

Abstract
The global population above 60 years has been growing exponentially in the last decades, which is accompanied by an increase in the prevalence of age-related chronic diseases, highlighting cardiovascular diseases (CVDs), such as hypertension, atherosclerosis, and heart failure. Aging is the main risk factor for these diseases. Such susceptibility to disease is explained, at least in part, by the increase of oxidative stress, in which it damages cellular components such as proteins, DNA, and lipids. In addition, the chronic inflammatory process in aging “inflammaging” also contributes to cell damage, creating a stressful environment which drives to the development of CVDs. Taken together, it is possible to identify the molecular connection between oxidative stress and the inflammatory process, especially by the crosstalk between the transcription factors Nrf-2 and NF-κB which are mediated by redox signalling and are involved in aging. Therapies that control this process are key targets in the prevention/combat of age-related CVDs. In this review, we show the basics of inflammation and oxidative stress, including the crosstalk between them, and the implications on age-related CVDs.
1. Introduction
The average life expectancy of the global population has been increasing rapidly [1]. According to the United Nations (UN), the world population over 60 years will keep growing exponentially in the next decades, rising from 12%, data from 2015, to 22% in 2050 [2]. In parallel, age-related diseases have also been increasing, highlighting cardiovascular diseases (CVDs) as the main cause of morbidity and mortality worldwide, aging being the main risk factor for these diseases [3–5].
Cardiovascular aging (cardiac and vascular aging) is characterized by a progressive decline in physiological processes at the molecular, cellular, and tissue levels [6–8]. Two factors play a key role in this process: the gradual and persistent increase in inflammation “inflammaging” and the oxidative stress, including the crosstalk between them [9, 10]. These processes are associated with the pathophysiology of aging-related CVDs, such as hypertension, acute myocardial infarction, and stroke (Figure 1) [11–13].
Figure 1.
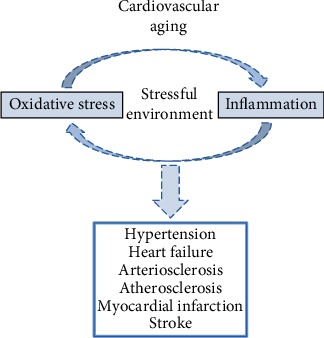
Aging and CVDs. The cardiac and arterial aging is characterized by a stressful environment to the cells, derived, at least in part, from the high levels of oxidative stress and chronic inflammation. The crosstalk between them is a vicious and gradual cycle, which accompanies the course of aging. These changes drive to the development of CVDs, such as hypertension, heart failure, arteriosclerosis, atherosclerosis, myocardial infarction, and stroke.
This review brings an integrated approach to the role of inflammation and oxidative stress associated with the pathophysiology of age-related CVDs, addressing molecular, cellular, and physiological aspects, with the potential source of antioxidants as pharmacological tools.
2. Cardiovascular Aging
Cardiovascular aging is a dynamic process caused by several mechanisms that include a progressive change in the function and structure, resulting in the impairment of cardiovascular homeostasis [14]. Briefly, the vessels and the heart become more rigid and more fibrotic as we age, a factor that predisposes the emergence of CVDs [15].
2.1. Cardiac Aging
Cardiac aging is characterized by cardiomyocyte hypertrophy, inflammation, and the gradual development of cardiac fibrosis [16]. In addition, there is decreased elasticity and increased stiffness [17]. These changes are consequences of molecular responses to cellular stressors, such as oxidative stress and inflammation, and involve mechanisms such as ventricular wall thickness, myocardial fibrosis, and fibrocalcification, which ultimately decrease cardiac output and ventricular compliance [14, 17].
Structurally, there is an increase in deposition of collagen fibers by crosslinking, which compromises function and promote fibrosis, plus increase myocardial stiffness [14]. This increased stiffness produces an increase in the incidence of cardiac hypertrophy, developed by the participation of key mediators such as ROS/RNS and inflammation [18].
Left ventricular hypertrophy, evidenced by thickening of the left ventricular wall, is a compensatory reaction to the cumulative loss of myocytes. However, in order to minimize the damage from this loss, hypertrophy minimizes myocardial wall stress and may help maintain cardiac function (Figure 2) [19].
Figure 2.
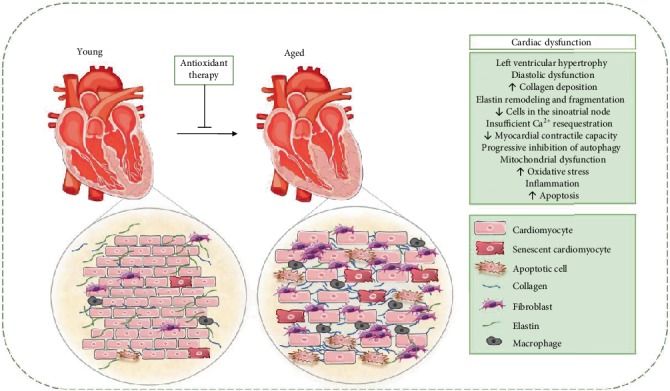
Cardiac structural and functional alterations during aging. In cardiac aging, there is a significant increase in myocardial thickness, characterized by an increase in heart size, whereas there is a decrease in the total number of cardiomyocytes. These changes alter the overall shape of the heart from elliptical to spheroid and generate greater stress on the heart wall and compromise the contractile efficiency of the heart. Fibrosis, one of the main determinants of cardiac remodelling, is characterized by increased collagen deposition, resulting in increased myocardial stiffness and, consequently, cardiac dysfunction. In the aged heart, there is diastolic dysfunction due to oxidative damage to the SERCA of the sarcoplasmic reticulum, decreasing its subsequent Ca2+ activity, prolonging diastolic relaxation. Finally, mitochondrial dysfunction, augmentation of inflammation and oxidative stress, and apoptotic and necrotic myocyte cell death are important determinants of the aging process, possibly mediating the occurrence of cardiac dysfunction in aging.
The renin angiotensin system seems to actively participate in the cardiac aging process [20]. This system mediated stimulation of ROS production via NADPH oxidase, which contributes in the stretching of myocytes and cardiac fibroblasts [21]. This triggers growth factor signalling (e.g., ANG-II/TGF-β), which has been shown in vitro to promote cell growth, matrix production, and increased apoptosis [22].
In addition, aging is accompanied by a decrease in the number and function of sinoatrial node pacemaker cells, with the concomitant increase in conduction abnormalities, resulting in a decrease in heart rate variability [12]. In addition, there is a variable degree of calcification and fibrosis on the left side of the cardiac skeleton. This can impact the atrioventricular node resulting in the blockade of the conduction [22].
2.2. Vascular Aging
Arterial aging is associated with failures in signalling pathways which are essential for the physiological functioning of the vascular system [23, 24]. These failures result from dynamic changes in the mechanical, structural, and functional properties of the vascular wall, promoting the development of endothelial dysfunction, inflammation, vascular remodelling, and arterial stiffness (Figure 3) [25, 26].
Figure 3.
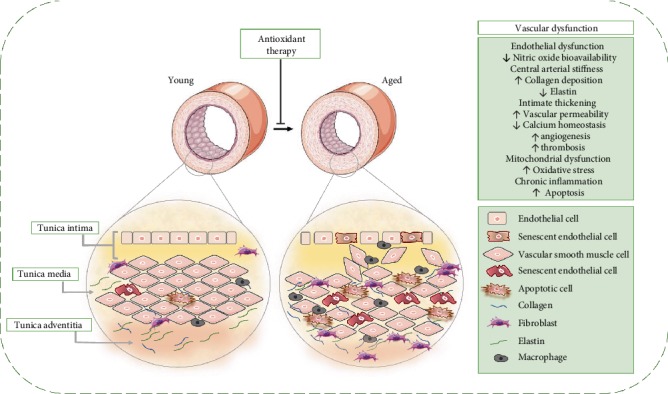
Vascular structural and functional alterations during aging. Vascular aging is associated with critical modifications of the vascular wall such as endothelial dysfunction and increased arterial thickness and stiffness. Endothelial dysfunction includes reduced vasodilatory and antithrombotic properties, with increased oxidative stress and inflammatory cytokines, increasing the risk of atherosclerosis and thrombosis. Furthermore, the endothelial barrier becomes porous and vascular smooth muscle cells migrate to subendothelial spaces and deposit extracellular matrix proteins resulting in the thickening of the tunica intima. Central arterial stiffness is related to the loss of elastic fibers and the increase of collagen accumulation in the vessel wall, which deteriorates vascular functionality. Endothelial dysfunction and arterial stiffness are mediators connected closely in vascular dysfunction during aging. If the artery is more rigid, greater will be the exposure of the endothelium to hemodynamic load, promoting endothelial activation, inflammation, and oxidative damage. Antioxidant therapies have been shown to attenuate aging-induced changes through endothelial dysfunction and changes in the extracellular arterial matrix that cause central arterial stiffness.
The arterial wall is a heterogeneous structure composed of three layers: an intima (consists essentially of endothelial cells), middle (smooth muscle cells and elastic fibers), and adventitial tunic [27]. Each layer exhibits specific histological, biochemical, and functional characteristics, and each contributes uniquely to maintain vascular integrity [28]. Under physiological conditions, the vascular endothelium synthesizes and releases substances to modulate the arterial structure and the vasodilatory, thrombolytic, and vasoprotective functions [29]. Nitric oxide (NO) is the major mediator of normal endothelial function, mainly due to its powerful vasodilator action [30].
However, in aging, the endothelium modulatory role is not totally preserved. The mechanisms associated with age-related endothelial dysfunction are basically related to decreased bioavailability of NO [31]. The lower bioavailability of NO results from decreased synthesis or increased NO degradation, which produces vasoconstriction, inflammation, and prothrombotic effects [32]. In addition, mediators such as endothelin-1 (ET-1), angiotensin II (ANG-II), and cyclooxygenase-derived (COX) eicosanoids also compromise endothelium-dependent vasodilation, resulting in increased vasoconstrictor tone, which eventually drives to CVDs, such as hypertension [33, 34].
As the vessels age, the balance between proteases and their inhibitors is impaired, and the release of proteases, such as matrix metalloproteinase (MMPs), increases [35]. Collagen and elastin are regulated by MMPs, which, in turn, are regulated by inflammatory mediators [25]. This chronic increase in the activation of MMP (-2/-7/-9/-14) in the vessels plays a central role in the vascular remodelling associated with age [36]. In addition, increased TGF-β activity contributes to increased vascular rigidity through the stimulation of interstitial collagen synthesis. Such structural and functional changes directly affect cardiac homeostasis and are intrinsically related to age-related CVDs [35].
3. Inflammaging: Implications on the Cardiovascular System
Acute and transient inflammation is vital to life by stimulating a beneficial immune response to harmful conditions such as tissue injury or pathogen invasion [37]. This process also allows cell elimination and renewal in several tissues, including the cardiovascular system [38, 39]. However, low levels and persistent inflammation lead to tissue deterioration, which is related to the pathophysiology of CVDs, such as hypertension and arteriosclerosis [13, 40]. In addition, this stage of chronic inflammation—characterized by the increase of proinflammatory cytokines in the circulation—is one of the hallmarks of aging “inflammaging” [6, 9], being considered an important risk factor for morbidity and mortality in the elderly population [41]. On the other hand, centenarians have high levels of systemic inflammation, however, without suffering from the deleterious effects of inflammation [42]. Scientists believe that suppression of inflammation is the most important drive of successful aging [43, 44].
3.1. Source of Inflammaging
There are several sources of immunological stimuli which contribute to the stage of chronic inflammation characteristic of aging [9, 37]. First, the accumulation of cell debris (e.g., proteins or organelles damaged) due to failure to clean up system in aging (e.g., deficiencies in the autophagy pathways) leads to a persistent alert status by which drives the activation of the innate immune system [37, 45]. A crucial part of the innate response is the assembly of the inflammasome a cytosolic complex of proteins that activates caspase-1 [46]. Among these inflammasomes, Nlrp3 has been under intensive investigation and has been considered a crucial driver of sterile inflammation during aging [47]. Nrlp3 mediates the maturation of IL-1β and IL-18, both proinflammatory cytokines, that are induced by several damage-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs) [46, 48]. An example of such a mechanism occurs when the mitochondria is damaged due to an increase in mtROS levels and mtDNA damage, which drives to Nlrp3 activation, due to the release of mitochondria-derived DAMPS [37, 49]. In addition, decreased NAD+ levels due to mitochondrial dysfunction are also implied in the Nlrp3 activation [50]. Moreover, autophagy and Nlrp3 activation are reversely correlated [51], which may explain the accumulation of mitochondrial dysfunction in aging [47]. On the other hand, Marín-Aguilar and colleagues showed that Nlrp3 suppression delays cardiac aging and improves longevity in mice [52].
The stage of cell senescence secretes a variety of proinflammatory cytokines, interleukins, and growth factors, known as SASP (secretory-associated senescence phenotype), being another source of inflammaging [53]. This phenomenon is explained by the senescent cells' attempt to attract cells of the immune system in order to be phagocytosed, promoting tissue regeneration [54]. However, in order for immunosenescence, the stimulus generated by the senescent cells is not able to recruit enough functional cells of the immune system, which decreases tissue homeostasis [55, 56], a long-term process that plays a negative effect on aging and age-related diseases [57]. In addition, the endothelial progenitor cells (EPC) suffer with the stem cell exhaustion process, which result in low regenerative capacity of these cells [58, 59]. The accumulation of senescent EC in aging and the dysfunction of progenitor lineages converge to vascular impairment in old age [60–62]. Recent studies have shown that senescent cell clearance from the body is a promising therapeutic target to delay aging and combat age-related CVDs [63, 64], which leads to increased health span [65].
Another source of chronic inflammation is the microbiota [66, 67]. The elderly present a decrease in microbiota diversity (decrease of commensal bacteria and increase of opportunistic bacteria) [68], which has been associated with frailty in the elderly [69]. The integrity of the gut barrier along with microbiota is the first defence against pathogens, and its deregulation may increase the infiltration to infectious agents [70, 71]. High levels of proinflammatory cytokines (e.g., TNF-α, IL-6, and IL-8) have been related to the changes in the microbiota [67], which is associated to a chronic systemic inflammation [72].
In addition, another source of chronic inflammation is based on an increased activation of the coagulation system in the elderly [73]. The coagulation system and inflammation have many shared pathways [74]. IL-1, IL-6, and TNF-α directly influence coagulation pathways, creating a reciprocal activation. They modify endothelial function, leading to a prothrombotic state with inhibition of fibrinolysis, increased production of platelet activation factors, and activation of the intrinsic and extrinsic coagulation pathways [75, 76]. Humans with exceptional longevity present also a hypercoagulation state in addition to the chronic inflammation [77]. The high incidence of thrombosis in the elderly population may be related to this characteristic phenomenon of aging [78].
Together, these factors generate continuous stimuli of the immune system, creating an unresolved inflammation stage, which is related to the pathophysiology of CVDs associated with aging (Figure 4) [79, 80].
Figure 4.
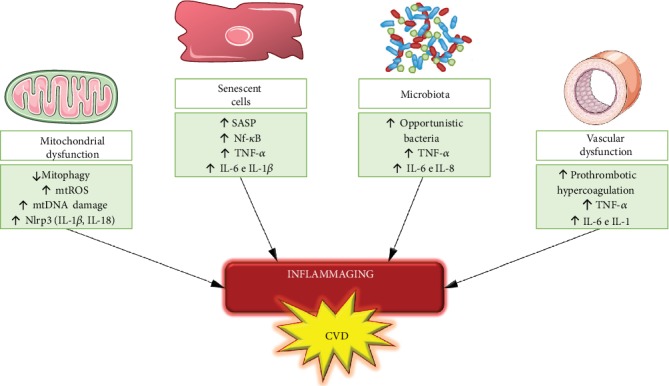
Different sources contribute to inflammation in aging. Aging is accompanied by mitochondrial dysfunction, increase in senescent cells numbers, dysregulated microbiota, and a hypercoagulation state that mediate the inflammatory processes that are characteristics of aging. Together, these processes play a key role on the development of age-related cardiovascular diseases.
3.2. Pro- and Anti-inflammatory Markers in Aging
The elderly have high levels of proinflammatory cytokines in the circulation, including IL-1β, IL-1 antagonist receptor (IL-1RN), IL-6, IL-8, TNF-α, IL-13, IL-18, IFN-γ, and C-reactive protein (CRP) [76]. Such cytokines mediate inflammation through the interaction with toll-like receptors (TLRs), IL-1R receptor, IL-6 receptor (IL-6R), and TNF receptor (TNFR) [81]. Activation of the receptors triggers intracellular signalling pathways, including mitogen-activated protein kinase (MAPK), nuclear kappa-B (NF-κB), Janus kinase (JAK), and transcriptional activator (STAT) pathways [82, 83]. Such signalling pathways have their expressions regulated by interleukins and inflammation products, generating a fast and efficient feeding loop. In addition, high levels of these markers are correlated with senescence, endothelial dysfunction, and cardiovascular aging [84, 85].
Anti-inflammatory cytokines and soluble protein antagonist receptors act to balance proinflammatory agents in the search for homeostasis. IL-10, IL-37, and transforming growth factor (TGF-β) are the main examples of these cytokines [76]. IL-10 acts by suppressing levels of IL-6, IL-8, and TNF-α. In addition, it has been reported as an endothelial protector [86]. However, it still has controversies about its actual role in vascular homeostasis. TGF-β acts on acute phase responses and is involved in postinjury repair to damage or infection [87]. IL-37 limits the action of innate inflammation, decreasing the production of proinflammatory cytokines, such as IL-1β and TNF-α [88].
3.3. Inflammaging: Molecular Pathways Implicated on the Cardiovascular System
The transcription factor NF-κB plays an important role in the control of several cellular processes such as immune response (innate and adaptive), inflammation, cell survival, proliferation, and apoptosis [89]. Although NF-κB is critical for physiological homeostasis, this transcription factor is superexpressed in aging, leading to a stage of chronic inflammation “inflammaging” [45], its overexpression being related to aging-related CVDs, such as hypertension and arteriosclerosis [90, 91].
NF-κB promotes the expression of various proinflammatory genes, including those coding of cytokines, chemokines, and adhesion molecules [92]. Cytokine production is often induced by molecular patterns associated with PAMPs and DAMPs, by which it acts through pattern recognition receptors (PRRs), such as TLRs and NOD-like receptors (NJRs) [93, 94]. DAMPS are released from the extracellular or intracellular spaces following cell death or tissue injury [95]. The most widely studied DAMPs are HMGB1, heat shock proteins (HSPs), and purine metabolites such as ATP and uric acid [96]. These DAMPs are recognized by macrophages, and inflammatory responses are triggered by different pathways, including TLRs and inflammasomes. Such a process is also involved in the pathogenesis of atherosclerosis due to macrophage recruitment as a result to arterial injury, which is rich in DAMPS [97].
Under physiological conditions, the activation of NF-κB in response to proinflammatory signals is short lived, and the reaction stops rapidly when the signal is finalized [92]. Briefly, the transcription factor NF-κB is bounded to its cytoplasmic inhibitor IκBα. PRRs use similar signal transduction mechanisms to activate IκB kinase (IKK), which is composed of two kinase subunits, IKKα and IKKβ, and a regulatory kinase subunit, IKKγ [98]. The IKK complex can be activated by several stimuli, such as growth or stress factors, which leads to the phosphorylation of IκBα, promoting its dissociation with NF-κB [99]. IκB is degraded via proteasome, while NF-κB is translocated to the nucleus, binding to its κB site for encoding specific genes [92].
Interestingly, many of the products encoded by NF-κB are activators of this transcription factor, generating a positive feedback, which means, the more inflammatory mediators produced, the greater is the expression of NF-κB [100]. This cycle is accompanied by an increase in ROS/RNS levels, generating a stress environment related to the development of age-related CVDs (Figure 5) [101, 102].
Figure 5.
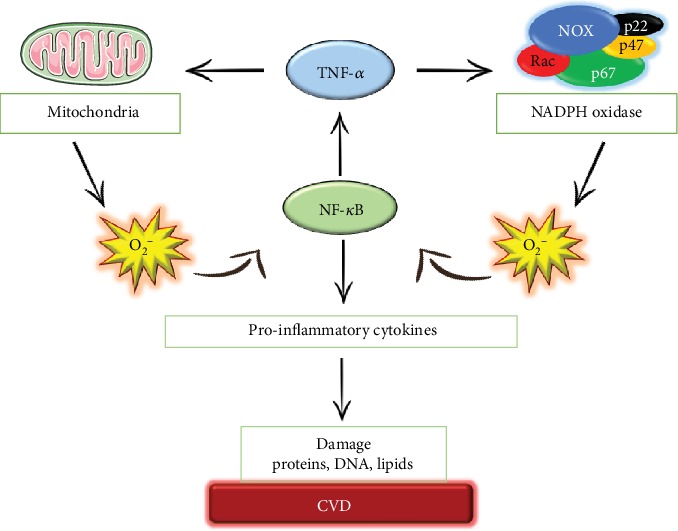
Many ROS/inflammation interactions are controlled by autoregulatory mechanisms. ROS produced by both NADPH oxidase and mitochondria induce proinflammatory release via NF-κB, such as TNF-α, which modulates an increase in NADPH oxidase and contributes to mitochondrial dysfunction, resulting in more ROS. The upregulation of these mechanisms contributes to cellular damage and eventually drives to cardiovascular diseases.
4. Reactive Oxygen/Nitrogen Species (ROS/RNS)
ROS/RNS are produced by all living organisms as a result of cellular metabolism [103]. Despite the organisms presenting their own antioxidant defences (e.g., enzymes, proteins, and vitamins) [104], ROS/RNS can accumulate with age due to its overproduction or failure in the antioxidant system, leading to the stage of “oxidative stress” [105]. Denham Harman, in 1956, proposed the “free radical theory of aging”, which argued that ROS derived from metabolism was the major cause of aging [106]. Since then, several publications have reported the deleterious effects of ROS/RNS on aging, as well as its relationship in the pathophysiology of CVDs [107–110].
4.1. Source of ROS/RNS
There are several sources of ROS/RNS present on the cardiovascular system, such as mitochondria and NADPH oxidases (NOX), the major sources of intracellular ROS/RNS [111]. Interestingly, both sources have interactions between them, which contribute to the gradual progression of oxidative stress [112, 113]. NOX also has its regulation increased by TNF-α [114], as well as activation of the AT1 receptor by the agonist angiotensin II [115], connecting, at least in part, oxidative stress with the inflammatory process and vascular dysfunction [116]. Other sources of ROS/RNS are uncoupled NO synthase, cytochrome p450, xanthine oxidase, the endoplasmic reticulum, peroxidases, cyclooxygenases, lipid oxidases, and some hemoproteins [117].
4.2. Examples of ROS/RNS
A range of molecules with oxidizing properties contributes to the regulation of cellular redox potential [118]. These ROS/RNS include superoxide anions (O2·−), radical hydroxyl (HO·), nitric oxide (NO·), and lipid radicals, which present unpaired electrons, being considered free radicals [119]. Other ROS/RNS such as hydrogen peroxide (H2O2), peroxynitrite (ONOO¯), and hypochlorous acid (HOCl) are not free radicals but have oxidizing effects that contribute to oxidative stress [120].
O2·− and H2O2 are produced enzymatically and are involved in physiological signalling as well as the pathologies associated with oxidative stress [121]. In addition, O2·− can be converted to H2O2 (less toxic) by superoxide dismutase (SOD) [122]. H2O2 acts as a second messenger, regulating redox signalling at homeostatic physiological levels [123–125]. Their levels ensure an adaptive response to stress, which is necessary for cellular survival [126, 127].
NO· is a potent vasodilator produced in the endothelium by eNOS activity [128, 129]. In addition, NO· has antiplatelet, antithrombotic, and anti-inflammatory functions [28]. Endothelial health is dependent of NO· levels [129, 130]. HO· and ONOO¯ are not considered signalling molecules due to their highly reactive nature, but they contribute significantly to oxidative stress and tissue damage [131]. While (HO·) is produced by the catalysis of H2O2, ONOO¯ is produced by the reaction between (O2·¯) and (NO·) [104]. High levels of ONOO¯ and depletion of NO· levels are related to endothelial dysfunction, an event present in the genesis of several CVDs [132].
4.3. Antioxidant System
In a homeostatic live system, ROS/RNS concentrations are controlled and preserved through enzymatic and nonenzymatic complexes of cellular detoxification, known as antioxidants [133, 134].
Nrf-2 (nuclear factor 2 related to erythroid factor 2), a transcription factor sensitive to redox potential, is the main regulator of the antioxidant system present on the cardiovascular system [135]. Under physiological conditions, Nrf-2 is downregulated in the cytoplasm by the Keap1 (Kelch-like ECH-associated protein-1) protein, leading to ubiquitination and consequent degradation via the proteasome [136–138]. Under stress conditions, Nrf-2 dissociates from the complex and translocates to the nucleus, binding to the AREs (antioxidant response elements), encoding the transcription of antioxidant enzymes and phase II detox, such as superoxide dismutase (SOD), catalase (CAT), glutathione peroxidase (GPx), glutathione reductase (GR), heme oxygenase-1 (HO-1), and NAD (P) H quinone oxidoreductase-1 (NQO1) [139, 140]. The cytoprotective effect of Nrf-2 is related to its antioxidant and anti-inflammatory properties [141, 142].
FOXO (Forkhead box O) is another family of transcription factors that present cytoprotective effects [143]. FOXO3, a member of the FOXO family, has been associated with the regulation of oxidative stress, attenuating ROS/RNS through the transcriptional activation of SOD and CAT [144]. Together, Nrf-2 and FOXO3 are the major transcription factors involved in cell protection [145, 146]. Thereby, they are very promising pharmacological targets for age-related CVDs [147–149].
5. Crosstalk between Nrf-2 and NF-κB
The change in the expression of Nrf-2/Keap1 and NF-κB is gradual and follow the course of cardiovascular aging [7]. In addition to acting individually on the redox signalling cascade, both interact with each other, guiding the communication between oxidative stress and inflammation (Figure 6) [123, 150].
Figure 6.

The crosstalk between Nrf-2 and NF-κB transcription factors. The stress environment caused by oxidative stress leads to functional modifications in both Nrf-2 and NF-κB transcription factors. Oxidation occurs at the thiol's sites of cysteine residues, allowing dissociation with their cytosolic inhibitors (Keap1 and IκB, respectively), leading to nucleus translocation and consequent encoding of target genes. Oxidative stress can interfere in both pathways indirectly by activating signalling pathways, such as ERK1/2, MAPK, JNK, and p38, which leads to the phosphorylation of these transcription factors. However, in basal situations, Nrf-2 and NF-κB are degraded via proteasome, which maintain homeostasis.
Transcription factors such as Nrf-2 and NF-κB are not directly oxidized but are translocated to the nucleus due to the oxidation of their cytosolic inhibitors (Keap1 and IκB, respectively), which are redox sensitive at their cysteine sites [151]. Keap1, which has its protein structure modified by the oxidation in the cysteine residues, loses the affinity for Nrf-2, allowing its accumulation in the cytoplasm and favouring its translocation to the nucleus [136, 137, 152].
HO-1, an antioxidant enzyme encoded by Nrf-2, mediates the inhibition of NF-κB [153]. Elevated levels of HO-1 in endothelial cells lead to decreased adhesion molecules mediated by NF-κB, such as E-selectin and VCAM-1 (vascular cell adhesion molecule 1) [150]. In addition, metabolites derived from HO-1 inhibit NF-κB translocation [154]. In contrast, excessive NF-κB expression showed lower HO-1 levels, revealing NF-κB suppressive activity, at least in part, on Nrf-2 transcription [155].
An oxidizing environment promotes the oxidation and degradation of IκB, favouring the activation of NF-κB [156]. On the other hand, a reducing environment has the opposite effect. This reducing environment is favoured by the action of GSH, an enzyme dependent on the Nrf-2 action. In fact, mice deficient in the Nrf-2 gene have a high susceptibility to high production of NF-κB-dependent proinflammatory agents, such as TNF-α, iNOS, and COX-2 [157].
Evidence shows the suppression of the transcriptional activity of Nrf-2 by NF-κB. The N-terminal region of the p65 (Rel-A) subunit of NF-κB binds physically to Keap1, promoting its translocation to the nucleus and consequent inhibition of the Nrf-2/ARE pathway [158]. In addition, NF-κB competes for CBP (CREB binding protein), a coactivator common to Nrf-2 near the ARE promoter region [159]. Thimmulappa and colleagues showed that knockdown mice for Nrf-2, in response to LPS, significantly increased the transcriptional activity of NF-κB [160]. Taken together, these studies suggest a tight connection between ROS/RNS and inflammation signalling, due in large part, to the many interactions evolving Nrf-2 and NF-κB pathways.
6. Age-Related Cardiovascular Diseases
Chronological age is the major risk factor for the development of CVDs [161]. In fact, this is due not only to the increase in conventional risk factors in the elderly but also to the independent and inevitable effect of aging itself [162]. The elderly, for example, are more susceptible to oxidative stress due to reduced efficiency of their endogenous antioxidant systems, favouring the activation of inflammatory pathways, which contributes to the gradual and chronic inflammation that accompanies aging [7]. These and other aging-induced changes reduce the functionality of the cardiovascular system by preventing stress response and cardioprotective interventions, making the individual more vulnerable to the development of CVDs [18].
6.1. Hypertension
Hypertension is characterized by persistent increase in blood pressure (BP), being one of the most common morbidities among the elderly [163]. It is a fact that this clinical condition increases due to age, which can be explained by the physiological and morphological changes in the cardiovascular system that occur during the aging process [164]. This clinical condition affects 1.13 million people worldwide and is considered one of the leading causes of death associated with chronic noncommunicable diseases [165] and a major risk factor for the development of cardiovascular complications such as stroke, AMI, heart failure, aortic aneurysm, and renal failure in this population [166]. A study by Mozaffarian and colleagues demonstrated that a high percentage of older people (70%) have hypertension compared with younger adults (32%) between 45 and 54 years [167].
The effect of aging on the vascular system is mainly marked by an increase in arterial thickness and stiffness, leading to a decrease in vascular tone [168]. These vascular changes contribute to increased procontractile response in vascular smooth muscle cells (CMLV), leading to increased BP [169].
Chronic inflammation and oxidative stress are common points between the biology of aging and CVDs [170]. Endothelial dysfunction—triggered by both processes—leads to a higher production of contracting factors (TXA2, PGH2, and ET-1) in detriment to the relaxing factors (NO, PGI2, and EDHF), contributing to a persistent vasoconstriction and consequent elevation of BP [171, 172]. This process leads to arterial stiffening due to collagen accumulation and a decrease in the amount of elastin, which decreases the ability of blood vessel to respond to stimuli [32].
Chaudhary and his group demonstrated that hypertensive patients over 60 years of age had a higher oxidative stress index and lower total blood antioxidant and mean arterial flow dilation rates than hypertensive patients under 60 years of age [173]. In addition, in EC, there is a change in proinflammatory phenotype triggering an increase in the expression of inflammatory cytokines, adhesion molecules, and chemokines that activate NF-κB signalling [60]. An increase in these factors leads to the infiltration of T cells and macrophages, which contributes to tissue damage [174]. In addition, increased cytokines such as interferon-γ, IL-1β, and TNF-α lead to increased oxidative stress in CMLV and EC [175]. Thus, it is noted that oxidative stress and the inflammatory process participate as essential components to affect the function of the vascular system, initiating a vicious cycle between increased BP, vascular remodelling, and stiffness, characterizing the state of continuous hypertension on the aging process [176].
6.2. Heart Failure
Heart failure (HF) is defined as a chronic and progressive clinical condition in which the heart cannot pump blood to meet the necessary tissue metabolic needs [177]. HF has an increased prevalence with age, with incidence rates <1% in individuals under 50 years, reaching 30% in individuals with advanced age (>80 years), being considered a disease of the elderly [178].
Morphological and functional changes in the heart due to aging explain the high rates of HF in old age [179]. The mechanisms involved behind these changes include (1) high levels of oxidative stress and inflammation, (2) high rate of apoptosis, (3) loss of regenerative capacity of cardiac progenitor cells, (4) hypertrophy of remaining cardiomyocytes, (5) loss in mitochondrial health, and (6) unbalance of calcium homeostasis. Such events are highly connected with HF [180–182].
Structural changes were observed regarding the reduction in the number of cardiomyocytes associated with their hypertrophy, collagen accumulation, and its metabolites, and functionally, it is possible to verify change in the maximum ejection fraction [183]. These hallmarks contribute to the aggravation of HF in elderly patients.
6.3. Arteriosclerosis-Atherosclerosis
Arteriosclerosis is defined by the formation and growth of plaques in the arterial lumen with consequent loss of vascular elasticity, leading to reduced blood flow in the affected vessel [184]. This clinical condition increases dramatically in the elderly, even in the absence of classical risk factors such as dyslipidemia [185]. Recently, atherosclerosis has come to be considered an inflammatory disease [186–188], affecting mainly the wall of large and medium arteries [189]. The formation and progression of atherosclerotic plaques support the “injury response” hypothesis of this disease, implying that plaque formation is a consequence of the local endothelial lesion associated with the inflammatory process [190, 191]. Metabolic syndrome, abdominal obesity, dyslipidemia, insulin resistance, hypertension, serum total and LDL cholesterol, smoking, and aging are related to the development of atherosclerotic lesions in arterial wall [184, 192].
The process to formation of atherosclerosis involves damage in vascular endothelial cells through oxidative stress, cytokines, and chemokines [193, 194]. These chemokines attract monocytes from blood circulation to the injured area which attach to the endothelium through interaction with adhesion molecules. Posteriorly, the monocytes enter in the subendothelial space, where it differentiate itself in macrophages who release cytokines. The high levels of low-density lipoprotein (LDL) infiltrate the vascular intima where it is oxidized. Then, the macrophages take up the oxidized LDL and form foam cells and atherogenesis. The vascular smooth muscle cells present in the media layer transform and migrate into the intima layer; after, these cells proliferate and produce extracellular matrix, where they also contribute to atherogenesis [195]. The rupture on this plaque leads to myocardial infarction and stroke.
ROS are involved in the development of atherosclerosis in a wide aspect. ROS facilitate inflammatory cell recruitment and lipid deposition in the intimal layer through the increase of adhesion molecules as ICAM-1 and VCAM-1. ROS also are related with the increase of proliferation of vascular smooth muscle cells [196, 197]. In addition, the ROS/RNS-rich environment associated with inflammation leads to elastin depletion, collagen accumulation, and immune recruitment, leading to disease progression [198, 199].
Proinflammatory cytokines also have an important role in atherosclerosis. They contribute to the formation of macrophages in the subendothelial space. Important cytokines such as TNF-α, IL-1, IL-6, IL-12, and NF-κB are related to atherosclerosis [193]. Kirii and colleagues evaluating the role of IL-1β, found, through experiments with mice, that the percentage of atherosclerotic lesions at aortic sinus was less in mice lacking IL-1β compared with wild mice [200]. Brånén and colleagues showed that mice lacking TNF-α had a reduction of 50% of the lesion compared to the control [201]. Childs and his group found in the experiments with mice that macrophages accumulate in the subendothelial space and increase the expression of inflammatory cytokines and chemokines [202]. Taken together, aging is an important factor to atherosclerosis because it accelerates the structural and compositional modifications of the vessel.
6.4. Myocardial Infarction
Myocardial infarction (MI) is the cardiovascular disease responsible for the highest incidence of mortality in old age [203]. MI occurs as a result of an interruption of blood supply to the heart, resulting in a lack of oxygen to the heart muscle (ischemia), which leads to the cardiomyocytes' death, triggered by coronary artery atherosclerosis [204]. Furthermore, this clinical issue highly increases with advancing of age. The mean age for the development of myocardial infarction is 65 years for men and 72 years for women [205].
Some studies show a tight connection between aging and its consequences for the genesis or aggravation of heart infarction. However, these studies are still scarce. It is established in the literature that aging is related to better heart vascularization, which prevents total organ ischemia in a patient from acute ischemic condition. On the other hand, aging leads the heart to damage, such as oxidative stress and inflammation, which have an influence on the development of ischemia and, ultimately, to MI.
The higher production of ROS in the cardiomyocytes is directly related to the sudden increase of Ca2+ in the cytoplasm, which directly contributes to the apoptosis and necrosis of ischemic cardiomyocytes, raising tissue damage. Waypa and colleagues demonstrated that hypoxia triggers increased intracellular Ca2+ concentration [206]. This effect is due to Ca2+ release from the sarcoplasmic reticulum and Kv inhibition, which triggers plasma membrane depolarization and Cav activation, allowing calcium influx to cytosol [207].
Both ROS and Ca2+ are capable of triggering the process of cellular apoptosis by opening the mitochondrial permeability transition pore (MPTP), which will lead to loss of mitochondrial shape, swelling, dissipation of membrane potential, and oxidative phosphorylation, in addition to the leakage of mitochondrial content [208, 209]. These factors together increase cell death in reperfusion, which, in short, increases the area of cardiac necrosis.
In animal infarction models, one of the most prominent findings is oxidative stress, which is detected by decreasing markers such as SOD and NO, plus the elevation of others such as serum MDA [210]. In addition to these classic markers, there is already evidence that other molecules are present as markers of oxidative stress in postinfarction patients, highlighting the decrease in serum thiol and disulphide levels in infarcted patients [211].
6.5. Stroke
Stroke is caused when there is an abrupt interruption in the blood supply to the brain that can lead to irreversible brain damage [212]. Age is the most important risk factor for developing a stroke [213] and considered the second leading cause of death worldwide [214]. With the aging of the population, as well as the increasing prevalence of underlying risk factors, there is an increased incidence of this disease, reaching about 69% of individuals over 65 years, and a prevalence of 34% in individuals over 75 years old [215].
The occurrence of acute stroke is closely related to atherosclerosis and hemodynamic changes. Blocking of the cerebral blood flow leads to hypoxia and glucose deprivation, activating different cascades of molecular signalling, which include depolarization of neurons, increased Ca2+ influx, ATP depletion, excitatory NT release [216], and increased expression of the hypoxia inducible factors (HIF) [217]. Increased activity of the glutamate receptor leads to an increase in intracellular Ca2+ levels, activation of NADPH oxidase signalling, mitochondrial dysfunction, and neuronal death [212]. The accumulation of HIF and ROS in neuronal cells generate different mechanisms involved in cell survival. HIF is related to neuroprotective effects in neuronal cells; however, in endothelial cells, it is related to the rupture of the blood brain barrier [217], whereas, ROS generate the activation of NF-κB, MAPK, and the upstream pathway MMPs, in addition to increasing the expression of VEGF and its receptors [218]. Thus, the increase of ROS is involved in different mechanisms in the pathophysiology of stroke, mainly affecting nutrient transport and tissue metabolism [219]. Neuronal cell death is the main injury caused by stroke, and its mechanism involves oxidative stress, inflammation, ischemia, hypoxia, calcium ion dysregulation, and apoptosis [220, 221].
7. Pharmacological Interventions
Research on therapies aimed at delaying aging and preventing age-related diseases, such as CVDs, have been a great interest [222] and are mainly related to the study of pharmacological tools used in clinical therapy as well as related to substances derived from natural products [223]. Many of them interact with oxidative stress and inflammation, delaying both conditions.
7.1. Antioxidants as Pharmacological Tools
Antioxidants usually scavenge ROS/RNS levels minimizing the oxidative damage caused by these molecules, plus reducing inflammation by downregulating the NF-κB pathway.
Exogenous antioxidants include ascorbic acid (vitamin C), which scavenges hydroxyl and superoxide radical anion, α-tocopherol (vitamin E), which is involved against lipid peroxidation of cell membranes, and phenolic antioxidants, which include stilbene derivatives (resveratrol, phenolic acids, and flavonoids), oil lectins, selenium, zinc, and drugs such as acetylcysteine [104].
Some vitamins have an important antioxidant function as vitamin A and its precursors β-carotene, vitamin C, and vitamin E [224]. Several large observational studies were conducted on the effect of intake of different vitamins and on the risk of CVDs, suggesting that higher intake of these vitamins significantly lowered the risk of these pathologic conditions [225]. Vitamin C is the major hydrophilic antioxidant and a powerful inhibitor of lipid peroxidation. In membranes, this molecule rapidly reduces 𝛼-tocopheroxyl radicals and LDL to regenerate 𝛼-tocopherol and inhibit propagation of free radicals. Vitamin E is the main hydrophobic antioxidant in cell membranes and circulating lipoproteins. Its antioxidant function is strongly supported by regeneration promoted by vitamin C. Vitamin E is thought to prevent atherosclerosis through inhibition of oxidative modification. Coenzyme Q (ubiquinol, CoQ) and lipoic acid in their reduced forms and melatonin are also efficient antioxidants [226].
In cardiovascular diseases, long-term treatment with antioxidants (vitamin C, vitamin E, coenzyme Q10, and selenium) significantly increased large and small artery elasticity in patients with multiple cardiovascular risk factors [227]. Li and colleagues showed the antioxidant benefits of individual and combined treatments of selenium, vitamin E, and purple carrot anthocyanins on D-galactose-induced oxidative damage in the blood, liver, heart, and kidney of rats [228].
Some polyphenols also play a key role in preventing CVDs. They are secondary metabolites derived from plants, characterized by the presence of an aromatic ring attached with a hydroxyl group [229]. Different studies have demonstrated the beneficial role of polyphenols, mainly related to the reduction of oxidative stress, which are implicated to the reduction of the incidence of age-related CVDs [10].
Quercetin, one of the most widely used polyphenols in the human diet, has been shown to be beneficial in preventing CVDs by acting on a wide variety of signalling pathways [230]. Studies have shown that quercetin has a protective effect on oxidative stress by reducing the activity of NADPH oxidase [231] and increase the levels of GSH, SOD, and catalase [232], in addition to reducing the expression of proinflammatory cytokines such as TNF-α and IL-1β [233].
Resveratrol also stands out as having a potent antioxidant action and has been proposed with a strong candidate in minimizing the effects associated with aging [234, 235]. Treatment with resveratrol in middle-aged rats has been shown to improve endothelial dysfunction associated with aging, probably by increasing NO and reducing ROS production [236]. In addition, resveratrol was able to reduce the production of inflammatory cytokines and chemokines through the inhibition of NF-κB, contributing to protection from age-related endothelial dysfunction [237]. Resveratrol supplementation in humans, aged 30 to 70 years, over a period of 8 weeks, showed a positive upregulation of the SOD and Nrf-2 genes [238]. Additional evidence shows that resveratrol promotes longevity and cardiac performance by causing hypoacetylation of proteins that control autophagy [239].
Curcumin, a yellow polyphenol found in Curcuma longa rhizome, has also been reported to have antiaging properties [240]. Recently, Santos-Parker and colleagues test 12-week supplementation with curcumin in healthy elderly. At the end of treatment, it was possible to observe the improvement of endothelial function due to an increase in NO bioavailability and reduction of oxidative stress [241]. Mechanistic studies have revealed superoxide dismutase, heme-oxygenase-1, and nuclear factor erythroid 2-related factor 2 as emerging targets for the beneficial effects of curcumin on the vasculature [242]. However, although different studies have already reported their antioxidant activity, additional studies are needed to elucidate the role of this polyphenol in the process of cardiovascular aging.
The literature studies have proven the molecular mechanisms of action of grape and red wine polyphenols against oxidative and inflammatory processes [229, 243, 244]. Polyphenols inhibit the phosphorylation of MAP kinases, causing a blocking effect on the transcription factors NF-κB and AP-1 and, consequently, blocking the synthesis of TNF-α, interleukins, chemokines, and molecule adhesion. Moreover, it can inhibit the activity of the cyclooxygenase and lipoxygenase enzymes [245]. Another proposed mechanism is the action of resveratrol on the activity of histone deacetylases, as SIRT-1 [246]. These actions together reduce the oxidation of LDL-c and the inflammatory process, attenuating CVDs [244, 247, 248].
Dietary supplementation with antioxidants has become popular. However, their biochemical mechanisms of protection against oxidative stress and antiaging effects are not fully understood. In addition, the antioxidants as pharmacological tools may be of great interest for future studies, especially in the promotion of healthy aging.
7.2. Metformin
Metformin is the most widely prescribed oral antidiabetic agent [249]. Mechanistically, metformin activates AMP-activated kinase (AMPK), a master regulator of metabolic homeostasis [250]. AMPK has been implicated in the control of autophagy, inflammation, mitochondrial dysfunction, and cell survival, being an important target for a healthy aging [251]. Regarding the cardiovascular system, treatment with metformin has been suggested as a potential target in the reversal of endothelial dysfunction, by inducing increased NO production mediated by MAPK through eNOS phosphorylation [252]. Moreover, metformin has been shown to improve endothelial function in vivo by reducing the production of superoxide anions through AMPK/PPARδ activation pathway [253]. Recent studies showed that activation of AMPK through metformin has also been shown to modulate the level of oxidative stress caused by hyperglycemia, which inhibits NADPH oxidase activation [254]. Moreover, metformin positively regulates the expression of GPx by reducing ROS formation through Nrf-2 activation [255].
In addition to the regulation of the oxidative stress, metformin is involved in the control of inflammation [256]. Its contribution to the reduction of inflammation is mediated by the release inhibition of IL-6 and IL-8 in smooth muscle cells, macrophages, and endothelial cells [257]. In addition, in smooth muscle cells receiving metformin treatment, the reduction in NF-κB activation and nuclear translocation was observed, as well as suppression of the proinflammatory phosphokinase AKT, p38, and ERK [257, 258].
7.3. Acetylsalicylic Acid (ASA)
ASA is considered one of the most promising substances for antiaging [222]. This molecule is indicated as antiplatelet therapy when used in low doses (75-100 mg daily) through the inhibition of hyperfunctional platelets, which contributes to prevent cardiovascular events such as thrombosis and atherosclerosis [259]. These effects are explained by COX-1 acetylation, leading to a reduction on thromboxane A2 production [260, 261]. In addition, ASA has an anti-inflammatory activity, through mechanisms involving COX inhibition and indirect modulation of the NF-κB pathway [260]. Bode-Böger and colleagues showed that ASA prevents endothelial senescence by improving NO release and reducing ROS levels [262].
From a clinical point of view, ASA has been demonstrated to reduce CVD mortality following long-term use and is currently being recommended for patients with postacute myocardial infarction [263, 264]. On the other hand, recent studies challenge the role of ASA on the prevention of cardiovascular events in aging [265]. Thus, futures studies are necessary to clarify the benefits of ASA in old age.
7.4. Statins
Statins are commonly used drugs for the treatment of dyslipidemia and, in particular, hypercholesterolemia due to their properties to reduce cholesterol synthesis and subsequently the LDL cholesterol level [266]. They are also recommended for primary and secondary prevention of cardiovascular disease. Clinical studies have shown that the use of statins reduces the risk of coronary syndrome and thromboembolic disease [267, 268]. The cardiovascular beneficial effect of statins has been attributable, at least in part, to their ability to reduce oxidative stress, to regulate the eNOS/NO pathway, and to reduce the level of inflammatory markers [269, 270]. Atorvastatin reduced also glyceraldehyde-derived formation of advanced glycation end products (AGEs) in patients with AMI, providing protection to the cardiovascular system [271]. Fluvastatin increased heme-oxygenase 1 (HO-1) expression and reduced AGE-induced proliferation of VSMC, probably by the upregulation of ERK5 through the Nrf-2 pathway [272]. Statins have also been shown to be important in reducing inflammatory markers [270]. Many of the pleiotropic properties of statins have been explained by their interference with the synthesis of isoprenoid intermediates [273]. The statin-induced inhibition of prenylation of Ras protein has been linked to the reduction of the inflammatory marker C-reactive protein (CRP) [274]. These effects of statins can contribute to reduce cardiovascular diseases, especially on the elderly population.
7.5. Rapamycin
Rapamycin is an inhibitor of mTOR, traditionally used as an immunosuppressant; however, recent studies show its use associated with cardioprotective effects related to the aging process [275, 276]. Lesniewski and colleagues showed that 6-8-week treatment with rapamycin in elderly mice has been shown to improve endothelial dysfunction by mTOR inhibition, plus the increase of NO bioavailability and reducing NADPH oxidase expression, which ameliorated oxidative stress [277]. Evaluating the effect of rapamycin treatment on middle-aged mice, rapamycin showed reduced mitochondrial ROS production and increased the gene expression of different endogenous antioxidants, such as SOD and GSH reductase [278, 279]. It was further noted that inhibition of mTOR mediated by rapamycin protected aged endothelial cells from oxidative stress and apoptosis induced by low shear stress [280]. In addition to the effects on oxidative stress, evidence shows that treatment with rapamycin can negatively modulate the inflammatory processes by reducing TNF-α expression and genes involved in NF-κB signalling [281]. Therefore, treatment with rapamycin has shown promise in the prevention of age-related CVDs [282].
8. Conclusions and Future Directions
With the increase in global life expectancy, studies on human aging have recently gained notoriety. Researchers have noted that activating longevity genes could prevent/combat many age-related diseases, increasing the health years of life “health span”. In fact, there is a bridge between the molecular mechanisms of aging and those that drive to chronic diseases, such as cardiovascular diseases. Two key points that circulate in the bridge are oxidative stress and persistent inflammation. Although its deleterious effects have been massively described, there is currently a search for understanding the reason for oxidative stress and persistent inflammation increase in aging and CVDs. In turn, persistent inflammation has several causes, which leads to damage on the cardiovascular system. An observed paradox occurs in the centenarians, which present high levels of persistent inflammation, but without suffering from its harmful effects. Such observations lead us to believe that the increase of oxidative stress and the inflammatory process has a beneficial proposal. However, it persistently leads to the disorders related with the development of CVDs in old age.
In this perspective, therapies that act in the control of oxidative stress and inflammation, however, without inhibiting their physiological functions are promising targets in the search for a healthy aging.
Acknowledgments
Financial supports from Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), Instituto UFPB de Desenvolvimento da Paraíba (IDEP), and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, Grant number 311711/2018-9) are gratefully acknowledged.
Disclosure
The authors are responsible for the content and writing of this paper.
Conflicts of Interest
The authors report no conflict of interests.
Authors' Contributions
The authors equally contributed to the composition of the manuscript.
References
- 1.Brown G. C. Living too long: The current focus of medical research on increasing the quantity, rather than the quality, of life is damaging our health and harming the economy. EMBO Reports. 2014;16(2):137–141. doi: 10.15252/embr.201439518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.UN. HelpAge, Global AgeWatch Index 2015: Insight Report. London, UK: HelpAge International; 2015. UNDESA Population Division, World Population Prospects: the 2015 Revision. [Google Scholar]
- 3.Franceschi C., Garagnani P., Morsiani C., et al. The continuum of aging and age-related diseases: common mechanisms but different rates. Frontiers in Medicine. 2018;5:p. 61. doi: 10.3389/fmed.2018.00061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jaul E., Barron J. Age-related diseases and clinical and public health implications for the 85 years old and over population. Frontiers in Public Health. 2017;5:335–335. doi: 10.3389/fpubh.2017.00335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Niccoli T., Partridge L. Ageing as a risk factor for disease. Current Biology. 2012;22(17):R741–R752. doi: 10.1016/j.cub.2012.07.024. [DOI] [PubMed] [Google Scholar]
- 6.López-Otín C., Blasco M. A., Partridge L., Serrano M., Kroemer G. The hallmarks of aging. Cell. 2013;153(6):1194–1217. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Almeida A. J. P. O. D., Ribeiro T. P., Medeiros I. A. . Aging: molecular pathways and implications on the cardiovascular system. Oxidative Medicine and Cellular Longevity. 2017;2017:19. doi: 10.1155/2017/7941563.7941563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Riera C. E., Merkwirth C., de Magalhaes Filho C. D., Dillin A. Signaling Networks Determining Life Span. Annual Review of Biochemistry. 2016;85(1):35–64. doi: 10.1146/annurev-biochem-060815-014451. [DOI] [PubMed] [Google Scholar]
- 9.Franceschi C., Campisi J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. The Journals of Gerontology Series A: Biological Sciences and Medical Sciences. 2014;69(Supplement 1):S4–S9. doi: 10.1093/gerona/glu057. [DOI] [PubMed] [Google Scholar]
- 10.Liguori I., Russo G., Curcio F., et al. Oxidative stress, aging, and diseases. Clinical Interventions in Aging. 2018;13:757–772. doi: 10.2147/CIA.S158513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fajemiroye J. O., Cunha L. C. ., Saavedra-Rodríguez R., et al. Aging-induced biological changes and cardiovascular diseases. BioMed Research International. 2018;2018:14. doi: 10.1155/2018/7156435.7156435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.North B. J., Sinclair D. A. The intersection between aging and cardiovascular disease. Circulation Research. 2012;110(8):1097–1108. doi: 10.1161/CIRCRESAHA.111.246876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ferrucci L., Fabbri E. Inflammageing: chronic inflammation in ageing, cardiovascular disease, and frailty. Nature Reviews Cardiology. 2018;15(9):505–522. doi: 10.1038/s41569-018-0064-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gude N. A., Broughton K. M., Firouzi F., Sussman M. A. Cardiac ageing: extrinsic and intrinsic factors in cellular renewal and senescence. Nature Reviews Cardiology. 2018;15(9):523–542. doi: 10.1038/s41569-018-0061-5. [DOI] [PubMed] [Google Scholar]
- 15.Laina A., Stellos K., Stamatelopoulos K. Vascular ageing: underlying mechanisms and clinical implications. Experimental Gerontology. 2018;109:16–30. doi: 10.1016/j.exger.2017.06.007. [DOI] [PubMed] [Google Scholar]
- 16.Meschiari C. A., Ero O. K., Pan H., Finkel T., Lindsey M. L. The impact of aging on cardiac extracellular matrix. GeroScience. 2017;39(1):7–18. doi: 10.1007/s11357-017-9959-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oh Y. J., Pau V. C., Steppan J., et al. Role of tissue transglutaminase in age-associated ventricular stiffness. Amino Acids. 2017;49(3):695–704. doi: 10.1007/s00726-016-2295-z. [DOI] [PubMed] [Google Scholar]
- 18.Boudina S. Cardiac aging and insulin resistance: could insulin/insulin-like growth factor (IGF) signaling be used as a therapeutic target? Current Pharmaceutical Design. 2013;19(32):5684–5694. doi: 10.2174/1381612811319320004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Matsushima S., Sadoshima J. The role of sirtuins in cardiac disease. American Journal of Physiology-Heart and Circulatory Physiology. 2015;309(9):H1375–H1389. doi: 10.1152/ajpheart.00053.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yoon H. E., Kim E. N., Kim M. Y., et al. Age-associated changes in the vascular renin-angiotensin system in mice. Oxidative Medicine and Cellular Longevity. 2016;2016:14. doi: 10.1155/2016/6731093.6731093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Conti S., Cassis P., Benigni A. Aging and the renin-angiotensin system. Hypertension. 2012;60(4):878–883. doi: 10.1161/HYPERTENSIONAHA.110.155895. [DOI] [PubMed] [Google Scholar]
- 22.Strait J. B., Lakatta E. G. Aging-associated cardiovascular changes and their relationship to heart failure. Heart Failure Clinics. 2012;8(1):143–164. doi: 10.1016/j.hfc.2011.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang M., Jiang L., Monticone R. E., Lakatta E. G. Proinflammation: the key to arterial aging. Trends in Endocrinology and Metabolism: TEM. 2014;25(2):72–79. doi: 10.1016/j.tem.2013.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu X., Wang B., Ren C., et al. Age-related impairment of vascular structure and functions. Aging and Disease. 2017;8(5):590–610. doi: 10.14336/AD.2017.0430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Harvey A., Montezano A. C., Touyz R. M. Vascular biology of ageing—Implications in hypertension. Journal of Molecular and Cellular Cardiology. 2015;83:112–121. doi: 10.1016/j.yjmcc.2015.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu X., Wang B., Ren C., et al. Recent progress in vascular aging: mechanisms and its role in age-related diseases. Aging and disease. 2017;8(4):486–505. doi: 10.14336/AD.2017.0507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mazurek R., Dave J. M., Chandran R. R., Misra A., Sheikh A. Q., Greif D. M. Chapter eight - Vascular cells in blood vessel wall development and disease. Advances in Pharmacology. 2017;78:323–350. doi: 10.1016/bs.apha.2016.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhao Y., Vanhoutte P. M., Leung S. W. S. Vascular nitric oxide: beyond eNOS. Journal of Pharmacological Sciences. 2015;129(2):83–94. doi: 10.1016/j.jphs.2015.09.002. [DOI] [PubMed] [Google Scholar]
- 29.Seals D. R., Jablonski K. L., Donato A. J. Aging and vascular endothelial function in humans. Clinical Science. 2011;120(9):357–375. doi: 10.1042/CS20100476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sepúlveda C., Palomo I., Fuentes E. Mechanisms of endothelial dysfunction during aging: predisposition to thrombosis. Mechanisms of Ageing and Development. 2017;164:91–99. doi: 10.1016/j.mad.2017.04.011. [DOI] [PubMed] [Google Scholar]
- 31.Herrera M. D., Mingorance C., Rodríguez-Rodríguez R., Alvarez de Sotomayor M. Endothelial dysfunction and aging: an update. Ageing Research Reviews. 2010;9(2):142–152. doi: 10.1016/j.arr.2009.07.002. [DOI] [PubMed] [Google Scholar]
- 32.Tesauro M., Mauriello A., Rovella V., et al. Arterial ageing: from endothelial dysfunction to vascular calcification. Journal of Internal Medicine. 2017;281(5):471–482. doi: 10.1111/joim.12605. [DOI] [PubMed] [Google Scholar]
- 33.Trindade M., Oigman W., Fritsch Neves M. Potential role of endothelin in early vascular aging. Current Hypertension Reviews. 2017;13(1):33–40. doi: 10.2174/1573402113666170414165735. [DOI] [PubMed] [Google Scholar]
- 34.Neves M. F., Cunha A. R., Cunha M. R., Gismondi R. A., Oigman W. The role of renin–angiotensin–aldosterone system and its new components in arterial stiffness and vascular aging. High Blood Pressure & Cardiovascular Prevention. 2018;25(2):137–145. doi: 10.1007/s40292-018-0252-5. [DOI] [PubMed] [Google Scholar]
- 35.Duca L., Blaise S., Romier B., et al. Matrix ageing and vascular impacts: focus on elastin fragmentation. Cardiovascular Research. 2016;110(3):298–308. doi: 10.1093/cvr/cvw061. [DOI] [PubMed] [Google Scholar]
- 36.Wang M., Kim S. H., Monticone R. E., Lakatta E. G. Matrix metalloproteinases promote arterial remodeling in aging, hypertension, and atherosclerosis. Hypertension. 2015;65(4):698–703. doi: 10.1161/HYPERTENSIONAHA.114.03618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sanada F., Taniyama Y., Muratsu J., et al. Source of chronic inflammation in aging. Frontiers in Cardiovascular Medicine. 2018;5:p. 12. doi: 10.3389/fcvm.2018.00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Frangogiannis N. G. The inflammatory response in myocardial injury, repair, and remodelling. Nature Reviews Cardiology. 2014;11(5):255–265. doi: 10.1038/nrcardio.2014.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Prabhu S. D., Frangogiannis N. G. The biological basis for cardiac repair after myocardial infarction. Circulation Research. 2016;119(1):91–112. doi: 10.1161/CIRCRESAHA.116.303577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Libby P. Inflammation and cardiovascular disease mechanisms. The American Journal of Clinical Nutrition. 2006;83(2):456S–460S. doi: 10.1093/ajcn/83.2.456S. [DOI] [PubMed] [Google Scholar]
- 41.Fulop T., Witkowski J. M., Olivieri F., Larbi A. The integration of inflammaging in age-related diseases. Seminars in Immunology. 2018;40:17–35. doi: 10.1016/j.smim.2018.09.003. [DOI] [PubMed] [Google Scholar]
- 42.Franceschi C., Ostan R., Santoro A. Nutrition and inflammation: are centenarians similar to individuals on calorie-restricted diets? Annual Review of Nutrition. 2018;38(1):329–356. doi: 10.1146/annurev-nutr-082117-051637. [DOI] [PubMed] [Google Scholar]
- 43.Arai Y., Martin-Ruiz C. M., Takayama M., et al. Inflammation, but not telomere length, predicts successful ageing at extreme old age: a longitudinal study of semi-supercentenarians. EBioMedicine. 2015;2(10):1549–1558. doi: 10.1016/j.ebiom.2015.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Storci G., De Carolis S., Papi A., et al. Genomic stability, anti-inflammatory phenotype, and up-regulation of the RNAseH2 in cells from centenarians. Cell Death & Differentiation. 2019;26(9):1845–1858. doi: 10.1038/s41418-018-0255-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Franceschi C., Garagnani P., Vitale G., Capri M., Salvioli S. Inflammaging and ‘Garb-aging’. Trends in Endocrinology & Metabolism. 2017;28(3):199–212. doi: 10.1016/j.tem.2016.09.005. [DOI] [PubMed] [Google Scholar]
- 46.He Y., Hara H., Núñez G. Mechanism and regulation of NLRP3 inflammasome activation. Trends in Biochemical Sciences. 2016;41(12):1012–1021. doi: 10.1016/j.tibs.2016.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Latz E., Duewell P. NLRP3 inflammasome activation in inflammaging. Seminars in Immunology. 2018;40:61–73. doi: 10.1016/j.smim.2018.09.001. [DOI] [PubMed] [Google Scholar]
- 48.Cordero M. D., Williams M. R., Ryffel B. AMP-activated protein kinase regulation of the NLRP3 inflammasome during aging. Trends in Endocrinology & Metabolism. 2018;29(1):8–17. doi: 10.1016/j.tem.2017.10.009. [DOI] [PubMed] [Google Scholar]
- 49.Zhou R., Yazdi A. S., Menu P., Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469(7329):221–225. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
- 50.Gurung P., Lukens J. R., Kanneganti T.-D. Mitochondria: diversity in the regulation of the NLRP3 inflammasome. Trends in Molecular Medicine. 2015;21(3):193–201. doi: 10.1016/j.molmed.2014.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shi C.-S., Shenderov K., Huang N.-N., et al. Activation of autophagy by inflammatory signals limits IL-1β production by targeting ubiquitinated inflammasomes for destruction. Nature Immunology. 2012;13(3):255–263. doi: 10.1038/ni.2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Marín‐Aguilar F., Lechuga‐Vieco A. V., Alcocer‐Gómez E., et al. NLRP3 inflammasome suppression improves longevity and prevents cardiac aging in male mice. Aging Cell. 2019;19(1, article e13050) doi: 10.1111/acel.13050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Freund A., Orjalo A. V., Desprez P.-Y., Campisi J. Inflammatory networks during cellular senescence: causes and consequences. Trends in Molecular Medicine. 2010;16(5):238–246. doi: 10.1016/j.molmed.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Burton D. G. A., Stolzing A. Cellular senescence: immunosurveillance and future immunotherapy. Ageing Research Reviews. 2018;43:17–25. doi: 10.1016/j.arr.2018.02.001. [DOI] [PubMed] [Google Scholar]
- 55.Fulop T., Larbi A., Dupuis G., et al. Immunosenescence and inflamm-aging as two sides of the same coin: friends or foes? Frontiers in Immunology. 2018;8, article 1960 doi: 10.3389/fimmu.2017.01960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vicente R., Mausset-Bonnefont A.-L., Jorgensen C., Louis-Plence P., Brondello J.-M. Cellular senescence impact on immune cell fate and function. Aging Cell. 2016;15(3):400–406. doi: 10.1111/acel.12455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Olivieri F., Prattichizzo F., Grillari J., Balistreri C. R. Cellular senescence and inflammaging in age-related diseases. Mediators of Inflammation. 2018;2018:6. doi: 10.1155/2018/9076485.9076485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rauscher F. M., Goldschmidt-Clermont P. J., Davis B. H., et al. Aging, progenitor cell exhaustion, and atherosclerosis. Circulation. 2003;108(4):457–463. doi: 10.1161/01.CIR.0000082924.75945.48. [DOI] [PubMed] [Google Scholar]
- 59.Goligorsky M. S. Endothelial progenitor cells: from senescence to rejuvenation. Seminars in Nephrology. 2014;34(4):365–373. doi: 10.1016/j.semnephrol.2014.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jia G., Aroor A. R., Jia C., Sowers J. R. Endothelial cell senescence in aging-related vascular dysfunction. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 2019;1865(7):1802–1809. doi: 10.1016/j.bbadis.2018.08.008. [DOI] [PubMed] [Google Scholar]
- 61.Rossman M. J., Kaplon R. E., Hill S. D., et al. Endothelial cell senescence with aging in healthy humans: prevention by habitual exercise and relation to vascular endothelial function. American Journal of Physiology-Heart and Circulatory Physiology. 2017;313(5):H890–H895. doi: 10.1152/ajpheart.00416.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Williamson K., Stringer S. E., Alexander M. Y. Endothelial progenitor cells enter the aging arena. Frontiers in Physiology. 2012;3:p. 30. doi: 10.3389/fphys.2012.00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Baker D. J., Wijshake T., Tchkonia T., et al. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature. 2011;479(7372):232–236. doi: 10.1038/nature10600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Childs B. G., Li H., van Deursen J. M. Senescent cells: a therapeutic target for cardiovascular disease. The Journal of Clinical Investigation. 2018;128(4):1217–1228. doi: 10.1172/JCI95146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.McHugh D., Gil J. Senescence and aging: causes, consequences, and therapeutic avenues. The Journal of Cell Biology. 2018;217(1):65–77. doi: 10.1083/jcb.201708092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kim S., Jazwinski S. M. The gut microbiota and healthy aging: a mini-review. Gerontology. 2018;64(6):513–520. doi: 10.1159/000490615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nagpal R., Mainali R., Ahmadi S., et al. Gut microbiome and aging: physiological and mechanistic insights. Nutrition and Healthy Aging. 2018;4(4):267–285. doi: 10.3233/NHA-170030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Santoro A., Ostan R., Candela M., et al. Gut microbiota changes in the extreme decades of human life: a focus on centenarians. Cellular and Molecular Life Sciences. 2018;75(1):129–148. doi: 10.1007/s00018-017-2674-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Claesson M. J., Jeffery I. B., Conde S., et al. Gut microbiota composition correlates with diet and health in the elderly. Nature. 2012;488(7410):178–184. doi: 10.1038/nature11319. [DOI] [PubMed] [Google Scholar]
- 70.Man A. L., Gicheva N., Nicoletti C. The impact of ageing on the intestinal epithelial barrier and immune system. Cellular Immunology. 2014;289(1-2):112–118. doi: 10.1016/j.cellimm.2014.04.001. [DOI] [PubMed] [Google Scholar]
- 71.Takiishi T., Fenero C. I. M., Câmara N. O. S. Intestinal barrier and gut microbiota: shaping our immune responses throughout life. Tissue Barriers. 2017;5(4, article e1373208) doi: 10.1080/21688370.2017.1373208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kumar M., Babaei P., Ji B., Nielsen J. Human gut microbiota and healthy aging: recent developments and future prospective. Nutrition and Healthy Aging. 2016;4(1):3–16. doi: 10.3233/NHA-150002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mari D., Mannucci P., Coppola R., Bottasso B., Bauer K., Rosenberg R. Hypercoagulability in centenarians: the paradox of successful aging. Blood. 1995;85(11):3144–3149. doi: 10.1182/blood.V85.11.3144.bloodjournal85113144. [DOI] [PubMed] [Google Scholar]
- 74.O'Brien M. The reciprocal relationship between inflammation and coagulation. Topics in Companion Animal Medicine. 2012;27(2):46–52. doi: 10.1053/j.tcam.2012.06.003. [DOI] [PubMed] [Google Scholar]
- 75.Michaud M., Balardy L., Moulis G., et al. Proinflammatory cytokines, aging, and age-related diseases. Journal of the American Medical Directors Association. 2013;14(12):877–882. doi: 10.1016/j.jamda.2013.05.009. [DOI] [PubMed] [Google Scholar]
- 76.Rea I. M., Gibson D. S., McGilligan V., McNerlan S. E., Alexander H. D., Ross O. A. Age and age-related diseases: role of inflammation triggers and cytokines. Frontiers in immunology. 2018;9:p. 586. doi: 10.3389/fimmu.2018.00586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tzoran I., Hoffman R., Monreal M. Hemostasis and thrombosis in the oldest old. Seminars in Thrombosis and Hemostasis. 2018;44(7):624–631. doi: 10.1055/s-0038-1657779. [DOI] [PubMed] [Google Scholar]
- 78.Kreidy R. Influence of acquired and genetic risk factors on the prevention, management, and treatment of thromboembolic disease. International Journal of Vascular Medicine. 2014;2014:5. doi: 10.1155/2014/859726.859726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nathan C., Ding A. Nonresolving inflammation. Cell. 2010;140(6):871–882. doi: 10.1016/j.cell.2010.02.029. [DOI] [PubMed] [Google Scholar]
- 80.Xia S., Zhang X., Zheng S., et al. An update on inflamm-aging: mechanisms, prevention, and treatment. Journal of Immunology Research. 2016;2016:12. doi: 10.1155/2016/8426874.8426874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Minciullo P. L., Catalano A., Mandraffino G., et al. Inflammaging and anti-inflammaging: the role of cytokines in extreme longevity. Archivum Immunologiae et Therapiae Experimentalis. 2016;64(2):111–126. doi: 10.1007/s00005-015-0377-3. [DOI] [PubMed] [Google Scholar]
- 82.Banerjee S., Biehl A., Gadina M., Hasni S., Schwartz D. M. JAK–STAT Signaling as a Target for Inflammatory and Autoimmune Diseases: Current and Future Prospects. Drugs. 2017;77(5):521–546. doi: 10.1007/s40265-017-0701-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Frazier W. J., Xue J., Luce W. A., Liu Y. MAPK signaling drives inflammation in LPS-stimulated cardiomyocytes: the route of crosstalk to G-protein-coupled receptors. PLoS One. 2012;7(11, article e50071) doi: 10.1371/journal.pone.0050071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Jurk D., Wilson C., Passos J. F., et al. Chronic inflammation induces telomere dysfunction and accelerates ageing in mice. Nature Communications. 2014;5(1, article 4172) doi: 10.1038/ncomms5172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.El Assar M., Angulo J., Vallejo S., Peiró C., Sánchez-Ferrer C. F., Rodríguez-Mañas L. Mechanisms involved in the aging-induced vascular dysfunction. Frontiers in Physiology. 2012;3:p. 132. doi: 10.3389/fphys.2012.00132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kinzenbaw D. A., Chu Y., Peña Silva R. A., Didion S. P., Faraci F. M. Interleukin-10 protects against aging-induced endothelial dysfunction. Physiological Reports. 2013;1(6, article e00149) doi: 10.1002/phy2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Krieglstein K., Miyazono K., ten Dijke P., Unsicker K. TGF-β in aging and disease. Cell and Tissue Research. 2012;347(1):5–9. doi: 10.1007/s00441-011-1278-3. [DOI] [PubMed] [Google Scholar]
- 88.Sapinsley Z. J., Ballak D. B., Brunt V. E., et al. Administration of anti-inflammatory interleukin-37 ameliorates age-related vascular, metabolic and physical dysfunction in mice. 2017;31(1_Supplement)1079.1073 [Google Scholar]
- 89.Taniguchi K., Karin M. NF-κB, inflammation, immunity and cancer: coming of age. Nature Reviews Immunology. 2018;18(5):309–324. doi: 10.1038/nri.2017.142. [DOI] [PubMed] [Google Scholar]
- 90.Tilstra J. S., Clauson C. L., Niedernhofer L. J., Robbins P. D. NF-κB in aging and disease. Aging and Disease. 2011;2(6):449–465. [PMC free article] [PubMed] [Google Scholar]
- 91.Lopez-Candales A., Hernández Burgos P. M., Hernandez-Suarez D. F., Harris D. Linking chronic inflammation with cardiovascular disease: from normal aging to the metabolic syndrome. Journal of Nature and Science. 2017;3(4):p. e341. [PMC free article] [PubMed] [Google Scholar]
- 92.Liu T., Zhang L., Joo D., Sun S.-C. NF-κB signaling in inflammation. Signal Transduction and Targeted Therapy. 2017;2(1, article 17023) doi: 10.1038/sigtrans.2017.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Balistreri C. R., Candore G., Accardi G., Colonna-Romano G., Lio D. NF-κB pathway activators as potential ageing biomarkers: targets for new therapeutic strategies. Immunity & Ageing. 2013;10(1):p. 24. doi: 10.1186/1742-4933-10-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kumar H., Kawai T., Akira S. Pathogen recognition by the innate immune system. International Reviews of Immunology. 2011;30(1):16–34. doi: 10.3109/08830185.2010.529976. [DOI] [PubMed] [Google Scholar]
- 95.Roh J. S., Sohn D. H. Damage-associated molecular patterns in inflammatory diseases. Immune Network. 2018;18(4):e27–e27. doi: 10.4110/in.2018.18.e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Feldman N., Rotter-Maskowitz A., Okun E. DAMPs as mediators of sterile inflammation in aging-related pathologies. Ageing Research Reviews. 2015;24(Part A):29–39. doi: 10.1016/j.arr.2015.01.003. [DOI] [PubMed] [Google Scholar]
- 97.Hansson G. K., Hermansson A. The immune system in atherosclerosis. Nature Immunology. 2011;12(3):204–212. doi: 10.1038/ni.2001. [DOI] [PubMed] [Google Scholar]
- 98.Zhao J., Li X., McGowan S., Niedernhofer L. J., Robbins P. D. NF-κB activation with aging: characterization and therapeutic inhibition. In: May M. J., editor. NF-kappa B. New York, NY USA: Springer New York; 2015. pp. 543–557. [DOI] [PubMed] [Google Scholar]
- 99.Hoesel B., Schmid J. A. The complexity of NF-κB signaling in inflammation and cancer. Molecular Cancer. 2013;12(1):86–86. doi: 10.1186/1476-4598-12-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lawrence T. The nuclear factor NF-κB pathway in inflammation. Cold Spring Harbor Perspectives in Biology. 2009;1(6, article a001651) doi: 10.1101/cshperspect.a001651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ruparelia N., Chai J. T., Fisher E. A., Choudhury R. P. Inflammatory processes in cardiovascular disease: a route to targeted therapies. Nature Reviews Cardiology. 2017;14(3):133–144. doi: 10.1038/nrcardio.2016.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wu J., Xia S., Kalionis B., Wan W., Sun T. The role of oxidative stress and inflammation in cardiovascular aging. BioMed Research International. 2014;2014:13. doi: 10.1155/2014/615312.615312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Schieber M., Chandel N. S. ROS function in redox signaling and oxidative stress. Current Biology. 2014;24(10):R453–R462. doi: 10.1016/j.cub.2014.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Pisoschi A. M., Pop A. The role of antioxidants in the chemistry of oxidative stress: a review. European Journal of Medicinal Chemistry. 2015;97:55–74. doi: 10.1016/j.ejmech.2015.04.040. [DOI] [PubMed] [Google Scholar]
- 105.Sies H. Oxidative stress: a concept in redox biology and medicine. Redox Biology. 2015;4:180–183. doi: 10.1016/j.redox.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Harman D. Aging: a theory based on free radical and radiation chemistry. Journal of Gerontology. 1956;11(3):298–300. doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- 107.Sena C. M., Leandro A., Azul L., Seiça R., Perry G. Vascular oxidative stress: impact and therapeutic approaches. Frontiers in Physiology. 2018;9, article 1668 doi: 10.3389/fphys.2018.01668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kattoor A. J., Pothineni N. V. K., Palagiri D., Mehta J. L. Oxidative stress in atherosclerosis. Current Atherosclerosis Reports. 2017;19(11):p. 42. doi: 10.1007/s11883-017-0678-6. [DOI] [PubMed] [Google Scholar]
- 109.Rahal A., Kumar A., Singh V., et al. Oxidative stress, prooxidants, and antioxidants: the interplay. BioMed Research International. 2014;2014:19. doi: 10.1155/2014/761264.761264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Panth N., Paudel K. R., Parajuli K. Reactive oxygen species: a key hallmark of cardiovascular disease. Advances in Medicine. 2016;2016:12. doi: 10.1155/2016/9152732.9152732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Holmström K. M., Finkel T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nature Reviews Molecular Cell Biology. 2014;15(6):411–421. doi: 10.1038/nrm3801. [DOI] [PubMed] [Google Scholar]
- 112.Daiber A., Di Lisa F., Oelze M., et al. Crosstalk of mitochondria with NADPH oxidase via reactive oxygen and nitrogen species signalling and its role for vascular function. British Journal of Pharmacology. 2017;174(12):1670–1689. doi: 10.1111/bph.13403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Dikalov S. Cross talk between mitochondria and NADPH oxidases. Free Radical Biology and Medicine. 2011;51(7):1289–1301. doi: 10.1016/j.freeradbiomed.2011.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Sriramula S., Francis J. Tumor necrosis factor - alpha is essential for angiotensin II-induced ventricular remodeling: role for oxidative stress. PLOS One. 2015;10(9, article e0138372) doi: 10.1371/journal.pone.0138372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Montezano A. C., Nguyen Dinh Cat A., Rios F. J., Touyz R. M. Angiotensin II and vascular injury. Current Hypertension Reports. 2014;16(6):p. 431. doi: 10.1007/s11906-014-0431-2. [DOI] [PubMed] [Google Scholar]
- 116.Konior A., Schramm A., Czesnikiewicz-Guzik M., Guzik T. J. NADPH oxidases in vascular pathology. Antioxidants & Redox Signaling. 2014;20(17):2794–2814. doi: 10.1089/ars.2013.5607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Cai H., Harrison D. G. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circulation Research. 2000;87(10):840–844. doi: 10.1161/01.RES.87.10.840. [DOI] [PubMed] [Google Scholar]
- 118.Sies H., Berndt C., Jones D. P. Oxidative Stress. Annual Review of Biochemistry. 2017;86(1):715–748. doi: 10.1146/annurev-biochem-061516-045037. [DOI] [PubMed] [Google Scholar]
- 119.Cervantes Gracia K., Llanas-Cornejo D., Husi H. CVD and oxidative stress. Journal of Clinical Medicine. 2017;6(2):p. 22. doi: 10.3390/jcm6020022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Phaniendra A., Jestadi D. B., Periyasamy L. Free radicals: properties, sources, targets, and their implication in various diseases. Indian Journal of Clinical Biochemistry: IJCB. 2015;30(1):11–26. doi: 10.1007/s12291-014-0446-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Taverne Y. J. H. J., Bogers A. J. J. C., Duncker D. J., Merkus D. Reactive oxygen species and the cardiovascular system. Oxidative Medicine and Cellular Longevity. 2013;2013:15. doi: 10.1155/2013/862423.862423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wang Y., Branicky R., Noë A., Hekimi S. Superoxide dismutases: dual roles in controlling ROS damage and regulating ROS signaling. Journal of Cell Biology. 2018;217(6):1915–1928. doi: 10.1083/jcb.201708007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Sies H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: oxidative eustress. Redox Biology. 2017;11:613–619. doi: 10.1016/j.redox.2016.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Sies H. Role of Metabolic H2O2 Generation: redox signaling and oxidative stress. Journal of Biological Chemistry. 2014;289(13):8735–8741. doi: 10.1074/jbc.r113.544635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Ray P. D., Huang B.-W., Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cellular Signalling. 2012;24(5):981–990. doi: 10.1016/j.cellsig.2012.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lennicke C., Rahn J., Lichtenfels R., Wessjohann L. A., Seliger B. Hydrogen peroxide - production, fate and role in redox signaling of tumor cells. Cell Communication and Signaling: CCS. 2015;13(1):p. 39. doi: 10.1186/s12964-015-0118-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Roy J., Galano J. M., Durand T., le Guennec J. Y., Chung-Yung Lee J. Physiological role of reactive oxygen species as promoters of natural defenses. The FASEB Journal. 2017;31(9):3729–3745. doi: 10.1096/fj.201700170R. [DOI] [PubMed] [Google Scholar]
- 128.Ahmad A., Dempsey S., Daneva Z., et al. Role of nitric oxide in the cardiovascular and renal systems. International Journal of Molecular Sciences. 2018;19(9, article 2605) doi: 10.3390/ijms19092605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Vanhoutte P. M. Nitric oxide: from good to bad. Annals of Vascular Diseases. 2018;11(1):41–51. doi: 10.3400/avd.ra.17-00134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Ribeiro T. P., Oliveira A. C., Mendes-Junior L. G., et al. Cardiovascular effects induced by northeastern Brazilian red wine: role of nitric oxide and redox sensitive pathways. Journal of Functional Foods. 2016;22:82–92. doi: 10.1016/j.jff.2015.12.032. [DOI] [Google Scholar]
- 131.Brown D. I., Griendling K. K. Regulation of signal transduction by reactive oxygen species in the cardiovascular system. Circulation Research. 2015;116(3):531–549. doi: 10.1161/CIRCRESAHA.116.303584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Bielli A., Scioli M. G., Mazzaglia D., Doldo E., Orlandi A. Antioxidants and vascular health. Life Sciences. 2015;143:209–216. doi: 10.1016/j.lfs.2015.11.012. [DOI] [PubMed] [Google Scholar]
- 133.Awad M. A., Aldosari S. R., Abid M. R. Genetic alterations in oxidant and anti-oxidant enzymes in the vascular system. Frontiers in Cardiovascular Medicine. 2018;5:p. 107. doi: 10.3389/fcvm.2018.00107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.He L., He T., Farrar S., Ji L., Liu T., Ma X. Antioxidants maintain cellular redox homeostasis by elimination of reactive oxygen species. Cellular Physiology and Biochemistry. 2017;44(2):532–553. doi: 10.1159/000485089. [DOI] [PubMed] [Google Scholar]
- 135.McSweeney S. R., Warabi E., Siow R. C. M. Nrf2 as an endothelial mechanosensitive transcription factor. Hypertension. 2016;67(1):20–29. doi: 10.1161/HYPERTENSIONAHA.115.06146. [DOI] [PubMed] [Google Scholar]
- 136.Handy D. E., Loscalzo J. Responses to reductive stress in the cardiovascular system. Free Radical Biology & Medicine. 2017;109:114–124. doi: 10.1016/j.freeradbiomed.2016.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Yamamoto M., Kensler T. W., Motohashi H. The KEAP1-NRF2 system: a thiol-based sensor-effector apparatus for maintaining redox homeostasis. Physiological Reviews. 2018;98(3):1169–1203. doi: 10.1152/physrev.00023.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Tu W., Wang H., Li S., Liu Q., Sha H. The anti-inflammatory and anti-oxidant mechanisms of the Keap1/Nrf2/ARE signaling pathway in chronic diseases. Aging and Disease. 2019;10(3):637–651. doi: 10.14336/AD.2018.0513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Sireesh D., Dhamodharan U., Ezhilarasi K., Vijay V., Ramkumar K. M. Association of NF-E2 related factor 2 (Nrf2) and inflammatory cytokines in recent onset type 2 diabetes mellitus. Scientific Reports. 2018;8(1, article 5126) doi: 10.1038/s41598-018-22913-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Chen B., Lu Y., Chen Y., Cheng J. The role of Nrf2 in oxidative stress-induced endothelial injuries. Journal of Endocrinology. 2015;225(3):R83–R99. doi: 10.1530/JOE-14-0662. [DOI] [PubMed] [Google Scholar]
- 141.Zakkar M., van der Heiden K., Luong L. A., et al. Activation of Nrf2 in endothelial cells protects arteries from exhibiting a proinflammatory state. Arteriosclerosis, Thrombosis, and Vascular Biology. 2009;29(11):1851–1857. doi: 10.1161/ATVBAHA.109.193375. [DOI] [PubMed] [Google Scholar]
- 142.Suzuki T., Yamamoto M. Stress-sensing mechanisms and the physiological roles of the Keap1-Nrf2 system during cellular stress. The Journal of Biological Chemistry. 2017;292(41):16817–16824. doi: 10.1074/jbc.R117.800169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Brown A. K., Webb A. E. Regulation of FOXO factors in mammalian cells. In: Ghaffari S., editor. Forkhead FOXO Transcription Factors in Development and Disease. Academic Press; 2018. pp. 165–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Klotz L.-O., Sánchez-Ramos C., Prieto-Arroyo I., Urbánek P., Steinbrenner H., Monsalve M. Redox regulation of FoxO transcription factors. Redox Biology. 2015;6:51–72. doi: 10.1016/j.redox.2015.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Bruns D. R., Drake J. C., Biela L. M., Peelor F. F., Miller B. F., Hamilton K. L. Nrf2 signaling and the slowed aging phenotype: evidence from long-lived models. Oxidative Medicine and Cellular Longevity. 2015;2015:15. doi: 10.1155/2015/732596.732596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Klotz L.-O., Steinbrenner H. Cellular adaptation to xenobiotics: Interplay between xenosensors, reactive oxygen species and FOXO transcription factors. Redox Biology. 2017;13:646–654. doi: 10.1016/j.redox.2017.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Wang Y., Zhou Y., Graves D. T. FOXO transcription factors: their clinical significance and regulation. BioMed Research International. 2014;2014:13. doi: 10.1155/2014/925350.925350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Ooi B. K., Chan K. G., Goh B. H., Yap W. H. The role of natural products in targeting cardiovascular diseases via Nrf2 pathway: novel molecular mechanisms and therapeutic approaches. Frontiers in Pharmacology. 2018;9, article 1308 doi: 10.3389/fphar.2018.01308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.de Freitas Silva M., Pruccoli L., Morroni F., et al. The Keap1/Nrf2-ARE pathway as a pharmacological target for chalcones. Molecules. 2018;23(7, article 1803) doi: 10.3390/molecules23071803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Wardyn J. D., Ponsford A. H., Sanderson C. M. Dissecting molecular cross-talk between Nrf2 and NF-κB response pathways. Biochemical Society Transactions. 2015;43(4):621–626. doi: 10.1042/BST20150014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Ferrer-Sueta G., Manta B., Botti H., Radi R., Trujillo M., Denicola A. Factors affecting protein thiol reactivity and specificity in peroxide reduction. Chemical Research in Toxicology. 2011;24(4):434–450. doi: 10.1021/tx100413v. [DOI] [PubMed] [Google Scholar]
- 152.Dinkova-Kostova A. T., Kostov R. V., Canning P. Keap1, the cysteine-based mammalian intracellular sensor for electrophiles and oxidants. Archives of Biochemistry and Biophysics. 2017;617:84–93. doi: 10.1016/j.abb.2016.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Ahmed S. M. U., Luo L., Namani A., Wang X. J., Tang X. Nrf2 signaling pathway: pivotal roles in inflammation. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 2017;1863(2):585–597. doi: 10.1016/j.bbadis.2016.11.005. [DOI] [PubMed] [Google Scholar]
- 154.Bellezza I., Tucci A., Galli F., et al. Inhibition of NF-κB nuclear translocation via HO-1 activation underlies α-tocopheryl succinate toxicity. The Journal of Nutritional Biochemistry. 2012;23(12):1583–1591. doi: 10.1016/j.jnutbio.2011.10.012. [DOI] [PubMed] [Google Scholar]
- 155.Soares M. P., Seldon M. P., Gregoire I. P., et al. Heme oxygenase-1 modulates the expression of adhesion molecules associated with endothelial cell activation. The Journal of Immunology. 2004;172(6):3553–3563. doi: 10.4049/jimmunol.172.6.3553. [DOI] [PubMed] [Google Scholar]
- 156.Morgan M. J., Liu Z.-G. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Research. 2011;21(1):103–115. doi: 10.1038/cr.2010.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Ganesh Yerra V., Negi G., Sharma S. S., Kumar A. Potential therapeutic effects of the simultaneous targeting of the Nrf2 and NF-κB pathways in diabetic neuropathy. Redox Biology. 2013;1(1):394–397. doi: 10.1016/j.redox.2013.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Yu M., Li H., Liu Q., et al. Nuclear factor p65 interacts with Keap1 to repress the Nrf2-ARE pathway. Cellular Signalling. 2011;23(5):883–892. doi: 10.1016/j.cellsig.2011.01.014. [DOI] [PubMed] [Google Scholar]
- 159.Bellezza I., Giambanco I., Minelli A., Donato R. Nrf2-Keap1 signaling in oxidative and reductive stress. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 2018;1865(5):721–733. doi: 10.1016/j.bbamcr.2018.02.010. [DOI] [PubMed] [Google Scholar]
- 160.Thimmulappa R. K., Lee H., Rangasamy T., et al. Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. The Journal of Clinical Investigation. 2006;116(4):984–995. doi: 10.1172/jci25790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Lakatta E. G. Arterial aging is risky. Journal of Applied Physiology. 2008;105(4):1321–1322. doi: 10.1152/japplphysiol.91145.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Izzo C., Carrizzo A., Alfano A., et al. The impact of aging on cardio and cerebrovascular diseases. International Journal of Molecular Sciences. 2018;19(2):p. 481. doi: 10.3390/ijms19020481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Pinto E. Blood pressure and ageing. Postgraduate Medical Journal. 2007;83(976):109–114. doi: 10.1136/pgmj.2006.048371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Donato A. J., Morgan R. G., Walker A. E., Lesniewski L. A. Cellular and molecular biology of aging endothelial cells. Journal of Molecular and Cellular Cardiology. 2015;89(Part B):122–135. doi: 10.1016/j.yjmcc.2015.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Brackmann L. K., Buck C., Nyangasa M. A., Kelm S., Sheikh M., Hebestreit A. Anthropometric and biochemical predictors for hypertension in a cross-sectional study in Zanzibar, Tanzania. Frontiers in Public Health. 2019;7:p. 338. doi: 10.3389/fpubh.2019.00338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Schulz E., Gori T., Münzel T. Oxidative stress and endothelial dysfunction in hypertension. Hypertension Research. 2011;34(6):665–673. doi: 10.1038/hr.2011.39. [DOI] [PubMed] [Google Scholar]
- 167.Mozaffarian D., Benjamin E. J., Go A. S., et al. Heart disease and stroke statistics—2015 update. Circulation. 2015;131(4):e29–322. doi: 10.1161/cir.0000000000000152. [DOI] [PubMed] [Google Scholar]
- 168.Farooq M. A., Gaertner S., Amoura L., et al. Intake of omega-3 formulation EPA:DHA 6:1 by old rats for 2 weeks improved endothelium-dependent relaxations and normalized the expression level of ACE/AT1R/NADPH oxidase and the formation of ROS in the mesenteric artery. Biochemical Pharmacology. 2020;173, article 113749 doi: 10.1016/j.bcp.2019.113749. [DOI] [PubMed] [Google Scholar]
- 169.Wirth A., Wang S., Takefuji M., et al. Age-dependent blood pressure elevation is due to increased vascular smooth muscle tone mediated by G-protein signalling. Cardiovascular Research. 2015;109(1):131–140. doi: 10.1093/cvr/cvv249. [DOI] [PubMed] [Google Scholar]
- 170.Sun Z. Aging, arterial stiffness, and hypertension. Hypertension. 2015;65(2):252–256. doi: 10.1161/HYPERTENSIONAHA.114.03617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Buford T. W. Hypertension and aging. Ageing Research Reviews. 2016;26:96–111. doi: 10.1016/j.arr.2016.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Higashi Y., Kihara Y., Noma K. Endothelial dysfunction and hypertension in aging. Hypertension Research. 2012;35(11):1039–1047. doi: 10.1038/hr.2012.138. [DOI] [PubMed] [Google Scholar]
- 173.Chaudhary P., Pandey A., Azad C. S., Tia N., Singh M., Gambhir I. S. Association of oxidative stress and endothelial dysfunction in hypertension. Analytical Biochemistry. 2020;590, article 113535 doi: 10.1016/j.ab.2019.113535. [DOI] [PubMed] [Google Scholar]
- 174.Wu J., Saleh M. A., Kirabo A., et al. Immune activation caused by vascular oxidation promotes fibrosis and hypertension. Journal of Clinical Investigation. 2016;126(1):50–67. doi: 10.1172/JCI80761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Schramm A., Matusik P., Osmenda G., Guzik T. J. Targeting NADPH oxidases in vascular pharmacology. Vascular Pharmacology. 2012;56(5-6):216–231. doi: 10.1016/j.vph.2012.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Guzik T. J., Touyz R. M. Oxidative stress, inflammation, and vascular aging in hypertension. Hypertension. 2017;70(4):660–667. doi: 10.1161/HYPERTENSIONAHA.117.07802. [DOI] [PubMed] [Google Scholar]
- 177.Kurmani S., Squire I. Acute heart failure: definition, classification and epidemiology. Current Heart Failure Reports. 2017;14(5):385–392. doi: 10.1007/s11897-017-0351-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Díez-Villanueva P., Alfonso F. Heart failure in the elderly. Journal of Geriatric Cardiology. 2016;13(2):115–117. doi: 10.11909/j.issn.1671-5411.2016.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Lazzarini V., Mentz R. J., Fiuzat M., Metra M., O'Connor C. M. Heart failure in elderly patients: distinctive features and unresolved issues. European Journal of heart Failure. 2013;15(7):717–723. doi: 10.1093/eurjhf/hft028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Loffredo F. S., Nikolova A. P., Pancoast J. R., Lee R. T. Heart failure with preserved ejection Fraction. Circulation Research. 2014;115(1):97–107. doi: 10.1161/CIRCRESAHA.115.302929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Chen W., Frangogiannis N. G. The role of inflammatory and fibrogenic pathways in heart failure associated with aging. Heart Failure Reviews. 2010;15(5):415–422. doi: 10.1007/s10741-010-9161-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Wong L. S. M., van der Harst P., de Boer R. A., Huzen J., van Gilst W. H., van Veldhuisen D. J. Aging, telomeres and heart failure. Heart Failure Reviews. 2010;15(5):479–486. doi: 10.1007/s10741-010-9173-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Osmanska J., Jhund P. S. Contemporary management of heart failure in the elderly. Drugs & Aging. 2019;36(2):137–146. doi: 10.1007/s40266-018-0625-4. [DOI] [PubMed] [Google Scholar]
- 184.Wang J. C., Bennett M. Aging and atherosclerosis. Circulation Research. 2012;111(2):245–259. doi: 10.1161/CIRCRESAHA.111.261388. [DOI] [PubMed] [Google Scholar]
- 185.Alenghat F. J. The Prevalence of Atherosclerosis in Those with Inflammatory Connective Tissue Disease by Race, Age and Traditional Risk Factors. Scientific Reports. 2016;6(1, article 20303) doi: 10.1038/srep20303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Libby P. Inflammation in atherosclerosis. Arteriosclerosis, Thrombosis, and Vascular Biology. 2012;32(9):2045–2051. doi: 10.1161/ATVBAHA.108.179705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Geovanini G. R., Libby P. Atherosclerosis and inflammation: overview and updates. Clinical Science. 2018;132(12):1243–1252. doi: 10.1042/CS20180306. [DOI] [PubMed] [Google Scholar]
- 188.Castellon X., Bogdanova V. Chronic inflammatory diseases and endothelial dysfunction. Aging and Disease. 2016;7(1):81–89. doi: 10.14336/AD.2015.0803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Yuan T., Yang T., Chen H., et al. New insights into oxidative stress and inflammation during diabetes mellitus- accelerated atherosclerosis. Redox Biology. 2019;20:247–260. doi: 10.1016/j.redox.2018.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Head T., Daunert S., Goldschmidt-Clermont P. J. The aging risk and atherosclerosis: a fresh look at arterial homeostasis. Frontiers in Genetics. 2017;8:p. 216. doi: 10.3389/fgene.2017.00216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Libby P. Once more unto the breach: endothelial permeability and atherogenesis. European Heart Journal. 2019;40(11):938–940. doi: 10.1093/eurheartj/ehz081. [DOI] [PubMed] [Google Scholar]
- 192.Tanianskii D., Pigarevsky P. V., Maltceva S. V., Denisenko A. D. Immunohistochemical detection of adiponectin in atherosclerotic plaque. Atherosclerosis. 2018;275:e134–e135. doi: 10.1016/j.atherosclerosis.2018.06.393. [DOI] [Google Scholar]
- 193.Li B., Li W., Li X., Zhou H. Inflammation: a novel therapeutic target/direction in atherosclerosis. Current Pharmaceutical Design. 2017;23(8):1216–1227. doi: 10.2174/1381612822666161230142931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.van der Vorst E. P. C., Daissormont I., Aslani M., et al. Interruption of the CXCL13/CXCR5 chemokine axis enhances plasma IgM levels and attenuates atherosclerosis development. Thrombosis and Haemostasis. 2020;120(2):344–347. doi: 10.1055/s-0039-3400746. [DOI] [PubMed] [Google Scholar]
- 195.Katakami N. Mechanism of development of atherosclerosis and cardiovascular disease in diabetes mellitus. Journal of Atherosclerosis and Thrombosis. 2018;25(1):27–39. doi: 10.5551/jat.RV17014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Kaneto H., Katakami N., Matsuhisa M., Matsuoka T.-A. Role of reactive oxygen species in the progression of type 2 diabetes and atherosclerosis. Mediators of inflammation. 2010;2010:11. doi: 10.1155/2010/453892.453892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Burtenshaw D., Kitching M., Redmond E. M., Megson I. L., Cahill P. A. Reactive oxygen species (ROS), intimal thickening, and subclinical atherosclerotic disease. Frontiers in Cardiovascular Medicine. 2019;6:p. 89. doi: 10.3389/fcvm.2019.00089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Hulsmans M., Holvoet P. The vicious circle between oxidative stress and inflammation in atherosclerosis. Journal of Cellular and Molecular Medicine. 2010;14(1-2):70–78. doi: 10.1111/j.1582-4934.2009.00978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Yang X., Li Y., Li Y., et al. Oxidative stress-mediated atherosclerosis: mechanisms and therapies. Frontiers in Physiology. 2017;8:p. 600. doi: 10.3389/fphys.2017.00600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Kirii H., Niwa T., Yamada Y., et al. Lack of interleukin-1β decreases the severity of atherosclerosis in ApoE-deficient mice. Arteriosclerosis, Thrombosis, and Vascular Biology. 2003;23(4):656–660. doi: 10.1161/01.ATV.0000064374.15232.C3. [DOI] [PubMed] [Google Scholar]
- 201.Brånén L., Hovgaard L., Nitulescu M., Bengtsson E., Nilsson J., Jovinge S. Inhibition of tumor necrosis Factor-α reduces atherosclerosis in apolipoprotein E knockout mice. Arteriosclerosis, Thrombosis, and Vascular Biology. 2004;24(11):2137–2142. doi: 10.1161/01.ATV.0000143933.20616.1b. [DOI] [PubMed] [Google Scholar]
- 202.Childs B. G., Baker D. J., Wijshake T., Conover C. A., Campisi J., van Deursen J. M. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science. 2016;354(6311):472–477. doi: 10.1126/science.aaf6659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Shih H., Lee B., Lee R. J., Boyle A. J. The aging heart and post-infarction left ventricular remodeling. Journal of the American College of Cardiology. 2011;57(1):9–17. doi: 10.1016/j.jacc.2010.08.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Thygesen K., Alpert J. S., Jaffe A. S., et al. Fourth universal definition of myocardial infarction (2018) Circulation. 2018;138(20):e618–e651. doi: 10.1161/CIR.0000000000000617. [DOI] [PubMed] [Google Scholar]
- 205.Go A. S., Mozaffarian D., Roger V. L., et al. Heart disease and stroke statistics--2014 update: a report from the American Heart Association. Circulation. 2014;129(3):e28–e292. doi: 10.1161/01.cir.0000441139.02102.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Waypa G. B., Marks J. D., Mack M. M., Boriboun C., Mungai P. T., Schumacker P. T. Mitochondrial reactive oxygen species trigger calcium increases during hypoxia in pulmonary arterial myocytes. Circulation Research. 2002;91(8):719–726. doi: 10.1161/01.RES.0000036751.04896.F1. [DOI] [PubMed] [Google Scholar]
- 207.McElroy G. S., Chandel N. S. Mitochondria control acute and chronic responses to hypoxia. Experimental Cell Research. 2017;356(2):217–222. doi: 10.1016/j.yexcr.2017.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Webster K. A. Mitochondrial membrane permeabilization and cell death during myocardial infarction: roles of calcium and reactive oxygen species. Future Cardiology. 2012;8(6):863–884. doi: 10.2217/fca.12.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Zhang Y., Jiao L., Sun L., et al. LncRNA ZFAS1 as a SERCA2a inhibitor to cause intracellular Ca2+ Overload and contractile dysfunction in a mouse model of myocardial infarction. Circulation Research. 2018;122(10):1354–1368. doi: 10.1161/CIRCRESAHA.117.312117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Hu J., Cheng P., Huang G. Y., et al. Effects of Xin-Ji-Er-Kang on heart failure induced by myocardial infarction: role of inflammation, oxidative stress and endothelial dysfunction. Phytomedicine. 2018;42:245–257. doi: 10.1016/j.phymed.2018.03.036. [DOI] [PubMed] [Google Scholar]
- 211.Kundi H., Ates I., Kiziltunc E., et al. A novel oxidative stress marker in acute myocardial infarction; thiol/disulphide homeostasis. The American Journal of Emergency Medicine. 2015;33(11):1567–1571. doi: 10.1016/j.ajem.2015.06.016. [DOI] [PubMed] [Google Scholar]
- 212.Seto S.-W., Chang D., Jenkins A., Bensoussan A., Kiat H. Angiogenesis in ischemic stroke and angiogenic effects of Chinese herbal medicine. Journal of Clinical Medicine. 2016;5(6):p. 56. doi: 10.3390/jcm5060056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Soriano-Tárraga C., Giralt-Steinhauer E., Mola-Caminal M., et al. Biological age is a predictor of mortality in ischemic stroke. Scientific Reports. 2018;8(1, article 4148) doi: 10.1038/s41598-018-22579-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Laso-García F., Diekhorst L., Gómez-de Frutos M. C., et al. Cell-based therapies for stroke: promising solution or dead end? Mesenchymal stem cells and comorbidities in preclinical stroke research. Frontiers in Neurology. 2019;10, article 332 doi: 10.3389/fneur.2019.00332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Lloyd-Jones D. M. Cardiovascular risk prediction. Circulation. 2010;121(15):1768–1777. doi: 10.1161/CIRCULATIONAHA.109.849166. [DOI] [PubMed] [Google Scholar]
- 216.Deb P., Sharma S., Hassan K. M. Pathophysiologic mechanisms of acute ischemic stroke: an overview with emphasis on therapeutic significance beyond thrombolysis. Pathophysiology. 2010;17(3):197–218. doi: 10.1016/j.pathophys.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 217.Engelhardt S., Huang S.-F., Patkar S., Gassmann M., Ogunshola O. O. Differential responses of blood-brain barrier associated cells to hypoxia and ischemia: a comparative study. Fluids and Barriers of the CNS. 2015;12(1):p. 4. doi: 10.1186/2045-8118-12-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Sarkar S., Chakraborty D., Bhowmik A., Ghosh M. K. Cerebral ischemic stroke: cellular fate and therapeutic opportunities. Frontiers in Bioscience. 2019;24(3):435–450. doi: 10.2741/4727. [DOI] [PubMed] [Google Scholar]
- 219.Forrester S. J., Kikuchi D. S., Hernandes M. S., Xu Q., Griendling K. K. Reactive oxygen species in metabolic and inflammatory signaling. Circulation Research. 2018;122(6):877–902. doi: 10.1161/CIRCRESAHA.117.311401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Newton D. F., Naiberg M. R., Goldstein B. I. Oxidative stress and cognition amongst adults without dementia or stroke: implications for mechanistic and therapeutic research in psychiatric disorders. Psychiatry Research. 2015;227(2-3):127–134. doi: 10.1016/j.psychres.2015.03.038. [DOI] [PubMed] [Google Scholar]
- 221.Narne P., Pandey V., Phanithi P. B. Interplay between mitochondrial metabolism and oxidative stress in ischemic stroke: an epigenetic connection. Molecular and Cellular Neuroscience. 2017;82:176–194. doi: 10.1016/j.mcn.2017.05.008. [DOI] [PubMed] [Google Scholar]
- 222.Piskovatska V., Strilbytska O., Koliada A., Vaiserman A., Lushchak O. Health benefits of anti-aging drugs. In: Harris J. R., Korolchuk V. I., editors. Biochemistry and Cell Biology of Ageing: Part II Clinical Science. Singapore: Springer Singapore; 2019. pp. 339–392. [DOI] [PubMed] [Google Scholar]
- 223.Vaiserman A., Lushchak O. Implementation of longevity-promoting supplements and medications in public health practice: achievements, challenges and future perspectives. Journal of Translational Medicine. 2017;15(1):p. 160. doi: 10.1186/s12967-017-1259-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Cochemé H. M., Murphy M. P. Can antioxidants be effective therapeutics? Current Opinion in Investigational Drugs. 2010;11(4):426–431. [PubMed] [Google Scholar]
- 225.Firuzi O., Miri R., Tavakkoli M., Saso L. Antioxidant therapy: current status and future prospects. Current Medicinal Chemistry. 2011;18(25):3871–3888. doi: 10.2174/092986711803414368. [DOI] [PubMed] [Google Scholar]
- 226.Sadowska-Bartosz I., Bartosz G. Effect of antioxidants supplementation on aging and longevity. BioMed Research International. 2014;2014:17. doi: 10.1155/2014/404680.404680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Dato S., Crocco P., D'Aquila P., et al. Exploring the role of genetic variability and lifestyle in oxidative stress response for healthy aging and longevity. International Journal of Molecular Sciences. 2013;14(8):16443–16472. doi: 10.3390/ijms140816443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Li J., Zhang C.-X., Liu Y.-M., Chen K.-L., Chen G. A comparative study of anti-aging properties and mechanism: resveratrol and caloric restriction. Oncotarget. 2017;8(39):65717–65729. doi: 10.18632/oncotarget.20084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Serino A., Salazar G. Protective role of polyphenols against vascular inflammation, aging and cardiovascular disease. Nutrients. 2018;11(1):p. 53. doi: 10.3390/nu11010053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Patel R. V., Mistry B. M., Shinde S. K., Syed R., Singh V., Shin H.-S. Therapeutic potential of quercetin as a cardiovascular agent. European Journal of Medicinal Chemistry. 2018;155:889–904. doi: 10.1016/j.ejmech.2018.06.053. [DOI] [PubMed] [Google Scholar]
- 231.Montenegro M. F., Neto-Neves E. M., Dias-Junior C. A., et al. Quercetin restores plasma nitrite and nitroso species levels in renovascular hypertension. Naunyn-Schmiedeberg's Archives of Pharmacology. 2010;382(4):293–301. doi: 10.1007/s00210-010-0546-1. [DOI] [PubMed] [Google Scholar]
- 232.Singh A., Naidu P. S., Kulkarni S. K. Reversal of aging and chronic ethanol-induced cognitive dysfunction by quercetin a bioflavonoid. Free Radical Research. 2009;37(11):1245–1252. doi: 10.1080/10715760310001616014. [DOI] [PubMed] [Google Scholar]
- 233.Liu H., Zhang L., Lu S. Evaluation of antioxidant and immunity activities of quercetin in isoproterenol-treated rats. Molecules. 2012;17(4):4281–4291. doi: 10.3390/molecules17044281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Labinskyy N., Csiszar A., Veress G., et al. Vascular dysfunction in aging: potential effects of resveratrol, an anti-inflammatory phytoestrogen. Current Medicinal Chemistry. 2006;13(9):989–996. doi: 10.2174/092986706776360987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Rouse M., Egan J. M. Resveratrol in Aging and Age-Related Diseases. In: Ram J. L., Conn P. M., editors. Conn's Handbook of Models for Human Aging (Second Edition) Academic Press; 2018. pp. 1133–1142. [DOI] [Google Scholar]
- 236.Xia N., Förstermann U., Li H. Resveratrol and endothelial nitric oxide. Molecules. 2014;19(10):16102–16121. doi: 10.3390/molecules191016102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Pearson K. J., Baur J. A., Lewis K. N., et al. Resveratrol delays age-related deterioration and mimics transcriptional aspects of dietary restriction without extending life span. Cell Metabolism. 2008;8(2):157–168. doi: 10.1016/j.cmet.2008.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Seyyedebrahimi S., Khodabandehloo H., Nasli Esfahani E., Meshkani R. The effects of resveratrol on markers of oxidative stress in patients with type 2 diabetes: a randomized, double-blind, placebo-controlled clinical trial. Acta Diabetologica. 2018;55(4):341–353. doi: 10.1007/s00592-017-1098-3. [DOI] [PubMed] [Google Scholar]
- 239.Mukherjee S., Ray D., Lekli I., Bak I., Tosaki A., Das D. K. Effects of Longevinex (modified resveratrol) on cardioprotection and its mechanisms of action. Canadian Journal of Physiology and Pharmacology. 2010;88(11):1017–1025. doi: 10.1139/Y10-082. [DOI] [PubMed] [Google Scholar]
- 240.Simioni C., Zauli G., Martelli A. M., et al. Oxidative stress: role of physical exercise and antioxidant nutraceuticals in adulthood and aging. Oncotarget. 2018;9(24):17181–17198. doi: 10.18632/oncotarget.24729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Santos-Parker J. R., Strahler T. R., Bassett C. J., Bispham N. Z., Chonchol M. B., Seals D. R. Curcumin supplementation improves vascular endothelial function in healthy middle-aged and older adults by increasing nitric oxide bioavailability and reducing oxidative stress. Aging. 2016;9(1):187–208. doi: 10.18632/aging.101149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Fleenor B. S., Carlini N. A., Campbell M. S. Curcumin and arterial function in health and disease. Current Opinion in Clinical Nutrition and Metabolic Care. 2019;22(6):459–464. doi: 10.1097/MCO.0000000000000598. [DOI] [PubMed] [Google Scholar]
- 243.Albuquerque J. G. F., Assis V. L., Almeida A. J. P. O., et al. Antioxidant and vasorelaxant activities induced by northeastern Brazilian fermented grape skins. BMC Complementary and Alternative Medicine. 2017;17(1):p. 376. doi: 10.1186/s12906-017-1881-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Oak M.-H., Auger C., Belcastro E., Park S.-H., Lee H.-H., Schini-Kerth V. B. Potential mechanisms underlying cardiovascular protection by polyphenols: role of the endothelium. Free Radical Biology and Medicine. 2018;122:161–170. doi: 10.1016/j.freeradbiomed.2018.03.018. [DOI] [PubMed] [Google Scholar]
- 245.Yahfoufi N., Alsadi N., Jambi M., Matar C. The immunomodulatory and anti-inflammatory role of polyphenols. Nutrients. 2018;10(11, article 1618) doi: 10.3390/nu10111618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Kim E. N., Lim J. H., Kim M. Y., et al. Resveratrol, an Nrf2 activator, ameliorates aging-related progressive renal injury. Aging. 2018;10(1):83–99. doi: 10.18632/aging.101361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Dyck G., Raj P., Zieroth S., Dyck J., Ezekowitz J. The effects of resveratrol in patients with cardiovascular disease and heart failure: a narrative review. International Journal of Molecular Sciences. 2019;20(4):p. 904. doi: 10.3390/ijms20040904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Bonnefont-Rousselot D. Resveratrol and cardiovascular diseases. Nutrients. 2016;8(5):p. 250. doi: 10.3390/nu8050250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Rojas L. B. A., Gomes M. B. Metformin: an old but still the best treatment for type 2 diabetes. Diabetology & Metabolic Syndrome. 2013;5(1):p. 6. doi: 10.1186/1758-5996-5-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Novelle M. G., Ali A., Diéguez C., Bernier M., de Cabo R. Metformin: a hopeful promise in aging research. Cold Spring Harbor Perspectives In medicine. 2016;6(3, article a025932) doi: 10.1101/cshperspect.a025932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Barzilai N., Crandall J. P., Kritchevsky S. B., Espeland M. A. Metformin as a tool to target aging. Cell Metabolism. 2016;23(6):1060–1065. doi: 10.1016/j.cmet.2016.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Davis B. J., Xie Z., Viollet B., Zou M.-H. Activation of the AMP-activated kinase by antidiabetes drug metformin stimulates nitric oxide synthesis in vivo by promoting the association of heat shock protein 90 and endothelial nitric oxide synthase. Diabetes. 2006;55(2):496–505. doi: 10.2337/diabetes.55.02.06.db05-1064. [DOI] [PubMed] [Google Scholar]
- 253.Cheang W. S., Tian X. Y., Wong W. T., et al. Metformin protects endothelial function in diet-induced obese mice by inhibition of endoplasmic reticulum stress through 5′ adenosine Monophosphate–Activated protein Kinase–Peroxisome Proliferator–Activated receptor δ pathway. Arteriosclerosis, Thrombosis, and Vascular Biology. 2014;34(4):830–836. doi: 10.1161/ATVBAHA.113.301938. [DOI] [PubMed] [Google Scholar]
- 254.Batchuluun B., Inoguchi T., Sonoda N., et al. Metformin and liraglutide ameliorate high glucose-induced oxidative stress via inhibition of PKC-NAD(P)H oxidase pathway in human aortic endothelial cells. Atherosclerosis. 2014;232(1):156–164. doi: 10.1016/j.atherosclerosis.2013.10.025. [DOI] [PubMed] [Google Scholar]
- 255.Fang J., Yang J., Wu X., et al. Metformin alleviates human cellular aging by upregulating the endoplasmic reticulum glutathione peroxidase 7. Aging Cell. 2018;17(4, article e12765) doi: 10.1111/acel.12765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Saisho Y. Metformin and inflammation: its potential beyond glucose-lowering effect. Endocrine, Metabolic & Immune Disorders-Drug Targets. 2015;15(3):196–205. doi: 10.2174/1871530315666150316124019. [DOI] [PubMed] [Google Scholar]
- 257.Isoda K., Young J. L., Zirlik A., et al. Metformin inhibits proinflammatory responses and nuclear factor-κB in human vascular wall cells. Arteriosclerosis, Thrombosis, and Vascular Biology. 2006;26(3):611–617. doi: 10.1161/01.ATV.0000201938.78044.75. [DOI] [PubMed] [Google Scholar]
- 258.Donato A. J., Machin D. R., Lesniewski L. A. Mechanisms of dysfunction in the aging vasculature and role in age-related disease. Circulation Research. 2018;123(7):825–848. doi: 10.1161/CIRCRESAHA.118.312563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Blagosklonny M. V. From rapalogs to anti-aging formula. Oncotarget. 2017;8(22):35492–35507. doi: 10.18632/oncotarget.18033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Ornelas A., Zacharias-Millward N., Menter D. G., et al. Beyond COX-1: the effects of aspirin on platelet biology and potential mechanisms of chemoprevention. Cancer Metastasis Reviews. 2017;36(2):289–303. doi: 10.1007/s10555-017-9675-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Warner T. D., Nylander S., Whatling C. Anti-platelet therapy: cyclo-oxygenase inhibition and the use of aspirin with particular regard to dual anti-platelet therapy. British Journal of Clinical Pharmacology. 2011;72(4):619–633. doi: 10.1111/j.1365-2125.2011.03943.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Bode-Böger S. M., Martens-Lobenhoffer J., Täger M., Schröder H., Scalera F. Aspirin reduces endothelial cell senescence. Biochemical and Biophysical Research Communications. 2005;334(4):1226–1232. doi: 10.1016/j.bbrc.2005.07.014. [DOI] [PubMed] [Google Scholar]
- 263.Raju N., Sobieraj-Teague M., Bosch J., Eikelboom J. W. Updated meta-analysis of aspirin in primary prevention of cardiovascular disease. The American Journal of Medicine. 2016;129(5):e35–e36. doi: 10.1016/j.amjmed.2015.10.046. [DOI] [PubMed] [Google Scholar]
- 264.Ittaman S. V., VanWormer J. J., Rezkalla S. H. The role of aspirin in the prevention of cardiovascular disease. Clinical Medicine & Research. 2015;12(3-4):147–154. doi: 10.3121/cmr.2013.1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Patrono C., Baigent C. Role of aspirin in primary prevention of cardiovascular disease. Nature Reviews Cardiology. 2019;16(11):675–686. doi: 10.1038/s41569-019-0225-y. [DOI] [PubMed] [Google Scholar]
- 266.Collins R., Reith C., Emberson J., et al. Interpretation of the evidence for the efficacy and safety of statin therapy. The Lancet. 2016;388(10059):2532–2561. doi: 10.1016/S0140-6736(16)31357-5. [DOI] [PubMed] [Google Scholar]
- 267.Chou R., Dana T., Blazina I., Daeges M., Jeanne T. L. statins for prevention of cardiovascular disease in adults. JAMA. 2016;316(19):2008–2024. doi: 10.1001/jama.2015.15629. [DOI] [PubMed] [Google Scholar]
- 268.Taylor F., Huffman M. D., Macedo A. F., et al. Reduced dietary salt for the prevention of cardiovascular disease. The Cochrane Database of Systematic Reviews. 2013;9 doi: 10.1002/14651858.CD009217.pub2. [DOI] [PubMed] [Google Scholar]
- 269.Costa S., Reina-Couto M., Albino-Teixeira A., Sousa T. Estatinas e stresse oxidativo na insuficiência cardíaca crónica. Revista Portuguesa de Cardiologia. 2016;35(1):41–57. doi: 10.1016/j.repc.2015.09.006. [DOI] [PubMed] [Google Scholar]
- 270.Gilbert R., Al-Janabi A., Tomkins-Netzer O., Lightman S. Statins as anti-inflammatory agents: a potential therapeutic role in sight-threatening non-infectious uveitis. Porto Biomedical Journal. 2017;2(2):33–39. doi: 10.1016/j.pbj.2017.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Shimomura M., Oyama J. I., Takeuchi M., et al. Acute effects of statin on reduction of angiopoietin-like 2 and glyceraldehyde-derived advanced glycation end-products levels in patients with acute myocardial infarction: a message from SAMIT (Statin for Acute Myocardial Infarction Trial) Heart and Vessels. 2016;31(10):1583–1589. doi: 10.1007/s00380-015-0773-y. [DOI] [PubMed] [Google Scholar]
- 272.Hwang A.-R., Han J.-H., Lim J. H., Kang Y. J., Woo C.-H. Fluvastatin inhibits AGE-induced cell proliferation and migration via an ERK5-dependent Nrf2 pathway in vascular smooth muscle cells. PloS One. 2017;12(5, article e0178278) doi: 10.1371/journal.pone.0178278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Liao J. K., Laufs U. Pleiotropic effects of statins. Annual Review of Pharmacology and Toxicology. 2005;45(1):89–118. doi: 10.1146/annurev.pharmtox.45.120403.095748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.An J., Shi F., Liu S., Ma J., Ma Q. Preoperative statins as modifiers of cardiac and inflammatory outcomes following coronary artery bypass graft surgery: a meta-analysis. Interactive CardioVascular and Thoracic Surgery. 2017;25(6):958–965. doi: 10.1093/icvts/ivx172. [DOI] [PubMed] [Google Scholar]
- 275.Flynn J. M., O'Leary M. N., Zambataro C. A., et al. Late-life rapamycin treatment reverses age-related heart dysfunction. Aging Cell. 2013;12(5):851–862. doi: 10.1111/acel.12109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Sciarretta S., Forte M., Frati G., Sadoshima J. New insights into the role of mTOR signaling in the cardiovascular system. Circulation Research. 2018;122(3):489–505. doi: 10.1161/CIRCRESAHA.117.311147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Lesniewski L. A., Seals D. R., Walker A. E., et al. Dietary rapamycin supplementation reverses age-related vascular dysfunction and oxidative stress, while modulating nutrient-sensing, cell cycle, and senescence pathways. Aging Cell. 2017;16(1):17–26. doi: 10.1111/acel.12524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Martínez-Cisuelo V., Gómez J., García-Junceda I., et al. Rapamycin reverses age-related increases in mitochondrial ROS production at complex I, oxidative stress, accumulation of mtDNA fragments inside nuclear DNA, and lipofuscin level, and increases autophagy, in the liver of middle- aged mice. Experimental Gerontology. 2016;83:130–138. doi: 10.1016/j.exger.2016.08.002. [DOI] [PubMed] [Google Scholar]
- 279.Kofman A. E., McGraw M. R., Payne C. J. Rapamycin increases oxidative stress response gene expression in adult stem cells. Aging. 2012;4(4):279–289. doi: 10.18632/aging.100451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Zhang J., Wang Z., Zhang J., et al. Rapamycin attenuates endothelial apoptosis induced by low shear stress via mTOR and Sestrin1 related redox regulation. Mediators of Inflammation. 2014;2014:9. doi: 10.1155/2014/769608.769608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Xu L., Brink M. mTOR, cardiomyocytes and inflammation in cardiac hypertrophy. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 2016;1863(7):1894–1903. doi: 10.1016/j.bbamcr.2016.01.003. [DOI] [PubMed] [Google Scholar]
- 282.Ming X.-F., Montani J.-P., Yang Z. Perspectives of targeting mTORC1–S6K1 in cardiovascular aging. Frontiers in Physiology. 2012;3:p. 5. doi: 10.3389/fphys.2012.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]