

Abstract
Prospective data of vedolizumab treatment for patients with inflammatory bowel disease (IBD) beyond 1 year of treatment is scarce but needed for clinical decision making. We prospectively enrolled 310 patients with IBD (191 with Crohn's disease (CD) and 119 patients with ulcerative colitis (UC)) with a follow‐up period of 104 weeks (interquartile range: 103–104) in a nationwide registry. The corticosteroid‐free clinical remission rate (Harvey Bradshaw Index ≤ 4, Short Clinical Colitis Activity index ≤ 2) at weeks 52 and 104 were 28% and 19% for CD and 27% and 28% for UC, respectively. Fifty‐nine percent maintained corticosteroid‐free clinical remission between weeks 52 and 104. Vedolizumab with concomitant immunosuppression showed comparable effectiveness outcomes compared with vedolizumab monotherapy (week 104: 21% vs. 23%; P = 0.77), whereas 8 of 13 severe infections occurred in patients treated with concomitant immunosuppression. To conclude, the clinical effect was 19% for CD and 28% for UC after 2 years of follow‐up regardless of concomitant immunosuppression.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ The efficacy and safety of vedolizumab therapy have been established in the GEMINI studies and confirmed in real‐life cohorts up to 1 year.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ Long‐term real‐world data are needed to determine the loss of response, dose escalation, and risk of infections beyond 52 weeks of treatment.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ The effectiveness of vedolizumab therapy between the first and second year of follow‐up remains fairly stable for patients with ulcerative colitis (UC), whereas it decreases over time for patients with Crohn's disease (CD). Concomitant use of immunosuppressive medication did not correlate with improved effectiveness outcomes but did impose a risk for severe infections. Dose optimization occurred frequently, also beyond the first year of treatment.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
☑ Long‐term maintenance of clinical remission on vedolizumab seems more feasible for UC than CD, whereas dose optimization may aid in improved outcomes. Based on our safety data, vedolizumab combination therapy should be carefully considered.
Vedolizumab is a humanized monoclonal IgG‐1 antibody inhibiting the interaction between the α4β7 integrin and mucosal addressin cell adhesion molecule‐1. This results in the prevention of lymphocyte homing to the inflamed gut tissue. Clinical efficacy of vedolizumab was demonstrated in the GEMINI trials1, 2 and vedolizumab was subsequently approved for the treatment of Crohn's disease (CD) and ulcerative colitis (UC) in the Netherlands in 2014. Vedolizumab is now implemented as biological therapy in daily inflammatory bowel disease (IBD) practice. However, patients in the initial phase III trials are not representative for the real‐life IBD population because a large proportion of patients in daily practice do not meet the predefined inclusion and exclusion criteria.3 Therefore, long‐term robust real‐world effectiveness and safety data are needed.
To date, the effectiveness and safety of vedolizumab in clinical practice has been described and confirmed in a number of cohorts up to 1 year.4, 5, 6, 7, 8, 9, 10 There is, however, only limited data available beyond the first year of treatment. Patients from the pivotal phase III clinical studies (GEMINI) could be included in the long‐term open‐label studies (GEMINI‐LTS) and these trials indicated that the effectiveness of vedolizumab remained fairly stable in patients who responded to vedolizumab induction.11, 12 However, only one study presented the safety and effectiveness results beyond 1 year in a real‐world population.13 In this study, the steroid‐free clinical remission rate for UC remained fairly stable beyond the first year of treatment, whereas the steroid‐free clinical remission rate of patients with CD tended to decrease over time. Long‐term prospective data on vedolizumab treatment in real‐life cohorts is of key importance to determine loss of response rates, treatment escalations, and risk of infections and malignancies beyond 52 weeks.
Using the Initiative on Crohn and Colitis (ICC) Registry, a prospective, nationwide, observational registry for novel IBD therapies, we aimed to evaluate the real‐world effectiveness, safety, and use of vedolizumab with a long‐term follow‐up of 2 years. Furthermore, we aimed to determine predictors of clinical remission and to describe different treatment strategies used in clinical practice.
Methods
Study design and setting
The ICC Registry is a nationwide, observational registry with prospective follow‐up of patients with IBD starting prespecified IBD therapies in the Netherlands. The design and rationale of the ICC Registry were described previously in more detail.14 Briefly, patients with IBD, ≥ 16 years, in 8 academic and 4 nonacademic hospitals are followed for 2 years after initiating prespecified therapies. Visits are scheduled at initiation of therapy (baseline), week 12, week 24, week 52, and at week 104 or until the medication is discontinued. Data collection is done using an electronic case report form with automated reminders to improve adherence to the protocol. We used the described ICC Registry to document effectiveness and safety of vedolizumab therapy for patients with IBD.
Participants
Following formal approval by the regulatory authorities, patients with IBD initiating vedolizumab were enrolled in the participating centers. Patients ≥ 16 years with an established IBD diagnosis starting vedolizumab in regular care were eligible. There were no exclusion criteria. The decision to start vedolizumab therapy was at the discretion of the treating physician. Patients received vedolizumab therapy i.v. according to the label with an induction infusion regimen of 300 mg vedolizumab at weeks 0, 2, and 6. In case of insufficient response, an additional vedolizumab infusion could be prescribed at week 10 at the discretion of the treating physician. The maintenance treatment consisted of 300 mg vedolizumab every 8 weeks, whereas the infusion interval could be shortened in case of inadequate response. Patients with combined clinical (e.g., Harvey Bradshaw Index (HBI) > 4 and Short Clinical Colitis Activity Index (SCCAI) > 2) and objective (endoscopy, radiology, or biochemical: C‐reactive protein > 5ml/L and a fecal calprotectin level > 200 µg/g) disease activity at baseline were used to determine the effectiveness outcomes while all enrolled patients, regardless of disease activity scores, were used to determine the safety outcomes.
Outcomes and definitions
The primary outcome was the proportion of patients in corticosteroid‐free clinical remission at week 104. Secondary effectiveness outcomes included: clinical response, clinical remission, biochemical remission, extra‐intestinal manifestations (EIM) remission, vedolizumab interval shortening, intestinal surgery, and vedolizumab discontinuation rate. Corticosteroid‐free clinical remission was compared between CD and UC and between patients on concomitant immunosuppressive therapies (thiopurines and methotrexate) and without concomitant immunosuppressive medication at baseline. Furthermore, predictors for corticosteroid‐free clinical remission at week 104 were determined. An HBI ≤ 4 and SCCAI ≤ 2 were considered as clinical remission. A decrease of HBI of SCCAI of ≥ 3 compared with baseline was considered as clinical response.15 Biochemical remission was defined as a CRP concentration ≤ 5 mg/L combined with a fecal calprotectin (FCP) level ≤ 200 µg/g when available for CD and only a FCP level of ≤ 200 µg/g for UC. EIM that were collected included: arthralgia, arthritis, uveitis, aphthous stomatitis, erythema nodosum, and pyoderma gangrenosum. EIM remission was assessed by the treating physician. Reason for discontinuation of vedolizumab was documented. The safety outcomes were presented as the number of medication‐related adverse events, infections, and disease‐related hospitalizations per 100 patient years. Adverse events were classified as possibly, probably, and not related. Serious adverse events were classified as reason for discontinuation of medication. Infections were classified as mild (no antibiotics or antiviral medication necessary), moderate (oral antibiotics or antiviral medication), or severe (hospitalization or i.v. administered antibiotics or antiviral medication).
Follow‐up time was determined based on the date of the first dose of vedolizumab until the last visit used in the analysis. Patients who discontinued vedolizumab due to primary or secondary nonresponse, adverse events, or patient request without long‐term sustained remission were considered treatment failure and were classified as nonresponders. Patients were considered censored cases when they discontinued vedolizumab because of pregnancy or long‐term sustained remission and were not included in the analysis. Because this registry still actively recruits patients, only patients with sufficient follow‐up time were used in the analysis at specific time points. For example, a patient with 70 weeks of follow‐up contributed to the 52‐week analysis, but not to the 104‐week analysis, independent of drug discontinuation.
Statistical methods
Patients were analyzed on an intention‐to‐treat basis. Continuous variables were presented as means with standard deviations (SDs) or as medians with interquartile ranges (IQRs) depending on the normality of the underlying distribution. Continuous variables were consequently compared using the independent t‐test or Mann–Whitney U test. Categorical variables were presented as percentages and compared by using the χ2 test. The overall drug survival was assessed using the Kaplan–Meier method. To compare the drug survival between patients with and without interval shortening during maintenance therapy, the time‐varying Cox regression was used. Variables associated with week 52 corticosteroid‐free clinical remission were explored using binary logistic regression. Multivariable analysis was performed on variables with a P value < 0.2 on univariable analysis using backward stepwise logistic regression. A twosided P value of 0.05 or less was considered statistically significant. All data analyses were performed using IBM SPSS Statistics for Windows, version 24.0 (IBM, Armonk, NY).
Ethical consideration
This study was reviewed and approved by the Committee on Research Involving Human Subjects at the Radboudumc (institutional review board: 4076).
Results
Baseline characteristics
Baseline characteristics are depicted in Table 1. In total, 310 patients with IBD were included: 191 patients with CD, 113 patients with UC, and 6 patients with IBD‐Unclassified (IBD‐U). The results of the patients with IBD‐U are presented with the results of the patients with UC due to the limited number of patients with IBD‐U. The 191 patients with CD were followed with a median follow‐up period of 104.0 weeks (IQR: 104.0–104.0). The patients with CD were predominately women (n = 122; 63.9%) with a median disease duration of 11.0 years (IQR: 6.4–19.7). At inclusion, an ileocolonic disease location was observed in 40.8% (n = 78), whereas 12.0% (n = 23) had a penetrating disease phenotype, and 12.0% (n = 23) had peri‐anal disease activity at maximum extent and behavior during their disease course. The majority of patients (95.9%) were previously exposed to thiopurines or methotrexate, 188 patients (98.4%) were exposed to at least 1 anti‐TNF agent, and 148 patients (77.5%) to 2 or more anti‐TNF agents. Eleven patients (5.8%) were previously exposed to ustekinumab. A history of intestinal resections was documented in 97 patients (50.8%).
Table 1.
Baseline characteristics
| CD N = 191 | UC N = 119 | ||
|---|---|---|---|
| Agea | Median (IQR) | 36.8 (27.0–52.5) | 40.4 (30.2–55.5) |
| Sex – male | N (%) | 69 (36.1) | 66 (55.5) |
| Body mass indexa | Mean (SD) | 23.8 (4.2) | 24.4 (4.7) |
| Current smoker | N (%) | 46 (24.1) | 4 (3.4) |
| Disease duration in years | Median (IQR) | 11.0 (6.4–19.7) | 7.8 (2.8–14.9) |
| Follow‐up duration | Median (IQR) | 104.0 (104.0–104.0) | 104.0 (96.9–104.0) |
| CD disease locationa | |||
| Ileum | N (%) | 57 (29.8) | |
| Colon | N (%) | 56 (29.3) | |
| Ileum and colon | N (%) | 78 (40.8) | |
| Upper GI involvement | N (%) | 19 (9.9) | |
| UC disease location | |||
| Proctitis | N (%) | 8 (6.7) | |
| Left‐sided | N (%) | 49 (41.2) | |
| Pancolitis | N (%) | 58 (48.7) | |
| Unknown | N (%) | 4 (3.4) | |
| Disease behaviora | |||
| Inflammatory disease | N (%) | 115 (60.2) | |
| Stricturing disease | N (%) | 50 (26.2) | |
| Penetrating disease | N (%) | 23 (12.0) | |
| Unknown | N (%) | 3 (1.6) | |
| Peri‐anal diseasea | N (%) | 23 (12.0) | |
| Prior intestinal resections | N (%) | 97 (50.8) | |
| Prior peri‐anal interventions | N (%) | 34 (17.8) | |
| Prior anti‐TNF therapy use | |||
| ≥ 1 | N (%) | 188 (98.4) | 104 (87.4) |
| ≥ 2 | N (%) | 148 (77.5) | 44 (36.9) |
| 3 | N (%) | 8 (4.2) | 6 (5.0) |
| Prior ustekinumab use | N (%) | 11 (5.8) | |
| Prior anti‐integrin trial participation | N (%) | 7 (3.7) | 1 (0.8) |
| Disease activitya | |||
| HBI/SCCAI | Median (IQR) | 7 (5–11) | 6 (3–9) |
| CRP, mg/L | Median (IQR) | 7 (3–19) | 4 (1–12) |
| Fecal calprotectin, µg/g | Median (IQR) | 689 (297–1800) | 1317 (527–2013) |
| Concomitant medication | |||
| No concomitant medication | N (%) | 72 (37.7) | 41 (34.5) |
| Corticosteroids | N (%) | 55 (28.8) | 33 (27.7) |
| Corticosteroids range | mg (IQR) | 25 (20–40) | 25 (18–40) |
| Immunosuppressants | N (%) | 39 (20.4) | 20 (16.8) |
| Both corticosteroids and immunosuppressants | N (%) | 25 (13.1) | 25 (21.0) |
| Corticosteroids range | mg (IQR) | 20 (15–30) | 30 (20–40) |
CD, Crohn's disease; HBI, Harvey Bradshaw Index; GI, gastrointestinal; IQR, interquartile range; SCCAI, short clinical colitis activity index; UC, ulcerative colitis.
At inclusion. bMaximum extent until inclusion.
In the UC cohort, 113 patients with UC and 6 patients with IBD‐U were followed with a median follow‐up period of 104.0 weeks (IQR: 96.9–104.0 weeks). The patients with UC were predominately men (n = 66; 55.5%) with a median disease duration of 7.8 years (IQR: 2.8–14.9). Pancolitis was seen in 48.7% (n = 58) at inclusion. The majority of patients (92.7%) were previously exposed to thiopurines or methotrexate and 104 (87.4%) had failed at least 1 anti‐TNF agent, whereas 44 (36.9%) had failed 2 or more anti‐TNF agents.
At baseline, combined clinical and objective disease activity was determined in 138 patients with CD with a median HBI of 9 (IQR: 6–12) and 94 patients with UC with a median SCCAI of 7 (IQR: 4–9). Of the 53 patients with CD and 25 patients with UC without both clinical and objective disease activity at baseline, 92.5% (n = 49/53) and 84.0% (n = 21/25) of patients had disease activity based on clinical disease activity, active draining fistula, inflammatory biomarkers, endoscopy, or radiology. These patients were not included in the effectiveness analyses.
Corticosteroid‐free clinical remission
For CD, the proportion of patients in corticosteroid‐free clinical remission at weeks 12, 24, 52, and 104 was 24.3% (n = 33/136), 30.6% (n = 41/134), 27.7% (n = 36/130), and 19.0% (n = 24/126), respectively (Figure 1). Of the 36 patients in corticosteroid‐free clinical remission at week 52, 55.6% remained in corticosteroid‐free clinical remission up to week 104. When analyzing all patients, including the 63 patients without both clinical and objective disease activity at baseline, the corticosteroid‐free clinical remission rate at weeks 0, 12, 24, 52, and 104 was 13.6% (n = 26/191), 30.3% (n = 57/188), 34.2% (n = 63/184), 28.3% (n = 51/180), and 22.5% (n = 39/173), respectively.
Figure 1.
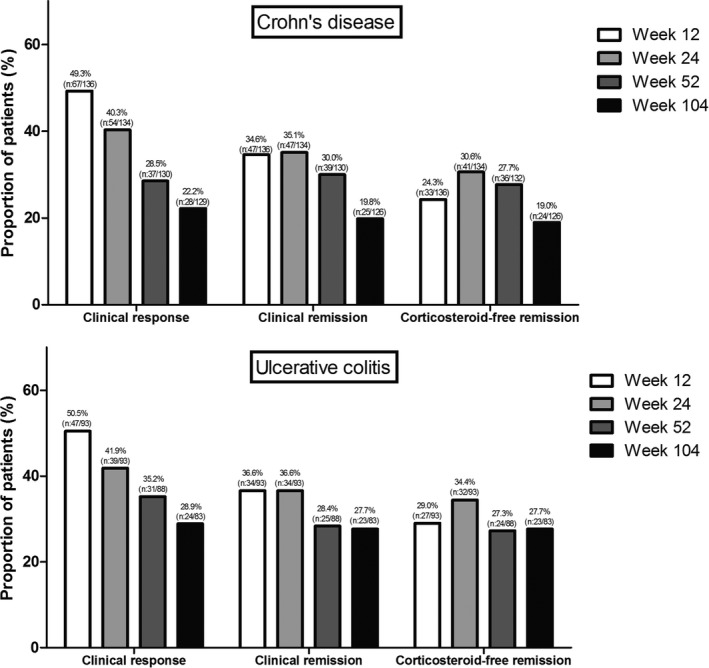
Clinical response, clinical remission, and corticosteroid‐free clinical remission for patients with Crohn's disease and patients with ulcerative colitis.
For UC, the proportion of patients in corticosteroid‐free clinical remission at weeks 12, 24, 52, and 104 was 29.0% (n = 27/93), 34.4% (n = 32/93), 27.3% (n = 24/88), and 27.7% (n = 23/83), respectively (Figure 1). Of the 24 patients in corticosteroid‐free clinical remission at week 52, 60.9% remained in corticosteroid‐free clinical remission up to week 104. When analyzing all patients, including the 26 patients in clinical remission at baseline, the corticosteroid‐free clinical remission rate at weeks 0, 12, 24, 52, and 104 was 8.4% (n = 10/119), 32.5% (n = 38/117), 36.2% (n = 42/116), 29.6% (n = 32/108), and 26.5% (n = 27/102), respectively.
Coadministration of immunosuppressive therapy (thiopurines or methotrexate, combination therapy) was documented in 82 patients with IBD (35.3%) with clinical and objective disease activity at baseline. When the latter group was compared with patients who received vedolizumab monotherapy, baseline characteristics, including clinical and biochemical disease activity, except CRP (monotherapy: 8 mg/L (IQR: 4–23) vs. combination therapy: 5 mg/L (IQR: 2–14), P = 0.02), were comparable between the subgroups. The proportion of patients in corticosteroid‐free clinical remission was 26.5% (monotherapy) vs. 25.6% (combination therapy) P = 0.88 at week 12, 32.4% vs. 31.7% P = 0.91 at week 24, 26.2% vs. 29.1% P = 0.65 at week 52, and 22.8% vs. 21.1% P = 0.77 at week 104 (Figure 2).
Figure 2.
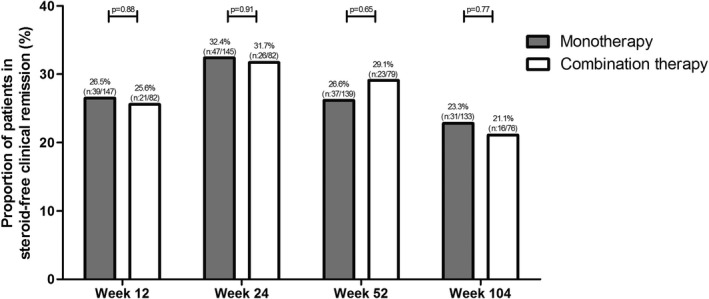
Corticosteroid‐free clinical remission of vedolizumab (VDZ) monotherapy vs. vedolizumab combination therapy (Combination therapy: vedolizumab therapy with an immunosuppressant (thiopurines or methotrexate)).
Clinical response and remission
For CD, the proportion of patients with a clinical response to vedolizumab therapy at weeks 12, 24, 52, and 104 was 49.3% (n = 67/136), 40.3% (n = 54/134), 28.5% (n = 37/130), and 22.2% (n = 28/129), respectively. The proportion of patients in clinical remission at weeks 12, 24, 52, and 104 was 34.6% (n = 47/136), 35.1% (n = 47/134), 30.0% (n = 39/130), and 19.8% (n = 25/126), respectively (Figure 1).
For UC, the proportion of patients with a clinical response to vedolizumab therapy at weeks 12, 24, 52, and 104 was 50.5% (n = 47/93), 41.9% (n = 39/93), 35.2% (n = 31/88), and 28.9% (n = 24/83), respectively. The proportion of patients in clinical remission at weeks 12, 24, 52, and 104 was 36.6% (n = 34/93), 36.6% (n = 34/93), 28.4% (n = 25/88), and 27.7% (n = 23/83), respectively (Figure 1).
Biochemical disease activity
For CD, the proportion of patients in biochemical remission at weeks 12, 24, 52, and 104 was 24.4% (n = 31/127), 33.1% (n = 40/121), 22.4% (n = 28/125), and 17.8% (n = 21/118), respectively (Figure 3 a). When missing data was imputed as nonresponder, the proportion of patients in biochemical remission was 22.8% (n = 31/136), 29.9% (n = 40/134), 21.2% (n = 28/132), and 16.3% (n = 21/129), respectively. The median CRP concentration of patients treated with vedolizumab at weeks 0, 12, 24, 52, and 104 was 8 mg/L (IQR: 4–23), 8 mg/L (IQR: 3–14), 5 mg/L (IQR: 2–11), 5 mg/L (IQR: 3–11), and 5 mg/L (IQR: 2–10), respectively. The median FCP level at weeks 0, 12, 24, 52, and 104 was 817 µg/g (IQR: 352–1800), 425 µg/g (IQR: 193–1,307), 160 µg/g (IQR: 54–755), 142 µg/g (IQR: 49–653), and 96 µg/g (IQR: 25–250), respectively.
Figure 3.
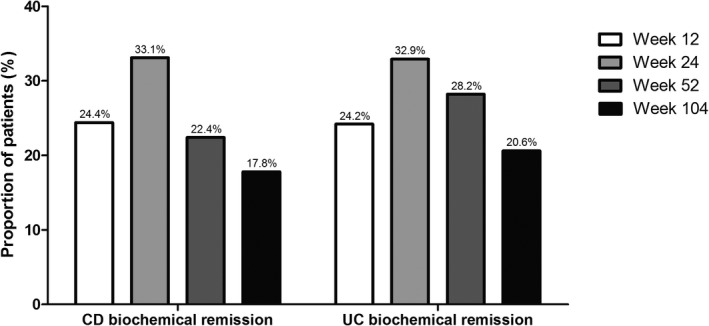
Biochemical remission (C‐reactive protein ≤ 5 mg/L, fecal calprotectin ≤ 200 µg/g) for patients with Crohn's disease (CD) and (fecal calprotectin ≤ 200 µg/g) for patients with ulcerative colitis (UC).
For UC, the proportion of patients in biochemical remission at weeks 12, 24, 52, and 104 was 24.2% (n = 16/66), 32.9% (n = 26/79), 28.2% (n = 20/71), and 20.6% (n = 14/68), respectively (Figure 3 b). When missing data were imputed as nonresponder, the proportion of patients in biochemical remission was 17.2% (n = 16/93), 28.0% (n = 26/93), 22.7% (n = 20/88), and 16.9% (n = 14/83), respectively. The median CRP concentration of patients treated with vedolizumab at weeks 0, 12, 24, 52, and 104 was 6 mg/L (IQR: 2–13), 4 mg/L (IQR: 1–7), 3 mg/L (IQR: 1–6), 3 mg/L (IQR: 1–6), and 3 mg/L (IQR: 1–6), respectively. The median FCP level at weeks 0, 12, 24, 52, and 104 was 1,317 µg/g (IQR: 588–2,004), 319 µg/g (IQR: 58–1,551), 85 µg/g (IQR: 10–510), 75 µg/g (IQR: 0–223), and 30 µg/g (IQR: 0–97), respectively.
Combined end point
The proportion of patients with CD in combined corticosteroid‐free clinical and biochemical remission at weeks 12, 24, 52, and 104 was 10.2% (n = 13/127), 18.2% (n = 22/121), 13.6% (n = 17/125), and 11.9% (n = 14/118), respectively.
The proportion of patients with UC in combined corticosteroid‐free clinical and biochemical remission at weeks 12, 24, 52, and 104 was 16.7% (n = 11/66), 24.1% (n = 19/79), 18.3% (n = 13/71), and 14.7% (n = 10/68), respectively.
Clinical factors associated with corticosteroid‐free clinical remission
Univariable and multivariable predictors of corticosteroid‐free clinical remission at week 104 are displayed in Table 2. There were no clinical or biochemical parameters predictive of corticosteroid‐free clinical remission at week 104 in the multivariable analysis.
Table 2.
Univariable and multivariable predictors for corticosteroid free remission at week 104
| Univariable analyses | Multivariable analyses | |||||
|---|---|---|---|---|---|---|
| OR | 95% CI | P value | OR | 95% CI | P value | |
| Age at inclusion | 1.00 | 0.98–1.02 | 0.93 | |||
| BMI per pointa | 1.01 | 0.92–1.10 | 0.91 | |||
| Sex | ||||||
| Male | ref | |||||
| Female | 0.95 | 0.49–1.84 | 0.88 | |||
| Disease duration | 0.99 | 0.95–1.02 | 0.46 | |||
| Disease location CDa | 0.16 | 0.25 | ||||
| Ileum | ref | ref | ||||
| Colon | 1.80 | 0.48–6.74 | 0.38 | 0.95 | 0.22–4.08 | 0.94 |
| Ileocolonic | 3.16 | 0.94–10.65 | 0.06 | 2.12 | 0.59–7.65 | 0.25 |
| Disease location UC | 0.90 | |||||
| Proctitis | Ref | |||||
| Left‐sided | 1.50 | 0.14–16.14 | 0.74 | |||
| Pancolitis | 1.03 | 0.10–10.97 | 0.98 | |||
| Upper GI involvementa | ||||||
| No | ref | |||||
| Yes | 0.27 | 0.03–2.19 | 0.22 | |||
| Disease behaviora | 0.29 | |||||
| Inflammatory disease | ref | |||||
| Stricturing disease | 0.25 | 0.05–1.17 | 0.08 | |||
| Penetrating disease | 1.36 | 0.42–4.35 | 0.61 | |||
| Peri‐anal disease | ||||||
| No | ref | |||||
| Yes | 1.01 | 0.99–1.03 | 0.37 | |||
| Prior intestinal resections | ||||||
| No | ref | ref | ||||
| Yes | 0.31 | 0.13–0.73 | <0.01 | 0.35 | 0.12–1.01 | 0.05 |
| Biochemical disease activitya | ||||||
| CRP per mg/L | 0.99 | 0.96–1.01 | 0.15 | |||
| Concomitant medicationa | ||||||
| Corticosteroids | 0.86 | 0.45–1.65 | 0.65 | |||
| Immunosuppressant | 0.88 | 0.44–1.74 | 0.88 | |||
Clinical parameters associated with corticosteroid‐free clinical remission at week 52 (BMI, GI, and CRP).
BMI, body mass index; CD, Crohn's disease; CI, confidence interval; GI, gastrointestinal; OR, odds ratio; UC, ulcerative colitis.
At inclusion. bMaximum extent until inclusion.
EIMs
At baseline, 32 patients with CD (16.8%) experienced active EIM: 29 patients reported arthralgia, 1 uveitis, 1 aphthous stomatitis, and 1 pyoderma gangrenosum. During the 2‐year follow‐up period, EIM remission was achieved in 13 patients (44.8%) with arthralgia, and all patients with uveitis (100%), aphthous stomatitis (100%), and pyoderma gangrenosum (100%). All but four patients with arthralgia were in clinical CD remission when EIM remission was achieved. During follow‐up, a total of 33 patients CD (17.3%; 28 patients with clinical disease activity) developed new EIM: 22 patients developed arthralgia, 2 uveitis, 6 aphthous stomatitis, 1 erythema nodosum, and 2 pyoderma gangrenosum. Of the newly developed EIMs, 6 arthralgia (25%), 2 uveitis (100%), 0 aphthous stomatitis (0%), 0 erythema nodosum (0%), and 0 pyoderma gangrenosum (0%), achieved remission during follow‐up.
Of all 119 patients with UC, 10 (8.4%) experienced EIM: 7 arthritis, 2 uveitis, and 1 pyoderma gangrenosum. During the entire follow‐up, four patients (57.1%) with arthritis and one (50%) patient with uveitis achieved remission. All but one patient with uveitis were in clinical UC remission when EIM remission was achieved. During follow‐up, a total of 21 patients with UC (17.6%; 13 patients with clinical disease activity) developed new EIMs: 13 arthritis, 4 uveitis, 2 aphthous stomatitis, and 2 erythema nodosum. Of the newly developed EIMs, 3 arthritis (37.5%), 2 uveitis (50%), 0 aphthous stomatitis (0%), and 0 erythema nodosum (0%) achieved remission during follow‐up.
Safety profile
The 310 patients included in our safety analysis were followed for 347.2 patient years and received in total 2,760 infusions. Ten patients (2.9 per 100 patient years) discontinued due to adverse events, including arthralgia (n = 4), infusion reactions (n = 2), infusion‐related adverse events (n = 2), headache (n = 1), and gastrointestinal infection (n = 1; Table 3). During follow‐up, we encountered 20 (5.8 per 100 patient years) probably related and 73 (21.0 per 100 patient years) possibly related adverse events. The most common adverse events were cutaneous lesions, infusion‐related adverse events, and arthralgia. Additionally, we documented 13 severe infections (3.7 per 100 patient years), 8 of these happened when the patient was on concomitant immunosuppressive therapies (thiopurines, corticosteroids, or both). Forty‐two (12.1 per 100 patient years) moderate infections and 78 (22.5 per 100 patient years) mild infections were documented. Most common infections affected the respiratory and gastrointestinal tracts. During follow‐up, two cases of malignancies were diagnosed and one progressed. Two patients discontinued vedolizumab therapy due to a malignancy (1 peritonitis carcinomatosa originating from the digestive tract and 1 progression of anaplastic oligodendroglioma). One 71‐year‐old patient developed prostate cancer with lymphoid metastasis but continued vedolizumab treatment. One patient died during follow‐up: a 51‐year‐old male patient with UC died due to a thrombosis in the basilar artery during vedolizumab treatment. In total, 84 (24.2 per 100 patient years) hospitalizations occurred during follow‐up. Fourteen patients with CD (7.3%) and 8 patients with UC (7.1%) required intestinal (sub)total colectomy.
Table 3.
Adverse events
| Possibly related | 73 (21.0 per 100 patient‐years) | |
| Cutaneous lesions | 23 | |
| Infusion related | 10 | |
| Arthralgia | 6 | |
| Headache | 8 | |
| Respiratory | 3 | |
| Other | 3 | |
| GI | 2 | |
| Exacerbation IBD | 2 | |
| Cardiac event | 2 | |
| Kidney/urinary tract | 2 | |
| Dizziness | 2 | |
| Malignancy | 2 | |
| Psychiatric | 2 | |
| Vascular | 2 | |
| Eye condition | 1 | |
| Fatigue | 1 | |
| Itch | 1 | |
| Transient tingling sensation | 1 | |
| Probably related | 20 (5.8 per 100 patient‐years) | |
| Infusion related | 8 | |
| Cutaneous lesions | 5 | |
| Arthralgia | 2 | |
| Nervous system | 2 | |
| Headache | 1 | |
| Dizziness | 1 | |
| Muscle cramp | 1 | |
| Serious adverse events | 10 (2.9 per 100 patient‐years) | |
| Arthralgia | 4 | |
| Infusion reaction | 2 | |
| Infusion related | 2 | |
| Headache | 1 | |
| GI infection | 1 | |
| Mild infections | 78 (22.5 per 100 patient‐years) | |
| Upper respiratory | 38 | |
| Flu‐like syndrome | 16 | |
| GI | 10 | |
| Fever (no focus) | 7 | |
| Cutaneous lesions | 3 | |
| Herpes zoster | 2 | |
| Soft tissue | 1 | |
| Cold sore | 1 | |
| Moderate infections | 42 (12.1 per 100 patient‐years) | |
| Upper respiratory | 15 | |
| GI | 6 | |
| Other | 5 | |
| Urinary tract | 4 | |
| Cutaneous lesions | 3 | |
| Pneumonia | 3 | |
| Herpes zoster | 2 | |
| Eye infection | 1 | |
| Gynecological | 1 | |
| Fever (no focus) | 1 | |
| Jaw/teeth | 1 | |
| Severe infections | 13 (3.7 per 100 patient‐years) | |
| GI | 5 | |
| Pneumonia | 5 | |
| Upper respiratory | 1 | |
| Other | 1 | |
| Musculoskeletal inflammation | 1 |
Adverse events during vedolizumab treatment (mild: no antibiotics or antiviral medication, moderate: oral antibiotics or antiviral medication, severe: hospitalization or intravenously administrated antibiotics or antiviral medication).
GI, gastrointestinal; IBD, inflammatory bowel disease.
Optimization of vedolizumab therapy
Of the 310 patients included in our cohort, 46 patients (14.8%) discontinued vedolizumab therapy before the fourth infusion (week 10 or 14). The extra induction infusion around week 10 was given to 34.5% (n = 107/310; CD: 47%, n = 89/191; UC: 15.1%, n = 18/119) of patients. During maintenance therapy, 23.1% (n = 61/264; CD: 45; UC: 16) received interval shortening (interval of ≤ 6 weeks) after a median treatment duration of 30.0 weeks (IQR: 14.1–48.6) for patients with CD and 46.1 weeks (IQR: 22.0–65.4) for patients with UC. The median duration between interval shortening and last visit or until treatment discontinuation was 30.0 weeks (IQR: 4.8–74.2) for patients with CD and 50.6 weeks (IQR: 24.6–62.8) for patients with UC. There was no significant difference in drug survival rate between patients on 8‐week infusions vs. patients who underwent interval shortening (hazard ratio: 1.18; 95% confidence interval (CI): 0.71–1.95).
Drug survival
Cumulative vedolizumab drug survival is demonstrated in Figure 4. Of 310 patients, 162 patients (52.3%; CD: 109; 57.1%; UC: 53 44.5%) discontinued vedolizumab treatment after a median treatment duration of 27.4 weeks (IQR: 17.2–51.8) for CD and 18.9 weeks (IQR: 11.8–40.9) for UC (Table 4). Main reasons for discontinuing treatment were: lack of response (CD: 61.5%; UC: 81.1%), loss of response (CD: 18.3%; UC: 9.4%), and adverse events (CD: 7.3%; UC: 3.8%). Five patients with CD discontinued vedolizumab treatment due to pregnancy. The probability of continuing receiving vedolizumab treatment after 52 and 104 weeks was 54.0% and 38.4% for CD and 60.8% and 51.3% for UC, respectively.
Figure 4.
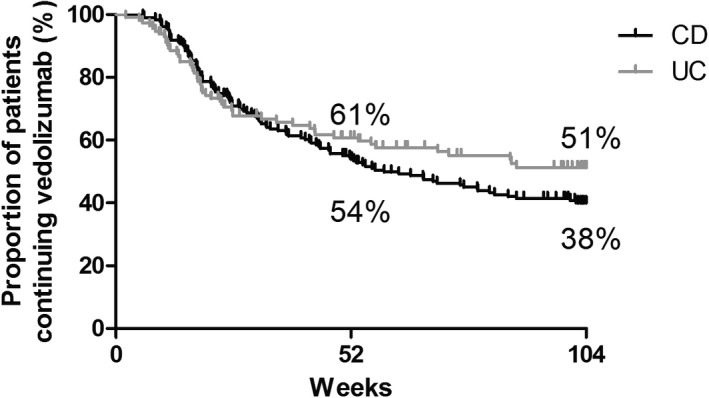
Cumulative drug survival. CD, Crohn's disease; UC, ulcerative colitis.
Table 4.
Discontinuation visit
|
CD N = 109 (57.1%) |
UC N = 53 (44.5%) |
||
|---|---|---|---|
| Treatment duration – weeks | Median (IQR) | 27.4 (17.2–51.8) | 18.9 (11.8–40.9) |
| Reason discontinuation | |||
| No response | N (%) | 67 (61.5) | 43 (81.1) |
| Loss of response | N (%) | 20 (18.3) | 5 (9.4) |
| Adverse events | N (%) | 8 (7.3) | 2 (3.8) |
| Malignancy | N (%) | 2 (1.8) | |
| Pregnancy | N (%) | 5 (4.6) | |
| Request patient | N (%) | 5 (4.6) | 1 (1.9) |
| Unknown | N (%) | 2 (1.8) | 2 (3.8) |
CD, Crohn's disease; IQR, interquartile range; UC, ulcerative colitis.
Discussion
In this real‐world prospective cohort of anti‐TNF exposed vedolizumab treated patients with IBD, the corticosteroid‐free clinical remission rate at week 104 was 19% for patients with CD and 28% for UC. Of the patients who were in corticosteroid‐free clinical remission at week 52, 59% remained in corticosteroid‐free clinical remission up to week 104. A relatively large proportion of patients received an additional dose on week 10 (35%) or dose escalation during maintenance therapy (23%). No new safety signals were observed, whereas the majority of severe infections (3.7 per 100 patients years) were seen in patients receiving concomitant treatment with immunosuppressive agents.
Here, we report the 104‐week corticosteroid‐free clinical remission rate in patients with IBD treated with vedolizumab. There is only one other study with this duration of follow‐up showing comparable results (week 108: CD: 24%, UC: 33%).13 Other available real‐life cohort studies used follow‐up periods up to 1 year. Here, we show corticosteroid‐free clinical remission rates of 28% for patients with CD and 27% for patients with UC at week 52. Differences in effectiveness rates between real‐world cohorts could, at least partly, be explained by the methods of analyzing patients who discontinued vedolizumab. Stallmach et al.6 (corticosteroid‐free clinical remission: 15% CD, 22% UC), the VICTORY consortium UC (corticosteroid‐free clinical remission: 37%, nonresponse imputation: 15%) used nonresponder imputation and intention‐to‐treat analysis comparable to our study. This conservative approach harbors the risk of underestimating treatment effect, but seems to be more appropriate for shared decision making in daily practice. Other cohorts excluded primary nonresponders before week 14 (GETAID, week 54 corticosteroid‐free clinical remission: 27% CD, 41% UC16), or only included in the effectiveness analysis a selection of patients who continued vedolizumab therapy after 52 weeks (corticosteroid‐free clinical remission: 54% CD, 59% UC8). The US VICTORY consortium for CD used the Kaplan–Meier method and reported cumulative rates of clinical remission of 35%. This method generally overestimates treatment effects as it ignores subjects who have discontinued treatment and does not consider loss of remission after initial response.17 Thus, clinical effectiveness at week 54 varies between 15% and 59% and differences in methodology, cohorts, and statistical analyses do not allow for a detailed comparison of corticosteroid‐free clinical remission rates.
Data on clinical benefit after 2 years of vedolizumab treatment and drug survival beyond the first year of treatment is scarce, but important for daily clinical practice. Due to the low rate of anti‐vedolizumab antibodies seen in clinical trials and observational cohorts (0–3% persistent positive1, 2, 18, 19), loss of response due to immunogenicity would be expected to be lower in vedolizumab‐treated patients compared with patients on anti‐TNF beyond the first year of therapy. Although the probability of continuing vedolizumab treatment decreased from 54% to 38% in patients with CD between the first and second year after treatment initiation, this probability remained fairly stable for UC (61% to 51%). The clinical benefit was maintained in 56% of patients with CD and 61% of patients with UC. These vedolizumab drug survival rates are comparable or possibly even better when compared with historic data on anti‐TNF treatment (13% discontinuation rate for both infliximab and adalimumab every year after the first year in CD20). However, clinical effect is not always maintained when remission is achieved. Additional real‐life cohorts with long‐term follow‐up are needed to assess and confirm these observations.
We assessed in exploratory analyses the impact of cotreatment with immunosuppressive medication (thiopurines and methotrexate) on clinical outcomes during vedolizumab treatment. Although this population was not randomized and these analyses may be at risk for selection bias, baseline characteristics were comparable between the subgroups. The comparable outcomes at week 104 (corticosteroid‐free clinical remission: vedolizumab monotherapy 23% vs. combination therapy 21% P = 0.77) suggest no clear clinical benefit of additional immunosuppressive therapy. From a safety perspective, 8 of 13 severe infections occurred in patients on combination therapy, suggesting that vedolizumab in patients with IBD might preferably be initiated as monotherapy.
A beneficial effect of vedolizumab treatment on EIM was seen in 21 of 42 patients (50%) who reported EIM at baseline, which is in line with the OBSERV‐IBD study.21 Although 21 patients achieved EIM remission in the present study, 54 patients developed EIM during follow‐up. Twelve of these EIMs (7 arthralgia, 2 uveitis, 2 aphthous stomatitis, and 1 arthritis) developed in patients who were in clinical remission. Only 2 of 12 patient (aphthous stomatitis and uveitis) were simultaneously in biochemical remission, however endoscopic disease activity was not ruled out. Due to the gut‐specific mechanism of action of vedolizumab, tapering of corticosteroids during the first months of vedolizumab treatment, and the relatively low probability of response to treatment (week 24: CD 40%, UC: 42%), insufficient anti‐inflammatory treatment of these specific patients with IBD may explain the development of new EIMs.
The safety profile of vedolizumab therapy observed in our study was similar to earlier studies (GEMINI trials and real‐world cohorts).1, 2, 11, 12, 22 The incidence of serious adverse events and severe infections was relatively low with 2.9 and 3.7 per 100 patient years, respectively. The most commonly reported infections in the current study were related to the gastrointestinal and respiratory tracts. One death occurred during follow‐up due to a basilar thrombosis in a male patient with UC with a disease duration of 5 years and with a history of atherosclerosis. Although we observed three malignancies, at least two and possibly all three were preexistent. The relatively low rate of serious adverse events, including thrombotic events and new malignancies, underscores the favorable safety profile of vedolizumab, but also underlines the importance to continue monitoring of long‐term safety outcomes in patients with IBD.
An additional week 10 infusion and interval shortening during maintenance therapy was frequently observed in our cohort (week 10 and optimization during maintenance treatment: 35% and 23%, respectively). Although an additional week 10 infusion is only suggested for patients with CD , according to the label and guidelines, a substantial proportion of patients with UC received this extra infusion (15.1%). The median treatment duration until interval shortening during maintenance treatment and between interval shortening and discontinuation or end of follow‐up, suggests that patients with an initial response to vedolizumab treatment may benefit from interval shortening following loss of response. This is in line with earlier work that reported loss of response rates of 47.9 (95% CI: 26.3–87.0) and 39.8 (95% CI: 35.0–45.3) per 100 person‐years with a 53.8% efficacy rate of dose intensification in secondary nonresponders.23 The latter and our results suggest that interval shortening may be a useful approach in secondary nonresponders in order to regain clinical effect.
Our study has several strengths. The systematic prospective follow‐up with a substantial cohort size, nationwide coverage, and long follow‐up period enabled us to create a representative cohort that reflects daily care. The characteristics of our cohort (anti‐TNF experienced) and intention‐to‐treat analyses of all patients initiating vedolizumab treatment allowed us to document clinically relevant effectiveness and safety outcomes useful for everyday practice. Our study also has some limitations. An important limitation lies in the lack of systematic information regarding mucosal healing. Because endoscopic evaluation was not mandatory in our design, not all centers performed endoscopy systematically or at predefined time points. Endoscopy was often performed when noninvasive markers were inconclusive, thereby creating selection bias when presenting these data. Furthermore, trough levels and anti‐vedolizumab antibodies were not available for all patients, limiting the interpretation of the relevance and effectiveness of nonresponse and dose escalation.
In conclusion, 19% (CD) and 28% (UC) of anti‐TNF exposed patients with IBD treated with vedolizumab were in corticosteroid‐free clinical remission after 2 years. Fifty‐nine percent of patients with IBD who were in corticosteroid‐free clinical remission at week 52 maintained corticosteroid‐free clinical remission up to 2 years. No new safety signals for vedolizumab treatment were found, whereas concomitant use of immunosuppressive therapy was associated with an increased risk of severe infections without an additional clinical benefit compared with vedolizumab monotherapy.
Funding
This study is, in part, supported by unrestricted grant from Takeda. This is an investigator‐initiated study; pharmaceutical companies played no role in study design, acquisition, analysis, interpretation, or presentation of the data.
Conflict of Interest
J.W. has served on advisory boards and/or received financial compensation from the following companies: MSD, Dr. FALK Pharma Benelux, Abbott Laboratories, Mundipharma Pharmaceuticals, Janssen, Takeda, and Ferring during the last 3 years. G.D. has unrestricted research grants from Abbvie, Takeda, and Ferring Pharmaceuticals, is on the advisory boards for Mundipharma and Pharmacosmos, and received speakers fees from Takeda, and Janssen Pharmaceuticals. A.E.M.‐d.J. has served on advisory boards, or as speaker or consultant for Takeda, Tramedico, and AbbVie, and has received grants from Takeda. B.O. is a speaker for Ferring, MSD, and Abbvie, serves on the advisory boards for Ferring, MSD, Abbvie, Takeda, Pfizer, and Janssen, has Research Grants from Abbvie, Ferring, Takeda, Pfizer, MSD, and Dr. FALK Pharma Benelux. N.K.H.B. has served as a speaker for AbbVie, Takeda, and MSD. He has served as consultant and principal investigator for Takeda and TEVA Pharma B.V. He has received (unrestricted) research grants from Dr. Falk Pharma Benelux and Takeda outside the submitted work. M.L. has served as speaker and/or principal investigator for: Abbvie, Celgene, Covidien, Dr. Falk Pharma Benelux, Ferring Pharmaceuticals, Gilead, GlaxoSmithKline, Janssen‐Cilag, Merck Sharp & Dohme, Pfizer, Protagonist Therapeutics, Receptos, Takeda, Tillotts, and Tramedico. He has received research grants from AbbVie, Merck Sharp & Dohme, Achmea Healthcare, and ZonMW. A.G.L.B. has served as speaker and/ or participant in advisory board for: Abbvie, Merck Sharp & Dohme, Takeda, Vifor Pharma, and Mundipharma. R.W. has served as speaker and/or participant in the advisory board for: Abbvie, Dr. FALK Pharma Benelux, and Janssen. J.M.J. has served on advisory boards or as speaker or consultant for Abbvie, Amgen, Ferring, Fresenius, Janssen, MSD, Pfizer, and Takeda. A.C.V. has participated in advisory board and/or received financial compensation from the following companies: Jansen, Takeda, Abbvie, and Tramedico. J.J.L.H. reports personal fees from the advisory board for Takeda Nederland B.V., personal fees from the advisory board for Lamepro B.V., outside the submitted work. D.J.J. received consulting fees from Synthon, Pharma, Abbvie, and MSD, and travel fees from Dr. FALK Pharma Benelux, Takeda, Abbvie, MSD, Ferring, Vifor Pharma, and Cablon Medical. M.J.P. reports grants from European Union, non‐financial support from Ferring, grants from Dr. FALK Pharma Benelux, Abbvie, and MSD, outside the submitted work. F.H. has served on advisory boards or as speaker or consultant for Abbvie, Celgene, Janssen‐Cilag, MSD, Takeda, Celltrion, Teva, Sandoz, and Dr. FALK Pharma Benelux, and has received unrestricted grants from Dr. FALK Pharma Benelux, Janssen‐Cilag, and Abbvie. All other authors declared no competing interests for this work.
Author Contributions
V.B., M.P., and F.H. wrote the manuscript. V.B., C.W., G.D., A.M., B.O., N.B., M.L., A.V., D.J., M.P., F.H., designed the study. V.B., C.W., G.D., A.M., B.O., N.B., M.L., N.S., A.B., R.W., J.J., A.V., J.H., D.J., M.P., and F.H. performed the research. V.B., M.P., and F.H. analyzed the data.
References
- 1. Feagan, B.G. et al Vedolizumab as induction and maintenance therapy for ulcerative colitis. N. Engl. J. Med. 369, 699–710 (2013). [DOI] [PubMed] [Google Scholar]
- 2. Sandborn, W.J. et al Vedolizumab as induction and maintenance therapy for Crohn's disease. N. Engl. J. Med. 369, 711–721 (2013). [DOI] [PubMed] [Google Scholar]
- 3. Ha, C. et al Patients enrolled in randomized controlled trials do not represent the inflammatory bowel disease patient population. Clin. Gastroenterol. Hepatol. 10, 1002–1007 (2012). [DOI] [PubMed] [Google Scholar]
- 4. Dulai, P.S. et al The real‐world effectiveness and safety of vedolizumab for moderate‐severe Crohn's disease: results from the US VICTORY consortium. Am. J. Gastroenterol. 111, 1147–1155 (2016). [DOI] [PubMed] [Google Scholar]
- 5. Narula, N. et al Vedolizumab for ulcerative colitis: treatment outcomes from the VICTORY consortium. Am. J. Gastroenterol. 113, 1345 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Stallmach, A. et al Vedolizumab provides clinical benefit over 1 year in patients with active inflammatory bowel disease – a prospective multicenter observational study. Aliment. Pharmacol. Ther. 44, 1199–1212 (2016). [DOI] [PubMed] [Google Scholar]
- 7. Allegretti, J.R. et al Predictors of clinical response and remission at 1 year among a multicenter cohort of patients with inflammatory bowel disease treated with vedolizumab. Dig. Dis. Sci. 62, 1590–1596 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Eriksson, C. et al Long‐term effectiveness of vedolizumab in inflammatory bowel disease: a national study based on the Swedish National Quality Registry for Inflammatory Bowel Disease (SWIBREG). Scand. J. Gastroenterol. 52, 722–729 (2017). [DOI] [PubMed] [Google Scholar]
- 9. Amiot, A. et al One‐year effectiveness and safety of vedolizumab therapy for inflammatory bowel disease: a prospective multicentre cohort study. Aliment. Pharmacol. Ther. 46, 310–321 (2017). [DOI] [PubMed] [Google Scholar]
- 10. Chaparro, M. et al Short and long‐term effectiveness and safety of vedolizumab in inflammatory bowel disease: results from the ENEIDA registry. Aliment. Pharmacol. Ther. 48, 839–851 (2018). [DOI] [PubMed] [Google Scholar]
- 11. Loftus, E.V. Jr et al Long‐term efficacy of vedolizumab for ulcerative colitis. J. Crohns Colitis 11, 400–411 (2017). [DOI] [PubMed] [Google Scholar]
- 12. Vermeire, S. et al Long‐term efficacy of vedolizumab for Crohn's disease. J. Crohns Colitis 11, 412–424 (2017). [DOI] [PubMed] [Google Scholar]
- 13. Amiot, A. et al Three‐year effectiveness and safety of vedolizumab therapy for inflammatory bowel disease: a prospective multi‐centre cohort study. Aliment. Pharmacol. Ther. 50, 40–53 (2019). [DOI] [PubMed] [Google Scholar]
- 14. Biemans, V.B.C. et al Ustekinumab for Crohn's disease: results of the ICC Registry, a nationwide prospective observational cohort study. J. Crohns Colitis. 14, 33–45 (2020). 10.1093/ecco-jcc/jjz119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Turner, D. et al A systematic prospective comparison of noninvasive disease activity indices in ulcerative colitis. Clin. Gastroenterol. Hepatol. 7, 1081–1088 (2009). [DOI] [PubMed] [Google Scholar]
- 16. Amiot, A. et al Effectiveness and safety of vedolizumab induction therapy for patients with inflammatory bowel disease. Clin. Gastroenterol. Hepatol. 14, 1593–1601.e2 (2016). [DOI] [PubMed] [Google Scholar]
- 17. Papoutsaki, M. et al The impact of methodological approaches for presenting long‐term clinical data on estimates of efficacy in psoriasis illustrated by three‐year treatment data on infliximab. Dermatology 221 (suppl. 1), 43–47 (2010). [DOI] [PubMed] [Google Scholar]
- 18. Williet, N. et al Association between low trough levels of vedolizumab during induction therapy for inflammatory bowel diseases and need for additional doses within 6 months. Clin. Gastroenterol. Hepatol. 15, 1750–1757.e3 (2017). [DOI] [PubMed] [Google Scholar]
- 19. Ungar, B. et al Association of vedolizumab level, anti‐drug antibodies, and alpha4beta7 occupancy with response in patients with inflammatory bowel diseases. Clin. Gastroenterol. Hepatol. 16, 697 – 705.e7 (2018). [DOI] [PubMed] [Google Scholar]
- 20. Ben‐Horin, S. & Chowers, Y. Review article: loss of response to anti‐TNF treatments in Crohn's disease. Aliment. Pharmacol. Ther. 33, 987–995 (2011). [DOI] [PubMed] [Google Scholar]
- 21. Tadbiri, S. et al Impact of vedolizumab therapy on extra‐intestinal manifestations in patients with inflammatory bowel disease: a multicentre cohort study nested in the OBSERV‐IBD cohort. Aliment. Pharmacol. Ther. 47, 485–493 (2018). [DOI] [PubMed] [Google Scholar]
- 22. Schreiber, S. et al Systematic review with meta‐analysis: real‐world effectiveness and safety of vedolizumab in patients with inflammatory bowel disease. J. Gastroenterol. 53, 1048–1064 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Peyrin‐Biroulet, L. et al Loss of response to vedolizumab and ability of dose intensification to restore response in patients with Crohn's disease or ulcerative colitis: a systematic review and meta‐analysis. Clin. Gastroenterol. Hepatol. 17, 838–846.e2 (2019). [DOI] [PubMed] [Google Scholar]