pocket (Figures 1c and S2).13 For compounds 1, 4, and 5, the carboxylic acid portion of the molecules superimposes completely and forms hydrogen bonds with the hydroxyl group of the ribose and Y158, while compound 3 only forms hydrogen bonds with the ribose hydroxyl group (Figure S2). Compound 3 presented a unique mode of binding as the Y158 residue showed a distinct conformation from all other X-ray crystal structures both in this study and in the literature (Figure 1d). This compound was observed to be sandwiched between the residues Y158 and F149, forming π-interactions with both (Figure S2). For compound 2, even though the carboxylic acid group forms similar interactions as compounds 1, 4, and 5, it sits in a different position with its benzo-moiety facing away from the ribose and occupying a similar area of the InhA site as the pyrazole group of compound 5.
The identified fragment hits were further evaluated by an InhA enzymatic assay using 2-trans-octanoyl-CoA as a substrate.22 The fragments were screened in triplicate at a final concentration of 2 mM using triclosan as a positive control. However, none of the fragments displayed any inhibitory activity against InhA at a concentration of 2 mM.
Fragment Growing
The examination of the X-ray structures of the fragments 1–5 supported the feasibility of a fragment growing approach. The carboxylate group in site I provided a good handle for elaboration toward site II of the InhA pocket (Figure 1c). On the basis of the novelty of the fragment hits and the vectors available to grow the fragments, fragment 1 was selected for further elaboration.
The X-ray crystal structure of triclosan was overlaid with the structure of fragment 1, and it showed a similar binding mode to the fragment in the enzyme–NAD+ complex (Figure 2a). The carboxylate of fragment 1 occupies the same region of the triclosan phenyl-ether linker.17 The carboxylic acid in fragment 1 (P1) was replaced by an isosteric sulfonamide as a linker to synthesize compound 6 (Figure 2b). Docking experiments with Glide XP23 (Schrodinger LLC), using the X-ray crystal structure of InhA with fragment 1 as a model, showed that the sulfonamide vector also provided the right geometry to grow the fragments into InhA site III, while also forming hydrogen bonds with Y158 and NAD+ (Figure 3a,b), with compound 6 showing a significant improvement of the activity (54% inhibition (100 μM)). All the synthesized compounds were tested at a concentration of 100 μM using the InhA enzymatic assay. The IC50 values were determined only for the compounds showing an inhibitory activity greater than 90% under these screening conditions.
Figure 2.
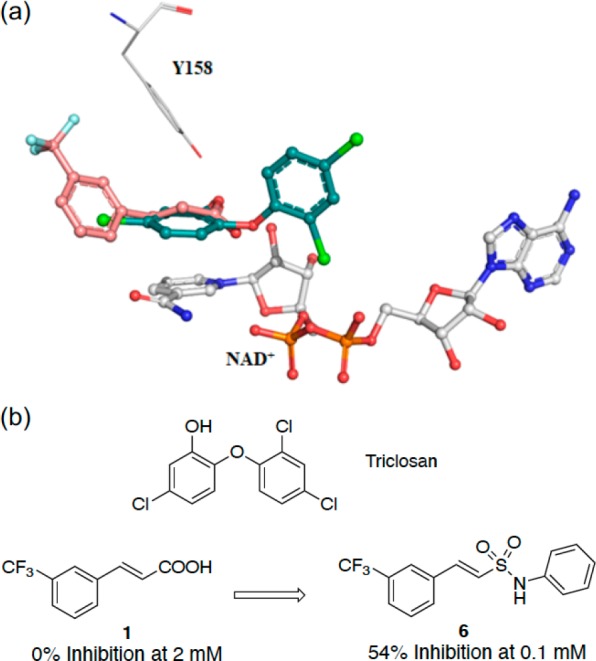
(a) X-ray crystal structure of fragments 1 (PDB code 6SQ5) overlaid with triclosan (PDB code 2B35) bound to the InhA–NAD+ complex with the “Y158-in” conformation. (b) Fragment growing strategy from fragment 1 to compound 6. The inhibition percentage of InhA using an enzymatic assay for each compound is shown.
Figure 3.

Docking poses for compounds 6 (A) and 19 (C) superposed with fragment 1 (green). Hydrogen bonds for compounds 6 and 19, shown as red dashes, respectively, in (B) and (D) with distances in Å. Compounds 6 and 19 were docked into the structure for fragment 1 (PDB code 6SQ5).
The phenyl ring was replaced by a piperidine saturated ring that could be useful to grow fragment 1 into site III. This modification led to the loss of potency in compound 7 (15% inhibition at 100 μM, Table 1). The introduction of a pyridine ring also displayed a lower percentage inhibition (17%) for compound 8. Therefore, the phenyl ring of compound 6 was maintained, and analogues of compound 6 were prioritized.
Table 1. Structure and Activities of Compounds 7–20 Evaluated by an Enzymatic Assay.
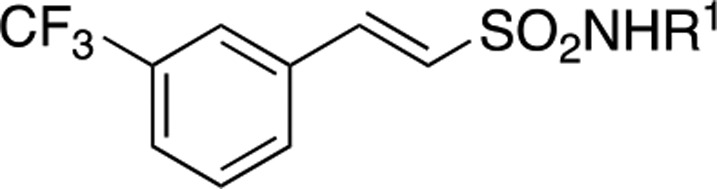

Inh %: inhibition percentage at 100 μM concentration.
IC50: measured in μM by an enzymatic assay from an average of three experiments. IC50 values were determined for compounds that showed >90% inhibition at 100 μM.
LE was calculated using the equation: LE = (1.37 × pIC50)/HA, where HA means heavy atom, i.e., a non-hydrogen atom. LE: compound 14 = 0.29; compound 19 = 0.31.
As shown in Table 1, compounds 9 and 10 did not add any improvement where a percentage inhibition of 0% and 11% was measured, respectively. Subsequent deprotection of compound 10 to afford the aniline 11 also did not show any improvement in the percentage inhibition (6%). This was also observed with the electron-donating methoxy group in compound 12. The benzyl ester 13 (% inhibition = 24%) showed a better potency than the previous analogues, although this was still low.
Interestingly a 10-fold increase of the potency from compound 6 was observed with the synthesis of benzylamine analogue 14 (Table 1). This compound showed an improved InhA inhibition (>90%) with an IC50 of 9 μM and a ligand efficiency (LE) of 0.29, which is at least a 200-fold more potent compound compared to the initial fragment hit 1 (IC50 > 1000 μM).
Subsequently, compounds 15 to 20 were synthesized as analogues of compound 14 to explore whether the primary amine could be replaced by other functional groups (Table 1; Scheme 1). In compounds 15 and 16, the amine of compound 14 was, respectively, monomethylated and acetylated. The amine was replaced by a carboxylic acid and an acetamide group in the analogues 17 and 18. All these modifications on this position of the methyleneamine of compound 14 led to a loss of potency with an InhA percentage inhibition between 0 and 25% at a ligand concentration of 100 μM. The inhibition of InhA was slightly improved (9 μM compared to 4 μM) by moving the methylamine to the meta-position, compound 19, and gave an IC50 of 4 μM and a LE of 0.31. The docking pose of compound 19 showed the amine group forms a hydrogen bond with the phosphate of NAD+ and forces the phenyl ring to move further away from the phosphates toward M161 (Figure 3C,D). This changes the orientation of the sulfonamide, now only forming a hydrogen bond with Y158, and it further alters the position of the trifluoromethylphenyl moiety when compared to compound 6 (Figure 3).
Scheme 1. Synthesis of Compounds Substituted on the Sulphonamide Group.

The activity was lost in compound 20 in which the amine is part of a closed 6-membered ring. The development of compounds 15–20 showed that the phenylmethanamine group was crucial to maintain affinity and was maintained for further development of a potent InhA inhibitor. Subsequently, the sulfonamide with the phenylmethanamine scaffold on P3 (Table 1) was maintained, and further modification on P2 was examined (Table 2).
Table 2. Structure and Activities of Compounds 21–26 Evaluated by the InhA Enzymatic Assay.
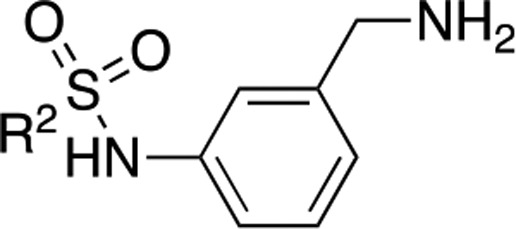

Inh %: inhibition percentage at a 100 μM concentration.
IC50: measured in μM by an enzymatic assay from an average of three experiments. IC50 values were determined for compounds that showed >90% inhibition at 100 μM.
LE was calculated using the equation LE = (1.37 × pIC50)/HA, where HA means heavy atom, i.e., a non-hydrogen atom.
Further Elaboration Strategies
The trifluoromethylstyrene moiety in compound 19 could potentially be metabolized to styrene oxide, and as a result, this scaffold was replaced to improve the drug-like properties. From the X-ray crystallography of the parent fragment 1 and from molecular docking, P2 of compound 19 should mainly occupy the hydrophobic site II of InhA (Figure 3C). However, docking studies of the derivatives of 1 showed that the trifluoromethylstyrene moiety could bind to different hydrophobic areas of site II (Figure 3A,C). Hydrophobic functional groups were chosen as modifications at P2 of compound 19 (Table 2). The tert-butylphenyl and chloro-naphthalene groups in compounds 21 and 22 were synthesized on the basis of similarities with fragment 4 and other fragments (Table S1). However, both compounds 21 and 22 showed a drop of percentage inhibition to 40% and 17%, respectively, at a screening concentration of 100 μM.
The introduction of a 5-chloro-3-methylbenzothiophene in compound 23 improved the InhA inhibition IC50 to a value of 310 nM with a ligand efficiency of 0.40. This is a 12-fold jump in potency in comparison to compound 19 (Table 2; Scheme 2). The replacement of the 5-chloro with a 5-fluoro substituent in compound 24 slightly improved the potency to 250 nM. The halogens and methyl substitutions prove to be essential for the activity as a significant drop in activity to 6 and 22 μM was observed, respectively, for compounds 25 and 26 (Table 2). This shows the importance of these functionalities in order to maintain a potency.
Scheme 2. Exploration of SAR Based on Compound 19.

The X-ray crystal structure for compound 23 was obtained (Figure 4A), and it shows, as predicted by docking (Figure S3), that the sulfonamide group forms a hydrogen bond with Y158 and has the correct orientation for the phenylmethanamine moiety to explore site III, forming π-interactions with the backbone of the protein at G96 and a sulfur−π interaction with M161 (Figure 4A,B). Furthermore, the amine in the meta-position of the phenyl ring interacts with the phosphates of NAD+ but also with a highly coordinated water that further interacts with the sulfonamide group and the NAD+ phosphate (Figure 4A). The benzothiophene group forms π-interactions with F149 and Y158 and sulfur−π interactions with M103, M155, and M199, while exploring a different hydrophobic area on site II (Figure 4B). The position of the phenylmethanamine group in relation to the sulfonamide is crucial for the binding affinity. In the earlier SAR examined in Table 1 when this was changed to a secondary amine (compounds 15 and 20) or an aniline (compound 11), there was little or no binding observed. This could be due to the increased sterics observed in compound 15 and 20, which would not allow the H-bonding interactions. With compound 11, the intramolecular H-bonding interactions through the water would not be possible due to the direct attachment of the NH2 group to the benzene ring.
Figure 4.

(A) X-ray crystal structure and interaction map of compound 23 (PDB Code 6SQL). NAD+ is shown in white, while compound 23 in shown in teal. Red disks represent hydrogen bonds; blue disks depict π–π interactions, and yellow disks show sulfur−π interactions. Interactions were calculated using Intermezzo plugin for Pymol (Ochoa B., et al. unpublished). (B) The substrate binding region of InhA divided into three sites. The X-ray structure of fragment 1 (violet) (PDB code 6SQ5) is overlaid with compound 23, showing the cavity surface.
Evaluation of the Effects of InhA Inhibitors on Mycolic Acid Synthesis in M. tuberculosis H37Ra
The IC50 values of the tested inhibitors against InhA protein were promising, and these compounds (14, 19, and 23–26) were screened to see whether they can inhibit the growth of M. tuberculosis. Further analysis of the effect on the synthesis of mycolic acids by metabolic labeling24,25 of the model strain M. tuberculosis H37Ra with 14C acetate was also explored. A 48 h cultivation in the presence of 200 μM of the compounds in the media led to 84% growth inhibition for compound 19; 40% and 33% growth inhibition for compounds 14 and 23, respectively; 38% growth inhibition for compound 24 and isoniazid; 23% growth inhibition for compounds 25 and 26. However, while the presence of isoniazid caused complete abolition of the synthesis of mycolic acids, only a slight inhibition was observed in the case of compound 19 and no inhibition, in the case of the other screened compounds (Figure 5A).
Figure 5.

(A) TLC of the metabolic labeling experiments of M. tuberculosis H37Ra with 14C acetate treated with the compounds 14, 19, and 23–26 for the analysis of mycolic acid inhibition. Fatty acid methyl esters, FAME; mycolic acid methyl esters, MAME; isoniazid, INH. (B) TLC of the metabolic labeling experiments of M. tuberculosis H37Ra with 14C acetate treated with the compounds 14, 19, and 23–26 for the analysis of the lipid inhibition. Trehalose monomycolates, TMM; trehalose dimycolates, TDM; phosphatidylethanolamine, PE; cardiolipin, CL; isoniazid, INH.
The analysis of lipid profiles revealed that the treatment with compound 19 led to the accumulation of trehalose monomycolates (TMM) and to the decrease of the amount of trehalose dimycolates (TDM; Figure 5B). None of other tested inhibitors affected the amounts of TMM and TDM in the mycobacterial cells, suggesting that, despite the potent IC50 values, the tested compounds possibly do not target InhA inside mycobacterial cells, and further experiments are needed in order to clarify this.
Conclusions
Fragment-based drug discovery is a robust and now widely used approach to identify drug-like molecules, and this methodology has led to the development of a number of drugs that have been approved by the FDA. In this work, we identified several fragment hits using a screening cascade consisting of DSF, ligand-based NMR, and X-ray crystallography. The initial fragment hits revealed a ligand having a unique binding mode and forcing Y158 to adopt a new conformation that “sandwiches” the compound between the residues F149 and Y158. However, the fragment hits had no detectable inhibitory activity. Using the available structural information, potent and novel nanomolar inhibitors of InhA were developed by applying a fragment-growing approach. The systematic exploration of chemical space in P3 and P1 after fixing P2 with a sulfonamide and helped by molecular docking led to the development of potency and an increase of ligand efficiency. The introduction of a benzothiophenene at P2 and the phenylmethanamine at P3 led to the development of compound 23, and this was shown to be a potent inhibitor of InhA. However, disappointingly, compound 23 was shown to be inactive against M. tuberculosis.
In the past decade, the TB drug discovery field has seen a shift from target-based approaches back to whole-cell phenotypic screens due to the problems faced by compounds derived from target-based approaches in translating their activity into cell-based assays.15 However, the number of lead compounds coming from these large-scale screens has been lower than expected. The compounds in this study illustrate the current challenge of obtaining new antitubercular compounds. The strong InhA inhibitory activity of compounds described herein is not reflected in antitubercular activity nor do we observe a significant reduction in the amount of mycolic acids produced by any of the compounds, suggesting that the compounds do not penetrate the cells in sufficient amounts, are being extruded by efflux mechanisms, or are metabolized. Further work is required to clarify the nature of the problem and to improve the cellular activity of the lead compounds.
Experimental Section
Protein Purification, Crystallization Data Collection, and Refinement
Mycobacterium tuberculosis InhA was purified as described previously.26 Briefly, E. coli BL21(DE3) containing a hexahistidine-SUMO tagged InhA construct in pET28a was grown to midexponential growth phase (OD610 = 0.8) in LB media (Invitrogen) containing 30 mg L–1 kanamycin at 37 °C. Gene expression was induced by adding isopropyl β-d-1-thiogalactopyranoside (IPTG) at a final concentration of 0.5 mM, and the temperature was lowered to 18 °C. Cells were lysed in 50 mM HEPES, pH 7.5, 0.5 M NaCl, 10% glycerol (w/v), and 20 mM imidazole, and recombinant InhA was purified with a HiTrap IMAC Sepharose FF column (GE-Healthcare) equilibrated in the same buffer. Elution was performed with 500 mM imidazole. The recovered protein was dialyzed into 50 mM HEPES, pH 7.5, 0.5 M NaCl, and 10% glycerol (w/v), and the SUMO tag was cleaved overnight at 4 °C by adding Ulp1 Protease at a 1:100 ratio. The SUMO tag, Ulp1 protease, and uncleaved SUMO-InhA were removed using the same column and equilibrated with 50 mM HEPES, pH 7.5, 0.5 M NaCl, 10% glycerol (w/v), and 20 mM imidazole. Flow through containing InhA was collected, concentrated, and loaded in a Superdex 200 column equilibrated with 50 mM HEPES, pH 7.5, 150 mM NaCl, and 10% glycerol (w/v). The fraction purity was determined by SDS-PAGE. The purest fractions were pooled, concentrated to ∼12 mg·mL–1, flash frozen in liquid nitrogen, and stored at −80 °C.
InhA was crystallized in the presence of 2 mM NAD at 18 °C using the sitting drop vapor diffusion method by mixing 1 μL of InhA at 12 mg·mL–1 in a 1:1 ratio with a reservoir solution containing 0.1 M HEPES, pH 7.0, 0.1 M sodium acetate, and 25–30% PEG 400. Fragment soaking was performed by mixing the compound solution at 200 mM in 100% DMSO with mother liquor to a final concentration of 20 mM. Crystals were soaked in these solutions overnight. Compound 24 and 2-trans-octanoyl-CoA were cocrystallized by mixing InhA 10 mg·mL–1 with 0.5 mM NAD+ and 0.5 mM compound 24 or 5 mM 2-trans-octanoyl-CoA in a 1:1 ratio with the reservoir solution containing 14% PEG 4k, 200 mM ammonium acetate, 100 mM HEPES, pH 7, for compound 23 and 10% PEG 8k, 100 mM TRIS, pH 8.5, for 2-trans-octanoyl-CoA. A cryogenic solution was prepared by adding 30% glycerol to the mother liquor, and crystals were briefly transferred to this solution, flash frozen in liquid nitrogen, and stored for data collection. All data sets were collected at stations i03 and i04-1 at Diamond Light Source (Oxford, UK). Data collection and refinement statistics are summarized in Table S2. Diffraction data were indexed, integrated, and reduced using autoPROC from Global Phasing Limited.27 Molecular replacement was performed with Phaser28 using PDB structure 2B35 as a search model. Refinement was carried out iteratively with PHENIX29 and Coot,30 and ligand and water fitting was performed with Coot.30
Molecular Docking
Glide XP23 (extra precision) included in the Schrodinger software package (Schrodinger, LLC, New York, NY, 2015 and 2016) was employed for docking of compounds using the X-ray crystal structure with fragment 1 as the receptor. Epik,31 included in the same software package, was used to prepare ligands. Docking was performed using default XP settings with flexible ligand sampling and postdocking minimization.
Differential Scanning Fluorimetry
Differential scanning fluorimetry was used to screen a library of 800 fragments. The assay was performed in a 96 well plate format using a CFX Connect (Bio-Rad). Each well contained a solution of 10 μM InhA, 1 mM NAD+, HEPES (50 mM), NaCl (150 mM), pH 7.5, 5× Sypro Orange, 5% DMSO, and fragments at 5 mM. A positive control with 0.5 mM Triclosan was included in all plates. Controls were made in quadruplicates for all the experiments.
Enzymatic Assay
InhA activity was followed by a colorimetric assay that measured the oxidation of NADH at 340 nm in the presence of 2-trans-octanoyl-CoA as described before in a buffer that contained 30 mM PIPES, pH 7.5, 50 mM NaCl, 0.1 mM EDTA, and 100 nM InhA.26 This was preincubated for 10 min at room temperature with 0.25 mM NADH and varying concentrations of the compounds with 1% (v/v) DMSO in a 150 μL reaction volume. The reaction was started by the addition of 2-trans-octanoyl-CoA at a final concentration of 1.5 mM, prepared as described previously.22 Reactions were followed for 20 min using a plate reader (CLARIOstar, BMG LABTECH).
Analysis of the Effects of Tested Compounds on Mycolic Acid Synthesis in M. tuberculosis H37Ra
The culture of M. tuberculosis H37Ra was grown statically at 37 °C in Middlebrook 7H9 broth (Difco) supplemented with albumin–dextrose–catalase and 0.05% Tween 80. When the culture reached the OD600nm value of 0.290, it was divided into 10 mL aliquots and tested compounds dissolved in DMSO were added at 200 μM final concentrations, while isoniazid that served as a control inhibitor was at 40 μM. The final concentration of DMSO in each culture was at 1%. After 24 h of cultivation with shaking (120 rpm) in the presence of tested inhibitors, 14C acetate (specific activity of 106 mCi/mmol) was added at a final concentration of 0.5 μCi/mL and the cells were cultivated for the next 24 h.
Lipids were extracted from whole cells as described earlier with minor modifications.24 Briefly, 3 mL of chloroform/methanol (1:2) was added to cells harvested from 4 mL culture aliquots; the mixtures were incubated at 56 °C for 1.5 h with intense mixing and centrifuged at 1000g, and the extracts were collected in glass tubes. This was followed by the extraction with chloroform/methanol (2:1) at the same conditions. Both extracts were combined together, dried under nitrogen, and subjected to biphasic wash with chloroform/methanol/water (4:2:1) as described earlier.25 The bottom organic phase was dried and dissolved in chloroform/methanol (2:1), 250 μL per 1 unit of OD600nm of harvested cells. Five μL of each lipid sample was loaded on the thin-layer chromatography (TLC) silica gel plates (F254, Merck); lipids were separated in chloroform/methanol/water [20:4:0.5] and detected by autoradiography.
Fatty acid methyl esters (FAMEs) and mycolic acids methyl esters (MAMEs) were prepared from whole cells harvested from 4 mL culture aliquots as previously described.32 Dried extracts were dissolved in chloroform/methanol (2:1) at a ratio of 250 μL per 1 unit (OD = 600) of harvested cells, and 5 μL of each sample was loaded on the TLC plates. Different forms of methylesters were separated by chromatography in n-hexane/ethyl acetate [95:5] 3× and detected by autoradiography.
Synthetic Chemistry
General Experimental Methods
Solvents were distilled prior to use and dried by standard methods. Unless otherwise stated, 1H and 13C NMR spectra were obtained in CDCl3, MeOD, or DMSO solutions using a Bruker 400 MHz AVANCE III HD Smart Probe, 400 MHz QNP cryoprobe, or 500 MHz DCH cryoprobe spectrometer. Chemical shifts (δ) are given in ppm relative to the residual solvent peak (CDCl3: 1H, δ = 7.26 ppm; 13C, δ = 77.16 ppm), and the coupling constants (J) are reported in hertz (Hz).
Reactions were monitored by TLC and LCMS to determine the consumption of the starting materials. Flash column chromatography was performed using an Isolera Spektra One/Four purification system and the appropriately sized Biotage SNAP column containing KP-silica gel (50 μm). Solvents are reported as volume/volume eluent mixture where applicable.
High resolution mass spectra (HRMS) were recorded using a Waters LCT Premier Time of Flight (TOF) mass spectrometer or a Micromass Quadrapole-Time of Flight (Q-TOF) spectrometer.
Liquid chromatography mass spectrometry (LCMS) was carried out using an Ultra Performance Liquid Chromatographic system (UPLC) Waters Acquity H-class coupled to a Waters SQ Mass Spectrometer detector. Samples were detected using a Waters Acquity TUV detector at 2 wavelengths (254 and 280 nm). Samples were run using an Acquity UPLC HSS column and a flow rate of 0.8 mL/min. The eluent consisted of 0.1% formic acid in water (A) and acetonitrile (B), gradient from 95% A to 5% A over a period of 4 or 7 min. All final compounds had a purity greater than 95% as determined by LCMS analysis.
(E)-2-(3-(Trifluoromethyl)phenyl)ethene-1-sulfonyl Chloride (27)
Sulfuryl chloride (3.8 mL, 46 mmol) was added dropwise at 0 °C to a solution of anhydrous DMF (10 mL) under N2. After the addition was completed, the mixture was warmed to r.t. and stirred further for 0.5 h. 1-(Trifluoromethyl)-3-vinylbenzene (1.71 mL, 11 mmol) was then added in three portions, and the reaction mixture was gradually heated at 90 °C for 3 h. The reaction mixture was cooled and then poured onto the crushed ice, and the separated oily layer was extracted with Et2O and dried. Evaporation of the solvent gave the desired compound 27 (77% yield) as a yellow solid. 1H NMR (400 MHz, CDCl3) δ 7.94–7.48 (m, 3H), 4.43–4.25 (m, J = 14.2, 5.5 Hz, 1H), 3.85–3.71 (m, 1H). LC/MS [mass of sulfonic acid – H] found 251.1.
(E)-N-Phenyl-2-(3-(trifluoromethyl)phenyl)ethene-1-sulfonamide (6)
To a solution of compound 27 (50 mg, 0.185 mmol) in DCM (5.0 mL), dry triethylamine (0.031 mL, 0.22 mmol) and aniline (17 mg, 0.18 mmol) were added. The reaction mixture stirred for 3 h at room temperature. The reaction was quenched with a saturated solution of NaHCO3, followed by extraction with DCM. The organic phase was dried on anhydrous MgSO4 and concentrated under reduced pressure, and the crude product was purified by silica flash column chromatography (column gradient of 20% to 50% EtOAc in Pet. Ether at 40–60 °C) to yield the desired product (32 mg, 53% yield) as a white powder. 1H NMR (400 MHz, CDCl3) δ 7.63 (s, 1H), 7.58–7.45 (m, 2H), 7.35–7.22 (m, 3H), 7.14 (t, J = 7.2 Hz, 1H), 6.92 (d, J = 15.5 Hz, 1H); HRMS (ESI) calcd. for [C15H12F3NO2S + H]+: 328.0619; Found: 328.0608. LC/MS [M – H]− found 326.1, purity >99%.
(E)-N-(Piperidin-4-yl)-2-(3-(trifluoromethyl)phenyl)ethene-1-sulfonamide (7)
Using the same procedure as compound 6, tert-butyl-4-aminopiperidine-1-carboxylate (0.100 g, 0.37 mmol) and 27 (0.100 mg, 0.37 mmol) yielded the Boc-protected intermediate tert-butyl-(E)-4-((2-(3-(trifluoromethyl)phenyl)vinyl)sulfonamido)piperidine-1-carboxylate (78 mg, 0.37 mmol, 78% yield) as a white powder. The crude product was purified by silica flash column chromatography (column gradient of 30% to 60% EtOAc/Pet. Ether at 40–60 °C). 1H NMR (400 MHz, CDCl3) δ 7.72 (s, 1H), 7.66 (d, J = 8.0 Hz, 2H), 7.55 (d, J = 7.7 Hz, 1H), 7.49 (d, J = 15.5 Hz, 1H), 6.86 (d, J = 15.4 Hz, 1H), 4.90 (d, J = 7.5 Hz, 1H), 3.94 (m, 2H), 3.52–3.26 (m, 1H), 3.02–2.69 (m, 2H), 1.92 (m, 2H), 1.51–1.43 (m, 2H), 1.42 (s, 9H). LC/MS [M – H] found 433.2, purity >99%.
To a solution of the Boc intermediate in dichloromethane (2 mL), trifluoroacetic acid (1 mL) was added dropwise, and the mixture was stirred for 0.5 h. The reaction was quenched with a saturated solution of NaHCO3. The organic phase was separated, and the solvent was removed under reduced pressure to yield the product (45 mg, 0.18 mmol, 75% yield) as a yellow oil. 1H NMR (400 MHz, DMSO-d6) δ 8.15 (s, 1H), 8.04 (d, J = 7.8 Hz, 1H), 7.79 (d, J = 7.8 Hz, 1H), 7.67 (t, J = 7.8 Hz, 1H), 7.47 (d, J = 1.7 Hz, 1H), 3.19 (m, 2H), 2.88 (m, 1H), 2.00–1.88 (m, 1H), 1.61 (m, 1H); HRMS (ESI) calcd. for [C14H17F3N2O4S + H]+: 335.1041; Found: 335.1053; LCMS: [M + H]+ found 335.1, purity >99%.
(E)-N-(Pyridin-3-yl)-2-(3-(trifluoromethyl)phenyl)ethene-1-sulfonamide (8)
To a solution of compound 27 (50 mg, 0.18 mmol) in DCM (5.0 mL), dry Et3N (0.03 mL, 0.220 mmol) and 3-aminopyridine (17 mg, 0.18 mmol) were added. The reaction mixture was stirred for 3 h at room temperature. The reaction was quenched with NaHCO3, followed by extraction with DCM. The organic phase was dried on anhydrous MgSO4 and concentrated under reduced pressure, and the crude product was purified by silica flash column chromatography (column gradient of 20% to 50% EtOAc/Pet. Ether at 40–60 °C) to yield the desired product (39 mg, 64% yield) as a white powder. 1H NMR (400 MHz, DMSO-d6) δ 8.76 (d, J = 3.3 Hz, 1H), 8.70 (dd, J = 4.8, 1.5 Hz, 1H), 8.25 (s, 1H), 8.10 (d, J = 8.0 Hz, 1H), 8.03 (ddd, J = 8.2, 2.6, 1.5 Hz, 1H), 7.93 (d, J = 15.3 Hz, 1H), 7.85 (d, J = 7.7 Hz, 1H), 7.77–7.64 (m, 2H), 7.59 (dd, J = 8.2, 4.0 Hz, 1H), 1.23 (s, 1H); LCMS: no molecular ion peak observed, purity 97% ; HRMS (ESI) calcd. for [C14H11N2O2F3S + H]+: 329.0572; Found: 329.0563.
Methyl-(E)-4-((2-(3-(trifluoromethyl)phenyl)vinyl)sulfonamido)benzoate (9)
Using the same procedure as with compound 6, methyl 3-aminobenzoate (0.168 g, 1.108 mmol) and compound 27 (0.300 g, 1.108 mmol) were added. The crude product was purified by silica flash column chromatography (column gradient of 20% to 50% EtOAc/Pet. Ether at 40–60 °C) which yielded the desired product 9 (0.281 g, 0.729 mmol, 66% yield) as a yellow powder. 1H NMR (400 MHz, CDCl3) δ 7.88 (d, J = 2.0 Hz, 1H), 7.82 (dd, J = 7.7, 1.3 Hz, 1H), 7.65 (d, J = 7.7 Hz, 2H), 7.63–7.54 (m, 1H), 7.55–7.46 (m, 2H), 7.41 (t, J = 7.9 Hz, 1H), 7.16 (s, 1H), 6.88 (d, J = 15.4 Hz, 1H), 3.91 (s, 3H), 3.50 (s, 1H); HRMS (ESI) calcd. for [C17H14F3NO4S + H]+: 386.0674; Found: 386.0688; LCMS: [M – H]− found 384.1, purity 94%.
tert-Butyl-(E)-(4-((2-(3-(trifluoromethyl)phenyl)vinyl)sulfonamido)phenyl)carbamate (10)
Using the same procedure as compound 6, tert-butyl (4-aminophenyl)carbamate (77 mg, 0.37 mmol) and compound 27 (100 mg, 0.37 mmol) were added. The crude product was purified by silica flash column chromatography (column gradient of 30% to 60% EtOAc/Pet. Ether at 40–60 °C). This yielded the desired product (50 mg, 0.050 mmol, 31% yield) as a clear oil. 1H NMR (400 MHz, CDCl3) δ 7.64 (d, J = 6.0 Hz, 2H), 7.57 (d, J = 8.2 Hz, 1H), 7.50 (t, J = 8.0 Hz, 1H), 7.43 (d, J = 15.5 Hz, 1H), 7.31 (d, J = 8.8 Hz, 2H), 7.20–7.11 (m, 2H), 6.90–6.80 (m, 2H), 6.54 (s, 1H), 1.49 (s, 9H); LCMS: [M – H]− found: 441.0, purity >99%.
(E)-N-(4-Aminophenyl)-2-(3-(trifluoromethyl)phenyl)ethene-1-sulfonamide (11)
Using the same procedure as compound 7, compound 10 (50 mg, 0.11 mmol) was deprotected to yield the desired product (26 mg, 0.070 mmol, 62% yield) as a brown oil. 1H NMR (500 MHz, DMSO-d6) δ 8.10 (s, 1H), 7.97 (d, J = 8.0 Hz, 1H), 7.75 (d, J = 7.1 Hz, 1H), 7.63 (t, J = 7.8 Hz, 1H), 7.53 (t, J = 7.2 Hz, 1H), 7.44 (t, J = 7.6 Hz, 2H), 7.35 (d, J = 11.8 Hz, 1H), 6.87 (d, J = 8.7 Hz, 1H), 6.46 (d, J = 8.7 Hz, 1H); LCMS: [M – H]− found 341.1, purity >99%.
(E)-N-(4-Methoxyphenyl)-2-(3-(trifluoromethyl)phenyl)ethene-1-sulfonamide (12)
Using the same procedure as compound 6, 4-methoxyaniline (37 mg, 0.28 mmol) and compound 27 (50 mg, 0.18 mmol) were added. The crude product was purified by silica flash column chromatography (column gradient of 30% to 70% EtOAc/Pet. Ether at 40–60 °C). This yielded the desired product (10 mg, 0.03 mmol, 15% yield) as a yellow oil. 1H NMR (400 MHz, DMSO-d6) δ 9.73 (s, 1H), 8.13 (s, 1H), 7.99 (d, J = 7.8 Hz, 1H), 7.86–7.57 (m, 2H), 7.19–7.02 (m, 2H), 6.98–6.80 (m, 2H), 6.71–6.58 (m, 1H), 6.55–6.24 (m, 1H), 3.68 (s, 2H); HRMS (ESI) calcd. for [C16H14NO3F3S + H]+: 358.0725; Found: 358.0735; LCMS: [M – H]− found 356.1, purity 96%
Methyl-(E)-2-(4-((2-(3-(trifluoromethyl)phenyl)vinyl)sulfonamido)phenyl)acetate (13)
Using the same procedure as compound 7, 2-(4-aminophenyl)acetate (46 mg, 0.28 mmol) and compound 27 (50 mg, 0.18 mmol) yielded the desired product methyl (11 mg, 0.025 mmol, 15% yield) as a white powder. The crude product was purified by silica flash column chromatography (column gradient of 30% to 60% EtOAc/Pet. Ether at 40–60 °C). 1H NMR (400 MHz, Chloroform-d) δ 7.65 (d, J = 6.6 Hz, 2H), 7.59 (d, J = 7.9 Hz, 1H), 7.54–7.46 (m, 2H), 7.23 (d, J = 8.5 Hz, 2H), 7.15 (d, J = 8.5 Hz, 2H), 6.86 (d, J = 15.4 Hz, 1H), 6.73 (s, 1H), 3.67 (s, 3H), 3.58 (s, 2H); HRMS (ESI) calcd. for [C18H16NO4F3S + H]+: 400.0830; Found: 400.0821; LCMS: [M – H]− found 398.1, purity >99%.
tert-Butyl-(E)-(4-((2-(3-(trifluoromethyl)phenyl)vinyl)sulfonamido)benzyl)carbamate (28)
Using the same procedure as compound 7, 4-[(N-Boc)aminomethyl]aniline (82 mg, 0.369 mmol) and compound 27 (100 mg, 0.369 mmol) yielded the Boc intermediate (138 mg, 0.302 mmol, 82% yield) as white crystals. The crude product was purified by silica flash column chromatography (column gradient of 30% to 60% EtOAc/Pet. Ether at 40–60 °C). 1H NMR (400 MHz, Chloroform-d) δ 7.65 (d, J = 6.3 Hz, 2H), 7.60 (d, J = 7.8 Hz, 1H), 7.56–7.43 (m, 2H), 7.23 (d, J = 8.3 Hz, 2H), 7.19–7.12 (m, 2H), 6.86 (d, J = 15.5 Hz, 1H), 6.63 (s, 1H), 4.83 (s, 1H), 4.26 (d, J = 5.7 Hz, 2H), 1.44 (s, 9H); LCMS: [M – H]− found 455.1; HRMS (ESI) calcd. for [C21H23N2O4F3S + Na]+: 479.1223; Found: 479.1232.
(E)-N-(4-(Aminomethyl)phenyl)-2-(3-(trifluoromethyl)phenyl)ethene-1-sulfonamide (14)
To a solution of the Boc intermediate 28 in dichloromethane (2 mL), trifluoroacetic acid (1 mL) was added dropwise, and the mixture was stirred for 0.5 h. The reaction was quenched with NaHCO3; the organic phase was isolated, and the final product was concentrated under reduced pressure to yield the desired product (84 mg, 0.24 mmol, 78% yield) as small yellow crystals. 1H NMR (500 MHz, DMSO-d6) δ 8.13 (s, 1H), 7.99 (d, J = 8.2 Hz, 1H), 7.74 (d, J = 9.0 Hz, 1H), 7.64–7.56 (m, 2H), 7.50 (d, J = 15.5 Hz, 1H), 7.36–7.30 (m, 2H), 7.23–7.13 (m, 2H), 3.88 (s, 2H), 2.33 (s, 1H); HRMS (ESI) calcd. for [C16H13F3NO2S + H]+: 340.0614; Found: 340.0600; LCMS: [M – H]− found 355.1, purity >99%.
(E)-N-(4-((Methylamino)methyl)phenyl)-2-(3-(trifluoromethyl)phenyl)ethene-1-sulfonamide (15)
Using the same procedure as compound 6, 4-((methylamino)methyl)aniline (25 mg, 0.18 mmol) and compound 27 (50 mg, 0.18 mmol) yielded the desired product (36 mg, 0.10 mmol, 53% yield) as yellow crystals. The crude product was purified by silica flash column chromatography (column gradient of 30% to 60% EtOAc/Pet. Ether at 40–60 °C). 1H NMR (400 MHz, DMSO-d6) δ 8.04 (d, J = 7.7 Hz, 1H), 7.79 (d, J = 7.9 Hz, 1H), 7.73–7.54 (m, 3H), 7.33–7.12 (m, 1H), 7.00 (d, J = 8.3 Hz, 2H), 6.55 (d, J = 8.3 Hz, 2H), 5.25(s, 1H), 4.05 (s, 2H), 2.60 (s, 3H); HRMS (ESI) calcd. for [C17H17F3N2O2S + H]+: 371.1041; Found: 371.1031; LCMS: [M + H]+ found 370.9, purity 87%.
(E)-N-(4-((2-(3-(Trifluoromethyl)phenyl)vinyl)sulfonamido)benzyl)acetamide (16)
Using the same procedure as compound 6, N-(4-aminobenzyl)acetamide (30 mg, 0.18 mmol) and compound 27 (50 mg, 0.18 mmol) yielded the desired product (20 mg, 0.050 mmol, 27% yield) as a colorless oil. The crude product was purified by silica flash column chromatography (column gradient of 30% to 60% EtOAc/Pet. Ether at 40–60 °C). 1H NMR (500 MHz, DMSO-d6) δ 10.08 (s, 1H), 8.24 (t, J = 5.9 Hz, 1H), 8.15 (s, 1H), 8.02 (d, J = 7.8 Hz, 1H), 7.76 (d, J = 7.8 Hz, 1H), 7.63 (t, J = 7.8 Hz, 1H), 7.56 (d, J = 15.5 Hz, 1H), 7.50 (d, J = 15.5 Hz, 1H), 7.14 (t, J = 2.6 Hz, 3H), 4.15 (d, J = 5.9 Hz, 2H), 1.82 (s, 3H); HRMS (ESI) calcd. for [C18H17F3N2O3S + H]+: 399.0990; Found: 399.1003; LCMS: [M – H]− found 397.1, purity >99%.
(E)-4-((2-(3-(Trifluoromethyl)phenyl)vinyl)sulfonamido)benzoic Acid (17)
Compound 9 (258 mg, 0.67 mmol) was stirred overnight in a 10% LiOH solution in methanol/water (1:1, 4 mL). Methanol was removed under reduced pressure, and then, cold water was added to the mixture. 1 N hydrochloric acid was used to precipitate the product, which was filtered and dried to give the desired product (174 mg, 0.47 mmol, 70% yield) as a yellow powder. 1H NMR (400 MHz, MeOD) δ 7.90 (t, J = 1.9 Hz, 1H), 7.85 (s, 1H), 7.80 (d, J = 7.8 Hz, 1H), 7.74 (dt, J = 7.6, 1.4 Hz, 1H), 7.68 (d, J = 8.3 Hz, 1H), 7.58 (d, J = 7.8 Hz, 1H), 7.53 (d, J = 15.4 Hz, 1H), 7.46 (ddd, J = 8.1, 2.3, 1.2 Hz, 1H), 7.40 (t, J = 7.8 Hz, 1H), 7.20 (d, J = 15.4 Hz, 1H); HRMS (ESI) calcd. for [C16H12F3NO4S + H]+: 372.0517; Found: 372.0502; LCMS: [M – H]− found 370.0, purity >99%.
(E)-4-((2-(3-(Trifluoromethyl)phenyl)vinyl)sulfonamido)benzamide (18)
To a solution of compound 17 (145 mg, 0.39 mmol), HOBt hydrate (66 mg, 0.43 mmol), and EDCl (82 mg, 0.43 mmol) in THF (3 mL), N,N-diisopropylethylamine (101 mg, 0.78 mmol) was added, and the mixture was stirred for 10 min at room temperature. 2 M ammonia in methanol (0.195 mL, 0.39 mmol) was added, and the reaction was stirred overnight. A NaHCO3/H2O solution (1:1, 3 mL) was added, and the reaction stirred for 2 h. The precipitate was filtered to yield the desired product 18 (149 mg, 0.389 mmol, 100% yield) as a cream colored powder. 1H NMR (400 MHz, MeOD) δ 7.86 (s, 1H), 7.81 (d, J = 7.7 Hz, 1H), 7.73 (t, J = 2.0 Hz, 1H), 7.69 (d, J = 7.9 Hz, 1H), 7.62–7.53 (m, 2H), 7.52 (s, 1H), 7.44–7.39 (m, 1H), 7.37 (d, J = 7.9 Hz, 1H), 7.22 (d, J = 15.5 Hz, 1H), 2.40 (s, 1H); HRMS (ESI) calcd. for [C16H13F3N2O3S + H]+: 371.0677; Found: 371.0681; LCMS: [M – H]− found 369.0, purity 95%.
tert-Butyl-(E)-(3-((2-(3-(trifluoromethyl)phenyl)vinyl)sulfonamido)benzyl)carbamate (29)
To a solution of compound 27 (50 mg, 0.55 mmol) in pyridine (5 mL), tert-butyl(3-aminobenzyl)carbamate (123 mg, 0.55 mmol) was added, and the reaction mixture was stirred under a nitrogen atmosphere at room temperature for 1 h. The crude product was concentrated under reduced pressure and purified by silica flash column chromatography (column gradient of 30% to 60% EtOAc/Pet. Ether at 40–60 °C) to yield the desired product 29 (175 mg, 0.38 mmol, 69% yield) as a pale yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.80 (s, 1H), 7.68–7.56 (m, 3H), 7.57–7.46 (m, 2H), 7.31–7.22 (m, 1H), 7.17 (d, J = 9.0 Hz, 2H), 7.05 (d, J = 7.5 Hz, 1H), 6.90 (d, J = 15.5 Hz, 1H), 5.03 (s, 1H), 4.28 (d, J = 6.1 Hz, 2H), 1.46 (s, 9H); LCMS: [M – Boc – H]− found 355.1, purity >99%.
(E)-N-(3-(Aminomethyl)phenyl)-2-(3-(trifluoromethyl)phenyl)ethene-1-sulfonamide (19)
Using the same procedure as compound 14, compound 29 (133 mg, 0.29 mmol) was deprotected to yield the desired product 19 (78 mg, 0.22 mmol, 76% yield) as pale yellow crystals. 1H NMR (400 MHz, DMSO-d6) δ 8.12 (s, 1H), 7.99 (d, J = 7.8 Hz, 1H), 7.75 (d, J = 7.8 Hz, 1H), 7.63 (t, J = 7.8 Hz, 1H), 7.55 (d, J = 15.5 Hz, 1H), 7.46 (d, J = 15.5 Hz, 1H), 7.20 (t, J = 7.8 Hz, 1H), 7.15 (d, J = 1.9 Hz, 1H), 7.05 (dd, J = 8.5, 2.1 Hz, 1H), 6.98 (d, J = 7.6 Hz, 1H), 3.72 (s, 2H); HRMS (ESI) calcd. for [C16H15F3N2O2S + H]+: 357.0885; Found: 357.0869; LCMS: [M – H]− found 355.1, purity >99%.
tert-Butyl-(E)-7-((2-(3-(trifluoromethyl)phenyl)vinyl)sulfonamido)-3,4-dihydroisoquinoline-2(1H)-carboxylate (30)
Using the same procedure as compound 28, tert-butyl 7-amino-3,4-dihydroisoquinoline-2(1H)-carboxylate (96 mg, 0.38 mmol) and compound 27 (104 mg, 0.38 mmol) yielded the desired product 30 (148 mg, 0.31 mmol, 80% yield) as pale yellow crystals. The crude product was purified by silica flash column chromatography (column gradient of 20% to 50% EtOAc/Pet. Ether at 40–60 °C). 1H NMR (400 MHz, CDCl3) δ 7.65 (d, J = 8.0 Hz, 2H), 7.61 (d, J = 7.8 Hz, 1H), 7.56–7.48 (m, 2H), 7.08 (d, J = 8.1 Hz, 1H), 6.98 (s, 2H), 6.87 (d, J = 15.5 Hz, 1H), 5.30 (s, 1H), 4.53 (s, 2H), 3.61 (s, 2H), 2.78 (t, J = 5.7 Hz, 2H), 1.47 (s, 9H); LCMS: [M – H]− found 481.2, purity >99%.
(E)-N-(1,2,3,4-Tetrahydroisoquinolin-7-yl)-2-(3-(trifluoromethyl)phenyl)ethene-1-sulfonamide (20)
Using the same procedure as compound 12, compound 30 (67.2 mg, 0.15 mmol) was deprotected to yield the desired product (104 mg, 0.28 mmol, 98% yield) as orange crystals. 1H NMR (400 MHz, DMSO-d6) δ 8.15 (s, 1H), 8.02 (d, J = 7.8 Hz, 1H), 7.77 (d, J = 7.9 Hz, 1H), 7.64 (t, J = 7.8 Hz, 1H), 7.58 (d, J = 15.5 Hz, 1H), 7.49 (d, J = 15.5 Hz, 1H), 7.14–7.01 (m, 2H), 6.97 (d, J = 2.0 Hz, 1H), 4.06 (s, 2H), 3.17 (t, J = 6.1 Hz, 2H), 2.78 (t, J = 6.1 Hz, 2H); HRMS (ESI) calcd. for [C18H17F3N2O2S + H]+: 383.1022; Found: 383.1036; LCMS: [M + H]+ found 383.1, purity >99%.
N-(3-(Aminomethyl)phenyl)-4-(tert-butyl)benzenesulfonamide (21)
Using the same procedure as compound 28, 4-(tert-butyl)benzenesulfonyl chloride (100 mg, 0.43 mmol) and tert-butyl (3-aminobenzyl)carbamate (96 mg, 0.43 mmol) yielded the Boc protected intermediate tert-butyl-(3-((4-(tert-butyl)phenyl)sulfonamido)benzyl)carbamate (81 mg, 45% yield) as a white powder. The crude product was purified by silica flash column chromatography (column gradient of 5% to 50% EtOAc/Pet. Ether at 40–60 °C. 1H NMR (400 MHz, CDCl3) δ 7.81–7.65 (m, 1H), 7.58 (s, 1H), 7.42 (d, J = 8.6 Hz, 1H), 7.16 (t, J = 7.8 Hz, 1H), 7.01 (dd, J = 17.0, 6.9 Hz, 1H), 4.88 (s, 1H), 4.88 (s, 1H), 4.21 (d, J = 5.5 Hz, 1H), 4.19 (t, J = 14.4 Hz, 1H), 1.28 (s, 5H). LC/MS [M – H]− found 416.8, purity >99%.
To a solution of the Boc intermediate in dichloromethane (2 mL), trifluoroacetic acid (1 mL) was added dropwise, and the mixture was stirred for 0.5 h. The reaction was quenched with a saturated solution of NaHCO3. The organic phase was isolated, and the final product was concentrated under reduced pressure to yield the desired compound 21 (55 mg, 89% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 7.69 (d, J = 8.5 Hz, 2H), 7.54 (d, J = 8.5 Hz, 2H), 7.11 (t, J = 7.8 Hz, 1H), 7.04 (d, J = 1.6 Hz, 1H), 6.93 (dd, J = 7.9, 1.9 Hz, 2H), 3.61 (s, 2H), 1.25 (s, 9H); HRMS (ESI) calcd. for [C17H22N2O2S + H]+: 319.1467; Found: 319.1475; LC/MS [M – H]− found 317, purity >99%.
N-(3-(Aminomethyl)phenyl)-2-(naphthalen-1-yl)ethane-1-sulfonamide (22)
Using the same procedure as compound 28, tert-butyl (3-aminobenzyl)carbamate (87 mg, 0.393 mmol) and 2-(naphthalen-1-yl)ethane-1-sulfonyl chloride (200 mg, 0.785 mmol, 2 equiv) yielded the Boc intermediate (36 mg, 0.081 mmol, 21% yield) as a white powder. The crude product was purified by silica flash column chromatography (column gradient of 20% to 50% EtOAc/Pet. Ether at 40–60 °C). 1H NMR (400 MHz, Chloroform-d) δ 7.86 (dd, J = 8.0, 1.4 Hz, 1H), 7.76 (t, J = 8.4 Hz, 2H), 7.54–7.32 (m, 4H), 7.20 (t, J = 7.8 Hz, 1H), 7.03 (d, J = 7.9 Hz, 1H), 6.97 (dd, J = 8.0, 2.2 Hz, 1H), 6.81 (s, 1H), 6.59 (s, 1H), 4.82 (s, 1H), 4.20 (d, J = 6.1 Hz, 2H), 3.64–3.53 (m, 2H), 3.51–3.40 (m, 2H), 1.44 (s, 9H); LCMS: [M – H]− found 439.3, purity >99%.
To a solution of the Boc intermediate (33 mg, 0.075 mmol) in dichloromethane (2 mL), trifluoroacetic acid (1 mL) was added dropwise, and the mixture was stirred for 0.5 h. The reaction was quenched with a saturated solution of NaHCO3. The organic phase was isolated, and the final product was concentrated under reduced pressure to yield the desired compound 22 (14.2 mg, 0.042 mmol, 56% yield) as colorless crystals. 1H NMR (400 MHz, Chloroform-d) δ 7.78 (d, J = 8.0 Hz, 1H), 7.69 (t, J = 7.1 Hz, 2H), 7.46–7.20 (m, 4H), 7.13 (t, J = 7.7 Hz, 1H), 7.06–6.93 (m, 3H), 5.30 (s, 1H), 5.04 (s, 2H), 3.80 (s, 2H), 3.58–3.45 (m, 2H), 3.43–3.33 (m, 2H); HRMS (ESI) calcd. for [C19H20N2O2S + H]+: 341.1324; Found: 341.1343; LCMS: [M – H]− found 339.1, purity >99%.
N-(3-(Aminomethyl)phenyl)-5-chloro-3-methylbenzo[b]thiophene-2-sulfonamide (23)
Using the same procedure as compound 28, tert-butyl (3-aminobenzyl)carbamate (79 mg, 0.356 mmol) and tert-butyl-(3-((5-chloro-3-methylbenzo[b]thiophene)-2-sulfonamido)benzyl)-carbamate (100 mg, 0.356 mmol) yielded the Boc intermediate (156 mg, 0.334 mmol, 94% yield) as a colorless oil. The crude product was purified by silica flash column chromatography (column gradient of 30% to 60% EtOAc/Pet. Ether at 40–60 °C). 1H NMR (400 MHz, CDCl3) δ 8.08 (s, 1H), 7.64 (d, J = 8.7 Hz, 2H), 7.38 (dd, J = 8.7, 2.0 Hz, 1H), 7.17 (t, J = 7.8 Hz, 1H), 7.10 (s, 1H), 7.06–7.00 (m, 2H), 4.90 (s, 1H), 4.21 (d, J = 6.1 Hz, 2H), 2.41 (s, 3H), 1.43 (s, 9H); 13C NMR (101 MHz, CDCl3); LC/MS [M – H]− found 465, purity 76%.
To a solution of the Boc intermediate (147 mg, 0.315 mmol) in dichloromethane (2 mL), trifluoroacetic acid (1 mL) was added dropwise, and the mixture was stirred for 0.5 h. The reaction was quenched with a saturated solution of NaHCO3. The organic phase was isolated, and the final product was concentrated under reduced pressure to yield the desired compound 23 (115 mg, 0.315 mmol, 100% yield) as a white powder. 1H NMR (400 MHz, DMSO-d6) δ 7.87 (dd, J = 8.5, 2.0 Hz, 1H), 7.72 (s, 1H), 7.35 (dd, J = 8.6, 2.1 Hz, 1H), 6.86–6.67 (m, 2H), 6.60 (s, 1H), 6.47 (d, J = 7.4 Hz, 1H), 6.39 (d, J = 7.3 Hz, 1H), 3.92 (d, J = 6.1 Hz, 2H), 3.48 (s, 2H), 2.46 (s, 3H); HRMS (ESI) calcd. for [C16H15ClN2O2S2 + H]+: 367.0338; Found: 367.0342; LCMS: [M – H]− found 365.1, purity >99%.
N-(3-(Aminomethyl)phenyl)-5-fluoro-3-methylbenzo[b]thiophene-2-sulfonamide (24)
Using the same procedure as compound 28, tert-butyl-(3-aminobenzyl)carbamate (84 mg, 0.378 mmol) and 5-fluoro-3-methylbenzo[b]thiophene-2-sulfonyl chloride (100 mg, 0.378 mmol) yielded the Boc intermediate (125 mg, 0.277 mmol, 73% yield) as a white powder. The crude product was purified by silica flash column chromatography (column gradient of 20% to 50% EtOAc/Pet. Ether of 40–60 °C). 1H NMR (400 MHz, Chloroform-d) δ 8.25 (s, 1H), 7.66 (dd, J = 8.8, 4.7 Hz, 1H), 7.36–7.28 (m, 1H), 7.22–6.94 (m, 5H), 4.92 (s, 1H), 4.20 (d, J = 5.7 Hz, 2H), 2.40 (s, 3H), 1.42 (s, 9H); LCMS: [M – H]− found 449.1, purity >99%.
To a solution of the Boc intermediate (161 mg, 0.358 mmol) in dichloromethane (2 mL), trifluoroacetic acid (1 mL) was added dropwise, and the mixture was stirred for 0.5 h. The reaction was quenched with a saturated solution of NaHCO3. The organic phase was isolated, and the final product was concentrated under reduced pressure to yield the desired compound 24 (127 mg, 0.361 mmol, 99% yield) as a white powder. 1H NMR (400 MHz, DMSO-d6) δ 7.90 (dd, J = 8.8, 5.1 Hz, 1H), 7.53 (dd, J = 10.1, 2.5 Hz, 1H), 7.24 (td, J = 8.9, 2.5 Hz, 1H), 7.03–6.81 (m, 3H), 6.61 (d, J = 7.2 Hz, 1H), 3.67 (s, 2H), 2.46 (s, 3H); HRMS (ESI) calcd. for [C16H15FN2O2S2Na]+: 373.0451; Found: 373.0437; LCMS: [M – H]− found 349.0, purity >99%.
N-(3-(Aminomethyl)phenyl)benzo[b]thiophene-2-sulfonamide (25)
Using the same procedure as compound 7, tert-butyl (3-aminobenzyl)carbamate (96 mg, 0.430 mmol) and benzo[b]thiophene-2-sulfonyl chloride (100 mg, 0.430 mmol) yielded the Boc intermediate (158 mg, 0.338 mmol, 95% yield) as a white powder. The crude product was purified by silica flash column chromatography (column gradient of 20% to 50% EtOAc/Pet. Ether at 40–60 °C). 1H NMR (400 MHz, Chloroform-d) δ 8.33 (s, 1H), 7.80–7.69 (m, 3H), 7.44–7.30 (m, 2H), 7.22–7.07 (m, 3H), 7.01 (d, J = 7.5 Hz, 1H), 4.96 (s, 1H), 4.22 (d, J = 6.1 Hz, 2H), 1.43 (s, 9H); LCMS: [M – H]− found 417.1, purity 93%.
Using the same procedure as compound 19, the Boc intermediate (118 mg, 0.282 mmol) was deprotected to yield compound 25 (90 mg, 0.282 mmol, 99% yield) as a white powder. 1H NMR (400 MHz, DMSO-d6) δ 7.98–7.89 (m, 1H), 7.89–7.81 (m, 1H), 7.73 (s, 1H), 7.39 (qd, J = 7.4, 3.7 Hz, 2H), 7.15–6.97 (m, 3H), 6.82 (d, J = 7.3 Hz, 1H), 3.83 (s, 2H), 2.68 (s, 1H), 2.07 (s, 2H); HRMS (ESI) calcd. for [C15H14N2O2S2 + H]+: 319.0569; Found: 319.0557; LCMS: [M – H]− found 317.0, purity >99%.
N-(3-(Aminomethyl)phenyl)benzofuran-2-sulfonamide (26)
Using the same procedure as compound 28, tert-butyl (3-aminobenzyl)carbamate (103 mg, 0.462 mmol) and benzofuran-2-sulfonyl chloride (100 mg, 0.462 mmol) yielded the Boc intermediate (128 mg, 0.320 mmol, 69% yield) as a white powder. The crude product was purified by silica flash column chromatography (column gradient of 20% to 50% EtOAc/Pet. Ether at 40–60 °C). 1H NMR (400 MHz, DMSO-d6) δ 10.93 (s, 1H), 7.75 (d, J = 7.7 Hz, 1H), 7.73–7.66 (m, 1H), 7.63 (s, 1H), 7.51 (ddd, J = 8.5, 7.2, 1.3 Hz, 1H), 7.37 (t, J = 7.1 Hz, 1H), 7.19 (t, J = 7.8 Hz, 1H), 7.10 (s, 1H), 7.03 (d, J = 7.9 Hz, 1H), 6.93 (d, J = 7.7 Hz, 1H), 4.03 (d, J = 6.0 Hz, 2H), 1.37 (s, 9H), 1.18 (s, 1H); HRMS (ESI) calcd. for [C20H21N2O5SNa + H]+: 425.1147; Found: 425.1146; LCMS: [M – H]− found 401.1, purity >99%.
Using the same procedure as compound 19, the Boc intermediate (103 mg, 0.256 mmol) was deprotected to yield compound 26 (51 mg, 0.169 mmol, 66% yield) as a cream powder. 1H NMR (400 MHz, DMSO-d6) δ 7.63 (d, J = 7.7 Hz, 1H), 7.55 (d, J = 8.3 Hz, 1H), 7.34 (t, J = 7.7 Hz, 1H), 7.24 (t, J = 7.5 Hz, 1H), 7.10 (s, 1H), 7.07–6.91 (m, 3H), 6.70 (d, J = 6.2 Hz, 1H), 3.83 (s, 2H); HRMS (ESI) calcd. for [C15H14N2O3S + H]+: 303.0798; Found: 303.0786; LCMS: [M – H]− found 301.1, purity >99%.
Glossary
Abbreviations Used
- ΔTm
change in melting temperature
- DSF
differential scanning fluorimetry
- IC50
half-maximal inhibitory concentration
- MIC
minimum inhibitory concentration
- Mtb
Mycobacterium tuberculosis
- TB
tuberculosis
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.0c00007.
Fragment hits identified by thermal shift (Table S1); X-ray crystallographic data collection and refinement statistics (Table S2); Fo–Fc “Omit” maps of compounds 1, 2, 3, 4, 5, and 23 (Figure S1), X-ray crystal structures of fragment hits 1, 2, 3, 4, and 5 (Figure S2); docking poses of compounds 23 and 24 (Figure S3); LCMS of all the compounds screened against InhA (Figure S4) (PDF)
Molecular formula strings (CSV)
Accession Codes
Atomic coordinates for the X-ray structures of compounds 1 (PDB code 6SQ5), 2 (PDB code 6SQ7), 3 (PDB code 6SQ9), 4 (PDB code 6SQB), 5 (PDB code 6SQD), and 23 (PDB code 6SQL) are available from the RCSB Protein Data Bank (www.rcsb.org).
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
V.M. and T.L.B. acknowledge the Bill and Melinda Gates Foundation [subcontract by the Foundation for the National Institute of Health (NIH)] (OPP1024021). V.M. further acknowledges the Bill and Melinda Gates Foundation [subcontract by the Foundation for the National Institutes of Health (FNIH)] (OPP1158806). V.M. and M.S. acknowledge the European Community’s Seventh Framework Programme (MM4TB, grant number 260872). D.M.G.D. was the recipient of an FCT PhD grant [SFRH/BD/81735/2011]. K.M. and J.K. acknowledge support by the Ministry of Education, Science, Research and Sport of the Slovak Republic (grant VEGA 1/0301/18), by the Slovak Research and Development Agency (contract DO7RP-0015-11), and by the Research and Development Operational Programme funded by the European Regional Development Fund (Contract ITMS 26240120027).
The authors declare no competing financial interest.
Supplementary Material
References
- Floyd K.; Glaziou P.; Zumla A.; Raviglione M. The global tuberculosis epidemic and progress in care, prevention, and research: an overview in year 3 of the End TB era. Lancet Respir. Med. 2018, 6, 299–314. 10.1016/S2213-2600(18)30057-2. [DOI] [PubMed] [Google Scholar]
- Vilcheze C.; Jacobs W. R. Resistance to Isoniazid and Ethionamide in Mycobacterium tuberculosis: genes, mutations, and causalities. Microbiol. Spectrum 2014, 2, MGM2-0014-2013. 10.1128/microbiolspec.MGM2-0014-2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abrahams K. A.; Chung C. W.; Ghidelli-Disse S.; Rullas J.; Rebollo-Lopez M. J.; Gurcha S. S.; Cox J. A.; Mendoza A.; Jimenez-Navarro E.; Martinez-Martinez M. S.; Neu M.; Shillings A.; Homes P.; Argyrou A.; Casanueva R.; Loman N. J.; Moynihan P. J.; Lelievre J.; Selenski C.; Axtman M.; Kremer L.; Bantscheff M.; Angulo-Barturen I.; Izquierdo M. C.; Cammack N. C.; Drewes G.; Ballell L.; Barros D.; Besra G. S.; Bates R. H. Identification of KasA as the cellular target of an anti-tubercular scaffold. Nat. Commun. 2016, 7, 12581. 10.1038/ncomms12581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W.; Lun S.; Wang S. H.; Jiang X. W.; Yang F.; Tang J.; Manson A. L.; Earl A. M.; Gunosewoyo H.; Bishai W. R.; Yu L. F. Identification of novel coumestan derivatives as polyketide synthase 13 inhibitors against Mycobacterium tuberculosis. J. Med. Chem. 2018, 61, 791–803. 10.1021/acs.jmedchem.7b01319. [DOI] [PubMed] [Google Scholar]
- Aggarwal A.; Parai M. K.; Shetty N.; Wallis D.; Woolhiser L.; Hastings C.; Dutta N. K.; Galaviz S.; Dhakal R. C.; Shrestha R.; Wakabayashi S.; Walpole C.; Matthews D.; Floyd D.; Scullion P.; Riley J.; Epemolu O.; Norval S.; Snavely T.; Robertson G. T.; Rubin E. J.; Ioerger T. R.; Sirgel F. A.; van der Merwe R.; van Helden P. D.; Keller P.; Bottger E. C.; Karakousis P. C.; Lenaerts A. J.; Sacchettini J. C. Development of a novel lead that targets M. tuberculosis polyketide synthase 13. Cell 2017, 170, 249–259.E25. 10.1016/j.cell.2017.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goins C. M.; Dajnowicz S.; Thanna S.; Sucheck S. J.; Parks J. M.; Ronning D. R. Exploring covalent allosteric inhibition of Antigen 85C from Mycobacterium tuberculosis by ebselen derivatives. ACS Infect. Dis. 2017, 3, 378–387. 10.1021/acsinfecdis.7b00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viljoen A.; Richard M.; Nguyen P. C.; Fourquet P.; Camoin L.; Paudal R. R.; Gnawali G. R.; Spilling C. D.; Cavalier J. F.; Canaan S.; Blaise M.; Kremer L. Cyclipostins and cyclophostin analogs inhibit the antigen 85C from Mycobacterium tuberculosis both in vitro and in vivo. J. Biol. Chem. 2018, 293, 2755–2769. 10.1074/jbc.RA117.000760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley S. A.; Kawate T.; Iwase N.; Shimizu M.; Clatworthy A. E.; Kazyanskaya E.; Sacchettini J. C.; Ioerger T. R.; Siddiqi N. A.; Minami S.; Aquadro J. A.; Grant S. S.; Rubin E. J.; Hung D. T. Diarylcoumarins inhibit mycolic acid biosynthesis and kill Mycobacterium tuberculosis by targeting FadD32. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 11565–11570. 10.1073/pnas.1302114110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sink R.; Sosic I.; Zivec M.; Fernandez-Menendez R.; Turk S.; Pajk S.; Alvarez-Gomez D.; Lopez-Roman E. M.; Gonzales-Cortez C.; Rullas-Triconado J.; Angulo-Barturen I.; Barros D.; Ballell-Pages L.; Young R. J.; Encinas L.; Gobec S. Design, synthesis, and evaluation of new thiadiazole-based direct inhibitors of enoyl acyl carrier protein reductase (InhA) for the treatment of tuberculosis. J. Med. Chem. 2015, 58, 613–624. 10.1021/jm501029r. [DOI] [PubMed] [Google Scholar]
- Manjunatha U. H.; Rao S. P. S.; Kondreddi R. R.; Noble C. G.; Camacho L. R.; Tan B. H.; Ng S. H.; Ng P. S.; Ma N. L.; Lakshminarayana S. B.; Herve M.; Barnes S. W.; Yu W.; Kuhen K.; Blasco F.; Beer D.; Walker J. R.; Tonge P. J.; Glynne R.; Smith P. W.; Diagana T. T. Direct inhibitors of InhA are active against Mycobacterium tuberculosis. Sci. Transl. Med. 2015, 7, 269ra3. 10.1126/scitranslmed.3010597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Encinas L.; O’Keefe H.; Neu M.; Remuinan M. J.; Patel A. M.; Guardia A.; Davie C. P.; Perez-Macias N.; Yang H.; Convery M. A.; Messer J. A.; Perez-Herran E.; Centrella P. A.; Alvarez-Gomez D.; Clark M. A.; Huss S.; O’Donovan G. K.; Ortega-Muro F.; McDowell W.; Castaneda P.; Arico-Muendel C. C.; Pajk S.; Rullas J.; Angulo-Barturen I.; Alvarez-Ruiz E.; Mendoza-Losana A.; Ballell Pages L.; Castro-Pichel J.; Evindar G. Encoded library technology as a source of hits for the discovery and lead optimization of a potent and selective class of bactericidal direct inhibitors of Mycobacterium tuberculosis InhA. J. Med. Chem. 2014, 57, 1276–1288. 10.1021/jm401326j. [DOI] [PubMed] [Google Scholar]
- Martinez-Hoyos M.; Perez-Herran E.; Gulten G.; Encinas L.; Alvarez-Gomez D.; Alvarez E.; Ferrer-Bazaga S.; Garcia-Perez A.; Ortega F.; Angulo-Barturen I.; Rullas-Trincado J.; Blanco Ruano D.; Torres P.; Castaneda P.; Huss S.; Fernandez Menendez R.; Gonzalez Del Valle S.; Ballell L.; Barros D.; Modha S.; Dhar N.; Signorino-Gelo F.; McKinney J. D.; Garcia-Bustos J. F.; Lavandera J. L.; Sacchettini J. C.; Jimenez M. S.; Martin-Casabona N.; Castro-Pichel J.; Mendoza-Losana A. Antitubercular drugs for an old target: GSK693 as a promising InhA direct inhibitor. EBioMedicine 2016, 8, 291–301. 10.1016/j.ebiom.2016.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozman K.; Sosic I.; Fernandez R.; Young R. J.; Mendoza A.; Gobec S.; Encinas L. A new ’golden age’ for the antitubercular target InhA. Drug Discovery Today 2017, 22, 492–502. 10.1016/j.drudis.2016.09.009. [DOI] [PubMed] [Google Scholar]
- Marchetti C.; Chan D. S.; Coyne A. G.; Abell C. Fragment-based approaches to TB drugs. Parasitology 2018, 145, 184–195. 10.1017/S0031182016001876. [DOI] [PubMed] [Google Scholar]
- Mendes V.; Blundell T. L. Targeting tuberculosis using structure-guided fragment-based drug design. Drug Discovery Today 2017, 22, 546–554. 10.1016/j.drudis.2016.10.003. [DOI] [PubMed] [Google Scholar]
- Prati F.; Zuccotto F.; Fletcher D.; Convery M. A.; Fernandez-Menendez R.; Bates R.; Encinas L.; Zeng J.; Chung C. W.; De Dios Anton P.; Mendoza-Losana A.; Mackenzie C.; Green S. R.; Huggett M.; Barros D.; Wyatt P. G.; Ray P. C. Screening of a novel fragment library with functional complexity against Mycobacterium tuberculosis InhA. ChemMedChem 2018, 13, 672–677. 10.1002/cmdc.201700774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan T. J.; Truglio J. J.; Boyne M. E.; Novichenok P.; Zhang X.; Stratton C. F.; Li H. J.; Kaur T.; Amin A.; Johnson F.; Slayden R. A.; Kisker C.; Tonge P. J. High affinity InhA inhibitors with activity against drug-resistant strains of Mycobacterium tuberculosis. ACS Chem. Biol. 2006, 1, 43–53. 10.1021/cb0500042. [DOI] [PubMed] [Google Scholar]
- Dalvit C.; Fogliatto G.; Stewart A.; Veronesi M.; Stockman B. WaterLOGSY as a method for primary NMR screening: practical aspects and range of applicability. J. Biomol. NMR 2001, 21, 349–359. 10.1023/A:1013302231549. [DOI] [PubMed] [Google Scholar]
- Mayer M.; Meyer B. Characterization of ligand binding by saturation transfer difference NMR spectroscopy. Angew. Chem., Int. Ed. 1999, 38, 1784–1788. . [DOI] [PubMed] [Google Scholar]
- Loria J. P.; Rance M.; Palmer A. G. A Relaxation-compensated Carr–Purcell–Meiboom–Gill sequence for characterizing chemical exchange by NMR spectroscopy. J. Am. Chem. Soc. 1999, 121, 2331–2332. 10.1021/ja983961a. [DOI] [Google Scholar]
- Shirude P. S.; Madhavapeddi P.; Naik M.; Murugan K.; Shinde V.; Nandishaiah R.; Bhat J.; Kumar A.; Hameed S.; Holdgate G.; Davies G.; McMiken H.; Hegde N.; Ambady A.; Venkatraman J.; Panda M.; Bandodkar B.; Sambandamurthy V. K.; Read J. A. Methyl-thiazoles: a novel mode of inhibition with the potential to develop novel inhibitors targeting InhA in Mycobacterium tuberculosis. J. Med. Chem. 2013, 56, 8533–8542. 10.1021/jm4012033. [DOI] [PubMed] [Google Scholar]
- He X.; Alian A.; Stroud R.; Ortiz de Montellano P. R. Pyrrolidine carboxamides as a novel class of inhibitors of enoyl acyl carrier protein reductase from Mycobacterium tuberculosis. J. Med. Chem. 2006, 49, 6308–6323. 10.1021/jm060715y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friesner R. A.; Murphy R. B.; Repasky M. P.; Frye L. L.; Greenwood J. R.; Halgren T. A.; Sanschagrin P. C.; Mainz D. T. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- Stadthagen G.; Kordulakova J.; Griffin R.; Constant P.; Bottova I.; Barilone N.; Gicquel B.; Daffe M.; Jackson M. p-Hydroxybenzoic acid synthesis in Mycobacterium tuberculosis. J. Biol. Chem. 2005, 280, 40699–40706. 10.1074/jbc.M508332200. [DOI] [PubMed] [Google Scholar]
- Folch J.; Lees M.; Sloane Stanley G. H. A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 1957, 226, 497–509. [PubMed] [Google Scholar]
- Mugumbate G.; Mendes V.; Blaszczyk M.; Sabbah M.; Papadatos G.; Lelievre J.; Ballell L.; Barros D.; Abell C.; Blundell T. L.; Overington J. P. Target identification of Mycobacterium tuberculosis phenotypic hits using a concerted chemogenomic, biophysical, and structural approach. Front. Pharmacol. 2017, 8, 681. 10.3389/fphar.2017.00681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonrhein C.; Flensburg C.; Keller P.; Sharff A.; Smart O.; Paciorek W.; Womack T.; Bricogne G. Data processing and analysis with the autoPROC toolbox. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, 67, 293–302. 10.1107/S0907444911007773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy A. J.; Grosse-Kunstleve R. W.; Adams P. D.; Winn M. D.; Storoni L. C.; Read R. J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams P. D.; Afonine P. V.; Bunkoczi G.; Chen V. B.; Davis I. W.; Echols N.; Headd J. J.; Hung L. W.; Kapral G. J.; Grosse-Kunstleve R. W.; McCoy A. J.; Moriarty N. W.; Oeffner R.; Read R. J.; Richardson D. C.; Richardson J. S.; Terwilliger T. C.; Zwart P. H. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 213–221. 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Lohkamp B.; Scott W. G.; Cowtan K. Features and development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 486–501. 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwood J. R.; Calkins D.; Sullivan A. P.; Shelley J. C. Towards the comprehensive, rapid, and accurate prediction of the favorable tautomeric states of drug-like molecules in aqueous solution. J. Comput.-Aided Mol. Des. 2010, 24, 591–604. 10.1007/s10822-010-9349-1. [DOI] [PubMed] [Google Scholar]
- Phetsuksiri B.; Baulard A. R.; Cooper A. M.; Minnikin D. E.; Douglas J. D.; Besra G. S.; Brennan P. J. Antimycobacterial activities of isoxyl and new derivatives through the inhibition of mycolic acid synthesis. Antimicrob. Agents Chemother. 1999, 43, 1042–1051. 10.1128/AAC.43.5.1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.