

Abstract
The objective of this present study is to focus on the in silico study to screen for an alternative drug that can block the activity of the angiotensin converting enzyme 2 (ACE2) as a receptor for SARS-CoV-2, potential therapeutic target of the COVID-19 virus using natural compounds (Isothymol, Thymol, Limonene, P-cymene and γ-terpinene) derived from the essential oil of the antiviral and antimicrobial plant Ammoides verticillata (Desf.) Briq. which is located in the occidental Algeria areas. This study reveals that Isothymol, a major component of this plant, gives the best docking scores, compared to, the co-crystallized inhibitor β-D-mannose of the enzyme ACE2, to Captropil drug as good ACE2 inhibitor and to Chloroquine antiviral drug also involved in other mechanisms as inhibition of ACE2 cellular receptor. In silico (ADME), drug-likeness, PASS & P450 site of metabolism prediction, pharmacophore Mapper showed that the compound Isothymol has given a good tests results compared to the β-D-mannose co-crystallized inhibitor, to Captopril and Chloroquine drugs. Also the other natural compounds gave good results. The Molecular Dynamics Simulation study showed good result for the Isotymol- ACE2 docked complex. This study revealed for the first time that Isothymol is a functional inhibitor of angiotensin converting enzyme 2 activity and the components of essential oils Ammoides verticillata can be used as potential inhibitors to the ACE2 receptor of SARS-CoV-2.
Communicated by Ramaswamy H. Sarma
Keywords: Ammoides verticillata, Angiotensin converting enzyme 2, COVID-19, isothymol, molcular docking
1. Introduction
Coronaviruses are a large virus’s family that causes illnesses ranging from a common cold (some seasonal viruses are coronaviruses) to more severe conditions such as SARS-CoV or Mers-CoV (Jabeer Khan et al., 2020; Enayatkhani et al., 2020; Elfiky & Azzam, 2020). This virus is from the family Coronaviridae and was detected in the city of Wuhan in China (Khan et al., 2020; Elmezayen et al., 2020). It was originally named 2019-nCoV. It is now called SARS-CoV-2 (Pant et al., 2020). The disease associated with this virus is COVID-19. The onset of the COVID-19 atypical pneumonia outbreak was reported on December 31, 2019 (Sarma et al., 2020; Gupta et al., 2020). Common symptoms include fever, cough, and shortness of breath . Other symptoms may include fatigue, muscle pain, diarrhea, sore throat, loss of smell, and abdominal pain (Hopkins, 2020). The time from exposure to onset of symptoms is typically around five days but may range from two to fourteen days (Velavan & Meyer, 2020). While the majority of cases result in mild symptoms, some progress to viral pneumonia and multi-organ failure (Hui et al., 2020; Muralidharan et al., 2020). As of 16 April 2020, more than 2.13 million cases have been reported across 210 countries and territories, resulting in more than 142,000 deaths. More than 540,000 people have recovered.
Coronaviruses have in common proteins designated by a letter indicating their location: S (protuberances), E (envelope), M (membrane) and N (nucleocapsid). Some, in particular those of subgroup A of the genus Betacoronavirus, have a characteristic HE protein (hemagglutinin esterase). The SARS coronavirus also presents on protein S a specific binding site for the angiotensin converting enzyme 2 (ACE2) which serves as an entry point into the host cell (Fang, 2005). Research suggests that the receptor is another potential target for vaccines or therapies. An inhibitor that blocks the receptor could make it more difficult for coronavirus to enter cells, which could at least significantly slow the epidemic until the virus disappears (Boopathy et al., 2020; Hasan et al., 2020).
Hence the interest of our study which consists in studying in silico the inhibition of ACE 2 by molecules derived from the essential oil of a medicinal plant which bears the name Ammoides verticillata (Desf.) Briq. This plant, is an aromatic plant widely used in western Algeria for its culinary and therapeutic purposes, it has proven antioxidant and antimicrobial activities of essential oil (Bekhechia et al., 2010). The different names of Ammoides verticillata (Desf.) Briq. are French name Ajowan (Bekhechia et al., 2010), Arabic name Nounkha Taleb El Koubs (Narayana et al., 1967), Nûnkha (Trabut, 1935a; Abdelouahid & Bekhechi, 2002). According to (Quezel & Santa, 1963), this species is an annual herb that grows spontaneously in the matorrals of the western region of Algeria. This thread-like Apiaceae, with branched stems of 10–40 cm, without rosette of basal leaves. Lower petiolate leaves have many whorled multifide segments, upper pennatifides have linear segments. The main umbels are 8 - 15 rays. Ovoid fruit is less than 1 mm long Figure 1. It is generally found in fields, lawns, mountains and forests.
Figure 1.

Ammoides verticillata (Desf.) Briq from western Algeria.
Studies have shown that the essential oil of Ammoides verticillata has very good antimicrobial activity against five bacterial strains (Bacillus cereus, Staphylococcus aureus, Escherichia coli, Listeria monocytogenes and Micrococcus luteus) and the yeast Candida albicans, with CIM values between 0.78 and 2.34 μl/ml. In addition, the essential oil can trap 50% of the free DPPH radical at a concentration of 15.37 μg/ml (Sandberg & Corrigon, 2001; Higley & Higley, 2005; Pole, 2013). Also other study show that aerial parts (leaves and flowers) of Ammoides verticillata (Desf.) Briq. collected in occidental Algeria areas, exhibited a very unusual composition. Isothymol was by far the major component (51.2%), accompanied by p-cymene (14.1%), thymol (13.0%), limonene (11.9%) and γ- terpinene (6.8%) 11 (Kambouche & El-Abed, 2003). According to (Lawrence, 2006), it is worth noting that this oil is the richest source of naturally occurring isothymol (Bekhechia et al., 2010). The aims of this study consist to establish a structure–activity relationship of actives principles of Ammoides verticillata compounds: Isothymol, Thymol, Limonene, P-Cymene and γ-Terpinene against ACE 2 activity compared to β-D- Mannose, co-cristallized inhibitor of this enzyme, this molecule is used in the RCSB data bank to block the ACE2 enzyme activity, and we compared also our results with 2 drugs which are used as good inhibitors to ACE2, the Captopril hypertension drug and Chloroquine antimalarials and amebicides drug which has also previously shown activity on coronavirus replication (Guastalegname & Vallone, 2020). In this work we will take it as a witness for the discussion of our results to lead to other inhibitors which can also block the ACE2 enzyme activity and used as potential treatment of COVID-19. For this purpose, a molecular docking study was applied for testing the type of interactions between active site residues and the principles actives compounds from Ammoides vericillata with ACE2, and calculation of the binding free energies (S-score, kcal/mol) and bonds in order to predict the binding model of the selected compound to the three-dimensional structure of the enzyme (Aanouz et al., 2020).
2. Material and methods
2.1. Ligands and protein preparation
The three-dimensional (3 D) structures of compounds Table 1 were pre-optimized by means of the Molecular Mechanics Force Field (MM+). After that, the resulted minimized structures were further refined using the semi-empirical AM1 (Stewart, 2007) with the Polak-Ribiere conjugate gradient algorithm of 0.01 kcal/(Åmol). All methods are implemented in Hyperchem 8.0.8 software (HyperChem v8, 2009), a database was created in which all the ligands were converted into their 3 D structures and was used as input MOE-docking.
Table 1.
2 D&3D representations of the all ligands used in the experiment. The ligands structures were taken from PubChem server (www.pubchem.ncbi.nlm.nih.gov).
| Compound name | 2D Structure | 3D Structure |
|---|---|---|
| Isothymol |  |
 |
| p-cymene |  |
 |
| Thymol |  |
 |
| limonene |  |
 |
| γ-terpinene | 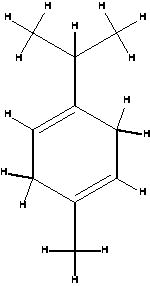 |
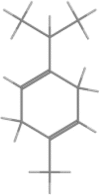 |
| beta-D-Mannose (Co-cristallized ligand) | 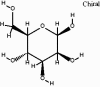 |
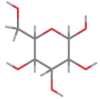 |
| Captopril |  |
 |
| Chloroquine |  |
 |
The X-ray crystal co-ordinates of ACE 2 (PDB ID: 6vw1) (Shang et al., 2020) in the bound state with β-D- Mannose (formula: C6H12O6) Table 2, were retrieved from RCSB Database (http://www.rcsb.org/pdb) Figures 2 and 3. This is selected for modeling studies. In the last, the energy of the proteins structures is minimized using the Energy minimization algorithm of MOE tool. These energy of proteins are calculated (in kcal/mol) by MOE using MMFF94x force field with conjugant gradient method.
Table 2.
Details related to the ACE2.
| Proteins | Methods | Resolution (Å) | R-Value | Residues number | Co-crystallized ligand | Docking Score Kcal/mol |
|---|---|---|---|---|---|---|
| ACE2 PDB ID:6vw1 | X-ray diffraction | 2.68 | 0.229 | 597 | β-D-Mannose | −5.4107 |
Figure 2.

3 D structure of 2019-nCoV chimeric receptor-binding domain complexed with its receptor human ACE2 enzyme (PDB ID: 6vw1). The structure was taken from Protein Data Bank online server (https://www.rcsb.org/) and Molecular Operating Environment (MOE).
Figure 3.

3 D structure of active site enzyme.
2.2. Molecular docking protocol
Docking calculations were carried out using standard default parameter settings in the MOE software package (Molecular Operating Environment (MOE),), 2013). In this molecular docking program, the flexibility of ligands is considered while the proteins are considered as a rigid structure. In this case β-D- Mannose is first re-docked into the binding site pocket of ACE2 enzyme, and the root-mean-square deviation (RMSD) values between the docking and initial poses were calculated. In general, the protein structure with a resolution between 1.5 and 2.5 Å have a good quality for further studies (Clément & Slenzka, 2006; Didierjean & Tête-Favier, 2016), whereas, the resolution value of ACE2 not far to this interval. At the end of molecular docking, the best conformations of the ligands were analyzed for their binding interactions and were evaluated by the binding free energies (S-score, kcal/mol) and bonds interactions between ligand atom and active site residues.
2.3. Ligand based druglikeness, ADME/toxicity properties and PASS prediction
Many potential therapeutic agents fail to reach the clinic trials because of their unfavorable absorption, distribution, metabolism, and elimination (ADME) parameters; also it is not checking the druglikeness. Lipinski’s rule of five (Lipinski et al., 2001) Veber’s rule (Veber et al., 2002), Egan’s rule (Veber et al., 2000) and Polar surface area (TPSA), number of rotatable (Daina et al., 2017), the ADME/Toxicity (pharmacological and pharmacodynamic) properties, the PASS prediction, the P450 Site of Metabolism (SOM) and the Molecular Target studies were calculated using respectively SwissADME properties calculation online, PASS-Way2Drug server, RS-WebPredictor 1.0 and swiss target prediction.
2.4. Phamacophore mapping
A pharmacophore consists of a pharmacologically active part of a molecule serving as a model. Pharmacophores are therefore sets of active atoms used in the design of drugs. The pharmacophore is an idealized geometric representation, only 3 D modeling can allow optimal use for the creation of new drugs.
A pharmacophore is the set of functional groups arranged in an adequate spatial arrangement, ensuring the fixation of the drug on the receptor and therefore capable of inducing the physiological response.
In this study the Phamacophore Mapping is carried for the Isothymol the best ligand among the selected ligands out using MOE software package (Molecular Operating Environment (MOE),), 2013).
2.5. Molecular dynamics simulation
The molecular dynamics simulation study was carried out for the ligand that was declared as the best among the selected molecules. From the analysis of the results, it was declared that, Isothymol was the best ligand among the selected ligand molecules. The molecular dynamics simulation study of Isothymol and ACE2 docked complex was performed by iMODS. It is a fast, user-friendly and effective molecular dynamics simulation tool that can be used efficiently to investigate the structural dynamics of the protein complexes. It provides the values of deformability, B-factor (mobility profiles), eigenvalues, variance, co-varience map and elastic network. For a complex or protein, the deformability depends on the ability to deform at each of its amino acid residues. The eigenvalue has relation with the energy that is required to deform the given structure and the lower the eigenvalue, the easier the deformability of the complex.
Moreover, the eigenvalue also represents the motion stiffness of the protein complex. IMODS is a fast and easy server for determining and measuring the protein flexibility (Awan et al., 2017; Prabhakar et al., 2016; López-Blanco et al., 2014; Lopéz-Blanco et al., 2011)
3. Results and discussion
3.1. Molecular docking simulation
3.1.1. The binding affinities of the ligands into ACE2 active site
The results calculations docking details received after dock all ligands with ACE2 target are listed in the Table 3.
Table 3.
The docking results (binding energy) of all ligands and the controls along their respective number of hydrogen bonds as well as interacting amino acids.
| Names of ligands | SP Docking Score (Binding Energy) (Kcal/mol) | Interacting residues of the target | Types of bonds | Distance (Å) | Energies (kcal-mol) |
|---|---|---|---|---|---|
| Chloroquine | −6.4530 | OE1-GLU402 OE1-GLU406 | H-donorIonic | 3.422.86 | -1.0-5.5 |
| Isothymol | −5.7853 | 6-ring-THR445 | Pi-H | 4.06 | -0.6 |
| beta-D-Mannose (Co-cristallisation ligand) | −5.4107 | O3-OE2 GLU 406 O4-OE2 GLU 406 O5-OE1 GLN 442 O5-NE2 GLN 442 O6-NZ LYS 441 | H-donorH-donorH-donorH-acceptorH-acceptor | 2.992.942.903.083.15 | -2.5 − 1.0 − 1.20.3-2.7 |
| Captopril | −5.2248 | NZ-LYS 441 | Ionic | 3.12 | -1.0 |
| Thymol | −4.7450 | O1-OE1 GLU 406 6-ring- CG2 THR445 | H-donorPi-H | 3.174.33 | -3.0-0.9 |
| limonene | −4.4067 | / | / | / | / |
| p-cymene | −4.3630 | / | / | / | / |
| γ-terpinene | −4.2320 | / | / | / | / |
The molecules that had the lowest binding energy of docking score were considered the best molecule and inhibiting the target receptor as the lower binding energy corresponds to higher binding affinity (Simon et al., 2017).
The ACE2-ligands complexes formed are shown in Figure 4.
Figure 4.

3 D representations of the best pose interactions between the ligands and their receptor. A. interaction between β-D-Mannose Co-cristallized ligand) and ACE2, B. interaction between Isothymol and ACE2, C. interaction between limonene and ACE2, D. interaction between p-cymene and ACE2, E. interaction between γ-terpinene and ACE2,F. interaction between Thymol and ACE2, G. interaction between Chloroquine and ACE2, H. interaction between Captopril and ACE2. The 3 D representations of the best pose interactions between the ligands and their respective receptors were visualized using Molecular Operating Environment (MOE).
The binding mode observed for all ligands with pocket of receptor is represented in the Figure 5.
Figure 5.

2 D representations of the best pose interactions between the ligands and their receptor. A. interaction between β-D-Mannose (Co-cristallized ligand) and ACE2, B. interaction between Isothymol and ACE2, C. interaction between limonene and ACE2, D. interaction between p-cymene and ACE2, E. interaction between γ-terpinene and ACE2,F. interaction between Thymol and ACE2, G. interaction between Chloroquine and ACE2, H. interaction between Captopril and ACE2. The 2 D representations of the best pose interactions between the ligands and their respective receptors were visualized using Molecular Operating Environment (MOE).
The Isothymol present score with -5.7853 Kcal/mol value and one hydrogen interaction with THR445 amino acids of 6vw1 target active site, the score β-D- Mannose (Co-crystallized ligand) is -5. 4107 Kcal/mol, 5 interactions with GLU406, GLU406, GLN442, GLN442, LYS441 residues target active site, the Thymol has -4, 7450 Kcal/mol, 2 interactions with GLU406 and THR445, the score values for the Limonene, P-Cymene and γ-Terpinene are respectively -4.4067, −4. 3630, −4. 2320 Kcal/mol and they have none interactions with residues target, Chloroquine has -6.4530 Kcal/mol score value, 2 interactions with OE1-GLU402 and OE1-GLU406 amino acids target and Captopril present -5.2248 Kcal/mol, one interaction with NZ-LYS 441 residues of 6vw1 target. We compared all the values score complexes; we found that complex formed by 6vw1-Isothymol has the lowest value energy and giving the best docking score compared to β-D- Mannose co-crystallized inhibitor and Captopril. On the other hand Isothymol has a lower molecular weight than Chloroquine and it gives almost the same values of the score (-5.7853 Kcal/mol and -6.4530 Kcal/mol respectively) therefore we can consider that the two complexes 6vw1-Isothymol and 6vw1-chloroquine are stables with higher binding affinity. According to these docking results we can classify Isothymol as the good inhibitor of the enzyme ACE2 compared to the all ligands studied.
3.2. In silico evaluation of druglikness, ADME/toxicity properties and PASS prediction
This test is performed to facilitate the creation of new drug molecules, the molecules of which must comply with the conditions of the following five Lipinski’s rule: molecular weight: ≤ 500, number of hydrogen bond donors: ≤ 5, number of hydrogen bond acceptors: ≤ 10, lipophilicity (expressed in LogP): ≤5 and molar refractivity from 40 to 130 (Lipinski et al., 2001). The results of this study are shown in the Table 4.
Table 4.
List of the results of the ligands druglikeness properties.
| Drug Likeness Properties | Chloroquine | Isothymol | beta-D-Mannose | Captopril | Thymol | limonene | p-cymene | γ-terpinene |
|---|---|---|---|---|---|---|---|---|
| Molecular weight g/mol | 319.87 | 220.31 | 180.16 | 217.29 | 150.22 | 136.23 | 134.22 | 136.23 |
| Concensus Log Po/w | 4.15 | 3.85 | −2.62 | 0.62 | 2.84 | 3.36 | 3.19 | 3.05 |
| Log S | −4.06 | −3.56 | 0.25 | −1.71 | −2.54 | −2.54 | −2.83 | −2.37 |
| Num. H-bond acceptors | 2 | 2 | 6 | 3 | 1 | 0 | 0 | 0 |
| Num. H-bond donors | 1 | 0 | 5 | 1 | 1 | 0 | 0 | 0 |
| Molar Refractivity | 97.41 | 67.10 | 35.74 | 59.97 | 48.01 | 47.12 | 45.99 | 47.12 |
| Lipinski | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Ghose | Yes | Yes | No | Yes | No | No | No | No |
| Veber | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Egan | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Muegge | Yes | Yes | No | Yes | No | No | No | No |
| Bioavailability score | 0.55 | 0.55 | 0.55 | 0.56 | 0.55 | 0.55 | 0.55 | 0.55 |
| Synthetic accessibility (SA) | 2.76 | 1.96 | 4.08 | 2.47 | 1.00 | 3.46 | 1.00 | 3.11 |
| TPSA (Ų) | 28.16 | 26.30 | 110.38 | 96.41 | 20.23 | 0.00 | 0.00 Ų | 0.00 |
| No of rotatable bonds | 8 | 4 | 1 | 4 | 1 | 1 | 1 | 1 |
| Druglikeness score | 7.39 | −1.37 | −3.78 | −0.96 | −3.02 | −21.85 | −5.63 | −2.82 |
| Drug-Score | 0.30 | 0.31 | 0.51 | 0.63 | 0.18 | 0.06 | 0.22 | 0.29 |
| Solubility | −4.06 | −3.56 | 0.25 | −1.71 | −2.54 | −2.54 | −2.83 | −2.37 |
| Reproductive effective | Yes | Yes | Yes | Yes | No | No | Yes | Yes |
| irritant | No | No | Yes | Yes | Yes | No | No | No |
| Tumorigenic | Yes | Yes | Yes | Yes | Yes | No | No | Yes |
| Mutagenic | No | Yes | Yes | Yes | No | No | Yes | Yes |
Chloroquine, Isothymol, β-D- Mannose, Captopril, Thymol, Limonene, P-Cymene and γ-Terpinene have respectively the following molecular weights: (319.87, 220.31, 180.16, 217.29, 150.22, 136.23, 134.22, 136.23) g/mol, they have all a molecular weight ≤ 500 g/mol, and also respectively present the following values of the topological polar surface (TPSA) (28.16, 26.30, 110 .38, 96.41, 20.23, 0.00, 0.00, 0.00) Å2. The lowest TPSA values always give good results, so we note that the molecules from the plant Ammoides verticillata are better behaved than the co-crystallized ligand and Captopril drug. By comparing the lipophilicity (LogP) values of our ligands series, we observe that they all have values less than 5 so they have shown very good results and can be easily absorbed in the body.
However, the LogS solubility values of the ligands tested are between -4.06 and 0.25. The Isotymol and chloroquine having the lowest values and β-D- Mannose having the greatest value, the lowest values of which are always appreciable. However all the ligands have a number of hydrogen bonding donors: ≤ 5, a number of hydrogen bonding acceptors: ≤ 10 and also reactivity values between 35.75 and 130. So we can say that the five Lipinski’s rule was verified for the ligands from the plant Ammoides verticillata. Veber’s rule which represents the oral bioavailability of a possible drug molecule; it is verified for all the list of ligands studied.
We also note that another Egan’s rule which defines the absorption of the drug molecule is verified for all ligands, on the other hand the 2 rules of Ghose’s and Muegge’s (Ghose et al., 1999; Veber et al., 2002; Egan et al., 2000; Muegge et al., 2001) are verified only for chloroquine drug and Isothymol therefore which means that Isothymol can be considered as a good drug. The ease of synthesizing a drug is given by the Synthetic Accessibility Score (SA). The molecule which gives a score 1 is easy to synthesize it, on the other hand the score 10 represents a difficulty of synthesis therefore according to our results the molecule Isothymol is easier to synthesize it by contribution to the Chloroquine drug. We also notice that some ligands gives the reproductive effects, the irritant, Tumorigenic, Mutagenic properties and all the ligands from the plant Ammoides verticillata showed a bioavailability score of 0.55, and also good values of Druglikness score, Solubility values and drug score. As conclusion the Isothymol which represents the highest yield in the plant Ammoides verticillata has followed the Lipinski’s, Veber’s, Egan’s, Ghose’s and Muegge’s rules, it also has an 1.96 SA value so it is easy to synthesize it and it can to be considered as successful drug.
The results of the ADME tests are listed in the Table 5 these tests were carried out to determine the pharmacological and pharmacodynamic properties of a drug in the biological system.
Table 5.
The ADME/T test results of ligands (various pharmacokinetic and pharmacodynamic properties).
| Class | Properties | Chloroquine | Isothymol | beta-D-Mannose | Captopril | Thymol | limonene | p-cymene | γ-terpinene |
|---|---|---|---|---|---|---|---|---|---|
| Absorption | Caco-2 permeability | Optimal | Optimal | Low | Optimal | Optimal | Optimal | Optimal | Optimal |
| Pgp-inhibitor | Non-inhibitor | Inhibitor | Non-inhibitor | Non-inhibitor | Non-inhibitor | Non-inhibitor | Non-inhibitor | Non-inhibitor | |
| Pgp-substrate | substrate | Non-substrate | Non-substrate | Non-substrate | Non-substrate | Non-substrate | Non-substrate | Non-substrate | |
| Human Intestinal Absorption (HIA) | HIA positive | HIA positive | HIA negative | HIA positive | HIA positive | HIA positive | HIA positive | HIA positive | |
| Distribution | Plasma Protein Binding | Good | Good | Low | Low | Good | Good | Low | Low |
| BBB (Blood–Brain Barrier) | BBB positive | BBB positive | BBB positive | BBB negative | BBB positive | BBB positive | BBB positive | BBB positive | |
| Metabolism | CYP450 1A2 inhibitor | Non-inhibitor | Inhibitor | Non-inhibitor | Non-inhibitor | Inhibitor | Non-inhibitor | Non-inhibitor | Non-inhibitor |
| CYP450 1A2 substrate | Substrate | Substrate | Non-substrate | Non-substrate | Substrate | Substrate | Substrate | Substrate | |
| CYP450 3A4 inhibitor | Non-inhibitor | Non-inhibitor | Non-inhibitor | Non-inhibitor | Non-inhibitor | Non-inhibitor | Non-inhibitor | Non-inhibitor | |
| CYP450 3A4 substrate | Substrate | Substrate | Non-substrate | Non-substrate | Non-substrate | Substrate | Non-substrate | Non-substrate | |
| CYP450 2C9 inhibitor | Non-inhibitor | Inhibitor | Non-inhibitor | Non-inhibitor | Non-inhibitor | Non-inhibitor | Non-inhibitor | Non-inhibitor | |
| CYP450 2C9 substrate | Non-substrate | Non-substrate | Non-substrate | Non-substrate | Substrate | Substrate | Substrate | Substrate | |
| CYP450 2C19 inhibitor | Non-inhibitor | Inhibitor | Non-inhibitor | Non-inhibitor | Inhibitor | Non-inhibitor | Non-inhibitor | Non-inhibitor | |
| CYP450 2C19 substrate | Substrate | Substrate | Non-substrate | Non-substrate | Substrate | Substrate | Substrate | Substrate | |
| CYP450 2D6 inhibitor | Inhibitor | Non-inhibitor | Non-inhibitor | Non-inhibitor | Non-inhibitor | Non-inhibitor | Non-inhibitor | Non-inhibitor | |
| CYP450 2D6 substrate | Substrate | Substrate | Non-substrate | Substrate | Substrate | Substrate | Substrate | Substrate | |
| Excretion | T1/2 (h) | 2.23 | 1.225 | 1.129 | 0.793 | 1.313 | 1.741 | 1.772 | 1.78 |
| Toxicity | hERG (hERG Blockers) | Blockers | Non-blockers | Non-blockers | Non-blockers | Non-blockers | Non-blockers | Non-blockers | Non-blockers |
| H-HT (Human Hepatotoxicity) | HHT positive | HHT negative | HHT positive | HHT positive | H-HT negative | H-HT negative | HHT negative | H-HT negative | |
| Ames (Ames Mutagenicity) | Ames positive | Ames negative | Ames negative | Ames negative | Ames negative | Ames negative | Ames negative | Ames negative | |
| DILI (Drug Induced Liver Injury) | DILI negative | DILI negative | DILI negative | DILI negative | DILI negative | DILI negative | DILI negative | DILI negative |
In the Absorption part, Isothymol, Thymol, Limonene, P-Cymene, γ-Terpinene, Chloroquine and Captopril present at optimal Caco-2 permeability except β-D- Mannose at low Caco-2 permeability. This test is done to check whether the drug will be easily absorbed in the intestine or not. However Isothymol, Thymol, Limonene, P-Cymene, γ-Terpinene, Chloroquine and Captopril show a positive HIA test so they will be well absorbed by the human intestine. The inhibition of the Pgp glycoprotein facilitates the transport of many drugs inside the cell, according to our results Isothymol is the only pgp inhibitor so it will be easily absorbed by the cells. Furthermore in the distribution part, the binding of drugs to plasma proteins is also an important pharmacological parameter; in our study the Captopril drug give a low capacity and negative test but Chloroquine drug and Isothymol show a good capacity and positive test for binding to plasma proteins so they are able to cross the blood-brain barrier. Also in the metabolism part, the cytochrome P450 is an enzyme family which metabolizes the drugs, so in our test only Isothymol is the inhibitor of CYP 450 A2, CYP4502C9, CYP4502C19, and also the substrate of CYP450A2, CYP4593A4, CYP4502C19, CYP4502D6, so the Isothymol can be metabolized by these enzymes . In the excretion part, the half-life of a drug describes the time required for the amount of a drug to be reduced in the body by half or 50% (Swierczewska et al., 2015; Smalling, 1996; Sahin & Benet, 2008). The series of ligands studied have a half-life between 1.129 h and 2.23 h of which the Isothymol ligand of the plant Ammoides verticillata has a significant half-life. That is to say, they give a potential effectiveness.
Regarding the toxicity part, HERG is a K + channel found in the heart muscle and ensures the correct rhythm of the heart, if HERG is blocked by certain drugs, it can cause cardiac arrhythmia and death (Sanguinetti et al., 1995; Aronov, 2005), the Chloroquine drug which is used for the treatment of COVID-19 is blocker to HERG so it can cause cardiac arrhythmia and death but the Isothymol is not HERG blocker which gives good result as drug. For the H-HT, AMES, Chloroquine drug gives the positive test but Isothymol present negative tests in the toxicity part, and all the molecules derived from the Ammoides verticillata present negative tests. In this ADMET part it can be suggested that the Isothymol molecule has better drug properties than the Chloroquine drug.
Prediction tests were also carried out for 10 biological activities, the results of which are in the Table 6. We note that all the ligands show some biological activities among the 10 tested.
Table 6.
The PASS prediction results of the biological activities of the ligands series.
| Sl no | Biological activities | Chloroquine |
Isothymol |
beta-D-Mannose |
Captopril |
Thymol |
limonene |
p-cymene |
γ-terpinene |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Predicted LD50: 1190mg/kg |
Predicted LD50: 2000mg/kg |
Predicted LD50: 1190mg/kg |
Predicted LD50: 1190mg/kg |
Predicted LD50: 1190mg/kg |
Predicted LD50: 1190mg/kg |
Predicted LD50: 1190mg/kg |
Predicted LD50: 1190mg/kg |
||||||||||
| Predicted Toxicity Class: 4 |
Predicted Toxicity Class: 4 |
Predicted Toxicity Class: 4 |
Predicted Toxicity Class: 4 |
Predicted Toxicity Class: 4 |
Predicted Toxicity Class: 4 |
Predicted Toxicity Class: 4 |
Predicted Toxicity Class: 4 |
||||||||||
| Pa | Pi | Pa | Pi | Pa | Pi | Pa | Pi | Pa | Pi | Pa | Pi | Pa | Pi | Pa | Pi | ||
| 01 | Membrane integrity agonist | 0,279 | 0,210 | 0,737 | 0,011 | 0,844 | 0,005 | – | – | 0,802 | 0,036 | 0,375 | 0,151 | 0,745 | 0,047 | – | – |
| 02 | HMOX1 expression enhancer | – | – | 0,335 | 0,079 | 0,383 | 0,064 | – | – | 0,635 | 0,015 | 0,391 | 0,062 | 0,509 | 0,034 | 0,368 | 0,068 |
| 03 | Chlordecone reductase inhibitor | – | – | 0,610 | 0,059 | – | – | – | – | 0,784 | 0,023 | 0,582 | 0,065 | 0,769 | 0,026 | 0,643 | 0,052 |
| 04 | HIF1A expression inhibitor | – | – | 0,534 | 0,047 | 0,514 | 0,053 | – | – | 0,808 | 0,011 | – | – | 0,815 | 0,011 | – | – |
| 05 | Histidine kinase inhibitor | – | – | 0,251 | 0,161 | 0,297 | 0,105 | – | – | 0,373 | 0,062 | – | – | – | – | – | – |
| 06 | Aldehyde oxidase inhibitor | 0,797 | 0,009 | – | – | – | – | 0,386 | 0,069 | 0,603 | 0,032 | – | – | 0,492 | 0,051 | 0,300 | 0,094 |
| 07 | Antimutagenic | – | – | – | – | – | – | – | – | – | – | 0,209 | 0,073 | – | – | – | – |
| 08 | Mucomembranous protector | – | – | 0,917 | 0,004 | – | – | 0,766 | 0,029 | 0,922 | 0,004 | – | – | 0,919 | 0,004 | 0,789 | 0,021 |
| 09 | TP53 expression enhancer | – | – | 0,479 | 0,100 | 0,765 | 0,015 | – | – | 0,592 | 0,052 | 0,627 | 0,042 | 0,541 | 0,071 | – | – |
| 10 | Chemopreventive | – | – | 0,305 | 0,036 | 0,573 | 0,011 | – | – | 0,360 | 0,027 | – | – | 0,234 | 0,054 | – | – |
Table 7 shows five toxic effects (Lagunin et al., 2000) tested for all the ligands of which it is noted that Chloroquine and captopril drugs presents the active Immunotoxicity test but Isothymol is inactive for the five toxic effects. However, according to Table 6, all the ligands Isothymol, Chloroquine and Captopril belong to toxicity class 4 with (300 <LD50 ≤ 2000), so they would be harmful if swallowed.
Table 7.
The PASS prediction results showing the adverse and toxic effects of the ligands series.
| Sl no | Target | Chloroquine |
Isothymol |
beta-D-Mannose |
Captopril |
Thymol |
limonene |
p-cymene |
γ-terpinene |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Probability | Prediction | Probability | Prediction | Probability | Prediction | Probability | Prediction | Probability | Prediction | Probability | Prediction | Probability | Prediction | Probability | Prediction | ||
| 01 | Immunotoxicity | 0.96 | Active | 0.89 | Inactive | 0.96 | Active | 0.96 | Active | 0.93 | Inactive | 0.96 | Active | 0.96 | Active | 0.96 | Active |
| 02 | Cytotoxicity | 0.93 | Inactive | 0.70 | Inactive | 0.93 | Inactive | 0.93 | Inactive | 0.89 | Inactive | 0.93 | Inactive | 0.93 | Inactive | 0.93 | Inactive |
| 03 | Mutagenicity | 0.97 | Inactive | 0.84 | Inactive | 0.97 | Inactive | 0.97 | Inactive | 0.99 | Inactive | 0.97 | Inactive | 0.97 | Inactive | 0.97 | Inactive |
| 04 | Carcinogenicity | 0.62 | Inactive | 0.62 | Inactive | 0.62 | Inactive | 0.62 | Inactive | 0.60 | Inactive | 0.62 | Inactive | 0.62 | Inactive | 0.62 | Inactive |
| 05 | Phosphoprotein (Tumor Supressor) p53 | 0.96 | Inactive | 0.97 | Inactive | 0.96 | Inactive | 0.96 | Inactive | 0.96 | Inactive | 0.96 | Inactive | 0.96 | Inactive | 0.96 | Inactive |
The possible sites of metabolism by CYPs 1A2, 2A6, 2B6, 2C19, 2C8, 2C9, 2D6, 2E1 and 3A4 of Isothymol are illustrated in Table 8. The possible sites of a chemical compound, where the metabolism by the isoforms of CYP450 enzymes may be taken place, are indicated by circles on the chemical structure of the molecule (Zaretzki et al., 2013). The P450 SOM predictions showed that Isothymol had 5 sites of metabolism (SOMs) for the CYP 450 2B6 enzyme, 4 sites for CYP 450 2A6, CYP 4502C8, CYP 450 2D6, and 3sites for CYP 4501A2, CYP 450 2C9, CYP 450 2C19 and CYP 450 2E1 .
Table 8.
List of the P450 sites of metabolism prediction study of the ligands molecules.
| Drug Likeness Properties | Isothymol |
|---|---|
| 1A2 | 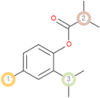 |
| 2A6 | 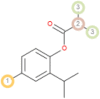 |
| 2B6 | 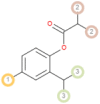 |
| 2C8 | 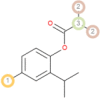 |
| 2C9 | 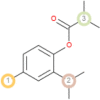 |
| 2C19 | 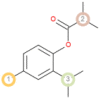 |
| 2D6 | 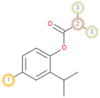 |
| 2E1 | 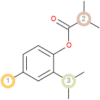 |
| 3A4 | 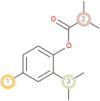 |
3.3. Target prediction
Molecular Target studies are important to find the phenotypical side effects or potential cross reactivity caused by the action of Isothymol the best ligand. The top 25 results are given in the Figure 6. The possible sites of target which the compound may bind to are mostly the targets of protease and oxidoreductase which stimulate the drug reaction accordingly.
Figure 6.

Top-25 of Target Predicted for Isothymol.
3.4. Pharmacophore mapping
The Pharmacophore Mapping is carried for the Isothymol best ligand of the Ammoides Verticilata. Isothymol showed 2 hydrogen acceptor bonds, 6 Hydrophobic groups and 3 Aromatic rings. It also generated a good number of good contacts with the Pharmacophore of ACE2 Figure 7. The Pharmacophore of Isothymol generates a hypothesis which can be used successfully in biological screening for further experiments (Dixon et al., 2006).
Figure 7.

Pharmacophore Mapping of Isothymol. Here, cyan color- hydrogen bond acceptor, orange color-aromatic, green color-hydrophobic.
3.5. Molecular dynamics simulation
The Molecular Dynamics Simulation results are showed in the Figure 8.
Figure 8.

Results of molecular dynamics simulation of Isothymol-ACE2 Receptor. (a) NMA mobility, (b) deformability, (c) B-factor, (d) eigenvalues, (e) variance (red color indicates individual variances and green color indicates cumulative variances), (f) co-variance map (correlated (red), uncorrelated (white) or anti-correlated (blue) motions) and (g) elastic network (darker gray regions indicate more stiffer regions) of the complex.
Figure 8a illustrates the normal mode analysis (NMA) of Isothymol-ACE2 complex. The deformability graph of the complex illustrates the peak in the graphs correspond to the regions in the protein with deformability Figure 8b. The B-factor graph of the complex gives easy visualization and understanding of the comparison between the NMA and the PDB field of the complex Figure 8c. The eigenvalue of the complex is illustrated in Figure 8d. The docked complex generated eigenvalue of 1.408491e-06. The variance graph indicates the individual variance by red colored bars and cumulative variance by green colored bars Figure 8e. Figure 8f illustrates the co-variance map of the complexes where the correlated motion between a pair of residues is indicated by red color, uncorrelated motion is indicated by white color and anti-correlated motion is indicated by blue color. The elastic map of the complex shows the connection between the atoms and darker gray regions indicate stiffer regions Figure 8g.
From the molecular dynamics study of Isothymol-ACE2 docked complex, it is clear that the complex had a very good amount of deformability Figure 8b as well as it had low eigenvalue of 1.408491e-06, for this reason, this lower eigenvalue, represent the easier the deformability of the complex Figure 8d and also represents the motion stiffness of the protein complex.
However, the variance map showed high degree of cumulative variances than individual variances Figure 8e. The co-variance and elastic network map also produced satisfactory results Figure 8f and 8g.
In vivo studies have shown that the therapeutic qualities of Ammoids verticillata are known to be the oldest in local folk medicine. Indeed, Ibn El Beithar, in his treatise on the simple, in the article Athrilal indicates that this plant was used in the treatment of leprosy by a section of the tribe of Oudjeham, near Bougie in 1220 (Trabut, 1935b). It also has major antibacterial activity with a broad spectrum of action, antifungal (Dubey & Mishra, 1990; Grosjean, 2004), antiviral (Grosjean, 2004), it is used for the treatment of cholera (Avesina, 1985; Schirner, 2004), asthma (Avesina, 1985; Boskabady & Shaikhi, 2000; Schirner, 2004), fever, typohoid fever and bronchopulmonary diseases (Bekhechi, 2002). Thymol and Isothymol molecules from this plant are also widely used in cough medicine, throat irritation and cholera (Bhargava et al., 1961; Joshi et al., 1963).
In our study and after the best results obtained for the Isothymol ligand of Ammoides verticilata it can be used as potential agents to treat COVID-19 if we compared with Choloroquine and it emerged as the most potent anti-ACE2 agent.
4. Conclusion
The aim of our study is to investigate the potent of new natural’s compounds such as Isothymol, Thymol, Limonene, P-Cymene and γ-Terpinene from Ammoides verticillata plant as inhibitors for ACE2 target. These compounds are tested in silico study for the inhibition to this enzyme. Molecular docking used to study interaction between new compounds and ACE2 with score energy investigation and druglikeness properties experiments, ADME/T tests, PASS prediction, the P450 Site of Metabolism (SOM), the Molecular Target, Phamacophore Mapping, Molecular dynamics simulation have been performed to verify in silico the drug properties of the best ligand.
The top ligand Isothymol which is the major component (51.2%), in Ammoides verticillata has high binding affinity (Score) and one almost stable interaction with ACE2 target.
Our study showed for these natural components quite similar and very good results in all the aspects into account. Hence, Isothymol can be further developed as drug candidates against ACE2 receptor of coronaviruses, notably that responsible for severe acute respiratory syndrome. Finally we can suggest from this in silico study that Ammoides verticillata (Desf.) Briq. essential oils components can blocks the receptor ACE2 and could make it more difficult for coronavirus to enter cells, which could at least significantly slow the epidemic until the virus disappears. Though these properties are appreciable in silico, further studies of in vitro and clinical studies dealing with SARS-CoV-2 should be considered for further studies.
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- Aanouz I., Belhassan A., Khatabi K. E., Lakhlifi T., Idrissi M. E., & Bouachrine M. (2020). Moroccan Medicinal plants as inhibitors of COVID-19: Computational investigations. Journal of Biomolecular Structure and Dynamics. doi: 10.1080/07391102.2020.1758790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdelouahid D. E., & Bekhechi C. (2002). Pouvoir antimicrobien de l’huile essentielle d’Ammoïdes verticillata (Nûnkha). Biologie et Santé, 4, 91–100. [Google Scholar]
- Aronov A. M. (2005). Predictive in silico modeling for hERG channel blockers. Drug Discovery Today, 10(2), 149–155. doi: 10.1016/S1359-6446(04)03278-7 [DOI] [PubMed] [Google Scholar]
- Avesina A. (1985). Law in medecine (2nd éd.). Soroush Press; pp 187. [Google Scholar]
- Awan F., Obaid A., Ikram A., & Janjua H. (2017). Mutation-structure-function relationship based integrated strategy reveals the potential impact of deleterious missense mutations in autophagy related proteins on hepatocellular carcinoma (HCC): A comprehensive informatics approach. International Journal of Molecular Sciences, 18(1), 139. doi: 10.3390/ijms18010139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekhechi C. (2002). Analyse de l’huile essentielle d’Ammoïdes verticillata (Nûnkha) de la région de Tlemcen et étude de son pouvoir antimicrobien. Mémoire de Magister, option Biologie Moléculaire et Cellulaire, université Abou Bah Belkaïd.
- Bekhechia C., Brice Botib J., Atik Bekkaraa F., Eddine Abdelouahida D., Casanovab J., & Tomib F. (2010). Isothymol in Ajowan essential oil. Naturel Product Communication, 5(7), 71107–71110. [PubMed] [Google Scholar]
- Bhargava H. (1961). Anoxidative effect of Ajowan in a model system. Journal of the American Oil Chemists' Society, 72(10), 1215–1218. in Mehta R. L., & Zayas J. F. (1995). doi: 10.1007/BF02540992 [DOI] [Google Scholar]
- Boopathy S., Poma A. B., & Kolandaivel P. (2020). Novel 2019 coronavirus structure, mechanism of action, antiviral drug promises and rule out against its treatment. Journal of Biomolecular Structure and Dynamics. doi: 10.1080/07391102.2020.1758788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boskabady M. H., & Shaikhi J. (2000). Inhibitory effect of Carum copticum on histamine (H1) receptors ofisolated guinea-pig. Tracheal chains. Journal of Ethnopharmacology, 69(3), 217–227. doi: 10.1016/S0378-8741(99)00116-6 [DOI] [PubMed] [Google Scholar]
- Clément G., &Slenzka K. (Eds.). (2006). Fundamentals of space biology: research on cells, animals, and plants in space. Springer Science & Business Media; pp. 18. [Google Scholar]
- Daina A., Michielin O., & Zoete V. (2017). SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Scientific Reports, 7(1), 42717. doi: 10.1038/srep42717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didierjean C., & Tête-Favier F. (2016). Introduction to protein science. architecture, function and genomics. 3rd ed. Lesk Arthur M. Oxford University Press; pp. 466. Paperback. Price GBP 39.99. ISBN 9780198716846. [Google Scholar]
- Dixon S. L., Smondyrev A. M., Knoll E. H., Rao S. N., Shaw D. E., & Friesner R. A. (2006). PHASE: A new engine for pharmacophore perception, 3D QSAR model development, and 3D database screening: 1. Methodology and preliminary results. Journal of Computer-Aided Molecular Design, 20(10–11), 647–671. doi: 10.1007/s10822-006-9087-6 [DOI] [PubMed] [Google Scholar]
- Dubey N. K., & Mishra A. K. (1990). Evaluation of some essential oils against dermatophytes. T Indian Drugs, 27, 529–531. [Google Scholar]
- Egan W. J., Merz K. M., & Baldwin J. J. (2000). Prediction of drug absorption using multivariate statistics. Journal of Medicinal Chemistry, 43(21), 3867–3877. doi: 10.1021/jm000292e [DOI] [PubMed] [Google Scholar]
- Elfiky A. A., & Azzam E. B. (2020). Novel Guanosine Derivatives against MERS CoVpolymerase: An in silico perspective. Journal of Biomolecular Structure and Dynamics. doi: 10.1080/07391102.2020.1758789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmezayen A. D., Al-Obaidi A., Tegin Şahin A., & Yelekçi K. (2020). Drug repurposing for coronavirus (COVID-19):in silico screening of known drugs againstcoronavirus 3CL hydrolase and protease enzymes. Journal of Biomolecular Structure and Dynamics. doi: 10.1080/07391102.2020.1758791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enayatkhani M., Hasaniazad M., Faezi S., Guklani H., Davoodian P., Ahmadi N., Ali Einakian M., Karmostaji A., & Ahmadi K. (2020). Reverse vaccinology approach to design a novel multi-epitope vaccine candidate against COVID-19: An in silico study. Journal of Biomolecular Structure and Dynamics. doi: 10.1080/07391102.2020.1756411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghose A. K., Viswanadhan V. N., & Wendoloski J. J. (1999). A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. Journal of Combinatorial Chemistry, 1(1), 55–68. doi: 10.1021/cc9800071 [DOI] [PubMed] [Google Scholar]
- Grosjean N. (2004). Huiles essentielles: Se soigner par l’aromathérapie. Ed. Eyrolles, pp 98.
- Guastalegname M., & Vallone A. (2020). Could chloroquine/hydroxychloroquine be harmful in Coronavirus Disease 2019 (COVID-19) treatment? Clinical Infectious Diseases, [Published online ahead of print, 2020 Mar 24]. doi: 10.1093/cid/ciaa321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta M. K., Vemula S., Donde R., Gouda G., Behera L., & Vadde R. (2020). In-silico approaches to detect inhibitors of thehuman severe acute respiratory syndromecoronavirus envelope protein ion channel. Journal of Biomolecular Structure and Dynamics.doi: 10.1080/07391102.2020.1751300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasan A., Ahamad Paray B., Hussain A., Ali Qadir F., Attar F., Mohammad Aziz F., Sharifi M., Derakhshankhah H., Rasti B., Mehrabi M., Shahpasand K., Akbar Saboury A., & Falahati M. (2020). A review on the cleavage priming of the spike protein on coronavirusby angiotensin-converting enzyme-2 and furin. Journal of Biomolecular Structure and Dynamics.doi: 10.1080/07391102.2020.1754293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higley C., & Higley A. (2005). Quick reference guide for using essential oils. 9th ed Abundant Health. Orange, pp. 6–10. [Google Scholar]
- Hopkins C. (2020). “Loss of sense of smell as marker of COVID-19 infection”. Ear, Nose and Throat surgery body of United Kingdom. Retrieved 28 March 2020.
- Hui D S., Azhar I. E., Madani T. A., Ntoumi F., Kock R., Dar O., Ippolito G., Mchugh T. D., Memish Z. A., Drosten C., Zumla A., & Petersen E. (2020). The continuing 2019-nCoV epidemic threat of novel coronaviruses to global health—The latest 2019 novel coronavirus outbreak in Wuhan, China. International Journal of Infectious Diseases, 91, 264–266. doi: 10.1016/j.ijid.2020.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- HyperChem v8 (2009). Molecular Modelling System, Hypercube Inc., 1115 NW 4th Street , Gainesville, FL 32601, USA.
- Jabeer Khan R., Jha R. K., Muluneh Amera G., Jain M., Singh E., Pathak A., Prabha Singh R., Muthukumaran J., & Singh A. K. (2020). Targeting SARS-CoV-2: A systematic drug repurposing approach to identifypromising inhibitors against 3C-like proteinase and 2-O-ribosemethyltransferase. Journal of Biomolecular Structure and Dynamics, 20 April. doi: 10.1080/07391102.2020.1753577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi B. S., Ramanujam S., & Sahena M. B. L. (1963). Improvement of some essential ou bearing spice plants. Bull. Regional Res. Lab. Ja, 1, 94–100. [Google Scholar]
- Kambouche N., & El-Abed D. (2003). Composition of the volatile oil from the aerial parts of Trachyspermum ammi (L.) Sprague from Oran (Algeria). Journal of Essential Oil Research, 15(1), 39–40. doi: 10.1080/10412905.2003.9712259 [DOI] [Google Scholar]
- Khan S. A., Zia K., Ashraf S., Uddin R., & Ul-Haq Z. (2020). Identification of chymotrypsin-like protease inhibitors of SARS-CoV-2viaintegrated computational approach. Journal of Biomolecular Structure and Dynamics. doi: 10.1080/07391102.2020.1751298 [DOI] [PubMed] [Google Scholar]
- Lagunin A., Stepanchikova A., Filimonov D., & Poroikov V. (2000). PASS: 2000. prediction of activity spectra for biologically active substances. Bioinformatics, 16(8), 747–748. doi: 10.1093/bioinformatics/16.8.747 [DOI] [PubMed] [Google Scholar]
- Lawrence B. M. (2006). Progress in essential oils. Perfumer & Flavorist, 31, 63–64. [Google Scholar]
- Li F., Li W., Farzan M., et., & Harrison S. C. (2005). Structure of SARS Coronavirus spike receptor-binding domain complexed with receptor. Science, 309 (5742), 1864–1868. doi: 10.1126/science.1116480 [DOI] [PubMed] [Google Scholar]
- Lipinski C. A., Lombardo F., Dominy B. W., & Feeney P. J. (2001). Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Advanced Drug Delivery Reviews, 46(1–3), 3–26. doi: 10.1016/j.addr.2012.09.019 [DOI] [PubMed] [Google Scholar]
- López-Blanco J. R., Aliaga J. I., Quintana-Ortí E. S., & Chacón P. (2014). iMODS: Internal coordinates normal mode analysis server. Nucleic Acids Research, 42(W1), W271–276. doi: 10.1093/nar/gku339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopéz-Blanco J. R., Garzón J. I., & Chacón P. (2011). iMod: Multipurpose normal mode analysis in internal coordinates. Bioinformatics, 27(20), 2843–2850. doi: 10.1093/bioinformatics/btr497 [DOI] [PubMed] [Google Scholar]
- Molecular Operating Environment (MOE) (2013). Chemical Computing Group Inc., 1010 Sherbooke St. West, Suite #910,. Montreal, QC, Canada, H3A 2R7, (2014).
- Muegge I., Heald S. L., & Brittelli D. (2001). Simple selection criteria for drug-like chemical matter. Journal of Medicinal Chemistry, 44(12), 1841–1846. doi: 10.1021/jm015507e [DOI] [PubMed] [Google Scholar]
- Muralidharan N., Sakthivel R., Velmurugan D., & Michael Gromiha M. (2020). Computational studies of drug repurposing and synergism of lopinavir,oseltamivir and ritonavir binding with SARS-CoV-2 protease against COVID-19. Journal of Biomolecular Structure and Dynamics.doi: 10.1080/07391102.2020.1752802 [DOI] [PubMed] [Google Scholar]
- Narayana C., Somayajulu B. A. R., & Thirumala S. D. (1967). Recovery of fatty oil from spent seeds of Ajowan. Trachyspermum Ammi Linn.). Indian Journal of Technology, 5, 268–269. [Google Scholar]
- Pant S., Singh M., Ravichandiran V., Murty U. S. N., & Srivastava H. (2020). Peptide-like and small-molecule inhibitors against Covid-19Peptide-like and small-molecule inhibitors against Covid-19. Journal of Biomolecular Structure and Dynamics.doi: 10.1080/07391102.1757510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pole S. (2013). Ayurvedic medicine the principles of traditional practice, edited by Dragon S., pp. 122–123. [Google Scholar]
- Prabhakar P. K., Srivastava A., Rao K. K., & Balaji P. V. (2016). Monomerization alters the dynamics of the lid region in Campylobacter jejuni CstII: An MD simulation study. Journal of Biomolecular Structure and Dynamics, 34(4), 778–791. doi: 10.1080/07391102.2015.1054430 [DOI] [PubMed] [Google Scholar]
- Quezel P., & Santa S. (1963). Nouvelle flore de l’Algérie et des régions désertiques méridoniales. Tome II. CNRS, pp. 671. [Google Scholar]
- Sahin S., & Benet L. Z. (2008). The operational multiple dosing half-life: A key to defining drug accumulation in patients and to designing extended release dosage forms. Pharmaceutical Research, 25(12), 2869–2877. doi: 10.1007/s11095-008-9787-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandberg F., & Corrigon D. (2001). Natural remedies, their origins and uses. Taylor & Francis. [Google Scholar]
- Sanguinetti M. C., Jiang C., Curran M. E., & Keating M. T. (1995). A mechanistic link between an inherited and an acquird cardiac arrthytmia: HERG encodes the IKr potassium channel. Cell, 81(2), 299–307. doi: 10.1016/0092-8674(95)90340-2 [DOI] [PubMed] [Google Scholar]
- Sarma P., Sekhar N., Prajapat M., Avti P., Kaur H., Kumar S., Singh S., Kumar H., Prakash A., Prasad Dhibar D., & Medhi B. (2020). In-silico homology assisted identificationof inhibitor of RNA binding against 2019-nCoV N-protein (N terminal domain). Journal of Biomolecular Structure and Dynamics. doi: 10.1080/07391102.2020.1753580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirner M. (2004). Huiles essentielles: Description de plus de 200 huiles essentielles et huiles végétales. Guy Trédaniel, 23. [Google Scholar]
- Shang J., Ye G., Shi K., Wan Y. S., Luo C. M., Aihara H., Geng Q. B., Auerbach A., & Li F. (2020). National Institutes of Health/National Institute of General Medical Sciences (NIH/NIGMS).
- Simon L., Imane A., Srinivasan K. K., Pathak L., & Daoud I. (2017). In silico drug-designing studies on flavanoids as anticolon cancer agents: Pharmacophore mapping, molecular docking, and Monte Carlo method-based QSAR Modeling. Interdisciplinary Sciences: Computational Life Sciences, 9(3), 445–458. doi: 10.1007/s12539-016-0169-4 [DOI] [PubMed] [Google Scholar]
- Smalling R. W. (1996). Molecular biology of plasminogen activators: What are the clinical implications of drug design? The American Journal of Cardiology, 78(12A), 2–7. doi: 10.1016/s0002-9149(96)00736-9 [DOI] [PubMed] [Google Scholar]
- Stewart J. J. (2007). Optimization of parameters for semiempirical methods V: Modification of NDDO approximations and application to 70 elements. Journal of Molecular Modeling, 13(12), 1173–1213. doi: 10.1007/s00894-007-0233-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swierczewska M., Lee K. C., & Lee S. (2015). What is the future of PEGylated therapies. Expert Opinion on Emerging Drugs, 20(4), 531–536. doi: 10.1517/14728214.2015.1113254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trabut L. (1935. a). Flore du Nord de l’Afrique: Répertoire des noms indigènes des plantes spontanées, cultivées et utilisées dans le Nord de l’Afrique. Collection du Centenaire de l’Algérie, Alger.
- Trabut L. C. (1935. b). Répertoire des noms indigènes des plantes spontanées, cultivées et utilisées dans le nord de l’Afrique. Alger. (Collection du centenaire de l’Algérie. Flore du nord de l’Afrique), 355.
- Veber D. F., Johnson S. R., Cheng H. Y., Smith B. R., Ward K. W., & Kopple K. D. (2002). Molecular properties that influence the oral bioavailability of drug candidates. Journal of Medicinal Chemistry, 45(12), 2615–2623. doi: 10.1021/jm020017n [DOI] [PubMed] [Google Scholar]
- Velavan T. P., & Meyer C. G. (2020). The COVID-19 epidemic. Tropical Medicine & International Health, 278–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaretzki J., Bergeron C., Huang T-w., Rydberg P., Swamidass S. J., & Breneman C. M. (2013). RS-WebPredictor: A server for predicting CYP-mediated sites of metabolism on drug-like molecules. Bioinformatics, 29(4), 497–498. doi: 10.1093/bioinformatics/bts705 [DOI] [PMC free article] [PubMed] [Google Scholar]