

Abstract
Physiologically‐based pharmacokinetic (PBPK) modeling has been extensively used to quantitatively translate in vitro data and evaluate temporal effects from drug–drug interactions (DDIs), arising due to reversible enzyme and transporter inhibition, irreversible time‐dependent inhibition, enzyme induction, and/or suppression. PBPK modeling has now gained reasonable acceptance with the regulatory authorities for the cytochrome‐P450‐mediated DDIs and is routinely used. However, the application of PBPK for transporter‐mediated DDIs (tDDI) in drug development is relatively uncommon. Because the predictive performance of PBPK models for tDDI is not well established, here, we represent and discuss examples of PBPK analyses included in regulatory submission (the US Food and Drug Administration (FDA), the European Medicines Agency (EMA), and the Pharmaceuticals and Medical Devices Agency (PMDA)) across various tDDIs. The goal of this collaborative effort (involving scientists representing 17 pharmaceutical companies in the Consortium and from academia) is to reflect on the use of current databases and models to address tDDIs. This challenges the common perceptions on applications of PBPK for tDDIs and further delves into the requirements to improve such PBPK predictions. This review provides a reflection on the current trends in PBPK modeling for tDDIs and provides a framework to promote continuous use, verification, and improvement in industrialization of the transporter PBPK modeling.
Relevance of transporters in drug pharmacokinetics
Membrane transporters are ubiquitously expressed in our body and facilitate transport of endogenous and xenobiotic substances. Thus, transporters play a critical role in governing a drug's disposition and cellular concentrations, which, in turn, drive pharmacological effect and/or toxicity.1 Activity or expression of these transporters is modulated by intrinsic factors, such as age, disease and genetic mutations, and presence of inhibiting/inducing drugs resulting in pharmacokinetic (PK) variability of substrate drugs. Transporters of clinical relevance, as suggested by the health authorities (US Food and Drug Administration (FDA), European Medicines Agency (EMA), Pharmaceuticals and Medical Devices Agency (PMDA), etc.), include P‐glycoprotein (P‐gp), breast cancer resistance protein (BCRP), organic anion transporting polypeptide (OATP)1B1, OATP1B3, organic anion transporter (OAT)1, OAT3, organic cation transporter (OCT)1, OCT2, and multidrug and toxic compound extrusion pumps (MATE)1 and MATE2‐K.2, 3 P‐gp and BCRP can limit oral absorption, brain distribution, and, thus efficacy of drugs.1, 4 OATP1B1/1B3 are hepatic uptake transporters and play a critical role in the disposition of high molecular weight acid and zwitterion drugs.5 Transporter polymorphisms can occur as noted with OATP1B1 (SLCO1B1 c.521T>C), which may lead to change in PKs and corresponding efficacy, and/or toxicity profiles.6 Role of renal transporters (predominantly OAT1, OAT3, MATE1, MATE2‐K, and OCT2) in drug clearance and the associated drug–drug interactions (DDIs) has also been well illustrated.1
Organ clearance of drugs often involves transporter‐enzyme/transporter interplay. The extended clearance model accounting for transporter‐enzyme interplay has been established to assess hepatic clearance of transporter substrates. This concept integrates hepatic uptake clearance PSinf (=PSinf,passive + PSinf,active), back‐flux clearance PSeff, and the sequestration processes (metabolic and biliary clearance, CLint, met, and CLint,sec).5, 7, 8
| (1) |
At steady state, the overall hepatic intrinsic clearance CLH,int can be described by the following equation.5, 7, 8. This can be further reduced to Eq. 3,
| (2) |
| (3) |
Here, “β” = CLint/(PSeff + CLint) value aids in readily understanding the rate‐determining process of CLH,int. For instance, when β is close to 1, CLH,int is determined by hepatic uptake, and when β 1, the CLH,int is thereby determined not only by hepatic uptake but also by CLint (intrinsic capacity of the hepatocytes to metabolize a drug).11 Although the β value can be a practical parameter for fitting, it is after all a composite of multiple, partly counteracting processes; hence it can be inaccurate and challenging to interpret due to identifiability issues. The overall hepatic intrinsic clearance incorporating these multiple processes can be evaluated using in vitro systems, such as suspension or cultured hepatocytes for transport rates, and human cryopreserved hepatocytes or liver microsomes for metabolism components.9, 12, 13 It is now well established that the in vitro–in vivo extrapolation (IVIVE) approach considering extended clearance model (Eq. 2) can be useful to estimate hepatic clearance of a hepatic uptake substrate drug.9, 12, 13
A similar concept can be applied for the prediction of renal clearance (CLR), integrating process clearances of renal uptake, metabolism, tubular secretion, and back‐flux from intracellular compartments across the basolateral membrane.14 Combining the predicted renal secretion clearance with glomerular filtration and the reabsorption rate allows the calculation of the in vivo CLR.5, 7, 8
Physiologically‐based pharmacokinetic modeling of transporter substrates
Integrating the various disposition characteristics in a mechanistic manner physiologically‐based pharmacokinetic (PBPK) modeling has become an essential part of drug development.15 Literature reports and recent implementations in an increasing number of regulatory filings established the utility of PBPK models for various purposes ranging from quantitative prediction of human PKs prior to first‐in‐human studies, to quantitatively evaluate/predict DDIs involving drug‐metabolizing enzymes and membrane transporters, to evaluate PK variability as a function of ethnicity, organ impairment, and pharmacogenomics. In this direction, the EMA and the FDA issued draft guidelines describing qualification of PBPK model platforms and reporting of PBPK modeling and simulations for regulatory submissions.16, 17 “Bottom‐up” PBPK models use model‐structure involving all relevant biological processes as system‐related parameters (e.g., human demographics and genetics, tissue volumes and blood flows, and enzyme and transporter expression levels), and drug‐related parameters derived from systematic preclinical studies (e.g., tissue partition coefficients, metabolism, or transport rates).15 Currently, the utility of preclinical data are confined to prediction of human plasma PK profiles and brain distribution driven by ABC transporters than informing human transporter‐mediated DDIs (tDDIs) due to differences in relative affinities of substrates and inhibitors for transporters across species. Nevertheless, ongoing efforts (Health and Environmental Sciences Institute initiative) of quantifying the absolute protein abundance and integrating protein data into PBPK platforms may lead to an improved translation of tDDI from animal to human. Abundance needs to be scaled to the activity specific for the transporter and species, which may not be feasible for all transporters.
Integration of such system‐related and drug‐related information along with the dynamic time‐dependent variables facilitates quantitative prediction of DDIs and additionally allow for simultaneous evaluation of multiple inhibition and induction mechanisms.18, 19, 20
Developing PBPK models for transporter substrates involve comprehensive characterization of the interplay between transporters and enzyme/transport to define the rate‐determining process.21, 22 Such models may need to be calibrated with both in vitro parameters (bottom‐up) and observed in vivo PK (middle‐out approach), and typically verified using additional clinical DDI data that can establish the quantitative role of individual mechanisms.15, 23, 24 The application and utility of PBPK models for DDIs involving hepatic transporters and transporter‐enzyme interplay are now well‐documented in the literature,18, 23, 26, 27 as well as in regulatory filings.29 Hepatic transport kinetics and enzymatic (CYPs) stability data of the victim drug obtained from in vitro systems like suspension or cultured hepatocytes and human liver microsomes are used as inputs along with a validated scaling factor (middle‐out approach) for active uptake for quantitative assessment of transporter‐mediated and complex‐DDI situations.19, 24, 25 In contrary, limited examples exist with respect to mechanistic PBPK modeling of transporter‐mediated intestinal and renal disposition.30, 31
A PBPK model can be built using general mathematical computing software (such as MATLAB or R). The latter has been the sandbox for modelers who were trying new ideas and extending existing models for examining their performance and consistency with observed data through various applications. However, not surprisingly, due to inability for version control and practical assessment of hundreds of line of model code, most investigations into tDDIs, either published or for regulatory submission, have been carried out using commercial PBPK software, such as Simcyp (Certara, UK), Gastroplus (Simulations Plus, PA), or PK‐Sim and MoBi (Bayer Technology Services, Germany; now open source). These systems often consist of elements that users cannot modify plus components that are under the control of the users (as input parameter or list of model selections). Although it has not been the case so far for all applications and all platforms, commercial PBPK platforms have the possibility of benefiting from provision of verified model structures, as per regulatory expectations. For the simulation of tDDIs, these include verification of the organs of primary interest (e.g., liver and intestine), preexisting system and drug data (e.g., for recommended transporter substrates and inhibitors), and embedded in IVIVE methodology and associated factors. Hence, the focus of regulatory assessment is on validity of the assumptions and selected options, input parameters and robustness, and adequacy of verification case examples.
Transporter inhibition can be incorporated into the intestine, liver, kidneys, and lungs and in all cases competitive inhibition is assumed. An important aspect for transporter inhibition is the consideration of the relevant inhibitor concentration at the site of interaction. For uptake transport, the free inhibitor concentration in extracellular fluid is used to drive inhibition, whereas in the case of the inhibition of efflux transporters, the unbound intracellular inhibitor concentration is used. However, when a mechanistic model is not built into an inhibitor model, the nearest surrogate concentration may be used.
PBPK modeling has now gained reasonable acceptance with the regulatory authorities for the CYP‐mediated DDIs.34 The goal of this cross‐industry collaborative effort is to reflect on the use of current PBPK tools to address the tDDIs and further delve in the requirements to improve these predictions with relevant gap analysis and future opportunities.
Available PBPK Model Files Within The Simcyp PBPK Simulator
Substrates
A summary of compound files for six substrates that have the active transport and passive permeability components and described using permeability‐limited models (i.e., ADAM, PerL, or Mech KiM) in V17 of the Simcyp Simulator are provided in Table 1. Due to the current challenges with IVIVE of drug transport, as discussed in the introduction, the intestinal and hepatic P‐gp‐mediated efflux components of the digoxin compound file are the only examples where active transport has been scaled via a “bottom‐up” approach. There were no quantitative data available for IVIVE of the renal basolateral uptake.35 For all other compounds, a “middle‐out” approach has been utilised, whereby an element of the transport component was optimized using clinical PK data. If suitable in vitro data, such as the transporter kinetic parameters and/or scaling factors, were available for a given transport process, most often these have been added to the model and the relative activity factor subsequently optimized. Alternatively, in the case of the hepatic uptake of repaglinide, the intrinsic clearance input was directly optimized using clinical data and parameter estimation module within the Simulator. Following the optimization of a transport process using clinical data, verification can be conducted via recovery of PKs under another scenario, which, in most cases, involves the simulation of data from individuals with a specific genetic polymorphism of a transporter or the simulation of DDIs associated with transporter inhibition, which are rarely specific to one transporter or metabolizing enzyme. For example, the intrinsic clearance for OATP1B1 mediated hepatic uptake of repaglinide was verified against PK data from OATP1B1 haplotypes “ultra‐rapid,” “intermediate,” and “poor expressers,” whereas the passive component (CLPD) was obtained from in vitro experiments in sandwich cultured human hepatocytes (SCHHs).24, 36
Table 1.
Substrates or victim drugs with established PBPK models within the Simcyp Simulator (V17)
| Substrate | Location | Input | Data source | Scaling | Optimised parameters (data) | Performance Verification | |||
|---|---|---|---|---|---|---|---|---|---|
| Doses | Population | DDIs | Details | ||||||
| Digoxin | |||||||||
| Transport: | 0.5–1 mg i.v. & p.o. SD; 0.125–0.25 mg p.o. q.d.; 0.25–0.5 mg p.o. b.i.d. | HV (Caucasian) | Ritonavir (hepatic and intestinal P‐gp); Verapamil / Norverapamil (hepatic and intestinal P‐gp) | ||||||
| P‐gp | Intestine | J max/K m | Caco‐2 | Default REF (Western Blot (WB) data) | |||||
| P‐gp | Liver | J max/K m | Caco‐2 | REF based on mRNA and WB data | |||||
| Passive: | |||||||||
| Intestine | P eff | Predicted (MechPeff) | Regional surface area | ||||||
| Liver | CLPD | Assumed (perfusion‐ limited) | HPGL | ||||||
| Metformin | |||||||||
| Transport: | 500, 1,000, 1,500 mg p.o.; 250, 1,000 mg i.v. | Caucasian | Cimetidine (OCTs and MATEs) | ||||||
| OCT1 | Liver | CLint | Cryo Hepatocytes | Optimised RAF |
RAFs PO (plasma and urine) |
MATEs REF assumed (no sensitivity with conventional model) | |||
| OCT2 | Kidney | CLint | OCT2‐HEK293 | Optimised RAF | |||||
| MATE1/2‐K | Kidney | CLint | MATE1‐HEK293 + MATE2‐K‐HEK293 | Assumed RAF | |||||
| Passive: | |||||||||
| Liver | CLPD | Cryo Heps | HPGL | ||||||
| Kidney | CLPD | Pampa P app | Estimated nephron surface area | ||||||
| Repaglinide | |||||||||
| Transport: | 0.25, 2 mg p.o. SD; 2 mg t.i.d. 7 days p.o. | HV (Caucasian) OATP1B1 phenotypes | Gemfibrozil (CYP2C8, CYP3A4, OATP1B1); Cyclosporine (OATP1B1) | ||||||
| OATP1B1 | Liver | CLint | Optimised | Assumed REF of 1 | CLint | ||||
| Passive: | |||||||||
| Liver | CLPD | SCHH | HPGL | ||||||
| Pravastatin | |||||||||
| Transport: | 20, 40, 60 mg p.o. SD; 20 mg b.i.d.; 40 mg q.d. | HV (Caucasian) OATP1B1 phenotypes | |||||||
| MRP2 | Intestine | CLint | SCHH | Hepatic abundance, assumed intestine / liver ratio | REF | ||||
| MRP2 | Liver | CLint | SCHH | Optimised REF | REF | ||||
| OAT3 | Kidney | CLint | Optimised | Assumed REF of 1 | CLint (using observed CLR) | ||||
| OATP1B1/1B3 | Liver | CLint | Optimised global CLint | OATP1B1 / OATP1B3 Relative contributions from in vitro expression data | CLint | ||||
| MATE1/2‐K | Kidney | CLint | Optimised | Assumed REF of 1 | CLint (using observed CLR) | ||||
| Passive: | |||||||||
| Liver | CLPD | SCHH | HPGL | ||||||
| Kidney | CLPD | Predicted (MechPeff) | Nephron SA | ||||||
| Intestine | P trans0 | Predicted (MechPeff) | Regional intestine SA | ||||||
| Rosuvastatin | |||||||||
| Transport: | 10, 20, 40, 80 mg p.o. SD; 10 mg p.o. q.d.; OATP1B1 phenotypes | HV (Caucasian) | Cyclosporine (OATP1B1/OATP1B3); Rifampicin | ||||||
| BCRP | Intestine | CLint | Intestinal abundance/activity of BCRP | CLint | |||||
| BCRP | Liver | CLint | SCHH | HPGL | |||||
| OATP1B1/OATP1B3/NTCP | Liver | CLint | Optimised ‐ Relative contribution assigned from in vitro OATP1B1/ NTCP transporter phenotyping experiments; remainder assigned to OATP1B3 | Assumed REF of 1 | Global hepatic Uptake CLint | ||||
| Passive: | |||||||||
| Liver | CLPD | SCHH | HPGL | ||||||
| Intestine | P app | Caco‐2 | P app → P eff correlation | ||||||
| Valsartan | |||||||||
| Transport: |
20 mg i.v 80, 160 mg p.o. SD; 160, 320 mg q.d. p.o. |
HV (Caucasian) | |||||||
| MRP2 | Intestine | Jmax/Km | Caco‐2 | Intestinal abundance/activity of MRP2 | |||||
| MRP2 | Liver | CLint | SCHH | REF | |||||
| OATP1B1 | Liver | CLint | OATP1B1‐HEK | REF (scalar obtained from abundance data) | Global hepatic uptake CLint | ||||
| OATP1B3 | Liver | CLint | OATP1B3‐HEK | REF (scalar obtained from abundance data) | |||||
| Inhibition: | |||||||||
| OATP1B1 | Liver | K i | OATP1B1‐HEK293 | ||||||
| OATP1B3 | Liver | K i | OATP1B3‐HEK293 | ||||||
| MRP2 | Intestine | K i | MRP2‐LLC‐PK1 | ||||||
| MRP2 | Liver | K i | MRP2‐LLC‐PK1 | ||||||
| Passive: | |||||||||
| Liver | CLPD | Predicted (MechPeff) | Sinusoidal SA | ||||||
| Intestine | P trans0 | Predicted (MechPeff) | Regional intestine SA | ||||||
CL, clearance; DDIs, drug–drug interactions; HEK, human embryonic kidney; HPGL, Hepatocytes per gram of liver, hepatocellularity; HV, healthy volunteers; MATEs, multidrug and toxic compound extrusions; OATP, organic anion transporting polypeptide; OCT, organic anion transporter; P‐gp, P‐glycoprotein; RAF, relative activity factor; REF, relative expression factor; SCHH, sandwich cultured human hepatocyte; WB, Western Blot.
In cases where a compound is a substrate for multiple transporters of similar function on the same membrane, such as hepatic sinusoidal uptake of rosuvastatin mediated by OATP1B1/3 and NTCP transporter, a global transport rate has been determined by fitting to clinical PK data and the available in vitro data were used to assign contributions from each transporter. In this case, clinical PK data in individuals with different OATP1B1 genotypes were subsequently used to verify the contribution from this transporter. A similar approach was taken to assign the relative contributions of OATP1B1/3 to the hepatic uptake of pravastatin.
In some cases, where the available data were limited at the time the model was created, transport processes have been included in a compound model without full verification. For example, BCRP‐mediated intestinal efflux within the current rosuvastatin model represents a net effect of intestinal transport, as individual uptake and efflux components cannot be de‐convoluted from the available data (in vitro and clinical). The BCRP‐mediated hepatic efflux of rosuvastatin was scaled from measured SCHH data37 and recovers the lowest reported rosuvastatin biliary clearance.38 The renal transport components of compounds secreted into urine are often difficult to verify. For example, although the OATP1B1 component of the pravastatin PBPK model has been fully verified (using clinical data in populations with different OATP1B1 phenotypes), its OAT3‐mediated renal basal uptake39 and renal apical efflux (currently included via a surrogate (MATE‐mediated) process in the absence of an identified transporter) can only be verified against observed CLR. Likewise, the simulated concentrations of metformin in plasma and urine have only limited sensitivity to the activity of MATE‐mediated apical efflux in the current model and kidney tissue concentrations, which would allow more robust verification, are not readily available. Currently, the relative activity factor value for this process is assumed to be like OCT2 (input data from transfected human embryonic kidney cells in both cases) in the metformin model. In the absence of clinical DDI data for each involved pathway, the individual transporter substrate and inhibition components of the valsartan model are difficult to verify.
Examples of transport processes that are known to occur for a given compound, but are not included in the current model, include renal digoxin transport (there are no quantitative data for the relevant renal basolateral uptake transporter currently available35), intestinal uptake of metformin (proposed to cause a “sponge‐effect” given the absence of a basolateral efflux transporter and significant paracellular permeability,40 and renal OAT‐mediated transport of rosuvastatin (known quantitative data for IVIVE are currently unavailable).
Inhibitors
A summary of the transport‐related components of the default Simcyp V17 for 10 transporter inhibitors are provided in Table 2. Although there can be evidence of active transport of an inhibitor (e.g., P‐gp mediated transport of nor‐verapamil, clarithromycin, and ritonavir), this is often not incorporated within the model, as the focus is primarily on recovering observed changes in PKs of a victim drug. Changes in plasma substrate concentrations are often the only clinical data available for the verification of an inhibitor model. Therefore, even if active transport can be incorporated within inhibitor models, verification of simulated tissue concentrations, which are used to drive the inhibition of efflux transporters, is difficult.
Table 2.
Inhibitors or perpetrator drugs with established PBPK models within the Simcyp Simulator (V17)
| Inhibitor | Location | Input | Data source | Scaling | Optimised parameters (data) | Performance Verification | |||
|---|---|---|---|---|---|---|---|---|---|
| Doses | Population | DDIs | Details | ||||||
| Cimetidine | |||||||||
| Inhibition: | 300, 400 mg p.o.; 400 mg b.i.d.; 400 mg t.i.d.; 300 mg i.v. | HV (Caucasian) | Metformin (OCTs and MATEs) | ||||||
| OCT2 | Kidney | K i | Optimised | N/A | OCT2 K i (Metformin DDI) |
Fitted OCT2 K i to recover metformin DDI. Alternative OCT2 kinetics required (Burt et al., 32). |
|||
| MATEs | Kidney | K i | MATE1‐HEK293 + MATE2‐K‐HEK293 | N/A | |||||
| Transport: | |||||||||
| OAT3 | Kidney | J max / K m | Cryo Heps | Optimised REF | |||||
| OCT2 | Kidney | J max / K m | OCT2‐HEK293 | Optimised REF | |||||
| MATE1/2‐K | Kidney | J max / K m | MATE1‐HEK293 + MATE2‐K‐HEK293 | Assumed REF | |||||
| Passive: | |||||||||
| Kidney | CLPD | Caco‐2 P app | Estimated nephron surface area | ||||||
| Gemfibrozil & 1‐O‐β Glucuronide | |||||||||
| Inhibition: | |||||||||
| OATP1B1 | Liver | K i | Optimised | N/A | OATP1B1 K i for both compounds (DDI rosuvastatin) | 600 mg SD and b.i.d. for 3 and 5 days p.o. | HV (Caucasian) | Repaglinide | |
| Cyclosporine | |||||||||
| Inhibition: | 180 and 200 mg p.o.; 1.5 and 2.5 mg/kg i.v. infusion (3 hours) | HV (Caucasian) | Repaglinide (OATP1B1) | ||||||
| OATP1B1 | Liver | K i | HEK‐OATP1B1 | N/A | OATP K i’s determined with preincubation | ||||
| OATP1B3 | Liver | K i | HEK‐OATP1B3 | N/A | |||||
| P‐gp | Intestine | K i | Membrane vesicles from MDR1‐ expressing Sf9 cells | N/A | P‐gp and BCRP K i’s are the lowest in vitro values | ||||
| P‐gp | Liver | K i | N/A | ||||||
| BCRP | Intestine | K i | N/A | ||||||
| BCRP | Liver | K i | N/A | ||||||
| Cyclosporine – M17 | |||||||||
| Inhibition: | 1.5 mg i.v. infusion (3 hours) | HV (Caucasian) | Repaglinide (OATP1B1) | ||||||
| OATP1B1 | Liver | K i | HEK‐OATP1B1 | N/A | OATP K i’s determined with preincubation | ||||
| OATP1B3 | Liver | K i | HEK‐OATP1B3 | N/A | |||||
| Clarithromycin | |||||||||
| Inhibition: |
250 and 500 mg PO BID |
HV (Caucasian) | Digoxin (intestinal and hepatic P‐gp) | ||||||
| P‐gp | Intestine | K i | Caco‐2 cells (IC50) | N/A | In vitro K i are same for gut and liver | ||||
| P‐gp | Liver | K i | Caco‐2 cells (IC50) | N/A | |||||
| Verapamil | |||||||||
| Inhibition: | 80 mg p.o. b.i.d. and t.i.d. | HV (Caucasian) | |||||||
| P‐gp | Liver | K i | Based on in vitro | N/A | Digoxin (intestinal and hepatic P‐gp) |
In vitro K i are same for gut and liver. Lowest in vitro IC50 values from multiple studies were converted to K i value. |
|||
| P‐gp | Intestine | K i | Based on in vitro | N/A | |||||
| Transport: | |||||||||
| P‐gp | Intestine | J max / K m | Based on in vitro | Fitted with SIVA | |||||
| MRP2 (apical efflux) | Intestine | CLint | N/A | Optimised apical efflux clearance | |||||
| Norverapamil | |||||||||
| Inhibition: | |||||||||
| P‐gp | Liver | K i | Based on in vitro | In vitro K i are same for gut and liver | 80 mg p.o. b.i.d and t.i.d of verapamil | HV (Caucasian) | Digoxin (intestinal and hepatic P‐gp) | The IC50 value from a report was scaled by the ratio between verapamil IC50 in this same study and P‐gp K i obtained from meta‐analysis. | |
| P‐gp | Intestine | K i | Based on in vitro | ||||||
| Rifampicin SD | |||||||||
| Transport | 600 mg i.v. (30 minutes infusion) and 600 mg p.o. | ||||||||
| OATP1B1 | Liver | K m | Optimized | In vitro K m / J max simultaneously optimized using dose‐dependent i.v. data | HV (Caucasian) | ||||
| OATP1B1 | Liver | J max | Optimized | ||||||
| Inhibition: | |||||||||
| OATP1B1 | Liver | K i | Optimized | In vitro K i reduced 7x to reproduce observed DDI with rosuvastatin or glyburide | HV (Caucasian) |
Rosuvastatin (OATP1B1/1B3/NTCP/BCRP) Glyburide (P‐gp/OATP1B1/2B1) |
|||
| OATP1B3 | Liver | K i | Optimized | ||||||
| OATP2B1 | Liver | K i | Optimized | ||||||
| NTCP | Liver | K i | Optimized | ||||||
| P‐gp | Liver | K i | Optimized | P‐gp induction is not covered but probably not relevant after single dose | |||||
| P‐gp | Intestine | K i | Optimized | ||||||
| BCRP | Liver | K i | Optimized | ||||||
| BCRP | Intestine | K i | Optimized | ||||||
| Ritonavir | |||||||||
| Inhibition: | 200 mg p.o. t.i.d. | HV (Caucasian) | Digoxin (intestinal and hepatic P‐gp) | Optimised K i as lowest in vitro value (LLC‐ PK1‐MDR1 cells) doesn’t recover DDI | |||||
| P‐gp | Intestine | K i | Optimised | In vitro K i are same for gut and liver | |||||
| P‐gp | Liver | K i | Optimised | ||||||
| Probenecid | |||||||||
| Inhibition: | |||||||||
| OAT1 | Kidney | K i | Based on in vitro | 500 mg i.v., 300 mg, 500 mg, 1,000 mg p.o. | HV (Caucasian) | Zidovudine (OAT1) | No OAT3 inhibition added. | ||
Compound summaries for all files are freely available to all users on the member's area (https://members.simcyp.com/account).
BCRP, breast cancer resistance protein; DDIs, drug–drug interactions; HV, healthy volunteer; IC50, half‐maximal inhibitory concentration; MATE, multidrug and toxin extrusion; N/A, not applicable; NTCP, sodium‐taurocholate co‐transporting polypeptide; OAT, organic anion‐transporter; OATP, organic anion‐transporting polypeptide; OCT, organic cation transporter; PBPK, physiologically‐based pharmacokinetic; P‐gp, P‐glycoprotein; REF, relative expression factor; SIVA, Simcyp in vitro data analysing tool kit.
The inhibition of transporters is assumed to be competitive and reversible in all cases. Where a Ki input is derived from an in vitro source, the value is usually converted from an half‐maximal inhibitory concentration (IC50) value with knowledge of the substrate concentration and corresponding Km value using the Cheng‐Prusoff equation.41
The general tendency for the simulation of transporter‐mediated DDIs is toward an underprediction of DDI magnitude when using in vitro derived Ki values. Therefore, in many cases, the input represents either the lowest of the available Ki estimate from literature (e.g., verapamil P‐gp inhibition) or is an optimized value to recover the clinical DDI (e.g., gemfibrozil OATP1B1 inhibition). The assumption inherent in the latter approach is that the attributed transporter and potency of inhibition are a true reflection of the clinical DDI/s used for optimization via middle‐out approach. In order to recover the extent of the DDI between the CYP3A4 and OATP1B1‐inhibitor cyclosporine and substrate repaglinide, the OATP1B1 In vitro Ki values after preincubation, which are significantly lower than without preincubation, are applied in the model.23 The mechanism responsible for this IC50 shift after preincubation is currently not clear, although some possible explanations are the effect of cyclosporin A (CsA) metabolites34 and the transinhibition.42 However, a 5‐fold to 20‐fold decrease in IC50 has been observed for some inhibitors, such as rifampin and cyclosporine, but not all OATP1B1/3 inhibitors. The OCT2 Ki value in the cimetidine compound file is optimized to match clinical profile.32, 43 The current value, which is ~ 500‐fold lower than the in vitro OCT2 Ki value in transfected human embryonic kidney cells, reflects the inability of the (Michaelis Menten) model to recover the indirect effect of MATE transporter inhibition (i.e., apical efflux transporter in the cimetidine PBPK model) on the activity of OCT2, and, in turn, on changes to plasma concentrations of the victim (i.e., metformin).32 To rectify the situation an electrochemical gradient driven transport model was evaluated developed for OCT2; however, this is currently not the default setting of the metformin compound file.32
Currently, (Simcyp V17) the P‐gp and BCRP inhibition components of the cyclosporine model are unverified with suitable DDI studies. There are several compounds, which have been developed within the simulator as inhibitors of CYPs that are known substrates (e.g., carbamazepine and quinidine) or inhibitors (e.g., ketoconazole and itraconazole) of P‐gp but currently do not have any transport components incorporated into the compound file. This would be continuously improved when new in vitro and in vivo data are available.
With respect to the transporter inducers there are no models in the current library of simulator due to lack of translational in vitro and in vivo data and gap in knowledge, as discussed in the later sections.
Recommendations For Verifying The Transporter PBPK Models
As with any PK modeling exercise, verifying a PBPK model that involves transporters is a required task. There are multiple ways of verifying transporter‐containing PBPK models and the selection of these depends on the intent of the model. For perpetrator models (inhibitors or inducers of transporters) it is essential to justify that the unbound concentrations at the site of the drug interaction (e.g., intestine, liver, or kidneys either from surrogate markers or from preclinical species) and that the interaction parameters (Ki and fuinc) are correct and/or predictive. For substrates models, it is necessary to verify that the active transport processes, the passive movement of drugs across membranes, and the metabolism processes are correctly captured by the model.
Model verification should be performed using data (e.g., plasma, urine, or bile concentrations) that were not used in the building process. Furthermore, data from studies containing different doses help to understand if the assumptions of linearity, or the input values for Km and V max are correct. Verification should not be done only against data from single‐dose studies, but also against data from multiple dose studies, given that these can help identify any changes in transporter‐mediated mechanisms caused by auto‐induction or inhibition.
As with enzymes, where the fraction metabolized can be verified, using available data from drug interaction studies, can help verify the assumptions about the fraction transported. Usually, the use of a strong inhibitor is recognized as the best approach for this task, although there are very few potent and selective inhibitors of transporters. Therefore, sometimes it becomes necessary to use a matrix of inhibitors to tease out the fraction of a drug that is absorbed or eliminated via a specific transporter.
Studies in individuals with different genotypes can also help identify the role of a transporter in the disposition of a drug when there are no selective inhibitors/inducers, such as studies reported for BCRP, OCT1, and OATP1B1, and have been used successfully for PBPK models; specifically the single‐nucleotide polymorphisms c.521T>C (Val174Ala) in the SLCO1B1 sequence in exon 5 and the c.388A>G in exon 4 and associated genotypes that have been shown to alter transporter activity (Repaglinide44). After the fraction transported has been assigned with confidence, power calculations can be performed to verify, in turn, used system data for these transporters.45 When polymorphism is involved, it can help to assign a transporter component; however, as best practice, a lack of effect of known phenotypes also needs to be explained. Depending on the individual transporter activity the interindividual PK profiles can be significantly different, questioning the report of mean profiles in reports.
The predictive performance of the models should be evaluated using the right type of data given that tDDI interactions are not always observed as changes in plasma concentrations. Such can be the case for the inhibition or induction of transporters in the apical membrane of the kidneys or the canalicular membrane in the liver, when the active uptake from the blood into the proximal tubular cells in the kidneys or into the hepatocytes, the liver is the limiting step in the disposition of a drug. In these cases, urine and/or bile collection becomes indispensable for testing the soundness of the model (pyrimethamine and MATE inhibition).46 If the pKa of the victim compound is within the physiological range of urine pH values and the renal elimination of the compound is significant, the change in passive and possibly active (e.g., MATE) secretion and reabsorption of the compound is altered. A good case example for this is memantine, where the influence of urine pH and urinary flow on the renal excretion has been well‐documented.47 To alkalinize the urine, the volunteers received doses of 4 g sodium bicarbonate, to acidify the urine the volunteers received doses of 1 g ammonium chloride. The plasma profiles at three different urine pH values (5.0, 7.4, and 8.0) were reported, as were the amount excreted by the kidneys under acidic, neutral, and alkaline urine conditions in the last dosage interval. These data were suitable to build a PBPK model for memantine, including the interplay between renal OCT2 and MATE1.48, 49 Like with enzymes, transporters can be colocalized in multiple tissues. For example, P‐gp, BCRP, and MRP2, are located in the intestine, the liver and the kidneys in the same membrane. Therefore, verification of models that include these transporters should include the comparison of multiple predictive PK parameters against observed ones (peak plasma concentration (Cmax), area under the curve (AUC), time of maximum plasma concentration (Tmax), and half‐life), given that changes in certain parameters can help recognize the location of the transporter affected by the drug interaction. This may be especially true for the DDIs driven by intestinal efflux transporters. For example, an increase/decrease in Cmax without a change in half‐life can suggest an increase/decrease in fraction absorbed due to inhibition/induction of intestinal efflux transporters (digoxin/rifampicin33). Although a reduced biliary clearance propagates as a change in AUC as part of enterohepatic recirculation and an inhibition of renal P‐gp propagates as decreased CLR and an increased AUC in the later elimination phase of the PK profile, specifically, if a basolateral renal efflux mechanism is indirectly activated as well (digoxin‐itraconazole DDI).51 Using drug interaction data after intravenous and oral dosing of the substrate makes the verification task simpler, although these studies are not very common (Supplementary Table from gap analysis section).
Charcoal studies can be useful in investigation of the involvement of enterohepatic recirculation or entero‐enteric recirculation (EER). A difference in the AUCs when a drug is administered alone vs. with charcoal suggests the presence of enterohepatic recirculation or EER, or both. For example, there was about a 28% decrease in the oral AUC of apixaban, a P‐gp and BCRP substrate, when activated charcoal was administered 6 hours after apixaban when the absorption of apixaban was largely complete.52 As biliary excretion was a relatively minor elimination pathway, the difference was mostly attributed to EER. The obvious drawback is that charcoal is not specific for any transporters, so the results need to be considered with other investigative in vitro and in vivo studies. Hence, a charcoal study may be considered for investigative purposes to further understand the absorptive mechanisms of a drug and refine the PBPK model.
Examples Of Transporter‐Mediated PBPK Submissions To Regulatory Health Authorities
Examples of transporter‐mediated DDI simulations using PBPK models that were either used to support submissions to regulatory agencies or played a crucial role in trial design or strategic internal decision making from the years 2013‐2018 have been summarized in Table 3 and Figure 1. The examples that were discussed in this section primarily related to tDDI cases (43% examples were for regulatory submissions and 50% were impacting the decision making within a company). Each example is presented with a brief background, key regulatory questions being addressed, and a summary of the qualification dataset and summary of regulatory interactions. Case examples from a sponsor or regulatory perspectives are presented in Table 3. A smaller portion (7% of the examples) of non‐tDDI comprised of understanding the nonlinear absorption of a molecule or understanding the brain penetration aspects of the drug, which were mainly used for an internal decision making are also captured. Examples in Table 3 were also classified as high impact (replace; provides inference that informs internal decisions without requiring a clinical study), medium impact (inform; provides inference that informs internal decisions), and low impact (describe; provides inference that has limited impact on internal decisions).
Table 3.
Examples of transporter‐mediated DDI PBPK analyses and their impact on drug development and regulatory decision
| Example number | Drug |
Key theme Transporter (location function) Inhibitor – inh Substrate ‐ sub |
Victim/perpetrator/ and question(s)? | Brief description | Impacta | Qualification dataset | FDA/EMA response |
|---|---|---|---|---|---|---|---|
| 1 | Simeprevir (marketed) |
Hepatic transporter: OATP (sinusoidal uptake) sub Intestinal and hepatic metabolism: CYP3A sub |
DDI potential as victim with OATP and CYP3A perpetrators and PK prediction of plasma and liver (at target site for efficacy) in specific populations (e.g., Asians, renal/hepatic impaired) |
Simeprevir PBPK model verified with human PK, DDI trial in HV met CYP3A inhibitor/inducers and OATP modulators (e.g., erythromycin, ritonavir, efavirenz, rifampicin, cyclosporine) Simcyp Version: 12 |
High Impact: Used to understand non‐linear PK characteristics and clinical trial design. Used to answer regulatory questions | Simeprevir PBPK model verified with human PK, DDI trial in HV with CYP3A inhibition/induction and OATP modulators (erythromycin, ritonavir, efavirenz, rifampicin, and cyclosporine) |
FDA: Accepted FDA commentary on usefulness of model verification and reporting was published along with sponsor manuscript (references) EMA: Submitted, but did not comment PMDA: Submitted, but did not comment |
| 2 | Ibrutinib (marketed) | Intestinal transporter: P‐gp (apical efflux) inhibitor | Does P‐gp inhibitor translate to clinical DDI liability? |
ADAM model was built to simulate ibrutinib concentrations in segments of GI tract Simcyp Version: 12 |
High Impact: FDA reviewer evaluated simulation output regarding predicted ibrutinib exposure in different segments of the GI tract to determine the potential for ibrutinib to inhibit P‐ gp. No formal DDI trial with P‐gp substrate is needed if dose staggering of ibrutinib and P‐gp substrate is applied | PBPK model verified with human PK and ketoconazole and Rifampicin DDI trial | FDA: Accepted |
| 3 | Apalutamide (marketed) |
Renal transporter: OAT3 (basolateral uptake) OCT2 (basolateral uptake) MATE (apical efflux) inhibition |
Does inhibition for these kidney transporters translate to clinical DDI liability (Cmax,u parent ~ 0.659 µM; Cmax,u metabolite ~ 0.568 µM) |
Apalutamide + metabolite PBPK models built to simulate plasma PK and kidney PK. Apalutamide + metabolite PBPK models built and verified with clinical PK data Simcyp Version: 16 |
High Impact: No DDI studies planned with these kidney transport substrates | Apalutamide + metabolite PBPK models built and verified with clinical PK data no DDI is expected with OAT3 substrates like e.g., penicillin. Minor interaction predicted with metformin using electrochemical gradient option within simulator |
FDA and Health Canada: Review published. Concerns raised on verification of metformin PBPK model and adequacy to predict OCT2/MATE mediated DDIs. No clinical metformin DDI study requested. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/210951Orig1s000MultidisciplineR.pdf |
| 4 | Axitinib (marketed) | Intestinal transporter: P‐gp (apical efflux) inhibitor | Does P‐gp inhibition in vitro translate to clinical DDI liability unbound Cmax of 0.0008 µM, | ACAT model using Gastroplus was built to simulate axitinib concentrations in segments of GI tract | High Impact: Agreement of HA that no formal DDI trial with P‐gp substrate is needed |
FDA: Accepted EMA: Not submitted |
|
| 5 | Naloxegol (marketed) |
Intestinal transporter: P‐gp (apical efflux) sub Intestinal and hepatic metabolism: CYP3A sub |
Because of Naloxegol in vitro, clinical DDI and PBPK package, FDA requested following information 1. Some CYP3A modulators are known to affect P‐gp. Therefore, full PBPK model accounting for P‐gp contribution should be developed for Naloxegol 2. Naloxegol CL considered predominately by CYP3A, What is the contribution of P‐gp to the biliary secretion of Naloxegol? 3. Incidence of headache doubled in a ketoconazole DDI study, a P‐gp inhibitor. Naloxegol may target receptors in the brain, we recommend you use your PBPK model to evaluate potential effect of P‐gp inhibition on brain drug exposure |
Perpetrators: Simcyp compound library files (quinidine, diltiazem and ketoconazole) No Naloxegol P‐gp kinetic parameters available, assumed to be same as digoxin. Fit‐for purpose model was required to capture the transporter mediated DDI with quinidine as the PBPK model was sensitive to distribution model used (2‐different model structures for CYP vs. tDDI) Simcyp Version: 12 |
Medium Impact Clinical study data of Naloxegol dosed with quinidine, diltiazem and ketoconazole, which are dual P‐gp and CYP3A inhibitors |
FDA accepted63 http://www.accessdata.fda.gov/drugsatfda_docs/nda/2014/204760Orig1s000ClinPharm.pdf |
|
| 6 | Olaparib (marketed) |
Intestinal, hepatic, and renal transporter P‐gp (apical efflux) inhibitor in intestine, kidney, and liver MATE1 (canalicular efflux) inhibitor OATP1B1 (sinusoidal uptake) BCRP (apical efflux) OCT1 (sinusoidal uptake) OCT2 (basal uptake) |
Regulator's asked to explain the applicant's view about necessity of conducting clinical studies to investigate PK interaction between olaparib and substrates of P‐gp, MATE1, OATP1B1 and OCT transporters EMA comment on BCRP inhibition potential by olparib: Without clinical data it is difficult to conclude whether a compound is a weak or a strong inhibitor because it depends of the magnitude of this effect in vivo |
ADAM and Full PBPK model were used to understand the tDDI risks for olaparib tablets in cancer patients Simcyp Version: 16 |
High Impact: Currently there are no clinical data of olaparib with any of the transporter substrates available. Used Simcyp compound files (which) and ran a sensitivity analysis to reflect the worst‐case scenario. |
PMDA accepted (all transporters) FDA: Accepted (P‐gp) EMA: Only P‐gp and BCRP simulations submitted EMA accepted P‐gp part55 |
|
| 7 | Osimertinib (marketed) | Intestinal and hepatic: BCRP (apical efflux) | To investigate the clinical impact of hepatic OATP1B1/BCRP inhibition by Osimertinib |
PBPK was used to understand the fit‐for‐purpose model vs. mechanistic model. Different model structure (for CYP DDI vs. tDDI) was needed to recover the DDI The potential of osimertinib to act as an inhibitor of BCRP was determined using Caco‐2 cells from concentration range of 1 to 300 μM using 1 μM [3H]‐rosuvastatin as in vitro probe and novobiocin as a positive control. From this assay BCRP IC50 was 2 μM for osimertinib. Good recovery of tDDI with higher follow‐up, gut compared to in silico predicted using SimCYP Simcyp Version: 16 |
Low Impact | Rosuvastatin (Simcyp in‐built file) used as a substrate of BCRP | Not used for regulatory submissions59 |
| 8 | Baricitinib (marketed) |
Renal transporter: OAT3 (basolateral uptake) substrate MATE2K, P‐gp, and BCRP (apical efflux) substrate |
Baricitinib can be administered with NSAIDS Baricitinib is a substrate of OAT3, MATE2K, P‐gp and BCRP. OAT3 Inhibitors: Probenecid (in house model built using clinical data and verified against clinical data) Ibuprofen (in house model verified using pemetrexed) Diclofenac (in house model verified using published clinical data) (Iu/IC50 < 0.1) not predicted to be an inhibitor |
Bottom‐up for renal data; and middle‐out (for Vss and F) using HV data. First order absorption and Full PBPK (Rodgers & Rowland) model For OAT3 and MATE2K Km and Vmax from in vitro were used in the model. P‐gp and BCRP in‐vitro CLint in the kidney values were used in the model. Simcyp Version: 14 |
High Impact. The interaction with probenecid was correctly predicted. (AUC ratio ~ 2) The exposure of baricitinib is not affected by NSAIDS. It showed IVIVE can be used to predict inhibition of OAT3 using in vitro IC50 for the OAT inhibitors |
Multiple clinical studies (at different doses) were used to test model performance. CLR values were taken from multiple studies |
30 |
| 9 | Pemetrexed (marketed) |
Renal transporters: OAT3 (basolateral uptake) OAT4 (apical efflux) |
Pemetrexed can be administered with NSAIDS |
Bottom‐up and middle‐out approach Pemetrexed: CL renal. Full PBPK (Rodgers & Rowland) model, Elimination (renal filtration and transport by OAT3 (basolateral membrane) and OAT4 (apical membrane). North European white population. For OAT3 Km and Vmax from in vitro were used in the model For OAT4 CLint value was used. Simcyp Version: 12 |
Low Impact: Bottom‐up PBPK model predicted 2‐fold lower CLR for pemetrexed. A middle‐out model for pemetrexed using a RAF value of 5.3 for OAT3 was used to predict the DDI |
Multiple clinical studies were used to test model performance for the victim drug and the perpetrator. Model recovered the observed clinical interaction with ibuprofen. All models built in house |
96 |
| 10 | Letermovir (marketed) |
Hepatic transporter: OATP1B (hepatic uptake) metabolising enzymes: UGT1A1, ‐1A3, and CYP3A |
Characterize unanticipated nonlinear human PK and to explain differences in letermovir PK in different populations including white HVs, Japanese healthy volunteers, and HSCT recipients |
First order absorption; Distribution: full PBPK model with a permeability‐limited liver model Elimination: Enzyme kinetics (UGT1A) Liver Transporter: OATP1B1 uptake kinetics and ASA for abundance of OATP1B1 |
High Impact: Provided mechanistic explanations for the nonlinear PK and observed differences in PK in selected populations; used to predict perpetrator DDIs with CYP2C8 substrates | The PBPK model was qualified by PK and plasma‐concentration profiles after multiple i.v. and p.o. doses of letermovir in white HVs |
FDA approved the PBPK model and requested to expand this modeling effort to include simulation in more populations, (e.g., hepatic/renal impairment) and to predict perpetrator DDI magnitude to CYP probe drugs EMA: Not accepted97 |
| 11 | Glecaprevir (GLE) + Pibrentasvir (PIB) (marketed)98 |
GLE: Hepatic transporter: OATP1B1/1B3 (sinusoidal uptake), P‐gp and BCRP (basolateral efflux) – sub + inh Intestinal transporters: P‐gp and BCRP (apical efflux) – sub + inh Intestinal and hepatic metabolism: CYP3A4 PIB: Hepatic transporter: P‐gp and BCRP (basolateral efflux) – sub + inh (only P‐gp) Intestinal transporters: P‐gp and BCRP (apical efflux) – sub + inh (only P‐gp) |
Interaction between GLE and PIB as combination therapy, DDI as victims with OATP, P‐gp, and CYP3A perpetrators (effect on plasma and liver exposures) |
GLE and PIB PBPK models separately verified with clinical PK (SAD, MAD) data and clinical DDI data with Ritonavir (CYP3A + P‐gp inhibitor). GLE and PIB models verified with clinical DDI data of GLE and PIB together, for the effect of one on the other. Simcyp Version: 16 |
High Impact: Used to understand non‐linear PK characteristics, effect of DDI on hepatic exposures, and oral absorption mechanisms |
GLE and PIB models in combination verified with perpetrator DDI data for rifampin and cyclosporine (CYP3A, P‐gp, BCRP, and OATP inhibitors). GLE and PIB models in combination verified with victim DDI data for digoxin (P‐gp), pravastatin (OATP1B1), and rosuvastatin (CYP3A, OATP1B1/1B2, BCRP) |
FDA: Not submitted EMA: Simulation results used to address regulatory queries on hepatic exposures with perpetrator DDI and DDI on victim comeds. EMA expressed reservations regarding qualification of the transporter capabilities in Simcyp PMDA: Not submitted |
| 12 | NVS‐X (phase III) |
Hepatic transporter: OATP1B3 (sinusoidal uptake) sub Intestinal transporter: P‐gp (apical efflux) sub |
1. Mechanistic understanding of drug disposition pathways by separate evaluation of contributions of P‐gp (intestine) and OATP1B3 (liver) to the net victim DDI potential 2. Victim DDI evaluation in different inhibition scenarios |
Active uptake by OATPs was indicated in cryopreserved human hepatocytes in the absence and presence of rifamycin (20 µM) and atorvastatin (10 µM). Predominant involvement of OATP1B3 in total active uptake was confirmed by OATP1B1/1B3/2B1‐HEK293 cells. Large accumulation in P‐gp‐LLC‐PK1 cells was observed in the presence of cyclosporine (10 µM). The in vitro incubations were performed in buffer (pH 7.4) w/o any protein components as HSA/BSA. Increase in AUC and Cmax of this drug by co‐administration of cyclosporine (175 mg p.o., b.i.d.) was not much affected by administration routes of the compound (i.v. or p.o.). The clinical DDI data indicated low effects of intestinal P‐gp inhibition on the net victim DDI potential. In addition, oral bioavailability and Vss (with and w/o cyclosporine) were measured. Using these parameters, a PBPK model was established based on middle‐out approach. Due to low contribution of intestinal P‐gp on the first pass, the first‐order absorption model was used. Kp, scalar was optimized to capture the measured Vss (full PBPK model). Contribution of UGT enzymes to CLh was estimated by in vitro enzyme phenotyping. CLr was estimated based on human mass balance data. Simulations were conducted in a HV population file. Simcyp Version: 16 |
Microdose i.v. and p.o. DDI with cyclosporine; SAD/MAD; and human mass balance. For a Simcyp cyclosporine model, Kp scalar values of tissues were changed based on Yoshikado et al.11 2016. A reported In vitro Ki of cyclosporine on OATP1B3 (with preincubation; Gertz et al.23 2013) was used |
Not submitted | |
| 13 | PF‐X (phase III) | Intestinal transporter: P‐gp (apical efflux) substrate | Intestinal absorption limitations and victim DDI liability with P‐gp inhibitors |
PBPK models were built for parent and major metabolite using in vitro and clinical data. In vitro studies in Caco‐2 cells were used to parameterize transport processes. Simcyp Version: 16 |
Supported design of clinical study | PBPK model was verified with FIH data, and DDI data with P‐gp inhibitor, verapamil | Internal impact and not submitted to health agencies |
| 14 |
Fostamatinib (marketed) |
Intestinal transporter: BCRP (apical efflux) inh hepatic transporters: OATP1B1 (sinusoidal uptake) inh BCRP (apical efflux) inh |
To investigate the clinical impact of hepatic OATP1B1/BCRP inhibition by Fostamatinib |
PBPK was built using public domain information (Washington DDI database) and used to recover the DDI Good recovery of tDDI with measured Ki value Simcyp Version: 17 |
Low Impact: Understanding the capability of PBPK in recovering the tDDI | Rosuvastatin (Simcyp in‐built file) used as a substrate of BCRP | |
| 15 | Asunaprevir (late stage) |
Hepatic transporter: OATP1B1 (sinusoidal uptake) sub Note: Approx. 15–20× increases of C max and AUC with single dose rifampin 600 mg dose |
FDA question on sponsor statement that inhibition of P‐gp alone but not CYP3A4 is unlikely to have clinically meaningful effect on asunaprevir's exposures; PMDA question on DDI as a victim drug with select concomitant medications; Internal question whether PBPK model could recover ethnic PK differences |
Model was developed using the middle‐out approach with ADAM, full PBPK distribution, CYP kinetics with biliary excretion, and transporter kinetics. The model was verified with clinical DDI studies and applied to address the various questions. The substrate studies were performed using stably transfected HEK‐293 cells that singly express human OATP1B1, OATP1B3, or OATP2B1. The validity of the transfected cell models was established by performing uptake studies with positive controls Simcyp Version: 13 |
Were able to waive several clinical DDI studies | Clinical DDI studies with ketoconazole, single and repeat dosing of rifampin, ritonavir, and midazolam. Simcyp ketoconazole, rifampin and ritonavir compound files were used with modifications. Simcyp midazolam compound file was used as is |
Not submitted to FDA and EMA PMDA: Simulation results are reflected in the dosing recommendations in the drug label |
| 16 | GSK‐X (phase III) |
Renal transporters: OAT1 (basolateral uptake) sub MRP4 (apical efflux) sub |
Tenofovir was used as a substrate for OAT1 and MRP4 Regulatory query: Concern on Tenofovir renal elimination by GSK drug especially due to inhibition of MRP4 and accumulation in the renal cells as clinically no difference in PK observed in a DDI study |
To investigate the clinical impact of OAT1 and especially MRP4 inhibition by GSK drug on renal elimination of Tenofovir. Simcyp Version: 12 |
Low Impact: Simcyp simulations predicted no risk of DDI predicted, including no accumulation of Tenofovir within the renal cells (PTC) due to MRP4 inhibition | Bespoke models built for both Tenofovir and GSK drug. Both models validated with clinical PK data | No further investigative work recommended |
| 17 | SA44121 (phase II) |
Renal transporter: OAT1 (basolateral uptake) inh OAT3 (basolateral uptake) inh |
DDI prediction for a compound as inhibitor of both transporters |
Model was developed using the middle‐out approach with a first order absorption model (Fa assumed = 1, Ka from ADAM), a minimal PBPK, and using CLiv and in vitro OAT1/3 Ki. Model qualification with plasma concentration after oral administration of 1,000 mg and DDI with ciprofloxacin and tenofovir Simcyp Version: 14.1 |
High Impact (support waiver for a clinical DDI study) |
Ciprofloxacin from Simcyp inhibitor library modified to include Mech KiM and OAT3 CLint Tenofovir. Verification with internal plasma conc data from DDI study |
64 |
| 18 | SA44121 (phase II) |
Renal transporter: OAT1 (basolateral uptake) OAT3 (basolateral uptake) |
DDI prediction for a compound as a substrate |
Model was developed using the middle‐out approach with a full PBPK distribution model (R&R), and a Mech KiM model (renal excretion (OAT1/3)) and CYP2C9 The model was qualified with urine and plasma concentration after i.v. bolus of 3 doses. Plasma and urine data obtained at the lowest dose was used to estimate the relevant model parameters (RAF, nonrenal clearance). During the model verification step, the observed plasma concentrations and renal clearance values obtained at the doses of medium and high doses were used to verify the predictability of the model Simcyp Version: 14.1 |
High Impact (support the clinical trial design and waiver for clinical DDI study) |
Probenecid from SimCYP inhibitor library refined to describe literature data obtained with the same administration schedule and simplified for absorption (ADAM‐>first order). Probenecid In vitro Ki optimized according to DDI result with S44121 |
64 |
| 19 |
Lilly X (phase II; discontinued) |
Renal transporters: OAT1 (basolateral uptake) OAT4 (apical efflux) |
Lilly X is a substrate of OAT1 Inhibitors used in modeling: Probenecid (in‐house model using clinical data and verified against clinical data and using baricitinib as substrate) Ibuprofen (in‐house model verified using pemetrexed). Diclofenac (in‐house model verified using published clinical data) |
Metabolite. Bottom‐up and middle‐out approach. First‐order absorption, full PBPK model, and renal transporters (OAT1 and OAT4). Simcyp Version: 16 V max and Km for OAT1 and OAT4 were used in the model for Drug X. HVs Simcyp Version: 16 |
No need of clinical study with ibuprofen. It showed successful IVIVE using in vitro IC50 values for renal OAT inhibitors |
Multiple clinical studies were used to test model performance. Probenecid predictions were used to verify the model. Bottom‐up model predicts CLR that is 1.5‐fold lower than observed CLR Middle‐out model recovers the clinical data using a scaling factor of 2 for OAT1 |
Not submitted |
| 20 | GSK‐Y (phase II) |
Hepatic transporters: OATP1B1 (sinusoidal uptake) inh BCRP (canalicular efflux) inh |
To investigate the clinical impact of hepatic OATP1B1/BCRP inhibition by GSK drug Regulatory query/internal assessment |
To investigate the clinical impact of hepatic OATP1B1/BCRP inhibition by GSK drug. Simcyp Version: 16 |
Simcyp simulations predicted no risk of DDI due to hepatic OATP1B1 or BCRP inhibition by GSK drug | Bespoke model built for GSK drug with a middle‐out approach | Simulation report used to address regulatory queries and no further investigation recommended |
| 21 | PF‐04991532 (phase II) |
Hepatic transporters: OATP1B (sinusoidal uptake) Renal transporters: OAT (basolateral uptake) BCRP (apical efflux) sub |
Victim DDI liability with OATP1B and BCRP inhibitors | Middle‐out PBPK model was developed with in vitro and preclinical data, then verified with SAD and MAD clinical data along with urine recovery. In vitro studies in cultured hepatocytes, Caco‐2 cells, and transfected OAT3 cells were used to parameterize transport processes | Used for design of clinical DDI study with cyclosporine. Model verified with the cyclosporine DDI data was applied to predict DDI liabilities with other perpetrator drugs | PBPK model was verified with SAD and MAD clinical data, and DDI data with OATP1B/BCRP inhibitor | Internal Impact and not submitted to health agencies |
| 22 | AZD‐Y (phase II) |
Hepatic transporters: OATP1B1 (sinusoidal uptake) sub |
PBPK based DDI prediction in lieu of clinical study |
Built‐in pravastatin model was used. FO and Full PBPK for AZD‐Y Simcyp Version: 15 |
Medium Impact: Virtual Clinical DDI Study enabled statin use during clinical trials and may help inform labelling |
In‐house PBPK model for AZD‐Y, verified with monotherapy clinical data. Simcyp built‐in model for pravastatin |
Scientific meeting with FDA. FDA agreed to include statins in a clinical study protocol |
| 23 | AZD‐X (phase I) | Intestinal transporter: P‐gp (apical efflux) sub |
Hypothesis for discrepancy: 1. Obs. CL or Vd ~2 fold higher than predicted 2. Lower Fabs due to P‐gp 3. Non‐CYP metabolic route/extra hepatic metabolism |
Bottom‐up approach including P‐gp kinetic parameters Simcyp Version: 15 |
Low Impact: (Go/no‐go decision) P‐gp Mediated Transporter‐Modeling for Understanding Absorption Results suggest that intestinal P‐gp do not play a role |
In‐house PBPK model for AZD‐X, verified with clinical SAD data | Not submitted |
| 24 | Gen‐X (phase I) |
Hepatic transporters: OATP1B1 (sinusoidal uptake) Inh OATP1B3 (sinusoidal uptake) inh OATP1b1/1B3 in vitro (IC50 ~ 0.3 and 0.9 uM, respectively). Clinical tDDI caused by Gen‐X OATP1B1/3 inhibition was predicted |
Prospective tDDI prediction for Gen‐X as a perpetrator to assess clinical risk and to support clinical development plan, including address potential regulatory questions |
Prospective tDDI prediction between (Gen‐X – pravastatin) PBPK model was built using middle‐out approach, and its PK prediction was verified using phase I clinical data. Absorption ‐1st order; Distribution –minimal PBPK + SAC; elimination – mainly enzymes (UGTs); In vitro OATP1B1 /1B3 inhibition data input into the model for tDDI prediction. HV population used for DDI simulation. The transporter DDI model was verified using known OATP1B1/1B3 inhibitors (rifampicin, cyclosporine, gemfibrozil) and substrate (pravastatin) prior to applying the model to compound X prediction. Pravastatin DDI caused by rifampicin and gemfibrozil OATP1B1/3 transporter inhibition was reasonably captured by the existing model, whereas DDI between cyclosporine and pravastatin in HV was underpredicted (> 2‐fold). Simcyp Version: 14/15 |
Medium Impact DDI prediction between (Gen‐X – pravastatin provided risk assessment to clinical team and assisted clinical development plan and tDDi study design |
For Gen x –phase I clinical PK used for model refinement and verification For pravastatin, Simcyp V15 default compound file was used (no modification), a few modifications were made for rifampicin, gemfibrozil and cyclosporine. Predicted C max ratio 1.01–2.05; predicted AUC ratio 1.04–2.23, depending on the f u,inc used to correct for inhibition Ki |
Not submitted to agency |
| 25 | JNJ‐001 |
Intestinal transporter: P‐gap (apical efflux) inhibition Renal transporter: OCT2 (basolateral uptake) MATE (apical efflux) inhibition |
Does inhibition for these intestinal or kidney transporters translate to clinical DDI liability |
JNJ‐001 PBPK model built to simulate plasma PK, intestinal and kidney PK. JNJ001 PBPK model built and verified with clinical PK data Simcyp Version: 16 |
High Impact No DDI studies planned with these intestinal and kidney transport substrates |
JNJ‐001 PBPK model built and verified with clinical PK data no DDI is expected with OCT2/MATE substrates (e.g., metformin). With P‐gp substrates (e.g., digoxin) increase in exposure simulated, which resulted in proposal to dose stagger JNJ‐001 and P‐gp substrates |
NA |
ADAM, advanced dissolution, absorption, and metabolism; ASA, automated sensitivity analysis; AUC, area under the curve; BCRP, breast cancer resistance protein; BSA, body surface area; CL, clearance; Cmax, maximal plasma concentration; DDI, drug–drug interaction; EMA, European Medicines Agency; FDA, US Food and Drug Administration; FIH, first in human; GI, gastrointestinal; GLE, glecaprevir; GSK, GlaxoSmithKlein; HEK, human embryonic kidney; HSA, human serum albumin; HSCT, hematopoietic stem‐cell transplant; HV, healthy volunteer; IC50, half‐maximal inhibitory concentration; IVIVE, in vitro‐in vivo extrapolation; MAD, multiple ascending dose; MATE, multidrug and toxin extrusion; NSAIDs, nonsteroidal anti‐inflammatory drugs; OATP, organic anion‐transporting polypeptide; OCT, organic cation transporter; PBPK, physiologically‐based pharmacokinetic; P‐gp, P‐glycoprotein; PK, pharmacokinetic; PMDA, Pharmaceuticals and Medical Devices Agency; RAF, relative activity factor; SAD, single ascending dose; tDDI, transporter‐mediated drug–drug interaction; Vmax, maximal rate of velocity; Vss, volume of distribution at steady state.
Impact classification: High impact: Replace; provides inference that informs internal decisions without requiring a clinical study; Medium impact: Inform; provides inference that informs internal decisions; and Low impact: Describe; Provides inference that has limited impact on internal decisions.
Figure 1.
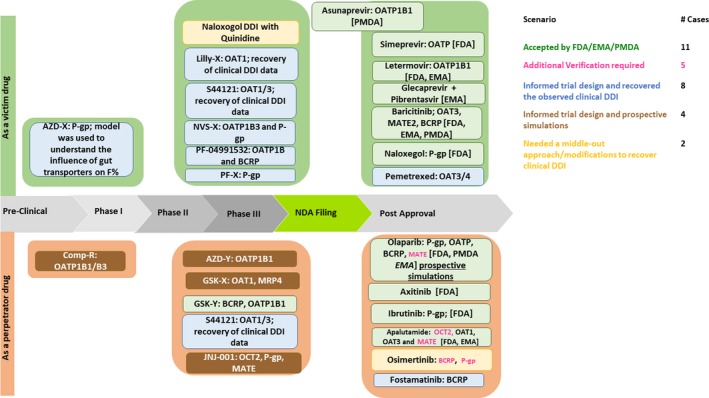
Summary of examples of transporter‐mediated drug–drug interaction (DDI) physiologically‐based pharmacokinetic analyses and their impact on drug development stages including regulatory decision outcomes. BCRP, breast cancer resistance protein; EMA, European Medicines Agency; FDA, US Food and Drug Administration; MATE, multidrug and toxin extrusion; OAT, organic anion transporter; OATP, organic anion‐transporting polypeptide; P‐gp, P‐glycoprotein; PMDA, Pharmaceuticals and Medical Devices Agency.
tDDI‐related cases accepted by regulatory agencies
In our dataset, six submissions were accepted by the FDA, two examples were accepted by the PMDA, one PBPK example was accepted by the EMA, and both the PMDA and the EMA did not comment on one each of the submissions. Six of the nine drugs with regulatory approval were related to P‐gp substrates or inhibition. The remaining accepted tDDI predictions included OAT3, BCRP, OATPs, and MATE2 either as substrates or inhibitors. So far, there are, to our knowledge, no cases of transporter PBPK induction that was a part of regulatory submission, although semimechanistic models have been successfully described in the literature.33 Most recently, a PBPK model of rifampicin for predicting interactions with drugs and an endogenous biomarker via complex mechanisms, including OATP1B induction, has been reported.53
Olaparib–inhibitor–renal, intestinal, and hepatic
Olaparib is a potent poly ADP ribose polymerase inhibitor that has demonstrated antitumor activity in patients with ovarian and breast cancer.54 In vitro, olaparib has been shown to be an inhibitor of P‐gp, BCRP, OATP1B1, OCT1, OCT2, OAT3, MATE1, and MATE2K, but not an inhibitor of OATP1B1/3, OAT1, or MRP2. The olaparib PBPK model was robustly defined to support the CYP3A4 mediated as well as tDDI predictions (specifically P‐gp).55 To address regulatory questions, the sponsor simulated the DDI of olaparib with substrates of P‐gp, OATP1B1, MATE1, OCT1, and BCRP transporters. PBPK modeling revealed no interaction (AUC‐ratio within 80–125%) for most cases except a weak interaction (AUC‐ratio 125–200%) with OATP1B1 (pravastatin). As part of the olaparib tablet formulation submission to the PMDA, the sponsor and PMDA discussed the rationale of suitable probe substrates and the necessity of conducting clinical studies for all involved transporters. PBPK analyses were conducted along with systematic sensitivity analysis by increasing and using a range of inhibitory potencies of olaparib against OATP1B1, OCT1, and MATE1. The OATP1B1 IC50 values, determined by using two different substrates (pravastatin/estradiol‐17β‐glucuronide), were similar, which gave confidence in the robustness of the in vitro values to be used in the PBPK model for OATP1B1.
Uncertainty in the measured IC50 values was investigated via sensitivity analyses by lowering Ki values (where Ki = IC50/2) of olaparib against P‐gp, OATP1B1, BCRP, OCT1, and MATE1 by 10‐fold to 100‐fold to quantitatively assess the impact of uncertainty associated with the inhibition parameter on an outcome variable, such as clearance or AUC. Such hypothetical Ki values suggested a clinically insignificant change in the exposure of substrates of the above‐mentioned transporters in the presence of the proposed olaparib therapeutic dose of 300 mg twice daily. With respect to OATP1B1 DDI prediction, AUC ratios were estimated to be 2‐fold and 3‐fold higher than the baseline model with 1/10‐fold and 1/100‐fold of In vitro Ki values, respectively. The PMDA accepted the PBPK simulations that showed a lack of clinical impact.
Prospective P‐gp mediated DDI simulation results (olaparib as perpetrator) were submitted to both the EMA and the FDA. PBPK simulations suggested no DDI and was accepted. It was evident from this example that a detailed verification with known probes and inhibitors along with the sensitivity analyses was essential for the acceptance by regulatory authorities for the prospective DDI simulations of a P‐gp, OATP1B1, MATE1, and OCT1 inhibitor.
Simeprevir–hepatic uptake substrate
Simeprevir is a protease inhibitor indicated for the treatment of chronic hepatitis C virus infection.56 In vitro data suggested simeprevir is a substrate for CYP3A4 and OATPs, hence, the mechanistic understanding of the interplay between these proteins is reflected in the PBPK model. Nonlinear PK was observed for this drug when dosed to steady‐state due to saturation of both CYP3A and OATPs. The verified PBPK model was used by the sponsor to understand whether CYP3A4 and/or OATP1B1/3 contributed to the observed nonlinear PK of simeprevir, the higher plasma exposure in Asian subjects vs. white subjects, to understand the mechanism for the higher exposure in patients with hepatitis C virus and to simulate the pharmacodynamic (PD) target organ exposure in various populations of interest.57 The described PBPK model for simeprevir was included in the FDA/EMA regulatory submissions and both agencies referred to the PBPK model in their Public Assessment new drug application (NDA) Reports.
Letermovir–hepatic uptake substrate
Letermovir is a marketed drug for the treatment of infection caused by human cytomegalovirus virus, which showed nonlinear human PK. Letermovir is a substrate of OATP1B1/1B3 and is metabolized by UGT1A1, UGT1A3, and CYP3A. A PBPK model suggested that nonlinear PK behavior was well described by the saturation of hepatic OATP1B‐mediated uptake. The PBPK model supported the hypothesis that the higher exposure observed in Japanese as compared to white healthy volunteers is likely due to differences in hepatic uptake transporter OATP1B activities as reported for other compounds58 in addition to known demographic and physiological differences between these two populations, such as body weight and consequently liver mass. In contrast to the Chinese patients, the liver weight is the same correlated to weight as white patients, however, as the body surface area of Japanese is an average smaller, the liver mass is smaller. This work was accepted by the FDA and details of this PBPK modeling are captured under the FDA's clinical pharmacology and biopharmaceutics review. In contrast, the same PBPK model was submitted to the EMA, but is currently not part of the European label.
Baricitinib–renal substrate
Baricitinib, an oral selective Janus kinase 1/2 inhibitor currently approved for the treatment of rheumatoid arthritis,30 is predominantly renally cleared and a substrate for OAT3, MATE2, P‐gp, and BCRP transporters. With PBPK modeling, the sponsor was interested to address if the exposure of baricitinib was influenced by inhibitors of the renal transporter OAT3.30 This model was built to understand only the OAT3‐mediated interactions. The model was verified with clinical DDI data of probenecid with baricitinib followed by a prospective DDI assessment with standard of care medicines, such as diclofenac and ibuprofen, were modeled. Results suggested no clinically relevant DDIs with ibuprofen and diclofenac driven by renal transporters. These results were accepted by the FDA and the PMDA.
Osimertinib–P‐gp and BCRP‐inhibitor
Osimertinib is an oral, irreversible EGFRm inhibitor used for treating patients with non‐small cell lung cancer. In vitro data suggests it inhibits BCRP (IC50 = 2 µM using Caco‐2 assay). The PBPK simulator was used prospectively to simulate the DDIs of osimertinib as a perpetrator drug with the BCRP substrate rosuvastatin library compound file without any modification. This was performed before the sponsor conducted a clinical study to understand the DDI risk when dosed with BCRP substrate, such as rosuvastatin, to adjust dosing if necessary.59 PBPK simulations suggested no DDI risk, but clinical study showed an AUC ratio change of 1.35‐fold and C max ratio change of 1.72‐fold. These simulation results were not used to inform the drug label as the clinical data was used for that purpose. Further post hoc analyses suggested that a fit‐for‐purpose tDDI model with Ki needed to be reduced by ×10‐fold (based on sensitivity analysis of observed DDI data). With these changes the sponsor could recover the clinical DDI data. On the contrary, for fostamatinib, a BCRP Ki value of 0.03 µM determined by vesicle assay60 could recover the DDI data without needing any adjustment for potency.61 A mechanistic model of fostamatinib with rosuvastatin recovers the clinical DDI data well, whereas a fit‐for purpose model was needed to recover the clinical DDI of osimertinib with rosuvastatin. As Pan et al.62 pointed out, one should run a sensitivity analysis or parameter estimation approach if clinical data available, or else it has been encouraged to use an uncertainty analysis to reflect the uncertainty in the measured Ki value, which could arise from several sources like assay method, assay conditions, laboratory‐to‐laboratory variations, etc.
Naloxegol–intestinal P‐gp substrate
A few examples that really helped understand the mechanism of drug absorption and disposition included Naloxegol, a P‐gp substrate, which poses intestinal absorption limitations and victim DDI liability with P‐gp inhibitors. Because of naloxegol in vitro, clinical DDI, and PBPK package, the FDA requested a few mechanistic questions such as: (i) Naloxegol is cleared predominately via CYP3A, what is the contribution of P‐gp to the biliary secretion of Naloxegol; (ii) some CYP3A modulators are known to affect P‐gp. Therefore, a full PBPK model accounting for P‐gp contribution should be developed for Naloxegol. PBPK modeling helped in answering these questions, but a PBPK model modification was required to recover the tDDI vs. CYP‐mediated DDI as the driving concentrations at the site of interaction are different when ADAM and full PBPK were used (Table S2 and Table S3 ). More details of questions and modeling approach taken can be found in Zhou et al.63
Based on the examples authors have collected so far (Table 3), it suggests that recovery of DDI relating to OAT3 with PBPK modeling using measured inhibition constant values is possible with confidence (as verified with clinical data of S44121,64 baricitinib,30 and Pemetrexed31, 62).
Table S4 shows the predictability of tDDI (i.e., the ratio of PBPK‐predicted and observed effect) where clinical data are available. To gain the broader acceptance across agencies, we have identified in this section several successful PBPK model examples for intestinal, hepatic, and renal transporters; however, in this relative new field of PBPK modeling, there are still gaps in predicting tDDIs and those are described in detail in the following section. Understanding and closing these gaps in our recent knowledge would allow sponsors to generate the required in vitro data for a more mechanistic understanding and consequently enable higher regulatory acceptance.
Gap Analysis And Fit For Purpose vs. Mechanistic PBPK Models
There has been considerable research effort in the area of transporters over the last decade in order to understand and build more robust IVIVE for transporters; however, some system information is still lacking or the activity of the transporter cannot be scaled via its expression over a wide range of abundance, which might be a factor for solute carriers and would explain the disconnect via in vitro (transfected cells with high abundance) and in vivo (even under induced conditions not as highly expressed as in vitro). Some specific examples focussing on the gaps in system data are shown in Table 4. In addition, mismatch between observed tDDI vs. simulated tDDI ratios could arise from in vitro experimental factors, as highlighted in the subsequent section under “Challenges and Opportunities” (Table 5).
Table 4.
Gaps in system data
| Parameter | Problem/open questions | Current solution | Future |
|---|---|---|---|
| Protein abundance used as surrogate for the transporter activity in vivo. |
Abundance data are not available for all transporters. Abundance data are not always a good representative of the in vivo activity. Is this correlation (abundance/activity) true for in vitro and in vivo, e.g., BCRP over the whole abundance range? Why are there differences between Caucasian and Japanese abundance/activity relationships (OATP1B1)? Has this been shown for other proteins? Is there a disease effect in activity of the transporter? |
A relative abundance approach is currently used for most published PBPK models accounting for transporters.
|
Correlations of protein abundance vs. activity over a relevant in vivo and in vitro protein concentration for drug molecules should be verified. Newly established transporter in vitro assays should aim for a comparable abundance of the transporter to in vivo and not a significant overexpression. |
| Localization of transporters |
Membrane and cell expression of transporter Abundance of transporter along the gut Abundance of transporter along the nephron Abundance of transporters within the liver |
The intestinal P‐gp is currently accounted for in all models as apical efflux transporter; however, recent staining data indicate that P‐gp is also expressed in the lateral membrane of the intestinal enterocytes. The function of the transporter in the lateral membrane has not been investigated so far. Localization of OATP2B1 has yet to be verified |
In vitro assays need to mimic the relevant localization of the transporter in vivo. Mechanisms explaining the alteration in localization of transporters (e.g., under disease conditions) need to be further investigated. |
| Age as covariate, ontogeny of transporters | Age (ontogeny profiles for several transporters under development, but not established with any convincing certainty; OCT1, P‐gp, and BCRP available; hepatic expressions and activity OATP1B data are contradictory) | Middle‐out approach for OCT1 has been shown successful.103 | Further research is required, but it needs to be shown that abundance data (mRNA, protein (Western blot, LC‐MS/MS)) is representative of the activity of that transporter. Activity data should be reported relative to the adult value. |
| Sex as covariate | Sex difference on mRNA and protein levels has been shown for rodents, but not that established in humans. | All transporters are currently handled as not sex‐specific in their abundance and/or activity. | Correlation matrixes in larger databases or from combining databases may help to find gender as covariate for human transporter. |
| Intestinal parameters |
|
|
Organ scalar comparable to those established for the liver (e.g., HPGL and MPPGL) need to be determined. |
| Renal parameters |
|
|
|
| Hepatic parameters |
|
|
|
| Brain parameter | Until recently, no reliable data for healthy white patients on brain transporter abundance and/or organ scalars like micro vessels per gram of brain was available106 | A relative scaling can be used (based on absolute abundance data, if available) and an overall empirical scalar can be set for the blood‐brain‐barrier and the blood‐CSF‐barrier transporter. | More data on the regional distribution (membrane expression as well as CSF expression) of brain transporter and on disease differences are required. |
BCRP, breast cancer resistance protein; CLPD, passive diffusion clearance; CsA, cyclosporine; CSF, cerebrospinal fluid; EGD, electrochemical gradient driven; LC‐MS/MS, liquid‐chromatography tandem mass spectrometry; mRNA, messenger RNA; OATP, organic anion‐transporting polypeptide; OCT, organic cation transporter; PBPK, physiologically‐based pharmacokinetic; P‐gp, P‐glycoprotein; SCHH, sandwich‐cultured human hepatocyte; SIVA, Simcyp in vitro data analysing tool kit.
Table 5.
Major challenges and areas of opportunities in supporting PBPK model development and verification involving drug transporters
|
In vitro methodologies:
|
|
In vivo methodologies:
|
|
Mechanistic studies:
|
|
In silico methodologies:
|
BCRP, breast cancer resistance protein; CLPD, passive diffusion clearance; DDIs, drug–drug interactions; EGD, electrochemical gradient driven; IC50, half‐maximal inhibitory concentration; IVIVE, in vitro‐in vivo extrapolation; Jmax, maximum flux; MATE, multidrug and toxin extrusion; OATP, organic anion‐transporting polypeptide; OCT, organic cation transporter; PBPK, physiologically‐based pharmacokinetic; PD, pharmacodynamic; PET, positron emission tomography; P‐gp, P‐glycoprotein.
For transporter substrates, PK predictions followed by DDI assessments based on bottom‐up approach are challenging due to insufficient predictability of CL i.v. (CLoral) and volume of distribution at steady state (V ss). The volume of distribution can be affected by active transport and V ss predictions, which assume perfusion‐limited behavior of the compounds, are, therefore, less reliable if transporters are involved. In addition, for hepatic transported compounds (with active transport > passive) with relative low clearance, i.v. PK data can still be misleading in deriving the Vss with traditional PK compartmental analysis (an underprediction of the real Vss with order of magnitude is possible).65
The middle‐out approach has been applicable showing the potential use of PBPK models for prediction of transporter DDI effects at several locations (e.g., intestine and liver). As an example, using i.v. and oral PK data as reference, P‐gp inhibition effects on the digoxin PK were well predicted. The magnitude of the DDI effects on the intestinal first pass could be separately evaluated from that on systemic clearance. The clearance value was calculated based on the clinical study results.33
For mechanistic prediction of CLH values of transporter substrates with PBPK modeling, adjustment of hepatobiliary and uptake clearance from SCHH was previously reported to capture the reference clinical PK data after i.v. administration, using empirical scaling factors (an average of 6 compounds: 0.061 and 58, respectively).24, 66 This could result in overestimation and under‐estimation of hepatobiliary and uptake clearance extrapolated using the in vitro data, respectively. Overestimation of hepatobiliary clearance is likely due to less reliable prediction of unbound hepatocellular concentrations. In addition, an acceleration of hepatic active uptake mediated by formation of a drug‐albumin complex was recently demonstrated.67 Similarly, CLr changes due to DDI effects on renal transporters may not be adequately achieved based on the bottom‐up approach.
For Vss prediction, Kp, scalar values for transporter substrates were potentially required to recover the measured Vss values (Table S5 ). This indicated that predictability of tissue distribution may be affected by active transport, besides physicochemical properties of a drug. This indicates difficulty with understanding DDI mechanism as, for example, Cmax and AUC increase of a victim drug due to decrease of Vss and/or increase of Fa.
In this regard, evaluation of the changes of i.v. and oral PK data (CLi.v., CLoral, Cmax, Vss, and F) in the absence and presence of perpetrator effects is strongly recommended. The data can be used as reference for model building, as exemplified by Bosgra et al.,68 Watanabe et al.,69 and others.68, 69 In Table S1 , i.v. and oral PK parameters for probe transporter substrates according to the FDA (2017) are summarized, which can be used for establishment and/or further refinement of PBPK models, potentially including nonlinear changes of Cmax and AUC values at a range of doses, irrespective of model building approaches.
Top‐down/bottom‐up/middle‐out approaches to recover clinical data when transporters are involved—current trends and any other better alternatives
Approaches to building PBPK models have been discussed in detail by Shebley et al.20 Briefly, the bottom‐up approach in large, provides the mechanistic understanding based on a in vitro data of the transporter of interest. However, this approach heavily depends on the quality of in vitro data generated, as well as the understanding of IVIVE data translation. Fit‐for‐purpose (top‐down or middle‐out) modeling approaches to support the clinical trial decisions by fitting to existing clinical data may require minimal in vitro and mechanistic knowledge compared to the bottom‐up approach (Table S6 ). There are many examples in the literature that suggest that the bottom‐up approach was successful in recovering the clinical tDDI (e.g., renal transporters involving OAT1, OAT3, and OAT4; Table 3 and Table S6 ), whereas fit‐for‐purpose modeling is needed to capture PK and tDDI data for intestinal and liver transporters (BCRP or OATP1B1‐mediated interactions). For a new chemical entity as transporter inhibitor (perpetrator), it is common that lower Ki values than measure need to be used to recover clinically observed tDDI. Thus, understanding perpetrator IC50 estimates seem to be a key factor for some of the examples that were discussed within this paper.
Challenges And Opportunities
Different methods and approaches are available for evaluating transporter kinetics in vitro. These include use of isolated primary cell cultures, transfected membrane vesicles, and cell lines as well as different culture and experimental conditions. Each approach has its advantages and limitations that need careful evaluation. For example, the use of transporter‐transfected cell lines requires inclusion of parental or “vector control” cells to consider endogenous transport activity background noise in the data, like endogenous transport activity and passive permeation. Primary cell systems (e.g., hepatocytes) undergo loss of transporter expression and/or activity on isolation, cryopreservation, and time in culture (e.g., ref. 70). In addition to these system‐dependent limitations, general factors, such as nonspecific binding to plastic ware and chemical instability in assay medium, may also be apparent. Without addressing these caveats, the quality of in vitro data may not be of the required standard. This might partly explain the issue with assessment of P‐gp inhibition, where > 700‐fold variability in apparent IC50 data between laboratories using comparable methods was reported.71 Such variability affects the validity of static model in IVIVE for digoxin‐related DDIs.72 The poor IVIVE for transport inhibition is also apparent when using dynamic PBPK modeling for OATP‐related DDIs.73 Furthermore, direct scaling of uptake clearance using primary hepatocytes to predict in vivo plasma clearance also typically results in several‐fold underprediction,24 which is not entirely explained by loss of transporter expression.74 Use of OATP transporter kinetic data generated using plateable human hepatocytes with human plasma showed improved in vitro‐to‐in vivo translation for human PK prediction,75 more substrates need to be tested using this approach.
The poor IVIVE for transport‐related clearance and DDI prediction may be addressed by ensuring that each in vitro system responds appropriately to test substrates and inhibitors. This requires addressing each experimental caveat, selection of optimum conditions, and careful method validation with appropriate reference substrates and inhibitors. One strategy to overcome these limitations is the modeling of the in vitro data,76, 77 where free concentrations and rate limiting mechanisms like an unstirred boundary layer can be accounted for and the kinetic data for the transporters are corrected for the bias of the in vitro system and the limitations of the experimental conditions (e.g., stirring rate, sampling time intervals, and volumes). These models allow the simultaneous and dynamic evaluation of different processes (active transport, metabolism, passive diffusion, and binding) and changes in drug concentration vs. time in extracellular and intracellular compartments. Because serial time and concentration points can be fitted simultaneously, relevant parameters can be estimated more robustly.
It is generally acknowledged, when determining intrinsic clearance via transporters from in vitro systems, that it is the free fraction of drug within the incubation that provides the relevant drug concentration.78 This gives a correction factor for the nominal concentration and sometimes losses due to binding to plastic or the material are relevant (e.g., felodipine and loperamide). Not including fuinc for the victim/perpetrator may sometimes lead to underprediction or overprediction of kinetic parameters.
Significant efforts are ongoing in searching for specific endogenous biomarkers for drug transporters that may aid DDI risk assessment during drug development.79 Biomarkers for transporter function, such as coproporphyrins I and III (CP‐I/III), tetradecanedioate, or glycochenodeoxycholate sulphate for hepatic uptake via OATP1B or N1‐methylnicotinamide for MATE–‐mediated renal secretion have been recently reviewed.79 Mechanistic modeling has been used to explore the predictive value of the biomarkers in the clinic.80 Recently, PBPK models explored utility of endogenous coproporphyrin‐I as an OATP1B biomarker involving inhibition of hepatic OATP1B1/OATP1B3.81 Its utility needs to be explored with further clinical estimations and IVIVC for appropriate validation.
Transporter‐mediated DDI can occur at tissue level that is not reflected from plasma exposure changes. In addition to positron emission tomography (PET) imaging studies, PDs, or clinical end points may be useful as surrogate markers for tissue‐level DDIs. For example, the hepatic uptake of metformin is mediated by OCT1. Because renal elimination is the main elimination pathway for metformin, DDI at hepatic OCT1 does not affect systemic exposures of metformin. However, changes of metformin concentrations in the liver due to hepatic OCT1 may affect the antihyperglycemic effect as liver is the main site of the PD by metformin.82
Among the mechanistic studies allowing assessment of DDI, cocktail studies have been widely used in recent years to screen out investigational drugs that are inhibitors of particular CYP enzymes; however, this approach has not been evaluated sufficiently yet for transporters. A valid cocktail requires lack of interaction between the probe substrates. Recently, the possibility of developing a cocktail‐based approach for the transporter was explored and investigated with four probe drugs, digoxin (P‐gp), furosemide (OAT1 and OAT3), metformin (OCT2, MATE1, and MATE2‐K), and rosuvastatin (OATP1B1/3 and BCRP).83 The first clinical study84 showed that after coadministration of the four substrate drugs, furosemide Cmax was 19% lower and rosuvastatin Cmax and AUC were 39% and 43% higher, respectively, compared with when administered alone. A follow‐up clinical study85 showed that reduction of metformin and furosemide could eliminate the interaction with rosuvastatin based on plasma data. However, all four compounds are predominantly renally eliminated, and the lack on interaction of changing Ae, CLR, or on PD has not been shown yet. As rosuvastatin is interacting also with renal OATs, maybe this compound should be replaced by another statin like pitavastatin. In addition, furosemide is a loop‐diuretic and, hence, from its mechanism of action furosemide is expected to interact with additional transporters in the kidneys. Hence, another OAT1/3 probe may be more reliable (Figure 3 b). Recently, a substrate cocktail consisting of pitavastatin (OATP1B), rosuvastatin (OATP1B/BCRP/OAT3), sulfasalazine (BCRP), and talinolol (P‐gp) was evaluated in cynomolgus monkey.86
Figure 3.
(a) Matrix approach for hepatic organic anion‐transporting polypeptides (OATPs). Three key OATPs, OATP1B1, OATP1B3, and OATP2B1, are expressed within the healthy liver of adults. Because there is a significant overlap in probe selectivity for OATPs and highly potent and selective probes are currently unknown for any specific OATP, a matrix approach is proposed. For the compounds in the box to the left, physiologically‐based pharmacokinetic (PBPK) models have been published for these substrates of OATPs, but all models are fit‐for‐purpose models using a lumped clearance for OATPs. For more mechanistic understanding of hepatic OATPs, kinetics for the transporters, and it should be included into the model as done for the compounds listed as “substrates.” The dotted line indicates that the clinical drug‐drug interaction (DDI) is not available for verification. (b) Matrix approach for renal organic anion transporters (OATs). Three OATs, OAT1, OAT3, and OAT4, are expressed within the healthy kidney of adults. Because there is a significant overlap in probe selectivity for OATs and highly potent and selective probes are currently unknown for any specific OAT, a matrix approach is proposed. For the compounds in the box to the left, PBPK models have been published for these substrates of OATs, but all models are fit‐for‐purpose models using a lumped clearance for OATs. For more mechanistic understanding of renal transporter‐mediated DDIs, kinetics for the transporters, and it should be included into the model as done for the compounds listed as “substrates." The dotted line indicates that the clinical DDI is not available for verification. BCRP, breast cancer resistance protein; MATE, multidrug and toxin extrusion; P‐gp, P‐glycoprotein. [Colour figure can be viewed at http://www.wileyonlinelibrary.com]
Recommendations
Schematics of strategy recommendation for using a PBPK model to address tDDI are shown Figure 2. Figure 2 a depicts the recommendations to build and verify a victim drug PBPK model, whereas Figure 2 b depicts the recommendations to build and verify perpetrator drug PBPK models.
Figure 2.
Schematics (workflow diagram) of strategy recommendation for using a physiologically‐based pharmacokinetic (PBPK) model (general) to address transporter‐mediated drug–drug interactions (DDIs). (a) Recommendations to build and verify a victim drug PBPK model. (b) Recommendations to build and verify a perpetrator drug PBPK model. AUC, area under the curve; CL, clearance; Cmax, peak plasma concentration; FIH, first‐in‐human; MAD, multiple‐ascending dose; PK, pharmacokinetic; SAD, single‐ascending dose.
Sensitivity analysis should account for different concentrations that a transporter will be exposed to in the intestine compared with the liver or kidneys. The boundaries used for the sensitivity analysis, therefore, depend on: (i) the location and function of the transporter (e.g., electrochemical gradient driven OCT2, pH‐gradient‐dependent MATE, and 2‐binding sites for OATPs), (ii) the quality of the IC50 values (preincubation, Km of the probe substrate), and (iii) the contribution of other transporters at that membrane also in relation to the passive flux across that membrane.
For instance, the apical efflux transporters, P‐gp, BCRP, and MRP2, are often colocalized and there are many compounds that are substrates for P‐gp as well as BCRP; however, both transporters require that the compound reaches the inner bilayer of the cell, so either the passive permeation of the compound needs to be enough or an active uptake into the cell is required. This interplay of uptake and efflux or between two efflux transporters along the gut needs to be understood to better estimate regional fractions of absorption. Although the overall fraction absorbed may not be different the regional fraction absorbed can be altered87 and this may be in the future a way to explain side effects along the gastrointestinal tract.
Currently, most PBPK models for transporters are built for fit‐for‐purpose and, hence, the hepatic uptake is lumped and assigned to the transporter of most relevance (i.e., often OATP1B1 as that can be “verified” by genotype/phenotype PK data), with the aim to mimic the worst case of a scenario. However, the more we learn about the interplay of enzymes and transporters within a cell or organ, but even more relevant between organs, the more we understand that oversimplified PBPK model and R‐value predictions are not capable of recovering interactions that are driven by several transporters/enzymes within different organs (e.g., inappropriate predictions for itraconazole for transporter compounds). Although the fit‐for‐purpose models are a good starting point to move forward with characterized ft, the luck of specific substrates and inhibitors only allows for limited verification of a single compound. A matrix approach is, therefore, the most likely success; however, it is unrealistic to aim for solving this “puzzle” alone, hence a combined effort is needed. Figure 3 a describes matrix approach for hepatic transporters, such as OATP1B1/1B3, whereas Figure 3 b describes matrix approaches for renal OATs.
Use of prospective PBPK modeling to assist clinical study design
Clinical DDI study design is based on the substrate‐inhibitor pair being tested. A new molecular entity (NME) may be tested as an inhibitor or a substrate of transporter/s. In cases where a tDDI is expected based on the NME absorption, distribution, metabolism, and excretion profile, population‐based PBPK models can be used for prospective predictions of interactions and a suitable design of such clinical tDDIs with either a clinically relevant substrate or inhibitor of a transporter depending on the transporter liability of the NME.
When a DDI study is being designed with the NME being an inhibitor of a transporter, the clinical DDI study is designed in such a way to achieve the highest possible interaction at the clinically relevant dose of the NME and using a dosage regimen equivalent with achieving the desired therapeutic exposure at a steady state. A clinically verified NME PBPK model enables predicting such exposure levels for the NME while incorporating the population‐based variability and, hence, giving an idea of the expected variation in the baseline exposure levels. This prospective exercise would not only help in selecting an appropriate dose range but may also help justify the selection of a suitable population recruitment based on the variability predicted from a verified PBPK model for the intended purpose.45 The PBPK model may also enable help in determining whether the clinical study should consider any prospective or retrospective genotyping of the transporter to avoid any bias in the expected DDI observations. Usually, a population with a null activity or less activity genotype should be avoided to account for the highest possible clinical inhibition of the transporter by the NME or a known inhibitor. Moreover, some NMEs or known inhibitors may show accumulation over a certain dosing regimen and, hence, a prospective PBPK simulation would enable determining a suitable dosing day for the transporter substrate being tested. Such predicted population‐based DDI also helps in deciding the dose of the NME tested as a transporter substrate or of a known clinically relevant transporter substrate. For example, when a DDI study is being designed to test the NME as an OATP1B1 inhibitor and the PBPK model may help in deciding the dose of a suitable statin tested as an OATP1B1 substrate as there is a range of commercially available statin formulations. A PBPK model further enables to design a suitable crossover study when more than one mechanism of DDIs are being tested. The tDDIs are commonly reversible interactions and usually a single dose of the transporter substrate (either known clinically relevant or NME) may be enough to test the DDI. Sometimes based on the PK profile of the known inhibitor or the NME, it may be required to dose the transporter substrate at two or more days at appropriate intervals (e.g., day 5 vs. day 11) and a PBPK model can help in designing such complex DDI study designs as well.
Future perspectives
One of the future perspectives for the utility of PBPK modeling is to estimate drug concentrations in tissues and accurate DDI predictions based on intracellular drug concentrations and not only systemic circulation. To date, it was demonstrated that transporter‐related DDIs or drug‐endogenous substance interactions could cause severe side effects in specific tissues, such as liver, kidneys, and central nervous system. For proper dose adjustments and deciding dosage regimens it is useful to understand the correlation among the PKs, efficacy, and toxicity for which the PBPK models can be an efficient prediction tool. Rose et al. have demonstrated the use of a PBPK model to understand the correlation between statin efficacy and OATP polymorphisms using rosuvastatin PBPK model and mevalonic acid as an efficacy marker.88
In liver toxicity, drug‐induced liver injury is key issue for drug candidates in preclinical and clinical stages. The association of elevated bile acid concentrations in liver with drugs, which could cause hepatotoxicity, led to the hypothesis that hepatotoxic drugs, including cyclosporine, bosentan, and troglitazone, inhibit hepatic bile acid transport by the bile salt efflux pump may be one mechanism of cholestasis and drug‐induced liver injury.89 Statin‐induced myopathy and rhabdomyolysis are also well known as the adverse event caused by transporter‐related DDI. Statins are typical substrates for OATP1B1/OATP1B3 and have potency for myopathy and rhabdomyolysis as a side effect. OATP inhibitors, such as cyclosporine, clarithromycin, and HIV protease inhibitors, increase systemic exposure of statins. The DDI could cause myopathy and rhabdomyolysis due to high accumulation of statins in muscle. One typical example for statin‐induced rhabdomyolysis is caused by the tDDI between cerivastatin and gemfibrozil.
In kidneys, MATEs are major contributors to secrete cation drugs from kidney proximal tubule cells into urine. Inhibition of MATEs could cause the high accumulation of drugs, MATEs substrates in renal tubule cells leading to nephrotoxicity. Cisplatin‐induced nephrotoxicity is one of the representative cases. Cisplatin is an anticancer platinum agent and secreted by OCT2 and MATEs at basolateral and apical membrane, respectively.90 It was reported that the renal accumulation and toxicity of cisplatin were caused in MATE1 knockout mice and the plasma concentration of pyrimethamine, a specific inhibitor of MATEs is sufficient to inhibit MATEs‐mediated efflux in humans,91 suggesting functional impairment of MATEs by pyrimethamine could cause cisplatin‐induced nephrotoxicity in humans.
In the brain, it was demonstrated that loperamide, an antidiarrheal agent and P‐gp substrate, caused respiratory depression after coadministration of quinidine. The side effect could appear to be because brain accumulation of loperamide would be increased by P‐gp inhibition of quinidine. When there was substantial impairment of ventilatory response to CO2 after quinidine combination, the plasma loperamide concentrations were identical between alone and combination with quinidine, suggesting that loperamide in the presence of quinidine produced respiratory depression due to high brain concentration of loperamide independent of changes in plasma concentrations.
Generally, human PK profiles and DDIs are discussed based on drug concentration in plasma and/or urine, because blood and urine collection are easy and less invasive procedures. However, PET imaging technology by which tissue accumulation of radiolabeled transporter substrates is detectable could fill the current gap for accurate DDI prediction in tissues and help identification of rate‐determining process in the elimination.57, 92 Ørntoft et al. 93 demonstrated that the use of bile acid tracer [11C]‐cholylsarcosine as an example for an imaging marker provided a proof‐of‐concept for the feasibility of visualizing transporter functions involved in bile formation in humans. When P‐gp function is inhibited at the human blood‐brain barrier, the brain concentration of [11C]‐loperamide, a P‐gp substrate, almost doubles, but the plasma concentration profile of [11C]‐loperamide is not affected by quinidine.94 In the kidneys, [11C]‐metformin was established to evaluate intracellular accumulation of MATEs substrate and the accumulation of [11C]‐metformin was increased with pyrimethamine, suggesting that [11C]‐metformin is useful to understand drug accumulation in proximal tubule cells by MATEs‐related DDIs.95
Another important aspect to understand transporter‐related DDIs in tissues is the development of detailed PBPK models, including drug disposition in tissues. Such models require tissue kinetics information of PET probes, which are transporter substrates, estimation of intracellular drug free concentration, and accurate knowledge of the protein abundance of targeted transporters in humans. The combined efforts using novel substrates for in vivo visualization of transporter functions and quantification of transporter protein abundance will achieve to a deep understanding of transporter‐related interaction for drug and endogenous substance accumulation in tissues and allow development of novel PBPK models, which permit quantitative prediction of drug concentration and liability for DDI‐related toxicity in tissues.
As a part of knowledge‐sharing and further advancing this field, after successful submissions and/or publications in peer‐reviewed journals, we recommend uploading key workspaces for the compounds and/or populations with verified updated systems parameters to a model database on the consortium members' area. An open‐source option via model‐sharing via uploading the workspaces to the members' area gives many advantages, as shown in Table S7 .
Concluding Remarks
Several examples listed in this collaborative review enable us to gauge the tremendous progress made in the science of PBPK modeling for transporter substrates and inhibitors. The use of PBPK platforms have enabled more representative predictions and advancing strategies to address tDDIs. As a first resort, regulatory DDI guidance and static models can be a good starting point to understand the tDDI liabilities of a compound. However, based on the flags from these conservative assessments, development of mechanistic PBPK models further help in quantitative evaluation of the tDDI liabilities and aid in the waiver, prioritization, and/or design of a suitable clinical DDI study.
Funding
No funding was received for this work.
Conflict of Interest
Howard Burt and Sibylle Neuhoff are employees of Simcyp Limited (a Certara Company). Yuichi Sugiyama is the member of the Simcyp Limited (a Certara Company) Scientific Advisory Board. The other authors have no conflicts of interest to declare.
Supporting information
Supplement Material: Tables S1–S7.
Acknowledgments
We are grateful for review and constructive input from Professor Amin Rostami‐Hodjegan, Professor of Systems Pharmacology at the University of Manchester, UK, and Senior Vice President of Research & Development and Chief Scientific Officer at Certara. We also thank Dr. An Van Den Bergh from Jensen Pharmaceutical for participating in early discussions.
References
- 1. Giacomini, K.M. et al Membrane transporters in drug development. Nat. Rev. Drug Discov. 9, 215–236 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. European Medical Agency. Guideline on the investigation of drug interactions (European Medical Agency, London, 2012). [Google Scholar]
- 3. US Food and Drug Administration . In Vitro Metabolism‐ and Transporter‐Mediated Drug‐Drug Interaction Studies – Guidance for Industry (Draft Guidance) (2017).
- 4. Pan, Y. et al The application of physiologically based pharmacokinetic modeling to predict the role of drug transporters: scientific and regulatory perspectives. J. Clin. Pharmacol. 56, S122–S131 (2016). [DOI] [PubMed] [Google Scholar]
- 5. Varma, M.V. , Steyn, S.J. , Allerton, C. & El‐Kattan, A.F. Predicting clearance mechanism in drug discovery: extended clearance classification system (ECCS). Pharm. Res. 32, 3785–3802 (2015). [DOI] [PubMed] [Google Scholar]
- 6. Galetin, A. , Zhao, P. & Huang, S.‐M. PBPK modelling of drug transporters to facilitate individualized dose prediction. J. Pharm. Sci. 106, 2204–2208 (2017). [DOI] [PubMed] [Google Scholar]
- 7. Shitara, Y. , Maeda, K. , Ikejiri, K. , Yoshida, K. , Horie, T. & Sugiyama, Y. Clinical significance of organic anion transporting polypeptides (OATPs) in drug disposition: their roles in hepatic clearance and intestinal absorption. Biopharm. Drug Dispos. 34, 45–78 (2013). [DOI] [PubMed] [Google Scholar]
- 8. Sirianni, G.L. & Pang, K.S. Organ clearance concepts: new perspectives on old principles. J. Pharmacokinet. Biopharm. 25, 449–470 (1997). [DOI] [PubMed] [Google Scholar]
- 9. Camenisch, G. & Umehara, K‐I. Predicting human hepatic clearance from in vitro drug metabolism and transport data: a scientific and pharmaceutical perspective for assessing drug–drug interactions. Biopharm. Drug Dispos. 33, 179–194 (2012). [DOI] [PubMed] [Google Scholar]
- 10. Asaumi, R. et al Comprehensive PBPK model of rifampicin for quantitative prediction of complex drug‐drug interactions: CYP3A/2C9 induction and OATP inhibition effects. CPT Pharmacometrics Syst. Pharmacol. 7, 186–196 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yoshikado, T. et al Quantitative analyses of hepatic OATP‐mediated interactions between statins and inhibitors using PBPK modeling with a parameter optimization method. Clin. Pharmacol. Ther. 100, 513–523 (2016). [DOI] [PubMed] [Google Scholar]
- 12. Watanabe, T. et al Investigation of the rate‐determining process in the hepatic elimination of HMG‐CoA reductase inhibitors in rats and humans. Drug Metab. Dispos. 38, 215–222 (2010). [DOI] [PubMed] [Google Scholar]
- 13. Varma, M.V. , Bi, Y.A. , Kimoto, E. & Lin, J. Quantitative prediction of transporter‐ and enzyme‐mediated clinical drug‐drug interactions of organic anion‐transporting polypeptide 1B1 substrates using a mechanistic net‐effect model. J. Pharmacol. Exp. Ther. 351, 214–223 (2014). [DOI] [PubMed] [Google Scholar]
- 14. Nishiyama, K. , et al Physiologically-based pharmacokinetic modeling analysis for quantitative prediction of renal transporter–mediated interactions between metformin and cimetidine. CPT Pharmacometrics Syst. Pharmacol. 8, 396–406 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rostami‐Hodjegan, A. Physiologically based pharmacokinetics joined with in vitro‐in vivo extrapolation of ADME: a marriage under the arch of systems pharmacology. Clin. Pharmacol. Ther. 92, 50–61 (2012). [DOI] [PubMed] [Google Scholar]
- 16. US Food and Drug Administration . Guidance for Industry: Physiologically Based Pharmacokinetic Analyses—Format and Content. December 2016 (2016).
- 17. European Medicines Agency . Guideline on the qualification and reporting of physiologically based pharmacokinetic (PBPK) modelling and simulation (2016).
- 18. Varma, M.V. , Lai, Y. , Kimoto, E. , Goosen, T.C. , El‐Kattan, A.F. & Kumar, V. Mechanistic modeling to predict the transporter‐ and enzyme‐mediated drug‐drug interactions of repaglinide. Pharm. Res. 30, 1188–1199 (2013). [DOI] [PubMed] [Google Scholar]
- 19. Varma, M.V. et al Quantitative prediction of repaglinide‐rifampicin complex drug interactions using dynamic and static mechanistic models: delineating differential CYP3A4 induction and OATP1B1 inhibition potential of rifampicin. Drug Metab. Dispos. 41, 966–974 (2013). [DOI] [PubMed] [Google Scholar]
- 20. Shebley, M. et al Physiologically based pharmacokinetic model qualification and reporting procedures for regulatory submissions: a consortium perspective. Clin. Pharmacol. Ther. 104, 88–110 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Watanabe, T. , Kusuhara, H. & Sugiyama, Y. Application of physiologically based pharmacokinetic modeling and clearance concept to drugs showing transporter‐mediated distribution and clearance in humans. J. Pharmacokinet. Pharmacodyn. 37, 575–590 (2010). [DOI] [PubMed] [Google Scholar]
- 22. Galetin, A. , Zhao, P. & Huang, S.M. Physiologically based pharmacokinetic modeling of drug transporters to facilitate individualized dose prediction. J. Pharm. Sci. 106, 2204–2208 (2017). [DOI] [PubMed] [Google Scholar]
- 23. Gertz, M. et al Cyclosporine inhibition of hepatic and intestinal CYP3A4, uptake and efflux transporters: application of PBPK modeling in the assessment of drug‐drug interaction potential. Pharm. Res. 30, 761–780 (2013). [DOI] [PubMed] [Google Scholar]
- 24. Jones, H.M. et al Mechanistic pharmacokinetic modeling for the prediction of transporter‐mediated disposition in humans from sandwich culture human hepatocyte data. Drug Metab. Dispos. 40, 1007–1017 (2012). [DOI] [PubMed] [Google Scholar]
- 25. Li, R. et al A "middle‐out" approach to human pharmacokinetic predictions for OATP substrates using physiologically‐based pharmacokinetic modeling. J. Pharmacokinet. Pharmacodyn. 41, 197–209 (2014). [DOI] [PubMed] [Google Scholar]
- 26. Varma, M.V. , Lin, J. , Bi, Y.A. , Kimoto, E. & Rodrigues, A.D. Quantitative rationalization of gemfibrozil drug interactions: consideration of transporters‐enzyme interplay and the role of circulating metabolite gemfibrozil 1‐O‐beta‐glucuronide. Drug Metab. Dispos. 43, 1108–1118 (2015). [DOI] [PubMed] [Google Scholar]
- 27. Varma, M.V. et al Mechanism‐based pharmacokinetic modeling to evaluate transporter‐enzyme interplay in drug interactions and pharmacogenetics of glyburide. AAPS J. 16, 736–748 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jamei, M. et al A mechanistic framework for in vitro‐in vivo extrapolation of liver membrane transporters: prediction of drug‐drug interaction between rosuvastatin and cyclosporine. Clin. Pharmacokinet. 53, 73–87 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Snoeys, J. , Beumont, M. , Monshouwer, M. & Ouwerkerk‐Mahadevan, S. Mechanistic understanding of the nonlinear pharmacokinetics and intersubject variability of simeprevir: a PBPK‐guided drug development approach. Clin. Pharmacol. Ther. 99, 224–234 (2016). [DOI] [PubMed] [Google Scholar]
- 30. Posada, M.M. et al Prediction of transporter‐mediated drug‐drug interactions for baricitinib. Clin. Transl. Sci. 10, 509–519 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Posada, M.M. et al Prediction of renal transporter mediated drug‐drug interactions for pemetrexed using physiologically based pharmacokinetic modeling. Drug Metab. Dispos. 43, 325–334 (2015). [DOI] [PubMed] [Google Scholar]
- 32. Burt, H.J. et al Metformin and cimetidine: physiologically based pharmacokinetic modelling to investigate transporter mediated drug‐drug interactions. Eur. J. Pharm. Sci. 88, 70–82 (2016). [DOI] [PubMed] [Google Scholar]
- 33. Neuhoff, S. , Yeo, K.R. , Barter, Z. , Jamei, M. , Turner, D.B. & Rostami‐Hodjegan, A. Application of permeability‐limited physiologically‐based pharmacokinetic models: part II – prediction of P‐glycoprotein mediated drug‐drug interactions with digoxin. J. Pharm. Sci. 102, 3161–3173 (2013). [DOI] [PubMed] [Google Scholar]
- 34. Zhang, L. , Huang, S.M. , Reynolds, K. , Madabushi, R. & Zineh, I. Transporters in drug development: scientific and regulatory considerations. Clin. Pharmacol. Ther. 104, 793–796 (2018). [DOI] [PubMed] [Google Scholar]
- 35. Neuhoff, S. , Yeo, K.R. , Barter, Z. , Jamei, M. , Turner, D.B. & Rostami‐Hodjegan, A. Application of permeability‐limited physiologically‐based pharmacokinetic models: part I‐digoxin pharmacokinetics incorporating P‐glycoprotein‐mediated efflux. J. Pharm. Sci. 102, 3145–3160 (2013). [DOI] [PubMed] [Google Scholar]
- 36. Burt, H.J. , Riedmaier, A.E. , Harwood, M.D. , Crewe, H.K. , Gill, K.L. & Neuhoff, S. Abundance of hepatic transporters in caucasians: a meta‐analysis. Drug Metab. Dispos. 44, 1550–1561 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Abe, K. , Bridges, A.S. & Brouwer, K.L. Use of sandwich‐cultured human hepatocytes to predict biliary clearance of angiotensin II receptor blockers and HMG‐CoA reductase inhibitors. Drug Metab. Dispos. 37, 447–452 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bergman, E. et al Biliary secretion of rosuvastatin and bile acids in humans during the absorption phase. Eur. J. Pharm. Sci. 29, 205–214 (2006). [DOI] [PubMed] [Google Scholar]
- 39. Nakagomi‐Hagihara, R. , Nakai, D. & Tokui, T. Inhibition of human organic anion transporter 3 mediated pravastatin transport by gemfibrozil and the metabolites in humans. Xenobiotica 37, 416–426 (2007). [DOI] [PubMed] [Google Scholar]
- 40. Proctor, W.R. , Bourdet, D.L. & Thakker, D.R. Mechanisms underlying saturable intestinal absorption of metformin. Drug Metab. Dispos. 36, 1650–1658 (2008). [DOI] [PubMed] [Google Scholar]
- 41. Cheng, Y. & Prusoff, W.H. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 22, 3099–3108 (1973). [DOI] [PubMed] [Google Scholar]
- 42. Shitara, Y. & Sugiyama, Y. Preincubation‐dependent and long‐lasting inhibition of organic anion transporting polypeptide (OATP) and its impact on drug‐drug interactions. Pharmacol. Ther. 177, 67–80 (2017). [DOI] [PubMed] [Google Scholar]
- 43. Tatrai, P. et al A systematic in vitro investigation of the inhibitor preincubation effect on multiple classes of clinically relevant transporters. Drug Metab. Dispos. 47, 768–778 (2019). [DOI] [PubMed] [Google Scholar]
- 44. Rowland Yeo, K. , Aarabi, M. , Jamei, M. & Rostami‐Hodjegan, A. Modeling and predicting drug pharmacokinetics in patients with renal impairment. Expert Rev. Clin. Pharmacol. 4, 261–274 (2011). [DOI] [PubMed] [Google Scholar]
- 45. Emami Riedmaier, A. , Burt, H. , Abduljalil, K. & Neuhoff, S . More power to OATP1B1: an evaluation of sample size in pharmacogenetic studies using a Rosuvastatin PBPK model for intestinal, hepatic, and renal transporter‐mediated clearances. J. Clin. Pharmacol. 56 (suppl. 7), S132–S142 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kusuhara, H. et al Effects of a MATE protein inhibitor, pyrimethamine, on the renal elimination of metformin at oral microdose and at therapeutic dose in healthy subjects. Clin. Pharmacol. Ther. 89, 837–844 (2011). [DOI] [PubMed] [Google Scholar]
- 47. Freudenthaler, S. , Meineke, I. , Schreeb, K.H. , Boakye, E. , Gundert‐Remy, U. & Gleiter, C.H. Influence of urine pH and urinary flow on the renal excretion of memantine. Br. J. Clin. Pharmacol. 46, 541–546 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Burt, H. , Neuhoff, S. , Gaohua, L. , Jamei, M. , Tucker, G.T. & Rostami‐Hodjegan, A. An assessment of the effect of urine pH on renal clearance using a mechanistic kidney model (Mech KiM). Gordon Conference, US, (2012).
- 49. Busch, A.E. et al Human neurons express the polyspecific cation transporter hOCT2, which translocates monoamine neurotransmitters, amantadine, and memantine. Mol. Pharmacol. 54, 342–352 (1998). [DOI] [PubMed] [Google Scholar]
- 50. Muller, F. et al Contribution of MATE1 to renal secretion of the NMDA receptor antagonist memantine. Mol. Pharm. 14, 2991–2998 (2017). [DOI] [PubMed] [Google Scholar]
- 51. Jalava, K.M. , Partanen, J. & Neuvonen, P.J. Itraconazole decreases renal clearance of digoxin. Ther. Drug Monit. 19, 609–613 (1997). [DOI] [PubMed] [Google Scholar]
- 52. Wang, X. et al Effect of activated charcoal on apixaban pharmacokinetics in healthy subjects. Am. J. Cardiovasc. Drugs 14, 147–154 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Asaumi, R. et al Expanded PBPK model of rifampicin for predicting interactions with drugs and an endogenous biomarker via complex mechanisms including OATP1B induction. CPT Pharmacometrics Syst. Pharmacol. 8, 845–857 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Robson, M. et al Olaparib for metastatic breast cancer in patients with a germline BRCA mutation. N. Engl. J. Med. 377, 523–533 (2017). [DOI] [PubMed] [Google Scholar]
- 55. Pilla Reddy, V. , Bui, K. , Scarfe, G. , Zhou, D. & Learoyd, M . Physiologically based pharmacokinetic modeling for olaparib dosing recommendations: bridging formulations, drug interactions, and patient populations. Clin. Pharmacol. Ther. 105, 229–241 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Snoeys, J. , Beumont, M. , Monshouwer, M. & Ouwerkerk‐Mahadevan, S. Elucidating the plasma and liver pharmacokinetics of simeprevir in special populations using physiologically based pharmacokinetic modelling. Clin. Pharmacokinet. 56, 781–792 (2017). [DOI] [PubMed] [Google Scholar]
- 57. Guo, Y. et al Advancing predictions of tissue and intracellular drug concentrations using in vitro, imaging and physiologically based pharmacokinetic modeling approaches. Clin. Pharmacol. Ther. 104, 865–889 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Tomita, Y. , Maeda, K. & Sugiyama, Y. Ethnic variability in the plasma exposures of OATP1B1 substrates such as HMG‐CoA reductase inhibitors: a kinetic consideration of its mechanism. Clin. Pharmacol. Ther. 94, 37–51 (2013). [DOI] [PubMed] [Google Scholar]
- 59. Pilla Reddy, V. , Walker, M. , Sharma, P. , Ballard, P. , Vishwanathan, K . Development, verification, and prediction of osimertinib drug‐drug interactions using PBPK modeling approach to inform drug label. CPT Pharmacometrics Syst. Pharmacol. 7, 321–330 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Elsby, R. , Martin, P. , Surry, D. , Sharma, P. & Fenner, K. Solitary inhibition of the breast cancer resistance protein efflux transporter results in a clinically significant drug‐drug interaction with rosuvastatin by causing up to a 2‐fold increase in statin exposure. Drug Metab. Dispos. 44, 398–408 (2016). [DOI] [PubMed] [Google Scholar]
- 61. Pilla Reddy, V . Transporter-mediated drug-drug interactions: prediction by physiologically based pharmacokinetic modelling: a pharmaceutical industry perspective. The 2nd Symposium on Transporters in Drug Discovery and Development, London, UK, May 15–16, 2017. https://www.maggichurchouseevents.co.uk/bmcs/Downloads/Archive/Transporters%20-%20Pilla%20Reddy%20Venkatesh.pdf
- 62. Pan, Y. et al The application of physiologically based pharmacokinetic modeling to predict the role of drug transporters: scientific and regulatory perspectives. J. Clin. Pharmacol. 56 (suppl. 7), S122–S131 (2016). [DOI] [PubMed] [Google Scholar]
- 63. Zhou, D. , Bui, K. , Sostek, M. & Al‐Huniti, N. Simulation and prediction of the drug‐drug interaction potential of naloxegol by physiologically based pharmacokinetic modeling. CPT Pharmacometrics Syst. Pharmacol. 5, 250–257 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ball, K. , Jamier, T. , Parmentier, Y. , Denizot, C. , Mallier, A. & Chenel, M. Prediction of renal transporter‐mediated drug‐drug interactions for a drug which is an OAT substrate and inhibitor using PBPK modelling. Eur. J. Pharm. Sci. 106, 122–132 (2017). [DOI] [PubMed] [Google Scholar]
- 65. Berezhkovskiy, L.M. The influence of hepatic transport on the distribution volumes and mean residence time of drug in the body and the accuracy of estimating these parameters by the traditional pharmacokinetic calculations. J. Pharm. Sci. 100, 5031–5047 (2011). [DOI] [PubMed] [Google Scholar]
- 66. Li, R. et al A "middle‐out" approach to human pharmacokinetic predictions for OATP substrates using physiologically‐based pharmacokinetic modeling. J. Pharmacokinet. Pharmacodyn. 41, 197–209 (2014). [DOI] [PubMed] [Google Scholar]
- 67. Miyauchi, S. et al The phenomenon of albumin‐mediated hepatic uptake of organic anion transport polypeptide substrates: prediction of the in vivo uptake clearance from the in vitro uptake by isolated hepatocytes using a facilitated‐dissociation model. Drug Metab. Dispos. 46, 259–267 (2018). [DOI] [PubMed] [Google Scholar]
- 68. Bosgra, S. et al Predicting carrier‐mediated hepatic disposition of rosuvastatin in man by scaling from individual transfected cell‐lines in vitro using absolute transporter protein quantification and PBPK modeling. Eur. J. Pharm. Sci. 65, 156–166 (2014). [DOI] [PubMed] [Google Scholar]
- 69. Watanabe, T. , Kusuhara, H. , Maeda, K. , Shitara, Y. & Sugiyama, Y. Physiologically based pharmacokinetic modeling to predict transporter‐mediated clearance and distribution of pravastatin in humans. J. Pharmacol. Exp. Ther. 328, 652–662 (2009). [DOI] [PubMed] [Google Scholar]
- 70. Ishida, K. , Ullah, M. , Toth, B. , Juhasz, V. & Unadkat, J.D. Successful prediction of in vivo hepatobiliary clearances and hepatic concentrations of rosuvastatin using sandwich‐cultured rat hepatocytes, transporter‐expressing cell lines, and quantitative proteomics. Drug Metab. Dispos. 46, 66–74 (2018). [DOI] [PubMed] [Google Scholar]
- 71. Bentz, J. et al Variability in P‐glycoprotein inhibitory potency (IC(5)(0)) using various in vitro experimental systems: implications for universal digoxin drug‐drug interaction risk assessment decision criteria. Drug Metab. Dispos. 41, 1347–1366 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Cook, J.A. et al Refining the in vitro and in vivo critical parameters for P‐glycoprotein, [I]/IC50 and [I2]/IC50, that allow for the exclusion of drug candidates from clinical digoxin interaction studies. Mol. Pharm. 7, 398–411 (2010). [DOI] [PubMed] [Google Scholar]
- 73. Duan, P. , Zhao, P. & Zhang, L. Physiologically based pharmacokinetic (PBPK) modeling of pitavastatin and atorvastatin to predict drug‐drug interactions (DDIs). Eur. J. Drug Metab. Pharmacokinet. 42, 689–705 (2017). [DOI] [PubMed] [Google Scholar]
- 74. Badee, J. , Achour, B. , Rostami‐Hodjegan, A. & Galetin, A. Meta‐analysis of expression of hepatic organic anion‐transporting polypeptide (OATP) transporters in cellular systems relative to human liver tissue. Drug Metab. Dispos. 43, 424–432 (2015). [DOI] [PubMed] [Google Scholar]
- 75. Mao, J. , Doshi, U. , Wright, M. , Hop, C. , Li, A.P. & Chen, Y. Prediction of the pharmacokinetics of pravastatin as an OATP substrate using plateable human hepatocytes with human plasma data and PBPK modeling. CPT Pharmacometrics Syst. Pharmacol. 7, 251–258 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Poirier, A. et al Design, data analysis, and simulation of in vitro drug transport kinetic experiments using a mechanistic in vitro model. Drug Metab. Dispos. 36, 2434–2444 (2008). [DOI] [PubMed] [Google Scholar]
- 77. Menochet, K. , Kenworthy, K.E. , Houston, J.B. & Galetin, A. Simultaneous assessment of uptake and metabolism in rat hepatocytes: a comprehensive mechanistic model. J. Pharmacol. Exp. Ther. 341, 2–15 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Grime, K. & Riley, R.J. The impact of in vitro binding on in vitro‐in vivo extrapolations, projections of metabolic clearance and clinical drug‐drug interactions. Curr. Drug Metab. 7, 251–264 (2006). [DOI] [PubMed] [Google Scholar]
- 79. Rodrigues, A.D. , Taskar, K.S. , Kusuhara, H. & Sugiyama, Y. Endogenous probes for drug transporters: balancing vision with reality. Clin. Pharmacol. Ther. 103, 434–448 (2018). [DOI] [PubMed] [Google Scholar]
- 80. Barnett, S. et al Gaining mechanistic insight into coproporphyrin i as endogenous biomarker for OATP1B‐mediated drug‐drug interactions using population pharmacokinetic modeling and simulation. Clin. Pharmacol. Ther. 104, 564–574 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Yoshikado, T. et al PBPK modeling of coproporphyrin I as an endogenous biomarker for drug interactions involving inhibition of hepatic OATP1B1 and OATP1B3. CPT Pharmacometrics Syst. Pharmacol. 7, 739–747 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Zamek‐Gliszczynski, M.J. , Giacomini, K.M. & Zhang, L. Emerging clinical importance of hepatic organic cation transporter 1 (OCT1) in drug pharmacokinetics, dynamics, pharmacogenetic variability, and drug interactions. Clin. Pharmacol. Ther. 103, 758–760 (2018). [DOI] [PubMed] [Google Scholar]
- 83. Ebner, T. , Ishiguro, N. & Taub, M.E. The use of transporter probe drug cocktails for the assessment of transporter‐based drug‐drug interactions in a clinical setting‐proposal of a four component transporter cocktail. J. Pharm. Sci. 104, 3220–3228 (2015). [DOI] [PubMed] [Google Scholar]
- 84. Stopfer, P. et al Pharmacokinetic evaluation of a drug transporter cocktail consisting of digoxin, furosemide, metformin, and rosuvastatin. Clin. Pharmacol. Ther. 100, 259–267 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Stopfer, P. et al Effects of metformin and furosemide on rosuvastatin pharmacokinetics in healthy volunteers: implications for their use as probe drugs in a transporter cocktail. Eur. J. Drug Metab. Pharmacokinet. 43, 69–80 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Kosa, R.E. et al Simultaneous assessment of transporter‐mediated drug‐drug interactions using a probe drug cocktail in cynomolgus monkey. Drug Metab. Dispos. 46, 1179–1189 (2018). [DOI] [PubMed] [Google Scholar]
- 87. Harwood, M.D. et al In vitro‐in vivo extrapolation scaling factors for intestinal P‐glycoprotein and breast cancer resistance protein: Part II. The impact of cross‐laboratory variations of intestinal transporter relative expression factors on predicted drug disposition. Drug Metab. Dispos. 44, 476–480 (2016). [DOI] [PubMed] [Google Scholar]
- 88. Rose, R.H. , Neuhoff, S. , Abduljalil, K. , Chetty, M. , Rostami‐Hodjegan, A. & Jamei, M. Application of a physiologically based pharmacokinetic model to predict OATP1B1‐related variability in pharmacodynamics of rosuvastatin. CPT Pharmacometrics Syst. Pharmacol. 3, e124 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Yang, K. , Woodhead, J.L. , Watkins, P.B. , Howell, B.A. & Brouwer, K.L. Systems pharmacology modeling predicts delayed presentation and species differences in bile acid‐mediated troglitazone hepatotoxicity. Clin. Pharmacol. Ther. 96, 589–598 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Yonezawa, A. & Inui, K. Organic cation transporter OCT/SLC22A and H(+)/organic cation antiporter MATE/SLC47A are key molecules for nephrotoxicity of platinum agents. Biochem. Pharmacol. 81, 563–568 (2011). [DOI] [PubMed] [Google Scholar]
- 91. Nakamura, T. , Yonezawa, A. , Hashimoto, S. , Katsura, T. & Inui, K. Disruption of multidrug and toxin extrusion MATE1 potentiates cisplatin‐induced nephrotoxicity. Biochem. Pharmacol. 80, 1762–1767 (2010). [DOI] [PubMed] [Google Scholar]
- 92. Billington, S. et al Positron emission tomography imaging of [(11) C]. Rosuvastatin hepatic concentrations and hepatobiliary transport in humans in the absence and presence of cyclosporin A. Clin. Pharmacol. Ther. 106, 1056–1066 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Orntoft, N. , Frisch, K. , Ott, P. , Keiding, S. & Sorensen, M. Functional assessment of hepatobiliary secretion by (11)C‐cholylsarcosine positron emission tomography. Biochim. Biophys. Acta Mol. Basis Dis. 1864, 1240–1244 (2018). [DOI] [PubMed] [Google Scholar]
- 94. Liu, L. et al Modulation of P‐glycoprotein at the human blood‐brain barrier by quinidine or rifampin treatment: a positron emission tomography imaging study. Drug Metab. Dispos. 43, 1795–1804 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Shingaki, T. et al Quantitative evaluation of mMate1 function based on minimally invasive measurement of tissue concentration using PET with [(11)C]. metformin in mouse. Pharm. Res. 32, 2538–2547 (2015). [DOI] [PubMed] [Google Scholar]
- 96. Posada, M.M. et al Prediction of renal transporter mediated drug‐drug interactions for pemetrexed using physiologically based pharmacokinetic modeling. Drug Metab. Dispos. 43, 325–334 (2015). [DOI] [PubMed] [Google Scholar]
- 97. Wang, Y.H. , Chen, D. , Hartmann, G. , Cho, C.R. & Menzel, K. PBPK modeling strategy for predicting complex drug interactions of letermovir as a perpetrator in support of product labeling. Clin. Pharmacol. Ther. 105, 515–523 (2018). [DOI] [PubMed] [Google Scholar]
- 98. Mukherjee, D. , Kosloski, M. , Liu, W. & Shebley, M. Physiologically based pharmacokinetic modeling of glecaprevir and pibrentasvir as a combination: mechanistic modeling of non-linear pharmacokinetics. Clin. Pharmacol. Ther. 103, S81 (2018). [Google Scholar]
- 99. Taipalensuu, J. , Tavelin, S. , Lazorova, L. , Svensson, A.C. & Artursson, P. Exploring the quantitative relationship between the level of MDR1 transcript, protein and function using digoxin as a marker of MDR1‐dependent drug efflux activity. Eur. J. Pharm. Sci. 21, 69–75 (2004). [DOI] [PubMed] [Google Scholar]
- 100. Kumar, V. et al Quantitative transporter proteomics by liquid chromatography with tandem mass spectrometry: addressing methodologic issues of plasma membrane isolation and expression‐activity relationship. Drug Metab. Dispos. 43, 284–288 (2015). [DOI] [PubMed] [Google Scholar]
- 101. Nies, A.T. et al Genetics is a major determinant of expression of the human hepatic uptake transporter OATP1B1, but not of OATP1B3 and OATP2B1. Genome Med. 5, 1 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Watanabe, C. , Kato, Y. , Ito, S. , Kubo, Y. , Sai, Y. & Tsuji, A. Na+/H+ exchanger 3 affects transport property of H+/oligopeptide transporter 1. Drug Metab. Pharmacokinet. 20, 443–451 (2005). [DOI] [PubMed] [Google Scholar]
- 103. Emoto, C. , Fukuda, T. , Johnson, T.N. , Neuhoff, S. , Sadhasivam, S. & Vinks, A.A. Characterization of contributing factors to variability in morphine clearance through PBPK modeling implemented with OCT1 transporter. CPT Pharmacometrics Syst. Pharmacol. 6, 110–119 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Tan, M.L. et al Use of physiologically based pharmacokinetic modeling to evaluate the effect of chronic kidney disease on the disposition of hepatic CYP2C8 and OATP1B drug substrates. Clin. Pharmacol. Ther. 105, 719–729 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Neuhoff, S. , Artursson, P. , Zamora, I. & Ungell, A.L. Impact of extracellular protein binding on passive and active drug transport across Caco‐2 cells. Pharm. Res. 23, 350–359 (2006). [DOI] [PubMed] [Google Scholar]
- 106. Billington, S. et al Interindividual and regional variability in drug transporter abundance at the human blood‐brain barrier measured by quantitative targeted proteomics. Clin. Pharmacol. Ther. 106, 228–237 (2019). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplement Material: Tables S1–S7.