

Abstract
Background
Orchardgrass (Dactylis glomerata L.) is a popular cool-season perennial grass with a high production value, and orchardgrass seed is the fourth top-selling forage grass seed in the world. However, its yield and quality are often affected by flooding. To date, the molecular responses of orchardgrass to flooding were poorly understood.
Results
Here, we performed mRNA-seq to explore the transcriptomic responses of orchardgrass to a short term flooding (8 h and 24 h). There were 1454 and 565 differentially expressed genes identified in the 8 h and 24 h of flooding, respectively, compared to well control. GO functional enrichment analysis showed that oxidoreductase activity and oxidation-reduction process were highly present, suggesting that flooding induced the response to oxygen stress. Pathways enrichment analysis highlights the importance of glutathione metabolism, peroxidase, glycolysis and plant hormone signal transduction in response to flooding acclimation. Besides, the ROS clearance system is activated by significantly expressed glutathione S-transferase and genes encoding SOD and CAT (CAT1 and CDS2). The significant positive correlation between RNA sequencing data and a qPCR analysis indicated that the identified genes were credible.
Conclusion
In the process of orchardgrass response to flooding stress, multiple differential genes and biological processes have participated in its acclimation to flooding, especially the biological processes involved in the removal of ROS. These results provide a basis for further research on the adaptation mechanism of orchardgrass to flood tolerance.
Keywords: Orchardgrass, Flooding, Transcriptome profiling, ROS scavenging
Background
Flooding is a major factor, limiting growth and production of plants on a global scale [1]. Agricultural development is hindered by flooding every year in many countries and regions around the world. For example, in 2012, flooding were devastating to economic development, and many indigenous fisheries and farming communities in the delta were damaged due to excess water covered in the area (Nigeria) [2]. In 2018, 24 provinces, autonomous regions, and municipalities suffered from floods in China. The area of crops affected by disasters reached 1304 thousand hectares, and the direct economic loss was up to 25.9 billion RMB. Therefore, flooding has considerably threatened the food security of developing countries and even has become a challenge for agricultural development in all countries of the world.
Plants suffer from various biotic and abiotic stresses throughout their life span. Higher plants are aerobic organisms. Flood is a kind of abiotic stress which have a devastating impact on crop growth and survival and thus on food production [3]. The main damage caused by flooding to plants is insufficient root oxygen supply caused by the poor gas exchange, affecting the synthesis of ATP and plant metabolism [4]. In addition, flood can also lead to plant mineral deficiency, nutrient imbalance or changes in nutrient distribution [5]. The physiological disturbances of plant roots caused by flooding can alter the concentration of macroscopic and trace elements in endogenous substances while changing the absorption of nutrients [6]. flooding can cause a variety of physiological disorders that affect plant growth, including root water flux reduction, hormone imbalance, carbohydrate distribution changes, nutrient absorption, early leaf senescence, and organ damage [7].
RNA-seq technology has higher sensitivity to capture low and high-level gene expression than array technologies [8]. RNA-seq can not only detect information about the relationship between a gene and its products, but also isolate genes that are difficult to handle by biochemistry [9]. The technique has been widely used to identify key pathways and differential gene expression in a variety of plant species involved in regulation during abiotic stress, such as drought stress [10], salt stress [11], heat stress [12], cold stress [13]. Recently, the use of transcriptome sequencing technology to study the response of plants to flooding stress is endless. Studies in Zea mays [14], Nymphoides peltata [15], Cucumis sativus [16], Persea americana [17], Sesamum indie [18], Taxodium [19], Cerasus sachalinensis [20] have revealed that antioxidative processes, carbohydrates, photosynthesis, glycolysis, phytohormone signal transduction, transcription factors (ERFs, MYB, HSP, and MAPK) and other metabolic processes are involved in the domestication of plants for flooding stress and play an important role. The results provide a better understanding for the adaptation mechanism of plants to flooding stress.
Orchardgrass (Dactylis glomerata L.) is one of the most important cool-season forage grasses [21]. Due to its high sugar and protein content, large biomass, and strong shade tolerance, it has been grown in East Asia, Europe, and North America for more than 100 years [22]. Orchardgrass seed is the fourth top-selling forage grass seed in the world [23]. It is widely planted in southwestern China for green feeding, hay or silage due to its strong adaptability and good palatability. In recent years, the research on orchardgrass has been increasing, and most of the research on orchardgrass resistance mainly focuses on drought stress [24], heat stress [25], and rust stress [26]. However, little is known about the flooding tolerance of orchardgrass and the transcriptome response of this species to flooding stress has not been reported. The objectives of this study were to identify the differentially expressed genes and analyze related pathways from transcriptome sequencing of orchardgrass. The results will provide a reference for further studying the molecular and genetic mechanisms of flooding tolerance in orchardgrass and other related perennial grass species.
Results
Transcriptome sequencing and mapping of Illumina reads
To comprehensively clarify the flooding tolerance of orchardgrass, the plants of orchardgrass seedlings were flooded at different times (0 h (CK), 8 h and 24 h). Sequencing results obtained raw data 427,211,572 and the raw reads were filtered obtain clean reads was 416,737,748 (Table S1). The orchardgrass genome was used as the reference genome. Approximately72% of the clean reads were total mapped to the reference genome, with more than 69% of them being uniquely mapped (Table 1). The raw data of this study was stored in the NCBI (National Coalition Building Institute) SAR (Sequence Read Archive) database, which can be viewed in BioProject (ID: PRJNA554779).
Table 1.
Analysis of statistical data RNA-seq of orchardgrass
| Samplea | WS_0h_1 | WS_0h_2 | WS_0h_3 | WS_8h_1 | WS_8h_2 | WS_8h_3 | WS_24h_1 | WS_24h_2 | WS_24h_3 |
|---|---|---|---|---|---|---|---|---|---|
| Total readsb | 44,643,436 | 51,100,544 | 41,539,096 | 48,916,078 | 46,175,616 | 39,333,770 | 39,625,568 | 48,857,684 | 56,545,956 |
| Total mapped (%)c | 32,213,341 | 36,722,643 | 29,484,126 | 34,653,721 | 32,228,040 | 27,919,523 | 28,219,174 | 33,577,514 | 39,533,008 |
| (72.16%) | (71.86%) | (70.98%) | (70.84%) | (69.79%) | (70.98%) | (71.21%) | (68.73%) | (69.91%) | |
| Multiple mapped (%)d | 1,120,609 | 1,233,315 | 946,216 | 1,225,851 | 1,073,156 | 899,453 | 852,050 | 1,147,239 | 1,276,249 |
| (2.51%) | (2.41%) | (2.28%) | (2.51%) | (2.32%) | (2.29%) | (2.15%) | (2.35%) | (2.26%) | |
| Uniquely mapped (%)e | 31,092,732 | 35,489,328 | 28,537,910 | 33,427,870 | 31,154,884 | 27,020,070 | 27,367,124 | 32,430,275 | 38,256,759 |
| (69.65%) | (69.45%) | (68.7%) | (68.34%) | (67.47%) | (68.69%) | (69.06%) | (66.38%) | (67.66%) | |
| Reads map to ‘+’ (%)f | 15,563,768 | 17,748,957 | 14,279,174 | 16,684,717 | 15,561,283 | 13,493,851 | 13,693,019 | 16,209,484 | 19,136,212 |
| (34.86%) | (34.73%) | (34.38%) | (34.11%) | (33.7%) | (34.31%) | (34.56%) | (33.18%) | (33.84%) | |
| Reads map to ‘-’ (%)g | 15,528,964 | 17,740,371 | 14,258,736 | 16,743,153 | 15,593,601 | 13,526,219 | 13,674,105 | 16,220,791 | 19,120,547 |
| (34.78%) | (34.72%) | (34.33%) | (34.23%) | (33.77%) | (34.39%) | (34.51%) | (33.2%) | (33.81%) |
aWS_0h_1, 2, 3 (flooding treated for 0 h), WS_8h_1, 2, 3 (flooding treated for 8 h), WS_24h_1, 2, 3 (flooding treated for 24 h); bThe amount of RNA-seq sequencing sequence filtered by sequencing data (Clean data);cTotal Mapped Reads and their percentage of clean reads; dMultiple Mapped Reads and their percentage of clean reads; eUniquely Mapped Reads and their percentage of clean reads; f, gSequencing sequence alignment to the total number of positive and negative strands on the genome (Total Mapped Reads) and its percentage of clean reads
Pearson correlation analysis was performed to validate the gene expression profiles of transcripts from nine different samples (Figure S1). The results showed that biological replicates from the same treatment group were highly correlated. In particular, ws_0h and ws_24h were highly correlated.
Differentially expressed gene identification in leaves of under flooding stress
To screen the differentially expressed genes (DEGs), we used DESeq software for differential gene analysis, and q-value (p-adj) < 0.05 was defined as DEGs. Integration the reproducibility of the biological replicates, we found 2019 DEGs in leaves following the flooding stress (Table S2, S3). Among them, 226 DEGs were identified in response to both 8 h and 24 h flooding stress (Fig. 1a). A total of 1454 DEGs were found in response to 8 h flooding stress, of which 715 DEGs were up-regulated and the remaining 739 DEGs were down-regulated (Fig. 1b). A total of 565 DEGs were found responding to 24 h flooding stress, of which 352 DEGs were up-regulated and 213 DEGs were down-regulated (Fig. 1c). There were more DEGs found in the 8 h flooding stress than in the 24 h, suggesting that the response mechanism of orchardgrass to the early flooding stress period was more dramatic and complicated.
Fig. 1.
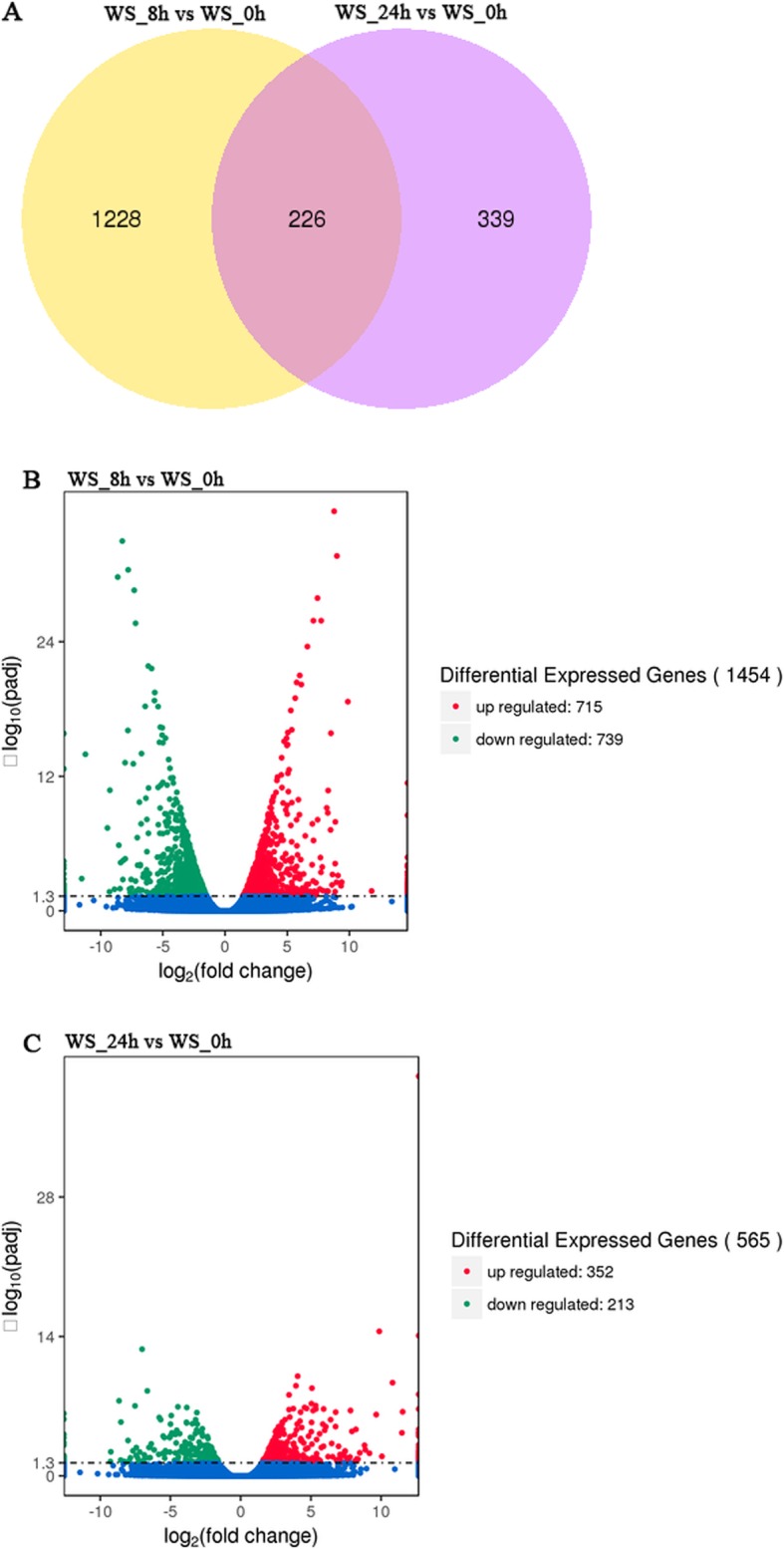
Venn diagrams and Volcano map of DEGs under flooding treatment in orchardgrass plantlets. a: Comparison of differentially expressed genes at 8 h and 24 h after flooding treatment with treatment for 0 h (CK); The numbers of DEGs with common were shown in the overlapping region. The total numbers of up- or down-regulated genes in each treatment were the sum of the numbers in each circle. b, c: Up-regulation and down-regulation of differential genes in 8 h, 24 h flooding treatment. The numbers of DEGs with common or opposite expression change tendencies between treatments were shown in the overlapping regions. The total numbers of up- or down-regulated genes in each treatment were the sum of the numbers in each circle
GO enrichment analysis of the DEGs
To better understand the response mechanism of orchardgrass to flooding stress, we performed GO enrichment analysis on the biological functions of all DEGs. Correction p-value < 0.05 can be used as an enrichment project for a certain function. DEGs GO enrichment histogram visually reflected the distribution of DEGs in GO terms of biological processes, cellular components, and molecular function enrichment.
Comparing DEGs in 8 h flooding stress with those in 0 h (CK) treatment, DEGs enriched in biological processes were the most abundant and significantly enriched, mainly involving, single-organism process (GO:0044699), transport (GO:0006810) and establishment of localization (GO:0051234); followed by membrane (GO:0016020) in the category of cell components. The DEGs enriched in molecular function were the least, mainly involving oxidoreductase activity (GO:0016491) and transporter activity (GO:0005215) (Fig. 2a, Table S4). Also, we found that the number of DEGs was down-regulated more than that of up-regulation (Figure S3A). In the down-regulated most enriched GO term, the DEGs have multiple enrichment terms, among which the DEGs in the biological process were the most abundant (Figure S2A). While in the up-regulated most enriched GO term, there was no DEG enrichment. We speculated that the DEGs were mostly inhibited by 8 h of flooding stress, especially for those enriched in the biological process.
Fig. 2.

GO functional classification of the control and flooding stress orchardgrass seedling. “*” was an enriched GO term. a: comparison between 8 h of flooding stress and 0 h stress (CK); b: comparison between 24 h of flooding stress and 0 h stress (CK)
Comparing 24 h flooding stress with those in 0 h (CK) treatment, the DEGs enriched in the biological process mainly involved the oxidation-reduction process (GO:0055114), and the DEGs enriched in the molecular function were more than the biological processes, including oxidoreductase activity (GO:0016491), co-enzyme binding (GO:0050662), and co-factor binding (GO:0048037) (Fig. 2b, Table S5). We found that after 24 h of flooding stress, the up-regulated DEGs had significant enrichment in GO terms (Figure S2B), while the down-regulated DEGs had no enrichment. Besides, most of the DEGs enriched in biological processes, molecular functions, and cellular components were up-regulated (Figure S3B). Interestingly, this is exactly the opposite of the enrichment mode of DEGs treated with 8 h of flooding stress.
Through a comprehensive comparison of two flooding stress (8 h and 24 h) DEGs most enriched GO Term (Figure S2A, B), we found that oxidoreductase activity, co-factor binding, and oxidation-reduction process were highly present in the 8 h and 24 h of flooding stress. The DEGs enriched in the oxidation-reduction process and oxidoreductase activity were down-regulated by 8 h of flooding stress. Under 24 h of flooding treatment stress, the DEGs enriched in oxidoreductase activity and oxidation-reduction process were promoted and up-regulated. In summary, it suggested that the DEGs enriched in these two GO terms (oxidoreductase activity, oxidation-reduction process) played a crucial role in the response to flooding stress.
KEGG pathways enrichment analysis of the DEGs
When plants encounter stress, different genes coordinate with each other to regulate biological functions. The significantly enriched pathway of DEGs can identify the most important biochemical metabolic pathways and signal transduction pathways in plant responses to stress. Here, we used KOBAS (2.0) for pathway enrichment analysis. FDR ≤ 0.05 indicated that the differential gene was significantly enriched in a certain pathway.
The key pathways of orchardgrass seedling in response to 8 h flooding stress were glutathione metabolism, selenocompound metabolism, peroxisome, tyrosine metabolism, isoquinoline alkaloid biosynthesis, biosynthesis of secondary metabolites, plant-pathogen interaction, thiamine metabolism, carotenoid biosynthesis, beta-alanine metabolism, glycerolipid metabolism (Fig. 3a, Table S6).
Fig. 3.

Differential gene KEGG enrichment scatter plot of the control and flooding stress orchardgrass seedling. The ordinate indicates the name of the path and the abscissa indicates the Rich factor. The size of the dot indicates how many DEGs were in the pathway, and the color of the dot corresponds to a different q-value range. a: comparison between 8 h of flooding stress and 0 h stress (CK); b: comparison between 24 h of flooding stress and 0 h stress (CK)
The key pathways in response to 24 h flooding stress were alpha-Linolenic acid metabolism, selenocompound metabolism, phenylalanine metabolism, ubiquinone and other terpenoid-quinone biosynthesis, phenylpropanoid biosynthesis, glutathione metabolism, fatty acid degradation, cutin suberine and wax biosynthesis, plant hormone signal transduction (Fig. 3b, Table S7).
The pathways of significant enrichment of DEGs under 8 h flooding stress were different from 24 h stress. Only glutathione metabolism and selenocompound metabolism were found in both (8 h and 24 h) flooding treatments. The DEGs enriched in the glutathione metabolism pathway after 8 h flooding stress was more abundant than that in the 24 h flooding, suggesting that glutathione metabolism more responded to 8 h of flooding stress than the 24 h.
Cluster analysis of the DEGs
KEGG enrichment analysis was performed on all DEGs at 8 h and 24 h compared with 0 h respectively. We screened the significantly enriched 58 DEGs from nine pathways (glutathione metabolism, selenocompound metabolisms, peroxisome, tyrosine metabolism, isoquinoline alkaloid biosynthesis, alpha-Linolenic acid metabolism, phenylalanine metabolism, ubiquinone and other terpenoid-quinone biosynthesis, phenylpropanoid biosynthesis).
The cluster heat map clearly showed that the expression patterns of 58 DEGs at different flooding time points were clustered into 6 clusters (Fig. 4). Among them, the expression patterns of DEGs clustered in Clusters1 and Clusters3 were the opposite. The main pathways involved in these two clusters are tyrosine metabolism and glutathione metabolism, as well as shared peroxisome. Compared with 0 h of flooding stress, the expression of DEGs was down-regulated, but both the 24 h stress DEGs and showed up-regulated patterns in Clusters1. This might indicate that under 8 h flooding stress, the expression of 9 DEGs in Cluster1 was inhibited. With the prolongation of flooding time, we hypothesized that the stressed plants could adjust themselves to cope with the stressful environment. Compared with the control group, the DEGs accumulated in Clusters2 were significantly down-regulated under flooding treatment, while the DEGs contained in Clusters4 and Clusters6 were significantly up-regulated. They were enriched in the pathways peroxisome, glutathione metabolism, selenocompound metabolism and phenylpropanoid biosynthesis, alpha-Linolenic acid metabolism, respectively (Table 2). In the Clusters5, Compared with 0 h (CK) flooding stress, the expression pattern of DEGs under 8 h was consistent with the control and down-regulated; while the expression pattern of DEGs was up-regulated at 24 h of flooding stress, contrary to the expression pattern of the control. In addition, we found that the DEGs contained in Cluster1, Cluster2, and Cluster3 were mainly derived from the 8 h treatment group, and the DEGs of Cluster4 were derived from 8 h and 24 h, while the DEGs of Cluster5 and Cluster6 were mainly derived from the enrichment pathway of the 24 h treatment group. This suggests that plants have a biological adaptation process in response to flooding stress to alleviate the damage caused by adversity. The flooding stress response of plants could be divided into three different stages, namely the alarm period, the acclimation period and the resistance period [27]. Studies have shown that the first stage of plant response to flooding stress is rapid induction of signals, the second stage is the adaptation stage activated by the first stage, and the third stage is the formation of aeration tissue at the root [28] .
Fig. 4.
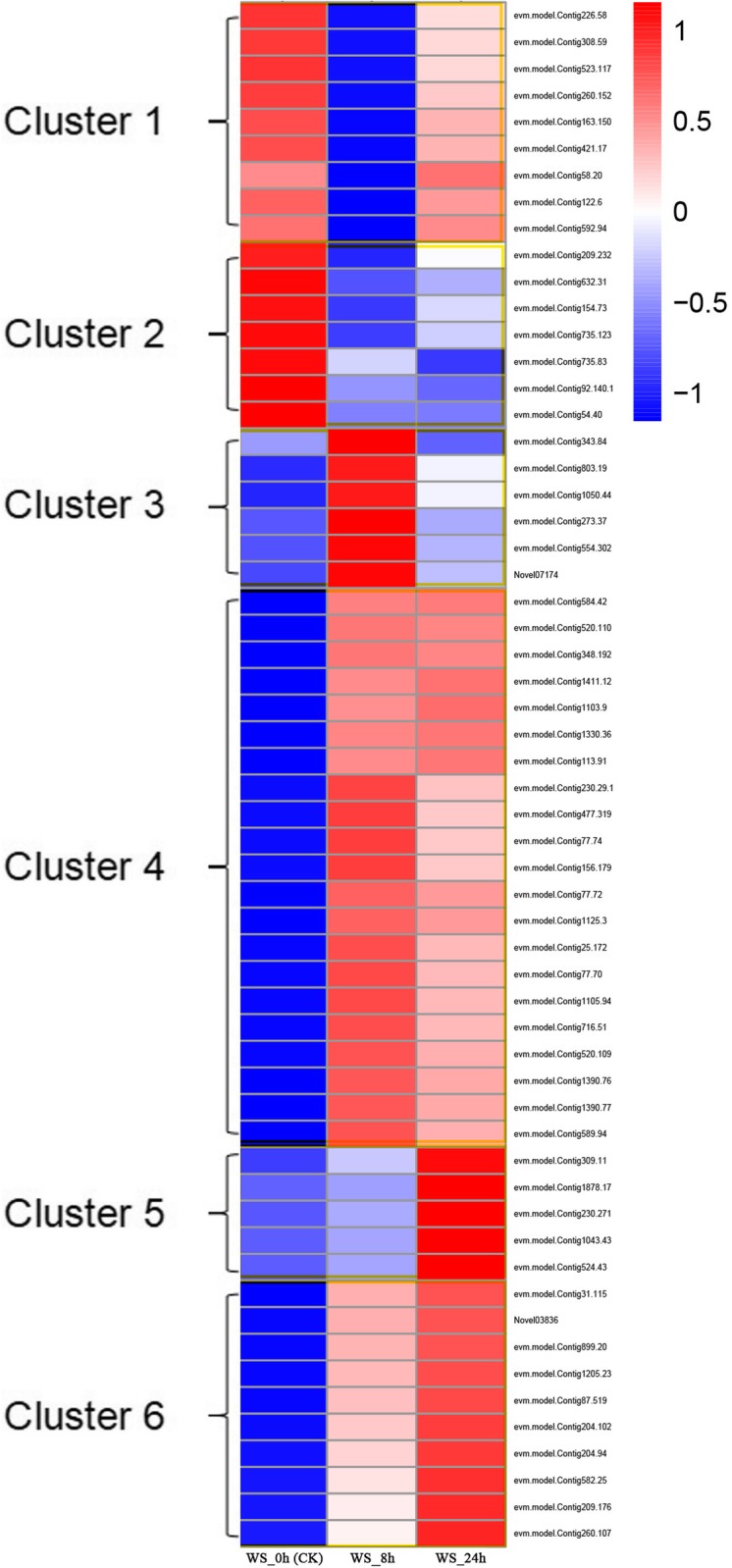
FPKM hierarchical clustering diagram of 58 DEGs in three-time points. The log10 (FPKM+ 1) values were scaled and clustered, with red indicating a highly expressed gene and blue indicating a low expressed gene. Colors from red to blue, indicating log10 (FPKM+ 1) from big to small. Cluster1, 2, 3, 4, 5, 6: Six patterns of 58 DEGs at different time points of flooding stress
Table 2.
Expression patterns of 58 DEGs under different flooding stress
| Cluster | Number of DEGs | DEGs expression mode | The main enriched pathway of DEGs | ||
|---|---|---|---|---|---|
| WS_0h | WS_8h | WS_24h | |||
| 1 | 9 | ↑ | ↓ | ↑ | Tyrosine metabolism, Peroxisome, |
| 2 | 7 | ↑ | ↓ | ↓ | Peroxisome |
| 3 | 6 | ↓ | ↑ | ↓ | Glutathione metabolism, Peroxisome |
| 4 | 21 | ↓ | ↑ | ↑ | Glutathione metabolism, Selenocompound metabolism |
| 5 | 5 | ↓ | ↓ | ↑ | alpha-Linolenic acid metabolism |
| 6 | 10 | ↓ | ↑ | ↑ | Phenylpropanoid biosynthesis, alpha-Linolenic acid metabolism |
Note: “↑” means that the expression of DEGs is up-regulated, and “↓” means that the expression of DEGs is down-regulated
Peroxidase is involved in the regulation of plant clearance of toxic ROS
Peroxisomes are important organelles equipped with a highly efficient reactive oxygen species (ROS) detoxification system, which is involved in many key metabolic processes such as oxidation of fatty acids, biosynthesis of ether lipids and detoxification of free radicals [29] (Fig. 5). It can enhance the adaptability of plants to stressful environments and plays a key role in plant redox signaling and lipid homeostasis.
Fig. 5.

“Peroxidase”, “glutathione metabolism”, “glycolysis” and “phytohormone signal transduction” in response to flooding stress. Red means that the expression of DEGs is up-regulated, and green means that the expression of DEGs is down-regulated
Eleven DEGs were significantly expressed in the peroxisome pathway, of which five were up-regulated and eight down-regulated at 8 h of flooding stress (Table S9). The DEGs, PEX10 were significantly up-regulated, indicating that it was promoted during peroxisome biogenesis. In abiotic stress, the total amount of intracellular peroxisomes could increase the total number of peroxisomes in addition to up-regulating ROS scavenging enzymes [30]. This increase was accompanied by the up-regulation of the peroxisome biosynthetic gene in Arabidopsis, including PEX10 [31], indicating that PEX10 was promoted during peroxisome biogenesis. Studies have shown that peroxidases proliferate in plants under stress conditions [32].
In recent years it has become apparent that peroxisomes play important roles in reactive oxygen metabolism [33]. Most reactive oxygen species metabolism is carried out in peroxisomes [34]. Excessive concentrations of ROS can cause oxidative damage to the peroxisome components exposed to it, but this can be eliminated by various ROS scavengers, such as non-enzymatic antioxidants: glutathione (GSH) and multiple antibodies. Oxidase: catalase (CAT), GSH reductase, ascorbate peroxidases (APX), peroxidase and superoxide dismutase (SOD) [35].
The differential gene-regulating MPV17 was significantly down-regulated during ROS metabolism, indicating that ROS metabolism was inhibited under 8 h of flooding stress (Table S9). This might be caused by water shortage in the flooded environment and oxygen stress. Under oxidative stress conditions, the production of peroxisome ROS was enhanced and the clearance of ROS was insufficient [35], resulting in an imbalance of ROS metabolism. The differential gene that regulated FAR5 was significantly up-regulated and promoted by stress. Differential genes encoding CAT1 and CSD2 were significantly up-regulated and down-regulated, respectively (Table S9). CAT was an important target for H2O2 scavengers. It is rich in plant peroxisomes while it has low substrate affinity. Regulating its catalytic activity might be the starting point to overcome this shortcoming, thereby making plant peroxisomes more effective in detoxification [36].
However, expressions of only five genes changed in the peroxidase pathway. Of them, four were up-regulated and one was down-regulated under 24 h of flooding stress (Table S9). PXN was a family of mitochondrial substrate carriers whose expression was promoted by flooding stress, while hypoxia was associated with decreased mitochondrial respiration [37]. Mitochondrial metabolism plays a key role in controlling the redox balance of whole plant cells [38]. The differential genes regulating ACX5 and FAR5 were significantly up-regulated, indicating that they responded positively to flooding stress to promote plant adaptation to stress.
Glutathione metabolism is involved in the response of plants to flooding stress
Glutathione is an important non-enzymatic antioxidant when plants encounter abiotic stress. It can protect the sulfhydryl and membrane systems of structural proteins in cells, effectively remove excessive ROS components produced by plants during stress, and reduce peroxidative damage [39]. In response to 8 h of flooding stress, fourteen differential genes were significantly expressed during glutathione metabolism (Table S9), in which two genes regulating GPX6 and SPDS3 were down-regulated and twelve genes were significantly up-regulated including GSTU7, ERD9, GSTU8, ATGSTF13, GSTL3 and GSH2. At 24 h of flooding stress, with a total of seven differential genes were associated with glutathione metabolism pathway (Table S9), including up-regulation of ERD9, GSTU8, ATGSTF13, GSTL3 and down-regulation of SPDS3 (Fig. 5). It has been reported that antioxidant genes such as GST and GPX are involved in flooding stress, suggesting that they activate the ROS elimination process [15].
Glycolysis is involved in the stress of plants on flooding stress
Glycolysis is one of the important ways of energy production under hypoxic conditions. Maintaining the growth of glycolysis may be the key to plant survival. When plants are exposed to flooding, they are also accompanied by oxygen stress, which causes rapid changes of gene transcription, protein synthesis and degradation, and cellular metabolism [40]. Under hypoxic conditions, glycolysis is mainly through the fermentation pathway (rather than aerobic respiration), which is necessary for cell survival to generate energy and to recover carbon for other pathways [38]. ADH accelerates ethanol fermentation and allows glycolysis to provide ATP to plants during flooding, thereby improving the ability of plant adaptation to stress [41].
There were ten differential genes involved in glycolysis, of which two genes were significantly up-regulated and nine genes were significantly down-regulated in to response 8 h of flooding stress (Table S9). The up-regulated functional genes were ALDH3I1 and galactose mutarotase-like superfamily protein. The down-regulated genes included galactose mutarotase-like superfamily protein, PFK2, FBA2, ALDH11A3, LOS2, PCK1, PKp3, and pyruvate kinase family protein. In response to 24 h of flooding stress, four DEGs were significantly up-regulated, and two DEGs were down-regulated (Table S9). Up-regulated genes were actate/malate dehydrogenase family protein, ADH1, and ALDH2B4. Galactose mutarotase-like superfamily protein and PFK2 were regulated by two DEGs, respectively, and they were down-regulated (Fig. 5).
Plant hormone-mediated pathways are involved in response to flooding stress
Hormones are important regulators of plant adaptation to environmental changes. Plant hormone signaling plays an important role in the stress resistance of many plants. In response to 8 h of flooding stress (Table S9), SAUR-like auxin-responsive protein family involved in the auxin-mediated tryptophan pathway was regulated by two differential genes, one of which was up-regulated and one down-regulated. In response to 24 h of flooding stress (Table S9), the differentially expressed genes regulating GID1C were significantly up-regulated during gibberellin-mediated diterpenoid biosynthesis. In the process of abscisic acid-mediated carotenoid biosynthesis, the four DEGs regulating PYL were up-regulated significantly, and one gene regulating HAB2 was down-regulated significantly. In the ethylene-mediated metabolism of Hemi-amino acid and methionine, a DEG that regulates EIN4 was up-regulated, and a DEG that regulates EBF1 was up-regulated. The SUB1 locus contains three clusters of ethylene response factor (ERF) genes, in which the SUB1A allele SUB1A-1 limits the growth of plants under flooding and confers flood tolerance [42].
Validation of the DEGs by qRT-PCR analysis
To validate the accuracy and reproducibility of the RNA-seq results, we randomly screened ten DEGs for qRT-PCR, which were up- and down-regulated. The relative expression of each gene was calculated using the 2-ΔΔCt method. The results showed that ten DEGs were differentially expressed between the control group (flooding for 0 h) and the stress-treated group (flooding for 8 h and 24 h) (Figure S4). The qRT-PCR verification results were highly correlated with the RNA-seq data (Figure S5), confirming the reproduci bility of the RNA-Seq data.
Discussion
In this study, high-throughput RNA sequencing technology was used to compare the expression profiles of DEGs in the leaves of orchardgrass cv. Dianbei in 8 h and 24 h under flooding stress. This study provided insights into a better understanding of the molecular mechanisms of flooding tolerance in orchardgrass.
By comparing the bioinformatics analysis of the obtained transcript data of orchardgrass, we found that many biological processes, cell components and molecular processes in the leaves of orchardgrass were affected by flooding stress. In response to stress, oxidoreductase activity in the GO enrichment term was significantly enriched at both time points, and it was also significantly up-regulated in the GO enrichment entry in cucumber exposed to flooding stress [16]. At 8 h and 24 h of stress, the differential genes were significantly expressed in the two GO terms entries of oxidation-reduction process and oxidoreductase activity, indicating that different groups of oxidoreductase genes were involved in maintaining redox balance or alleviating sputum-induced oxidative stress [1] suggesting that oxidoreductase activity played an important role in plant response to flooding stress. In the study, in the GO enriched molecular function category, binding, catalytic activity and transporter activity were highly present, which was consistent with the analysis of the short-term smear stress on the rhizome transcriptome of Taxodium [19].
In this experiment, a total of 102 pathways were found in the list of KEGG differential gene enrichment pathways in the 8 h of flooding treatment, and 77 pathways in the 24 h of flooding treatment. The pathways on the top list were glutathione metabolism, peroxisome, selenium compound metabolism, tyrosine metabolism, biosynthesis of secondary metabolites, plant-pathogen interaction, plant hormone signal transduction, suggesting that flooding impacted a wide range of physiological processes. We have found that these processes have been reported in studies of other plant flooding stresses. Flooding stress should be considered as a compound stress consisting of several potential changes in the internal and external sources of ethylene, carbon dioxide, O2, ROS and plant toxins [43].
ROS is a by-product of basic cellular processes such as plant photosynthesis and respiration, and is produced by the joint action of several enzymes [44]. One of the main sources of active oxygen in plants is a reaction mediated by coagulin oxidase, which is responsible for the conversion of O2− to superoxide anion, resulting in the production of hydrogen peroxide [45]. ROS are important signals in plants and key regulators of a variety of processes including metabolism, growth and development, response to abiotic and biotic stresses, solute transport, autophagy and programmed cell death [46].
Accumulation of ROS is an important indicator of abiotic stress at the molecular level. Once the ROS removal system is disrupted, the plant will suffer from oxidative stress. In general, plants can detoxify ROS by producing different types of antioxidants. Antioxidants are divided into enzymatic and non-enzymatic antioxidants. Enzyme antioxidants include superoxide dismutase (SOD), peroxidase (POD), catalase (CAT) and glutathione reductase (GR), and ascorbic acid, glutathione, tocopherol, and carotenoids are non-enzymatic antioxidants [47]. Many antioxidant enzymes are the key to the survival of many plants under varying degrees of flooding stresses [19]. In the study, down-regulation of CSD2 encoding SOD and up-regulation of ROS scavenging genes (CAT1 and glutathione S-transferase) were observed under flooding stress. SOD is the first line of defense for removing active oxygen in plants [48]. At the core of the enzyme protection system, the flood tolerance of plants is directly related to the strength of SOD activity. Its main function is to catalyze the disproportionation reaction of superoxide anion into hydrogen peroxide, which is further decomposed to water and oxygen by conversion of POD, CAT, GR and APX [49]. Down-regulation of CSD2 might increase ROS levels in the leaves of flooded orchardgrass seedlings. Catalase may be involved in the control of hydrogen peroxide (H2O2) levels by converting H2O2 to O2.This may be the reason why Dianbei is resistant to flooding. Similar to this study, the ROS scavenging genes (encoding SOD, CAT and glutathione-S-transferase) were significantly expressed in rape. The difference is that the gene encoding CAT is down-regulated and the gene encoding SOD is down-regulated [50]. Similar findings were found in cucumber [45], Maize [2], and A. philoxeroides [51] flooding studies.
Glycolysis is one of the important ways of energy production under hypoxic conditions. Maintaining the growth of glycolysis may be the key to plant survival [20]. The glycolysis and fermentation processes are enhanced under flooding conditions, including ethanol fermentation (PDC and ADH catalysis) and lactic acid fermentation (LDH catalysis). ADH accelerates ethanol fermentation and allows glycolysis to provide ATP to plants during flooding, thereby improving the ability of plants to adapt to adversity [41]. Under flooding conditions, plants regulate mitochondrial respiration to glycolysis by fermentation of pyruvate decarboxylase and alcohol dehydrogenase [52]. In this study, up-regulated expression of ADH verified that ADH plays an important role in plant response to flooding. Comparative transcriptome analysis of other plants has also identified glycolysis and upregulation of fermentation genes as common responses to flooding, such as Jatropha [53], Sesbania cannabina [54], soybean [55]. Our results supported the longstanding notion that flooding promotes anaerobic respiration, as observed by the up-regulation of DEGs encoding enzymes involved in glycolysis and fermentation.
Plant hormones play an important role in plant resistance to stress. Plant growth regulators are induced to participate in a signal cascade that affects cellular responses under flooding stress, including reduction of ethylene (C2H4), abscisic acid (ABA), gibberellin (GA), auxin (IAA), and cytokinin (CK). Many genes involved in hormone production and signaling were found to be affected in both roots and shoots of flooded plants [40]. Ethylene has long been known to be involved in responses to hypoxia and is thought to contribute to adventitious root production and aerenchyma formation [56]. The Ethylene response factor (ERF) represented the highest number of significantly expressed TFs under hypoxic conditions. In this study, the expression of EBF1/2 in orchardgrass leaves was significantly increased under flooding conditions, suggesting that flooding is related to ethylene synthesis and perception. This proves that ethylene plays an important role in plant flooding stress. The phytohormone ethylene mediates physiological, developmental, and stress responses by activating ethylene-responsive factors (ERFs) belonging to the multi-gene family of transcription factors [57]. In Arabidopsis, ERF1 plays a positive role in salt, drought, and heat stress tolerance by regulating stress-specific gene, and by integrating jasmonic acid, ethylene, and abscisic acid signals [58]. This has also appeared in other studies of plant flooding stress. These results indicate that plants need to induce genes associated with hypoxia response through ERF regulation, but further studies are needed to test other ERFs. ABA has long been recognized as a key factor in abiotic stress response in plants [59]. The major ABA receptor (PYR / PYL / RCAR) responsible for stomatal closure is a soluble protein [60]. In this study, PYL2, PYL5, and RCAR3 were significantly expressed in the abscisic acid-mediated carotenoid biosynthesis. This also suggests that ABA plays an important role in responding to flooding stress. IAA is the main biologically active auxin in plants [61]. In the present study, the gene IAA18 encoding indole-3-acetic acid was up-regulated. The abundance of IAA activates the MAPK signal cascade at intervals, leading to the formation of adventitious roots, which is very important for plants in flooded environments [62]. The formation of adventitious roots is an important morphological change in plants in response to flooding or hypoxia.
By qRT-PCR analysis, it was found that the expression patterns of genes encoding GSTL3, ATGSTF13, CaMCML, GSTU7, GSTU8, CPNIFS, HMGCL, MPV17, and DND1 were significantly expressed under flooding stress, indicating that they play an important role in flood resistance of orchardgrass. Among them, we found that compared to the control group, genes encoding GST (GSTL3, ATGSTF13, GSTU7, GSTU8) were significantly induced under water flooding, indicating that these genes regulate GST in an important role in the ROS clearance system. Similar research conclusions have also appeared in the study of resistance to rape [17], cucumber [45] and tall fescue [63].
Conclusion
This study is the first time that a transcriptome analysis of orchardgrass leaves in response to short-term flooding stress has identified a total of 2019 differentially expressed genes. Oxidoreductase activity, cofactor bling and oxidation -reduction process are highly present in GO-enriched terms, suggesting that the leaves of orchardgrass have a complex oxidoreductase process in response to flooding stress. This study also found that peroxidase, glutathione, glycolysis and phytohormone signal transduction play important roles in the domestication of orchardgrass. This study laid a foundation on a further study of genetic control of flooding tolerance for orchardgrass.
Materials and methods
Plant materials and stress conditions
The test material is “Dianbei”, which is an orchardgrass (Dactylis glomerata L.) variety with strong flood tolerance.
Orchardgrass seeds of uniform size and fullness were screened and sown in pots containing sand and soil in a ratio of 1: 2 (V: V) (diameter 10 cm, height 8 cm) (Chongqing, China). After sowing, the pots were placed in a light incubator. The relative humidity in the light incubator is constant, the temperature is set to 25 °C/20 °C (day / night), the photoperiod is 14 h / 10 h (day / night), and the photon flux density is about 300 μmol m − 2 s − 1. Huguelan solution was supplied regularly during shoot growth. Huguelan solution was supplied regularly during the seedling period.
Three months after sowing, select well-grown orchard grasses for testing. The pot was placed in a water tank (length 80 cm; width 57 cm; height 50 cm) filled with room temperature water, and the water level was kept higher than the highest part of the plant as the treatment group, and the control group was a normal-growing orchardgrass plant. The 24 h samples were first flooded, and after 16 h, the 8 h samples were flooded, with three biological replicates per treatment. When all treatments were completed, the leaves of the orchard grass from three treatments (0 h, 8 h, and 24 h) were cut at the same time, put into sterile tubes, quickly frozen with liquid nitrogen, and stored at − 80 °C.
RNA extraction and library construction
Total RNA was extracted from the samples using an RNA prep pure plant kit (Tiangen Biochemical Technology Company, Beijing, China). (1) Analysis of the degree of RNA degradation and contamination by running agarose gel electrophoresis; (2) Nanodrop testing (OD260/280 ratio); (3) qubit accurately quantification of RNA concentration; (4) Agilent 2100 detecting RNA integrity. Finally, only RNA samples that passed the quality test were selected for RNA-Seq analysis. Transcriptome sequencing was conducted on an Illumina Hiseq 4000 (Illumina, San Diego, CA, USA) at the Novogene Bioinformatics Institute (Beijing, China).
Approximately 3 μg of RNA for each sample was used for library construction, using the NEBNext® Ultra™ Directional RNA Library Prep Kit for Illumina® (NEB, USA). Transcriptome sequencing was conducted on an Illumina Hiseq 4000 (Illumina, San Diego, CA, USA) at the Novogene Bioinformatics Institute (Beijing, China). The orchardgrass genome reference was used for read mapping (http://orchardgrassgenome.sicau.edu.cn/). All of the sequencing data was deposited into the NCBI SRA database.
Reads mapping
Filtering the original sequencing sequence obtained by sequencing: 1) removing the reads with adapters;(2) removing N (N means that the base information cannot be determined) the ratio of reads greater than 10%;(3) Remove low-quality reads (Qphred < = 20 bases account for more than 50% of the entire read length of reads). We selected HISAT [64] software to perform genomic localization analysis on the filtered sequence. In the analysis process, the software default parameters were used. Reference to the genome and gene model of orchardgrass [65]. The HISAT algorithm is mainly divided into three parts: (1) the entire sequence of the sequencing sequence was aligned to the single exon of the genome; (2) the sequencing sequence was segmentally aligned to the two exons of the genome; (3) the sequencing sequences into more than three (including three) exons in the genome.
In RNA-seq analysis, we can estimate gene expression levels by counting the number of sequencing reads that are located in the genomic region or exon region. In this experiment, each sample was analyzed for gene expression level using HTSeq [66] software, and the model used was union. To make the estimated gene expression levels of different genes and different experiments comparable, the concept of FPKM was introduced. FPKM is currently the most commonly used method for estimating gene expression levels. The input data of gene differential expression analysis was the readcount data obtained from the gene expression level analysis. The analysis was mainly divided into three parts: 1) first normalized the readcount; 2) then calculated the hypothesis test probability (p-value) according to the model 3) Finally, multiple hypothesis test corrections were performed to obtain the FDR value (error discovery rate). Analysis of gene differential expression using DESeq [67] software.
GO and KEGG enrichment analysis of differentially expressed genes
The software method used in our analysis of GO enrichment analysis was GOseq, which is based on the Wallenius non-central hyper-geometric distribution. Compared to the ordinary hyper-geometric distribution, this distribution is characterized by the fact that the probability of extracting an individual from a certain category is different from extracting an individual from a outside certain category and the probability is different. It is estimated by the length of the gene, so that the probability of GO term being enriched by differential genes can be calculated more accurately. Pathway Significant Enrichment Analysis Using Pathway in the KEGG database, hypergeometric tests were used to find Pathway that was significantly enriched in differentially expressed genes compared to the entire genome background.
Validation of RNA-Seq data by qRT-PCR
Ten genes were randomly selected from the DEGs to verify differential gene expression levels with three biological repeats. Gene-specific qRT-PCR primers were designed using Primer3 software (http://primer3.ut.ee/), for ten selected genes with the sequence data in 3’UTR (Table S8). PrimeScript RT reagent Kit with gDNA Eraser (Takara, Chongqing, China) is a dedicated reverse transcription kit for RT-PCR reactions, following the manufacturer’s protocol. qPCR was performed using TB Green™ Premix Ex Taq™ II (Takara) on Applied Biosystems 7500 Fast Real-Time PCR System. The volume of the qRT-PCR reaction was 10 μl, containing 1 μl cDNA, 5 μl TB Green Premix Ex Taq II (Tli RNaseH Plus) (2×), 0.4 μl ROX Reference Dye II (50×), 0.8 μl of the forward and reverse primers, and 7 μl ddH2O. Actin was used as the endogenous reference gene. The reaction had three phases. Stage 1 (hold): 95 °C 30 s 1 Cycle, Stage 2 (PCR reaction): 95 °C 5 s,60 °C 20 s,40 Cycles, Stage 3 (melting curve analysis): 95 °C 15 s, 60 °C 1 h,95 °C 15 s. All PCR reactions were normalized using the Ct value corresponding to the endogenous reference gene (translationally controlled tumor protein [TCTP] [68]). Three biological replicates were generated and three measurements were performed on each replicate.
Statistical analysis
Data analysis and mapping were performed using IMS SPSS Statistics and OriginPro8.5, respectively. All values are expressed as mean ± SD. All the values were expressed as mean ± SD. Comparison between control and flooding groups was estimated with One-way ANOVA followed by LSD post hoc test, and P-value < 0.05 was considered to be statistically significant.
Supplementary information
Additional file 1: Table S1. Summary of RNA-seq sequence a nalysis of orchardgrass. Table S2. Differential analysis results of all DEGs suffering flooding at WS_8h vs WS_0h in leaves. Table S3. Differential analysis results of all DEGs suffering flooding at WS_24h vs WS_0h in leaves. Table S4. GO enrichment analysis of all DEGs suffering flooding at WS_8h vs WS_0h in leaves. Table S5. GO enrichment analysis of all DEGs suffering flooding at WS_24h vs WS_0h in leaves. Table S6. KEGG Pathway enrichment analysis of all DEGs suffering flooding at WS_8h vs WS_0h in leaves. Table S7. KEGG Pathway enrichment analysis of all DEGs suffering flooding at WS_24h vs WS_0h in leaves. Table S8. The primers used in the qRT-PCR analysis.
Additional file 2: Table S9. Analysis of 4 key pathways in response to flooding stress.
Additional file 3: Figure S1. Correlation analysis under different time flooding stress. Figure S2. GO enrichment analysis of down-regulated DEGs suffering flooding at WS_8h vs WS_0h and up-regulated DEGs suffering flooding at WS_24h vs WS_0h in leaves. Figure S3. GO functional classification of all DEGs suffering flooding at WS_8h vs WS_0h and WS_24h vs WS_0h in leaves. Figure S4. qRT-PCR verification results of ten DGEs in control and treatment groups. Figure S5. Correlation between qRT-PCR and RNA sequencing for the ten selected genes.
Acknowledgements
Not applicable.
Abbreviations
- DEGs
Differentially expressed genes
- ROS
Reactive oxygen species
- PEX10
Peroxin 10
- MPV17
Peroxisome membrane 22 KDA
- FAR5
Fatty acid reductase 5
- CAT1
Catalase 1
- CSD2
Copper/zinc superoxide dismutase 2
- PXN
Mitochondrial substrate carrier
- ACX5
Acyl-CoA oxidase 5
- GPX6
Glutathione peroxidase 6)
- SPDS3
Spermidine synthase 3
- GSTU7
Glutathione S-transferase tau 7
- ERD9
Glutathione S-transferase family protein
- GSTU8
Glutathione S-transferase TAU 8
- ATGSTF13
Glutathione S-transferase family protein
- GSTL3
Glutathione S-transferase family protein
- GSH2
Glutathione synthetase 2
- ADH
Alcohol dehydrogenase
- ALDH3I1
aldehyde dehydrogenase 3I1
- PFK2
Phosphfructokinase 2
- FBA2
Fructose-bisphosphate aldolase 2
- ALDH11A3
Aldehyde dehydrogenase 11A3
- LOS2
Enolase2
- PCK1
Phosphoenolpyruvate carboxykinase 1
- PKp3
Plastidial pyruvate Kinase 3
- ALDH2B4
Aldehyde dehydrogenase 2B4
- GID1C
Alpha/beta-Hydrolases superfamily protein
- PYL
Polyketide cyclase/dehydrase and lipid transport superfamily protein
- HAB2
Homology to ABI2
- EIN4
Signal transduction histidine kinase, hybrid-type, ethylene sensor
- EBF1
EIN3-binding F box protein 1
- qRT-PCR
quantitative real-time PCR
Authors’ contributions
DDQ, BZ, HL, YJZ participated designed the experiment. XMX, MYL, KC performed the experiments. DDQ, YJZ analyzed the data. DDQ, BZ, HL wrote the paper. All authors read and approved the manuscript.
Funding
The research was funded by project supported by the National Natural Science Foundation of China (NSFC 31771866), Chongqing University Innovation Team Building Plan Projects in 2016 “Beef cattle modern production technology (No: CXTDG201602003).
Availability of data and materials
All data produced by the study are disclosed in the manuscript and the additional files. The raw data of this study was stored in the NCBI (National Coalition Building Institute) SAR (Sequence Read Archive) database, which can be viewed in BioProject (ID: PRJNA554779).
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Dandan Qiao, Email: qdyiweng@163.com.
Yajie Zhang, Email: zyj1534248684@126.com.
Xuemei Xiong, Email: 15213040905@163.com.
Mingyang Li, Email: jiladahe1995@163.com.
Kai Cai, Email: cktlxhyqs111@163.com.
Hui Luo, Email: luohui2629@126.com.
Bing Zeng, Email: zengbing1978@swu.edu.cn.
Supplementary information
Supplementary information accompanies this paper at 10.1186/s41065-020-00134-0.
References
- 1.Muhammad AA. Waterlogging stress in plants: A review. Afr J Agric Res. 2012;7:1976–1981. [Google Scholar]
- 2.Vwioko E, Adinkwu O, El-Esawi MA. Comparative physiological, biochemical, and genetic responses to prolonged waterlogging stress in okra and maize given exogenous ethylene priming. Front Physiol. 2017;8:632. doi: 10.3389/fphys.2017.00632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Normile D, Agricultural Research Reinventing Rice to feed the world. Science. 2008;321:330–333. doi: 10.1126/science.321.5887.330. [DOI] [PubMed] [Google Scholar]
- 4.Mary-Rus Martínez-Cuenca AQ, Primo-Millo E, Forner-Giner MÁ. Flooding impairs Fe uptake and distribution in citrus due to the strong down-regulation of genes involved in strategy I responses to Fe deficiency in roots. PLoS One. 2015;10:e0123644. doi: 10.1371/journal.pone.0123644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pezeshki SR. Wetland plant responses to soil flooding. Environ Exp Bot. 2001;46:299–312. [Google Scholar]
- 6.Schaffer BD, Frederick S, Crane Jonathan H. Responses of subtropical and tropical fruit trees to flooding in calcareous soil. Hortscience. 2006;41:549–555. [Google Scholar]
- 7.Colmer TDV, L. A. C. J. Flooding tolerance:suites of plant traits in variable environments. Funct Plant Biol. 2009;36:665–681. doi: 10.1071/FP09144. [DOI] [PubMed] [Google Scholar]
- 8.John C, Marioni CEM, Shrikant M, Mane MS, Gilad Y. RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome Res. 2008;18:1509–1517. doi: 10.1101/gr.079558.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Malik VS. RNA sequencing as a tool for understanding biological complexity of abiotic stress in plants. J Plant Biochem Biotechnol. 2016;25:1–2. [Google Scholar]
- 10.Ling Hu YX, Fan S, Wang Z, Wang F, Zhang B, Li H, et al. Comparative analysis of root transcriptome profiles between drought-tolerant and susceptible wheat genotypes in response to water stress. Plant Sci. 2018;272:276–293. doi: 10.1016/j.plantsci.2018.03.036. [DOI] [PubMed] [Google Scholar]
- 11.Hui Li Jing L, Yang Q-S, Li X-G, Chang Y-H. Comprehensive analysis of differentially expressed genes under salt stress in pear (Pyrus betulaefolia) using RNA-Seq. Plant Growth Regul. 2017;82:409–420. [Google Scholar]
- 12.Jing Sun LR, Cheng Y, Gao J, Dong B, Chen S, Chen F, et al. Identification of differentially expressed genes in Chrysanthemum nankingense (Asteraceae) under heat stress by RNA Seq. Gene. 2014;552:59–66. doi: 10.1016/j.gene.2014.09.013. [DOI] [PubMed] [Google Scholar]
- 13.Chunxiu Shen DL, He R, Fang Z, Xia Y, Gao J, Shen H, et al. Comparative transcriptome analysis of RNA-seq data for cold-tolerant and cold-sensitive rice genotypes under cold stress. J Plant Biol. 2014;57:337–348. [Google Scholar]
- 14.Nepolean Thirunavukkarasu FH, Mohan S, Shiriga K, Mittal S, Sharma R, Singh RK, et al. Genome-wide expression of transcriptomes and their co-expression pattern in subtropical maize ( Zea mays L.) under waterlogging stress. PLoS One. 2013;8:e70433. doi: 10.1371/journal.pone.0070433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jinwei Wu H-BZ, Yu D, Xinwei X. Transcriptome profiling of the floatingleaved aquatic plant Nymphoides peltata in response to flooding stress. BMC Genomics. 2017;18:119. doi: 10.1186/s12864-017-3515-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xuewen Xu MC, Ji J, Xu Q, Qi X, Chen X. Comparative RNA-seq based transcriptome profiling of waterlogging response in cucumber hypocotyls reveals novel insights into the de novo adventitious root primordia initiation. BMC Plant Biol. 2017;17:129. doi: 10.1186/s12870-017-1081-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reeksting BJ, Coetzer N, Mahomed W, Engelbrecht J, van den Berg N. De novo sequencing, assembly, and analysis of the root transcriptome of Persea americana (mill.) in response to Phytophthora cinnamomi and flooding. PLoS One. 2014;9:e86399. doi: 10.1371/journal.pone.0086399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang L, Zhang Y, Qi X, Li D, Wei W, Zhang X. Global gene expression responses to waterlogging in roots of sesame (Sesamum indicum L.) Acta Physiol Plant. 2012;34:2241–2249. [Google Scholar]
- 19.Baiyan Qi YY, Yin Y, Xu M, Li H. De novo sequencing, assembly, and analysis of the Taxodium ‘Zhongshansa’ roots and shoots transcriptome in response to short-term waterlogging. BMC Plant Biol. 2014;14:201. doi: 10.1186/s12870-014-0201-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang P, Lyu D, Jia L, He J, Qin S. Physiological and de novo transcriptome analysis of the fermentation mechanism of Cerasus sachalinensis roots in response to short-term waterlogging. BMC Genomics. 2017;18:649. doi: 10.1186/s12864-017-4055-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xie W, Zhang X, Cai H, Liu W, Peng Y. Genetic diversity analysis and transferability of cereal EST-SSR markers to orchardgrass (Dactylis glomerata L.) Biochem Syst Ecol. 2010;38:740–749. [Google Scholar]
- 22.Casler MD, Fales SL, AR ME, Hall MH, Hoffman LD, Leath KT. Genetic Progress from 40 years of Orchardgrass breeding in North America measured under Hay management. Crop Sci. 2000;40:1019–1025. [Google Scholar]
- 23.Stewart AV, Ellison NW. Wild crop relatives: genomic and breeding resources. 2011. Dactylis; pp. 73–87. [Google Scholar]
- 24.Yang Ji PC, Chen J, Pennerman KK, Zhang X, Hu Y, Huang L, et al. Combinations of small RNA, RNA, and Degradome sequencing uncovers the expression pattern of microRNA–mRNA pairs adapting to drought stress in leaf and root of Dactylis glomerata L. Int J Mol Sci. 2018;19:3114. doi: 10.3390/ijms19103114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang LK, Yan HD, Zhao XX, Zhang XQ, Wang J, Frazier T, et al. Identifying differentially expressed genes under heat stress and developing molecular markers in orchardgrass (Dactylis glomerata L.) through transcriptome analysis. Mol Ecol Resour. 2015;15:1497–1509. doi: 10.1111/1755-0998.12418. [DOI] [PubMed] [Google Scholar]
- 26.Yan H, Yu Zhang BZ, Yin G, Zhang X, Ji Y, Huang L, et al. Genetic diversity and association of EST-SSR and SCoT markers with rust traits in Orchardgrass (Dactylis glomerata L.) Crop Sci. 2000;40:1019–1025. doi: 10.3390/molecules21010066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Setsuko Komatsu NS. And Katsumi Sakata. ‘Omics’ techniques for identifying flooding–response mechanisms in soybean. J Proteome. 2013;93:169–178. doi: 10.1016/j.jprot.2012.12.016. [DOI] [PubMed] [Google Scholar]
- 28.Dennis ES, Dolferus R, Ellis M, Rahman M, Wu Y, Hoeren FU, et al. Molecular strategies for improving waterlogging tolerance in plants. J Exp Bot. 2000;51:89–97. [PubMed] [Google Scholar]
- 29.Fahy D, Sanad MNME, Duscha K, Lyons M, Liu F, Bozhkov P, et al. Impact of salt stress, cell death, and autophagy on peroxisomes_ quantitative and morphological analyses using small fluorescent probe N-BODIPY. Sci Rep. 2017;7:39069. doi: 10.1038/srep39069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Charlton WL, Matsui K, Johnson B, Graham IA, Ohme Takagi M, Baker A. Salt-induced expression of peroxisome-associated genes requires components of the ethylene, jasmonate and abscisic acid signalling pathways. Plant Cell Environ. 2005;28:513–524. [Google Scholar]
- 31.Foyer CH, Noctor G. Redox sensing and signalling associated with reactive oxygen in chloroplasts, peroxisomes and mitochondria. Physiol Plant. 2003;19:355–364. [Google Scholar]
- 32.Kessel-Vigelius SK, Wiese J, Schroers MG, Wrobel TJ, Hahn F, Linka N. An engineered plant peroxisome and its application in biotechnology. Plant Sci. 2013;210:232–240. doi: 10.1016/j.plantsci.2013.06.005. [DOI] [PubMed] [Google Scholar]
- 33.Sparkes IA, Brandizzi F, Slocombe SP, El-Shami M, Hawes C, Baker A. An Arabidopsis pex10 null mutant is embryo lethal, implicating peroxisomes in an essential role during plant embryogenesis. Plant Physiol. 2003;133:1809–1819. doi: 10.1104/pp.103.031252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Smertenko A. Can peroxisomes inform cellular response to drought? Trends Plant Sci. 2017;22:1005–1007. doi: 10.1016/j.tplants.2017.09.021. [DOI] [PubMed] [Google Scholar]
- 35.Del RL, Corpas FJ, Sandalio LM, Palma JM, Gomez M, Barroso JB. Reactive oxygen species, antioxidant systems and nitric oxide in peroxisomes. J Exp Bot. 2002;53:1255–1272. [PubMed] [Google Scholar]
- 36.Mhamdi A, Noctor G, Baker A. Plant catalases Peroxisomal redox guardians. Arch Biochem Biophys. 2012;525:181–194. doi: 10.1016/j.abb.2012.04.015. [DOI] [PubMed] [Google Scholar]
- 37.Palma JM, Jiménez A, Sandalio LM, Corpas FJ, Lundqvist M, Gomez M, et al. Antioxidative enzymes from chloroplasts, mitochondria, and peroxisomes during leaf senescence of nodulated pea plants. J Exp Bot. 2006;57:1747–1758. doi: 10.1093/jxb/erj191. [DOI] [PubMed] [Google Scholar]
- 38.Locato V, Cimini S, De Gara L. ROS and redox balance as multifaceted players of cross-tolerance-epigenetic and retrograde control of gene expression. J Exp Bot. 2018;69:3373–3391. doi: 10.1093/jxb/ery168. [DOI] [PubMed] [Google Scholar]
- 39.Alscher RG. Biosynthesis and antioxidaiit function of glutathione in plants. Physiol Plant. 1989;77:457–464. [Google Scholar]
- 40.Christianson JA, Llewellyn DJ, Dennis ES, Wilson IW. Global gene expression responses to waterlogging in roots and leaves of cotton (Gossypium hirsutum L.) Plant Cell Physiol. 2010;51:21–37. doi: 10.1093/pcp/pcp163. [DOI] [PubMed] [Google Scholar]
- 41.Ismond KP, Dolferus R, De Pauw M, Dennis ES, Good AG. Enhanced low oxygen survival in Arabidopsis through increased metabolic flux in the fermentative pathway. Plantphysiol. 2003;132:1292–1302. doi: 10.1104/pp.103.022244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kuroha T, Keisuke N, Rico G, Diane RW, Furuta T, Masanari N, et al. Ethylene-gibberellin signaling underlies adaptation of rice to periodic flooding. Science. 2018;361:181–186. doi: 10.1126/science.aat1577. [DOI] [PubMed] [Google Scholar]
- 43.Perata PAW, Voesenek LACJ. Plants and flooding stress. Plants and flooding stress. New Phytol. 2011;190:269–273. doi: 10.1111/j.1469-8137.2011.03702.x. [DOI] [PubMed] [Google Scholar]
- 44.Nguyen T-N, Tuan PA, Mukherjee S, Son S, Ayele BT. Hormonal regulation in adventitious roots and during their emergence under waterlogged conditions in wheat (Triticum aestivum L.) J Exp Bot. 2018;69:4065–4082. doi: 10.1093/jxb/ery190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xiao-Hua Qi X-WX, Lin X-J, Zhang W-J, Chen X-H. Identification of differentially expressed genes in cucumber (Cucumis sativus L.) root under waterlogging stress by digital gene expression profile. Genomics. 2012;99(3):160–168. doi: 10.1016/j.ygeno.2011.12.008. [DOI] [PubMed] [Google Scholar]
- 46.Turkan I. ROS and RNS: key signalling molecules in plants. J Exp Bot. 2018;69:3313–3315. doi: 10.1093/jxb/ery198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gupta KJ, Stoimenova M, Kaiser WM. In higher plants, only root mitochondria, but not leaf mitochondria reduce nitrite to NO, in vitro and in situ. J Exp Bot. 2005;26:2601–2609. doi: 10.1093/jxb/eri252. [DOI] [PubMed] [Google Scholar]
- 48.Mittal S, Kumari N, Sharma V. Differential response of salt stress on Brassica juncea :photosynthetic performance, pigment, proline, D1 and antioxidant enzymes. Plant Physiol Biochem. 2012;54:17–26. doi: 10.1016/j.plaphy.2012.02.003. [DOI] [PubMed] [Google Scholar]
- 49.Oueslati S, Karray-Bouraoui N, Attia H, Rabhi M, Ksouri R, Lachaal M. Physiological and antioxidant responses of Mentha pulegium (pennyroyal) to salt stress. Acta Physiol Plant. 2010;32:289–296. [Google Scholar]
- 50.Lee YH, Kim KS, Jang YS, Hwang JH, Lee DH, Choi IH. Global gene expression responses to waterlogging in leaves of rape seedlings. Plant Cell Rep. 2014;33:289–299. doi: 10.1007/s00299-013-1529-8. [DOI] [PubMed] [Google Scholar]
- 51.Elaine Yeung JB-S. And Rashmi Sasidharan. After the deluge:plant revival post-flooding. Trends Plant Sci. 2019;24:443–454. doi: 10.1016/j.tplants.2019.02.007. [DOI] [PubMed] [Google Scholar]
- 52.ZL ZOU X-l, Guang-yuan LU, Yong CHENG, Jin-song XU, Xue-kun ZHANG. Comparison of transcriptomes undergoing waterlogging at the seedling stage between tolerant and sensitive varieties of Brassica napus L. J Integr Agric. 2015;14:1723–1734. [Google Scholar]
- 53.Juntawong P, Sirikhachornkit A, Pimjan R, Sonthirod C, Sangsrakru D, Yoocha T, et al. Elucidation of the molecular responses to waterlogging in Jatropha roots by transcriptome profiling. Front Plant Sci. 2014;5:658. doi: 10.3389/fpls.2014.00658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cheng-Gang Ren C-CK, Yan K, Zhang H, Luo Y-M, Xie Z-H. Elucidation of the molecular responses to waterlogging in Sesbania cannabina roots by transcriptome profiling. Sci Rep. 2017;7:9256. doi: 10.1038/s41598-017-07740-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nanjo Y, Maruyama K, Yasue H, Yamaguchi-Shinozaki K, Shinozaki K, Komatsu S. Transcriptional responses to flooding stress in roots including hypocotyl of soybean seedlings. Plant Mol Biol. 2011;77:129–144. doi: 10.1007/s11103-011-9799-4. [DOI] [PubMed] [Google Scholar]
- 56.Sairam RK, Kumutha D, Ezhilmathi K, Deshmukh PS, Srivastava GC. Physiology and biochemistry of waterlogging tolerance in plants. Biol Plant. 2008;52:401–412. [Google Scholar]
- 57.Imen Klay SG, Liu M, Mila I, Khoudi H, Bernadac A, Mondher Bouzayen JP. Ethylene response factors (ERF) are differentially regulated by different abiotic stress types in tomato plants. Plant Sci. 2018;274:137–145. doi: 10.1016/j.plantsci.2018.05.023. [DOI] [PubMed] [Google Scholar]
- 58.Mei-Chun Cheng P-ML, Kuo W-W, Lin T-P. The Arabidopsis ETHYLENE RESPONSE FACTOR1 regulates abiotic stress-responsive gene expression by binding to different cis-acting elements in response to different stress signals. Plant Physiol. 2013;162:1566–1582. doi: 10.1104/pp.113.221911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yamaguchi-Shinozaki K, Shinozaki K. Transcriptional regulatory networks in cellular responses and tolerance to dehydration and cold stresses. Annu Rev Plant Biol. 2006;57:781–803. doi: 10.1146/annurev.arplant.57.032905.105444. [DOI] [PubMed] [Google Scholar]
- 60.Takashi TDK, Seo M, Shinozaki K. ABA-mediated transcriptional regulation in response to osmotic stress in plants. Trends Plant Sci. 2018;23:513–522. [Google Scholar]
- 61.Kasahara H. Current aspects of auxin biosynthesis in plants. Biosci Biotechnol Biochem. 2015;80:1–9. doi: 10.1080/09168451.2015.1086259. [DOI] [PubMed] [Google Scholar]
- 62.Pagnussat GC, Lanteri ML, Lombardo MC, Lamattina L. Nitric oxide mediates the indole acetic acid induction activation of a mitogen-activated protein kinase Cascade involved in adventitious root development. Plant Physiol. 2004;135:279–286. doi: 10.1104/pp.103.038554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12:357–360. doi: 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Huang L, Feng G, Yan H, Zhang Z, Bushman BS, Wang J, et al. Genome assembly provides insights into the genome evolution and flowering regulation of orchardgrass. Plant Biotechnol J. 2019;17:1183–1470. doi: 10.1111/pbi.13205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Anders S, Pyl PT, Huber W. HTSeq-a python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang L, Feng Z, Wang X, Wang X, Zhang X. DEGseq:an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics. 2010;26:136–138. doi: 10.1093/bioinformatics/btp612. [DOI] [PubMed] [Google Scholar]
- 67.Huang L, Yan H, Jiang X, Zhang Y, Zhang X, Ji Y, et al. Reference gene selection for quantitative real-time reverse-transcriptase PCR in orchardgrass subjected to various abiotic stresses. Gene. 2014;553:158–165. doi: 10.1016/j.gene.2014.10.017. [DOI] [PubMed] [Google Scholar]
- 68.Huihui Zhua B, Honglian A, Liwen C, Ran S, Hengpeng Y, Dongyun D, et al. Transcriptome analysis providing novel insights for cd-resistant tall fescue responses to cd stress. Ecotox Environ Safe. 2018;160:349–356. doi: 10.1016/j.ecoenv.2018.05.066. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Table S1. Summary of RNA-seq sequence a nalysis of orchardgrass. Table S2. Differential analysis results of all DEGs suffering flooding at WS_8h vs WS_0h in leaves. Table S3. Differential analysis results of all DEGs suffering flooding at WS_24h vs WS_0h in leaves. Table S4. GO enrichment analysis of all DEGs suffering flooding at WS_8h vs WS_0h in leaves. Table S5. GO enrichment analysis of all DEGs suffering flooding at WS_24h vs WS_0h in leaves. Table S6. KEGG Pathway enrichment analysis of all DEGs suffering flooding at WS_8h vs WS_0h in leaves. Table S7. KEGG Pathway enrichment analysis of all DEGs suffering flooding at WS_24h vs WS_0h in leaves. Table S8. The primers used in the qRT-PCR analysis.
Additional file 2: Table S9. Analysis of 4 key pathways in response to flooding stress.
Additional file 3: Figure S1. Correlation analysis under different time flooding stress. Figure S2. GO enrichment analysis of down-regulated DEGs suffering flooding at WS_8h vs WS_0h and up-regulated DEGs suffering flooding at WS_24h vs WS_0h in leaves. Figure S3. GO functional classification of all DEGs suffering flooding at WS_8h vs WS_0h and WS_24h vs WS_0h in leaves. Figure S4. qRT-PCR verification results of ten DGEs in control and treatment groups. Figure S5. Correlation between qRT-PCR and RNA sequencing for the ten selected genes.
Data Availability Statement
All data produced by the study are disclosed in the manuscript and the additional files. The raw data of this study was stored in the NCBI (National Coalition Building Institute) SAR (Sequence Read Archive) database, which can be viewed in BioProject (ID: PRJNA554779).