

Abstract
Natural products have long played a pivotal role in the development of therapeutics for a variety of diseases. Traditionally, soil and marine environments have provided a rich reservoir from which diverse chemical scaffolds could be discovered. Recently, the human microbiome has been recognized as a promising niche from which secondary metabolites with therapeutic potential have begun to be isolated. In this Review, we address how the expansive history of identifying bacterial natural products in other environments is informing the approaches being brought to bear on the study of the human microbiota. We also touch on how these tools can lead to insights about microbe-microbe and host-microbe interactions and help generate biological hypotheses that may lead to developments of new therapeutic modalities.
Introduction
A rapidly growing number of studies suggest a role for the human microbiome in complex pathophysiological processes ranging from the regulation of the immune system to the development of the brain and the central nervous system (Kau et al., 2011; Smith, 2015). Mouse models highlight the necessity of native bacterial ecology for normal physiologic functions and demonstrate that dysbiosis is associated with diseases like obesity, cancer, diabetes, and colitis among others (Bongers et al., 2014; Garrett et al., 2007; Ridaura et al., 2013; Smith et al., 2013). The potential future impact of human microbiome research on human health is evident by the recent surge of clinical trials and venture capital spending in the field (Gormley, 2016; Marchesi et al., 2016). Currently, more than 1,200 clinical trials can be found in the National Institutes of Health clinical trials database using the search query for “gut microbiome.” Just over half of these are active or are actively recruiting patients (https://clinicaltrials.gov).
Despite mounting evidence linking host-associated bacteria to normal development and disease in animal models and correlative evidence in humans, the mechanisms by which specific bacterial functions affect mammalian or microbiome physiology (i.e., effector functions) remain largely undefined. The central dogma of molecular biology, traditionally outlined as the transfer of information from DNA to RNA to protein, stops short of the ultimate end point of much of the information flow in a biological system. In a significant fraction of cellular processes, the end point of information flow is not a protein but is, instead, a small molecule. This is especially true for bacteria which rely heavily on low-molecular-weight compounds (i.e., small molecules or natural products) to interact with their surroundings. The systematic characterization of small molecules produced by human-associated bacteria will undoubtedly be key to deciphering the mechanistic details of the role the human microbiome plays in our health and disease.
The study of biologically active natural products made by bacteria associated with other ecosystems (e.g., soils and marine environments) has traditionally been a very effective gateway to identify small molecules that have proved useful as therapeutics and as tools for modulating complex biological systems (Cragg et al., 1997; Harvey et al., 2015; Knight et al., 2003; Newman et al., 2000; Schreiber et al., 2002). In fact, approximately 80% of medicines identified up to 1996 were either directly derived from or inspired by natural products (Sneader, 1996), and half of all drugs approved since 1994 have their origins in natural products (Butler et al., 2017; Newman and Cragg, 2016). The exploration of human-associated bacteria for the production of small molecules with therapeutic potential is still very much in its infancy. Whether these bacteria will prove to be a gold mine of novel therapeutic molecules, as has been the case for bacteria from most other ecosystems, remains to be seen.
In addition to providing a source for new natural products discovery, studying the metabolites produced by human-associated bacteria can reveal the language of bacteria-host communication. Such knowledge has scientific merit in itself but can also be used to guide new therapies that seek to modulate the human microbiota. The development of therapeutic prebiotics, which are dietary compounds not digestible by human enzymes that serve as substrates for beneficial microbial conversions (Holscher, 2017), could prosper from a more intricate understanding of bacterial metabolic processes in the gut (Martens et al., 2011). Furthermore, therapeutic administration of live bacteria into patients has been used since the mid-20th century for a handful of diseases with limited mechanistic understanding (O’Toole et al., 2017). Therapeutic preparations containing live microorganisms, which are now referred to as live biotherapeutic products (LBPs) by the U.S. Food and Drug Administration (FDA), have been subject to increased regulatory measures over the last decade, signifying a concerted effort to more strictly define how these bacteria function in the human body (Dreher-Lesnick et al., 2017). Thus, defining the chemical crosstalk of human-associated bacteria could promote an informed application of alternative therapies, such as prebiotics and LBPs, in addition to the discovery of discrete therapeutic natural products.
In this Review, we will summarize the methodologies that have been used to mine the human microbiome for metabolites demonstrating biological function (functional metabolites) that might ultimately have therapeutic utility. This is not intended to be a comprehensive review of the small molecules characterized from the microbiome, which have been extensively reviewed elsewhere (Donia and Fischbach, 2015; Mousa et al., 2017; Sharon et al., 2014). Notably, two of the most studied bacterial molecules, colibactin and short-chain fatty acids (SCFAs), have been extensively reviewed (Balskus, 2015; Tan et al., 2014) and are not discussed here. Instead, we hope to broadly describe the approaches being used by researchers to bridge the gap between bacteria and host pathophysiology through the discovery of functional bacterial metabolites. Systematically identifying small molecules produced by host-associated bacteria will be fundamental to the characterization of the human microbiome. Currently, we have only a glimpse of the full scope of metabolites; however, the studies below suggest a bright future for discovery.
Mining for Therapeutic Leads
The mining of the human microbiome for bioactive small molecules has largely come about in the post-genomics era. Consequently, the approaches being most heavily used in these studies tend to first leverage either molecular biology methods or sequence data rather than analytical chemistry to identify natural products or natural product gene clusters of interest. The availability of rapid and inexpensive sequencing of entire bacterial genomes and the development of targeted metagenomic sequencing approaches have enabled researchers to access molecules even if their biosynthetic genes are silent or their bacterial source is recalcitrant to cultivation (Milshteyn et al., 2014; Rutledge and Challis, 2015). Here, we present examples of the various complementary microbiome mining approaches that have led to the characterization of discrete bioactive natural products with potential therapeutic applications. These approaches fall into the broad categories of functional metagenomics, which is aimed at identifying metabolites from metagenomic libraries that show biological activity in functional assays (Figure 1), and sequence-based metagenomics (Figure 2), which relies on (meta)genomic sequence information to guide the discovery of biologically active metabolites. After presenting these approaches, we will highlight alternative paths that fall outside of the distinction of (meta)genomics, particularly those that rely on chemical or functional analysis of secreted metabolites from laboratory cultivation of bacteria without a priori genetic hypotheses (Figure 4).
Figure 1. Functional Metagenomics Workflow.
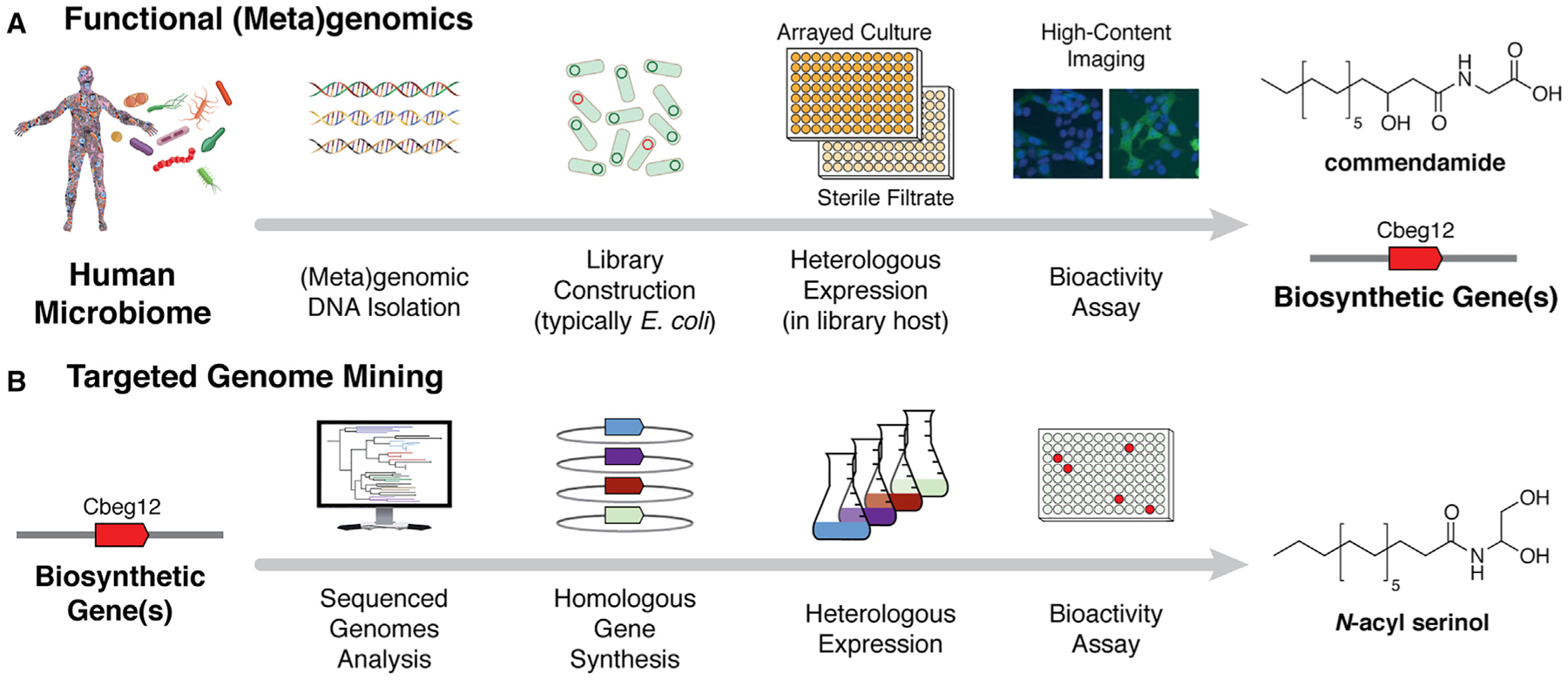
(A) Schematic outline of the functional (meta)genomics mining approach used in the discovery of commendamide, a GPCR agonist.
(B) Functional (meta)genomics provides direct access to the biosynthetic gene(s) responsible for metabolite production, which can then be used to mine available sequence data for similar biosynthetic pathways. This can lead to the discovery of chemically similar metabolites with related function, as was the case with the commendamide-inspired discovery of a second N-acyl GPCR agonist, N-acyl serinol.
Figure 2. Sequence-Based (Meta)genomics Workflows.
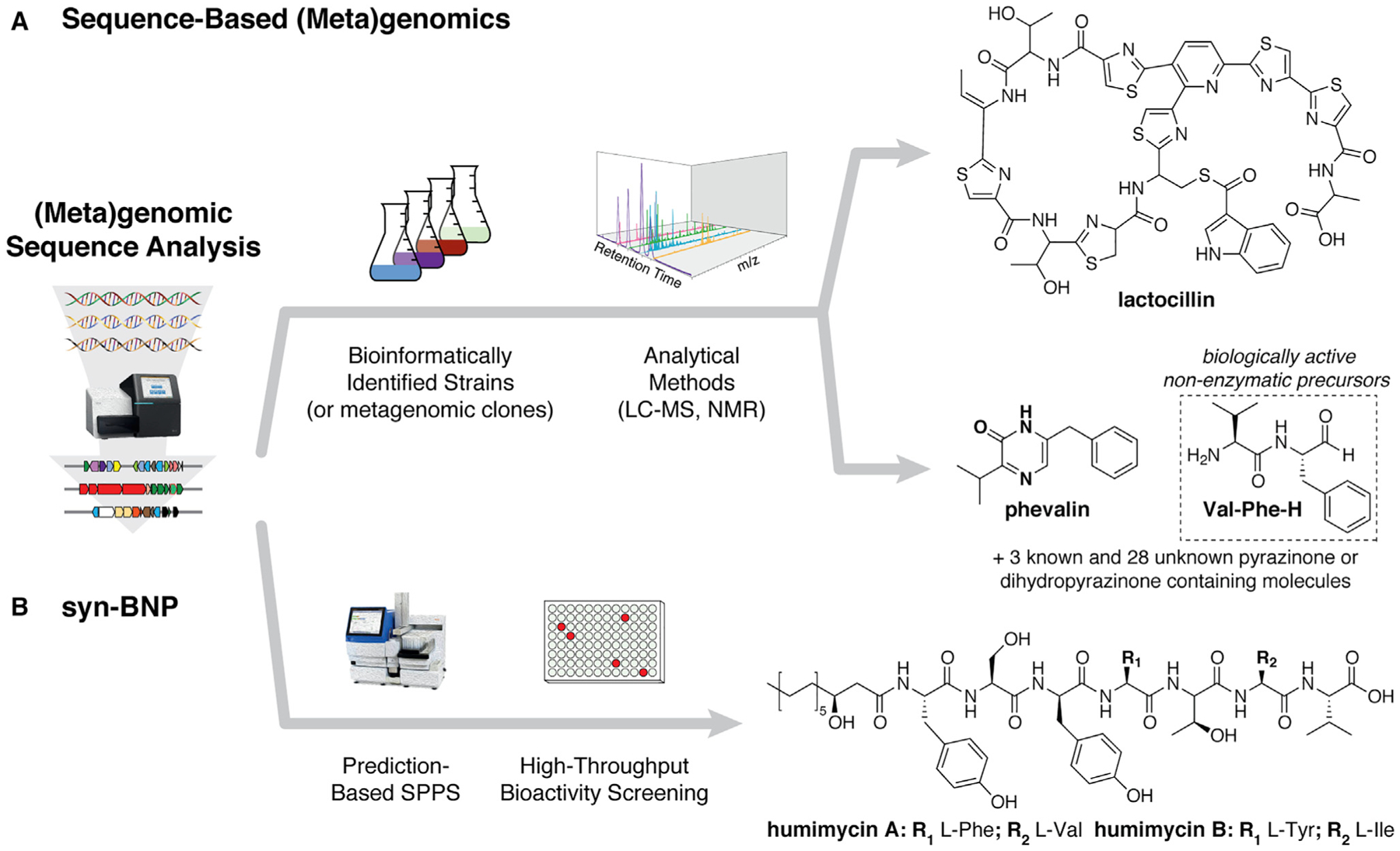
(A) Sequence-based (meta)genomics leverages the power of predictive bioinformatics tools to identify biosynthetic gene clusters in the available sequence data and guide the targeted discovery of bacterial metabolites. This approach was used to discover the novel antibiotic lactocillin and various dipeptide aldehydes with implications in mammalian protease inhibition.
(B) In the synthetic-bioinformatic natural product (syn-BNP) workflow, specific chemical structures of natural products are predicted from analyses of biosynthetic gene clusters, and these molecules are chemically synthesized and subsequently tested for biological activity, such as antibiosis in the case of the humimycins.
Figure 4. Chemistry-Forward Discovery Workflow.
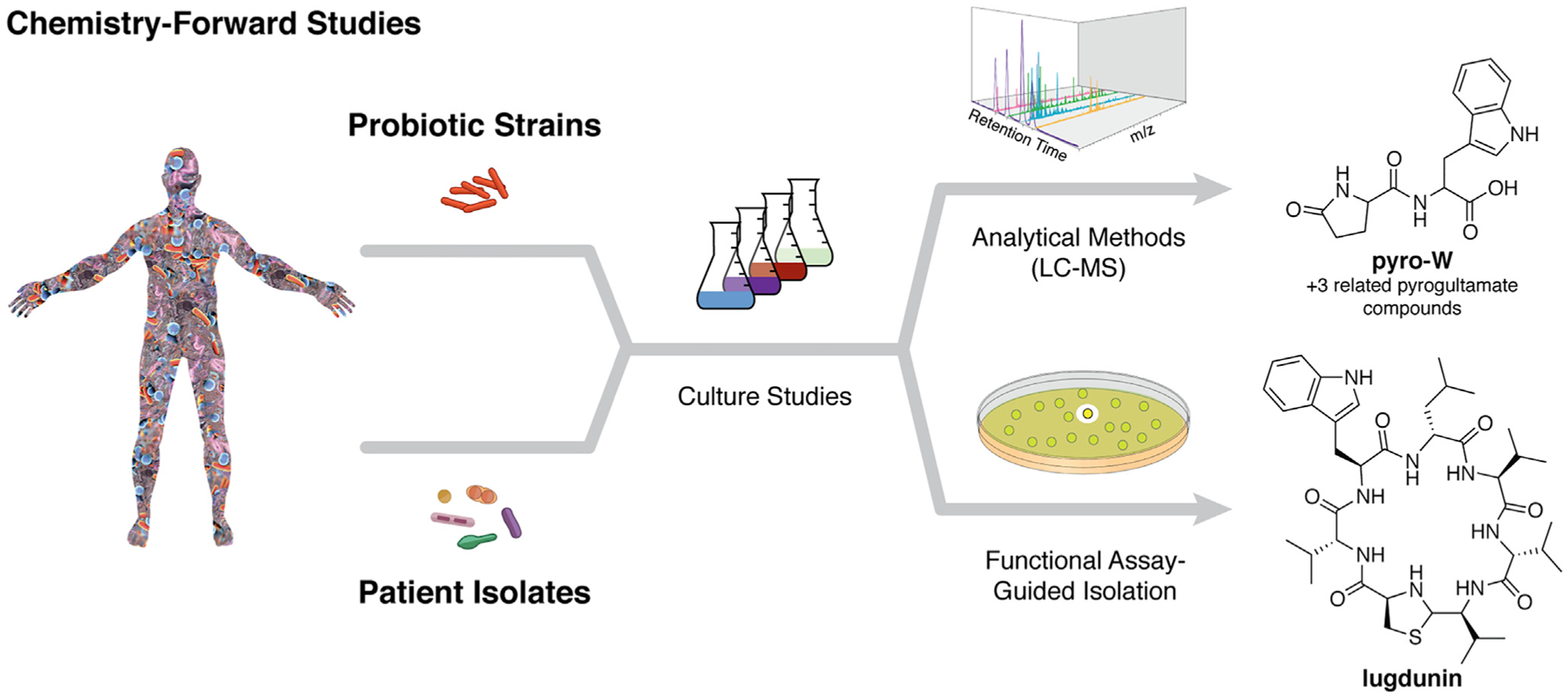
Traditional, chemistry-forward approaches do not rely on biosynthetic gene sequence information but instead use analytical techniques (e.g., pyroglutamate containing compounds) or functional assays (e.g., lugdunin) to identify metabolites of interest from bacterial strains after laboratory cultivation.
Functional Metagenomics: Hunting for Biological Activity in Metagenomic Libraries
The development of sequencing technologies that revolutionized how we study bacteria has also brought with it the recognition that culture-based studies have barely scratched the surface of the bacterial diversity in the environment, with the vast majority of bacteria not being amenable to culturing techniques (Rappé and Giovannoni, 2003). To overcome this limitation, methods have been developed to capture total bacterial DNA from an environment or a biological sample in the form of metagenomic cosmid or fosmid DNA libraries. These methods enable culture-independent studies of the secondary metabolite biosynthesis pathways encoded by bacteria represented in the metagenome (Brady, 2007). Individual clones in these libraries can then be screened for the production of clone-specific metabolites in diverse assays (Brady and Clardy, 2000; Lim et al., 2005; MacNeil et al., 2001; Owen et al., 2012; Wang et al., 2000).
The functional metagenomic approach aims to provide access to bacterial metabolites by screening metagenomic libraries for bioactivities of interest in a high-throughput fashion (Figure 1A). Once a bioactive library clone is identified in a functional screen, it can be isolated and sequenced to identify the gene, or a collection of genes, responsible for the production of the bioactive metabolite. This ability to connect the metabolite directly with its biosynthetic components is one of the major strengths of functional metagenomics. In the context of the human microbiome, where a large fraction of the bacterial species has been fully sequenced, this can also allow the researchers to simultaneously connect the bioactive metabolite with its function in the host and its producer in the microbiome.
Although there are as many ways to employ functional metagenomics as there are screening methods, the most productive screening approaches to date using human microbiome metagenomic libraries have involved screening culture broth filtrates from individually arrayed metagenomic clones against human cell reporter assays. In the earliest such effort, Lakhdari and co-workers used a nuclear factor-kB (NF-kB) activation screen to identify 171 clones in a 2,640-clone human gut microbiome fosmid library that appeared to modulate the activity of the NF-kB reporter (Lakhdari et al., 2010). NF-kB is a rapidly inducible transcription factor that plays a broad role in innumerable cellular responses (Zhang et al., 2017). Importantly, it is intimately involved in the immuno-inflammatory response in the gut, making it a good candidate to detect a broad range of microbe-host interactions (Hayden et al., 2006).
Commendamide: High-Content Imaging Screen Reveals GPCR Modulator
In a more recent study, Cohen et al. (2015) used an NF-kB-driven GFP reporter construct to identify a number of host-associated bacteria effector genes (Cbegs) from cosmid metagenomic libraries constructed using DNA extracted from human stool samples. For this study, a total ~ of 75,000 individually arrayed cosmid clones hosted in E. coli were grown in lysogeny broth medium, and filter-sterilized spent culture broth was then applied to human cells carrying an NF-kB reporter. Library clones that triggered the reporter were then subjected to transposon mutagenesis to identify the genes responsible. Identified effector genes fell into three broad categories: hydrolases (catabolic), transferases (anabolic), and binding proteins. An in-depth investigation of one identified effector gene revealed that it encoded the production of a novel long-chain N-acyl amide, commendamide (Figure 1A), which is structurally similar to endogenous human metabolites that are known to activate G-protein-coupled receptors (GPCRs) (Hanus et al., 2014). Fittingly, commendamide was found to activate a single receptor, GPR132/G2A, in a screen of 242 GPCRs using a cell-based assay. This receptor is believed to be activated endogenously by lysophosphatidylcholine and oxidized long-chain fatty acids and has been implicated in a variety of immune cell functions (Kabarowski, 2009).
N-Acyl Amides: Follow-Up Metagenome Mining Expands Known GPCR Modulators
In a follow-up study, Cohen et al. used bioinformatics and targeted gene synthesis to systematically heterologously express phylogenetically diverse N-acyl amide synthase genes found the Human Microbiome Project (HMP) datasets (Figure 1B) (Cohen et al., 2017; Human Microbiome Project Consortium, 2012). Detailed analysis of extracts derived from E. coli cultures transformed with these constructs revealed six distinct N-acyl amide families that differed by amine head group and fatty acid tail. Individual N-acyl amides were found to interact with specific GPCRs known to regulate gastrointestinal tract physiology, and it was observed that human microbiota-encoded N-acyl amides bear structural similarity to endogenous GPCR-active ligands (Cohen et al., 2015). The clearest overlap in structure and function between bacterial and human ligands was for the activators of endocannabinoid receptor GPR119. The N-acyl serinol structures isolated in this study only differed from 2-oleoyl glycerol by the presence of an amide instead of an ester and from oleoylethanolamide by the presence of an additional ethanol substituent (Figure 1B). Mouse- and cell-based models demonstrate that bacterial GPR119 agonists regulate metabolic hormones and glucose homeostasis as efficiently as the endogenous GPR119 ligands. Colonization of mice with E. coli engineered to produce N-acyl serinol altered glucose homeostasis in these animals at levels similar to what is seen for synthetic GPR119 agonists. This, and other examples, suggests that mimicry of endogenous signaling molecules may be common in host-associated bacteria and that manipulation of microbiota biosynthetic genes provides a new small-molecule therapeutic modality—microbiome-biosynthetic gene therapy (Cohen et al., 2015).
Taken together, these two studies provide a clear example of how functional metagenomics can not only uncover novel molecular mechanisms underlying microbiome-host interactions, but also lead to identification of new therapeutic targets and strategies. Furthermore, using patient-specific source material for metagenomic library construction allows one to take a targeted approach to intelligently design screening strategies. Functional metagenomics still faces a number of challenges, including the ability to capture large biosynthetic gene clusters (BGCs) on a single clone and the expression efficiency of heterologous hosts (Gabor et al., 2004). These two challenges have hindered the application of functional metagenomics to environmental microbiomes and should not be overlooked in the context of mining the human microbiome. However, they are mitigated somewhat by the fact that a large fraction of the known, bacterially produced effectors of host pathophysiology are encoded by individual genes or small sets of genes that can be captured on individual clones and expressed in E. coli (Donia and Fischbach, 2015). We also know that by varying the hosts employed for heterologous expression, a greater variety of metagenomic clones that actively express secondary metabolites can be identified (Craig et al., 2010). As much as 35% to 65% of the bacteria comprising the human microbiome have been amenable to culture. This pool provides many potential heterologous hosts for functional metagenomic studies of the human microbiome (Lagkouvardos et al., 2017).
Approaches for Sequence-Guided (Meta)genome Mining of Natural Products
Genome mining is a computationally driven small-molecule discovery approach that is based on the predictive power of algorithms developed to identify BGCs in sequence data. Existing algorithms largely leverage data from the conserved sequences of well-characterized classes of BGCs to identify new gene clusters in these families (e.g., antiSMASH, eSNaPD, NP.searcher, ClustScan, MutliGeneBlast) (Li et al., 2009; Medema and Fischbach, 2015; Medema et al., 2013; Reddy et al., 2014; Starcevic et al., 2008). More recently, efforts have been made to develop algorithms, such as ClusterFinder, capable of identifying as yet unknown BGC classes (Cimermancic et al., 2014). The computational aspects of sequence-guided natural product discovery are covered in detail in a recent review and will not be addressed here (Medema and Fischbach, 2015). Instead, we will focus on the applications of sequence-based bacterial metabolite discovery methods as they pertain to metabolite discovery from the human microbiome.
Over the past decade, extensive efforts to sequence and annotate bacteria in the human microbiome coupled with intense interest in how the microbiota affects human health have produced an impressive amount of sequence data specific to the human microbiome. This includes more than 3,000 complete and partial reference genomes and thousands of (meta)genomic shotgun sequencing datasets from both healthy and diseased patients (Aagaard et al., 2013; Donia et al., 2014). These data are now being used to query for new secondary metabolite BGCs in the human microbiome. The ultimate goal of these bioinformatics-guided discovery efforts is the expression and functional characterization of metabolites encoded by newly uncovered natural product BGCs.
Lactocillin: Gene Cluster in Human-Associated Bacterium Reveals Novel Thiopeptide Antibiotic
One of the first efforts to define the global network of putative BGCs in the human microbiome was performed using the ClusterFinder algorithm (Cimermancic et al., 2014). ClusterFinder uses Pfam frequency and neighboring gene relationships learned from a large training set of BGCs to identify known and new BGCs in sequence data. Using the ClusterFinder algorithm to analyze the reference genomes produced by the HMP, Donia et al. identified >14,000 BGCs predicted to encode a broad range of small molecule families, including ribosomally encoded and post-translationally modified peptides (RiPPs), saccharides, polyketides (PKs), nonribosomal peptides (NRPs), nonribosomal peptide synthetase (NRPS)-independent sider phores, and a variety of hybrid BGCs (Cimermancic et al., 2014; Donia et al., 2014). By cross referencing these BGCs with metagenomic shotgun sequencing data generated from healthy participants of the HMP and focusing on gene clusters that were widely distributed across body sites and samples, a subclass of highly modified RiPPs, the thiopeptides, was identified. Based on their prevalence in HMP sequence data, their known activity against Gram-positive bacteria, and the existence of a semisynthetic member of the class in phase II clinical trials for treatment of C. difficile infection, Mullane et al. (2015) postulated that thiopeptides would be an interesting target for therapeutic discovery in the microbiome.
One thiopeptide BGC-containing bacterium, the vaginal isolate Lactobacillus gasseri JV-V03, was found to produce the novel natural product lactocillin (Figure 2A). Consistent with known thiopeptides, lactocillin demonstrated nanomolar-level antibiotic activity against pathogenic bacteria, including Staphylococcus aureus and Enterococcus faecalis. Lactocillin was not, however, active against other related Lactobacillus species common to the vaginal microbiome, suggesting the presence of resistance within the normal niche occupied by the producing species. Interestingly, analysis of publicly available metatran scriptomics data revealed that, while the lactocillin BGC was not widely distributed, related thiopeptide BGCs were actively expressed in a large number of samples originating from the oral microbiome. These data led Donia et al. (2014) to propose that lactocillin and other thiopeptides may play a role in mediating microbe-microbe interactions, including protection of the niches occupied by their producers from pathogen invasion. Notably, this study identified a widely distributed class of antibiotics produced by the human microbiota that demonstrated a narrow spectrum of activity. The microcin colicin V, which demonstrates a similarly narrow antibacterial profile, was recently identified from gut bacteria using a functional metagenomics approach (Cohen et al., 2018). The RiPP family of molecules has been identified from chemistry-forward approaches (Donia and Fischbach, 2015; Mousa et al., 2017) and appears to be an important mechanism by which closely related bacterial species compete in the human body.
Humimycins: Discovery of Novel Syn-BNP Antibiotics
In a new twist on sequence-guided natural product discovery, Chu and coworkers recently used sequence data from the HMP to demonstrate a new bioactive molecule discovery method that combines the predictive power of bioinformatics algorithms with chemical synthesis (Chu et al., 2016). In this approach, natural product-like structures are bioinformatically predicted from BGCs and then chemically synthesized in the lab, resulting in small molecules that are inspired by bacterial biosynthetic systems but not physically derived from bacteria (Figure 2B). Such molecules have been termed synthetic-bioinformatic natural products or syn-BNPs. The major highlight of this methodology is that it bypasses the need to isolate metabolites from large-scale bacterial fermentations, a process that can be arduous for many reasons, including poorly expressed or silent BGCs, low natural titers of metabolites, and time-consuming analytical methodology.
NRPs are some of the most common and diverse bacterial natural products in the environment (Charlop-Powers et al., 2015; Doroghazi et al., 2014). Unsurprisingly, large numbers of NRPS BGCs were identified in the global human microbiome biosynthetic analysis generated using ClusterFinder as described above (Donia et al., 2014). Following bioinformatic prediction of a pool of NRP structures from human microbiome-associated BGCs and production of these peptides by solid-phase peptide synthesis (Minowa et al., 2007; Röttig et al., 2011; Stachelhaus et al., 1999; Weber et al., 2015), Chu et al. (2016) screened the resulting molecules for antibiotic activity against a panel of human commensal and pathogenic bacteria. This screen led to the identification of two novel antibiotics, humimycins A and B (human microbiome mycins) (Figure 2B), which mapped back to NRPS gene clusters encoded by closely related species Rhodococcus equi and Rhodococcus erythropolis. These antibiotics were broadly active against Firmicutes and some Actinobacteria, with particularly potent activity against S. aureus and Streptococcus pneumoniae, including a number of common methicillin-resistant S. aureus isolates. This initial application of the syn-BNP approach focused, once again, on the identification of antibiotics. However, NRPs have an extremely broad range of biological and pharmacological activities, including antifungal (bacillomycin D), cytostatic (bleomycin), and immunosuppressive (cyclosporine), and are also known to be toxins (HC-toxin), siderophores (enterobactin), surfactants (surfactin), and pigments (indigoidine) (Caboche et al., 2008). Thus, the application of the syn-BNP method could extend beyond antibiotic discovery to yield NRPs that interact directly with the host.
Dipeptide Aldehydes: Genome Mining for Widespread Gene Clusters of Unknown Function
The previous stories have touched on how specific genes related to known bioactive classes of natural products have been intentionally sought for their potential to produce molecules with therapeutically useful bioactivities. Genome mining can also be used to gain insight into BGCs that are interesting because of other characteristics, such as the frequency or unique distribution pattern across human-associated bacteria. Drawing on their earlier analysis of the HMP sequence data (Donia et al., 2014) and extending it to include the most recent data from the HMP, Guo et al. (2017) identified a family of 47 NRPS BGCs of unknown function that were present in >90% of HMP stool samples. What made this family interesting was the fact that nearly all of these BGCs were identified in anaerobic Firmicutes from the Clostridia class residing in the guts of humans and other mammals, with very few instances observed in non-gut inhabiting, related organisms. This analysis suggested that the small-molecule product of these BGCs may play a role in the biology of host colonization. Heterologous expression studies revealed that this family of BGCs encodes production of a large family of pyrazinones and dihydropyrazinones (Figure 2A), and analysis of a publicly available transcriptomics dataset suggested that these gene clusters were actively expressed under host colonization conditions (Franzosa et al., 2014). Guo et al. (2017) suggest that these cyclic metabolites are derived via a non-enzymatic mechanism in which the terminal aldehyde is attacked by the primary amine on the opposite terminus to form a heterocycle that is prone to oxidation. Furthermore, they suggest that the precursor to this non-enzymatic step is biologically active as an electrophilic acceptor of proteinaceous nucleophiles. Electrophilic substrates, particularly aldehydes, have been known to covalently inhibit the active sites of serine and cysteine proteases and the proteasome (Siklos et al., 2015). The peptide aldehydes in this study showed preferential inhibition of cathepsin-type cysteine proteases in vitro. Cathepsins are implicated in diverse human diseases (Kramer et al., 2017). They are known to play a role in immune surveillance, particularly in the lysosome, leading Guo et al. (2017) to propose that these peptide aldehydes could modulate immune responses to a subset of bacteria.
Indoleacrylic Acid: Genome Mining for Mucin Utilizers Generates IBD Hypotheses
Finally, we describe an example of how genome mining was used in an effort to better understand how the gut microbiota is affected by physiological changes to the gut, which led directly to a mechanistic hypothesis regarding a specific bacterial metabolic pathway and inflammatory bowel disease (IBD). An omnipresent layer of complexity in the gastrointestinal tract is the network of highly O-glycosylated mucin proteins, particularly mucin 2 (MUC2), between the epithelial cells apical surface and the dynamic flow of the lumen (Birchenough et al., 2015). Knowing that decreased intestinal mucus layers at the interface between bacteria and host epithelium are common in IBD, Wlodarska et al. (2017) reasoned that populations of mucin-utilizing bacteria could be important to IBD pathology. By compiling a dataset of genes that are likely to be involved in utilization of mucin and using it to query the existing metagenomic sequencing datasets from healthy participants in the HMP, Wlodarska and colleagues found that predicted mucin utilizers were more abundant in Bacteroidales and Clostridiales orders than was previously thought. A particularly robust mucin utilizer, Peptostreptococcus russellii, was found to have protective properties in the dextran sodium sulfate (DSS)-induced colitis model in mice. Differential analysis of metabolic genes in the genomes of P. russellii and a related strain that did not protect from colitis led to the identification of a specific aromatic amino acid metabolic pathway in P. russellii. This cluster, fldAIBC, encoded enzymes that produced indoleacrylic acid, 3-indolepropionic acid, and 3-(4-hydroxyphenyl)propionic acid from amino acid substrates (Figure 3). Wlodarska et al. (2017) used cytokine profiling and gene expression analysis to interrogate the in vitro anti-inflammatory effect of these three metabolites on various immune cell lines in traditional or spheroid cultures and patient-derived peripheral blood mononuclear cells. Most importantly, stool metagenomic samples from patients with IBD were analyzed, and the fldAIBC cluster was found in a lower percentage of patients with either ulcerative colitis or Crohn’s disease, relative to healthy patients. These data indicate that a healthy mucus layer can promote the colonization of bacteria that secrete anti-inflammatory aromatic amino acids and that dysbiosis caused by decreased mucus layers could eliminate an ecological niche for these protective species.
Figure 3. Simple Aromatic Amino Acids with Demonstrated Effects in Microbe-Host Relationship.
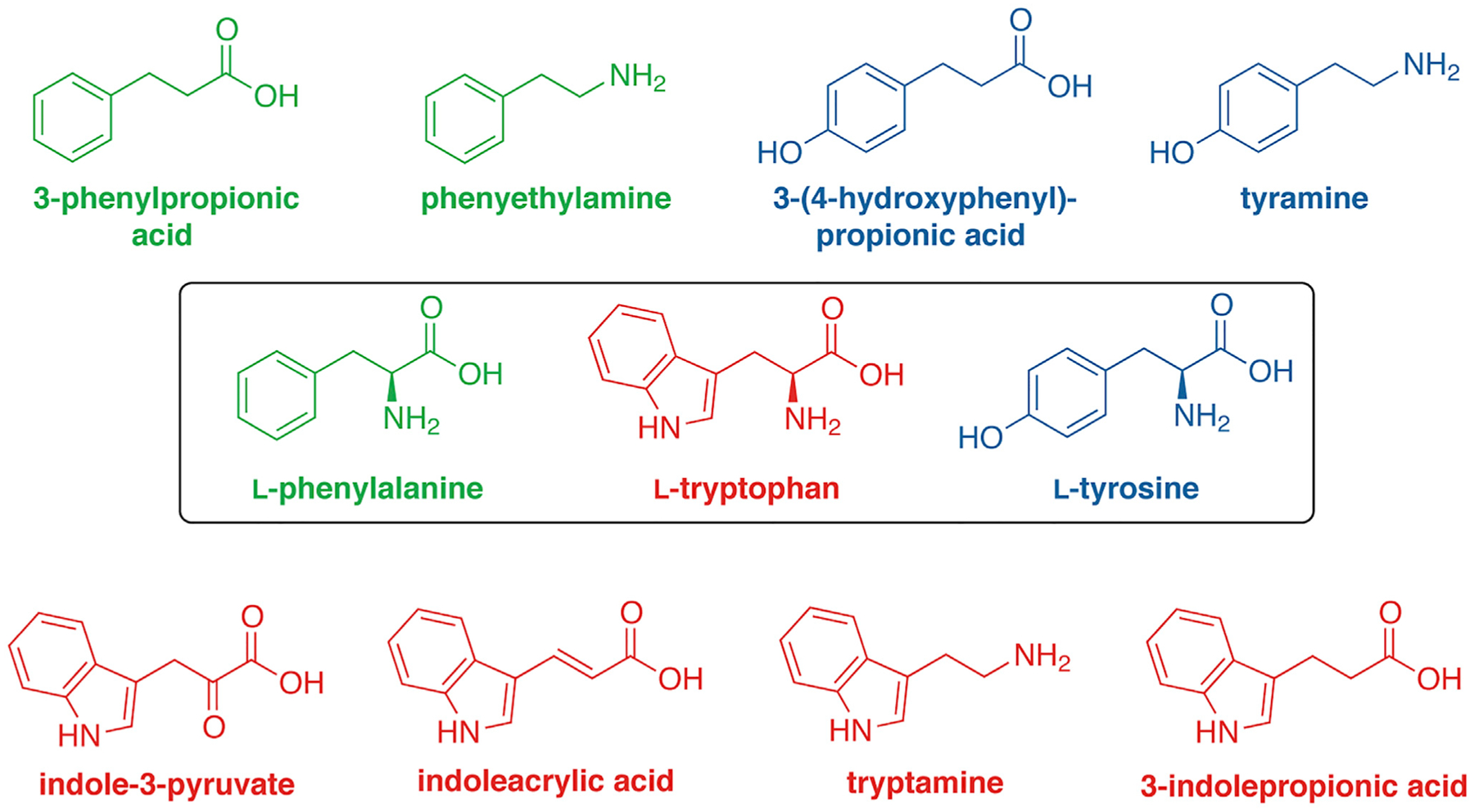
A growing number of simple derivatives of aromatic amino acids (inside the box) have been found to not only be produced by commensal bacteria but elicit functional responses during in vitro or in vivo biological experiments. These studies suggest that bacteria take advantage of simple enzymatic processes using abundant substrates to interact with their environment. Color of derivatives indicates matching amino acid source.
(Meta)genome mining is an approach that has revitalized small-molecule discovery and is indispensable in the present, post-genomic age of studying microbial natural products. A key advantage of mining DNA over cultivating bacteria is that biosynthetic pathways are revealed even when the metabolites they encode cannot be detected in the broth of a laboratory fermentation. These types of analysis have been particularly powerful for finding novel incorporations of known chemical motifs (Ju et al., 2015). However, newer algorithms are starting to address the reliance on sequence homology and accurate annotation, offering prospects for discovery of new classes of useful bacterial metabolites (Medema and Fischbach, 2015). As bioinformatics tools used to predict natural product biosynthetic gene clusters from (meta)genome sequences continue to advance and the number of sequenced bacteria that can be explored continues to rapidly grow, (meta)genome mining will continue to be an increasingly powerful methodology.
Primary Metabolite Derivatives: Putative Functions of Simple Molecules
It is worth noting that the story of indoleacrylic acid adds to a growing body of evidence that suggests a role for simple metabolites produced by the human microbiota (Figure 3) (Devlin et al., 2016; Marcobal et al., 2013). One such class is the biogenic amines, which are chemically simple, amine-containing molecules that are produced from amino acid precursors, namely, the aromatic amino acids phenylalanine, tryptophan, tyrosine, and histidine (Figure 3) (Fan et al., 2017). This class of metabolites is produced by both eukaryotes and prokaryotes and consists of the major neurotransmitters serotonin, dopamine, epinephrine, norepinephrine, and histamine, as well as functionally diverse trace amines, which includes many of the biosynthetic precursors or derivatives of the listed neurotransmitters. Included in this latter group are the decarboxylated aromatic amino acids: tryptamine, tyramine, and phenylethylamine (Figure 3). These metabolites can be further diversified with functionalization, such as hydroxylation (e.g., octopamine), methylation (e.g., N-methyltyramine), or a variation of aromatic substitution pattern (e.g., m-octopamine).
With knowledge that simple metabolites (1) are produced in both bacteria and humans via simple enzymatic pathways, (2) have myriad effects upon binding their cognate receptor, and (3) are easily accessible from the large quantities of amino acid precursors taken in through diet, it is no surprise that they have been some of the most oft-reported gut microbiome-associated metabolites in recent years (Gao et al., 2018). While their structural complexity does not compare to that of many of the well-known biomedically relevant natural products isolated from soil bacteria, their inferred biological relevance derived from their chemical similarity to important human signaling molecules, particularly neurotransmitters, has made them an intriguing subset of metabolites to explore in the human microbiome, particularly in the context of the gutbrain axis (Sarkar et al., 2016). Tryptophan derivatives, including tryptamine, 3-indolepropionic acid, indoleacrylic acid, and indole itself, have been widely described by the distribution of their biosynthetic genes within the human microbiome (Luqman et al., 2018; Williams et al., 2014), their systemic distribution in the circulatory system known to be solely due to gut bacteria (Dodd et al., 2017), their putative use as biomarkers for neurological disorders (Coppen et al., 1965; Herkert and Keup, 1969; Smith and Kellow, 1969), and agonism of important gastrointestinal receptors, such as the serotonin receptors (5-HTRs) (Jones, 1982), as well as newly identified receptors (Khan and Nawaz, 2016). In many of the studies of tryptophan-based metabolites, similar molecules resembling other aromatic amino acids are detected, such as phenylalanine and tyrosine, which can be derivatized similarly to tryptophan by both substrate specific and promiscuous enzymes (Luqman et al., 2018). While it remains to be seen whether modulation of simple metabolic pathways can be used to guide the creation of probiotics, their prominence as functional metabolites is undeniable.
Chemistry-Forward Approaches for Natural Product Discovery from Human Microbiome
So far, we have highlighted studies using advanced genomicsbased tools for discovering secondary metabolites with therapeutically relevant bioactivities. Nonetheless, there are innumerable additional examples that did not rely on these genome-centric approaches. In fact, at the most fundamental level, secondary metabolites from pathogenic and nonpathogenic bacteria alike have been characterized for decades primarily via laboratory cultivation and analytical chemistry. These in vitro interrogations of individual bacteria have resulted in the accumulation of a substantial list of molecules, with varying complexity and biological implication, that have been systematically considered throughout recent reviews (Donia and Fischbach, 2015; Mousa et al., 2017). As the human microbiome is currently undergoing intense scientific scrutiny, there is now a desire to seek deeper biological context for secondary metabolites discovered in vitro, a component that has not always been at the forefront of classical natural product discoveries. Strong foundations of chemistry-forward discovery techniques are now more regularly being used to generate interesting biological hypotheses based on the identified natural products.
Pyroglutamic Acids: Modern Chemical Analysis of Targeted Bacteria
The following study is one of many examples in which a classical analytical chemistry platform, featuring liquid chromatography mass spectrometry (LC-MS), is used to examine the metabolites produced by a human-associated bacterium in vitro. However, this study also demonstrates how supplemental in vivo experiments can be used to generate biological hypotheses to explain the benefit of a common probiotic bacterium. In 2009, multiple studies reported that the beneficial probiotic Lactobacillus plantarum could decrease inflammation in vivo and that the spent media from L. plantarum culture could do so by modulating NF-kB in vitro (Bansal et al., 2010; Petrof et al., 2009; van Baarlen et al., 2009). In an effort to pinpoint the mechanism of this bacterial-induced phenotype, Zvanych et al. (2014) applied principal component analysis to the data generated by LC-MS to identify metabolic fluctuations throughout the growth stages of a L. plantarum culture. Their analyses resulted in the discovery of four dipeptide molecules (pyro-phenylalanine, pyro-leucine, pyro-isoleucine, and pyro-tryptophan) each containing a unique pyroglutamic ring (Figure 4). Introduction of pyro-phenylalanine and pyro-tryptophan into the peritoneum of mice led to decreased splenic production of IFN-g, suggesting that these molecules may be an important part of the mechanism by which L. plantarum inhibits inflammatory pathways in vivo. While the mechanistic details of this inhibition must still be defined, this study highlights the use of comparative chemical analysis to provide novel insights into the chemistry of known, biologically relevant microbes. Many of the bacteria used in probiotics contain Lactobacillus species (O’Toole et al., 2017); however, the mechanisms by which they elicit a benefit, if they do at all, are severely lacking in detail. Systematically interrogating the biological activities of the molecules that Lactobacillus and other probiotic species secrete into the gastrointestinal milieu would undoubtedly bring a more detailed molecular understanding to these probiotic therapies.
Lugdunin: Functional Screening of Nasal Bacteria Reveals a Novel Antibiotic
Functional screening of secreted bacterial metabolites, a mainstay of studies screening the environmental microbiota for many years, has also proved to be a successful method for discovering novel natural products from the human microbiome. A typical experimental setup requires the in vitro cultivation of bacteria followed by the direct, or indirect via chemical extraction, application of the resulting secreted metabolite mixture to a biological assay with a robust readout. Queried bacteria with bioactivity of interest can then be fermented in large-scale format, after which the secreted molecules are collected, concentrated, and submitted to bioassay-guided purification using the original antibiotic activity as a guide for serial chromatography methods to ultimately arrive at a pure, active compound. At this point, a variety of techniques, including infrared and ultraviolet-visible spectroscopy, low-resolution and high-resolution mass spectrometry (HRMS), and nuclear magnetic resonance (NMR), can be used to deduce molecular structure.
In the world of pathogenic infections, the most problematic bacteria are commonly opportunistic pathogens that survive in a dormant state in the body until a traumatic physiological event, such as surgery or immunosuppression, causes maleficent expansion (Arias and Murray, 2009; Wertheim et al., 2005). S. aureus is one such opportunistic pathogenic species of bacteria that resides in the nasal cavities of 30% of the population and is prone to be multi-drug resistant (DeLeo et al., 2010; Wertheim et al., 2005). The diverse flora in the human body suggests that host-associated bacteria are proficient in controlling other bacterial species (Hibbing et al., 2010)—a suggestion that has been gaining evidence, as described throughout this Review. In a functional, chemistry-forward approach to find antibiotics produced by nasal microbiota against methicillin-resistant S. aureus (MRSA), Zipperer et al. (2016) assayed a collection of bacteria isolated from the nasal passages of healthy volunteers using a simple agar plate diffusion assay (Figure 4). They found that the S. lugdunensis IVK28 strain produced a novel antibiotic, lugdunin, which demonstrated a broad range of activity across Gram-positive bacteria, including MRSA, vancomycin, and glycopeptide-intermediate resistant strains. Not only could the pure compounds reduce skin infections in murine models, but inoculation of rat models with IVK28 reduced S. aureus levels, while an IVK28 strain with a mutated lugdunin biosynthetic gene cluster did not. Finally, Zipperer et al. (2016) found, in the nasal swabs of 187 patients, that there was a strong inverse relationship between S. lugdunensis and S. aureus, suggesting a putative inhibitory effect of lugdunin. The therapeutic value of antibiotics, such as lugdunin, active against drug-resistant pathogens cannot be overstated; however, an even more exciting conclusion is that application of functional screening, which has been perfected since the golden age of antibiotics, may be ripe for more discoveries of novel antibiotics when applied to culturable, human-associated bacteria.
Metabolomics as a Tool to Determine Chemical Fingerprints of the Human Microbiota
Arguably, the most powerful tools to examine the chemical fingerprint of the human microbiota are metabolomics platforms featuring HRMS. While NMR has been used to measure bacterial metabolites in the past (Jacobs et al., 2008), technological advances allowing for molecular deconvolution of complex biological samples have increasingly made MS the technique of choice in more laboratories (Cameron and Takáts, 2018). There are a variety of ways in which targeted metabolomics has been used to interrogate the chemical fingerprint of the human microbiota (Lamichhane et al., 2018). Promising results have compared primary metabolic derivatives between germ-free and colonized mice (Wikoff et al., 2009), provided biomarkers for cardiovascular health (Brown and Hazen, 2017), and discovered molecules capable of partially resolving neurological disease in mouse models (Hsiao et al., 2013). A full scope of metabolomics in the context of the gut microbiota has been thoroughly reviewed elsewhere (Holmes et al., 2012; Lamichhane et al., 2018). Overall, MSbased metabolomics platforms have been successful in characterizing known metabolites for which reference samples can be used to generate a searchable feature; however, untargeted metabolomics for unknown complex secondary metabolites, such as the antibiotics discussed previously, has yet to yield therapeutic leads. Front-end (data collection) and back-end (data analysis) strategies have been proposed to overcome these challenges (Peisl et al., 2017), which provides hope that mining for novel chemistry using HRMS may be a possibility in the future.
The chemistry-forward approaches outlined above are representative of traditional natural products discovery process, in which bacterial isolates are cultured in the laboratory and their secreted molecules are assessed analytically (e.g., LC/MS, NMR) and assayed for a desired biological effect (e.g., anticancer, antibiotic, antifungal). The strength of these methods is that they do not rely on prediction but instead start with the identification of a physical, active molecule. However, the total biosynthetic potential of the cultured bacterium may not be realized in the cultivation flask, as not all BGCs are highly expressed under laboratory conditions (Rutledge and Challis, 2015). Additionally, the inability to culture fastidious bacteria under laboratory conditions can hamper these efforts; albeit, this caveat is not exclusive to chemistry-forward approaches. Perhaps, the biggest challenge of such approaches is the rediscovery of known molecules. This pitfall could be alleviated with earlier genetic insight. Although, in the context of the human microbiome, rediscovery becomes less relevant because what matters is linking the molecule to its biological function in the microbe-host relationship and not the novelty of its chemical structure.
Conclusion
We separated the approaches outlined in this Review into categories based on the methodologies driving them. It is clear, however, that the studies described here, and others that were not, which have yielded small molecules from the human microbiota, have benefited greatly from multi-disciplinary approaches. Large datasets continue to accumulate, including taxonomy-driven lists of bacteria in defined patient sets, genomic sequences of known host-associated bacteria, metagenomes of human-derived microbiome samples, and metabolic signatures generated by metabolomics of biological samples from patients. In the future, laboratories will hopefully use these diverse resources to generate additional testable hypotheses that, as a whole, will paint a more complete picture of microbe-host interactions mediated through the bacterial metabolome.
At present, the human microbiome appears to be an inexhaustible source of biological hypotheses regarding interactions between the microbes, the host, and the constant flux of xenobiotics. The methodologies outlined here suggest that we are gradually building a toolbox capable of interrogating these complex networks and finding novel chemical effectors. While the therapeutic potential of the natural products that have been discovered to date remains to be seen, accumulating (meta) genomic data suggest that there is likely to be a plethora of both known and new chemical motifs within the human microbiome whose functions have yet to be described. Metabolic investigations are uncovering not only complex bioactive natural products, like those commonly associated with bacteria isolated from many other environments, but also, and perhaps more frequently, simple molecules whose functions appear to dictate the benefit or pathogenicity of the producing bacteria. These findings are opening the door to the potential development of not only small molecules as therapies, but also alternative therapeutic paths, including LBPs and prebiotics, which are under less stringent regulatory oversight than traditional therapeutics and have experienced meteoric rise in interest over the last decade. Clearly, the future is promising for natural products discovery from the human microbiome.
ACKNOWLEDGMENTS
This publication was made possible by grant number R01AT009562 from the National Center for Complementary and Integrative Health (NCCIH) at the National Institutes of Health. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of NCCIH.
REFERENCES
- Aagaard K, Petrosino J, Keitel W, Watson M, Katancik J, Garcia N, Patel S, Cutting M, Madden T, Hamilton H, et al. (2013). The Human Microbiome Project strategy for comprehensive sampling of the human microbiome and why it matters. FASEB J. 27, 1012–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arias CA, and Murray BE (2009). Antibiotic-resistant bugs in the 21st century–a clinical super-challenge. N. Engl. J. Med 360, 439–443. [DOI] [PubMed] [Google Scholar]
- Balskus EP (2015). Colibactin: understanding an elusive gut bacterial genotoxin. Nat. Prod. Rep 32, 1534–1540. [DOI] [PubMed] [Google Scholar]
- Bansal T, Alaniz RC, Wood TK, and Jayaraman A (2010). The bacterial signal indole increases epithelial-cell tight-junction resistance and attenuates indicators of inflammation. Proc. Natl. Acad. Sci. USA 107, 228–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birchenough GM, Johansson ME, Gustafsson JK, Bergström JH, and Hansson GC (2015). New developments in goblet cell mucus secretion and function. Mucosal Immunol. 8, 712–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bongers G, Pacer ME, Geraldino TH, Chen L, He Z, Hashimoto D, Furtado GC, Ochando J, Kelley KA, Clemente JC, et al. (2014). Interplay of host microbiota, genetic perturbations, and inflammation promotes local development of intestinal neoplasms in mice. J. Exp. Med 211, 457–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady SF (2007). Construction of soil environmental DNA cosmid libraries and screening for clones that produce biologically active small molecules. Nat. Protoc 2, 1297–1305. [DOI] [PubMed] [Google Scholar]
- Brady SF, and Clardy J (2000). Long-chain N-acyl amino acid antibiotics isolated from heterologously expressed environmental DNA. J. Am. Chem. Soc 122, 12903–12904. [Google Scholar]
- Brown JM, and Hazen SL (2017). Targeting of microbe-derived metabolites to improve human health: the next frontier for drug discovery. J. Biol. Chem 292, 8560–8568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler MS, Blaskovich MA, and Cooper MA (2017). Antibiotics in the clinical pipeline at the end of 2015. J. Antibiot 70, 3–24. [DOI] [PubMed] [Google Scholar]
- Caboche S, Pupin M, Leclère V, Fontaine A, Jacques P, and Kucherov G (2008). NORINE: a database of nonribosomal peptides. Nucleic Acids Res. 36, D326–D331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron SJ, and Takáts Z (2018). Mass spectrometry approaches to metabolic profiling of microbial communities within the human gastrointestinal tract. Methods. . Published online April 25, 2018. 10.1016/j.ymeth.2018.04.027. [DOI] [PubMed] [Google Scholar]
- Charlop-Powers Z, Owen JG, Reddy BV, Ternei MA, Guimarães DO, de Frias UA, Pupo MT, Seepe P, Feng Z, and Brady SF (2015). Global biogeographic sampling of bacterial secondary metabolism. Elife 4, e05048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu J, Vila-Farres X, Inoyama D, Ternei M, Cohen LJ, Gordon EA, Reddy BV, Charlop-Powers Z, Zebroski HA, Gallardo-Macias R, et al. (2016). Discovery of MRSA active antibiotics using primary sequence from the human microbiome. Nat. Chem. Biol 12, 1004–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimermancic P, Medema MH, Claesen J, Kurita K, Wieland Brown LC, Mavrommatis K, Pati A, Godfrey PA, Koehrsen M, Clardy J, et al. (2014). Insights into secondary metabolism from a global analysis of prokaryotic biosynthetic gene clusters. Cell 158, 412–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen LJ, Kang HS, Chu J, Huang YH, Gordon EA, Reddy BV, Ternei MA, Craig JW, and Brady SF (2015). Functional metagenomic discovery of bacterial effectors in the human microbiome and isolation of commendamide, a GPCR G2A/132 agonist. Proc. Natl. Acad. Sci. USA 112, E4825–E4834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen LJ, Esterhazy D, Kim SH, Lemetre C, Aguilar RR, Gordon EA, Pickard AJ, Cross JR, Emiliano AB, Han SM, et al. (2017). Commensal bacteria make GPCR ligands that mimic human signalling molecules. Nature 549, 48–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen LJ, Han S, Huang YH, and Brady SF (2018). Identification of the colicin V bacteriocin gene cluster by functional screening of a human microbiome metagenomic library. ACS Infect. Dis 4, 27–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppen A, Shaw DM, Malleson A, Eccleston E, and Gundy G (1965). Tryptamine metabolism in depression. Br. J. Psychiatry 111, 993–998. [DOI] [PubMed] [Google Scholar]
- Cragg GM, Newman DJ, and Snader KM (1997). Natural products in drug discovery and development. J. Nat. Prod 60, 52–60. [DOI] [PubMed] [Google Scholar]
- Craig JW, Chang FY, Kim JH, Obiajulu SC, and Brady SF (2010). Expanding small-molecule functional metagenomics through parallel screening of broad-host-range cosmid environmental DNA libraries in diverse proteobacteria. Appl. Environ. Microbiol 76, 1633–1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLeo FR, Otto M, Kreiswirth BN, and Chambers HF (2010). Community-associated meticillin-resistant Staphylococcus aureus. Lancet 375, 1557–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devlin AS, Marcobal A, Dodd D, Nayfach S, Plummer N, Meyer T, Pollard KS, Sonnenburg JL, and Fischbach MA (2016). Modulation of a circulating uremic solute via rational genetic manipulation of the gut microbiota. Cell Host Microbe 20, 709–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodd D, Spitzer MH, Van Treuren W, Merrill BD, Hryckowian AJ, Higginbottom SK, Le A, Cowan TM, Nolan GP, Fischbach MA, and Sonnenburg JL (2017). A gut bacterial pathway metabolizes aromatic amino acids into nine circulating metabolites. Nature 551, 648–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donia MS, and Fischbach MA (2015). HUMAN MICROBIOTA. Small molecules from the human microbiota. Science 349, 1254766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donia MS, Cimermancic P, Schulze CJ, Wieland Brown LC, Martin J, Mitreva M, Clardy J, Linington RG, and Fischbach MA (2014). A systematic analysis of biosynthetic gene clusters in the human microbiome reveals a common family of antibiotics. Cell 158, 1402–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doroghazi JR, Albright JC, Goering AW, Ju KS, Haines RR, Tchalukov KA, Labeda DP, Kelleher NL, and Metcalf WW (2014). A roadmap for natural product discovery based on large-scale genomics and metabolomics. Nat. Chem. Biol 10, 963–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreher-Lesnick SM, Stibitz S, and Carlson PE Jr. (2017). U.S. regulatory considerations for development of live biotherapeutic products as drugs. Microbiol. Spectr 5, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan P, Song P, Li L, Huang C, Chen J, Yang W, Qiao S, Wu G, Zhang G, and Ma X (2017). Roles of biogenic amines in intestinal signaling. Curr. Protein Pept. Sci 18, 532–540. [DOI] [PubMed] [Google Scholar]
- Franzosa EA, Morgan XC, Segata N, Waldron L, Reyes J, Earl AM, Giannoukos G, Boylan MR, Ciulla D, Gevers D, et al. (2014). Relating the metatranscriptome and metagenome of the human gut. Proc. Natl. Acad. Sci. USA 111, E2329–E2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabor EM, Alkema WB, and Janssen DB (2004). Quantifying the accessibility of the metagenome by random expression cloning techniques. Environ. Microbiol 6, 879–886. [DOI] [PubMed] [Google Scholar]
- Gao J, Xu K, Liu H, Liu G, Bai M, Peng C, Li T, and Yin Y (2018). Impact of the gut microbiota on intestinal immunity mediated by tryptophan metabolism. Front. Cell. Infect. Microbiol 8, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett WS, Lord GM, Punit S, Lugo-Villarino G, Mazmanian SK, Ito S, Glickman JN, and Glimcher LH (2007). Communicable ulcerative colitis induced by T-bet deficiency in the innate immune system. Cell 131, 33–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gormley B (2016). Microbiome companies attract big investments. Wall Street Journal, September 18, 2016 https://www.wsj.com/articles/microbiome-companies-attract-big-investments-1474250460. [Google Scholar]
- Guo CJ, Chang FY, Wyche TP, Backus KM, Acker TM, Funabashi M, Taketani M, Donia MS, Nayfach S, Pollard KS, et al. (2017). Discovery of reactive microbiota-derived metabolites that inhibit host proteases. Cell 168, 517–526. e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanus L, Shohami E, Bab I, and Mechoulam R (2014). N-acyl amino acids and their impact on biological processes. Biofactors 40, 381–388. [DOI] [PubMed] [Google Scholar]
- Harvey AL, Edrada-Ebel R, and Quinn RJ (2015). The re-emergence of natural products for drug discovery in the genomics era. Nat. Rev. Drug Discov 14, 111–129. [DOI] [PubMed] [Google Scholar]
- Hayden MS, West AP, and Ghosh S (2006). NF-kappaB and the immune response. Oncogene 25, 6758–6780. [DOI] [PubMed] [Google Scholar]
- Herkert EE, and Keup W (1969). Excretion patterns of tryptamine, indoleacetic acid, and 5-hydroxyindoleacetic acid, and their correlation with mental changes in schizophrenic patients under medication with alpha-methyldopa. Psychopharmacology (Berl.) 15, 48–59. [DOI] [PubMed] [Google Scholar]
- Hibbing ME, Fuqua C, Parsek MR, and Peterson SB (2010). Bacterial competition: surviving and thriving in the microbial jungle. Nat. Rev. Microbiol 8, 15–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes E, Kinross J, Gibson GR, Burcelin R, Jia W, Pettersson S, and Nicholson JK (2012). Therapeutic modulation of microbiota-host metabolic interactions. Sci. Transl. Med 4, 137rv6. [DOI] [PubMed] [Google Scholar]
- Holscher HD (2017). Dietary fiber and prebiotics and the gastrointestinal microbiota. Gut Microbes 8, 172–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao EY, McBride SW, Hsien S, Sharon G, Hyde ER, McCue T, Codelli JA, Chow J, Reisman SE, Petrosino JF, et al. (2013). Microbiota modulate behavioral and physiological abnormalities associated with neuro developmental disorders. Cell 155, 1451–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Human Microbiome Project Consortium (2012). Structure, function and diversity of the healthy human microbiome. Nature 486, 207–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs DM, Deltimple N, van Velzen E, van Dorsten FA, Bingham M, Vaughan EE, and van Duynhoven J (2008). (1)H NMR metabolite profiling of feces as a tool to assess the impact of nutrition on the human microbiome. NMR Biomed. 21, 615–626. [DOI] [PubMed] [Google Scholar]
- Jones RS (1982). Tryptamine: a neuromodulator or neurotransmitter in mammalian brain? Prog. Neurobiol 19, 117–139. [DOI] [PubMed] [Google Scholar]
- Ju KS, Gao J, Doroghazi JR, Wang KK, Thibodeaux CJ, Li S, Metzger E, Fudala J, Su J, Zhang JK, et al. (2015). Discovery of phosphonic acid natural products by mining the genomes of 10,000 actinomycetes. Proc. Natl. Acad. Sci. USA 112, 12175–12180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabarowski JH (2009). G2A and LPC: regulatory functions in immunity. Prostaglandins Other Lipid Mediat. 89, 73–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kau AL, Ahern PP, Griffin NW, Goodman AL, and Gordon JI (2011). Human nutrition, the gut microbiome and the immune system. Nature 474, 327–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan MZ, and Nawaz W (2016). The emerging roles of human trace amines and human trace amine-associated receptors (hTAARs) in central nervous system. Biomed. Pharmacother 83, 439–449. [DOI] [PubMed] [Google Scholar]
- Knight V, Sanglier JJ, DiTullio D, Braccili S, Bonner P, Waters J, Hughes D, and Zhang L (2003). Diversifying microbial natural products for drug discovery. Appl. Microbiol. Biotechnol 62, 446–458. [DOI] [PubMed] [Google Scholar]
- Kramer L, Turk D, and Turk B (2017). The future of cysteine cathepsins in disease management. Trends Pharmacol. Sci 38, 873–898. [DOI] [PubMed] [Google Scholar]
- Lagkouvardos I, Overmann J, and Clavel T (2017). Cultured microbes represent a substantial fraction of the human and mouse gut microbiota. Gut Microbes 8, 493–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakhdari O, Cultrone A, Tap J, Gloux K, Bernard F, Ehrlich SD, Lefèvre F, Doré J, and Blottière HM (2010). Functional metagenomics: a high throughput screening method to decipher microbiota-driven NF-kB modulation in the human gut. PLoS One 5, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamichhane S, Sen P, Dickens AM, Oresic M, and Bertram HC (2018). Gut metabolome meets microbiome: A methodological perspective to understand the relationship between host and microbe. Methods. . Published online April 30, 2018. 10.1016/j.ymeth.2018.04.029. [DOI] [PubMed] [Google Scholar]
- Li MH, Ung PM, Zajkowski J, Garneau-Tsodikova S, and Sherman DH (2009). Automated genome mining for natural products. BMC Bioinformatics 10, 185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim HK, Chung EJ, Kim JC, Choi GJ, Jang KS, Chung YR, Cho KY, and Lee SW (2005). Characterization of a forest soil metagenome clone that confers indirubin and indigo production on Escherichia coli. Appl. Environ. Microbiol 71, 7768–7777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luqman A, Nega M, Nguyen MT, Ebner P, and Götz F (2018). SadAexpressing Staphylococci in the human gut show increased cell adherence and internalization. Cell Rep. 22, 535–545. [DOI] [PubMed] [Google Scholar]
- MacNeil IA, Tiong CL, Minor C, August PR, Grossman TH, Loiacono KA, Lynch BA, Phillips T, Narula S, Sundaramoorthi R, et al. (2001). Expression and isolation of antimicrobial small molecules from soil DNA libraries. J. Mol. Microbiol. Biotechnol 3, 301–308. [PubMed] [Google Scholar]
- Marchesi JR, Adams DH, Fava F, Hermes GDA, Hirschfield GM, Hold G, Quraishi MN, Kinross J, Smidt H, Tuohy KM, et al. (2016). The gut microbiota and host health: a new clinical frontier. Gut 65, 330–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcobal A, Kashyap PC, Nelson TA, Aronov PA, Donia MS, Spormann A, Fischbach MA, and Sonnenburg JL (2013). A metabolomic view of how the human gut microbiota impacts the host metabolome using humanized and gnotobiotic mice. ISME J. 7, 1933–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens EC, Lowe EC, Chiang H, Pudlo NA, Wu M, McNulty NP, Abbott DW, Henrissat B, Gilbert HJ, Bolam DN, and Gordon JI (2011). Recognition and degradation of plant cell wall polysaccharides by two human gut symbionts. PLoS Biol. 9, e1001221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medema MH, and Fischbach MA (2015). Computational approaches to natural product discovery. Nat. Chem. Biol 11, 639–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medema MH, Takano E, and Breitling R (2013). Detecting sequence homology at the gene cluster level with MultiGeneBlast. Mol. Biol. Evol 30, 1218–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milshteyn A, Schneider JS, and Brady SF (2014). Mining the metabiome: identifying novel natural products from microbial communities. Chem. Biol 21, 1211–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minowa Y, Araki M, and Kanehisa M (2007). Comprehensive analysis of distinctive polyketide and nonribosomal peptide structural motifs encoded in microbial genomes. J. Mol. Biol 368, 1500–1517. [DOI] [PubMed] [Google Scholar]
- Mousa WK, Athar B, Merwin NJ, and Magarvey NA (2017). Antibiotics and specialized metabolites from the human microbiota. Nat. Prod. Rep 34, 1302–1331. [DOI] [PubMed] [Google Scholar]
- Mullane K, Lee C, Bressler A, Buitrago M, Weiss K, Dabovic K, Praestgaard J, Leeds JA, Blais J, and Pertel P (2015). Multicenter, randomized clinical trial to compare the safety and efficacy of LFF571 and vancomycin for Clostridium difficile infections. Antimicrob. Agents Chemother 59, 1435–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman DJ, and Cragg GM (2016). Natural products as sources of new drugs from 1981 to 2014. J. Nat. Prod 79, 629–661. [DOI] [PubMed] [Google Scholar]
- Newman DJ, Cragg GM, and Snader KM (2000). The influence of natural products upon drug discovery. Nat. Prod. Rep 17, 215–234. [DOI] [PubMed] [Google Scholar]
- O’Toole PW, Marchesi JR, and Hill C (2017). Next-generation probiotics: the spectrum from probiotics to live biotherapeutics. Nat. Microbiol 2, 17057. [DOI] [PubMed] [Google Scholar]
- Owen JG, Robins KJ, Parachin NS, and Ackerley DF (2012). A functional screen for recovery of 4´-phosphopantetheinyl transferase and associated natural product biosynthesis genes from metagenome libraries. Environ. Microbiol 14, 1198–1209. [DOI] [PubMed] [Google Scholar]
- Peisl BYL, Schymanski EL, and Wilmes P (2017). Dark matter in host-microbiome metabolomics: tackling the unknowns–a review. Anal. Chim. Acta . Published online December 30, 2017. 10.1016/j.aca.2017.12.034. [DOI] [PubMed] [Google Scholar]
- Petrof EO, Claud EC, Sun J, Abramova T, Guo Y, Waypa TS, He SM, Nakagawa Y, and Chang EB (2009). Bacteria-free solution derived from Lactobacillus plantarum inhibits multiple NF-kappaB pathways and inhibits proteasome function. Inflamm. Bowel Dis 15, 1537–1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappé MS, and Giovannoni SJ (2003). The uncultured microbial majority. Annu. Rev. Microbiol 57, 369–394. [DOI] [PubMed] [Google Scholar]
- Reddy BV, Milshteyn A, Charlop-Powers Z, and Brady SF (2014). eSNaPD: a versatile, web-based bioinformatics platform for surveying and mining natural product biosynthetic diversity from metagenomes. Chem. Biol 21, 1023–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridaura VK, Faith JJ, Rey FE, Cheng J, Duncan AE, Kau AL, Griffin NW, Lombard V, Henrissat B, Bain JR, et al. (2013). Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science 341, 1241214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Röttig M, Medema MH, Blin K, Weber T, Rausch C, and Kohlbacher O (2011). NRPSpredictor2–a web server for predicting NRPS adenylation domain specificity. Nucleic Acids Res. 39, W362–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutledge PJ, and Challis GL (2015). Discovery of microbial natural products by activation of silent biosynthetic gene clusters. Nat. Rev. Microbiol 13, 509–523. [DOI] [PubMed] [Google Scholar]
- Sarkar A, Lehto SM, Harty S, Dinan TG, Cryan JF, and Burnet PWJ (2016). Psychobiotics and the manipulation of bacteria-gut-brain signals. Trends Neurosci. 39, 763–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber SL, Nicolaou KC, and Davies K (2002). Diversity-oriented organic synthesis and proteomics. New frontiers for chemistry & biology. Chem. Biol 9, 1–2. [DOI] [PubMed] [Google Scholar]
- Sharon G, Garg N, Debelius J, Knight R, Dorrestein PC, and Mazmanian SK (2014). Specialized metabolites from the microbiome in health and disease. Cell Metab. 20, 719–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siklos M, BenAissa M, and Thatcher GRJ (2015). Cysteine proteases as therapeutic targets: does selectivity matter? A systematic review of calpain and cathepsin inhibitors. Acta Pharm. Sin. B 5, 506–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith PA (2015). The tantalizing links between gut microbes and the brain. Nature 526, 312–314. [DOI] [PubMed] [Google Scholar]
- Smith I, and Kellow AH (1969). Aromatic amines and Parkinson’s disease. Nature 221, 1261. [DOI] [PubMed] [Google Scholar]
- Smith MI, Yatsunenko T, Manary MJ, Trehan I, Mkakosya R, Cheng J, Kau AL, Rich SS, Concannon P, Mychaleckyj JC, et al. (2013). Gut microbiomes of Malawian twin pairs discordant for kwashiorkor. Science 339, 548–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sneader W (1996). Drug Prototypes and Their Exploitation (Wiley; ). [Google Scholar]
- Stachelhaus T, Mootz HD, and Marahiel MA (1999). The specificity conferring code of adenylation domains in nonribosomal peptide synthetases. Chem. Biol 6, 493–505. [DOI] [PubMed] [Google Scholar]
- Starcevic A, Zucko J, Simunkovic J, Long PF, Cullum J, and Hranueli D (2008). ClustScan: an integrated program package for the semi-automatic annotation of modular biosynthetic gene clusters and in silico prediction of novel chemical structures. Nucleic Acids Res. 36, 6882–6892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan J, McKenzie C, Potamitis M, Thorburn AN, Mackay CR, and Macia L (2014). The role of short-chain fatty acids in health and disease. Adv. Immunol 121, 91–119. [DOI] [PubMed] [Google Scholar]
- van Baarlen P, Troost FJ, van Hemert S, van der Meer C, de Vos WM, de Groot PJ, Hooiveld GJ, Brummer RJ, and Kleerebezem M (2009). Differential NF-kappaB pathways induction by Lactobacillus plantarum in the duodenum of healthy humans correlating with immune tolerance. Proc. Natl. Acad. Sci. USA 106, 2371–2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G-Y-S, Graziani E, Waters B, Pan W, Li X, McDermott J, Meurer G, Saxena G, Andersen RJ, and Davies J (2000). Novel natural products from soil DNA libraries in a streptomycete host. Org. Lett 2, 2401–2404. [DOI] [PubMed] [Google Scholar]
- Weber T, Blin K, Duddela S, Krug D, Kim HU, Bruccoleri R, Lee SY, Fischbach MA, Müller R, Wohlleben W, et al. (2015). antiSMASH 3.0-a comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids Res 43 (W1), W237–W243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wertheim HF, Melles DC, Vos MC, van Leeuwen W, van Belkum A, Verbrugh HA, and Nouwen JL (2005). The role of nasal carriage in Staphylococcus aureus infections. Lancet Infect. Dis 5, 751–762. [DOI] [PubMed] [Google Scholar]
- Wikoff WR, Anfora AT, Liu J, Schultz PG, Lesley SA, Peters EC, and Siuzdak G (2009). Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc. Natl. Acad. Sci. USA 106, 3698–3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams BB, Van Benschoten AH, Cimermancic P, Donia MS, Zimmermann M, Taketani M, Ishihara A, Kashyap PC, Fraser JS, and Fischbach MA (2014). Discovery and characterization of gut microbiota decarboxylases that can produce the neurotransmitter tryptamine. Cell Host Microbe 16, 495–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wlodarska M, Luo C, Kolde R, d’Hennezel E, Annand JW, Heim CE, Krastel P, Schmitt EK, Omar AS, Creasey EA, et al. (2017). Indoleacrylic acid produced by commensal Peptostreptococcus species suppresses inflammation. Cell Host Microbe 22, 25–37.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Lenardo MJ, and Baltimore D (2017). 30 years of NF-kB: a blossoming of relevance to human pathobiology. Cell 168, 37–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zipperer A, Konnerth MC, Laux C, Berscheid A, Janek D, Weidenmaier C, Burian M, Schilling NA, Slavetinsky C, Marschal M, et al. (2016). Human commensals producing a novel antibiotic impair pathogen colonization. Nature 535, 511–516. [DOI] [PubMed] [Google Scholar]
- Zvanych R, Lukenda N, Kim JJ, Li X, Petrof EO, Khan WI, and Magarvey NA (2014). Small molecule immunomodulins from cultures of the human microbiome member Lactobacillus plantarum. J. Antibiot 67, 85–88. [DOI] [PubMed] [Google Scholar]