

Abstract
Recently, we have developed a coronavirus-specific gene-finding system, ZCURVE_CoV 1.0. In this paper, the system is further improved by taking the prediction of cleavage sites of viral proteinases in polyproteins into account. The cleavage sites of the 3C-like proteinase and papain-like proteinase are highly conserved. Based on the method of traditional positional weight matrix trained by the peptides around cleavage sites, the present method also sufficiently considers the length conservation of non-structural proteins cleaved by the 3C-like proteinase and papain-like proteinase to reduce the false positive prediction rate. The improved system, ZCURVE_CoV 2.0, has been run for each of the 24 completely sequenced coronavirus genomes in GenBank. Consequently, all the non-structural proteins in the 24 genomes are accurately predicted. Compared with known annotations, the performance of the present method is satisfactory. The software ZCURVE_CoV 2.0 is freely available at http://tubic.tju.edu.cn/sars/.
Keywords: Coronavirus, Severe acute respiratory syndrome, SARS-coronavirus, Polyprotein, Cleavage site
1. Introduction
Due to the severity of a life-threatening disease, referred to as severe acute respiratory syndrome (SARS), the World Health Organization (WHO) has issued a global alert for the illness. SARS apparently began in Guangdong province of China in November 2002, and has spread to Hong Kong, Singapore, Vietnam, Canada, the USA and several European countries [1], [2], [3], [4], [5], [6]. By early June 2003, more than 700 SARS-related deaths were recorded by WHO (http://www.who.int/csr/sars/country/en/).
A novel coronavirus, called SARS-coronavirus or SARS-CoV, has been proved to be the cause of SARS. The coronaviruses (order Nidovirales, family Coronaviridae, genus Coronavirus) are members of a family of large, enveloped, positive-stranded RNA viruses that replicate in the cytoplasm of animal host cells [7]. There are three groups of coronaviruses; groups I and II contain mammalian viruses, while group III contains only avian viruses. The viruses are associated with a variety of diseases in humans and domestic animals, including gastroenteritis and diseases of the upper and lower respiratory tract. Many researchers have analyzed the phylogeny of SARS-CoV and concluded that it is not closely related to any of the previously characterized coronaviruses and forms a distinct group (group IV) within the genus Coronavirus [7], [8]. At the time this paper was written, there were 12 strains of SARS-CoV complete genome sequences available from GenBank [7], [8], [9]. Among these genomes, six have been annotated manually, and the remaining six have not been annotated yet. The genomic organization of SARS-CoV is that of a typical coronavirus, with the order of the characteristic genes being replicase [rep], spike [S], envelope [E], membrane [M], nucleocapsid [N] from the 5′ to the 3′ terminus. SARS-CoV also encodes a number of non-structural proteins located between S and E, between M and N, or downstream of N with unknown functions. We have developed a coronavirus-specific gene-finding system ZCURVE_CoV 1.0 [10], which is especially suitable for gene recognition in SARS-CoV genomes. The software has the advantages of simplicity, reliability, high accuracy and quickness and can be obtained freely at the website http://tubic.tju.edu.cn/sars/. The system ZCURVE_CoV 1.0 has been run for each of the 12 SARS-CoV genomes. In addition to the polyprotein chains Orf1a and Orf1b and the four genes encoding the major structural proteins, S, E, M and N, respectively, ZCURVE_CoV 1.0 also predicts five to six putative proteins between 39 and 274 amino acids in length, with unknown functions in SARS-CoV genomes. However, the cleavage sites of viral proteinase in replicases are not predicted in ZCURVE_CoV 1.0.
The coronavirus replicases are encoded by two large, 5′-proximal open reading frames (ORFs) that comprise approximately two-thirds of the genome. Polyproteins ORF1a and ORF1b are connected by a ribosomal frameshift site, which is believed to occur at the conserved ‘slippery sequence’, UUUAAAC. It results in the translation of an ORF1a protein and a carboxyl-extended ORF1ab frameshift protein, which are also known as replicase polyproteins pp1a and pp1ab [11]. The ORF1a and ORF1ab translation products are polyprotein precursors, which are cleaved by viral proteinases, resulting in a minimum of 13 non-structural proteins, including a 3C-like proteinase, an RNA-dependent RNA polymerase, an ATPase/helicase and other function-unknown non-structural proteins [11]. These proteins in turn are responsible for replicating the viral genome as well as generating nested transcripts that are used in the synthesis of viral proteins. In this paper, all the putative non-structural proteins resulting from the cleavage by viral proteinases in the polyproteins are precisely predicted using ZCURVE_CoV 2.0.
2. Materials and methods
Seven genomic sequences of coronaviruses and the annotation information were downloaded from the NCBI RefSeq project. These coronaviruses include avian infectious bronchitis virus (IBV) (NC_001451), bovine coronavirus (BCoV) (NC_003045), human coronavirus 229E (HCoV-229E) (NC_002645), murine hepatitis virus (MHV) (NC_001846), porcine epidemic diarrhea virus (PEDV) (NC_003436), SARS coronavirus TOR2 (TOR2) (NC_004718) and transmissible gastroenteritis virus (TGEV) (NC_002306). The above genomes have been annotated by NCBI and the sequences of mature peptides are available. According to the annotation, a total of 77 sites cleaved by the 3C-like proteinase and 17 sites cleaved by the papain-like proteinase were extracted from the above seven genomes. Octapeptides cleaved by the 3C-like proteinase and 12-mer peptides cleaved by the papain-like proteinase were used to train the corresponding positional weight matrix (PWM) [12]. The cleavage site is at the center of the octapeptide or 12-mer peptide. The length distribution of non-structural proteins within ORF1ab was also derived from the annotated genomes. At the time this paper was written, there were 24 complete sequences of coronavirus genomes available in the GenBank database, of which 12 are SARS-CoVs and 12 are other groups of coronaviruses. The former comprises SARS-CoV TOR2 (NC_004718), Urbani (AY278741), HKU-39849 (AY278491), CUHK-W1 (AY278554), BJ01 (AY278488), CUHK-Su10 (AY282752), SIN2500 (AY283794), SIN2748 (AY283797), SIN2679 (AY283796), SIN2774 (AY283798), SIN2677 (AY283795) and TW1 (AY291451), whereas the latter comprises IBV (NC_001451), BCoV (NC_003045), bovine coronavirus strain Mebus (BCoVM) (U00735), bovine coronavirus isolate BCoV-LUN (BCoVL) (AF391542), bovine coronavirus strain Quebec (BCoVQ) (AF220295), HCoV-229E (NC_002645), MHV (NC_001846), murine hepatitis virus strain ML-10 (MHVM) (AF208067), murine hepatitis virus strain 2 (MHV2) (AF201929), murine hepatitis virus strain Penn 97-1 (MHVP) (AF208066), PEDV (NC_003436) and TGEV (NC_002306).
The mature peptides cleaved by the 3C-like proteinase are highly conserved in length among different groups of coronaviruses, while others cleaved by the papain-like proteinase are not so conserved. The lengths of all the non-structural proteins cleaved by the 3C-like proteinase within polyprotein 1ab are listed in Table 1 , while the lengths for the non-structural proteins cleaved by the papain-like proteinase are listed in Table 2 . The average length and standard deviation for each kind of non-structural proteins are calculated. As shown in Table 1, Table 2, the lengths of the non-structural proteins cleaved by the 3C-like proteinase are highly conserved, while the lengths and the number of the papain-like cysteine proteinase cleavage products (abbreviated as PCP CP) appear to be irregular. Since the NCBI annotations are not always correct, the annotations of cleavage products of the papain-like proteinase may be incomplete. It is observed that the size of the annotated PCP CP3 of SARS-CoV, MHV and IBV is approximately the sum of the sizes of PCP CP3 and PCP CP4 of other mammalian coronaviruses listed in Table 2. Therefore, the PCP CP3 of SARS-CoV, MHV and IBV may be further cleaved, i.e. it is possible that another papain-like proteinase cleavage site is present in the PCP CP3 of SARS-CoV, MHV and IBV. Based on the above analysis, a cleavage model of the papain-like proteinase is presented schematically in Fig. 1 . According to this model, all coronaviruses have four non-structural proteins cleaved by the papain-like proteinase. Consequently, the cleavage products of the papain-like proteinase predicted by this model show the conservation in both their length and number. The average length and standard deviation for each papain-like proteinase cleavage product are estimated based on the genomes of BCoV, HCoV-229E, TGEV and PEDV, in which four of the papain-like proteinase cleavage products are annotated (see Table 2). Fig. 2A,B shows the conservation sites cleaved by the 3C-like proteinase and papain-like proteinase, respectively. It can be seen that both the 3C-like proteinase and papain-like proteinase have conserved cleavage sites. The same arrangement order of the cleavage products in polyprotein 1ab, similar sizes of non-structural proteins and the conserved residues in the cleavable peptides form the basis of the present algorithm to predict cleavage sites of polyproteins. Here, the method is described briefly as follows.
Table 1.
The lengths for 11 non-structural proteinsa cleaved by the 3C-like proteinase
| Genome | The length of non-structural proteins (aa) |
||||||||||
| nsp2 | nsp3 | nsp4 | nsp5 | nsp6 | nsp7 | nsp9 | nsp10 | nsp11 | nsp12 | nsp13 | |
| TOR2 | 306 | 290 | 83 | 198 | 113 | 139 | 932 | 601 | 527 | 346 | 298 |
| HCoV-229Eb | 302 | 279 | 83 | 195 | 109 | 135 | 927 | 597 | 518 | 348 | 300 |
| MHVb | 303 | 287 | 92 | 194 | 110 | 137 | 928 | 600 | 521 | 374 | 299 |
| BCoV | 303 | 287 | 89 | 197 | 110 | 137 | 928 | 603 | 521 | 374 | 299 |
| IBVb | 307 | 293 | 83 | 210 | 111 | 145 | 940 | 600 | 521 | 338 | 302 |
| TGEV | 302 | 294 | 83 | 195 | 111 | 135 | 929 | 599 | 519 | 339 | 300 |
| PEDV | 302 | 280 | 83 | 195 | 108 | 135 | 927 | 597 | 517 | 339 | 301 |
| Average lengthc | 304 | 287 | 85 | 198 | 110 | 138 | 930 | 600 | 521 | 351 | 300 |
| Standard deviation | 2.07 | 5.87 | 3.76 | 5.59 | 1.60 | 3.60 | 4.67 | 2.15 | 3.26 | 16.07 | 1.35 |
These proteins are cleaved by the 3C-like proteinase within polyprotein 1ab derived from the seven coronavirus genomes annotated by NCBI.
The cleavage sites have been confirmed by experimental evidence in these genomes.
The genomes that have maximum lengths for nsp2–13 except nsp8 are IBV, TGEV, MHV, IBV, TOR2, IBV, IBV, BCoV, TOR2, MHV (BCoV) and IBV respectively. The genomes that have the minimum lengths for nsp2–13 except nsp8 are HCoV-229E (TGEV, PEDV), HCoV-229E, TOR2 (HCoV-229E, IBV, TGEV, PEDV), MHV, PEDV, HCoV-229E (TGEV, PEDV), HCoV-229E (PEDV), HCoV-229E (PEDV), PEDV, IBV and TOR2, respectively.
Table 2.
The lengths for the non-structural proteinsa cleaved by the papain-like proteinase
| Genome | Length (aa) |
|||
| PCP CP1 | PCP CP2 | PCP CP3 | PCP CP4 | |
| IBV | – | 673b | 2106 | – |
| TOR2 | 179 | 639 | 2422 | – |
| MHV | 247b | 585b | 2501 | – |
| BCoV | 246 | 605 | 1899 | 496 |
| HCoV-229E | 111b | 786 | 1587b | 481 |
| TGEV | 108 | 771 | 1509 | 490 |
| PEDV | 110 | 785 | 1622 | 480 |
| Average lengthc | 144 | 737 | 1654 | 487 |
| Standard deviationc | 68.18 | 88.10 | 169.87 | 7.63 |
These proteins are cleaved by the papain-like proteinase within polyprotein 1ab derived from the seven coronavirus genomes annotated by NCBI.
These cleavage products have been confirmed by experimental evidence.
The average length and standard deviation are calculated based on the genomes of BCoV, HCoV-229E, TGEV and PEDV.
Fig. 1.
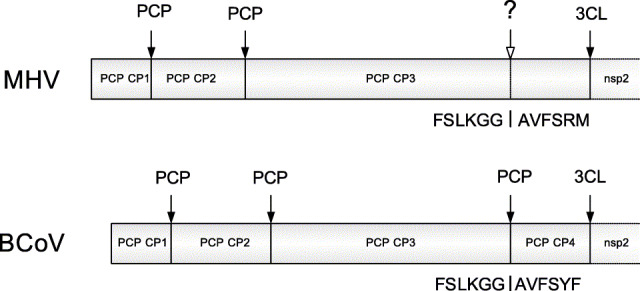
Comparison between the N-terminal sequences of the polyprotein 1abs in MHV and BCoV is shown schematically. The additional cleavage site in the annotated PCP CP3 predicted by the present method for MHV is situated at the corresponding position where the PCP CP3 and PCP CP4 are cleaved in BCoV. Cleavage sites that have been annotated by NCBI are indicated by black arrows, while the cleavage site predicted by the present method is indicated by an open arrow.
Fig. 2.
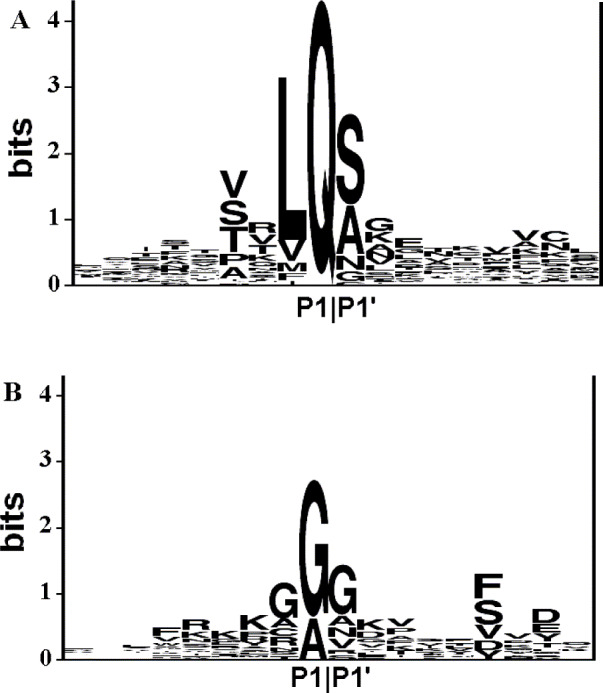
Conservation of the sites cleaved by coronavirus proteinases. Two separate multiple, gap-free alignments around the P1|P1′ positions of the sites cleaved by the 3C-like proteinase (A) and papain-like proteinase (B) in the training set are converted to logo presentations in which the size of an amino acid is proportional to its conservation at the specific position and the sampling size. The amino acid conservation is measured in bits of information plotted on a vertical axis whose upper limit is determined by the natural diversity of amino acids (20) expressed as a logarithm of 2 [16]. Seventy-seven sites cleaved by the 3C-like proteinase were used to generate the logo in A, and 17 sites cleaved by the papain-like proteinase were used to generate the logo in B.
First, ORF1ab and the slippery sequences are identified using ZCURVE_CoV 1.0. Subsequently, the predicted ORF1ab is translated into amino acid sequence. Starting from the C-terminus of the predicted ORF1ab polyprotein, the candidate cleavage site of nsp13 is searched within a particular region using the sliding-window technique. The distance between the scanning region center and the C-terminus of polyprotein 1ab should be equal to the average length of nsp13. Denoting the center position by c, a window with an octapeptide size slides from the positions c−3δ to c+3δ, where δ is the standard deviation of the length distribution for nsp13 (see Table 1). Given an octapeptide within the region S=X4X3X2X1X1′X2′X3′X4′, where Xi (i=4, 3, 2, 1, 1′, 2′, 3′, 4′) represents the amino acid at the position Pi, the score of the octapeptide is computed as
| Score (X4X3X2X1X1′X2′X3′X4′)=∏i=44′ f(i, Xi) | (1) |
where f(i, Xi) (i=4, 3, 2, 1, 1′, 2′, 3′, 4′) is the frequency of amino acid Xi occurring at the position Pi, which is an element in the corresponding positional weight matrix. The site with maximum score is selected as a candidate site. Consequently, the cleavage site of nsp12|13 is determined and nsp13 is found.
Prediction of other cleavage sites is performed in a recurrent way. Once the cleavage site of nsp12|13 is determined, the next cleavage site to be predicted is nsp11|12, then nsp10|11, and so forth until nsp1|2. Generally, if the site of nspk|(k+1) is determined, the next target is to predict the site of nsp(k−1)|k, where k=12, 11, …, 2, but k≠8 (see the explanation below). For clarity, take k=6 as an example, where the site of nsp6|7 is known. First, the center position and the sliding window used for identifying the site of nsp5|6 need to be determined. The center position c is situated upstream of the site of nsp6|7. The distance between the center position c and the site of nsp6|7 should be equal to l 6, which is the average length of nsp6. In Table 1, we find l 6=110 aa and the standard deviation δ of the length distribution for nsp6 is 1.6. A window with an octapeptide size thus slides from the position c−3δ≈c−5 to c+3δ≈c+5. Second, the site with the highest score is predicted to be the candidate site of nsp5|6. Note that in some cases the scores may be zero because of the limited training samples. In this case, a very small quantity (0.001) is assigned to the zero elements in the positional weight matrix. Also note that the nsp7|8 site is cleaved in polyprotein 1a, while the nsp7|9 site is cleaved in polyprotein 1ab. Therefore, the cleavage sites of nsp7|8 and nsp7|9 are in fact the same, leading to the result of k≠8. Furthermore, if the following two conditions are satisfied, besides the site with the maximum score, the site with the second maximum score is also taken into account: (i) Gln and Leu are found at the P1 and P2 positions, respectively; (ii) the distance between the two sites is less than five amino acid residues. This procedure considers the prediction of two adjacent cleavage sites in the scanning window. Consequently, two alternative cleavage sites annotated by NCBI are also found in the genomes of MHV and BCoV. Note that such cases occur rarely in the genomes studied.
Repeating the above procedure 11 times, all of the mature peptides cleaved by the 3C-like proteinase are identified one by one. Then, the papain-like proteinase cleavage products are searched within the remaining regions of polyprotein 1ab. A similar recurrent procedure is performed to search for the papain-like proteinase cleavage sites. The scores of 12-mer peptides are calculated as described above. The center position and the size of the sliding window used to search for the papain-like proteinase cleavage sites are determined in a way similar to that used for the 3C-like proteinase. The sites associated with the maximum scores in the corresponding scanning regions are predicted to be cleavage sites. Consequently, three papain-like proteinase cleavage sites are predicted for each genome.
3. Results and discussion
Replicase polyprotein processing is carried out by two or three ORF1a-encoded viral proteinases. Coronaviruses encode a chymotrypsin-like proteinase, 3C-like proteinase, which is analogous to the main picornaviral proteinase, 3C proteinase [11]. As mentioned above, the cleavage sites of the 3C-like proteinase are highly conserved. As shown in Fig. 2A, the P1 position of the peptide sequence is exclusively occupied by Gln. Leu is dominant at the P2 position (more than 75%) and Val, Ser, Thr and Pro are clearly favored at the P4 position. At the P1′ position, small, aliphatic residues (Ser, Ala, Asn, Gly and Cys) are found, of which the content of Ser is more than 50%. There are no highly favored residues at the P3, P2′, P3′ and P4′ positions. The length distributions of each of the 11 non-structural proteins cleaved by the 3C-like proteinase in the annotated genomes are listed in Table 1. Of these non-structural proteins, nsp2 is the putative 3C-like proteinase; nsp3 contains a hydrophobic domain; nsp7 is known as a growth-factor-like protein; nsp9 is the putative RNA-dependent RNA polymerase; nsp10 contains a metal ion-binding domain and NTPase/helicase domain. Recently the mRNA cap-1 methyltransferase function has been assigned to nsp13 [13]. The functions of other non-structural proteins are unknown. Moreover, coronaviruses also encode one (group III) or two (groups I and II) papain-like proteinases, which are analogous to the foot and mouth disease virus leader proteinase. SARS-CoV appears to contain only one papain-like proteinase domain in the predicted gene product of ORF1a [7]. For the papain-like proteinase, the cleavage sites are also conserved, but not as conserved as those of the 3C-like proteinase. Gly and Ala are found at the P1 position and Gly accounts for more than 75%. At the P2 and P1′ positions, Gly is also the dominant residue, which accounts for more than 45% and 50%, respectively. No residues exceed 40% at other positions. In this study, similar sizes of non-structural proteins and conserved cleavage sites form the basis of the present algorithm.
The performance of the algorithm is satisfactory by comparing the predicted results with known annotations. Although all the SARS genomes have been annotated by in silico analysis so far, some annotations for other coronaviruses, such as IBV, MHV and HCoV-229E, are supported by experimental evidence [11]. The jack-knife (leave-one-out) test has been performed here to ensure the validation of the prediction results for the cleavage sites of the 3C-like proteinase. By the jack-knife test, each genome out of the seven genomes under study is singled out in turn, and used as a testing genome. The remaining six genomes are used as the training set. Based on the data derived from the six training genomes, the cleavage sites of the 3C-like proteinase in the testing genome are predicted and evaluated. The jack-knife test was finished by repeating the above procedure seven times. Consequently, the predicted results by the jack-knife test are found to be as good as those by a self-consistency test mentioned previously, suggesting that the prediction results are reliable.
The prediction results for TGEV and PEDV, which are different from the annotations of NCBI RefSeq projects, are listed in Table 3 . The prediction results for other genomes can be obtained from the supplementary materials (http://tubic.tju.edu.cn/sars/). The coronavirus −1 frameshift site [14] is believed to occur at the ‘slippery sequence’, UUUAAAC. This assumption has been supported by experimental evidence [15]. But the annotated frameshift sites are not always consistent with this pattern, as in the case of PEDV, whose frameshift site lies upstream of the UUUAAAC sequence according to the annotation. This may be due to the questionable annotation. For example, the genomes of MHV and BCoV were originally annotated by the authors as the ones having a non-standard frameshift site, however, these conclusions were then corrected by the re-annotations of NCBI as the ones having standard frameshift sites. In light of this, we adopt UUUAAAC as the standard slippery sequence.
Table 3.
Comparison of the predicted results for TGEV and PEDV with those annotated by NCBIa
| Number | Genome | Location (bp) |
Location (aa) |
Length (aa) | Cleavable peptide | Feature | ||
| Start | Stop | Start | Stop | |||||
| 1 | TGEV | 315 | 638 | 1 | 108 | 108 | – | PCP CP1 |
| NC_002306 | 639 | 2 951 | 109 | 879 | 771 | KIARTG|RGAIYV | PCP CP2 | |
| 2 952 | 7 478 | 880 | 2 388 | 1 509 | YNKMGG|GDKTVS | PCP CP3 | ||
| 7 479 | 8 948 | 2 389 | 2 878 | 490 | VSPKSG|SGFFDV | PCP CP4 | ||
| 8 949 | 9 854 | 2 879 | 3 180 | 302 | STLQ|SGLR | nsp2 | ||
| 9 855 | 10 736 | 3 181 | 3 474 | 294 | VNLQ|AGKV | nsp3 | ||
| 10 737 | 10 985 | 3 475 | 3 557 | 83 | STVQ|SKLT | nsp4 | ||
| 10 986 | 11 570 | 3 558 | 3 752 | 195 | TILQ|SVAS | nsp5 | ||
| 11 571 | 11 903 | 3 753 | 3 863 | 111 | TKLQ|NNEI | nsp6 | ||
| 11 904 | 12 308 | 3 864 | 3 998 | 135 | VRLQ|AGKP | nsp7 | ||
| 12 309 | 15 094 | 3 999 | 4 927 | 929 | TSMQ|SFTV | nsp9b | ||
| 15 095c | 16 891c | 4 928 | 5 526 | 599 | TVLQ|AAGM | nsp10 | ||
| 16 892c | 18 448c | 5 527 | 6 045 | 519 | IGLQ|AKPE | nsp11 | ||
| 18 449c | 19 465c | 6 046 | 6 384 | 339 | KALQ|SLEN | nsp12 | ||
| 19 466c | 20 365c | 6 385 | 6 684 | 300 | PQLQ|SAEW | nsp13 | ||
| 2 | PEDV | 297 | 626 | 1 | 110 | 110 | – | PCP CP1 |
| NC_003436 | 627 | 2 981 | 111 | 895 | 785 | FGRRGG|NIVPVD | PCP CP2 | |
| 2 982 | 7 847 | 896 | 2 517 | 1 622 | FKKKGG|GDVKFS | PCP CP3 | ||
| 7 848 | 9 287 | 2 518 | 2 997 | 480 | ANKKGA|GLPSFS | PCP CP4 | ||
| 9 288 | 10 193 | 2 998 | 3 299 | 302 | STLQ|AGLR | nsp2 | ||
| 10 194 | 11 033 | 3 300 | 3 579 | 280 | VNLQ|GGYV | nsp3 | ||
| 11 034 | 11 282 | 3 580 | 3 662 | 83 | SSVQ|SKLT | nsp4 | ||
| 11 283 | 11 867 | 3 663 | 3 857 | 195 | SMLQ|SVAS | nsp5 | ||
| 11 868 | 12 192 | 3 858 | 3 965 | 108 | VKLQ|NNEI | nsp6 | ||
| 12 191 | 12 596 | 3 966 | 4 100 | 135 | VRLQ|AGKQ | nsp7 | ||
| 12 597 | 15 376 | 4 101 | 5 027 | 927 | SIMQ|STDM | nsp9d | ||
| 15 377 | 17 167 | 5 028 | 5 624 | 597 | AVLQ|SAGL | nsp10 | ||
| 17 168 | 18 718 | 5 625 | 6 141 | 517 | SDLQ|ANEG | nsp11 | ||
| 18 719 | 19 735 | 6 142 | 6 480 | 339 | NNLQ|GLEN | nsp12 | ||
| 19 736 | 20 638 | 6 481 | 6 781 | 301 | PQLQ|ASEW | nsp13 | ||
Note that of the 24 coronavirus genomes, the predicted results by ZCURVE_CoV 2.0 are in complete agreement with those annotated by NCBI, except for the genomes of TGEV and PEDV, in which the predicted results are different from those annotated by NCBI. In this table the reasons for these conflicts are analyzed.
This conflict with the annotation is caused by the problematic annotation.
The locations are different from the annotation, which is caused by a questionable additional insertion of an amino acid residue in nsp9.
This conflict with the annotation is caused by the non-standard frameshift.
Using the present method, only few false positive predictions exist in the prediction results. The tedious calculations for deriving the cutoff value can be avoided by restricting the sizes of the scanning regions and only selecting the site with the maximum score within this region. The annotated cleavage sites often correspond to the highest scores measured by the PWM method. However, the sites scored high by the PWM method do not always correspond to the cleavage sites and vice versa. Restricting the scanning regions for each of the cleavage sites is more efficient to reduce the false positive prediction rate. For the prediction of the 3C-like proteinase cleavage sites, there are only two conflicts between the predicted results and the annotations, which are marked in Table 3. The first conflict lies in the locations of non-structural proteins downstream of nsp9 in TGEV, which may be due to the problematic annotation. The length of amino acid sequences for ORF 1ab (315–20 368 bp) should be 6684 aa, instead of 6685 aa, which is annotated by NCBI. The questionable additional insertion of an amino acid residue in nsp9 causes one conflict of location errors. The second is caused by a non-standard frameshift site in PEDV, which causes the difference of five amino acid residues between the non-standard frameshift site and the standard frameshift site. For this reason, the octapeptide predicted by the present method is SIMQ|STDM instead of the annotated SIMQ|STDY.
Using the cleavage model of the papain-like proteinase presented here, the additional cleavage sites in the annotated PCP CP3 predicted by this method for SARS-CoV TOR2, MHV and IBV are ISLKGG|KIVSTC, FSLKGG|AVFSRM and VEKKAG|GIVSGT, respectively. The predicted cleavable peptides are similar to those annotated by NCBI, for example, the cleavable peptide FSLKGG|AVFSRM in MHV is different from the annotated peptide FSLKGG|AVFSYF in BCoV only at the P5′ and P6′ positions. Comparison between the N-terminal sequences of the polyprotein 1abs in MHV and BCoV is shown in Fig. 1. The additional cleavage site in the annotated PCP CP3 predicted by this method for MHV is situated at the corresponding position where the PCP CP3 and PCP CP4 are cleaved in BCoV. Cleavage sites that have been annotated by NCBI are indicated by black arrows, whereas that predicted by the present method is indicated by the open arrow. Therefore, the annotated PCP CP3 of SARS-CoV TOR2, MHV and IBV may be a precursor, which can be cleaved further.
Based on the present method, the genomes without annotation have been annotated. To save printing space, only the results of BCoVL and SARS-CoV BJ01 are summarized in Table 4 . The detailed annotations for other coronavirus genomes are accessible at http://tubic.tju.edu.cn/sars/.
Table 4.
The predicted results by the present method for BCoVL and SARS-CoV BJ01
| Number | Genome | Location (bp) |
Location (aa) |
Length (aa) | Cleavable peptide | Feature | ||
| Start | Stop | Start | Stop | |||||
| 1 | BCoVL | 211 | 948 | 1 | 246 | 246 | – | PCP CP1 |
| AF391542 | 949 | 2 763 | 247 | 851 | 605 | IRGYRG|VKPLLY | PCP CP2 | |
| 2 764 | 8 460 | 852 | 2 750 | 1 899 | WRVPCA|GRRVTF | PCP CP3 | ||
| 8 461 | 9 948 | 2 751 | 3 246 | 496 | FSLKGG|AVFSYF | PCP CP4 | ||
| 9 949 | 10 857 | 3 247 | 3 549 | 303 | SFLQ|SGIV | nsp2 | ||
| 10 858 | 11 718 | 3 550 | 3 836 | 287 | IKLQ|SKRT | nsp3 | ||
| 11 719 | 11 985 | 3 837 | 3 925 | 89 | SQFQ|SKLT | nsp4 | ||
| 11 986 | 12 576 | 3 926 | 4 122 | 197 | TVLQ|ALQSa | nsp5 | ||
| 12 577 | 12 906 | 4 123 | 4 232 | 110 | TVLQ|NNEL | nsp6 | ||
| 12 907 | 13 317 | 4 233 | 4 369 | 137 | VRLQ|AGTA | nsp7 | ||
| 13 318 | 16 100 | 4 370 | 5 297 | 928 | TTVQ|SKDT | nsp9 | ||
| 16 101 | 17 909 | 5 298 | 5 900 | 603 | AVMQ|SVGA | nsp10 | ||
| 17 910 | 19 472 | 5 901 | 6 421 | 521 | TRVQ|CSTN | nsp11 | ||
| 19 473 | 20 594 | 6 422 | 6 795 | 374 | TKLQ|SLEN | nsp12 | ||
| 20 595 | 21 491 | 6 796 | 7 094 | 299 | PRLQ|AASD | nsp13 | ||
| 2 | BJ01 | 246 | 782 | 1 | 179 | 179 | – | PCP CP1 |
| AY278488 | 783 | 2 699 | 180 | 818 | 639 | TRELNG|GAVTRY | PCP CP2 | |
| 2 700 | 8 465 | 819 | 2 740 | 1 922 | FRLKGG|APIKGV | PCP CP3 | ||
| 8 466 | 9 965 | 2 741 | 3 240 | 500 | ISLKGG|KIVSTCb | PCP CP4 | ||
| 9 966 | 10 883 | 3 241 | 3 546 | 306 | AVLQ|SGFR | nsp2 | ||
| 10 884 | 11 753 | 3 547 | 3 836 | 290 | VTFQ|GKFK | nsp3 | ||
| 11 754 | 12 002 | 3 837 | 3 919 | 83 | ATVQ|SKMS | nsp4 | ||
| 12 003 | 12 596 | 3 920 | 4 117 | 198 | ATLQ|AIAS | nsp5 | ||
| 12 597 | 12 935 | 4 118 | 4 230 | 113 | VKLQ|NNEL | nsp6 | ||
| 12 936 | 13 352 | 4 231 | 4 369 | 139 | VRLQ|AGNA | nsp7 | ||
| 13 353 | 16 147 | 4 370 | 5 301 | 932 | PLMQ|SADA | nsp9 | ||
| 16 148 | 17 950 | 5 302 | 5 902 | 601 | TVLQ|AVGA | nsp10 | ||
| 17 951 | 19 531 | 5 903 | 6 429 | 527 | ATLQ|AENV | nsp11 | ||
| 19 532 | 20 569 | 6 430 | 6 775 | 346 | TRLQ|SLEN | nsp12 | ||
| 20 570 | 21 463 | 6 776 | 7 073 | 298 | PKLQ|ASQA | nsp13 | ||
The alternative cleavage site predicted by the present method is at QALQ|SEFV (Gln-3928|Ser-3929).
Compared with the annotation, this cleavage site is predicted additionally by the present method.
4. Conclusion
SARS is an extremely severe disease, which has spread to many countries around the world. Evidence shows that SARS is caused by a new coronavirus, i.e. SARS-CoV. A system, called ZCURVE_CoV 1.0, has been developed previously to recognize protein-coding genes in coronavirus genomes, especially suitable for SARS-CoV genomes [10]. Here an improved version of the system, ZCURVE_CoV 2.0, has been developed to identify all the non-structural proteins cleaved by viral proteinases in the polyproteins. Consequently, all the non-structural proteins in the 24 completely sequenced coronavirus genomes are predicted. Compared with the known annotations, including those based on experimental evidence, the performance of the present method is satisfactory.
Acknowledgements
We are indebted to Prof. Jingchu Luo of Peking University for the timely updated SARS-related information provided. We are also grateful to both referees for their constructive comments, which are very useful to improve the quality of the paper. Invaluable assistance from Ren Zhang is gratefully acknowledged. The present study was supported in part by the 973 Project of China (Grant 1999075606).
References
- 1.Peiris J.S. Lancet. 2003;361:1319–1325. doi: 10.1016/S0140-6736(03)13077-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ksiazek T.G. New Engl. J. Med. 2003;348:1953–1966. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- 3.Drosten C. New Engl. J. Med. 2003;348:1967–1976. doi: 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]
- 4.Tsang K.W. New Engl. J. Med. 2003;348:1977–1985. doi: 10.1056/NEJMoa030666. [DOI] [PubMed] [Google Scholar]
- 5.Lee N. New Engl. J. Med. 2003;348:1986–1994. doi: 10.1056/NEJMoa030685. [DOI] [PubMed] [Google Scholar]
- 6.Poutanen S.M. New Engl. J. Med. 2003;348:1995–2005. doi: 10.1056/NEJMoa030634. [DOI] [PubMed] [Google Scholar]
- 7.Rota P.A. Science. 2003;300:1394–1398. [Google Scholar]
- 8.Marra M.A. Science. 2003;300:1399–1404. doi: 10.1126/science.1085953. [DOI] [PubMed] [Google Scholar]
- 9.Qin E’d. Chin. Sci. Bull. 2003;48:941–948. doi: 10.1007/BF03184203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen L.L., Ou H.Y., Zhang R., Zhang C.-T. Biochem. Biophys. Res. Commun. 2003;307:382–388. doi: 10.1016/S0006-291X(03)01192-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ziebuhr J., Snijder E.J., Gorbalenya A.E. J. Gen. Virol. 2000;81:853–879. doi: 10.1099/0022-1317-81-4-853. [DOI] [PubMed] [Google Scholar]
- 12.von Heijne G. Nucleic Acids Res. 1986;14:4683–4690. doi: 10.1093/nar/14.11.4683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.von Grotthuss M., Wyrwicz L.S., Rychlewski L. Cell. 2003;113:701–702. doi: 10.1016/S0092-8674(03)00424-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brierley I., Jenner A.J., Inglis S.C. J. Mol. Biol. 1992;227:463–479. doi: 10.1016/0022-2836(92)90901-U. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nam S.H., Copeland T.D., Hatanaka M., Oroszlan S. J. Virol. 1993;67:196–203. doi: 10.1128/jvi.67.1.196-203.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schneider T.D., Stephens R.M. Nucleic Acids Res. 1990;18:6097–6100. doi: 10.1093/nar/18.20.6097. [DOI] [PMC free article] [PubMed] [Google Scholar]