

Abstract
Neural cell adhesion molecule 2 (NCAM2) is involved in the development and plasticity of the olfactory system. Genetic data have implicated the NCAM2 gene in neurodevelopmental disorders including Down syndrome and autism, although its role in cortical development is unknown. Here, we show that while overexpression of NCAM2 in hippocampal neurons leads to minor alterations, its downregulation severely compromises dendritic architecture, leading to an aberrant phenotype including shorter dendritic trees, retraction of dendrites, and emergence of numerous somatic neurites. Further, our data reveal alterations in the axonal tree and deficits in neuronal polarization. In vivo studies confirm the phenotype and reveal an unexpected role for NCAM2 in cortical migration. Proteomic and cell biology experiments show that NCAM2 molecules exert their functions through a protein complex with the cytoskeletal-associated proteins MAP2 and 14-3-3γ and ζ. We provide evidence that NCAM2 depletion results in destabilization of the microtubular network and reduced MAP2 signal. Our results demonstrate a role for NCAM2 in dendritic formation and maintenance, and in neural polarization and migration, through interaction of NCAM2 with microtubule-associated proteins.
Keywords: dendritogenesis, MAP2, microtubules, NCAM2, neuronal differentiation, 14-3-3
Introduction
The precise regulation of neuronal migration and morphogenesis, including polarization, branching and maturation of dendrites, axonal outgrowth, and synaptogenesis, is essential for correct brain development (Namba et al. 2015). Within these different steps, the correct transduction of extracellular and membrane bound signaling proteins is essential to stereotyped cytoskeletal rearrangement and dynamics that ultimately lead to the sustaining or destabilization of these processes (Dent et al. 2011; Takano et al. 2015; Kon et al. 2017; Schelski and Bradke 2017). Among these molecules, cell adhesion molecules (CAMs) play a fundamental role (Missaire and Hindges 2015; Sytnyk et al. 2017; Zinn and Ozkan 2017).
The mammalian NCAM family has two members, NCAM1 and NCAM2 (Pebusque et al. 1998; Makino and McLysaght 2010). The two proteins bear a similar ectodomain with five immunoglobulin domains and two fibronectin type III domains, the latter responsible for binding with fibroblast growth factor receptor (FGFR) (Kiselyov et al. 2003; Rasmussen et al. 2018). NCAM1 has three different isoforms (180, 140, and 120 KDa), and NCAM2 has two isoforms: NCAM2.1 bearing transmembrane and cytoplasmic domains and the shorter NCAM2.2 isoform, which is GPI-anchored (Alenius and Bohm 1997; Yoshihara et al. 1997).
While NCAM1 has been extensively studied, playing a fundamental role in neuronal migration, neurite development and synaptogenesis, and adult plasticity (Jorgensen and Bock 1974; Angata et al. 2007; Muller et al. 2010; Sheng et al. 2013; Leshchyns'ka and Sytnyk 2016), NCAM2 is known to be widely expressed in the central nervous system (CNS), although investigation has been focused on the olfactory system where it is important for the formation and maintenance of dendritic and axonal compartmentalization (Alenius and Bohm 1997; von Campenhausen et al. 1997; Kulahin and Walmod 2010; Winther et al. 2012). Furthermore, the role of NCAM2 in other CNS regions remains largely unknown. Only recently, it has been shown that in the cerebral cortex NCAM2 regulates neurite outgrowth and synapse formation via Src and local calcium spikes (Sheng et al. 2015, 2018) and that at later stages it plays a role in synapse maintenance (Leshchyns'ka et al. 2015).
Genetic analyses provide evidence that deletions and single nucleotide polymorphism in the NCAM2 gene are found in patients with autism spectrum disorder (Molloy et al. 2005; Hussman et al. 2011; Scholz et al. 2016) and in disorders linked to abnormal neurodevelopment (Petit et al. 2015). Moreover, NCAM2 has been proposed as a candidate for the intellectual disability phenotype in Down syndrome since the NCAM2 gene is located on chromosome 21 (Paoloni-Giacobino et al. 1997; Winther et al. 2012). Taken together, these data suggest a relationship of NCAM2 with cortical brain development, although the molecular and cellular mechanisms involved are poorly understood.
In this study, we investigated the role of NCAM2 in neuronal differentiation and morphogenesis using gain- and loss-of-function approaches to modulate NCAM2 expression both in vitro and in vivo. We found that NCAM2 depletion has dramatic effects on neurite development and polarization, with retraction of dendritic arbors and increased numbers of primary dendrites with aberrant morphology and dynamics. These observations were confirmed in vivo, and we were also able to demonstrate the role of NCAM2 in cortical neuronal migration. Furthermore, proteomic, biochemical, and functional rescue experiments provide evidence that NCAM2 exerts the above functions at least in part by modulating microtubule dynamics and by interacting with the microtubule-associated protein MAP2 and some 14-3-3 protein family members. These data reveal novel functions of NCAM2 protein in neural development through a previously unknown NCAM2/cytoskeletal protein complex.
Materials and Methods
All experimental procedures were carried out following the guidelines of the Committee for the Care of Research Animals of the University of Barcelona, in accordance with the directive of the Council of the European Community (2010/63 y 86/609/EEC) on animal experimentation. The experimental protocol was approved by the local University Committee (CEEA-UB, Comitè Ètic d’Experimentació Animal de la Universitat de Barcelona) and by the Catalan Government (Generalitat de Catalunya, Departament de Territori I Sostenibilitat).
Animals and Cell Lines
Hippocampal neurons were obtained from E15-17 CD1 mouse embryos. Brains were dissected in PBS containing 3% glucose, and hippocampi were excised. After trypsin (GIBCO) and DNAse (Roche Diagnostics) treatments, hippocampi were dissociated with gentle sweeping. Cells were counted and seeded onto poly-d-lysine-coated dishes 105 cells/cm2 or 2 × 104 cells/cm2 for low-density cultures. Cells were plated in neurobasal medium containing 2% B27 supplement (GIBCO), penicillin/streptomycin (Life technologies), and Glutamax (Life technologies) and conditioned media from matured glia culture. HEK293T cell lines were grown in DMEM containing 10% FBS (Life technologies) and penicillin/streptomycin (Life technologies). All cells were kept at 37°C in a humidified atmosphere containing 5% CO2.
Antibodies and Reagents
The following commercial primary antibodies were used for immunohistochemistry: anti-GFP (A11122, Invitrogen, 1:2000); anti-ChFP (ab167453, Abcam, 1:300); anti-MAP2 (MA1406, Sigma, 1:2000); anti-NCAM2 (AF778, R&D Systems, 1:250); anti-NCAM2.1 (EB06991, Everest, 1:250); anti-Neurofilament (837 904 SMI-312, Biolegend, 1/300); anti-acetylated α-tubulin (T6793, Sigma, 1/50000); anti-detyrosinated α-Tubulin (AB3201, Sigma, 1/200); and anti-β-III-tubulin (801 201, Biolegends, 1/2500). For western blot (WB), we used anti-NCAM2 (AF778, R&D Systems, 1:500); anti-actin (MAB1501, Chemicon, 1:5000); anti-GFP (A11122, Invitrogen, 1:2000); anti-MAP2 (M9942 clone HM-2, Sigma, 1:1000); anti-14-3-3 (1657, SantaCruz, 1:2000); and anti-Tau (3-repeat isoform RD3, 05-803, Sigma, 1:1000). Alexa Fluor fluorescent secondary antibodies were from Invitrogen. To counterstain nuclei, the tissue and cells were incubated in 2-(4-amidinophenyl)-1H-indole-6-carboxamidine (DAPI, D-6564, Sigma, 1:1000). For actin staining, the cells were incubated in phalloidin-TRICT (P-1951, Sigma, 1:200). Biotinylated secondary antibodies were from Vector Labs; streptavidin-biotinylated/HRP complex and ECL were from GE Healthcare. The HRP-labeled secondary antibodies used for WB were from DAKO. Diaminobenzidine reagent (DAB) and Eukitt mounting media were from Sigma-Aldrich. Mowiol was from Calbiochem. BCA protein assay was from Thermo Fisher.
Plasmids
The target sequences for depletion of both Ncam2 isoforms were GCACCTGGACATCGAATAT (ShNcam2N) and GAAGGTACAGGGAAATAAA (ShNcam2), corresponding to nucleotides 1395-1414 and 2142-2161, respectively, of mouse Ncam2 mRNA. The ShNcam2 sequence was 5′-CGCGTCCCCGAAGGTGCAGGGAAATAAATTCAAGAGATTTATTTCCCTGCACCTTCTTTTTGGAAAT-3′; the ShNcam2N sequence was 5′-CGCGTCCCCGCACCTGGACATCGAATATTTCAAGAGAATATTCGATGTCCAGGTGCTTTTTGGAAAT-3′; and the ShCnt sequence was 5′-GATCCCCGCAGTGCAATATCGGAAACTTCAAGAGAGTTTCCGATATT-GCACTGCTTTT-3. They were cloned into pLVTHM within the MluI and ClaI sites (Plasmid #12247, #Addgene).
The cDNA of Ncam2.2 was amplified with 5′-ACTGGAATTCGTGGCAGCGGAAGGTTCTC-3′ and 5′-ACTGTCTAGAAATTCAGGGGGAAGGCGAAT-3′ from pCR4-TOPO (Mouse Ncam2 cDNA, ABIN4003230) and cloned into pCDNA3.1 (Addgene) using EcoRI/XbaI sites, pCNcam2.2. The c-terminal of Ncam2.1 cDNA was amplified with 5′-ACTGGAATTCGTGGCAGCGGAAGGTTCTC-3′ and 5′-GTGGCTAGAGAAGAAGGTAC-3′ from hippocampal mRNA and cloned into pCNcam2.2 using PstI/XbaI sites, pCNcam2.1.
The cDNA of Ncam2.1 was amplified with 5′-ACCATGAGCCTCCTCCTCTCC-3′ and 5′-CTGACCAAGGTGCTGAAACT-3′and cloned into pWPI (Plasmid #12254, Addgene) within PmeI site. The cDNA of Ncam2.2 was amplified with 5′-ACCATGAGCCTCCTCCTCTCC-3′ and 5′-TCTCTGATCAGGGAGTACCA-3′ and cloned into pWPI (Plasmid #12254, Addgene) within PmeI site.
For the rescue experiment, pCNcam2.1 and pCNcam2.2 vectors were amplified using PCR reaction (QuickChange II, Agilent Technologies); in the first reaction with 5′-AGAGAAGAAGGTACAGGGAAACAAGGACCACATTATCTTGGAGC-3′ and 5′-AGATCAGTGGCTAGAGAAGAAAGTTCAGGGTAACAAGGACCA-3′ and the second reaction with 5′-GCTCCAAGATAATGTGGTCCTTGTTTCCCTGTACCTTCTTCTCT-3′ and 5′-GATAATGTGGTCCTTGT TACCCTGAACTTTCTTCTCTAGCCACTGATCT-3′, Mt-NCAM2.1 and Mt-NCAM2.2 were obtained.
CamKIIα-mCherry and CamKIIβ-mCherry were a generous gift from Miquel Bosch (IDIBAPS, Institute for Research in Biomedicine August Pi i Sunyer). MAP2B-GFP and MAP2C-GFP plasmids were a generous gift from Casper Hoogenraad (University of Utrecht) (Farah et al. 2005; Gumy et al. 2017).
Neuronal Culture Treatments
Hippocampal cultures were transfected at 3 days in vitro (DIV) using Lipofectamine 2000 (Life Technologies) according to the manufacturer’s instructions; cultures were fixed at 7 DIV for analysis of neuronal morphology. In MT-stabilization experiments, hippocampal cultures were treated at 4 DIV with 3 μM Taxol (Sigma) or DMSO for 72 h and fixed for analysis of neuronal morphology. In FGFR inhibition experiments, hippocampal cultures were treated at 4 DIV with 25 μM PD173074 (Sigma) or DMSO for 72 h and fixed for analysis of neuronal morphology. To perform live-imaged of EB3-labeled comets, cultures were transfected at 4 DIV. The HEK293T cells were transfected with Lipofectamine Plus (Life Technologies) according to the manufacturer’s instruction.
Immunoprecipitation Mass Spectrometry and Western Analysis
Brains were dissected from P12–P15 and P3 mice for the mass spectrometry experiment and the western blotting, respectively. Hippocampus and cortex regions were homogenized in isotonic buffer (Tris 10 mM a pH 7.4, KCl 10 mM, MgCl2 1.5 mM, EGTA 1 mM) and protease inhibitors (Complete, Roche), using a polytron. Membrane fraction was selected by centrifugation at 15 000 rpm for 15 min at 4°C and the supernatant was discarded. Membrane fraction was homogenized in lysis buffer (Hepes 50 mM pH 7.5, 150 mM NaCl, 1.5 mM MgCl2, 1 mM EGTA, 10% glycerol, and 1% Triton X-100) in orbital agitation for 45 min. The supernatant was selected by centrifugation at 15 000 rpm for 15 min at 4°C.
For the mass spectrometry, the magnetic beads (Dynabeads Antibody Coupling Kit, Lifetechnologies) were conjugated with an antibody against Ncam2.1 (EB06991, Everest) or against both Ncam2 isoforms (AF778, R&D Systems) according to the manufacturer’s instructions. Membrane fraction supernatant was incubated with the magnetic beads o/n at 4°C. After washing with lysis buffer three times, the proteins were eluted by 30 μL Urea buffer (urea 8 M, Tris 50 mM a pH 7.5, DTT 60 mM) for 15 min at room temperature (RT). The samples were processed and analyzed at the Proteomic facility of UB (CCiTUB, Cientific Parc of Barcelona). Briefly, the samples were digested with trypsin (2 μg, pH 8, 32.5°C, o/n). Peptides were separated (HPLC NanoAcquity, Waters) and detected (Orbitrap Velos, Thermo Scientific), with resolution of 60 000, ratio 400 m/z, and acquisition 300–1800 m/z. Protein lists were obtained with FDR ≤ 0.01%. String 10.0 was using for the bioinformatics analysis.
For immunoprecipitation and western blotting, membrane fraction supernatant was incubated with 2 μg of antibodies overnight (Anti-NCAM2 [AF778, R&D Systems]; Anti-NCAM2.1 [EB06991, Everest]; Anti-MAP2 [M9942 clone HM-2, Sigma], and Anti-14-3-3 [1657, SantaCruz]). Protein G-Sepharose beads (GE Healthcare) were added for 2 h. After washing with lysis buffer, the proteins were eluted by boiling in 20 μL of loading buffer (0.5 M Tris-HCl, pH 6.8; 2.15 M β-mercaptoethanol; 10% SDS; 30% glycerol; and 0.012% bromophenol blue), boiled for 5 min at 95°C, and processed for WB.
Immunofluorescence Microscopy
Cultured neurons and cell lines were fixed in PBS 4% paraformaldehyde (PFA) for 15 min at RT. In the analysis of fluorescence intensities, neuronal cultures were simultaneously permeabilized and fixed in 4% PFA/4% sucrose/0.25% glutaraldehyde/0.1% Triton X-100 diluted in PHEM buffer (60 mM Pipes, 25 mM Hepes pH 7.4, 5 mM EGTA, 1 mM MgCl2) as previously described (Sanchez-Huertas et al. 2016). Cells were permeabilized with PBS 0.1% Triton X-100 for 5 min, blocked with 10% serum, and incubated in 5% serum overnight with primary antibodies. Images or mosaics for dendritic and axonal parameters (number of primary dendrites, branching, total length or longest length) and Sholl analysis were taken with an inverted microscope AF6000 (Leica Microsystem) equipped with a 20× objective (1344 × 1024 pixel resolution). For analysis of neuronal stainings, single-plane images of somatodendritic regions were acquired with an inverted confocal Leica TCS SP5 microscope (Leica) equipped with a 63×/1.40 Oil objective (1344 × 1024 pixel resolution).
In Utero Electroporation
In utero microinjection and electroporation were performed at E14.5 essentially as described (Simo 2010), using timed pregnant CD-1 mice (Charles River Laboratories). Briefly, DNA solutions (pCAG-EGFP and a short-hairpin RNA [shRNA] against the indicated gene) were mixed in 10 mM Tris (pH 8.0) with 0.01% Fast Green. Needles for injection were pulled from Wiretrol II glass capillaries (Drummond Scientific) and calibrated for 1 μL injections. Forceps-type electrodes (Nepagene) with 5 mm pads were used for electroporation (five 50 ms pulses of 45 V at E14.5). Embryos were collected at E19.5. Brains were dissected and successful electroporations identified by epifluorescence microscopy. Positive brains were fixed in 4% formalin/PBS and cryoprotected in 30% sucrose/PBS overnight at 4°C. Brains were frozen in O.C.T. compound before 14-μm-thick brain cross-sections were obtained with a cryostat and placed on slides. Sections were antigen-retrieved by immersion of the slides in 0.01 M sodium citrate buffer, pH 6.0, at 95°C for 20 min. Sections were blocked for 2 h with 10% normal goat serum (NGS), 10 mM glycine, and 0.3% Triton X-100 in PBS at RT. Primary antibodies (anti-GFP) were incubated overnight at 4°C. Slides were washed four times for 10 min in 0.1% Triton X-100/PBS. Secondary antibodies were added for 2 h at RT. Slides were washed as before and coverslipped with Prolong Gold anti-fade reagent (Molecular Probes). Most images were obtained with epifluorescent illumination and 10× objective (Leica 760). Positions of GFP-positive neurons were recorded from several sections per embryo. Data were collected from the lateral part of the anterior neocortex. The cortex was divided into “bins” as follows: The distance from the pial surface to the bottom of the SVZ was measured and divided into 10 equal-sized bins. The percentage of GFP-labeled neurons in each bin for each embryo was then calculated. The graphs plot the mean and standard error of % neurons in each bin for the N embryos in a group. These values were also averaged across the N embryos in a group. P values are from two-way ANOVA, Bonferroni comjparison post hoc test.
Western Blot
Mice were sacrificed by decapitation and the brain was quickly removed from the skull. The brain was dissected into different brain regions (hippocampus, cortex [anterior and posterior], cerebellum, striatum, and olfactory bulb), which were frozen in liquid nitrogen and stored at −80°C until use. Brains were homogenized in lysis buffer (Tris base 125 mM and 2% SDS) using a polytron. Samples were frozen, 5 min in dry ice, and unfrozen, 2 min at 45°C, for three times. Samples were sonicated for 30 s in 0.5 cycles and 80% of amplitude and centrifuged to remove insoluble debris. Supernatant was collected and stored at −20°C until use. For the cell culture, lysates were prepared with lysis buffer at 95°C and processed as brain samples.
Samples were resolved by SDS-polyacrylamide gels and transferred onto nitrocellulose membranes. Membranes were blocked for 1 h at RT in TBST (Tris 10 mM [pH 7.4], sodium chloride 140 mM [TBS] with 0.1% Tween 20) containing 5% nonfat milk. Primary antibodies were incubated for 90 min in TBST–0.02% azide. After incubation with Horseradish Peroxidase (HRP)-labeled secondary antibodies for 1 h at RT in TBST, membranes were developed with the ECL system (GE Healthcare).
Histological Staining
Animals were anesthetized and perfused for 20 min with PBS 4% PFA. Brains were removed, post-fixed overnight with PBS 4% PFA, cryoprotected with PB-30% sucrose, and frozen. Brains were sectioned coronally (30 μm) and sections were blocked for 2 h at RT with PBS containing 10% NGS and 0.2% gelatin. Primary antibody was incubated overnight at 4°C with PBS-5% NGS. For immunohistofluorescence, sequential incubation was with a secondary antibody (AlexaFluoro, Invitrogen), and the sections were mounted (Mowiol, Calbiochem). For immunohistochemistry, sequential incubation with biotinylated secondary antibody (2 h at RT) and streptavidin-HRP (1:400; 2 h at RT) was performed in PBS–5% NGS; bound antibodies were visualized by reaction using DAB and H2O2 as peroxidase substrates; the sections were dehydrated and mounted (Eukitt).
Neuron Time Course and Time-Lapse Microscopy
Hippocampal cultures were plated in gridded glass bottom dishes (35 mm dish/No. 1.5 gridded coverslip/14 mm glass diameter, MatteK) and transduced at 3 DIV with shRNAs and pEGFP-N3, ratio 1:3. Beginning 1 day after transfection, neurons were imaged every 24 h using an Olympus IX81 microscope equipped with Yokogawa CSU-X1 spinning disc and a temperature controlled CO2 incubation chamber. Image stacks were acquired with a 60×/1.4 Oil immersion objective and an iXon EMCCD Andor DU-897 camera, using iQ2 software. At 7 DIV, images were taken at 30-s interval for 30 min. Cultures were fixed and immunostaining was performed, using the previously described protocols. Immunofluorescence images were taken with an Olympus CellR-Xcellence inverted microscope equipped with a Hamamatsu Orca-ER camera with a 60×/1.42 Oil objective.
Comet Time-Lapse Microscopy
Live imaging of EB3-labeled comets was carried out as previously described (Sanchez-Huertas et al. 2016). Briefly, hippocampal cultures were plated in glass bottom dishes (MatTek) transduced at 4 DIV with shRNAs and EB3-Tomato and imaged 48 h later. Live imaging of EB3 comets and of mitochondria was performed in the dendrite and/or within the proximal axons of random transfected cells, using an Olympus IX81 microscope equipped with Yokogawa CSU-X1 spinning disc and a temperature-controlled CO2 incubation chamber. Image stacks were acquired with a 100×/1.4 Oil immersion objective and an iXon EMCCD Andor DU-897 camera, using iQ2 software. Fluorescent images with a pixel size of 0.14 μm were taken at 1-s interval for 2.5 min.
Image Analysis
All images were processed and quantified using the ImageJ software (NIH). Axonal and dendritic EB3 comet analysis was made using the kymograph macro (ImageJ software), with lines drawn on the trajectories of comets. The average fluorescence intensity staining (β-tubulin, acetylated α-tubulin, detyrosinated α-tubulin, MAP2, and actin) was measured within the somatodendritic area delimited by the GFP signal. Whole axon and dendrite length were measured using the NeuronJ macro (ImageJ software).
Statistical Analysis
Statistical analysis was carried out using the Prism 5 software. Two-tailed unpaired t-tests or analysis of variance tests were performed to compare the experimental groups.
Results
NCAM2 Is Widely Expressed during Brain Development
We first investigated the pattern of NCAM2 expression in the developing and adult brain. At adult stages, NCAM2.1 and NCAM2.2 isoforms were detected in most of the regions analyzed by WB, with the expression of NCAM2.1 protein being higher (Supplementary Fig. 1A). In the developing cerebral cortex (including hippocampus), the expression of both isoforms increased progressively, again with higher levels for the NCAM2.1 isoform (Supplementary Fig. 1B). Immunohistochemistry showed that at early embryonic stages (E14), NCAM2 protein was mainly expressed in the marginal zone, subplate, and ventricular zone, as well as in some fiber systems. At perinatal stages, the staining increased and was particularly high in cortical pyramidal neurons and in several plexiform layers. From P15 onward, NCAM2 expression was enriched in the cortical neuropil (Supplementary Fig. 1C). The pattern of NCAM2 expression was confirmed by in situ hybridization studies (Supplementary Fig. 1D–F). We thus concluded that NCAM2 protein is widely expressed in both the developing and adult brain.
NCAM2 Is Required for Proper Neuronal Morphogenesis
To better understand the role of NCAM2 in neuronal differentiation, we altered the expression of ncam2 in hippocampal cultures by inducing the overexpression of NCAM2 isoforms during stage 4 of Banker in vitro development (Bentley and Banker 2016). The increased levels of NCAM2 expression in transfected cells, identified by the reporter gene GFP, were confirmed by immunolocalization (Fig. 1A). We found that the overexpression of both the NCAM2.1 and NCAM2.2 isoforms did not substantially alter neuronal polarization or the overall morphology of axons and dendrites compared to controls (Fig. 1A–C). Indeed, quantification of the dendritic and axonal compartments showed similar numbers of primary dendrites, distribution of branching points, and total extension of dendritic and axonal arbors in all conditions (Fig. 1D,E). We found only a slight reduction in the length of the longest axonal branch and a tendency toward change in axonal branching frequency (Fig. 1E). We thus concluded that the overexpression of NCAM2 does not result in marked neuronal morphologic alterations in dendrites or axons.
Figure 1.

NCAM2 overexpression does not alter dendritic and axonal arborization. (A) Immunostaining detection of NCAM2 and the reporter gene GFP in primary hippocampal neurons transfected with the overexpression plasmids NCAM2.1 OE and NCAM2.2 OE or the control plasmid. Arrowheads point to neurons transfected with control and overexpression plasmids. NCAM2-specific signal is increased upon individual overexpression of both isoforms in transfected neurons. (B) Low magnification of GFP labeling shows the whole dendritic and axonal arborization in transfected neurons. (C) Sholl analysis of the dendritic tree of neurons transfected with NCAM2 and control vectors does not reveal significant differences between conditions. (D, E) Quantification of dendritic (D) and axonal (E) arborization parameters (i.e., number of primary dendrites, total dendritic and axonal tree lengths, longest dendrite and axon lengths, and density of branching points in dendrites and axon) in neurons with NCAM2.1 OE, NCAM2.2 OE, and in control transfection. Only NCAM2.1 overexpression induces a slight decrease of the longest axonal length in hippocampal neurons. For the analysis of dendritic tree parameters, N = 28, N = 30, and N = 28 neurons from three independent experiments were analyzed for Control, NCAM2.1 OE, and NCAM2.2 OE, respectively. For the analysis of axonal parameters, n = 17, n = 18, and n = 19 neurons were analyzed, respectively, for Control, NCAM2.1 OE, and NCAM2.2 OE. Data are represented as mean ± SEM; *P < 0.05; one-way ANOVA, Tukey’s multiple comparison post hoc test. Scale bars: (A) 20 μm; (B) 50 μm.
We next performed knockdown experiments using Ncam2-specific shRNA (ShNcam2) to silence simultaneously the expression of both NCAM2.1 and NCAM2.2 isoforms. We first corroborated that the designed ShNcam2 decreased NCAM2.1 and NCAM2.2 protein levels by 81% in transfected cell lines, while scramble shRNA (ShCnt) did not alter NCAM2 levels (Supplementary Fig. 2A–D). In neuronal cultures, NCAM2 expression levels were also dramatically reduced by ShNcam2, as determined with immunocytochemistry (Fig. 2A). We found that knockdown of NCAM2 protein led to dramatic alterations in the morphology and overall organization of the neuritic tree of the transfected hippocampal neurons (Fig. 2A,B). The most prominent alteration in NCAM2-silenced neurons was a marked increase in the number of primary dendrites with a decrease in the maximum lengths of individual dendrite tracts arising from cell somas and an overall reduction in total dendritic tree length (Fig. 2B); dendritic alterations were supported by Sholl analysis (Fig. 2B). Similar branching alterations were found in axons, although the total axonal extension was not altered (Fig. 2C,D). Also, we found that about 20% of NCAM2-silenced neurons displayed two or more neurites with the morphological characteristics of axons (see the example in Fig. 2C, right panel). In addition to the fact that these processes were of constant, thin diameter and considerably longer than dendrites, immunocytochemical studies revealed that they were positive for Tau (an axonal marker) (Supplementary Fig. 3A). Live imaging tracking experiments (EB3 comets; Supplementary Fig. 3B, see also Fig. 7) in two neurons demonstrated that a large percentage of their microtubule-associated comets had an anterograde orientation (Supplementary Fig. 3B; range 71–100%). Thus, these findings indicate that these two processes are axonal-like in nature.
Figure 2.

NCAM2 silencing produces aberrant development of dendritic and axonal arborization of hippocampal neurons in vitro. (A) Immunostaining detection of NCAM2 and the reporter gene GFP in neurons transfected with the NCAM2-specific shRNA (ShNcam2) or the control vector ShCnt. Arrowheads point to neurons transfected with ShCnt and ShNcam2 plasmids. NCAM2-specific signal is reduced upon silencing of NCAM2 in transfected neurons. GFP labeling reveals numerous short neurites arising from ShNncam2-transfected neurons (right panels). (B) Sholl analysis and quantification of different arborization parameters of dendritic trees in neurons transfected with ShNcam2 and ShCnt show a drastic alteration of dendritic trees upon NCAM2 silencing. N = 32 and N = 32 neurons from three independent experiments were analyzed for ShCnt and ShNcam2, respectively. (C) Low magnifications of GFP-labeled ShNcam2- and ShCnt-transfected neurons showing the whole axonal and dendritic trees. Arrows points to axons as determined by morphological parameters; note, in the right panel, an example of a NCAM2-silenced neuron displaying three axons. (D) Quantification of axonal arborization parameters show alterations in longest axon extension and axonal branching in NCAM2-silenced neurons. N = 36 and N = 37 neurons from three independent experiments were analyzed for ShCnt and ShNcam2, respectively. Data are represented as mean ± SEM; *P < 0.05, **P < 0.01, ***P < 0.001; Student’s t-test. Scale bars: (A) 50 μm; (B) 50 μm; (D) 50 μm.
Figure 7.

NCAM2 depletion alters tubulin polymerization in dendrites and axons. (A) Immunostaining of MAP2 and GFP in neurons transfected with ShCnt or ShNcam2 from 3 DIV to 7 DIV and treated with Taxol or DMSO from 4 DIV to 7 DIV. The aberrant morphology induced by ShNCam2 is partially recovered after Taxol treatment. (B) Quantification of the number of primary dendrites in neurons from three independent experiments with Taxol assay. N = 50, N = 52, N = 48, and N = 49 neurons were analyzed, respectively, for ShCnt+DMSO, ShCnt+Taxol, ShNcam2 + DMSO, and ShNcam2 + Taxol. (C) Representative kymographs of EB3-fluoresecence from the time-lapse imaging in 25-μm dendritic tracts from neurons cotransfected with recombinant EB3-tomato protein and ShCnt or ShNcam2 from 4 DIV to 6 DIV. (D) Quantification of different parameters in tracked EB3-comets from the kymographs depicted in (C) indicates a reduction in density and an increase in length of the comets upon depletion of NCAM2. N = 25 ShCnt and N = 27 ShNcam2 neurons from two independent experiments. (E) Representative kymographs of EB3-fluoresecence in 25 μm axonal tracts from neurons cotransfected with recombinant EB3-tomato protein and ShCnt or ShNcam2 from 4 DIV to 6 DIV. (F) Quantification of different parameters in tracked EB3-comets from kymographs depicted in (E) indicates a reduction in density and an increase in length of the comet upon depletion of NCAM2. N = 30 ShCnt and N = 27 ShNcam2 neurons from three independent experiments. *P < 0.05; ***P < 0.001; one-way ANOVA, Tukey’s multiple comparison post hoc test and Student’s t-test. Scale bars: 10 μm.
Taken together, our results show that NCAM2 protein is necessary for the correct differentiation and morphogenesis of the dendritic and axonal compartments and suggest that this adhesion protein may also be involved in neuronal polarization.
NCAM2.1 and NCAM2.2 Isoforms Cooperate in Dendrite Development
In order to identify the NCAM2-isoform(s) responsible for the dendritic and axonal alterations described above, we performed rescue experiments by coexpressing the ShNcam2 sequence and ncam2.1- or ncam2.2-harpin-resistant cDNAs (Mt-NCAM2.1 and Mt-NCAM2.2). The specificity of this approach was tested in cell lines cotransfected with shRNA- and cDNA-expressing vectors (Fig. 3A). Our results confirmed that ShNcam2 does not alter the expression of either Mt-NCAM2.1 or Mt-NCAM2.2 as expected (Fig. 3A). Then, we cotransfected hippocampal cultures with ShNcam2 and Mt-NCAM2.1/Mt-NCAM2.2 and analyzed the morphology of neurons as above. We found that the aberrant dendritic phenotype induced by the silencing of the ncam2 gene was moderately rescued by coexpression with the Mt-NCAM2.2 isoform in terms of the number of primary dendrites and the longest dendrite length (Fig. 3B–D). Cotransfection with the transmembrane Mt-NCAM2.1 isoform led to a substantial rescue of the dendritic phenotype, as shown by Sholl analysis and the quantification of primary dendrites, longest dendritic length, and dendritic branching (Fig. 3B–D). Both Ncam2 isoforms cooperate in the NCAM2-deficient phenotype, although the rescue produced by the NCAM2.1 isoform was stronger.
Figure 3.

Both NCAM2.1 and NCAM2.2 contribute to correct dendritic tree development of hippocampal neurons in vitro. (A) HEK293 cells were cotransfected with different constructs to test the efficiency of ShNcam2 silencing vector and the resistance of mutated NCAM2 forms (Mt-NCAM2.1 and Mt-NCAM2.2) designed for rescue experiments. WB detection of NCAM2 in protein extracts showed that ShNcam2 efficiently targets overexpression of NCAM2.1 and NCAM2.2 isoforms while mutated forms are resistant. WB detection of actin was used as loading control; WB detection of GFP was used to detect expression of the reporter gene. (B) Representative images of GFP immunostaining in neurons transfected with ShCnt, ShNcam2, ShNcam2 + Mt-NCAM2.1, and ShNcam2 + Mt-NCAM2.2 vectors suggesting partial recovery upon overexpression of resistant forms of NCAM2. Sholl analysis (C) and quantification of dendritic tree parameters (D) in neurons corresponding to the rescue experiments. Rescue experiments with NCAM2.1 isoform rescue most of the alterations in dendritic arborization. NCAM2.2 isoform only partially rescues the number of primary dendrites and the longest dendritic length. N = 44, N = 36, N = 42, and N = 37 neurons from three independent experiments were analyzed, respectively, for ShCnt, ShNcam2, ShNcam2 + Mt-NCAM2.1, and ShNcam2 + Mt-NCAM2.2. Data are represented as mean ± SEM; *P < 0.05, ***P < 0.001; one-way ANOVA, Tukey’s multiple comparison post hoc test. Scale bar: 50 μm.
Finally, to investigate the possible role of FGFR signaling in the above processes, we performed similar experiments but including a FGFR inhibitor (PD173074) (Skaper et al. 2000). Our results show that blocking FGFR signaling does not alter the dendritic rescue induced by NCAM2 overexpression (Supplementary Fig. 4).
NCAM2 Is Necessary for the Maintenance of the Dendritic Tree
In the above experiments, hippocampal cultures were transfected at 3 DIV, when cultured neurons already have well-developed dendrites and axons. In order to determine how the complex dendritic phenotype described above arises, we performed time-lapse imaging experiments in which selected neurons were imaged daily from 4 to 7 DIV. Neurons transfected with a shRNA exhibited relatively stable dendrites around the cell body throughout the experiment (Fig. 4A, upper panels). In contrast, NCAM2-silenced neurons already exhibited dendritic alterations at 48 h after transfection. ShNcam2-transfected neurons showed a progressive thinning of primary dendrites, increased numbers of branching points, and retraction of some of these (up to 80%), which was simultaneous to the formation of a large number of new primary neurites arising from the cell bodies (Fig. 4A, lower panels). These new dendrites were very thin and highly branched. To substantiate these findings, we quantified the number of primary dendrites over time. The data confirm the progressive increase in primary dendrites per cell body after NCAM2 silencing (Fig. 4B).
Figure 4.

Depletion of NCAM2 in cultured neurons produces dendritic retraction. (A) Time-course imaging of dendritic morphology in vitro from 4 DIV to 7 DIV upon transfection of neurons at 3 DIV with ShCnt (a–d) or ShNcam2 (e–h). Repeated imaging of living neurons was performed upon transfection by detecting the reporter gene GFP. ShNcam2 transfection induced an increased number of dendrites in retraction (black arrowheads) and a reduction of growing dendrites (blue arrowheads) and dendrites bearing growth cones (red arrowheads). Axons in the figure are labeled with an asterisk. (B) Quantification shows a steady increase in the number of primary dendrites in NCAM2-depleted neurons. N = 14 and N = 15. Neurons from three independent experiments were analyzed, respectively, for ShCnt and ShNcam2. (C) Time-lapse imaging was performed at 7 DIV and selected dendritic terminals are shown in higher magnification from the squares marked in (d) and (h), from ShCnt and ShNcam2 neurons, respectively. Images were taken every 30 s for 30 min (complete sequence of images in Supplementary Video 1). Data are represented as mean ± SEM; ***P < 0.001; two-way ANOVA, Bonferroni comparison post hoc test. Scale bars: (A) 10 μm; (C) 5 μm.
Finally, we also made high-resolution live-imaging videos (30 min at intervals of 30 s) in 7 DIV transfected neurons in order to investigate the dynamic characteristics of dendrites/neurites (Fig. 4C and Supplementary Video 1). Whereas control dendrites were highly dynamic, especially at their tips displaying highly motile growth cones, NCAM2-silenced neurons did not display typical growth cones at their endings but elongation characteristic of motility, exhibiting continuous cycles of advancement and retraction (Supplementary Video 1). These data, together with the above experiments, suggest that NCAM2 silencing leads to a disruption of dendritic growth cones and a retraction of already formed dendrites, which is concomitant with the formation of a large number of new primary neurites arising from the cell body. These data suggest that NCAM2 is necessary not only for the development of the dendritic tree but also for its maintenance once it has been formed.
NCAM2 Regulates Neuronal Migration and Dendritic Development In Vivo
The above results show that NCAM2 protein is essential for the correct neuronal development of the axonal and dendritic compartments in in vitro experiments. In order to determine the effect of NCAM2 protein depletion in these processes in vivo, we performed in utero electroporation experiments. Mouse embryos were electroporated at E14 with either ShNcam2 or control vectors and analyzed at P0 and P5 using GFP as reporter gene. First, we analyzed the radial distribution of E14-electroporated neurons. At P0, most of the E14-generated control neurons were present in the upper portion of the cortical plate, as expected. E14-generated NCAM2-silenced neurons displayed a wider distribution in the cortical plate at P0, with additional neurons located in the lower layers of the cortical plate (BINs 7–8) (Fig. 5A,B). At P5, E14-born control neurons were correctly positioned in the lower part of layers II–III. NCAM2-silenced neurons showed a wider distribution in layers II–III persisting as in P0, which was especially evident in the lower aspect of layers II–III (Fig. 5C,D). We next examined the morphology of E14-GFP-labeled neurons. Already at P0, most electroporated neurons displayed a typical immature pyramidal neuron shape, with a main apical dendrite directed at the marginal zone. In contrast, many NCAM2-silenced neurons did not have a clear apical dendrite and displayed many primary dendrites, often offering a stellate-like morphology (Fig. 5E). Indeed, this aberrant phenotype was more evident at P5 when control neurons displayed a typical pyramidal shape (including a prominent apical dendrite, triangular cell body shapes, and a few basal dendrites), whereas NCAM2-silenced neurons lacked a clear main apical dendrite and displayed numerous primary dendrites arising from any position in the cell body (thereby again displaying a stellate-like morphology) (Fig. 5F,G). Quantification of the number of primary dendrites and of their orientation confirmed these aberrant phenotypes at P5 (Fig. 5G). These results indicated that the NCAM2 protein is necessary for correct cortical migration and for the appropriate development of dendritic trees in vivo in cortical pyramidal neurons.
Figure 5.
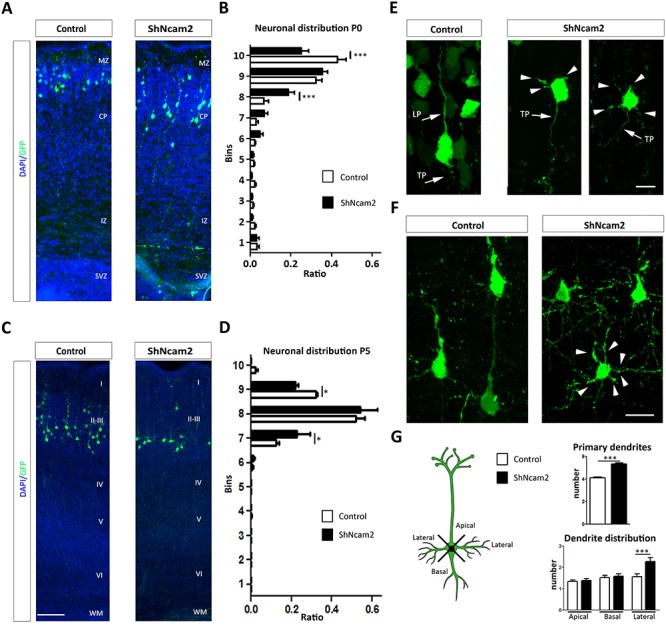
NCAM2 regulates neuronal migration and dendritic tree formation in vivo. (A) Representative images of GFP-electroporated neurons in cortical sections for P0 mice, which were electroporated with the ShNcam2 and ShCnt vectors at E14. Distribution of cells within cortical layers was determined in sections counterstained with DAPI. (B) Quantification of neuronal distribution at P0 of transfected cells within cortical layers was determined by dividing cortical thickness in 10 Bins. ShNcam2-transfected cells are distributed differently from ShCnt cells and occupy lower BIN levels. Data are presented as the ratio of neurons with somas located in each BIN; N = 7 and N = 7, animals electroporated with ShCnt and ShNcam2 constructs; ***P < 0.001; two-way ANOVA, Bonferroni comparison post hoc test. (C) Representative images from the reporter gene GFP immunodetection in cortical sections for P5 mice, which were electroporated with the ShNcam2 and ShCnt vectors at E15. (D) Quantification of neuronal distribution at P5 confirmed differences between control and NCAM2-silenced neurons, with the latter occupying lower positions in cortical layer also in P5. Data are presented as the ratio of neurons with somas located in each Bin; N = 3 and N = 2, animals electroporated with ShCnt and ShNcam2 constructs; *P < 0.05; two-way ANOVA, Bonferroni comparison post hoc test. (E) Higher magnification of representative images from electroporated neurons at P0 during neuronal migration. Neurons with NCAM2 silencing show increased stellate phenotype. (F) Higher magnification of representative images from transfected neurons at P5. Neurons with NCAM2 silencing show stellate phenotype into dendrites arising from all sides of cell body (arrowheads). (G) Neurons electroporated with ShNcam2 construct show increased numbers of primary dendrites with an increased amount of lateral primary dendrites. N = 122 and N = 87 neurons were analyzed to determine number of primary dendrites in ShCnt and ShNcam2, respectively. Dendrite distribution, N = 41 and N = 44 neurons were analyzed to determine distribution of primary dendrites in ShCnt and ShNcam2, respectively. ***P < 0.001; Student’s t-test. CP, cortical plate; IZ, intermediate zone; LP, leading process; MZ, marginal zone; SVZ, subventricular zone; TP, tailing process; I-VI, cortical layers. Scale bars: (A, D) 200 μm; (C) 10 μm; (F) 15 μm.
NCAM2 Is Not Required for Adult Dendritic Maintenance
NCAM2 protein is also highly expressed in the adult brain (Supplementary Fig. 1A). To determine whether the NCAM2 is also required for the maintenance of adult dendritic cortical architecture, we performed lentiviral injections of ShNcam2 constructs coexpressing GFP in CA1 hippocampal neurons and in granule cells of adult mice; animals were analyzed 4 weeks after injections. Lentiviral ShNcam2 effectiveness on endogenous ncam2 was corroborated in primary neuronal cultures showing a high reduction in both NCAM2.1 and NCAM2.2 protein levels, while control ShCnt lentivirus did not affect them (Supplementary Fig. 2E). Both in control and NCAM2-silenced neurons, pyramidal cells displayed a typical morphology including a prominent apical dendrite with horizontal dendrites and a prominent apical dendritic tuft, as well as many basal dendrites directed toward the ventricular surface. Branching alterations and abnormally retracted dendritic morphology were not observed in NCAM2-silenced pyramidal neurons (Supplementary Fig. 5). These results indicate that, unlike in developing neurons, the NCAM2 protein is not necessary for the maintenance of dendritic tree morphology in adult neurons already integrated in circuitry.
NCAM2 Silencing Generates Alterations in Microtubule Polymerization and Cytoskeleton Proteins
Because dendritic and axonal stability largely depend on cytoskeletal components, we next explored whether knockdown of NCAM2 alters cytoskeletal composition/distribution in neurons. As a first step, we analyzed the distribution of several cytoskeletal proteins in NCAM2-silenced neurons by immunofluorescence, focusing on the dendritic compartment. Whereas the distribution and immunofluorescence signal for actin, Tau, or neurofilaments were not affected by NCAM2 depletion (Supplementary Fig. 6). These studies also showed that the short dendrites arising from NCAM2-silenced neurons were neurofilament- and Tau-negative (Supplementary Fig. 6). We found a decrease in the immunofluorescence intensity signal for tubulin, acetylated tubulin (AcTub), and detyrosinated tubulin (DetyrTub) in the somatodendritic region of ShNcam2-transfected neurons (Supplementary Fig. 7A,B). Moreover, acetylated tubulin immunofluorescence signal was reduced in the axon (Supplementary Fig. 7C).
Next, we investigated the distribution of MAP2 protein, a dendritic microtubule-associated protein. MAP2 immunofluorescence signals were dramatically reduced in NCAM2-silenced neurons and virtually absent in distal dendritic segments (Fig. 6A,B). A similar decrease was found in live imaging tracked neurons in retracting dendrites in NCAM2 knocked down at 3 DIV, once early dendrites had been formed (Fig. 6C). These findings show that the depletion of NCAM2 leads to a marked decrease in MAP2 protein in both emerging and already formed dendrites (Fig. 6).
Figure 6.

NCAM2 depletion alters the MAP2 protein levels in neurons with a dendrite retraction process. (A) ShCnt- or ShNcam2-transfected neurons were immunostained for MAP2 (red) and GFP (green). Quantification of MAP2 staining in GFP-labeled neurons transfected with ShCnt or ShNcam2 (N = 27 ShCnt and N = 27 ShNcam2 neurons). Quantifications indicate a reduction of MAP2 content upon depletion of NCAM2. (B) Higher magnification of a representative dendritic shaft in the immunostaining of MAP2 marker. A cross-section of each dendrite was selected for quantification (white lines). Plot profile of the section in dendritic shafts depicted. (C) ShCnt- or ShNcam2-tracked neurons from the live-imaging experiment, Figure 5, were immunostained for GFP (green), MAP2 (red), and ACTIN (blue). The images confirm a reduction in MAP2 levels in ShNcam2-transfected neurons. Scale bar 10 μm. Magnification images of the ShCnt and ShNcam2 (a–d). Arrow and arrowheads label the same neurites stained with different antibodies. Scale bar: 5 μm.
Because tubulin acetylation and detyrosination are post-translational modifications typically found on more stable microtubules, the above results suggest that NCAM2 may control stability of the microtubule network, resulting in a decreased stability upon NCAM2 knockdown. To determine whether microtubule destabilization mediates NCAM2 silencing-induced dendritic alterations, we performed experiments using taxol to stabilize microtubules and reduce catastrophe rates (Witte et al. 2008). We found that with taxol treatment the NCAM2-silenced hippocampal neurons exhibited robust MAP2 staining similar to that in control neurons (Fig. 7A), indicating that microtubule stabilization can rescue the silencing phenotype. Quantification of the number of primary dendrites confirmed that treatment with taxol partially recovered the aberrant dendritic phenotype in NCAM2-silenced neurons (Fig. 7B). These findings indicate that the dendritic alterations in NCAM2-silenced cells are at least in part mediated by destabilization of the microtubule network.
Next, we performed microtubule polymerization assays, using the recombinant EB3-tomato protein as a marker for growing microtubule plus ends in time-lapse imaging (Sanchez-Huertas et al. 2016). ShCnt and ShNcam2 were transfected at 4 DIV, and 2 days later, EB3-comet number, trajectories, and velocity in dendritic tracts were tracked and represented in kymographs (Fig. 7C). We found that NCAM2 silencing did not significantly alter the average speed and mixed orientation of the comets, although it induced a significantly reduced density of comets and slightly modulated the speed of retrograde comets. (Fig. 7D). We also explored the dynamics of growing microtubule plus ends in axons, in which microtubules have an almost exclusively anterograde orientation (Fig. 7E). We found reduced comet density and increased average length (Fig. 7F). Together, our data show that NCAM2 regulates the stability of the microtubule network and suggest that NCAM2 depletion has a destabilizing effect, resulting in a reduced number of dendritic microtubules.
NCAM2 Interacts with 14-3-3, CaMKII, and MAP2, and These Interactions Are Essential for Proper Dendrite Development
Having proved that dendritic alterations caused by NCAM2 knockdown involve changes in the stability of the microtubule network, we sought to identify the putative molecular mediators of this process. We addressed this aim with a proteomic approach by immunoprecipitating NCAM2-associated proteins and performing a subsequent mass spectrometry analysis. Briefly, P10–12 cortical extracts were enriched in cell membranes and immunoprecipitated with anti-NCAM2 antibodies. Bound proteins were identified using mass spectrometry, with a total of 103 proteins found, out of which 54 were identified by two or more different peptide sequences. Among these, potential NCAM2 interactors included six proteins annotated in four different gene ontology (GO) terms linked to cytoskeleton (i.e., MAP2, YWHAE [14-3-3ξ], CALM1, DINLL1, TUBB4A, and CAMKIIβ proteins, which are annotated in the following GO terms: cell projection [GO:0042995], cytoskeleton [GO:0005856], cytoskeletal part [GO:0044430], and microtubule cytoskeleton [GO:0015630]). Microtubule-associated protein 2 (MAP2) was the main NCAM2 interactor, since it was detected by 24 different single peptides corresponding to both MAP2B and MAP2C isoforms (20 specific peptides for MAP2B and 4 peptides with shared sequences between both isoforms) (Supplementary Fig. 8). We identified peptides corresponding to 14-3-3ζ (6 specific peptide sequences), 14-3-3γ (4 specific peptide sequences), and 14-3-3 ξ (1 peptide sequences), as well as two additional peptides with shared sequences between the three isoforms (Supplementary Fig. 8). Additionally, we also identified CaMKIIα and CaMKIIβ isoforms, with six and five specific peptides for each isoform respectively, as well as six shared peptides between the two isoforms (Supplementary Fig. 8). To validate these putative interactions, we performed reverse coimmunoprecipitation assays for MAP2 and 14-3-3 proteins. WBs revealed that immunoprecipitation of protein extracts from P3 forebrains with NCAM2 antibodies yielded MAP2B, MAP2C, and 14-3-3 bands identified with specific antibodies (Fig. 8A,B). Reverse immunoprecipitation with MAP2 and 14-3-3 antibodies led to the identification of weak bands compatible with the MW of the NCAM2.1 isoform (Supplementary Fig. 8). These data indicate that NCAM2 molecules interact with the pool of cytoskeletal proteins (e.g., MAP2 and 14-3-3).
Figure 8.

MAP2, CaMKII, and 14-3-3 isoforms interact with NCAM2 and rescue the NCAM2-silenced phenotype. (A, B) Western blot detection of NCAM2.1 with either MAP2 isoforms (A) or 14-3-3 isoforms (B) in different coimmunoprecipitation experiments from P12–15 mouse cortex and hippocampal protein extracts. Immunoprecipitation using NCAM2 antibodies confirmed the presence of MAP2 and 14-3-3 proteins in the WBs. (C) Representative images of the somatodendritic compartment in neurons cotransfected with shRNAs (ShCnt or ShNcam2) and MAP2 isoforms (B, C), 14-3-3 isoforms (ξ, ζ and γ), and CaMKII isoforms (α and β) evidencing rescue phenotype in most of the cases. (D) Quantifications of number of primary dendrite neurons cotransfected with 14-3-3ξ, 14-3-3γ, 14-3-3ζ, CaMKIIα, CaMKIIβ, MAP2B, and MAP2C; with either ShCnt or ShNCam2 the strongest rescue was found with 14-3-3 ζ and γ and with MAP2C. N = 28–34 neurons were analyzed, respectively, in each group from 2 to 3 independent experiments per condition. (E) Schematic representation of interactions and phenotype in NCAM2 normal or NCAM2 knockdown neurons. *P < 0.05, **P < 0.01; ***P < 0.001; two-way ANOVA using Tukey’s comparison post-test. Scale bar: 50 μm.
We next investigated whether these cytoskeletal proteins were necessary for the dendritic phenotype seen in NCAM2-deficient neurons. We cotransfected hippocampal neurons with the ShNcam2-silencing vector together with CaMKIIα, CaMKIIβ 14-3-3ξ, 14-3-3γ, 14-3-3ζ, MAP2B, or MAP2C overexpressing cDNA. While silencing of the NCAM2 gene resulted in the aberrant dendritic phenotype described above, with many short neurites arising from the cell bodies, cotransfection with either 14-3-3γ, 14-3-3ζ, or MAP2C encoding cDNAs resulted in a marked rescue of this aberrant phenotype, with neurons displaying long dendrites and few primary dendrites, similar to control neurons (Fig. 8C). Quantitative analyses of the number of primary dendrites confirmed these observations (Fig. 8D). A partial rescue was found when 14-3-3ξ, MAP2B, and CaMKIIβ proteins were overexpressed in NCAM2-silenced neurons while cotransfection with CaMKIIα did not lead to any significant rescue (Fig. 8C,D). We conclude that the overexpression of the NCAM2-associated cytoskeletal CaMKII, MAP2, and 14-3-3 protein isoforms is sufficient to rescue the dendritic phenotype of NCAM2-deficient neurons. Together with our previous findings on microtubules, these observations suggest that NCAM2 in complexes with cytoskeletal-associated proteins promotes the stability of the microtubule network in dendrites and that loss of this regulation is responsible for the dendritic alterations reported upon NCAM2 depletion (Fig. 8E).
Discussion
The present study reveals that the NCAM2 gene is essential not only for dendritic development and morphogenesis but also for dendritic maintenance. In addition, our data provide evidence for a role of NCAM2 in neuronal migration, and they point to microtubule-associated proteins as important components in NCAM2 functions.
Whereas the expression and role of the NCAM2 protein have been extensively studied in the olfactory system (Alenius and Bohm 1997; Paoloni-Giacobino et al. 1997), to date NCAM2 expression has remained largely unknown in other CNS areas including the cerebral cortex. Our data show wide expression of the NCAM2 gene in cortical areas, from E14 onward, present in both the dendritic and fiber compartments. Moreover, the NCAM2.1 isoform is more abundant than the truncated NCAM2.2 isoform, with the latter detectable by WB only after P10.
Recent studies have shown an interesting role of NCAM2 in the formation and maintenance of excitatory glutamatergic synapses (Leshchyns'ka et al. 2015; Sheng et al. 2018). Here, we show that whereas the overexpression of NCAM2 leads to moderate alteration of axonal tree, the downregulation of NCAM2 gene results in dramatic changes in both the dendritic and the axonal architectures of hippocampal neurons in vitro. Thus, NCAM2-deficient neurons show decreased, and shorter, dendritic and axonal trees, as well as a dramatic increase in short primary dendrites arising from the cell bodies. To unravel the cellular process involved in this phenotype, we made time-lapse video recordings after NCAM2 depletion. These experiments revealed that upon NCAM2 depletion, already well-developed dendritic shafts became thinner and retracted, concomitantly to the emergence of narrower, short neurites arising from the cell body. Thus, although some of these primary, aberrant neurites are formed “de novo” (Fig. 4), the fact that most are retracting dendrites, are faintly stained for MAP2, are Tau-negative, and are considerably longer than typical filopodial neural extensions (up to 10 μm) (Qu et al. 2017) leads us to consider them as dendrites. However, some of the features of these processes resemble the so-called proto-dendrites (Kahn et al. 2015).
Interestingly, this in vitro phenotype was mimicked in in vivo experiments, in which NCAM2-deficient cortical pyramidal neurons exhibit a radial phenotype, thinner dendrites, and the lack of a prominent apical dendrite. We thus conclude that NCAM2 is necessary for proper dendritic development and maintenance and for the correct cytoarchitecture of pyramidal neurons. Interestingly, we were unable to detect changes in dendritic architecture when the NCAM2 gene was silenced in adult hippocampal neurons, suggesting that the role of NCAM2 in dendritic formation is developmental stage-dependent. Our rescue experiments with both the NCAM2.1 and NCAM2.2 isoforms indicate that although both are able to partially rescue the NCAM2-knockdown phenotype, such rescue was more efficient in NCAM2.1-overexpressing neurons (e.g., Sholl analysis, Fig. 3C and dendritic tree parameters, Fig. 3D). This is consistent with the fact that the longest NCAM2.1 form is highly expressed during development, whereas the shorter NCAM2.2 isoform appears to be expressed only after P10. Our data thus suggest that the endogenous NCAM2.1 isoform is the main protein responsible for the described phenotype. We can only speculate about how the NCAM2.2 isoform, lacking the intracellular domain, may partially rescue the dendritic aberrant phenotype. One possibility is that the effects of the NCAM2.2 isoform are mediated by transmembrane interacting proteins, such as the FGF receptor (Rasmussen et al. 2018), which may activate the specific intracellular signaling cascades. However, although a contribution of FGFR to NCAM2 functions could not be completely ruled out, our experiments using FGFR inhibitors suggest that, at least for dendritic differentiation and morphogenesis, NCAM2 effects are largely independent of FGFR signaling. Also, and in contrast to NCAM1 (Soroka et al. 2003, 2010), synergy in cis of coexpressed NCAM2.1 and NCAM2.2 isoforms in the same neurons seems unlikely, due to the rigid structure of the immunoglobulin domains of NCAM2, which may favor trans interactions (Kulahin et al. 2011; Rasmussen et al. 2018). It has been described that the domains responsible for NCAM2–NCAM2 interactions (immunoglobulin domains) present a rigid structure that may prevent a potential cis and favor trans interactions (Kulahin et al. 2011). Thus, the analysis of synergistic effects in cis configuration (coexpression within the same cell) is unlikely to be informative. In contrast, NCAM1 presents a quite more flexible structure that permits cis interactions (Soroka et al. 2003, 2010). Nevertheless, our data suggest that NCAM2.1 and NCAM2.2 isoforms have, at least in part, overlapping functions.
We also describe an unknown role of NCAM2 in cortical migration; embryonic, cell-autonomous NCAM2 depletion of cortical precursors results in the mispositioning of cortical pyramidal neurons, which is likely to be permanent, as neuron misposition remains at P5, once cortical migration has been completed (Marin 2013). A direct role of NCAM2 in neuronal migration is consistent with the expression of the NCAM2 gene in embryonic cortical precursors. However, we cannot exclude the possibility that the observed migration phenotype is secondary to the alterations in dendritic orientation, which may potentially interfere with the function of the leading edge of migrating neurons (Demyanenko et al. 2004).
Our experiments also suggest a putative role of the NCAM2 gene in neuronal polarization. The determination and differentiation of early neurites in dendrites and axons is a complex process in which many proteins and pathways have been implicated including TAG-1 or small GTPases (Jossin and Cooper 2011; Caceres et al. 2012; Namba et al. 2014; Shah and Puschel 2014; Takano et al. 2015). Our experiments first reveal that a small, but substantial, number of NCAM2-depleted neurons (20%) exhibits two or more axons. Second, NCAM2 depletion results in the retraction of already formed primary dendrites and the appearance of newly formed thin neurites with clearly lacking dendritic markers (MAP2, Fig. 6) or axonal markers (Tau or neurofilaments, Supplementary Fig. 6). Whereas they display normal actin abundance, these neurites were extremely motile and displayed numerous growing and retracting filopodia-like structures. Although the molecular mechanisms involved in this process remain to be determined, it is likely that the NCAM2-dependent stabilization of the microtubule network (see below) is one of the mechanisms involved.
One of the most relevant findings of the present study involves the changes in cytoskeletal proteins caused by NCAM2 deficiency. First, we found that NCAM2 depletion led to a decrease in microtubule markers, including tubulin, as well as acetylated and detyrosinated tubulin, suggesting a role of NCAM2 in the stabilization of microtubules. Consistent with this, our microtubule comet tracking studies reveal that NCAM2-deficient dendrites display decreased comet density. In axons, NCAM2 depletion led to a more severe comet phenotype. These findings are consistent with the notion that NCAM2 depletion results in destabilization of the microtubule network. In contrast, the actin network appeared to remain unaltered. Most importantly, microtubule stabilization by taxol treatment markedly rescued NCAM2-depletion effects, strongly suggesting that a compromised microtubule network is underlying the aberrant dendritic phenotype in NCAM2-deficient neurons. The intensity of MAP2 staining upon taxol treatment was markedly increased, suggesting that microtubule stabilization by taxol may in turn stabilize MAP2 protein in dendrites (Witte et al. 2008).
Consistent with these data, we show that the depletion of NCAM2 results in a dramatic reduction in the amount of the dendritic protein MAP2, a microtubule-associated protein fundamental for dendritic cytoarchitecture and maintenance (Huber and Matus 1984; Caceres et al. 1992; Harada et al. 2002) and that MAP2B/C overexpression rescues the NCAM2 phenotype. Further, our mass spectrometry and coimmunoprecipitation studies reveal that NCAM2 protein interacts with MAP2B and MA2C isoforms and that overexpression of either MAP2B or MAP2C isoforms almost completely rescues the dendritic phenotype caused by NCAM2 depletion. As it is likely that only a small pool of MAP2 protein is actually directly interacting with NCAM2 protein at the cell membrane, we can only speculate about the possible mechanisms implicated in the depletion of MAP2 protein throughout the dendrite and the parallel disorganization of the dendritic network. In many CAMs, “cortical” membrane-linked cytoskeletal proteins have a profound effect on the organization of the remaining cytoskeleton (Maness and Schachner 2007). It is thus conceivable that the direct interaction of NCAM2 with the outermost located MAP2 protein is sufficient to control both the outermost and central microtubular networks (Kapitein and Hoogenraad 2015; Leshchyns'ka and Sytnyk 2016). Also, microtubule arrays in dendrites were shown to spatially segregate into bundles based on their post-translational modifications and their orientation. While it is not known what microtubule properties are recognized by MAP2, it may selectively associate with a subset of dendritic microtubules (Tas et al. 2017). In addition, the effect of NCAM2 could be mediated by specific signaling pathways (e.g., 14-3-3, CaMKII or Src) which, when activated close to the membrane, may regulate the phosphorylation levels of MAP2 throughout the dendrite. Indeed, it is known that CaMKII, Src, or 14-3-3 controls the microtubule dynamics via MAP2 phosphorylation levels (Sanchez et al. 2000; McVicker et al. 2015; Jansen et al. 2017; Sheng et al. 2019). Interestingly, the above two possible mechanisms may in turn regulate MAP2 protein instability. Finally, it is possible that NCAM2 may regulate gene expression (in this case MAP2) similarly to what has been observed for other cell-adhesion molecules. For instance, L1CAM, which directly interacts with MAP2, has also been described to control the expression levels of MAP2 (Poplawski et al. 2012).
Although MAP2B and MAP2C have a different expression pattern during neuronal morphogenesis, both isoforms present the microtubule-binding domain. This domain is important for the microtubule bundling, protrusion, and formation and also binds different signaling proteins that modulate microtubule organization and stability (Chen et al. 1992; Belanger et al. 2002; Melkova et al. 2019). Since the lack of the projection domain in MAP2C does not impede this isoform to rescue the phenotype of NCAM2-depletion, we hypothesize that the mechanism of rescuing the proper number of primary dendrites may be directly attributable to the microtubule-binding domain and stabilization of microtubules. However, additional, distinct properties of MAP2B and MAP2C may contribute to the rescue of the NCAM2 phenotype. For instance, MAP2B has been shown to lower microtubule dynamics, leading to increased diameter of dendrites and consolidated dendritic branches (Belanger et al. 2002; Dehmelt and Halpain 2005). Together, these data suggest that the NCAM2 protein contributed to the stabilization of dendritic microtubules; upon NCAM2 downregulation, MAP2 protein content decreases concomitantly with the destabilization and decrease of the dendritic microtubule network.
In addition, our mass spectrometry data and coimmunoprecipitation experiments reveal interaction of NCAM2 with other cytoskeletal proteins, such as 14-3-3 ξ, γ, and ζ isoforms and the CaMKII α and β isoforms. 14-3-3 proteins form either heterodimers or homodimers and have been described as interacting with both the microtubule and actin cytoskeletons participating in several neuronal developmental processes (Berg et al. 2003; Sarmiere and Bamburg 2004; Sluchanko and Gusev 2010; Brandwein and Wang 2017). Among other developmental and plasticity processes, CaMKII protein isoforms are also known to regulate neuritogenesis (Sogawa et al. 2001; Fink et al. 2003). Similar to overexpression of MAP2 isoforms, the 14-3-3 γ and ζ isoforms also efficiently rescued dendritic abnormalities induced by NCAM2 deficiency, and a partial rescue was observed for the 14-3-3 ξ and CaMKII β proteins. These data suggest that NCAM2/MAP2/14-3-3 protein complexes may contribute to the stability of the microtubule network in dendrites, whose disruption leads to severe dendritic malformation. Although the molecular details of this mechanism remain to be investigated, such regulation may be similar to what has been proposed for L1CAM, whose depletion results in regulated MAP2 degradation (Poplawski et al. 2012).
In summary, the present study provides evidence for previously unknown roles of the NCAM2 proteins in distinct important corticogenesis processes, namely dendritic development and maintenance, neuronal migration, and neuronal polarization (Fig. 8E). Furthermore, we have uncovered a molecular network (MAP2 and 14-3-3 proteins) by which NCAM2 regulates the stability of dendritic microtubules. Given the proposed role of the NCAM2 gene in several neurodevelopmental disorders including autism, Down syndrome, macro- and microcephaly (Molloy et al. 2005; Petit et al. 2015; Lin et al. 2016; Scholz et al. 2016), as well as the contribution of MAP2 and 14-3-3 genes to autism and developmental epilepsy (Mukaetova-Ladinska et al. 2004; Xu et al. 2015; Pagan et al. 2017; Westphal et al. 2018), future analysis of the interplay between NCAM2-organized protein complexes with the dendritic microtubule cytoskeleton may improve our understanding of the molecular and cellular mechanisms involved in these pathologies.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Notes
We thank N. Masachs for technical assistance in viral injections; E. de Oliveira and M.A. Odena for mass spectrometry experiments; L. Bardia and A. LLadó (Microscopy facility of the IRB-Barcelona); A. Bosch, E. Coll and M. Calvo (Microscopy facility of the University of Barcelona), and members of the Soriano lab for experimental help and comments. Conflict of Interest: The authors declare no competing financial interests.
Funding
MINECO (Ministerio de Economía y Competitividad) grants SAF2016-76340-R to E.S. and L.P., and BFU2015-69275-P and PGC2018-099562-B-I00 to J.L.; Department of Economy and Knowledge of the Generalitat de Catalunya (Predoctoral fellowship to A.P.); Centro de Investigación Biomédica en Red para Enfermedades Neurodegenerativas (CIBERNED-ISCIII) collaborative intramural projects to E.S.; The Marató de TV3 Foundation to L.P.; NIH grants R01 NS109176 to S.S.; and R21 NS101450 to S.S. and A.L.T.; and IRB Barcelona (Institut de Recerca Biomédica) intramural funds.
Authors’ Contributions
E.S., L.P., and A.P. conceived and designed the study. A.P. performed most of the experiments and analyzed the data. A.O. and B.P.-A. contributed to NCAM2 depletion and rescue experiments in in vitro cultures. R.V. contributed to comet experiments. K.H., A.T., and S.S. contributed to the design and performed in utero electroporation and in situ hybridization. J.F.-S. and R.T. contributed to design and produced the shRNAs. J.L. designed and supervised the cytoskeletal network characterization and microtubule dynamics. A.P., L.P., and E.S. wrote the manuscript. All authors read, discussed, and corrected the manuscript.
References
- Alenius M, Bohm S. 1997. Identification of a novel neural cell adhesion molecule-related gene with a potential role in selective axonal projection. J Biol Chem. 272:26083–26086. [DOI] [PubMed] [Google Scholar]
- Angata K, Huckaby V, Ranscht B, Terskikh A, Marth JD, Fukuda M. 2007. Polysialic acid-directed migration and differentiation of neural precursors are essential for mouse brain development. Mol Cell Biol. 27:6659–6668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belanger D, Farah CA, Nguyen MD, Lauzon M, Cornibert S, Leclerc N. 2002. The projection domain of MAP2b regulates microtubule protrusion and process formation in Sf9 cells. J Cell Sci. 115:1523–1539. [DOI] [PubMed] [Google Scholar]
- Bentley M, Banker G. 2016. The cellular mechanisms that maintain neuronal polarity. Nat Rev Neurosci. 17:611–622. [DOI] [PubMed] [Google Scholar]
- Berg D, Holzmann C, Riess O. 2003. 14-3-3 proteins in the nervous system. Nat Rev Neurosci. 4:752–762. [DOI] [PubMed] [Google Scholar]
- Brandwein D, Wang Z. 2017. Interaction between Rho GTPases and 14-3-3 proteins. Int J Mol Sci. 18(10), 2148; https://doi.org/10.3390/ijms18102148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caceres A, Mautino J, Kosik KS. 1992. Suppression of MAP2 in cultured cerebellar macroneurons inhibits minor neurite formation. Neuron. 9:607–618. [DOI] [PubMed] [Google Scholar]
- Caceres A, Ye B, Dotti CG. 2012. Neuronal polarity: demarcation, growth and commitment. Curr Opin Cell Biol. 24:547–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Kanai Y, Cowan NJ, Hirokawa N. 1992. Projection domains of MAP2 and tau determine spacings between microtubules in dendrites and axons. Nature. 360:674–677. [DOI] [PubMed] [Google Scholar]
- Dehmelt L, Halpain S. 2005. The MAP2/tau family of microtubule-associated proteins. Genome Biol. 6:204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demyanenko GP, Schachner M, Anton E, Schmid R, Feng G, Sanes J, Maness PF. 2004. Close homolog of L1 modulates area-specific neuronal positioning and dendrite orientation in the cerebral cortex. Neuron. 44:423–437. [DOI] [PubMed] [Google Scholar]
- Dent EW, Gupton SL, Gertler FB. 2011. The growth cone cytoskeleton in axon outgrowth and guidance. Cold Spring Harb Perspect Biol. 3(3), a001800; doi:10.1101/cshperspect.a001800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farah CA, Liazoghli D, Perreault S, Desjardins M, Guimont A, Anton A, Lauzon M, Kreibich G, Paiement J, Leclerc N. 2005. Interaction of microtubule-associated protein-2 and p63: a new link between microtubules and rough endoplasmic reticulum membranes in neurons. J Biol Chem. 280:9439–9449. [DOI] [PubMed] [Google Scholar]
- Fink CC, Bayer KU, Myers JW, Ferrell JE Jr, Schulman H, Meyer T. 2003. Selective regulation of neurite extension and synapse formation by the beta but not the alpha isoform of CaMKII. Neuron. 39:283–297. [DOI] [PubMed] [Google Scholar]
- Gumy LF, Katrukha EA, Grigoriev I, Jaarsma D, Kapitein LC, Akhmanova A, Hoogenraad CC. 2017. MAP2 defines a pre-axonal filtering zone to regulate KIF1- versus KIF5-dependent cargo transport in sensory neurons. Neuron. 94:347–362. [DOI] [PubMed] [Google Scholar]
- Harada A, Teng J, Takei Y, Oguchi K, Hirokawa N. 2002. MAP2 is required for dendrite elongation, PKA anchoring in dendrites, and proper PKA signal transduction. J Cell Biol. 158:541–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber G, Matus A. 1984. Differences in the cellular distributions of two microtubule-associated proteins, MAP1 and MAP2, in rat brain. J Neurosci. 4:151–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussman JP, Chung RH, Griswold AJ, Jaworski JM, Salyakina D, Ma D, Konidari I, Whitehead PL, Vance JM, Martin ER et al. 2011. A noise-reduction GWAS analysis implicates altered regulation of neurite outgrowth and guidance in autism. Mol Autism. 2:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen S, Melkova K, Trosanova Z, Hanakova K, Zachrdla M, Novacek J, Zupa E, Zdrahal Z, Hritz J, Zidek L. 2017. Quantitative mapping of microtubule-associated protein 2c (MAP2c) phosphorylation and regulatory protein 14-3-3zeta-binding sites reveals key differences between MAP2c and its homolog tau. J Biol Chem. 292:6715–6727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen OS, Bock E. 1974. Brain specific synaptosomal membrane proteins demonstrated by crossed immunoelectrophoresis. J Neurochem. 23:879–880. [DOI] [PubMed] [Google Scholar]
- Jossin Y, Cooper JA. 2011. Reelin, Rap1 and N-cadherin orient the migration of multipolar neurons in the developing neocortex. Nat Neurosci. 14:697–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn OI, Sharma V, Gonzalez-Billault C, Baas PW. 2015. Effects of kinesin-5 inhibition on dendritic architecture and microtubule organization. Mol Biol Cell. 26:66–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapitein LC, Hoogenraad CC. 2015. Building the neuronal microtubule cytoskeleton. Neuron. 87:492–506. [DOI] [PubMed] [Google Scholar]
- Kiselyov VV, Skladchikova G, Hinsby AM, Jensen PH, Kulahin N, Soroka V, Pedersen N, Tsetlin V, Poulsen FM, Berezin V et al. 2003. Structural basis for a direct interaction between FGFR1 and NCAM and evidence for a regulatory role of ATP. Structure. 11:691–701. [DOI] [PubMed] [Google Scholar]
- Kon E, Cossard A, Jossin Y. 2017. Neuronal polarity in the embryonic mammalian cerebral cortex. Front Cell Neurosci. 11:163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulahin N, Kristensen O, Rasmussen KK, Olsen L, Rydberg P, Vestergaard B, Kastrup JS, Berezin V, Bock E, Walmod PS et al. 2011. Structural model and trans-interaction of the entire ectodomain of the olfactory cell adhesion molecule. Structure. 19:203–211. [DOI] [PubMed] [Google Scholar]
- Kulahin N, Walmod PS. 2010. The neural cell adhesion molecule NCAM2/OCAM/RNCAM, a close relative to NCAM. Adv Exp Med Biol. 663:403–420. [DOI] [PubMed] [Google Scholar]
- Leshchyns'ka I, Liew HT, Shepherd C, Halliday GM, Stevens CH, Ke YD, Ittner LM, Sytnyk V. 2015. Aβ-dependent reduction of NCAM2-mediated synaptic adhesion contributes to synapse loss in Alzheimer's disease. Nat Commun. 6:8836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leshchyns'ka I, Sytnyk V. 2016. Reciprocal interactions between cell adhesion molecules of the immunoglobulin superfamily and the cytoskeleton in neurons. Front Cell Dev Biol. 4:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YC, Frei JA, Kilander MB, Shen W, Blatt GJ. 2016. A subset of autism-associated genes regulate the structural stability of neurons. Front Cell Neurosci. 10:263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makino T, McLysaght A. 2010. Ohnologs in the human genome are dosage balanced and frequently associated with disease. Proc Natl Acad Sci U S A. 107:9270–9274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maness PF, Schachner M. 2007. Neural recognition molecules of the immunoglobulin superfamily: signaling transducers of axon guidance and neuronal migration. Nat Neurosci. 10:19–26. [DOI] [PubMed] [Google Scholar]
- Marin O. 2013. Cellular and molecular mechanisms controlling the migration of neocortical interneurons. Eur J Neurosci. 38:2019–2029. [DOI] [PubMed] [Google Scholar]
- McVicker DP, Millette MM, Dent EW. 2015. Signaling to the microtubule cytoskeleton: an unconventional role for CaMKII. Dev Neurobiol. 75:423–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melkova K, Zapletal V, Narasimhan S, Jansen S, Hritz J, Skrabana R, Zweckstetter M, Ringkjobing Jensen M, Blackledge M, Zidek L. 2019. Structure and functions of microtubule associated proteins tau and MAP2c: similarities and differences. Biomolecules. 2019, 9, 105; doi:10.3390/biom9030105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Missaire M, Hindges R. 2015. The role of cell adhesion molecules in visual circuit formation: from neurite outgrowth to maps and synaptic specificity. Dev Neurobiol. 75:569–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molloy CA, Keddache M, Martin LJ. 2005. Evidence for linkage on 21q and 7q in a subset of autism characterized by developmental regression. Mol Psychiatry. 10:741–746. [DOI] [PubMed] [Google Scholar]
- Mukaetova-Ladinska EB, Arnold H, Jaros E, Perry R, Perry E. 2004. Depletion of MAP2 expression and laminar cytoarchitectonic changes in dorsolateral prefrontal cortex in adult autistic individuals. Neuropathol Appl Neurobiol. 30:615–623. [DOI] [PubMed] [Google Scholar]
- Muller D, Mendez P, Deroo M, Klauser P, Steen S, Poglia L. 2010. Role of NCAM in spine dynamics and synaptogenesis. Adv Exp Med Biol. 663:245–256. [DOI] [PubMed] [Google Scholar]
- Namba T, Funahashi Y, Nakamuta S, Xu C, Takano T, Kaibuchi K. 2015. Extracellular and intracellular signaling for neuronal polarity. Physiol Rev. 95:995–1024. [DOI] [PubMed] [Google Scholar]
- Namba T, Kibe Y, Funahashi Y, Nakamuta S, Takano T, Ueno T, Shimada A, Kozawa S, Okamoto M, Shimoda Y et al. 2014. Pioneering axons regulate neuronal polarization in the developing cerebral cortex. Neuron. 81:814–829. [DOI] [PubMed] [Google Scholar]
- Pagan C, Goubran-Botros H, Delorme R, Benabou M, Lemiere N, Murray K, Amsellem F, Callebert J, Chaste P, Jamain S et al. 2017. Disruption of melatonin synthesis is associated with impaired 14-3-3 and miR-451 levels in patients with autism spectrum disorders. Sci Rep. 7:2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paoloni-Giacobino A, Chen H, Antonarakis SE. 1997. Cloning of a novel human neural cell adhesion molecule gene (NCAM2) that maps to chromosome region 21q21 and is potentially involved in Down syndrome. Genomics. 43:43–51. [DOI] [PubMed] [Google Scholar]
- Pebusque MJ, Coulier F, Birnbaum D, Pontarotti P. 1998. Ancient large-scale genome duplications: phylogenetic and linkage analyses shed light on chordate genome evolution. Mol Biol Evol. 15:1145–1159. [DOI] [PubMed] [Google Scholar]
- Petit F, Plessis G, Decamp M, Cuisset JM, Blyth M, Pendlebury M, Andrieux J. 2015. 21q21 deletion involving NCAM2: report of 3 cases with neurodevelopmental disorders. Eur J Med Genet. 58:44–46. [DOI] [PubMed] [Google Scholar]
- Poplawski GH, Tranziska AK, Leshchyns'ka I, Meier ID, Streichert T, Sytnyk V, Schachner M. 2012. L1CAM increases MAP2 expression via the MAPK pathway to promote neurite outgrowth. Mol Cell Neurosci. 50:169–178. [DOI] [PubMed] [Google Scholar]
- Qu Y, Hahn I, Webb SE, Pearce SP, Prokop A. 2017. Periodic actin structures in neuronal axons are required to maintain microtubules. Mol Biol Cell. 28:296–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen KK, Falkesgaard MH, Winther M, Roed NK, Quistgaard CL, Teisen MN, Edslev SM, Petersen DL, Aljubouri A, Christensen C et al. 2018. NCAM2 Fibronectin type-III domains form a rigid structure that binds and activates the fibroblast growth factor receptor. Sci Rep. 8:8957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Huertas C, Freixo F, Viais R, Lacasa C, Soriano E, Luders J. 2016. Non-centrosomal nucleation mediated by augmin organizes microtubules in post-mitotic neurons and controls axonal microtubule polarity. Nat Commun. 7:12187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez C, Diaz-Nido J, Avila J. 2000. Phosphorylation of microtubule-associated protein 2 (MAP2) and its relevance for the regulation of the neuronal cytoskeleton function. Prog Neurobiol. 61:133–168. [DOI] [PubMed] [Google Scholar]
- Sarmiere PD, Bamburg JR. 2004. Regulation of the neuronal actin cytoskeleton by ADF/cofilin. J Neurobiol. 58:103–117. [DOI] [PubMed] [Google Scholar]
- Schelski M, Bradke F. 2017. Neuronal polarization: from spatiotemporal signaling to cytoskeletal dynamics. Mol Cell Neurosci. 84:11–28. [DOI] [PubMed] [Google Scholar]
- Scholz C, Steinemann D, Malzer M, Roy M, Arslan-Kirchner M, Illig T, Schmidtke J, Stuhrmann M. 2016. NCAM2 deletion in a boy with macrocephaly and autism: cause, association or predisposition? Eur J Med Genetics. 59:493–498. [DOI] [PubMed] [Google Scholar]
- Shah B, Puschel AW. 2014. In vivo functions of small GTPases in neocortical development. Biol Chem. 395:465–476. [DOI] [PubMed] [Google Scholar]
- Sheng L, Leshchyns'ka I, Sytnyk V. 2013. Cell adhesion and intracellular calcium signaling in neurons. Cell Commun Signal. 11:94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng L, Leshchyns'ka I, Sytnyk V. 2015. Neural cell adhesion molecule 2 promotes the formation of filopodia and neurite branching by inducing submembrane increases in Ca2+ levels. J Neurosci. 35:1739–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng L, Leshchyns'ka I, Sytnyk V. 2019. Neural cell adhesion molecule 2 (NCAM2)-induced c-Src-dependent propagation of submembrane Ca2+ spikes along dendrites inhibits synapse maturation. Cereb Cortex. 29:1439–1459. [DOI] [PubMed] [Google Scholar]
- Simo S, Jossin Y, Cooper JA. 2010. Cullin 5 regulates cortical layering by modulating the speed and duration of Dab1-dependent neuronal migration. The Journal of neuroscience: the official journal of the Society for Neuroscience. 30:5668–5676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skaper SD, Kee WJ, Facci L, Macdonald G, Doherty P, Walsh FS. 2000. The FGFR1 inhibitor PD 173074 selectively and potently antagonizes FGF-2 neurotrophic and neurotropic effects. J Neurochem. 75:1520–1527. [DOI] [PubMed] [Google Scholar]
- Sluchanko NN, Gusev NB. 2010. 14-3-3 proteins and regulation of cytoskeleton. Biochemistry. 75:1528–1546. [DOI] [PubMed] [Google Scholar]
- Sogawa Y, Yoshimura Y, Yamauchi T. 2001. Investigation of the Ca(2+)-independent form of Ca(2+)/calmodulin-dependent protein kinase II in neurite outgrowth. Brain Res Brain Res Protocol. 8:159–169. [DOI] [PubMed] [Google Scholar]
- Soroka V, Kasper C, Poulsen FM. 2010. Structural biology of NCAM. Adv Exp Med Biol. 663:3–22. [DOI] [PubMed] [Google Scholar]
- Soroka V, Kolkova K, Kastrup JS, Diederichs K, Breed J, Kiselyov VV, Poulsen FM, Larsen IK, Welte W, Berezin V et al. 2003. Structure and interactions of NCAM Ig1-2-3 suggest a novel zipper mechanism for homophilic adhesion. Structure. 11:1291–1301. [DOI] [PubMed] [Google Scholar]
- Sytnyk V, Leshchyns'ka I, Schachner M. 2017. Neural cell adhesion molecules of the immunoglobulin superfamily regulate synapse formation, maintenance, and function. Trends Neurosci. 40:295–308. [DOI] [PubMed] [Google Scholar]
- Takano T, Xu C, Funahashi Y, Namba T, Kaibuchi K. 2015. Neuronal polarization. Development. 142:2088–2093. [DOI] [PubMed] [Google Scholar]
- Tas RP, Chazeau A, Cloin BMC, Lambers MLA, Hoogenraad CC, Kapitein LC. 2017. Differentiation between oppositely oriented microtubules controls polarized neuronal transport. Neuron. 96:1264–1271e1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campenhausen H, Yoshihara Y, Mori K. 1997. OCAM reveals segregated mitral/tufted cell pathways in developing accessory olfactory bulb. NeuroReport. 8:2607–2612. [DOI] [PubMed] [Google Scholar]
- Westphal DS, Andres S, Makowski C, Meitinger T, Hoefele J. 2018. MAP2—a candidate gene for epilepsy, developmental delay and behavioral abnormalities in a patient with microdeletion 2q34. Front Genet. 9:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winther M, Berezin V, Walmod PS. 2012. NCAM2/OCAM/RNCAM: cell adhesion molecule with a role in neuronal compartmentalization. Int J Biochem Cell Biol. 44:441–446. [DOI] [PubMed] [Google Scholar]
- Witte H, Neukirchen D, Bradke F. 2008. Microtubule stabilization specifies initial neuronal polarization. J Cell Biol. 180:619–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Jaehne EJ, Greenberg Z, McCarthy P, Saleh E, Parish CL, Camera D, Heng J, Haas M, Baune BT et al. 2015. 14-3-3zeta deficient mice in the BALB/c background display behavioural and anatomical defects associated with neurodevelopmental disorders. Sci Rep. 5:12434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshihara Y, Kawasaki M, Tamada A, Fujita H, Hayashi H, Kagamiyama H, Mori K. 1997. OCAM: a new member of the neural cell adhesion molecule family related to zone-to-zone projection of olfactory and vomeronasal axons. J Neurosci. 17:5830–5842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinn K, Ozkan E. 2017. Neural immunoglobulin superfamily interaction networks. Curr Opin Neurobiol. 45:99–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.