

Abstract
We have evaluated antisense design and efficacy of locked nucleic acid (LNA) and DNA oligonucleotide (ON) mix‐mers targeting the conserved HIV‐1 dimerization initiation site (DIS). LNA is a high affinity nucleotide analog, nuclease resistant and elicits minimal toxicity. We show that inclusion of LNA bases in antisense ONs augments the interference of HIV‐1 genome dimerization. We also demonstrate the concomitant RNase H activation by six consecutive DNA bases in an LNA/DNA mix‐mer. We show ON uptake via receptor‐mediated transfection of a human T‐cell line in which the mix‐mers subsequently inhibit replication of a clinical HIV‐1 isolate. Thus, the technique of LNA/DNA mix‐mer antisense ONs targeting the conserved HIV‐1 DIS region may provide a strategy to prevent HIV‐1 assembly in the clinic.
Keywords: LNA, Antisense, RNase H, HIV, Dimerization initiation site
1. Introduction
Antisense oligonucleotides (ONs) have become a major tool for functional genomics and there are many compounds in clinical trials for a variety of human disorders [1]. One antisense ON (Vitravene, Isis Pharmaceuticals, Carlsbad) has been approved for the clinic to inhibit cytomegalovirus. The antisense ON recognizes its target by Watson‐Crick base‐pairing. The inhibitory functions of antisense ONs are believed to be degradation of the target RNA by RNase H, steric hindrance of the translation machinery or prevention of other protein interactions with the target RNA. RNase H is a cellular enzyme that cleaves the RNA strand in a DNA/RNA hybrid [2, 3].
For antisense applications, increased ON stability towards nuclease degradation is important. Other desired properties are high affinity for the target, RNase H activation, and low toxicity along with good solubility and uptake. A promising and recent technology is the development of locked nucleic acid (LNA, Fig. 1 A) [4, 5]. LNA shows minimal toxicity, enhanced binding to its target sequence and is stable to nuclease degradation [6, 7]. LNA alone cannot activate RNase H, but in a mix‐mer combination with DNA increased RNase H activation can be achieved [7, 8, 9, 10]. LNA mix‐mers have very high potency compared to other antisense chemistries and their downregulating capacity also close in on that of siRNA [11].
Figure 1.

(A) Locked nucleic acid (LNA) structure. (B) HIV‐1 subtype A dimerization initiation site (DIS). The six self‐complementary bases are marked in bold.
We have applied a LNA/DNA mix‐mer technology on a well‐conserved target in HIV‐1, the dimerization initiation site (DIS) [12, 13]. The HIV genome is a homo‐dimer of two sense RNA single strands. The DIS is a stem loop structure with six self‐complementary nucleotides at the top (Fig. 1B, bold). There are two major motifs in HIV‐1; GUGCAC in subtypes A and C, and GCGCGC in subtypes B and D [14, 15] (see also current alignments http://www.hiv.lanl.gov base 881–887). The loop is located between the primer binding site and the splice donor site at the end of the long terminal repeat (LTR) [16] and is involved in the dimerization of the HIV‐1 genome, packaging and proviral synthesis [17, 18, 19, 20, 21]. The HIV‐1 subtype A motif is a good antisense target in in vitro cell free system where the HIV‐1 genome dimerization is inhibited by DIS targeting ONs. It has been shown that a 9‐mer RNA ON is a good inhibitor, whereas the corresponding DNA ON does not affect the dimerization [22, 23]. These studies also indicate that sense ONs work as competitive inhibitors to this self‐complimentary target. Here, we aim at increasing the potency of the DIS‐targeting ONs by exchanging DNA nucleotides for high affinity, nuclease resistance LNA in the DNA sequence creating LNA/DNA mix‐mers.
We demonstrate that a mix‐mer design can enhance the HIV‐1 inhibiting properties. In vitro, the LNA/DNA mix‐mers enhance the inhibition of HIV‐1 genome dimerization and activate RNase H when the mix‐mer has a DNA stretch of at least six bases. We subsequently show good uptake of the LNA/DNA mix‐mers in a T‐cell line and that they can inhibit replication of a clinical HIV‐1 isolate.
2. Materials and methods
2.1. Antisense oligonucleotides
All ONs used in this study are listed in Table 1 . LNA/DNA mix‐mers were synthesized by Exiqon A/S (Copenhagen, Denmark) and Proligo (Boulder, CO) under license from Exiqon A/S. Regular DNA oligonucleotides were synthesized by ThermoHybaid (Ulm, Germany). The ONs cover the HIV‐1 subtype A DIS loop; 9‐mers (DNA, RNA, LNA and LNA/DNA mix‐mers) and 18‐mers (LNA/DNA mix‐mers with either 3′ or 5′ DNA tails).
Table 1.
Oligonucleotides used in this study
HIV‐1 subtype A, DIS  (reversed): (reversed): |
3′‐agcggagaacgac
 gucguucggcuc‐5′
gucguucggcuc‐5′ |
|
| Name | Length | Sequence |
| asDIS 1 | 9 nt | 5′‐ugugcaccu‐3′ |
| asDIS 2 | 9 nt | 5′‐tgtgcacct‐3′ |
| asDIS 3 | 9 nt | 5′‐TgtgcacCT‐3′ |
| asDIS 4 | 9 nt | 5′‐TGTgcACCT‐3′ |
| asDIS 5 | 9 nt | 5′‐TGTGCACCT‐3′ |
| sDIS 6 | 9 nt | 5′‐AGGTgcACA‐3′ |
| asDIS 7 mm | 18 nt | 5′‐TG Gt cAC A Tc c gcaagcc‐3′ |
| asDIS 8 | 18 nt | 5′‐ TGTgcACCTcagcaagcc‐3′ |
| asDIS 9 | 18 nt | 5′‐ctcttgctgTGTgcACCT‐3′ |
| sDIS 10 | 18 nt | 5′‐AGGTgcACAcagcaagag‐3′ |
| sDIS 11 | 18 nt | 5′‐ggcttgctgAGGTgcACA‐3′ |
as: antisense, s: sense, mm: mismatch control. Upper case bold: LNA, lower case: DNA, lower case italics: RNA, underlined: mismatch.
2.2. Virus and cells
Virus was multiplied in the Jurkat‐Tat T‐cell line [24, 25], cultured in RPMI‐1640 with Glutamax I, 10% fetal bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin and 0.29 mg/ml l‐glutamine (LifeTechnologies, Sweden). The virus stock has 200 TCID50 on Jurkat‐Tat cells. The HIV strain used was a clinical subtype A designated #2102 (GenBank Accession No. http://AY817688). The virus was isolated on human PBL, amplified on PBMC according to the standard protocol.
2.3. Plasmid construction
pDIS2102 was constructed by RT‐PCR amplification of the first 620 bases of the #2102 genome. The amplificate was ligated into pGem‐T (Promega, WI) downstream the T7 promoter. RNA for the reverse transcriptase reaction was isolated from the #2102 viral supernatants [26]. Primers for the PCR amplification were 5′‐atatatgcatgctctctcttgttagac‐3′ ∼260 nt upstream DIS including an SphI site for directional cloning and 5′‐tgtcttttacctctatccgttgatg‐3′ ∼360 nt downstream DIS, corresponding to the region 1–615 of HIV‐1 MAL described previously [27]. The cloned fragment from #2102 was sequenced to confirm the DIS region.
2.4. In vitro transcribed RNA and purification
pDIS2102 was linearized with NotI and transcribed with T7 RNA polymerase (Promega) generating a 630 nucleotide long transcript. The transcribed product was purified on an agarose gel, the appropriate band was excised and electro‐eluted into 7.5 M ammonium acetate, precipitated by ethanol, washed and re‐suspended in nuclease free water.
2.5. In vitro dimerization
The dimerization was performed as described previously [22, 23]. In vitro transcribed RNA was denatured at 96 °C for 2 min, put on ice and mixed with dimerization buffer (5×: 250 mM sodium cacodylate, 1.5 μM KCl and 25 mM MgCl2) and ON. The final RNA concentration was 100–200 nM and ON concentration 1–2 μM, a molar ratio of 1:10. The mix was incubated at 37 °C for 30 min and analyzed on a 1.2% agarose gel in TBM5 buffer (45 mM Tris borate, pH 8.3, 5 mM MgOAc2) containing ethidium bromide. The separation was performed at +4 °C. The gel bands were quantified with GelDoc and QuantityOne software (BioRad, CA).
2.6. RNase H activation
100 μg RNA (final concentration 20 nM) was mixed with antisense ON to a final concentration of 200 nM, 2.5 U rRNasin (Promega), RNase H buffer and 2.5 U RNase H (Fermentas AB, Vilnius, Lithuania), and incubated at 37 °C for 30 min. The RNA was analyzed on a 1.5% glyoxal denaturing agarose gel (Northern Max Gly kit, Ambion, Huntingdon, UK), stained with SYBR gold (Molecular Probes, Leiden, The Netherlands) and documented with GelDoc and Quantity One software (BioRad).
2.7. Receptor‐mediated transfection
Transferrin linked to polyethyleneimine (PEI) [28, 29], sold as DuoFect from Qbiogene (IIkirch, France), was used to transfect Jurkat‐Tat cells. Jurkat‐Tat cells were kept in exponential phase and split the day before transfection into medium with 50 μM deferrioxamine but without antibiotics. At the day of transfection, the cells were seeded in flat bottom 96‐well plates, 45 000 cells per well in 130 μl medium without antibiotics and deferrioxamine. A N/P (nitrogen/phosphate) ratio (see Duofect protocol, Qbiogene) of 4.8 was used, since it gave the highest frequency of transfected cells (data not shown). 1.9 μl of 50 μM antisense ON stock was mixed with 40 μl HBS (20 mM HEPES, pH 7.3 and 150 μM NaCl). 0.17–0.49 μl Duofect was added to another 40 μl of HBS (volume depending on weight of the ON; 0.17 μl for 9‐mers and 0.34 μl for 18‐mers to keep a N/P ratio of 4.8). The two solutions were combined and incubated at room temperature for 20–30 min and then added to the cells. Transfection mix was prepared for four wells and 20 μl of the mix was used per well (final ON concentration 160 nM).
Uptake of antisense ONs into the Jurkat‐Tat cell line was confirmed by transfection of a FITC labeled 16 nucleotides long LNA/phosphorothioate mix‐mer. The cells were subsequently analyzed by fluorescence microscopy and digital camera operated by OpenLab software (Improvision, Coventry, UK).
2.8. Inhibition of HIV replication
Cells were transfected with the LNA/DNA mix‐mers in 96 wells the day prior to infection (see transfection above). 100 μl transfected cell suspension was transferred to a new 96 well U‐bottom plate and each well was infected with 100 μl of undiluted virus supernatant (200 TCID50). At day one post infection (PI), cells were spun down and the medium changed to fresh medium with antibiotics. At day four PI, 100 μl of supernatant was removed and 100 μl fresh medium was added. Supernatants were harvested on day seven PI. The amount of HIV in the supernatants was quantified by p24 ELISA and reverse transcriptase activity assay (Lenti‐RT kit, CavidiTech, Uppsala, Sweden).
3. Results
3.1. Dimerization inhibition and RNase H activation by 9‐mer ONs
The inhibition of HIV dimerization by nine nucleotides long mix‐mer ONs shows improvement by exchanging DNA for LNA. A 630 nucleotide in vitro transcribed stretch of the #2102 isolate ltr/gag RNA was denatured and allowed to dimerize in the presence or absence of ONs, including LNA/DNA mix‐mers (Fig. 2 A). The RNA 9‐mer asDIS 1 reduced dimer formation by 61% compared to no ON, while the DNA 9‐mer asDIS 2 only induced 14% reduction (Fig. 2B). By exchanging three DNA bases (1 + 2) to LNA at the ends of the DNA 9‐mer (asDIS 3), the inhibitory capacity was enhanced to the levels of the RNA ON (asDIS 1), 62% reduction. asDIS 4 with seven exchanged bases and asDIS 5, all LNA, reduced dimerization with 42% and 45%, respectively. The sense 9‐mer sDIS 6 reduced the dimerization with 50% compared to the sample without ON.
Figure 2.
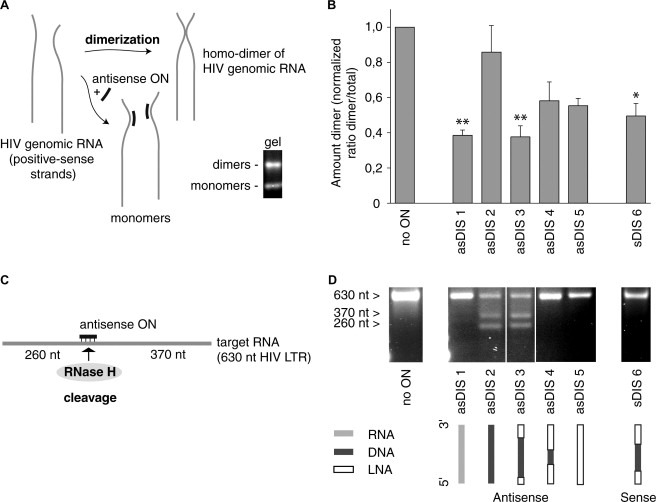
Assay schemes and 9‐mer performance. (A) Schematic of HIV‐1 dimerization and appearance of dimer and monomer on a gel. (B) Inhibition of in vitro dimerization by LNA/DNA mix‐mers. In vitro transcribed HIV DIS RNA has been allowed to dimerize in the absence or presence of oligonucleotides. Dimers and monomers are separated by gel electrophoresis and the bands are quantified and plotted as a ratio of the amount of dimer to the total amount of dimer and monomer together. The ratio has been normalized to the ratio where no oligonucleotide is present. Mean and standard deviation are from two to three replicates (*P < 0.05, **P < 0.01 and feb2s0014579304013791P < 0.001, as compared to DNA ON asDIS2). (C) Schematic of RNase H activation. (D) RNase H activation by LNA/DNA mix‐mers. In vitro transcribed HIV RNA (630 nucleotides) has been subjected to RNase H digestion in the absence or presence of oligonucleotides. The digested/undigested RNA has been separated by gel electrophoresis. The full‐length transcript is 630 nucleotides long (no ON), cut transcript at target site generates two fragments, 260 nt and 370 nt long. asDIS 2 also shows full‐length undigested transcript as with no ON.
LNA/DNA mix‐mers activate RNase H mediated RNA cleavage. The 630 nucleotide in vitro transcribed #2102 RNA was incubated with RNase H and the different ONs. The DIS target region is located roughly in the middle of the transcript and RNase H digestion at the target site would generate two cleavage products of around 260 and 370 nucleotides, depending on where the DNA is located in the ON (Fig. 2C). Incubation with RNase H but without ON did not mediate cleavage and showed the full‐length transcript only (Fig. 2D, no ON). The RNA transcript incubated with RNase H and the DNA antisense 9‐mer asDIS 2 generated the two expected bands, while the RNA ON asDIS 1 did not activate RNase H. RNase H was also activated by the antisense LNA/DNA mix‐mer asDIS 3, containing six consecutive DNA bases. asDIS 4 and asDIS 5 antisense 9‐mers containing two consecutive and no DNA bases, respectively, did not activate RNase H. The sense 9‐mer sDIS 6 did not activate RNase H.
3.2. Dimerization inhibition and RNase H activation by 18‐mer ONs
LNA/DNA 18‐mers contained either 3′ or 5′ nine nucleotides long DNA tails while keeping the 9 bases covering the DIS loop constant as in the 9‐mers asDIS 4 or sDIS 6. The nine nucleotide DNA tail should be sufficient for activating RNase H, while the loop spanning nine bases should give strong binding.
The dimerization inhibition using the 18‐mers was performed as with the 9‐mers. The antisense 18‐mers asDIS 8 and asDIS 9, with 3′‐ and 5′ DNA tail, respectively, reduced dimer formation with 59% and 54% (Fig. 3 A). They performed slightly better than the corresponding sense ONs (sDIS 10 and sDIS 11), which showed 36% and 40% reduction, respectively. asDIS 8, with 59% reduction, was similar in performance to the 9‐mers asDIS 1 and asDIS 3. The mismatch control asDIS 7 mm corresponding to asDIS 8 did not show noteworthy inhibition (10% reduction).
Figure 3.

18‐mer performance. (A) Inhibition of in vitro dimerization by different 18‐mer ONs in analogy with Fig. 2B. Mean and standard deviation are from two to three replicates (*P < 0.05, **P < 0.01 and feb2s0014579304013791P < 0.001, as compared to mismatch control asDIS 7 mm). (B) RNase H activation by different 18‐mer ONs as in Fig. 2D.
In the case of RNase H activation, the antisense 18‐mers asDIS 8 and asDIS 9 activated RNase H, whereas sDIS 10 and sDIS 11 did not (Fig. 3B). There was, however, weak non‐specific cleavage with sDIS 10 as well as with asDIS 9. The mismatch control asDIS 7 mm also showed non‐specific activation.
3.3. Receptor‐mediated transfection
Delivering mix‐mers by receptor‐mediated transfection showed high percentage of uptake, a prerequisite for antisense ON inhibition of HIV replication. PEI condenses and protects the ON, while the transferrin binds CD‐71 (transferrin receptor, on T‐cells for example) and causes receptor‐mediated endocytosis. The transfected ONs were taken up by most Jurkat‐Tat cells and accumulated in the endosomal compartments (Fig. 4 A). ONs could be detected up to eight days in some cells (data not shown). Other commercial transfection agents, including PEI, were also tried. However, all gave low percentage of transfection even though in some cases the few transfected cells displayed more ONs per cell (i.e., stronger fluorescent signal) (data not shown).
Figure 4.

(A) Receptor mediated delivery of FITC labeled LNA/phosphorothioate mix‐mer to Jurkat‐Tat cells. FITC has a green color and the DAPI nuclear stain is blue. Uptake in close to 100% of the cells is shown (low magnification). FITC labeled oligonucleotides are accumulated in endosomal compartments (high magnification). (B) Inhibition of HIV replication by 18‐mer LNA/DNA mix‐mers. The amount of reverse transcriptase in cell supernatants was measured seven days post infection by reverse transcriptase activity assay. Mean and standard deviation are from two separate experiments, each in which three to four separate infections per oligonucleotide were made (*P < 0.05, **P < 0.01 and feb2s0014579304013791P < 0.001, as compared to mismatch control asDIS 7 mm).
3.4. Inhibition of virus replication
LNA/DNA mix‐mers inhibit HIV‐1 replication. The antisense 18‐mer with a 3′ DNA tail asDIS 8 had a clear effect in inhibiting the HIV replication. HIV‐1 RT activity was reduced with 64% compared to untreated cells (Fig. 4B). HIV‐1 inhibition by asDIS 8 was initially seen by ELISA detecting the HIV‐1 p24 antigen (data not shown). asDIS 9 and sDIS 10 reduced the virus production with 33% and 50%, respectively. The sense ON sDIS 11 surprisingly displayed a 59% reduction. The mismatch control (asDIS 7 mm) slightly affected the virus production. The 9‐mers did not inhibit virus replication per se (data not shown).
4. Discussion
The current study shows the benefit of the nucleotide analog LNA in a biological context and is the first study that demonstrates that the HIV‐1 dimerization site is a potential antisense target that can combat the virus replication. This is of importance for implication of the LNA/DNA mix‐mer technology in development of antisense therapeutics against HIV‐1 and other diseases.
Here, we have shown that high affinity antisense LNA/DNA mix‐mers enhance inhibition of HIV dimerization and that they can induce RNase H degradation of the target RNA. In addition, the LNA modification promotes stability but not toxicity [7]. To deliver the ONs, we have employed a receptor‐mediated transfection system, which may be clinically applicable and has given a good uptake in the transfected T‐cell line.
LNA has high binding affinity to RNA [4, 30]. The antisense RNA 9‐mer asDIS 1 showed inhibition and the corresponding DNA asDIS 2 did not, as has been shown previously [22]. When exchanging three flanking DNA nucleotides (one 5′ and two 3′) bases for LNA (asDIS 3), the same potent inhibitory effect as with the RNA was seen. These results showed that it was indeed possible to confer DNA ON with better DIS targeting properties by including the enhanced binding capacity of LNA. Further increase in the LNA content (asDIS 4 and asDIS 5) also showed improved dimerization inhibitory effects compared to DNA alone. Interestingly, but not surprisingly, the sense LNA/DNA mix‐mer (sDIS 6) also had a good inhibitory effect. DIS is self‐complementary and the dimerization interaction occurs between two RNA sense strands. Hence, the sense ON can bind DIS and there are indications that sense ONs work as competitive inhibitors to the dimerization process [22, 23].
In the RNase H assays asDIS 3, the LNA/DNA mix‐mer with a 6 DNA nucleotide window, activated RNase H mediated cleavage as effectively as the DNA ON (asDIS 2). This is in analogy with previous studies [7, 10, 31]. Kurreck et al. showed that eight consecutive DNA bases in an LNA/DNA mix‐mer antisense ON give full RNase H activation, even though six bases will still be sufficient to induce activation. Shen et al. have shown that four consecutive DNA bases activate RNase H even if there is a nucleotide mismatch [32].
The antisense 18‐mer with a 3′ DNA tail, asDIS 8, was a good inhibitor of dimerization and a good activator of RNase H. asDIS 8 subsequently also inhibited HIV‐1 replication. The sense version sDIS 11 was also a good inhibitor of virus replication without activating RNase H. This is in accordance with the discussion concerning the sense 9‐mer sDIS6. HIV genome has two RNA sense strands coming together in the dimerization process. The 18‐mer sense ON sDIS 11 can pair to DIS and likely function as a competitive inhibitor to the dimerization process. The sense ON sDIS 10 also showed a slight downregulation of HIV‐1 replication. Even though asDIS 9 inhibited in vitro dimerization and activated RNase H, it was not an efficient ON in inhibiting HIV‐1 replication. This indicates that target site availability also may be different in vitro and in vivo for the different ONs. Seemingly, inhibition of dimerization is necessary for HIV‐1 downregulation of virus replication when targeting DIS, while RNase H activation is not. Hence, interruption of the natural HIV‐1 genome processing is more important than targeting the genome for RNase H degradation when targeting DIS. RNase H also induced some non‐specific target cleavage for some of the 18‐mers, which also might contribute to reduce specificity. Additionally, we believe that the seen inhibition of virus replication is due to antisense effects and exclude side‐effects like cytotoxicity. Neither the “no ON” nor the mismatch control (asDIS 7 mm) inhibited virus replication, which indicates that there are no side‐effects due to the presence of control ON per se that cause inhibition of virus replication. The ONs have also been checked for immunostimulatory CpG motifs, which could interfere and there are none.
The 9‐mer ONs did not inhibit HIV replication (data not shown). This could be due to unspecific binding of the ONs to cellular RNA molecules and might not play a practical role for cell culture or in vivo use.
We have relative modest inhibitory effects on HIV replication. However, this is performed with a clinical HIV‐1 subtype A isolate, not adapted for assaying and replication in a T‐cell line. The target has been cloned from this isolate and adapted for evaluation in vitro and finally tested for virus replication, with the objective to use materials as close to clinical relevance. The ON concentration used (160 nM) is also very low. This makes the LNA/DNA mix‐mers competitive also to siRNA. For example, the SARS virus replication is inhibited by siRNA in the 100 nM range [33].
The mix‐mer design from this study could be used to target other important secondary structures along with other efficient antisense targets like trans‐activating region (TAR), which has in vitro been successfully targeted with LNA mix‐mers [34, 35]. In conclusion, introduction of LNA improves antisense inhibition of HIV‐1 dimerization. A stretch of six consecutive DNA bases in an LNA/DNA mix‐mer is necessary and sufficient to activate RNase H degradation of the target RNA. Receptor‐mediated transfection is suitable for ON delivery to a T‐cell line and the LNA/DNA mix‐mers are capable of inhibiting HIV‐1 replication at low concentrations. We hope these data would stimulate the introduction of LNA/DNA mix‐mer technology against HIV in in vivo models and possibly in clinical settings.
Acknowledgments
This work was supported by the PhD Program in Biotechnology with an Industrial Focus at Karolinska Institutet and Pharmacia Corporation. Plasmids for an initial study were kindly provided by Roland Marquet and Jean‐Christophe Paillart (IBMC, Strasbourg, France). We thank Kajsa Aperia for viral stocks, Karin Mattsson for microscopy and Ola Larsson for statistical analysis.
Elmén Joacim,Zhang Hong-Yan,Zuber Bartek,Ljungberg Karl,Wahren Britta,Wahlestedt Claes and Liang Zicai(2004), Locked nucleic acid containing antisense oligonucleotides enhance inhibition of HIV-1 genome dimerization and inhibit virus replication, FEBS Letters, 578, doi: 10.1016/j.febslet.2004.11.015
References
- 1. Dove A., Antisense and sensibility. Nat. Biotechnol., 20, (2002), 121– 124. [DOI] [PubMed] [Google Scholar]
- 2. Opalinska J.B., Gewirtz A.M., Nucleic-acid therapeutics: Basic principles and recent applications. Nat. Rev. Drug Discov., 1, (2002), 503– 514. [DOI] [PubMed] [Google Scholar]
- 3. Stein C.A., The experimental use of antisense oligonucleotides: a guide for the perplexed. J. Clin. Invest., 108, (2001), 641– 644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Koshkin A.A., Nielsen P., Meldgaard M., Rajwanshi V.K., Singh S.K., Wengel J., LNA (Locked Nucleic Acid): An RNA mimic forming exceedingly stable LNA:LNA duplexes. J. Am. Chem. Soc., 120, (1998), 13252– 13253. [Google Scholar]
- 5. Singh S.K., Nielsen P., Koshkin A.A., Wengel J., J. Chem. Commun., (1998), 455– 456. [Google Scholar]
- 6. Braasch D.A., Corey D.R., Locked nucleic acid (LNA): fine-tuning the recognition of DNA and RNA. Chem. Biol., 8, (2001), 1– 7. [DOI] [PubMed] [Google Scholar]
- 7. Wahlestedt C., Potent and nontoxic antisense oligonucleotides containing locked nucleic acids. Proc. Natl. Acad. Sci. USA, 97, (2000), 5633– 5638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Braasch D.A., Liu Y., Corey D.R., Antisense inhibition of gene expression in cells by oligonucleotides incorporating locked nucleic acids: effect of mRNA target sequence and chimera design. Nucleic Acids Res., 30, (2002), 5160– 5167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Crinelli R., Bianchi M., Gentilini L., Magnani M., Design and characterization of decoy oligonucleotides containing locked nucleic acids. Nucleic Acids Res., 30, (2002), 2435– 2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kurreck J., Wyszko E., Gillen C., Erdmann V.A., Design of antisense oligonucleotides stabilized by locked nucleic acids. Nucleic Acids Res., 30, (2002), 1911– 1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Grunweller A., Wyszko E., Bieber B., Jahnel R., Erdmann V.A., Kurreck J., Comparison of different antisense strategies in mammalian cells using locked nucleic acids, 2′-O-methyl RNA, phosphorothioates and small interfering RNA. Nucleic Acids Res., 31, (2003), 3185– 3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Paillart J.C., Shehu-Xhilaga M., Marquet R., Mak J., Dimerization of retroviral RNA genomes: an inseparable pair. Nat. Rev. Microbiol., 2, (2004), 461– 472. [DOI] [PubMed] [Google Scholar]
- 13. Jakobsen M.R., Damgaard C.K., Andersen E.S., Podhajska A., Kjems J., A genomic selection strategy to identify accessible and dimerization blocking targets in the 5′-UTR of HIV-1 RNA. Nucleic Acids Res., 32, (2004), e67– [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Laughrea M., Jetté L., Mak J., Kleiman L., Liang C., Wainberg M.A., Mutations in the kissing-loop hairpin of human immunodeficiency virus type 1 reduce viral infectivity as well as genomic RNA packaging and dimerization. J. Virol., 71, (1997), 3397– 3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. St. Louis D., Gotte D., Sanders-Buell E., Ritchey D.W., Salminen M.O., Carr J.K., McCutchan F.E., Infectious molecular clones with the nonhomologous dimer initiation sequences found in different subtypes of human immunodeficiency virus type 1 can recombine and initiate a spreading infection in vitro. J. Virol., 72, (1998), 3991– 3998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Skripkin E., Paillart J.C., Marquet R., Ehresmann B., Ehresmann C., Identification of the primary site of the human immunodeficiency virus type 1 RNA dimerization in vitro. Proc. Natl. Acad. Sci. USA, 91, (1994), 4945– 4949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shen N., Jette L., Liang C., Wainberg M.A., Laughrea M., Impact of human immunodeficiency virus type 1 RNA dimerization on viral infectivity and of stem-loop B on RNA dimerization and reverse transcription and dissociation of dimerization from packaging. J. Virol., 74, (2000), 5729– 5735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. McBride M.S., Panganiban A.T., The human immunodeficiency virus type 1 encapsidation site is a multipartite RNA element composed of functional hairpin structures. J. Virol., 70, (1996), 2963– 2973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Paillart J.-C., Berthoux L., Ottmann M., Darlix J.-L., Marquet R., Ehresmann B., Ehresmann C., A dual role of the putative RNA dimerization initiation site of human immunodeficiency virus type 1 in genomic RNA packaging and proviral DNA synthesis. J. Virol., 70, (1996), 8348– 8354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Clever J.L., Parslow T.G., Mutant human immunodeficiency virus type 1 genomes with defects in RNA dimerization or encapsidation. J. Virol., 71, (1997), 3407– 3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Berkhout B., van Wamel J.L., Role of the DIS hairpin in replication of human immunodeficiency virus type 1. J. Virol., 70, (1996), 6723– 6732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Skripkin E., Paillart J.-C., Marquet R., Blumenfeld M., Ehresmann B., Ehresmann C., Mechanisms of inhibition of in vitro dimerization of HIV type 1 by sense and antisense oligonucleotides. J. Biol. Chem., 271, (1996), 28812– 28817. [DOI] [PubMed] [Google Scholar]
- 23. Lodmell J.S., Paillart J.C., Mignot D., Ehresmann B., Ehresmann C., Marquet R., Oligonucleotide-mediated inhibition of genomic RNA dimerization of HIV-1 strains MAL and LAI: a comparative analysis. Antisense Nucleic Acid Drug Dev., 8, (1998), 517– 529. [DOI] [PubMed] [Google Scholar]
- 24. Caputo A., Sodroski J.G., Haseltine W.A., Constitutive expression of HIV-1 tat protein in human Jurkat T cells using a BK virus vector. J. Acquir. Immune Defic. Syndr., 3, (1990), 372– 379. [PubMed] [Google Scholar]
- 25. Asjo B., Albert J., Chiodi F., Fenyo E.M., Improved tissue culture technique for production of poorly replicating human immunodeficiency virus strains. J. Virol. Methods, 19, (1988), 191– 196. [DOI] [PubMed] [Google Scholar]
- 26. Chomczynski P., Sacchi N., Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem., 162, (1987), 156– 159. [DOI] [PubMed] [Google Scholar]
- 27. Marquet R., Paillart J.C., Skripkin E., Ehresmann C., Ehresmann B., Dimerization of human immunodeficiency virus type 1 RNA involves sequences located upstream of the splice donor site. Nucleic Acids Res., 22, (1994), 145– 151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Boussif O., Lezoualc'h F., Zanta M.A., Mergny M.D., Scherman D., Demeneix B., Behr J.-P., A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: Polyethyleneimine. Proc. Natl. Acad. Sci. USA, 92, (1995), 7297– 7301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kircheis R., Kichler A., Wallner G., Kursa M., Ogris M., Felzmann T., Buchberger M., Wagner E., Coupling of cell-binding ligands to polyethyleneimine for targeted gene delivery. Gene Therapy, 4, (1997), 409– 418. [DOI] [PubMed] [Google Scholar]
- 30. Crook S.T., Antisense Drug Technology: Principles, Strategies and Applications. (2001), Marcel Dekker, Inc ; New York: [Google Scholar]
- 31. Malchère C., Verheijen J., Van Der Laan S., Bastide L., Van Boom J., Lebleu B., Robbins I., A short phosphodiester window is sufficient to direct RNase H-dependent RNA cleavage by antisense peptide nucleic acid. Antisense Nucleic Acid Drug Dev., 10, (2000), 463– 468. [DOI] [PubMed] [Google Scholar]
- 32. Shen L.X., Kandimalla E.R., Agrawal S., Impact of mixed-backbone oligonucleotides on target binding and target cleaving specificity and selectivity by Escherichia coli RNase H. Bioorg. Med. Chem., 6, (1998), 1695– 1705. [DOI] [PubMed] [Google Scholar]
- 33. Elmén J., Wahlestedt C., Brytting M., Wahren B., Ljungberg K., SARS Virus Inhibited by siRNA. Preclinica, 2, (2004), 135– 142. [Google Scholar]
- 34. Arzumanov A., Walsh A.P., Liu X., Rajwanshi V.K., Wengel J., Gait M.J., Oligonucleotide analogue interference with the HIV-1 Tat protein-TAR RNA interaction. Nucleosides Nucleotides Nucleic Acids, 20, (2001), 471– 480. [DOI] [PubMed] [Google Scholar]
- 35. Arzumanov A., Walsh A.P., Rajwanshi V.K., Kumar R., Wengel J., Gait M.J., Inhibition of HIV-1 Tat-dependent trans activation by steric block chimeric 2′-O-methyl/LNA oligoribonucleotides. Biochemistry, 40, (2001), 14645– 14654. [DOI] [PubMed] [Google Scholar]