

Abstract
Purpose:
OX40 agonist-based combinations are emerging as a novel avenue to improve the effectiveness of cancer immunotherapy. To better guide its clinical development, we characterized the role of the OX40 pathway in tumor-reactive immune cells. We also evaluated combining OX40 agonists with targeted therapy to combat resistance to cancer immunotherapy.
Experimental Design:
We utilized patient-derived tumor infiltrating lymphocytes (TILs) and multiple preclinical models to determine the direct effect of anti-OX40 agonistic antibodies on tumor-reactive CD8+ T cells. We also evaluated the antitumor activity of an anti-OX40 antibody plus PI3Kβ inhibition in a transgenic murine melanoma model (Braf-mutant, PTEN null), which spontaneously develops immunotherapy-resistant melanomas.
Results:
We observed elevated expression of OX40 in tumor reactive CD8+ TILs upon encountering tumors; activation of OX40 signaling enhanced their cytotoxic function. OX40 agonist antibody improved the antitumor activity of CD8+ T cells and the generation of tumor-specific T cell memory in vivo. Furthermore, combining anti-OX40 with GSK2636771, a PI3Kβ selective inhibitor, delayed tumor growth and extended the survival of mice with PTEN-null melanomas. This combination treatment did not increase the number of TILs, but it instead significantly enhanced proliferation of CD8+ TILs and elevated the serum levels of CCL4, CXCL10, and IFN-γ, which are mainly produced by memory and/or effector T cells.
Conclusion:
These results highlight a critical role of OX40 activation in potentiating the effector function of tumor-reactive CD8+ T cells and suggest further evaluation of OX40 agonist-based combinations in patients with immune-resistant tumors.
Keywords: OX40, PI3K, cancer immunotherapy
INTRODUCTION
Several immunomodulatory agents that target T cell co-inhibitory receptors, such as PD-1 and CTLA-4, have been developed to boost T cell-mediated antitumor immune responses in cancer patients. These immunotherapies have demonstrated durable clinical benefit in many types of cancer, and immune checkpoint blockade has become a standard front-line treatment in multiple solid cancers, including melanoma, lung cancer, bladder cancer, and kidney cancer (1,2). This new clinical paradigm has shifted research efforts in tumor immunology to prioritize the identification of additional immunoregulatory targets and rational combinatorial treatments to further increase the rate of potent and durable antitumor immune responses.
T cell activation is tightly regulated by two sets of signals via T cell receptors (TCR) and T cell co-signaling receptors. Positive (co-stimulatory) and negative (co-inhibitory) signals from T cell co-signaling receptors direct T cell function in response to TCR stimulation. Several studies have demonstrated that activating T cell co-stimulatory receptors, such as OX40 and 4–1BB, can facilitate T cell-mediated antitumor immunity (3,4). Moreover, disrupting T cell co-inhibitory signaling pathways, such as PD-1 and CTLA-4, has been reported to reinvigorate tumor-reactive T cells and stem tumor development in patients with a variety of tumors (5). However, a durable and effective antitumor immune response only can be achieved in a small percentage of cancer patients treated with immune checkpoint blockade (ICB) (6). One mechanism of primary resistance to ICB is insufficient tumor-reactive T cells in patients with non-immunogenic tumors (7). Under the notion that activation of T cell co-stimulatory signaling pathways can augment the generation of effector and memory T cells (8), more studies are focused on targeting T cell co-stimulatory receptors to overcome primary resistance to ICB therapy in cancer patients. One such prominent T cell co-stimulatory molecule is OX40. Indeed, early phase clinical trials evaluating agonist antibodies targeting the OX40 pathway alone or in combination with ICB in cancer patients are ongoing, such as NCT02221960 (formerly of MedImmune), NCT02528357 (GlaxoSmithKline) and NCT02554812 (Pfizer). While these trials have begun, an improved understanding of the impact of OX40 activation on immune effector cells may help to optimize the clinical evaluation of OX40-based immunotherapy and develop novel combinatorial approaches to treat cancer patients with primary resistance to ICB.
Here, by utilizing melanoma patient-derived cell lines and multiple preclinical models, we sought to determine the role of the OX40 pathway in regulating the effector function of tumor-reactive T cells and evaluate the therapeutic potential of combining OX40 agonist antibody with cancer targeted therapy. Our results describe the value of an OX40 agonist antibody to augment T cell-mediated antitumor response by directly enhancing proliferation and cytotoxicity of CD8+ tumor-reactive T cells. This study also provides critical rationale for the clinical evaluation of the combination of an OX40 agonist antibody and a selective PI3Kβ inhibitor in patients with immunoresistant PTEN loss tumors.
Material and Methods
Cell lines and Mice
Human Mel2399, Mel2559, and their autologous TILs were established from metastatic melanoma patients enrolled in the adoptive T cell therapy (ACT) trial at MD Anderson Cancer Center (MDACC) as previously described (9). The murine MC38/gp100 cell line was generated in our previous study (10). All tumor cell lines were maintained in RPMI 1640 complete medium supplemented with 10% heat-inactivated fetal bovine serum (Atlanta Biologicals) and normocin (InvivoGen). TIL cell lines were maintained in RPMI 1640 with 10% human type AB serum (GEMINI), 3000 U/ml IL-2 (Prometheus Laboratories), and normocin. Cells were routinely monitored for mycoplasma contamination by using the MycoAlert kit (Lonza). Short tandem repeat (STR) profiling was used to confirm the identity of patient-derived cell lines. The maximum length of time of in vitro cell culture between thawing and use in the described experiments was two weeks.
C57BL/6 mice and C57BL/6 albino mice were purchased from Charles River Frederick Research Model Facility. Tyr:CreER; PTENlox/lox; BRAF V600E/+ (BP) mice bred onto a C57BL/6 background were kindly provided by Dr. Bosenberg (Yale University School of Medicine). Pmel-1 TCR/Thy1.1 mice were from in-house breeding colonies. All mice were maintained in a specific pathogen-free barrier facility at MDACC. All studies were conducted in accordance with the MDACC and GlaxoSmithKline (GSK) Policy on the Care, Welfare and Treatment of Laboratory Animals. All animal experiment protocols were reviewed by the Institutional Animal Care and Use Committee either at GSK or at MDACC, the institution where the work was performed.
Caspase-3 based T cell killing assay
Patient-derived tumor cells were labelled with DDAO dye (Thermo Fisher) according to the manufacturer’s instructions. Peripheral blood mononuclear cells (PBMCs) from healthy donors were isolated from buffy coats (Gulf Coast Regional Blood Center) and irradiated with 5000 rad of gamma-radiation. Irradiated PBMCs were washed with PBS and then incubated with 10 μg/ml full length or Fc-fragment deleted anti-human OX40 (GSK3174998, GlaxoSmithKline) at 37°C for 1 hour. Antibody-pulsed PBMCs were mixed with DDAO-labelled tumor cells and autologous TILs at 37°C for an additional 3 hours. The ratio of T cells to PBMCs used in this assay was 1:1. To evaluate the effect of the activation of the OX40 pathway in murine CD8+ T cells, we cross-linked anti-mouse OX40 antibody by pretreating bone marrow derived dendritic cells (DCs) from C57BL/6 mice with 10 μg/ml anti-murine OX40 antibody at 37°C for 1 hour. After washing with PBS, antibody-pulsed DCs were mixed with gp100-specific CD8+ Pmel-1 T cells and MC38/gp100 tumor cells for an additional 3 hours. The ratio of DCs to T cells was 1:1. The cell mixtures were then permeabilized with Fix/Perm solution (BD Biosciences) for 20 min at room temperature and stained with a PE-conjugated anti-cleaved caspase-3 monoclonal antibody (BD Biosciences) as previously described (11). Samples were analyzed using a FACSCanto II (BD Biosciences). The percentage of cleaved caspase-3+ tumor cells was calculated and used to determine the extent of T-cell induced tumor apoptosis.
Retroviral transduction of pmel-1 T cells
Full-length human OX40 was amplified and cloned into a retroviral vector, pMXs-IG, which was kindly provided by Dr. Kitamura (University of Tokyo, Japan)(12). The retroviral vector expressing an enhanced firefly luciferase was generated in our previous study (13). Retroviral vectors and the packaging vectors were transiently co-transfected into the packaging cell line, Plate-E, using Lipofectamine 2000 (Invitrogen). Supernatants containing viral particles were used to infect pre-activated splenocytes from pmel-1 mice as previously described (10). Three days after transduction, transduced pmel-1 T cells were sorted using a FACSAria (BD Biosciences) based on the expression of appropriate reporter genes embedded in the expression vectors.
Tumor and vaccination models
To determine the in vivo effect of targeting OX40 on the function of tumor-reactive CD8+ T cells, luciferase-expressing pmel-1 T cells were transferred into C57BL/6 albino mice bearing MC38/gp100 tumor as previously described (10). One hundred micrograms of anti-mouse OX40 (OX-86, BioXcell) or mouse anti-human OX40 (Kindly provided Dr. Voo) (14) was intraperitoneally administered to tumor-bearing mice (twice per week). Tumor size was monitored every two days, and in vivo bioluminescence imaging analyses were performed by using an IVIS 200 system (Xenogen) on day 6 after T cell transfer.
To evaluate the antitumor activity of anti-OX40 alone and in combination with PI3Kβ inhibition, Tyr:CreER; PTENlox/lox; BRAF V600E/+ mice (BP mice, 6–8 weeks of age) were treated with 4-hydroxytamoxifen to induce tumor formation. Tumor-bearing mice were randomized into four groups to receive anti-OX40 and/or GSK2636771. GSK2636771 (GlaxoSmithKline) was suspended in 1% (w/v) methylcellulose and administered to mice daily by oral gavage at a dose of 30 mg/kg. Anti-OX40 (OX-86, BioXcell) was administered at a dose of 50 μg/per mouse. The relevant solvent and control rat IgG antibody (Sigma) were administered to animals in the control group.
Because our previous study showed that CD40 agonistic antibody can promote in vivo proliferation and activation of T cells (15), when we evaluated the in vivo effect of anti-OX40 monotherapy and in combination with selective PI3Kβ inhibition on antigen-specific T cells, we re-designed our vaccine model to eliminate the possible confounding effects of a CD40 antibody-containing immunoadjuvant on the antitumor activity of an OX40 agonist (11,16). Briefly, C57BL/6 mice received 1,000 naive pmel-1 T cells intravenously (i.v.) and were vaccinated with two distinct subcutaneous (s.c.) injections in each flank with 100 μl of saline containing 100 μg of human gp10025–33. In addition, vaccinated mice received 100,000 IU rhIL-2 protein intraperitoneally (i.p.) once on the day of vaccination and twice daily on the next 2 days and were topically treated with 50 mg of 5% imiquimod cream (Aldara, Fougera) on the vaccination site once after each vaccination.
Luminex assay and profiling of tumor infiltrating immune cells
Serum, spleen, and tumor tissue samples were collected from BP mice on day 6 after treatment. Twenty-five microliters of serum from each mouse was assayed using the MILLIPLEX mouse cytokine/chemokine panels I, II, and III according to the manufacturer’s protocol (EMD Millipore). The concentration of each cytokine/chemokine present in the serum samples was measured using a Luminex 200 system (Luminex Corporation). Fresh tumor tissues were incubated with RPMI medium containing 1 mg/ml collagenase, 100 μg/ml hyaluronidase (Sigma-Aldrich) at 37°C for 60 min, and manually dissociated to generate single cell suspensions. Single cell suspensions from tumor or spleen tissues were then washed twice with staining buffer and incubated with a cocktail of antibodies targeting surface markers at 4°C for 30 min. Cells were then fixed and permeabilized using the Foxp3 fix and permeabilization kit according to manufacturer’s protocol (eBioscience), and then incubated with a cocktail of antibodies against intracellular markers. Stained samples were analyzed with a FACSCanto II or a Helios mass cytometer (Fluidigm). Antibody details for the flow cytometry and mass cytometry staining panels used in this study are provided in Supplementary Table 1.
Statistical analyses
Summary statistics (e.g., mean, SEM) of the data are reported. Assessments of differences in continuous measurements between two groups were made using two-sample t-test posterior to data transformation (typically logarithmic, if necessary), or Wilcoxon rank-sum test. Differences in tumor size and T cell numbers among several treatments were evaluated using analysis of variance (ANOVA) models. The Kaplan-Meier method and log-rank test were used to compare survival between groups. P<0.05 was considered statistically significant. Graph generation and statistical analyses were performed using GraphPad Prism (version 7) and R software programming language (version 3.1.0).
RESULTS
The OX40 pathway plays a critical role in regulating the antitumor function of tumor-infiltrating T cells (TILs) in melanoma patients
To determine the importance of the OX40 pathway in regulating the effector function of TILs in melanoma patients, we first assessed the expression of OX40 on TILs before and after re-stimulation with autologous tumors. We used two tumor-reactive TIL cell lines previously generated from advanced melanoma patients (11,17). Cryopreserved TILs were thawed and cultured in fresh culture medium in the presence of IL-2 for 3–4 days. Revived TILs were incubated with autologous tumors at the varying ratios of T cells to tumor cells (E:T ratio), and the expression level of OX40 on the surface of TILs over time was determined by flow cytometry analysis (Fig. 1A). The majority of resting CD4+ TILs, but less than 10% of CD8+ TILs, expressed OX40 before re-stimulation. The percentage of OX40+ CD4+ and CD8+ TILs peaked at 6 hours after TCR stimulation with autologous tumors. Seventy-two hours after stimulation, the percentage of OX40+ TILs returned to baseline levels. To validate that the OX40 expression on re-stimulated TILs is tumor dependent, we assessed the percentage of OX40+ TILs after a 12-hour co-incubation with tumors at E:T ratios ranging from 0.1:1 to 3:1. The results showed that increasing the E:T ratio can enhance the percentage of both OX40+ CD4+ and CD8+ TILs (Supplementary Fig 1A). These results demonstrate that OX40 expression on patient-derived TILs is inducible and under the control of TCR signaling. Notably, OX40 was expressed on a significant portion of CD8+ TILs (>50%) at 6 hours after encountering autologous tumors (Fig 1A), suggesting that activation of the OX40 pathway had the potential to directly modulate the function of CD8+ T cells at tumor sites. To test this hypothesis, we used a cytotoxicity assay based on the expression of cleaved caspase-3 in tumor cells to evaluate whether activation of the OX40 signaling can alter cytotoxicity of patient-derived TILs against autologous tumors. Given that immobilization of anti-OX40 antibody is required to activate the OX40 pathway in T cells (14), gamma-irradiated PBMCs were pulsed with anti-human OX40 (hOX40) antibody (GSK3174998) for one hour and used to stimulate TILs in the presence of autologous tumors. When compared with Fc-fragment deleted anti-hOX40 antibody, full-length anti-hOX40-pulsed PBMCs significantly increased TIL-induced apoptosis of tumors (Fig. 1B). In contrast, treatment with anti-hOX40-pulsed PBMCs alone had no impact on tumor apoptosis. Additionally, anti-mouse OX40-pulsed dendritic cells enhanced murine tumor apoptosis induced by tumor-reactive CD8+ T cells (Supplementary Fig 1B). Although OX40 was not highly expressed on resting cytotoxic CD8+ TILs, our results suggest that tumor exposure induced the expression of OX40 on CD8+ TILs and that activation of OX40 signaling enhances the cytotoxic function of CD8+ TILs.
Figure 1. Kinetics of OX40 expression and in vitro effect of OX40 agonist antibody on tumor infiltrating T cells (TILs) from melanoma patients.
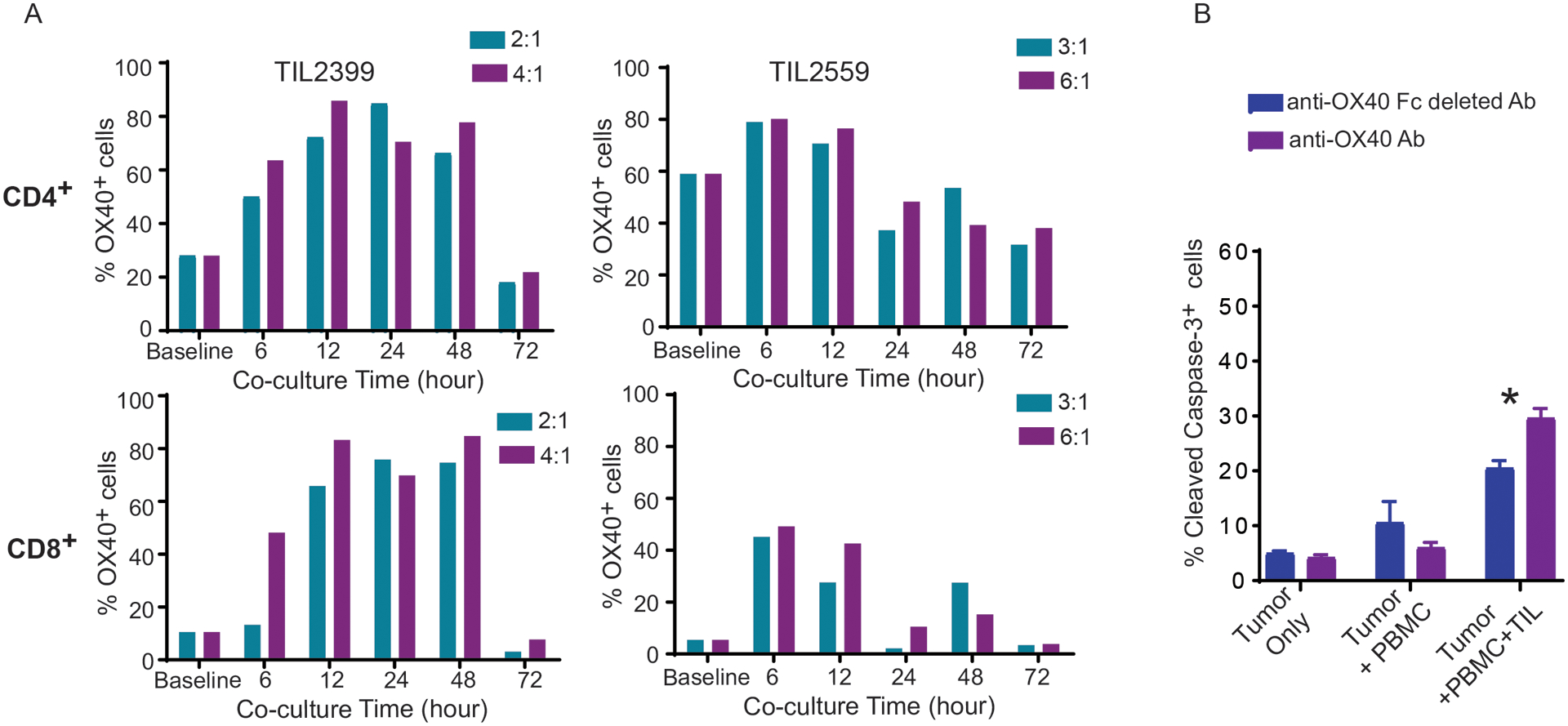
(A) Increased OX40 expression on both CD4+ and CD8+ TILs upon re-stimulation by autologous tumor cells. Two patient-derived TIL lines, TIL2399 and TIL2559, were cultured in T cell growth medium in the presence of IL-2 for at least 3 days. Revived TILs were then co-cultured with autologous tumor cells at different ratios of effector to target cells (E:T). The percentage of TILs expressing OX40 was determined by flow cytometry analysis at indicated the time points. (B) Cross-linked anti-OX40 antibody enhanced the cytotoxicity of TILs against autologous tumors. Irradiated PBMCs from healthy donors were pulsed with 10 μg/ml of full length or Fc-fragment-deleted anti-human OX40 (GSK3174998) at 37°C for 1 hour. After washing with PBS, antibody-pulsed PBMCs were mixed with DDAO-labelled tumor cells (MEL2559) and autologous TILs (TIL2599) at 37°C for an additional three hours. The cell mixtures were stained intracellularly with an anti-cleaved caspase-3 antibody. The cytotoxicity of TILs against tumors was evaluated by flow cytometry analysis based on the percentage of cleaved caspase-3+ DDAO-labeled tumor cells. One-way ANOVA demonstrated statistical significance (*P<0.05). Representative data from three independent experiments are shown.
Activation of the OX40 pathway improves antitumor activity of CD8+ T cells and facilitates the generation of tumor-specific T cell memory
To characterize the in vivo effects of OX40 agonist antibody on antitumor activity of CD8+ T cells, we adoptively transferred luciferase-expressing tumor-reactive CD8+ T cells (pmel-1) into tumor-bearing mice one day after sublethal irradiation, which is required for expansion of transferred T cells. These mice were then treated with either a control antibody or an anti-mouse OX40 (mOX40) antibody as shown in Figure 2A. The gp100-expressing MC38 tumors in mice treated with pmel-1 T cells grew significantly slower than those not treated with T cells (P<0.0001, Fig. 2B). Importantly, anti-mOX40 treatment significantly delayed tumor growth in all T cell-treated mice (P<0.001, Fig. 2B). We also used bioluminescence imaging analysis to determine the change in tumor trafficking of transferred tumor-reactive T cells in response to anti-mOX40 treatment. Although the average bioluminescence intensity at the tumor site in mice treated with anti-mOX40 was higher than the control group on day 6 after T-cell transfer, the difference between these two group was not statistically significant (P=0.053, Fig 2C).
Figure 2. In vivo effect of OX40 activation on antitumor activity and memory generation of tumor-reactive T cells.
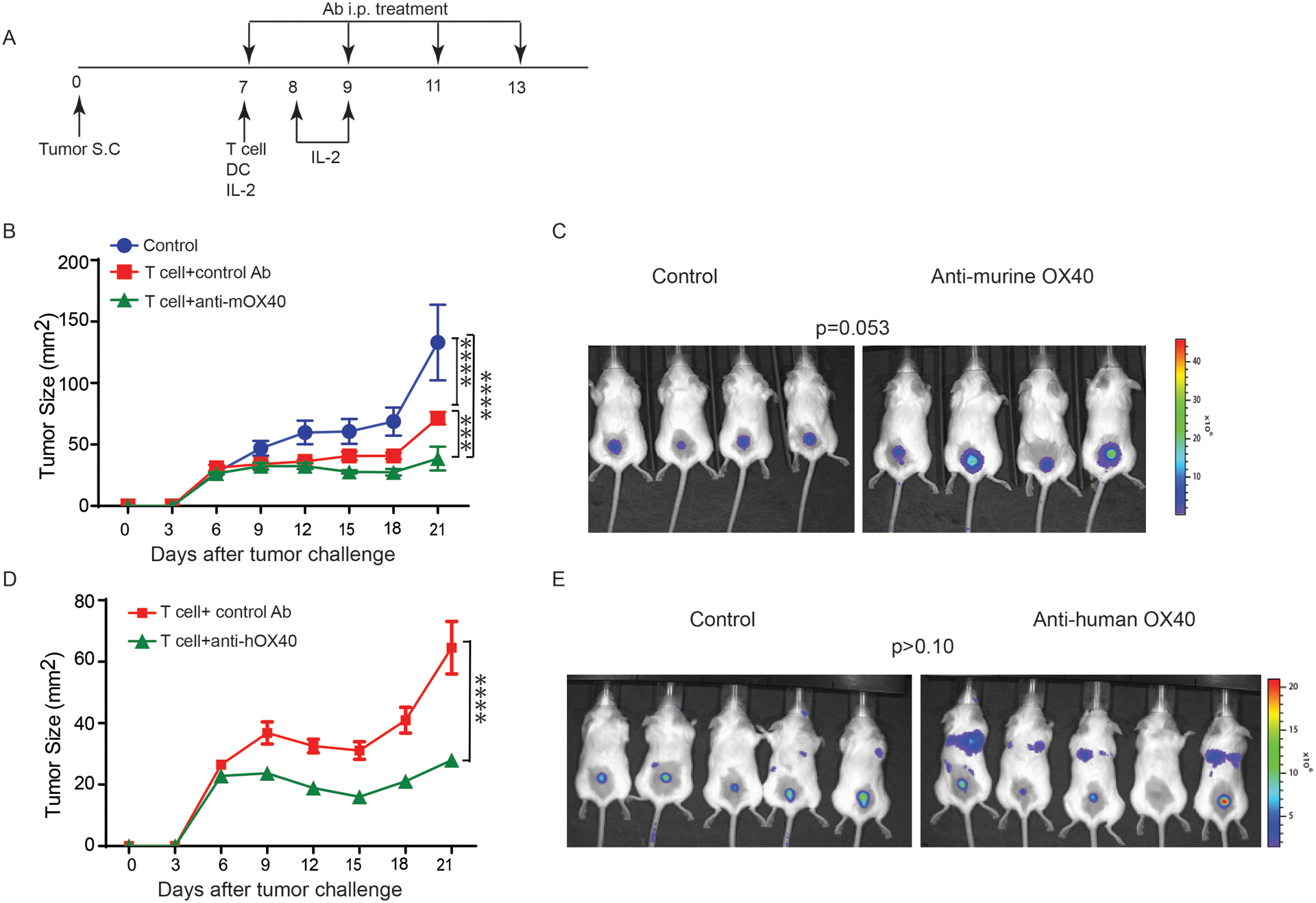
(A) Experimental setup of a murine ACT protocol to evaluate in vivo effect of the activation of OX40 signaling on tumor-reactive T cells. (B) Increased antitumor activity of tumor-reactive T cells in the presence of anti-mouse OX40 antibody. Pmel-1 T cells that express a TCR specifically recognizing a melanoma tumor antigen (gp100) were modified to express luciferase for in vivo monitoring of tumor trafficking. Luciferase-expressing pmel-1 T cells were transferred into mice bearing gp100-expressing MC38 tumors (N=4 per group). All experimental mice were then treated with DC vaccine and IL-2 as described previously (41). One hundred microgram per dose of anti-OX40 or control antibody was used to treat mice twice weekly for two weeks. Tumor size was monitored every two days. (C) Luciferase signaling intensity at tumor sites in mice with ACT. Tumor tracking of transferred pmel-1 T cells was evaluated on day 6 after T-cell transfer. Quantitative imaging analysis revealed that anti-mOX40 did not significantly alter tumor tracking of transferred T cells. (D) Anti-human OX40 antibody facilitated human OX40-expressing pmel-1 T cells control of the growth of MC38/gp100 tumors (N= 5 per group). (E) In vivo tumor tracking of human OX40-expressing T cells in response to anti-human OX40 antibody treatment. Quantitative imaging analysis revealed that anti-hOX40 did not significantly alter tumor trafficking of transferred T cells. The pairwise multiple comparisons after two-way ANOVA test and the t-test were used to evaluate the statistical significance of the difference in tumor growth and tumor trafficking, respectively. *** P<0.001 and ****P<0.0001. Representative data from two independent experiments are shown.
Given that an OX40 agonist antibody has been reported to enhance the proliferation of CD4+ T helper (Th) cells and suppress the function of T regulatory (Treg) cells (14), the OX40 agonist-enhanced antitumor activity of transferred CD8+ T cells observed in this model may have been achieved by indirect regulation via CD4+ T cells (18). To test whether anti-OX40 treatment can directly promote CD8+ T cell function, we modified our murine ACT model to ensure that the OX40 agonist antibody only targeted OX40-expressing transferred CD8+ T cells. We first transduced full-length human OX40 (hOX40) cDNA into murine pmel-1 T cells, and adoptively transferred hOX40-expressing CD8+ pmel-1 T cells into tumor-bearing mice. Instead of using the mOX40 agonist, we treated all experimental mice with either isotype control antibody or anti-human OX40 antibody. Reduced tumor size was observed in mice from the anti-hOX40 group as early as two days after the first dose of antibody treatment (Fig 2D). Bioluminescence imaging analysis of mice on day 6 after T cell transfer revealed that the number of transferred CD8+ T cells in the tumors was comparable between the control and anti-hOX40 groups (Fig 2E). Although OX40 agonist antibody treatment had limited impact on directly regulating tumor trafficking of CD8+ T cells, data from both models supports that activation of OX40 signaling promotes the effector function of tumor reactive CD8+ T cells.
We then examined the impact of anti-OX40 on the generation of antigen-specific CD8+ memory T cells using a murine vaccine model (Fig. 3A) (11,16). The composition of adjuvants in our previously described murine vaccination model was simplified to avoid confounding effects of the anti-CD40 antibody on anti-OX40 antibody activity. Briefly, fresh splenocytes from pmel-1 mice were adoptively transferred into C57BL/6 mice. Experimental mice were then vaccinated with the gp100 peptide on day 0 and day 28, and treated with anti-mOX40 on days 0, 4, 7, 12, 28, 31, 35, and 40. One week after the last anti-OX40 treatment, experimental mice were challenged with gp100-expressing MC38 tumor cells. By monitoring the percentage of transferred pmel-1 T cells in PBMCs, we found that anti-OX40 enhanced the proliferation of pmel-1 T cells after initial antigen stimulation (on day 5), and this positive effect of anti-OX40 was dose-dependent (Fig 3B). Five days after the booster vaccine (on day 33), the percentage of antigen-specific T cells in peripheral blood CD8+ T cells in mice is also significantly higher than the rest of vaccinated mice (Fig 3B). Moreover, administration of 200 μg of anti-OX40 to gp100-vaccinated mice before tumor inoculation successfully suppressed the development of gp100-expressing tumors, indicating that OX40 agonists can induce the generation of tumor-reactive memory T cells (Fig 3C).
Figure 3. In vivo OX40 agonist antibody treatment enhanced the proliferation of tumor-reactive T cells upon TCR stimulation and induced the generation of tumor-specific T cell memory.
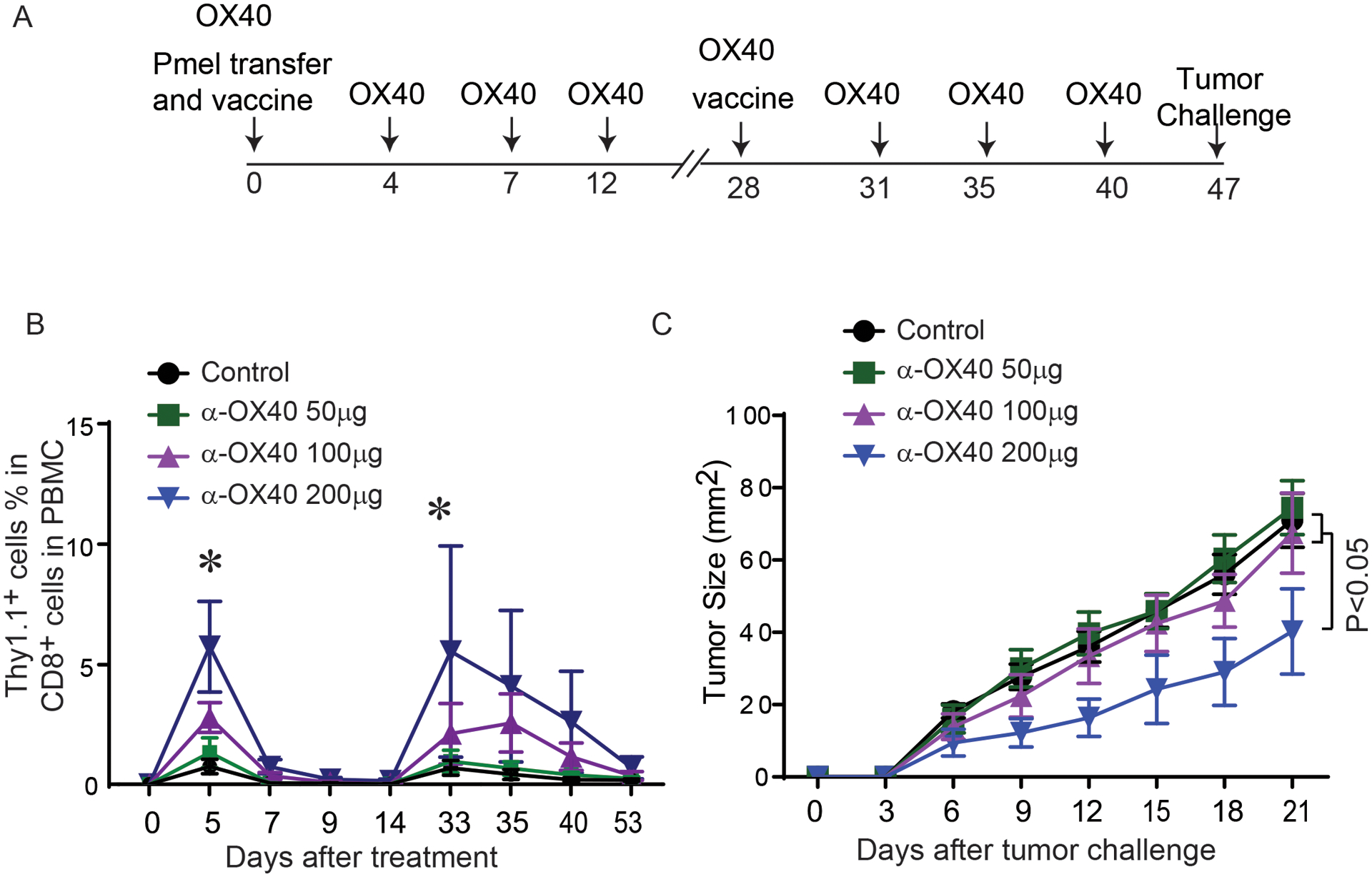
(A) Schematic representation of a murine vaccine model used to evaluate the in vivo effect of the activation of OX40 signaling on tumor-reactive T cells. C57BL/6 mice were transferred with the splenocytes from pmel-1 mice and vaccinated with gp100 peptide. Vaccinated mice received either control antibody or anti-mouse OX40 antibody. After 4 weeks, mice received a second gp100 peptide vaccine (booster). Gp100-expressing MC38 tumors were subcutaneously injected into vaccinated mice on day 47. (B) The percentage of gp100-specific T cells in CD8+ T cells in the peripheral blood of mice treated with OX40 agonist antibody. Thy1.1, a congenic marker for transferred pmel-1 T cells, was used to determine the number of gp100-specific T cells in peripheral blood after antigen stimulation. The pairwise multiple comparisons after two-way ANOVA test demonstrated statistical significance (*P<0.05): control/ α-OX40 50 μg versus α-OX40 100 μg/ α-OX40 200 μg on day 5; control/ α-OX40 50 μg versus α-OX40 200 μg on day 33. (C) The growth curves of MC38/gp100 tumors in vaccinated mice treated with anti-OX40 (N=8 per group). Representative data from two independent experiments are shown.
OX40 agonist antibody synergizes with GSK2636771 in controlling the development of PTEN-null melanoma
Previously, we demonstrated that oncogenic activation of the PI3K pathway by PTEN loss promotes tumor-associated immunosuppressive mechanisms and is associated with poor clinical outcomes in melanoma patients treated with anti-PD-1(11). We also found that Braf-mutant, PTEN-null melanomas developed in Tyr:CreER; BRAFV600E/+; PTENlox/lox mice (BP mice) are resistant to immune checkpoint inhibitory antibodies. However, improved tumor growth inhibition was observed with the combination of anti-PD-1 antibodies with GSK2636771, a PI3Kβ selective inhibitor, which was selected based on data supporting a role for this PI3K isoform selective inhibition in cells with loss of PTEN (11,19,20). The combination of pembrolizumab and GSK2636771 is now being evaluated in a phase I/II clinical trial (NCT03131908).
To evaluate the effectiveness of combining T cell co-stimulatory receptor-based immunotherapy and targeted therapy, we combined anti-OX40 with GSK2636771 in the BP model (Fig. 4A). BP mice bearing measurable melanoma lesions were randomized and treated with isotype control antibody, GSK2636771, anti-OX40 antibody, or a combination of the two agents. Single agent anti-OX40 and single-agent GSK2636771 both failed to significantly inhibit tumor growth, but the combination was highly effective and markedly extended the survival of BP tumor-bearing mice (the median survival times of control, GSK2636771, anti-OX40 and combination groups are 14.5 days, 18 days, 14 days and 30 days respectively; p= 0.0021) (Fig. 4B and 4C). A linear mixed model (21) determined that the anti-tumor effect of GSK2636771 with anti-OX40 was synergistic (P=0.0004, Supplementary Fig 2).
Figure 4. OX40 agonist antibody synergized with PI3Kβ selective inhibition to control the growth of PTEN loss tumors.
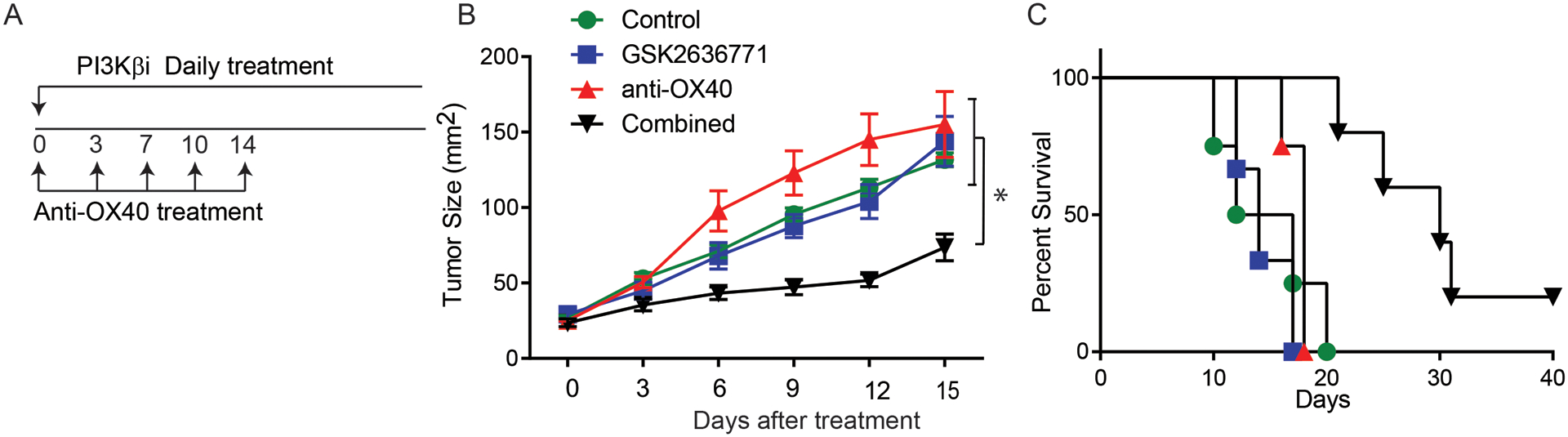
(A) The treatment schedule of antibody and the PI3Kβ inhibitor (GSK2636771) is shown. Melanoma was induced in a group of Tyr:CreER; PTENlox/lox; BRAFV600E/+ mice. Mice with measureable tumors were randomized and treated with control, GSK2636771 (30 mg/kg/d), anti-mouse OX40 (50 μg/dose), and the combination of both reagents. (B) Tumor size was monitored in each of the treatment groups every two days. The pairwise multiple comparisons after two-way ANOVA test were used to determine statistical significance. *P<0.05. (D) Kaplan-Meier survival curves of mice treated with GSK2636771 and/or anti-mouse OX40. Log-rank test demonstrated statistical significance (P<0.05): GSK2636771+anti-OX40 vs control, GSK2636771, anti-OX40 (N=4–7). Data presented are a summary of two independent experiments.
Importantly, no overt adverse effects or toxicities were observed with the combination treatment. We further tested whether GSK2636771, anti-OX40, or the combination affected the proliferation of antigen-specific T cells upon in vivo antigen stimulation. These experiments showed that the combination treatment did not significantly reduce whole blood cell counts or inhibit the proliferation of gp100-specific T cells upon gp100 peptide immunization, (Supplementary Fig 3). Taken together, these data suggest that combining anti-OX40 with GSK2636771 is another potentially effective strategy to overcome immune resistance in melanomas with PTEN loss.
Combining anti-OX40 with a PI3Kβ inhibitor enhances T cell-mediated antitumor immune activity
We then explored the underlying mechanisms by which PI3Kβ inhibition synergizes with anti-OX40 to control Braf-mutant, PTEN-null melanomas. Additional tumor-bearing BP mice were treated with anti-OX40 and/or GSK2636771 as described above. On day 6, serum, spleen, and tumor tissue samples were collected for immune profiling. We measured the serum concentrations of a broad set of chemokines/cytokines in each experimental mouse using Luminex assays. Among 43 tested chemokines/cytokines, the combination treatment significantly increased the serum concentrations of CCL2, CCL4, CCL15, CXCL10, and G-CSF in comparison to the monotherapy or control treatments (Fig 5A and Supplementary Fig 4). Two of these factors, CCL4 and CXCL10, are mainly produced by memory and/or effector T cells. In addition, the serum levels of IFN-γ, another important antitumor cytokine produced by T cells, were significantly higher in mice who received the combination treatment than in mice treated with the control antibody or PI3Kβ inhibitor alone (Fig 5A). Next, we characterized the function and phenotype of immune cells in spleens from mice in the different treatment groups using a 24-channel mass cytometry (CyTOF) panel (Supplementary table 1). High-dimensional analysis using SPADE was performed to examine the changes in immune cells in the treatment groups. Anti-OX40 monotherapy reduced the percentage of M2 macrophages, which are immunosuppressive immune cells found in peripheral lymphoid organs. In comparison to anti-OX40 monotherapy, combination treatment further reduced the percentage of M2 macrophages and significantly increased the expression of Ki-67 in T cells, dendritic cells, and M1 macrophages, suggesting enhanced proliferation of antitumor immune cells (Fig 5B). Due to poor tumor infiltration of immune cells in this tumor model, the number of immune cells at the tumor site was insufficient to perform CyTOF analysis. Therefore, we evaluated the changes of T cell compartments at the tumor sites by a 5-channel flow cytometry panel (Supplementary table 1). Although GSK2636771 significantly increased the number of CD8+ T cells within tumors, there was no significant difference in the number of tumor infiltrating CD8+ T cells in the GSK2636771 monotherapy group versus the combination treatment group (Fig 6A). In addition, neither monotherapy treatment nor the combination significantly altered the total number of tumor infiltrating CD4+ T cells (Fig 6A). In addition, the number of Tregs in the tumors from the combination treatment group was comparable to those of the monotherapy-treated tumors (Fig 6B). By using the expression of Ki-67 to determine T cell function at tumor sites, we observed that a significant increase in the percentage of Ki-67+ CD8+ T cells, but not in the percentage of Ki-67+ CD4+ T cells, was detected with the combination versus each of the other treatment groups (Fig 6C). These results suggest that combining GSK2636771 with anti-OX40 promotes T cell-mediated antitumor immune responses by inducing robust proliferation of CD8+ tumor infiltrating T cells.
Figure 5. OX40 agonist antibody in combination with PI3Kβ selective inhibition altered the immune cell profile and the serum concentration of cytokine/chemokines produced by T cells.
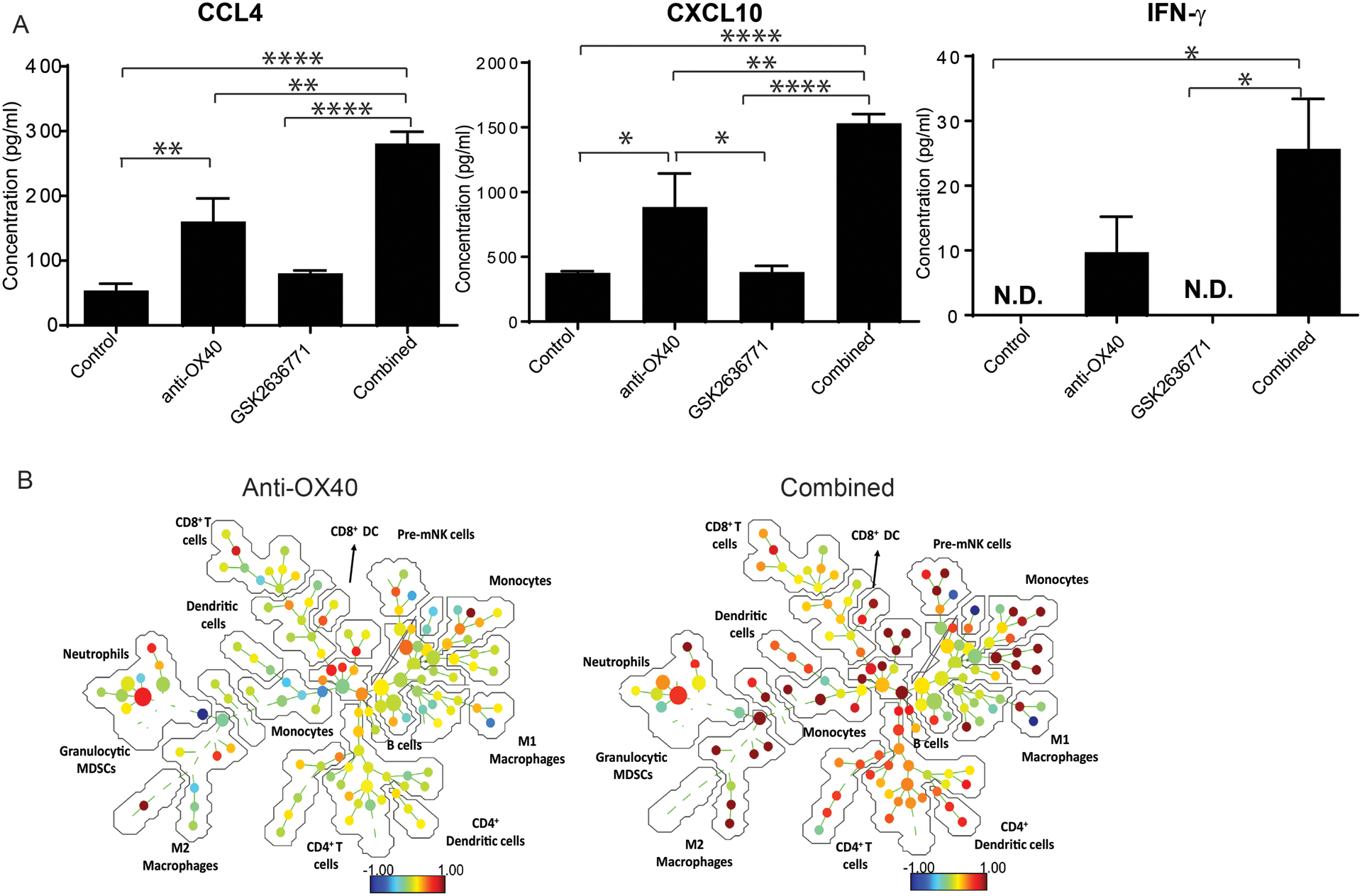
Tyr:CreER; PTENlox/lox; BRAFV600E/+ mice with measureable tumors were treated with control, GSK2636771, anti-mouse OX40 , and the combination of both reagents. Mice were euthanized on day 6 after treatment and used to characterize the changes in the immune profile of mice in the different treatment groups. (A) The serum levels of T cell-associated cytokines/chemokines. Serum from each experimental mouse was collected and used to measure the concentration of 43 cytokines/chemokines using the MILLIPLEX MAP mouse cytokine/chemokine panels. The average of the serum cytokine/chemokine concentration in each group is shown. *P < 0.05, ** P<0.01, ****P<0.0001 (N=3). (B) The results of CyTOF analysis revealed the systemic effects of anti-OX40 alone or in combination with PI3Kβi. Spleens were collected from mice in the different treatment groups and processed into single cell suspensions at the concentration of 20 million cells/ml. Equal amounts of single cell suspensions from experimental mice in each group were pooled (N=3). Pooled samples of control, anti-OX40, and the combination groups were analyzed by CyTOF to determine the percentages of different immune cell subsets and their proliferation (measured by Ki-67 expression). High-dimensional visualization of changes in Ki-67 expression in response to treatment was generated using SPADE. The ratio of Ki-67 (OX40 alone or combination group): Ki-67 (control group) is represented by the color scale, with blue indicating a reduced level of Ki-67 after treatment. The number and size of nodules in each immune cell subset represents the percentage of the indicated immune cell subset in spleens. Data presented are a summary of two independent experiments.
Figure 6. OX40 agonist antibody in combination with PI3Kβ selective inhibition promoted the proliferation of tumor infiltrating CD8+ T cells in mice with PTEN-loss tumor.
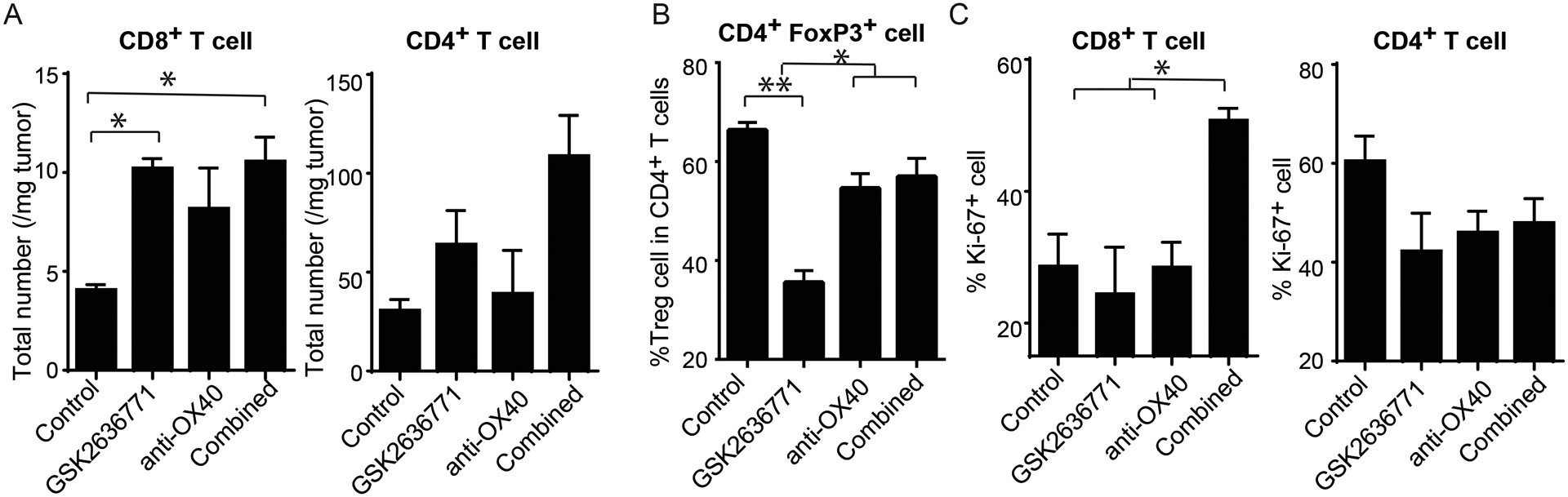
Tyr:CreER; PTENlox/lox; BRAFV600E/+ mice with measureable tumors were treated with control, GSK2636771, anti-mouse OX40, and the combination of both reagents (N=3). On day 6 after treatment, tumor tissues were harvested, weighted, and stained with antibodies for flow cytometry analysis. (A) The total number of CD4+ and CD8+ T cells in tumors from mice treated with anti-OX40 and/or GSK2636771. (B) The percentage of Treg cells (CD25+FOXp3+) in CD4+ T cells in tumors from mice treated with anti-OX40 and/or GSK2636771. (C) The percentage of Ki-67+ CD4+ and CD8+ T cells in tumors from mice treated with anti-OX40 and/or GSK2636771. One-Way ANOVA test demonstrated statistical significance (P<0.05): *P < 0.05 and ** P<0.01. Data presented are a summary of two independent experiments.
DISCUSSION
In this article, we examined the expression of OX40 on tumor infiltrating lymphocytes (TILs) derived from melanoma patients and tested whether stimulating OX40 signaling can promote cytotoxicity of TILs against autologous tumor cells. Data from preclinical tumor models confirmed that OX40 agonist antibody can improve T cell-mediated antitumor immune responses by OX40 receptor engagement on CD8+ T cells and inducing a protective tumor-specific T cell memory. To develop effective therapeutic approaches in cancer patients who fail to respond to immune checkpoint blockade, we used a transgenic Braf-mutant and PTEN loss murine model, which can spontaneously develop immune-resistant melanomas, to evaluate the efficacy of combining OX40 agonist antibody with targeted therapy. The combination of an OX40 agonist antibody and a selective PI3Kβ inhibitor successfully potentiated the proliferation of antitumor immune cells and suppressed tumor development in mice bearing Braf-mutant and PTEN loss melanoma.
OX40, also known as TNFRSF4 or CD134, belongs to the tumor necrosis factor receptor superfamily (TNFRSF) (22). The engagement of three molecules of OX40 and trimeric OX40 ligand (OX40L) initiates the signaling cascade through TNF receptor-associated factors (TRAFs) and eventually drives NF-κB activation (8). Although OX40 expression can be induced by TCR activation in both CD4+ and CD8+ T cells, the expression of OX40 in CD4+ TILs is significantly higher than in CD8+ TILs (23,24). In addition, in vitro and in vivo studies using viral infection and autoimmune disease models have demonstrated that the effect of OX40 activation on CD8+ T cells is largely indirect and is mediated by OX40 regulation of CD4+ T helper cell function (25–27). The activation of the OX40 pathway in regulatory CD4+ T (Treg) cells has also been reported to blunt the immunosuppressive function of Treg cells (14). Therefore, the current working model of OX40 agonist antibody function in tumors mainly focuses on its effect on CD4+ T cells. In our studies, we evaluated the OX40 expression levels in established TIL lines from patients with advanced melanoma under different culture conditions. Consistent with the results from other types of cancer, OX40 is predominantly expressed on resting CD4+ TILs. However, upon encountering autologous tumor cells, the OX40 expression on CD8+ TILs is significantly upregulated and is restored to the baseline 72 hours after TCR stimulation. These results prompted us to test whether OX40 agonist antibody can directly potentiate the function of tumor-reactive CD8+ TILs.
This hypothesis was first supported by data demonstrating that in vitro, anti-OX40 antibody can promote the proliferation of naïve CD8+ T cells after anti-CD3 stimulation (14). In our study, by using patient-derived TILs and paired autologous tumor cells, we found that cross-linked OX40 agonist antibody facilitated tumor apoptosis induced by autologous TILs in vitro. Given that the tumor-specific cytotoxicity of TILs used in this study has been previously shown to be largely dependent on the expression of MHC class I molecules (11), our results suggest that OX40 signaling can directly regulate cytotoxicity of tumor-reactive CD8+ TILs. Additionally, we examined the changes in CD8+ T cells in response to anti-OX40 treatment using multiple murine tumor models. Our in vivo data further demonstrated that anti-OX40 treatment enhances antitumor activity of tumor-reactive CD8+ T cells. Furthermore, we used a vaccination model established in our previous studies to evaluate T cell memory formation (16) and found that anti-OX40 treatment promoted CD8+ T cell-mediated antitumor memory induced by antigen vaccination. In particular, when treating tumor-bearing mice with anti-human OX40 antibody without cross-reactivity to mouse OX40, we consistently observed improved tumor suppression by human OX40-expressing CD8+ T cells. These results imply that the direct role of OX40 signaling in tumor-reactive CD8+ T cells should not be overlooked.
In clinic, the potential of OX40 as a target for cancer immunotherapy was initially tested by using a murine anti-human OX40 IgG1 monoclonal antibody, 9B12 (28). Although no patients achieved a clinical response based on the Response Evaluation Criteria in Solid Tumors (RECIST), 12 of 30 treated patients had at least one regressed metastatic nodule. Multiple fully human or humanized OX40 agonist antibodies have been generated in the last two decades. At least five different antibodies have entered clinical development, including GSK3174998 (GlaxoSmithKline), INCAGN01949 (Agenus), MEDI0562 (formerly of MedImmune), MOXR0916 (Genentech), and PF-04518600 (Pfizer) (29–33). Similar to the preclinical results from murine tumor models (34), the early data from two phase I clinical trials showed anti-OX40 monotherapy was well tolerated in cancer patients, with only one serious treatment-related grade 3 adverse event (pneumonitis responsive to corticosteroids) out of 71 patients reported (29,30). These results suggest that OX40 agonist antibody treatment in cancer patients is generally well-tolerated. In addition, up to 200 μg of anti-OX40 per dose for two weeks had no adverse effect on the health and well-being of experimental mice in this study.
The antitumor effect of OX40 agonist antibody in cancer patients has not been fully elucidated. However, primary and acquired resistance to anti-OX40 monotherapy are expected in cancer patients due to a wide variety of tumor associated immunosuppressive factors. Therefore, combining anti-OX40 therapy with other treatments targeting these tumor-associated immunosuppressive factors may result in better response rates and improved overall survival in cancer patients. Additionally, eradication of well-established tumors has been reported in mice treated with anti-OX40 in combination with other immune reagents, such as TLR9 agonist in a spontaneous breast cancer murine model (35). However, when evaluating the combination of anti-OX40 and anti-PD-1, two research groups independently demonstrated that concurrent treatment using these two agents induced T cell apoptosis and produced antagonistic antitumor responses. Enhanced antitumor effect was only observed in mice sequentially treated with anti-OX40 and anti-PD-1 (36,37). A similar potentially antagonistic antitumor effect has been observed with the combination of immunotherapy and targeted therapy. In that, although both CpG-based tumor vaccine and BRAF inhibitor have therapeutic benefit as monotherapies in cancer patients, combining CpG with BRAF inhibitor negates the antitumor effect of BRAF inhibitor in Braf-mutant tumors in a B-cell dependent manner (38). Therefore, to develop potent therapeutic strategies for OX40-based cancer treatment, we need to not only rationally choose combination partners with complementing effects, but also optimize treatment schedules to maximize the antitumor effect of anti-OX40.
Melanomas that spontaneously develop in transgenic mice bearing the Braf V600E mutation and PTEN loss in melanocytes, display primary resistance to cancer immunotherapy, due to lack of tumor-associated antigens and upregulated immunosuppressive factors induced by oncogenic activation of the PI3K pathway (11,39,40). In this study, we used this immune-resistant tumor model to evaluate the therapeutic efficacy of anti-OX40 in combination with PI3K inhibition. Although anti-OX40 monotherapy did not effectively control tumor development, concurrent treatment of anti-OX40 and PI3Kβ selective inhibition significantly delayed tumor growth and extended the survival of mice bearing Braf-mutant and PTEN loss melanomas. Unlike the combination of PI3Kβ inhibition and ICB, this combinatorial approach did not significantly increase the number of CD4+ and CD8+ T cells at the tumor sites but promoted the proliferation of CD8+ T cells at the tumor sites. Anti-OX40 plus PI3Kβ inhibitor treatment also systemically enhanced the proliferation of antitumor immune cells but reduced the number of immunosuppressive M2 macrophages. We also found elevated serum levels of cytokine/chemokines, which were predominantly produced by effector T cells in mice treated with the combination therapy. In addition, our studies showed that this combination did not increase the susceptibility of T cells to activation-induced apoptosis. Overall, our results offer the first preclinical evidence demonstrating that combining anti-OX40 with PI3Kβ inhibitor could be an effective treatment for patients with PTEN loss tumors. These results also provide the rationale to clinically test this combination in patients with immunoresistant PTEN loss tumors.
Taken together, our studies suggest that OX40 agonist-based combination treatment can induce a robust and durable antitumor immune response by promoting effector T cell function and the generation of memory T cells.
Supplementary Material
Translational Relevance:
Cancer immunotherapy is revolutionizing cancer treatment. However, most patients still fail to respond to currently available immunomodulatory agents. Thus, there remains a critical need to identify novel immunoregulatory targets and rational combinatorial strategies to induce robust and durable antitumor immune responses. Here, we used patient samples and clinically relevant animal models to evaluate the immunological and antitumor effects of OX40 agonist-based immunotherapy. Our results add to the growing body of evidence that OX40 agonists can boost antitumor immune responses by modulating T cell effector function and tumor-specific memory. Our results also identify a novel therapeutic strategy of combining OX40 agonist antibodies with targeted therapy in cancer patients, particularly those with tumors with loss of the tumor suppressor PTEN.
Acknowledgments
This work was supported in part by the following National Cancer Institute grants: R01CA187076 (PH&MD), P50CA093459 (UT M.D. Anderson Cancer Center SPORE in Melanoma), T32CA009666-21(MD) and P30CA016672 (UT MDACC CCSG for the Flow Cytometry & Cellular Imaging facility), by philanthropic contributions to MDACC Melanoma Moon Shots Program; Melanoma Research Alliance Young Investigator Award (WP, 558998); Dr. Miriam and Sheldon G. Adelson Medical Research Foundation; Aim at Melanoma Foundation, Miriam and Jim Mulva Research Fund; and by Cancer Prevention and Research Institute of Texas (PH, RP170401; JAM, RP140106 and RP170067).
The authors would like to thank the past and present TIL lab members at MDACC: Orenthial J. Fulbright, Arely Wahl, Esteban Flores, Shawne T. Thorsen, René J. Tavera, Renjith Ramachandran, Audrey M. Gonzalez, Christopher Toth, Seth Wardell and Rahmatu Mansaray as well as Drs Chantale Bernatchez, Cara Haymaker, Marie-Andrée Forget and Shruti Malu for generation of research TIL and tumor lines.
Footnotes
Conflict of Interest:
The authors of this publication have research support from GlaxoSmithKline (GSK). The terms of this agreement have been reviewed and approved by the University of Texas MD Anderson Cancer Center (MDACC) in accordance with its policy on objectivity in research. W. Peng received honoraria and travel support from Bristol-Myers Squibb (BMS). H. Jackson, A. Hoos, J. Smothers, R. Srinivasan, E. Paul and N. Yanamandra are full-time employees of GSK. P. Hwu is a consultant/an advisory board member for Immatics, Dragonfly, Sanofi, and GSK. M.A. Davies is an advisory board member for BMS, GSK, Novartis, Roche/Genentech, Array, Sanofi, and Vaccinex. M.A. Davies is also the PI of research funding to MDACC from GSK, AstraZeneca, Roche/Genentech, Myriad, Oncothyreon, and Sanofi-Aventis. No potential conflicts of interest were disclosed by other authors.
Reference
- 1.Sharma P, Allison JP. The future of immune checkpoint therapy. Science 2015;348(6230):56–61 doi 10.1126/science.aaa8172. [DOI] [PubMed] [Google Scholar]
- 2.Park YJ, Kuen DS, Chung Y. Future prospects of immune checkpoint blockade in cancer: from response prediction to overcoming resistance. Exp Mol Med 2018;50(8):109 doi 10.1038/s12276-018-0130-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jensen SM, Maston LD, Gough MJ, Ruby CE, Redmond WL, Crittenden M, et al. Signaling through OX40 enhances antitumor immunity. Semin Oncol 2010;37(5):524–32 doi 10.1053/j.seminoncol.2010.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chester C, Sanmamed MF, Wang J, Melero I. Immunotherapy targeting 4–1BB: mechanistic rationale, clinical results, and future strategies. Blood 2018;131(1):49–57 doi 10.1182/blood-2017-06-741041. [DOI] [PubMed] [Google Scholar]
- 5.Callahan MK, Postow MA, Wolchok JD. CTLA-4 and PD-1 Pathway Blockade: Combinations in the Clinic. Frontiers in oncology 2014;4:385 doi 10.3389/fonc.2014.00385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Darvin P, Toor SM, Sasidharan Nair V, Elkord E. Immune checkpoint inhibitors: recent progress and potential biomarkers. Exp Mol Med 2018;50(12):165 doi 10.1038/s12276-018-0191-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jenkins RW, Barbie DA, Flaherty KT. Mechanisms of resistance to immune checkpoint inhibitors. Br J Cancer 2018;118(1):9–16 doi 10.1038/bjc.2017.434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Croft M, So T, Duan W, Soroosh P. The significance of OX40 and OX40L to T-cell biology and immune disease. Immunological reviews 2009;229(1):173–91 doi 10.1111/j.1600-065X.2009.00766.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chacon JA, Wu RC, Sukhumalchandra P, Molldrem JJ, Sarnaik A, Pilon-Thomas S, et al. Co-stimulation through 4–1BB/CD137 improves the expansion and function of CD8(+) melanoma tumor-infiltrating lymphocytes for adoptive T-cell therapy. PLoS One 2013;8(4):e60031 doi 10.1371/journal.pone.0060031 PONE-D-12–34281 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peng W, Ye Y, Rabinovich BA, Liu C, Lou Y, Zhang M, et al. Transduction of tumor-specific T cells with CXCR2 chemokine receptor improves migration to tumor and antitumor immune responses. Clin Cancer Res 2010;16(22):5458–68 doi 10.1158/1078-0432.CCR-10-07121078-0432.CCR-10–0712 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Peng W, Chen JQ, Liu C, Malu S, Creasy C, Tetzlaff MT, et al. Loss of PTEN Promotes Resistance to T Cell-Mediated Immunotherapy. Cancer Discov 2016;6(2):202–16 doi 10.1158/2159-8290.CD-15-0283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kitamura T, Koshino Y, Shibata F, Oki T, Nakajima H, Nosaka T, et al. Retrovirus-mediated gene transfer and expression cloning: powerful tools in functional genomics. Exp Hematol 2003;31(11):1007–14. [PubMed] [Google Scholar]
- 13.Rabinovich BA, Ye Y, Etto T, Chen JQ, Levitsky HI, Overwijk WW, et al. Visualizing fewer than 10 mouse T cells with an enhanced firefly luciferase in immunocompetent mouse models of cancer. Proc Natl Acad Sci U S A 2008;105(38):14342–6 doi 10.1073/pnas.08041051050804105105 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Voo KS, Bover L, Harline ML, Vien LT, Facchinetti V, Arima K, et al. Antibodies targeting human OX40 expand effector T cells and block inducible and natural regulatory T cell function. J Immunol 2013;191(7):3641–50 doi 10.4049/jimmunol.1202752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu C, Lewis CM, Lou Y, Xu C, Peng W, Yang Y, et al. Agonistic antibody to CD40 boosts the antitumor activity of adoptively transferred T cells in vivo. J Immunother 2012;35(3):276–82 doi 10.1097/CJI.0b013e31824e7f4300002371-201204000-00008 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hailemichael Y, Dai Z, Jaffarzad N, Ye Y, Medina MA, Huang XF, et al. Persistent antigen at vaccination sites induces tumor-specific CD8(+) T cell sequestration, dysfunction and deletion. Nat Med 2013;19(4):465–72 doi 10.1038/nm.3105nm.3105 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cascone T, McKenzie JA, Mbofung RM, Punt S, Wang Z, Xu C, et al. Increased Tumor Glycolysis Characterizes Immune Resistance to Adoptive T Cell Therapy. Cell Metab 2018;27(5):977–87 e4 doi 10.1016/j.cmet.2018.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gough MJ, Ruby CE, Redmond WL, Dhungel B, Brown A, Weinberg AD. OX40 agonist therapy enhances CD8 infiltration and decreases immune suppression in the tumor. Cancer Res 2008;68(13):5206–15 doi 10.1158/0008-5472.CAN-07-6484. [DOI] [PubMed] [Google Scholar]
- 19.Mateo J, Ganji G, Lemech C, Burris HA, Han SW, Swales K, et al. A First-Time-in-Human Study of GSK2636771, a Phosphoinositide 3 Kinase Beta-Selective Inhibitor, in Patients with Advanced Solid Tumors. Clin Cancer Res 2017;23(19):5981–92 doi 10.1158/1078-0432.CCR-17-0725. [DOI] [PubMed] [Google Scholar]
- 20.Schmit F, Utermark T, Zhang S, Wang Q, Von T, Roberts TM, et al. PI3K isoform dependence of PTEN-deficient tumors can be altered by the genetic context. Proc Natl Acad Sci U S A 2014;111(17):6395–400 doi 10.1073/pnas.1323004111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Slinker BK. The statistics of synergism. J Mol Cell Cardiol 1998;30(4):723–31 doi 10.1006/jmcc.1998.0655. [DOI] [PubMed] [Google Scholar]
- 22.Buchan SL, Rogel A, Al-Shamkhani A. The immunobiology of CD27 and OX40 and their potential as targets for cancer immunotherapy. Blood 2018;131(1):39–48 doi 10.1182/blood-2017-07-741025. [DOI] [PubMed] [Google Scholar]
- 23.Lai C, August S, Albibas A, Behar R, Cho SY, Polak ME, et al. OX40+ Regulatory T Cells in Cutaneous Squamous Cell Carcinoma Suppress Effector T-Cell Responses and Associate with Metastatic Potential. Clin Cancer Res 2016;22(16):4236–48 doi 10.1158/1078-0432.CCR-15-2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Montler R, Bell RB, Thalhofer C, Leidner R, Feng Z, Fox BA, et al. OX40, PD-1 and CTLA-4 are selectively expressed on tumor-infiltrating T cells in head and neck cancer. Clin Transl Immunology 2016;5(4):e70 doi 10.1038/cti.2016.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Serghides L, Bukczynski J, Wen T, Wang C, Routy JP, Boulassel MR, et al. Evaluation of OX40 ligand as a costimulator of human antiviral memory CD8 T cell responses: comparison with B7.1 and 4–1BBL. J Immunol 2005;175(10):6368–77. [DOI] [PubMed] [Google Scholar]
- 26.Sitrin J, Suto E, Wuster A, Eastham-Anderson J, Kim JM, Austin CD, et al. The Ox40/Ox40 Ligand Pathway Promotes Pathogenic Th Cell Responses, Plasmablast Accumulation, and Lupus Nephritis in NZB/W F1 Mice. J Immunol 2017;199(4):1238–49 doi 10.4049/jimmunol.1700608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kopf M, Ruedl C, Schmitz N, Gallimore A, Lefrang K, Ecabert B, et al. OX40-deficient mice are defective in Th cell proliferation but are competent in generating B cell and CTL Responses after virus infection. Immunity 1999;11(6):699–708. [DOI] [PubMed] [Google Scholar]
- 28.Curti BD, Kovacsovics-Bankowski M, Morris N, Walker E, Chisholm L, Floyd K, et al. OX40 is a potent immune-stimulating target in late-stage cancer patients. Cancer Res 2013;73(24):7189–98 doi 10.1158/0008-5472.CAN-12-4174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Infante JR, Ahlers CM, Hodi FS, Postel-Vinay S, Schellens JHM, Heymach J, et al. ENGAGE-1: A first in human study of the OX40 agonist GSK3174998 alone and in combination with pembrolizumab in patients with advanced solid tumors. Journal of Clinical Oncology 2016;34(15_suppl):TPS3107-TPS doi 10.1200/JCO.2016.34.15_suppl.TPS3107. [DOI] [Google Scholar]
- 30.Glisson BS, Leidner R, Ferris RL, Powderly J, Rizvi N, Norton JD, et al. Phase 1 study of MEDI0562, a humanized OX40 agonist monoclonal antibody (mAb), in adult patients (pts) with advanced solid tumors. Annals of Oncology 2016;27(suppl_6):1052PD-PD doi 10.1093/annonc/mdw378.07. [DOI] [Google Scholar]
- 31.Gonzalez AM, Manrique ML, Breous E, Savitsky D, Waight J, Gombos R, et al. Abstract 3204: INCAGN01949: an anti-OX40 agonist antibody with the potential to enhance tumor-specific T-cell responsiveness, while selectively depleting intratumoral regulatory T cells. Cancer Research 2016;76(14 Supplement):3204- doi 10.1158/1538-7445.Am2016-3204. [DOI] [Google Scholar]
- 32.Infante JR, Hansen AR, Pishvaian MJ, Chow LQM, McArthur GA, Bauer TM, et al. A phase Ib dose escalation study of the OX40 agonist MOXR0916 and the PD-L1 inhibitor atezolizumab in patients with advanced solid tumors. Journal of Clinical Oncology 2016;34(15_suppl):101- doi 10.1200/JCO.2016.34.15_suppl.101. [DOI] [Google Scholar]
- 33.Diab A, El-Khoueiry A, Eskens FA, Ros W, Thompson JA, Konto C, et al. A first-in-human (FIH) study of PF-04518600 (PF-8600) OX40 agonist in adult patients (pts) with select advanced malignancies. Annals of Oncology 2016;27(suppl_6):1053PD-PD doi 10.1093/annonc/mdw378.08. [DOI] [Google Scholar]
- 34.Aspeslagh S, Postel-Vinay S, Rusakiewicz S, Soria JC, Zitvogel L, Marabelle A. Rationale for anti-OX40 cancer immunotherapy. Eur J Cancer 2016;52:50–66 doi 10.1016/j.ejca.2015.08.021. [DOI] [PubMed] [Google Scholar]
- 35.Sagiv-Barfi I, Czerwinski DK, Levy S, Alam IS, Mayer AT, Gambhir SS, et al. Eradication of spontaneous malignancy by local immunotherapy. Sci Transl Med 2018;10(426) doi 10.1126/scitranslmed.aan4488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shrimali RK, Ahmad S, Verma V, Zeng P, Ananth S, Gaur P, et al. Concurrent PD-1 Blockade Negates the Effects of OX40 Agonist Antibody in Combination Immunotherapy through Inducing T-cell Apoptosis. Cancer Immunol Res 2017;5(9):755–66 doi 10.1158/2326-6066.CIR-17-0292. [DOI] [PubMed] [Google Scholar]
- 37.Messenheimer DJ, Jensen SM, Afentoulis ME, Wegmann KW, Feng Z, Friedman DJ, et al. Timing of PD-1 Blockade Is Critical to Effective Combination Immunotherapy with Anti-OX40. Clin Cancer Res 2017;23(20):6165–77 doi 10.1158/1078-0432.CCR-16-2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huang L, Wang Z, Liu C, Xu C, Mbofung RM, McKenzie JA, et al. CpG-based immunotherapy impairs antitumor activity of BRAF inhibitors in a B-cell-dependent manner. Oncogene 2017;36(28):4081–6 doi 10.1038/onc.2017.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hooijkaas A, Gadiot J, Morrow M, Stewart R, Schumacher T, Blank CU. Selective BRAF inhibition decreases tumor-resident lymphocyte frequencies in a mouse model of human melanoma. Oncoimmunology 2012;1(5):609–17 doi 10.4161/onci.20226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cooper ZA, Juneja VR, Sage PT, Frederick DT, Piris A, Mitra D, et al. Response to BRAF inhibition in melanoma is enhanced when combined with immune checkpoint blockade. Cancer Immunol Res 2014;2(7):643–54 doi 10.1158/2326-6066.CIR-13-0215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Peng W, Liu C, Xu C, Lou Y, Chen J, Yang Y, et al. PD-1 blockade enhances T-cell migration to tumors by elevating IFN-gamma inducible chemokines. Cancer Res 2012;72(20):5209–18 doi 10.1158/0008-5472.CAN-12-11870008-5472.CAN-12-1187 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.