

Abstract
Ferroptotic death is the penalty for lost control over three processes – iron metabolism, lipid peroxidation and thiol regulation – common for pro-inflammatory environment where professional phagocytes fulfill their functions yet survive. We hypothesized that redox reprogramming of 15-lipoxygenase (15-LOX) during generation of pro-ferroptotic signals, 15-hydroperoxy-eicosa-tetra-enoyl-phosphatidylethanolamine (15-HpETE-PE), modulates the ferroptotic endurance. Here, we discovered that iNOS/NO•-enrichment of activated M1 (but not alternatively activated M2) macrophages/microglia modulates susceptibility to ferroptosis. Genetic or pharmacologic depletion/inactivation of iNOS confers sensitivity on M1 cells whereas NO• donors empower resistance of M2 cells to ferroptosis. In vivo, M1 (vs M2) phagocytes exert higher resistance to pharmacologically induced ferroptosis; this resistance is diminished in iNOS-deficient cells in pro-inflammatory conditions of brain trauma or tumor microenvironment. The nitroxygenation of ETE-PE intermediates and oxidatively-truncated species by NO• donors and/or suppression of NO• production by iNOS inhibitors represent a novel redox mechanism of regulation of ferroptosis in pro-inflammatory conditions.
INTRODUCTION
Ferroptosis is a newly identified type of redox-driven cell death program 1, 2. The regulatory metabolic cornerstones of the program include Fe-dependent generation of (phospho)lipid hydroperoxides and their reduction to alcohols by glutathione peroxidase 4 (GPX4), 3. The latter prevents the decay of hydroperoxy-lipids to oxidatively truncated reactive electrophilic species 4, 5 that attack yet to be identified critical protein targets leading to the cell demise 6, 7.
While several components of the specific enzymatic machinery coordinating the process of ferroptosis have been identified, a number of regulatory pathways are still unknown. Among the former, are lipid metabolizing enzymes engaged in the formation of oxidation substrates, arachidonoyl-phosphatidylethanolamines (ETE-PE), such as ACSL4, LPCAT3 and a dioxygenase, 15-LOX, catalyzing the formation of 15-HpETE-PE 8–10. While ferroptosis has been documented in many different mammalian cells as well as in plants 11, some selected types of cells, particularly cells of innate immune system 12 (e.g., macrophages and microglia), display significant defiance against pro-ferroptotic stimulation. We hypothesized that the decision between resilience or vulnerability to ferroptosis may stem from different phenotypic features maximized under activated (M1) or alternatively activated (M2) states of macrophages.
RESULTS
iNOS/NO• determine the resistance of M1 and sensitivity of M2 macrophages and microglia to ferroptosis.
We endeavored to test the sensitivity of macrophages (RAW 264.7 cells, bone marrow-derived macrophages (BMDM)) and microglial cells (EOC 20 cells and primary mouse microglial cells) to a standard pro-ferroptotic treatment with a specific GPX4 inhibitor, RSL3 (1S,3R)-2-(2-chloroacetyl)-2,3,4,9-tetra-hydro-1-[4-(methoxycarbonyl)phenyl]-1H-pyrido[3,4-b]indole-3-carboxylic acid). These experiments revealed striking and uniform differences: M1 activated cells exerted high resistance to RSL3; whereas M0 and alternatively activated M2 cells were vulnerable and consistently responded by the loss of viability effectively preventable by a selective ferroptosis inhibitor, Ferrostatin-1 (Fer-1) (Fig. 1a and Supplementary Fig. 1a). Given the reported RSL3 vulnerability to metabolic transformations 13, we developed an LC-MS protocol, estimated the concentrations of RSL3 and determined that the same amounts of the reagent were present in resistant M1 macrophages and more vulnerable M0 and M2 phenotypes (Fig. 1b and Supplementary Fig. 1b).
Fig. 1. Differential sensitivity of activated M1 and alternatively activated M2 macrophages and microglia to RSL3-induced ferroptosis.
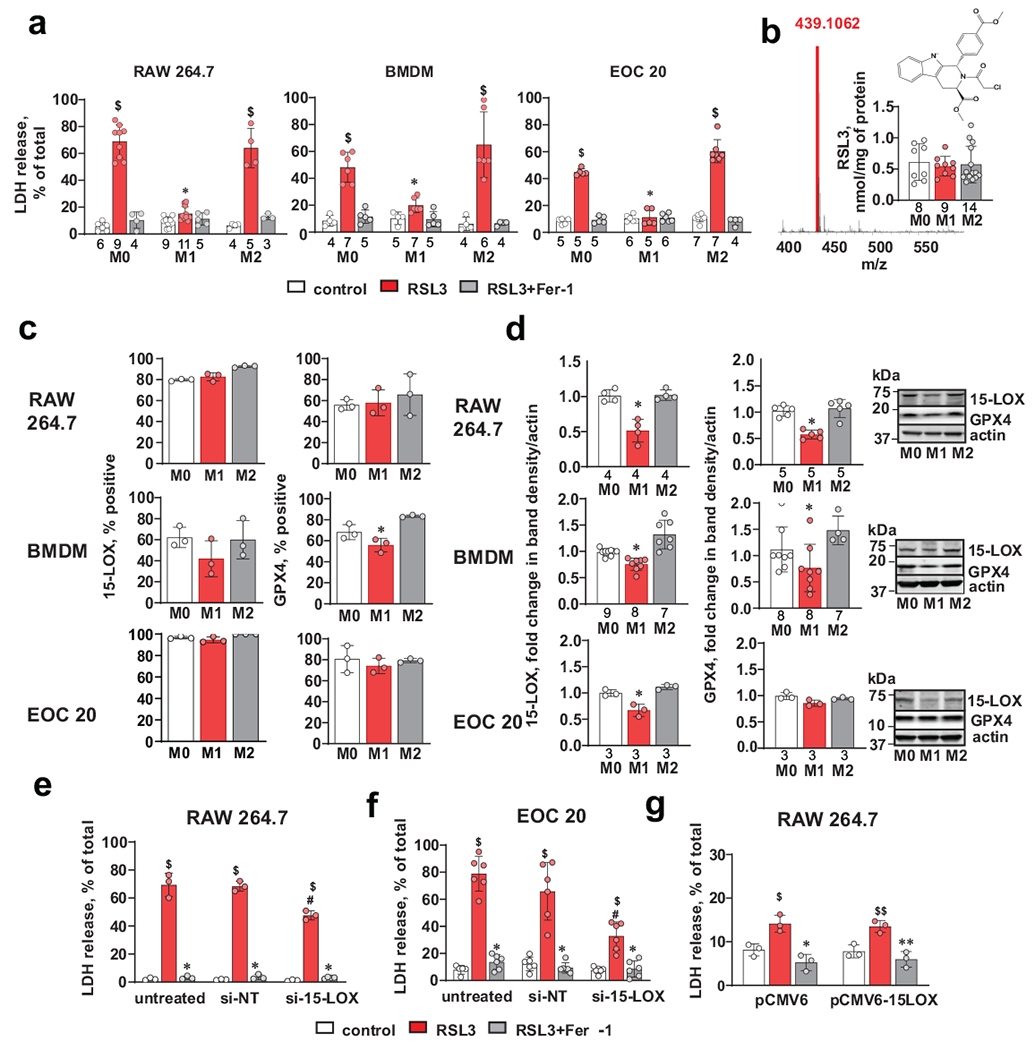
(a) Activated (M1) cells display resistance to RSL3 induced ferroptosis. RAW 264.7 macrophages, bone marrow derived macrophages (BMDM) and EOC 20 microglial cells were treated with RSL3 (500 nM) in the presence or absence of Fer-1 (400 nM) for 5 h. Data are means ± s.d.; the number of biologically independent samples are indicated on the X-axis below each bar. $p<0.0001 vs. corresponding control, *p< 0.0001 vs M2/RSL3, 2-way ANOVA, Tukey’s multiple comparison.
(b) LC-MS quantification of RSL3 in different macrophage phenotypes incubated for 5 hrs with RSL3 (500 nM) demonstrating similar amounts of the inhibitor in macrophages with different phenotypes. The number of biologically independent samples are indicated on the X-axis below each bar.
(c) Multicolor flow cytometry-based evaluation of 15-LOX and GPX4 positive cells. Data are means ± s.d., n=3 biologically independent samples, *p = 0.0308 vs corresponding M2, one-way ANOVA, Tukey’s multiple comparison.
(d) Western blots with densitometry-based quantitative assessments of mean relative intensity for 15-LOX and GPX4 in activated (M1) and alternatively activated (M2) RAW 264.7 macrophages, bone marrow derived macrophages (BMDM) and EOC 20 cells. The intensities of 15-LOX and GPX4 were normalized to those of actin and are reported as fold change to M0. Data are means ± s.d.; the number of biologically independent samples are indicated on the X-axis below each bar. P-values: RAW 264.7/15-LOX, *p=0.0004 vs M2; BMDM/15-LOX, *p< 0.0001 vs M2; EOC 20/15-LOX, *p = 0.0013 vs M2; RAW 264.7/GPX4, *p < 0.0001 vs M2; BMDM/GPX4, *p = 0.0308 vs M2; one-way ANOVA, Tukey’s multiple comparison.
(e, f) 15-LOX KD decreases sensitivity to RSL3-induced ferroptosis in alternatively activated (M2) RAW 264.7 macrophages (e) and EOC 20 cells (f). Cells were transfected with scrambled si-RNA (si-NT) or 15-LOX siRNA (si-15-LOX). Data are means ± s.d., n=3 and 6 biologically independent samples for RAW 264.7 and EOC 20 respectively. $p<0.0001 vs corresponding control, *p<0.0001 vs corresponding RSL3; #p < 0.0001 vs corresponding si-NT/RSL3.
(g) Overexpression of 15-LOX does not sensitize activated (M1) RAW264.7 macrophages to RSL3 induced ferroptosis. Cells were transfected with vector only (pCMV6) or vector containing 15-LOX (pCMV6-15-LOX). Data are means ± s.d., n=3 biologically independent samples. $p = 0.0082 vs pCMV6/control, *p = 0.0003 vs pCMV6/RSL3, $$p = 0.0120 vs pCMV6-15LOX/control; **p = 0.0013 vs pCMV6-15-LOX /RSL3, two-way ANOVA, Tukey’s multiple comparisons test.
To identify the mechanisms underlying the robust differences in the ferroptotic responses, we further explored several known anti-ferroptotic pathways of the cells and quantitated the expression levels of 15-LOX, ACSL4 and LPCAT3 (Fig. 1c,d and Supplementary Fig. 1c,d). The mechanisms linked to the availability of pro-ferroptotic substrates for 15-LOX controlled by ACSL4 and LPCAT3 were not markedly different between M1 and M2 states of RAW 264.7 macrophages: the amounts of ACSL4 were not different and the content of LPCAT3 was higher in M1 vs M2 state (Supplementary Fig. 1d). The levels of GPX4 – controlling pro-ferroptotic 15-HpETE-PE – were lower or similar in M1 vs M2 cells (Fig. 1c,d) suggesting this regulator is not the reason for the differences in M1 vs M2 sensitivity to pro-ferroptotic stimulation.
We next examined the amounts of the catalyst of pro-ferroptotic signal formation, 15-LOX, and found that its levels were markedly suppressed in M1 activated cells vs M2 alternatively activated cells (Fig. 1c,d). This might explain, at least in part, the higher pro-ferroptotic sensitivity of M2 cells vs M1 cells. Indeed, knocking down (KD) of 15-LOX in M2 RAW 264.7 macrophages and EOC 20 cells resulted in significantly reduced sensitivity to ferroptosis triggered by RSL3 (Figs. 1e,f and supplementary Fig. 1e, f). These results are compatible with the previously established role of 15-LOX in generating 15-HpETE-PE as a specific and predictive pro-ferroptotic oxidation product 8. To further explore the role of 15-LOX, we transfected M1 RAW 264.7 macrophages with a 15-LOX plasmid; this substantially (2.5-fold) increased the 15-LOX contents but did not change the resistance of M1 macrophages to RSL3 (Fig. 1g and Supplementary Fig. 1g). Overall, these results are suggestive of the involvement of other factor(s) as influential determinants of the much higher sensitivity of M2 vs M1 cells to pro-ferroptotic stimulation by RSL3.
In search for these yet to be identified mechanism(s), we note that M1 macrophages are characterized by a high content of inducible NO synthase (iNOS or NOS2) and consequently high NO• production 14. The latter has been shown to act as an inhibitor of 15-LOX catalyzed oxygenation reactions 15. With this in mind, we evaluated the iNOS expression and NO• levels in different cell types. Both macrophages and microglial cells all had markedly (~30-fold) higher levels of iNOS protein in M1 activation state than in M0 and M2 (Fig. 2a and Supplementary Fig. 2a–c). The results of Western-blotting were confirmed by fluorescence microscopy that demonstrated high levels of iNOS-positivity in M1 microglial cells and its strongly suppressed expression in M2 cells (Supplementary Fig. 2d). Moreover, live cell microscopy assessments of NO• production with a cell-permeable fluorescent probe, diaminorhodamine-4M (DAR-4M) revealed significantly higher levels of NO• in activated M1 vs. alternatively activated M2 RAW 264.7 cells (Fig. 2b and Supplementary Fig. 2e).
Fig. 2. Sensitivity of activated (M1) macrophages and microglial cells to RSL3-induced ferroptosis depends on the levels of iNOS expression.
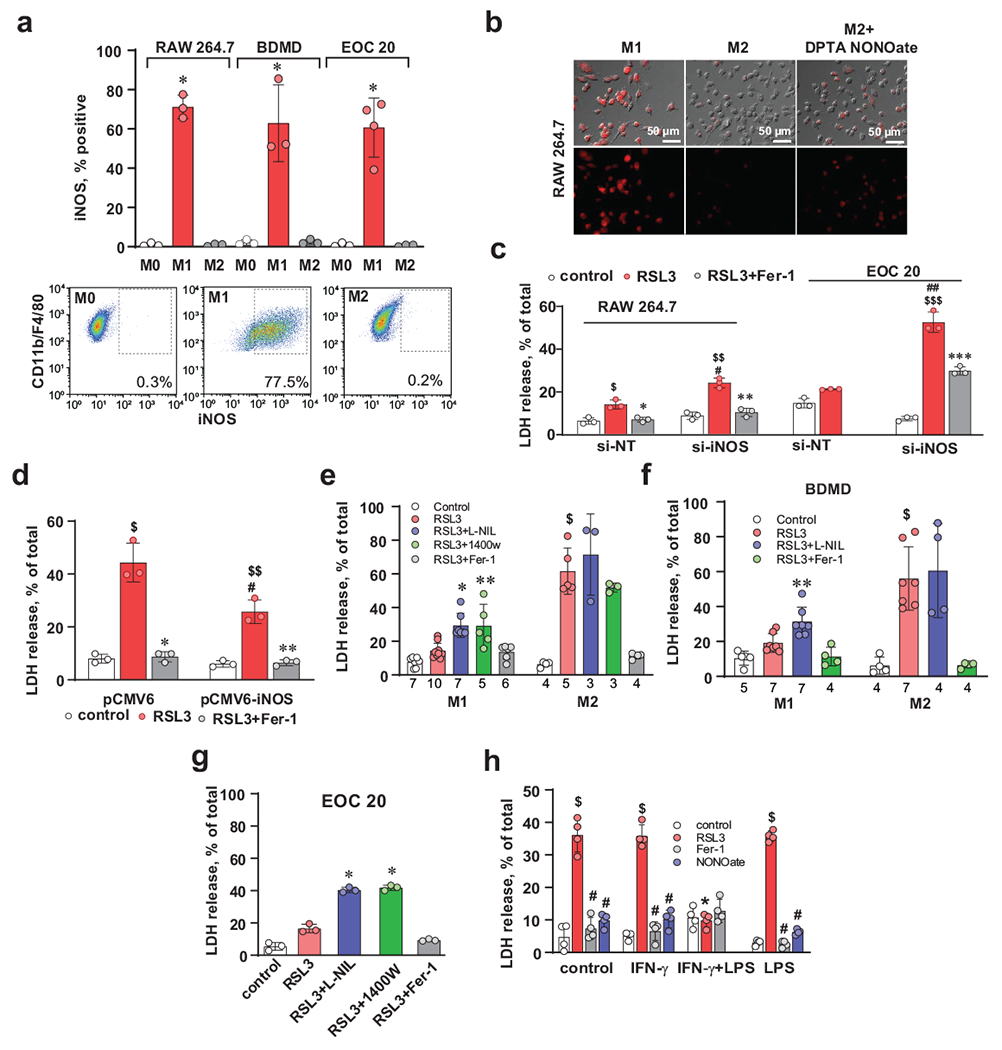
(a) Representative multicolor flow cytometry image and evaluation of the percentage of iNOS-positive cells. Data are means ± s.d., n=4 biologically independent samples for M1/EOC 20 and 3 for all other samples. *p < 0.0001 vs corresponding M2, two-way ANOVA, Tukey’s multiple comparison).
(b) Raw 264.7 macrophages activated into M1 phenotype produced high amount of NO• as assessed by live cell microscopy. Representative images from 3 independent experiments.
(c) Knock down of iNOS sensitizes activated (M1) RAW 264.7 macrophages and EOC 20 cells to RSL3-induced ferroptosis. IFN-γ treated RAW 264.7 and EOC 20 macrophages were transfected with scrambled si-RNA (si-NT) or iNOS siRNA (si-iNOS). Data are means ± s.d.; n=3 biologically independent samples. $p = 0.0022 vs si-NT/control, *p = 0.0044 vs si-NT/RSL3, $$p<0.0001 vs si-iNOS/control, **p<0.0001 vs si-iNOS/RSL3, #p = 0.0002 vs si-NT/RSL3, $$$p < 0.0001, vs si-NT/RSL3; ##p < 0.0001 vs si-iNOS/control; ***p < 0.0001 vs si-iNOS/RSL3, two-way ANOVA, Tukey’s multiple comparison.
(d) Transfection of M2 macrophages with iNOS expressing plasmid increased resistance to ferroptosis. M2 macrophages transfected with vector only (pCMV6) or iNOS expressing plasmid (pCMV6-iNOS). Data are means ± s.d.; n=3 biologically independent samples. $p < 0.0001 vs pCMV6/control; *p < 0.0001 vs pCMV6/RSL3; $$p = 0.0003 vs pCMV6-iNOS/control; **p = 0.0004 vs pCMV6-iNOS/RSL3; #p = 0.0006 vs pCMV6/RSL3, two-way ANOVA, Tukey’s multiple comparisons test. Representative Western blots of iNOS and densitometry-based quantitative assessments of mean relative intensity for the protein. *p = 0.0002, two-tailed, unpaired student’s t-test.
(e, f) iNOS inhibitors enhance RSL3-induced ferroptosis in activated (M1) RAW 264.7 macrophages (e) and bone marrow derived macrophages (f) but had no effect on alternatively activated (M2) cells. Data are means ± s.d. The number of biologically independent samples are indicated on the X-axis below each bar. $p < 0.0001 vs corresponding control, *p = 0.0283 vs M1/RSL3, **p < 0.0001 vs M1/RSL3, two-way ANOVA, Tukey’s multiple comparisons test.
(g) iNOS inhibitors enhance RSL3-induced ferroptosis in EOC 20 cells activated into M1 state. Cells were treated with RSL3 (500 nM) in the presence or absence of iNOS inhibitors (L-NILn 1400W) or Fer-1 (400 nM). Data are means ± s.d., n=3 biologically independent samples, *p < 0.0001 vs RSL3, two-way ANOVA, Tukey’s multiple comparison.
(h) RAW 264.7 gamma NO(−) macrophages expressing iNOS are resistant to RSL3-induced ferroptosis. Macrophages were activated with (IFN-γ (100 ng/ml) or IFN-γ + LPS) or LPS (10 ng/ml) for 48h. Ferroptosis was induced by treatment with RSL3 (500 nM) in the presence or absence of Fer-1 (400 nM) for 5 h. Data are means ± s.d.; $p < 0.0001 vs corresponding controls, #p < 0.0001 vs corresponding RSL3, *p < 0.0001 vs Control/RSL3, IFN-γ/RSL3, IFN-γ+LPS/RSL3, and LPS/RSL3 groups, two-way ANOVA, Tukey’s multiple comparisons test.
We hypothesized that iNOS expression may represent a regulatory pathway controlling pro-inflammatory resistance of M1 macrophages and microglial cells to ferroptotic death program. To experimentally test this, we genetically manipulated iNOS expression and assessed the sensitivity to RSL3-induced ferroptosis. Knockdown (KD) of iNOS caused increased sensitivity of M1 RAW 264.7 macrophages and EOC 20 cells to RSL3 induced death that was effectively suppressed by Fer-1 (Fig. 2C and Supplementary Fig. 2f). Conversely, transfection of M2 cells with iNOS plasmid caused elevated levels of iNOS and increased resistance of cells to ferroptosis (Fig. 2d and Supplementary Fig. 2g). Following this chain of logic, we tested iNOS inhibitors, which decreased the amount of NO• in M1 macrophages measured by a fluorescent probe DAR 4M (Supplementary Fig. 2h). We found that iNOS inhibitors enhanced sensitivity of activated RAW 264.7 cells (Fig. 2e), BMDM (Fig. 2f), EOC 20 microglial cells (Fig. 2g) and primary microglial cells (Supplementary Fig. 2i) to RSL3-induced death in M1 state but had no effect on M2 cells.
To further investigate the contribution of iNOS/NO• to the resistance of macrophages to ferroptosis, we utilized a RAW 264.7 derived-cell line, RAW 264.7 gamma NO(−) cells, which compared to the parental macrophages, requires co-stimulation by IFN-γ and LPS for expression of iNOS 16. Treatment with IFN-γ or LPS alone did not induce iNOS, while co-treatment (IFN-γ + LPS) had a higher expression of iNOS (Fig. 2h and Supplementary Fig. 3a, b). We found that only (IFN-γ + LPS) co-stimulated RAW 264.7 gamma NO(−) cells expressing iNOS were resistant to RSL3 induced ferroptosis (Fig. 2h and Supplementary Fig. 3b). Intracellular availability of iron, controlled by transferrin receptors, is an important ferroptosis regulating factor , particularly in macrophages 17, 18. In RAW 264.7 gamma NO(−) cells, expression of transferrin receptors was similar in macrophages activated by either IFN-γ alone or IFN-γ + LPS (Fig. 2h and Supplementary Fig. 3b). As cells activated by IFN-γ or LPS alone were more sensitive to RSL3-induced ferroptosis than those activated by (IFN-γ + LPS), we concluded that expression of iNOS/NO• was sufficient to inhibit RSL3 induced ferroptosis and transferrin receptors were not involved in this process (Fig. 2h and Supplementary Fig. 3b).
NO• donors suppress ferroptosis in M2 macrophages and microglial cells.
We further reasoned that NO• donors may act as small molecule regulators of ferroptosis. Using flow cytometry and live cell microscopy, we detected that incubation of cells labeled with an NO• probe, DAR-4M, in the presence of an NO•-donor, DTPA NONOate, significantly increased fluorescence of cells, demonstrating accumulation of NO• (Supplementary Fig. 4a). Testing several NO• donors on RAW 264.7 macrophages, EOC 20 cells or primary microglial cells showed that they caused concentration-dependent inhibition of RSL3 induced ferroptosis (Figs. 3a and Supplementary Fig. 4b, c). To ensure that the anti-ferroptotic effects of NO• are not RSL3-specific, we used a different GPX4 inhibitor, ML162, as well as two other reagents affecting the GPX4/GSH system - erastin and imidazole ketone erastin (IKE) - belonging to class 1 ferroptosis inducers and acting by depleting GSH 19. An NO•-donor, DETA NONOate, inhibited ferroptosis induced by these alternative pro-ferroptotic reagents (Fig. 3b).
Fig. 3. NO• protects cells against RSL3-induced ferroptosis.
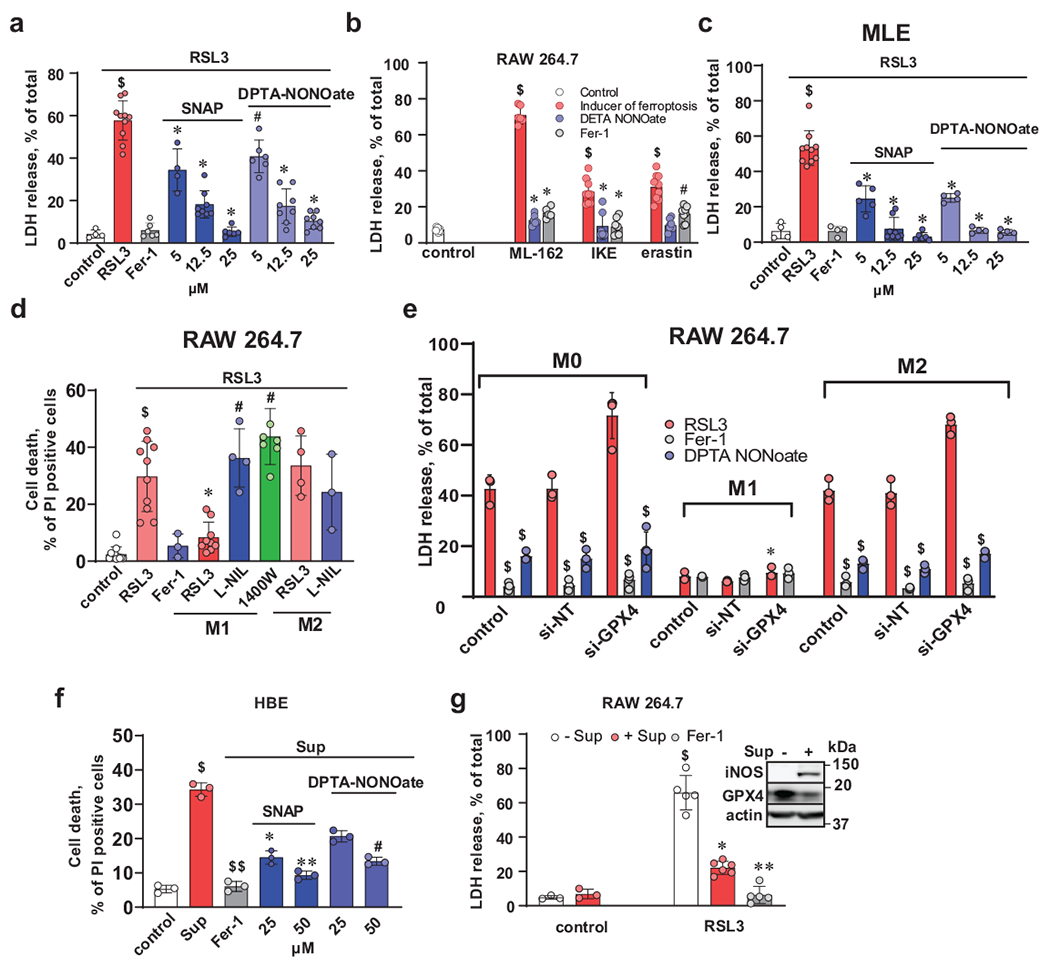
(a) NO• donors (SNAP and DPTA NONOate) inhibit RSL3-induced ferroptosis in RAW 264.7 macrophages. Cells were treated with RSL3 (500 nM) in the presence of different concentrations of NO•-donors for 5 h. Cell death was estimated by LDH release assay. Fer-1 (400 nM) was used as a positive control. Data are mean ± s.d.; For RAW 264.7 macrophages, number of biologically independent samples in: control (n =4), RSL3 (n=10), RSL3+Fer-1 (n=6), DPTA-NONOate (n=6, 8, 8) for 5 μM, 12.5 μM and 25 μM, respectively, SNAP (n=4, 8, 6) for 5 μM, 12.5 μM and 25 μM, respectively. $p < 0.0001 vs control, *p < 0.0001 vs RSL3, #p = 0.0004 vs RSL3, two-way ANOVA, Tukey’s multiple comparisons test.
(b) NO• protects RAW 264.7 macrophages against ferroptosis induced by ML162, IKE or erastin. Cells were treated with ML162 (2 μM), IKE (2 μM) or erastin (20 μM) in the presence of 25 μM of DETA NONOate for 18 h. Cell death was estimated by LDH release assay. Fer-1 (400 nM) was used as a positive control. Data are means ± s.d.; Number of biologically independent samples in control (n=9), ML-162( n=6, 6, 9) IKE (n=9, 8, 7) erastin (n=9, 9, 8) for inducer of ferroptosis, RSL3, RSL3+Fer-1 groups, respectively. $p < 0.0001 vs control, *p < 0.0001 vs ferroptosis inducer, #p < 0.0001 vs ferroptosis inucer, two-way ANOVA, Tukey’s multiple comparisons test.
(c) NO• donors (SNAP and DPTA NONOate) inhibit RSL3-induced ferroptosis in MLE-cells. Cells were treated with RSL3 (500 nM) in the presence of different concentrations of NO•-donors for 5 h. Cell death was estimated by LDH release assay. Fer-1 (400 nM) was used as a positive control. Data are mean ± s.d.; n=4 biologically independent samples per group, $p < 0.0001 vs control, *p < 0.0001 vs RSL3, two-way ANOVA, Tukey’s multiple comparisons test.
(d) NO• released by iNOS overexpressing M1 activated RAW 264.7 (but not M2 alternatively activated macrophages) inhibit RSL3-induced ferroptosis in MLE cells. MLE cells labeled with Cell Tracker Green were incubated with or without unlabeled M1 or M2 RAW 264.7 macrophages (macrophages to MLE ratio, 2:1). Ferroptosis was induced by treatment with RSL3 (500 nM) for 5 h in the absence or in the presence of Fer-1 (500 nM). Cell death was estimated by PI-staining using flow cytometer and calculated as percentage of double positive cells (Cell Tracker Green and PI) to total amount of Cell Tracker Green-positive cells. Data are means ± s.d.; Number of biologically independent samples: control=11, RSL3+Fer-1=3, RSL3=10, 9, 4 for MLE, MLE+M1 and MLE+M2, respectively, RSL3+L-NIL=4, 3 for MLE+M1 and MLE+M2, respectively, 1400W =7. $p < 0.0001 vs control, *p < 0.0001 vs RSL3, #p < 0.0001 vs MLE cells treated with RSL3 and incubated with M1 macrophages, two-way ANOVA, Tukey’s multiple comparisons test.
(e) GPX4 knock down increased sensitivity to RSL3-induced ferroptosis in non-activated (M0) and alternatively activated (M2) RAW 264.7 macrophages but not in M1 macrophages. (1) Densitometry-based assessments of mean relative intensity for the protein demonstrating efficiency of GPX4 knock-down. Data are means ± s.d.; n=3 biologically independent samples. *p = 0.0311 vs M0/Control, $p = 0.0023 vs. M0/si-NT, **p = 0.0328 vs M1/Control, $$p = 0.0168 vs. M1/si-NT, ***p = 0.0432 M2/Control and $$$p = 0.0165 vs. M2/si-NT, two-way ANOVA, Tukey’s multiple comparisons test.
(f) NO• donors prevented ferroptosis induced in human bronchial epithelial (HBE) cells by Pseudomonas aeruginosa supernatants. HBE cells were incubated with P. aeruginosa supernatant (Sup) alone or with two different types of NO• donors (SNAP or DETA NONOate) for 20 h. Cell death was estimated by PI-staining using flow cytometry. Fer-1 was used as a positive control. Data are means ± s.d., n = 3 biologically independent samples, $p = 0.0125 vs control, $$p = 0.0162 vs Sup., *p = 0.0435 vs Sup., **p = 0.0039 vs Sup., #p = 0.0154 vs Sup., one-way ANOVA, Tukey’s multiple comparisons test.
(g) Pre-treatment of RAW 264.7 macrophages with P. aeruginosa supernatant induced iNOS expression, depleted GPX4 and rendered macrophages resistant to ferroptosis. RAW 264. Macrophages were pre-treated with P. aeruginosa supernatants (Sup) overnight and then ferroptosis was triggered by addition of RSL3 (500 nM) for 5 hrs. Fer-1 was used as a positive control. Data are means ± s.d.; Number of biologically independent samples: control (n=3 each), RSL3/-Sup (n=5), RSL3/+Sup (n=6) , RSL3/Fer-1 (n=5). $p < 0.0001 vs control/-Sup and control/+Sup, *p = 0.0011 vs. RSL3/-Sup., **p < 0.0001 vs. RSL3/-Sup., two-way ANOVA, Tukey’s multiple comparisons test. Inset: Representative Western blots of iNOS and GPX4.
Distant suppression of ferroptosis in epithelial cells by M1 macrophages.
Assuming that NO• readily diffuses through and across membranes 20, we reasoned that high expression of iNOS leading to its massive production in M1 activated (but not in M2 alternatively activated) macrophages may exert distant protective effects against ferroptosis triggered in other remotely located cells. To test this, we used RSL3 to trigger ferroptosis in mouse lung epithelial (MLE) cells in the presence and absence of macrophages activated to M1 state with high levels of iNOS/NO•. MLE cells readily responded to RSL3, and the cell death was inhibited by NO•-donors (Fig. 3c). When we co-incubated MLE cells with M1 RAW 264.7 macrophages, no ferroptotic death of MLE cells was detected. Notably, M2 macrophages did not confer resistance against RSL3 induced death onto MLE cells (Fig. 3d). Addition of iNOS inhibitors, L-NIL and 1400W, eliminated the protective effect of M1 macrophages (Fig. 3d).
iNOS/NO• can substitute GPX4 as an anti-ferroptotic defense.
To study the relationship between NO• and GPX4 in regulation of ferroptosis we genetically manipulated GPX4 levels in M0, M1 and M2 macrophages using GPX4 siRNA. In iNOS-deficient M0 and M2 macrophages, knockdown of GPX4 expectedly increased sensitivity of cells to RSL3-induced ferroptosis (Fig. 3e and Supplementary Fig. 4d, e). Importantly, macrophages activated into iNOS-enriched M1 state still exerted low sensitivity to ferroptosis in spite of the markedly decreased levels of GPX4 in KD cells. In M0 and alternatively activated M2 cells, NO• donors caused a robust suppression of ferroptosis independently of substantially lowered (due to KD) GPX4 levels (Fig. 3e and Supplementary Fig. 4d, e).
NO• donors inhibit ferroptosis triggered by bacterial (P. aeruginosa) 15-LOX (pLOXA).
To further scrutinize the role of the enzymatic LOX-catalyzed PE peroxidation in protective effects of NO• against ferroptosis, we employed extracellular membrane vesicles produced and released into the environment by a Gram-negative bacterium, Pseudomonas aeruginosa. The vesicles containing many intracellular proteins, including a bacterium-specific 15-LOX, pLoxA, could be isolated as supernatants after centrifugation of growing cultures. Our previous work established that the supernatants obtained from P. aeruginosa and incubated with human bronchio-epithelilal cells (HBE) caused depletion of GPX4 and ferroptosis 21. P. aeruginosa supernatants with vesicles containing pLoxA effectively induced ferroptosis in HBE cells. Both NO• donors, SNAP and DETA NONOate, showed high efficiency in protecting HBE cells against pLoxA induced ferroptosis (Fig. 3f). These results directly support the15-LOX-association of the protective mechanism of NO• donors against ferroptosis.
We further used the P. aeruginosa supernatants to examine the ability of iNOS to regulate ferroptosis as we previously established that the supernatants cause degradation of GPX4 21. We pretreated RAW 264. macrophages with P. aeruginosa supernatants overnight and then added RSL3 for 5 hrs. While the GPX4 depletion effect was reproduced, no ferroptotic death was observed (Fig. 3g). Notably, the supernatants activated the macrophages to M1 state and characteristically enhanced expression of iNOS (Fig. 3g). Overall, these experiments demonstrate the strong ability of iNOS/NO• or NO• donors to regulate sensitivity of RAW 267.4 macrophages to ferroptosis independently of GPX4 contents.
iNOS/NO• inhibit pro-ferroptotic lipid peroxidation.
To get a better insight in the effects of NO• on the formation of lipid hydroperoxides as pro-ferroptotic signals, we employed a fluorogenic probe, Liperfluo, which selectively reduces hydroperoxy-lipids to respective alcohols to yield a fluorescent product 22. Incubation of M2 macrophages in the presence of RSL3 induced a time-dependent increase in the fluorescence signal from Liperfluo, indicating the accumulation of hydroperoxy-lipids. In contrast, fluorescence of M1 macrophages or M2 macrophages incubated with an NO• donor, DPTA NONOate, was at the level of noise (Fig. 4a and Supplementary Fig. 5a).
Fig. 4. NO• suppresses RSL-3-induced accumulation of oxidatively modified PE species in alternatively activated (M2) RAW 264.7 macrophages.
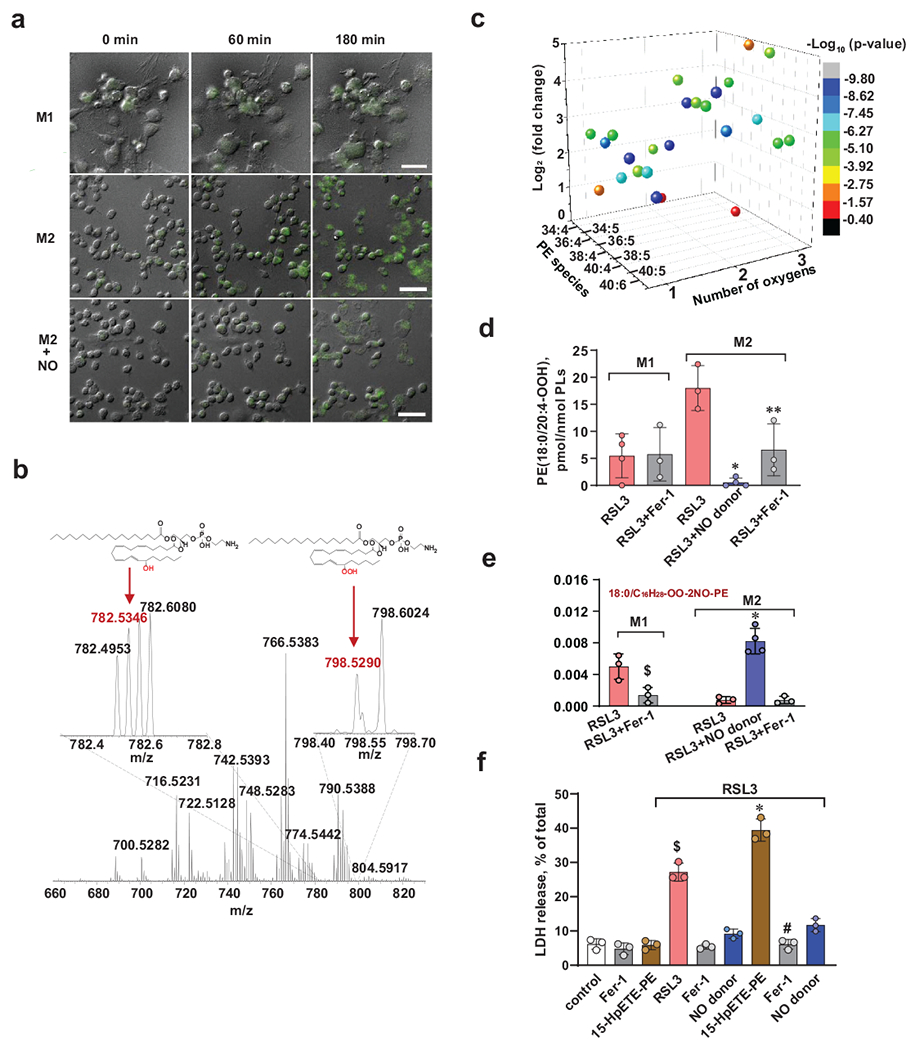
(a) Live cell fluorescence imaging of RAW 264.7 macrophages with Liperfluo. Activated (M1) or alternatively activated (M2) RAW 267.4 macrophages were treated with Liperfluo (10 μM) and RSL3 (500 nM) in the presence and absence of DPTA NONOate (25 μM). Fluorescence of Liperfluo was registered using inverted Nikon TiE fluorescent microscope (Nikon Inc.) for 3 h. Typical changes in fluorescence in one stage position (of 10) at three time points (0, 60 min, and 180 min) are shown. Representative images from 3 independent experiments
(b) Representative full MS spectra of phosphatidylethanolamines (PE) from alternatively activated (M2) RAW 264.7 macrophages. Inserts: Mass spectra of PE in the range of m/z from 782.2 to 782.8 and from 798.4 to 798.7. Spectra demonstrate the presence of peaks with m/z 782.5346 and m/z 798.5290 corresponding to 15-HETE and15HpETE-PE species, respectively. Structures of the major ferroptotic signal, C18:0/C20:4-OOH-PE (15-HpETE-PE) species, and its reduced form C18:0/15-HETE-PE are shown as well. Representative image from 3 independent experiments.
(c). 3D volcano-plot of major oxygenated arachidonoyl-PE species induced by RSL3 in M2 RAW 264.7 macrophages.
(d) Contents of ferroptotic cell death signal, 18:0/20:4-OOH-PE (15-HpETE-PE) species, in activated (M1) and alternatively activated (M2) RAW 264.7 macrophages exposed to RSL3 (500 nM) in the presence or absence of Fer-1 (400 nM) or DPTA NONOate (25 μM) for 5 h vs RSL3. Number of biologically independent samples: 4 each for M1/RSL3 and M2/RSL3+NO donor, and 3 for all other groups. *p = 0.0006 vs M2/RSL3, **p = 0.0244 vs M2/RSL3, one way ANOVA, Tukey’s multiple comparisons test.
(e) Bar graph showing the relative amounts of nitroxygenated/oxidatively truncated 15-HpETE-PE species (m/z 804.480) in activated (M1) and alternatively activated (M2) RAW 264.7 macrophages. Number of biologically independent samples: 4 for M2/RSL3+NO donor and 3 for all other groups. $p = 0.0141 vs M1/RSL3, *p < 0.0001 vs M2/RSL3, two-way ANOVA, Tukey’s multiple comparisons test.
(f) NO• protects alternatively activated (M2) RAW 264.7 cells from 15-HpETE-PE induced ferroptosis. RAW 264.7 cells were pre-treated with RSL3 (150 nM) for 3 h and then incubated with 15-HpETE-PE (1 μM) with or without NO• donor, DPTA NONOate, (25 μM) for further 3 h. Cell death was measured by LDH release assay. Fer-1 was used as a positive control. Data are means ± s.d.; n = 3 biologically independent samples per group. $p < 0.0001 vs control *p < 0.0001 vs RSL3, #p < 0.0001 vs RSL3+15-HpETE-PE, one-way ANOVA, Tukey’s multiple comparisons test.
As our previous work has identified 15-HpETE-PE as predictive biomarkers of ferroptosis 8, we further used LC-MS redox lipidomics analysis and determined the contents of these products and the effects of an NO•-donor in RSL3-treated M2 activated RAW 264.7 cells (Fig. 4b–e and Supplementary Fig. 5b). RSL3 treatment caused accumulation of 73 species of oxidized PE, particularly various AA-containing species, including the 15-HpETE-PE (18:0/20:4-OOH-PE species) (Fig. 4b and Supplementary Fig. 5c). In the presence of the NO• donor, DTPA NONOate, the RSL3-dependent accumulation of this product was markedly suppressed (Fig. 4d). In contrast, no significant accumulation of this 15-HpETE-PE species was found in activated (M1) RAW 264.7 macrophages upon exposure to RSL3 (vs non-treated controls) (Fig. 4d). To test whether the elevated levels of 15-HpETE-PE in M2 macrophages could be associated with higher levels of polyunsaturated phospholipid precursors as substrates of 15LOX catalysis, we performed LC-MS analysis of major classes of polyunsaturated phospholipids. We noted that the contents of oxidizable species of PE and PC with arachidonoyl- and adrenoyl-residues were slightly higher in M1 macrophages compared to M0 and M2 macrophages (Supplementary Fig. 5d). This implies that the availability of pro-ferroptotic substrates could not be the cause of observed ferroptotic resistance in M1 macrophages.
NO• can inhibit LOX activity via different mechanisms, including the reduction of catalytic non-heme iron to non-productive ferrous state, and reaction with radical intermediates leading to the formation of nitroxygenated lipid products. To examine these mechanisms we conducted tandem LC-MS/MS identifications of the products formed upon RSL3 stimulation of the M1 cells vs. M2 cells in the presence and absence of DPTA NONOate (Supplementary Fig. 5b). Low levels of nitroxygenated PE products were detectable in M2 RAW 264.7 macrophages treated with RSL3 in the presence of DPTA NONOate (Supplementary Fig. 5b). The level of nitroxygenated PE products was estimated as 1.2+0.5 pmol/μmol phospholipids. This product was not found in any samples generated from M2 RAW 264.7 macrophages in any other incubation conditions. Given the inhibitory effects of the NO• donor on ferroptosis, these results indicate that NO• could interact with the enzyme’s reactive intermediates or lipid radical intermediates triggered by RSL3 thus suppressing the execution of the ferroptotic program.
Interaction of NO• with lipid radical intermediates may occur during functioning of 15-LOX, as well as downstream of this stage, when 15-LOX-generated 15-HpETE-PE are cleaved to yield oxidatively truncated (shorter-chain) species 4, 5, 23. Indeed, we were able to detect elevated levels of the nitroxygenated oxidatively truncated product derived from the 18:0/20:4-OOH-PE species in LC-MS spectra of lipids from the M2 cells treated with RSL3 + DPTA NONOate as well as in M1 cells treated with RSL3 (Fig. 4e and Supplementary Fig. 5e). Thus NO• scavenging of lipid radicals can occur not only during the enzymatic production of 15-HpETE-PE, but also in the subsequent reaction(s) of their oxidative decomposition leading to shorter chain derivatives. To further explore this mechanism, we studied the effect of DPTA NONOate on ferroptosis of M2 RAW264.7 macrophages induced directly by exogenously added 15-HpETE-PE, i.e. independently of 15-LOX (Fig. 4f). In line with our previously published results 8, 21, 15-HpETE-PE was effective in inducing RSL3 supported ferroptosis (Fig. 4f). The NO• donor inhibited ferroptosis almost completely suggesting it could directly interact with the radical intermediates generated during the decomposition of 15-HpETE-PE (Fig. 4f).
Mathematical modeling of NO• dependent inhibition of ferroptosis.
To improve our understanding of the NO• effects on the ferroptotic program, we constructed and analyzed a mathematical model composed of a network of the major ferroptotic interactions and regulatory pathways involving NO• reactions (Supplementary Fig. 6a, Supplementary Tables 1 and 2). The model is composed of 18 components and its dynamics is represented by a system of ordinary differential equations (Supplementary Table 2). We utilized the data on ferroptotic cell death of M1- and M2-activated cells in response to various doses of RSL3 stimulation under normal and 15-LOX KD conditions to estimate the unknown parameters in the model. Supplementary figures 6b and c display the excellent agreement achieved between the model predictions (gray bars) and the experimental data (blue bars), using the optimized set of parameters. We further validated our model using two additional datasets. Comparison of experimental and computational results in supplementary figure 6c demonstrated that the model reproduced the experimentally observed ferroptotic cell death of M2 cells in response to RSL3, as well as the dose-dependent protection by NO• donors. The model also reflected the increased sensitivity of M1 macrophages to ferroptosis in the presence of an iNOS inhibitor, L-NIL (Supplementary Fig. 6c). These results suggest that the calibrated model has both descriptive and predictive power.
We next performed in silico experiments to quantitatively assess the role of iNOS-derived NO• in regulating ferroptotic cell death. Increased expression of iNOS decreased sensitivity of macro-phages to RSL3 stimulation (Supplementary Fig. 6d). In terms of controlling ferroptosis, the efficacy of iNOS and GPX4 were on the same level. iNOS-dependent suppression of RSL3-stimulated ferroptosis in M1 cells likely depends on two inhibitory mechanisms of NO•: (i) reduction of 15-LOX iron (Fe3+ to Fe2+), and (ii) scavenging of lipid radical intermediates generated during the formation of HOO-AA-PE and its cleavage leading to the production of oxidatively-truncated species (Supplementary Fig. 6e). Sensitivity analysis highlighted the significance of the kinetic parameters associated with both mechanisms in regulating ferroptotic death, while the second mechanism was slightly more important than the first one (Supplementary Fig. 6f). We then perturbed the reaction rate constant of lipid radicals scavenging and simulated the influences of iNOS and 15-LOX on the cell fate decision (Supplementary Fig. 5g). The results suggest that, via a positive feedback loop in the network, the lipid radicals scavenging reactions can contribute to the robustness of the iNOS-regulated resistance of M1 cells and sensitivity of M2 cells to ferroptosis in noisy microenvironment.
In vivo effects of iNOS/NO• on feroptotic death and contents of M1 and M2 macrophages and microglia.
To test whether iNOS/NO• driven mechanisms of regulation of ferroptosis operate in vivo, we employed three different models. In a zymosan-induced peritonitis mouse model 24, we found that M2 macrophages (CD11b+/CD45+/CD206+) were the predominant cell type compared to M1 at 72h after zymosan administration (Fig. 5a, b). Intraperitoneal injection of RSL3 at 72h after zymosan administration markedly decreased the fraction of M2 macrophages, while treatment with a NO• donor, DETA NONOate, suppressed this decrease (Fig. 5a, b).
Fig. 5. iNOS/NO• driven mechanisms of ferroptosis regulation in vivo.
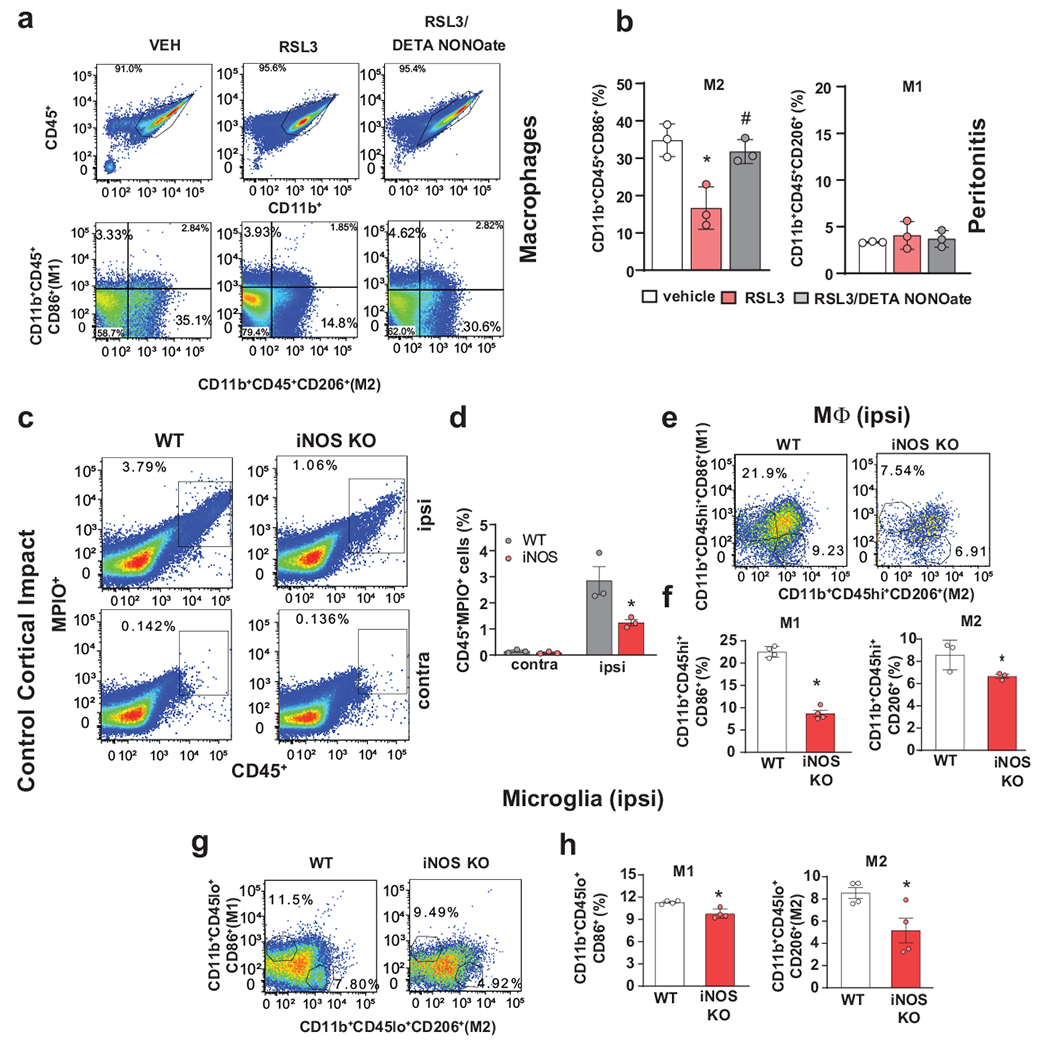
(a,b) DETA NONOate rescued CD11b+CD45+CD206+ peritoneal macrophages (M2) in a mouse model of zymosan-peritonitis plus RSL3. (a) Representative flow cytometry images (from 3 independent experiments) show CD206+ or CD86+ cells gated from CD11b+CD45+ population. Representative images from 3 biologically independent samples. (b) DETA NONOate significantly increased the number of CD11b+CD45+CD206+ macrophages (M2, left panel) compared to treatment with RSL3, there were no changes in CD11b+CD45+CD86+ macrophages (M1, right panel). Data are mean ± s.d., n = 3 biologically independent samples, *p = 0.0064 vs Vehicle, $p = 0.0151 vs RSL3, one-way ANOVA, Tukey’s multiple comparisons test.
(c, d) Typical flow cytometry images show percentages of CD45+MPIO+ cells from ipsilateral and contralateral cortices, respectively, of WT and iNOS KO mice 96h after CCI. Representative images from 3 biologically independent samples (c). Quantitative analysis shows significantly increased contents of CD45+MPIO+ cells in the ipsilateral sites of WT vs iNOS KO mice, but the lack of changes in the contralateral sites (d). Data are means ± s.e., n = 3 biologically independent samples, *p = 0.0139 vs WT, two-way ANOVA, Tukey’s multiple comparisons test.
(e, f) Flow cytometry images show percentages of M1(CD86+)/M2(CD206+) macrophages, which were gated from CD11b+ and CD45 high(hi)+ population, from ipsilateral cortices of WT and iNOS KO mice, respectively. Representative images from 4 biologically independent samples (e). Increase of M1 and M2 macrophages in ipsilateral cortex of WT mice compared to iNOS KO mice 96h after CCI (f). Data are means ± s.d., n = 4 and 3, biologically independent samples in M1 and M2, respectively. *p < 0.0001 vs WT in M1, *p = 0.0357 vs WT in M2, unpaired one-tailed Student’s t-test.
(g, h) Flow cytometry images show percentages of M1(CD86+)/M2(CD206+) microglia, which were gated from CD11b+ and CD45 low(lo)+ population, respectively. Representative images from 4 biologically independent samples (g). Increase of M1 and M2 microglia in ipsilateral cortex of WT mice vs iNOS KO mice at 96h (h). Data are means ± s.d., n = 4 biologically independent samples, *p = 0.0022 vs WT in M1, *p = 0.0011 vs WT in M2, unpaired one-tailed Student’s t-test.
To examine the availability of RSL3 in vivo, we directly assessed its amounts in peritoneal macrophages using our newly developed LC-MS/MS protocol. We found that after the peritoneal injection, RSL3 content in macrophages was 3.6+0.3 nmoles/mg of protein. This is about 6-fold higher than the amounts of RSL3 required for the induction of ferroptosis in macrophages in vitro (Fig. 1b and Supplementary Fig. 7a). As an alternative approach to examine whether the injected RSL3 - acting as a GPX4 inhibitor - was efficiently integrated into peritoneal macrophages, we assessed the effectiveness of GPX4 in enzymatic reduction of PE-OOH. As a surrogate marker of GPX4 inhibition by RSL3 we used the ratio of PE-alcohols (PE-OH, produced by the reduction of PE-OOH by GPX4) to PE-OH+PE-OOH. The ratio of PE-OH/( PE-OH+PE-OOH) was significantly reduced in samples from RSL3 treated animals (Supplementary Fig 7b). This suggests that the amount of RSL3 available in peritoneal macrophages following in vivo treatment was sufficient to inhibit GPX4 activity.
To further explore the inhibitory role of iNOS/NO• in in vivo model, we repeated experiments with zymosan-induced peritonitis mouse model using another activator of ferroptosis, imidazole ketone erastin (IKE), that has been reported as having a higher metabolic stability than erastin, and acceptable pharmacokinetic profiles 13. Intraperitoneal injection of IKE at 72h after zymosan administration markedly decreased the amount of M2 macrophages (CD11b+/CD45+/CD206+), while it did not change the number of M1 macrophages (CD11b+/CD45+/CD86+). Treatment with an NO•-donor, DETA NONOate eliminated the effect of IKE on M2 macrophages (Supplementary Fig. 7c, d).
We employed another model in which the tumor microenvironment is enriched with immunosuppressive cells, including myeloid cells, and assessed the role of iNOS/NO• in regulation of myeloid cell survival in tumors using bone marrow (BM) chimera approach. Lethally irradiated recipient mice were reconstituted with 2x106 bone marrow cells from wild-type (WT) and nos2−/−mice (iNOS KO). Six weeks later Lewis Lung Carcinoma was established subcutaneously. Three weeks after that, mice were euthanized, and cell content of bone marrow, spleen and tumors was evaluated. The lack of iNOS (NOS2) expression in BM chimeras was confirmed in macrophages by using qRT-PCR (Supplementary Fig. 7e). Mice reconstituted with NOS2 deficient BM cells demonstrated significantly lower tumor burden than mice reconstituted with BM from WT mice (Supplementary Fig. 7f). Total population of tumor associated macrophages (TAM) (CD45+CD11b+Ly6G−Ly6ClowF4/80hi) was slightly reduced in mice with NOS2 BM as compared to mice WT BM. However, those differences were not significant (Supplementary Fig. 7E). Similarly, no differences were observed in the population of M2 polarized macrophages – (CD206highMHC class IIlow cells gated out of total macrophages) (Supplementary Fig. 7f). In contrast, population of M1 polarized macrophages (CD206lowMHC class IIhigh) was significantly reduced in mice with NOS2 KO BM as compared to mice with WT BM Supplementary Fig. 7f).
To comparatively examine the role of GPX4 vs NIOS/NO• in the regulation of sensitivity of different phenotypic states of macrophages to ferroptotic death in vivo, we performed experiments using GPX4-depleted bone marrow derived monocytes isolated from Cre-(Wild Type) and Cre+/GPX4 fl fl (S100A8 Cre+) mice (Supplementary Fig. 7g). Monocytes were differentiated for 5 days in the presence of M-CSF, and polarized into M1 or M2 states by treatment with INF-γ+LPS or IL-4+IL1, respectively. The results revealed that polarization of GPX4-deficient cells into M2 state was associated with their lower survival compared to WT cells (Supplementary Fig. 7f). These experiments support the notion that the protective anti-ferroptotic effects of iNOS/NO• realized in M1 macrophages can compensate for GPX4 depletion in vivo.
Finally, we utilized a model of acute brain injury for which engagement of ferroptosis has been documented 9, 25 and in which the contusion environment with increased generation of ROS, lipid peroxidation and released hemoglobin represent a death threat to arriving macrophages and residential microglia. It has also been established that genetic deficiency or pharmacological inhibition of iNOS worsens histological and neurocognitive outcome after traumatic brain injury 26. Peripheral monocyte derived macrophage activation and polarization influences the extent of tissue damage and repair 27. While neutrophil accumulation in the contusional (ipsilateral) cerebral cortex peaks within the first 24h after controlled cortical impact (CCI), macrophages and microglia are the predominant inflammatory cell types 72h after impact 28. To examine the role of NO• in macrophage accumulation and loss after CCI we employed in situ magnetic resonance imaging protocols using paramagnetic iron oxide (MPIO) particles as described previously 29. Discrete, punctuate, areas of hypo-intensity indicating one or a few MPIO-labeled macrophages were seen in the trauma region 72h after CCI. Quantitative analysis showed a significantly larger area of MPIO positivity in the ipsilateral cortex and hippocampus of WT vs. iNOS KO mice (Supplementary Fig. 8a) though contusion and edema volumes did not differ between the two groups (Supplementary Fig. 8b). Flow cytometry analysis further confirmed phagocytic nature of the cells containing MPIO (Fig. 5c, d). There was no difference in the MPIO volume in the uninjured (contralateral) cortex between the WT and iNOS KO mice (Supplementary Fig. 8a).
We next examined the effect of NO• on the cortical/hippocampus contents of macrophages (CD11b+/CD45hi+) and microglia (CD11b+/CD45lo+) in M1 and M2 polarization states after CCI. While both M1 macrophages (CD11b+/CD45hi/CD86+) and microglia (CD11b+/CD45lo/CD86+) as well as M2 macrophages (CD11b+/CD45hi/CD206+) and microglia (CD11b+/CD45lo/CD206+) were decreased in the ipsilateral cortex/hippocampus in iNOS KO vs WT mice after CCI, these differences were markedly more pronounced for M1 than for M2 cells (Figs. 5e–h). There was no difference between groups for the uninjured cortex/hippocampus (Supplementary Figs. 8c, d). These data are compatible with the proposed mechanism of iNOS/NO• dependent protection of M1 activated phagocytosing cells against ferroptotic cell death triggered by CCI.
DISCUSSION
Life and biology of professional phagocytes (macrophages, microglial cells) is associated with perilous pro-oxidant environments 30, yet they are notoriously lacking from the lists of cells readily undergoing ferroptotic cell death 1, 11. Given that their genomes contain sequences encoding for the known components of ferroptotic machinery 31, 32, we assumed that additional, yet to be identified mechanisms, may be responsible for the inability of phagocytes to execute the ferroptotic program, at least under some circumstances. Macrophages can be activated into the phenotypes often divided between two broad categories: M1 and M2. In response to inflammatory signals, such as IFNγ and LPS, macrophages polarize into the classical, or M1, state with characteristic transcriptional, morphological and secretory profiles 14, 33, 34 including strong upregulation of NOS2 (iNOS), and pro-inflammatory signals, such as interleukin 6 (IL6) and IL12 34. By contrast, alternatively activated M2 macrophages are polarized by anti-inflammatory signals, such as IL4 and IL13 14, and upregulate genes, such as Arg1, Mrc1 and Cd163. In spite of the fact that this simplified distinction fails to account for the vast diversity – subcategories of macrophage phenotypes 32 – this binary classification of polarized macrophages can be useful 14, 33, 35.
Lipid peroxidation has been recognized as a fundamental part of ferroptosis 10–13. In spite of this prominent role, its specific mechanisms engaged in the execution of the program remain elusive . Two major Fe-dependent pathways of lipid peroxidation – non-enzymatic random free radical chemical reactions and highly selective and specific enzymatic lipoxygenase-controlled process – have been considered 36. A number of natural or synthetic free radical scavengers – from members of the vitamin E family to a variety of aromatic amines and phenolic compounds – with well-established or previously unknown biological functions have been shown to act as inhibitors of ferroptosis 8, 23, 37. Two enzymatic mechanisms, ACSL4 and GPX4, are strong regulators of ferroptosis via the control of: i) lipid peroxidation substrates, arachidonoyl-phospholipids, or ii) lipid peroxidation products, hydroperoxy-arachidonoyl phospholipids dictates ferroptosis sensitivity by shaping cellular lipid composition 8, 38. Intriguingly, a pleiotropic regulator of many biological processes, iNOS generated NO•, representing a reactive radical capable of interacting with other radicals, including lipid radicals 39 has not been considered as a potential regulator of ferroptosis. Assuming that iNOS-derived NO• can interact with the 15-LOX-generated lipid intermediates, here we report, for the first time, that iNOS/NO•, indeed, represent a potent regulator of ferroptotic death leading to high resistance of M1 polarized macrophages and microglia to the initiators of ferroptosis. In contrast, M2 phenotype, lacking iNOS, displayed high sensitivity to pro-ferroptotic stimulation which, however, was completely abolished by exogenously added NO• donors. Notably, iNOS generated NO• of M1 macrophages can diffuse and exert long-distance protective effects against ferroptosis in MLE cells, otherwise sensitive to pro-ferropptotic stimulation.
The anti-ferroptotic effects of NO• are likely due to its ability to react with: i) reactive intermediates of 15-LOX; ii) lipid radicals generated by 15-LOX, and iii) secondary lipid radicals formed during cleavage and oxidative truncation of hydroperoxy-PE. This interpretation is supported by direct LC-MS detection of nitroxygenated species of pro-ferroptotic 15-HpETE-PE as well as nitroxygenated oxidatively truncated derivatives formed from 15-HpETE-PE. Of note, the amounts of these nitroxygenated species were very low, markedly less than the amounts of 15-HpETE-PE. Given the small size of NO• molecule, one can consider that NO• may be delivered to the 15-LOX catalytic site through the “oxygen channel” 40 and interfere with the turnover of the AA-PE intermediates.
Notably, molecular dynamics (MD) simulations of NO• interactions with 15-LOX indeed revealed the ability of NO• to insert into the oxygen channel via two “entrances” on the protein surface to reach the immediate proximity of Fe2+ at the 15-LOX-2 catalytic site (labelled as Entrances 1 and 2, Fig. 6). Simulations showed a competition between NO• and O2 molecules to bind the catalytic site, evidenced by the swapping between bound NO• and O2 molecules near the catalytic residues and Fe2+ ion during the course of simulations (see Supplementary Movie 1). The NO•/O2 binding site was localized near two leucines, L374 and L379, in direct contact with the catalytic residues H373 and H378 (Fig. 6). We note that these two leucines, as well as two other residues, N413 and R417, that coordinate NO• and/or O2 at the catalytic site, are highly conserved among LOXs. Earlier simulations of O2 binding to 12/15-LOX-1 40 revealed four possible oxygen channels. Our current simulations (of > 100 ns) corroborate three of these paths: Entrance 1 corresponds to path 3; Entrance 2 to paths 1 and 2. Furthermore L367 (15-LOX-1) which is the counterpart of L379 (15-LOX-2) has been reported to play a key role in catalytic activity 40 as a site of maximal oxygen occupation, further lending support to the significance of the site observed here for competitively binding NO• or O2.
Fig. 6. NO•/O2 interactions with 15-LOX-2 and entry/exit pathways observed in MD simulations of 15-LOX-2 and 15-LOX-2-PEBP1 complex.
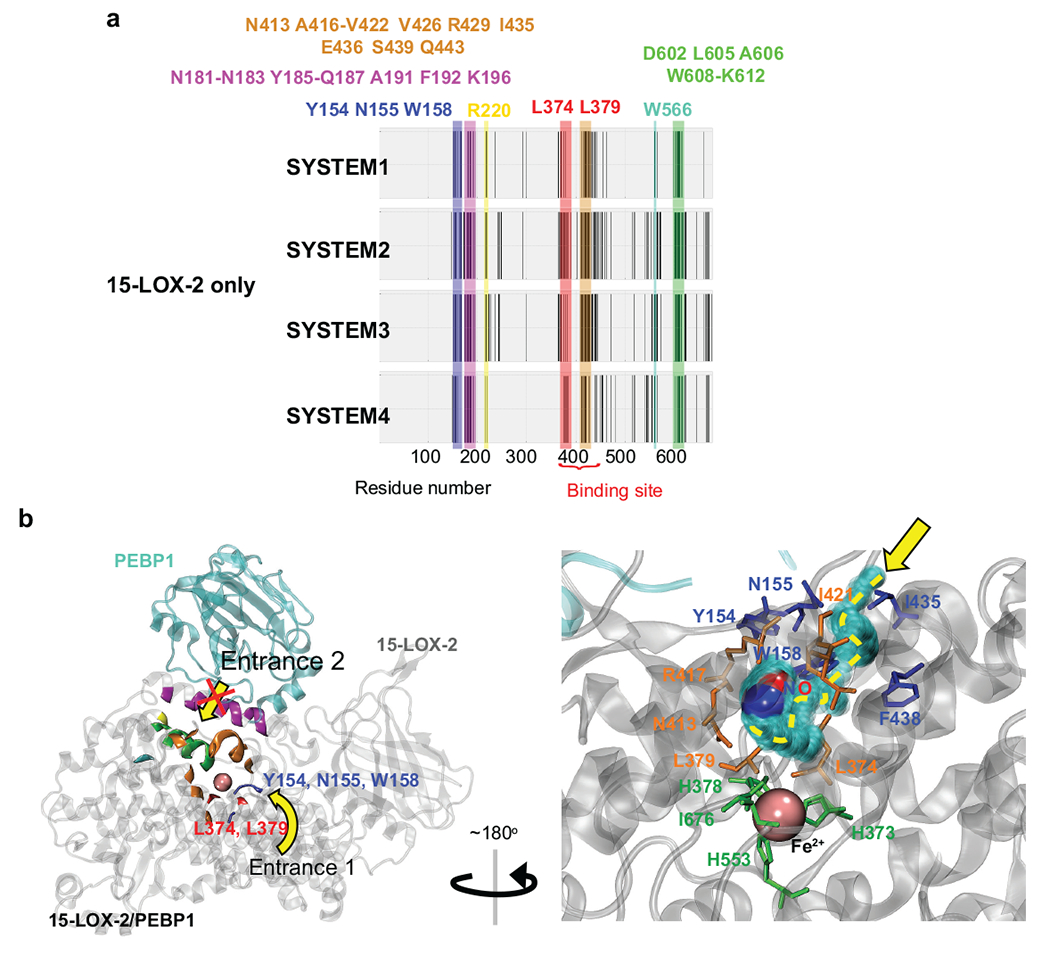
(a) 15-LOX-2 residues that are observed to make contacts with NO•/O2 molecules during four independent MD simulations (SYSTEM1-4). Residues consistently observed to make contacts with NO•/O2 in all runs are highlighted in blue (Y154, N155, W158), pink (N181-N183, Y185-Q187 A191, F192, K196), yellow (R220), red (L374, L379), orange (N413, A416-V422, V426, R429, I435, E436, S439, Q443), cyan (W566), and green (D602, L605, A606, W608-K612).
(b) Crystal structure of 15-LOX-2 (PDB code: 4nre) in the complex formed with PEBP1 (PDB code: 1beh). Colored elements of 15-LOX-2 denote the residues displayed in panel a. Yellow arrows indicate two entry paths shared by NO• and O2 revealed in the MD simulations of 15-LOX-2 in the presence of NO• and O2 molecules: Entrance 1 (from the side) near Y154, N155, W158, I435, and F438 on the surface, and Entrance 2 (from the top), with residues color-coded as in panel a. The red cross indicates the obstruction of Entrance 2 upon complexation with PEBP1. The inset (lower right panel) displays the architecture near Entrance 1 and the migration path of small molecules to the binding site at the catalytic region, accesible to NO• in 15-LOX-2-PEBP1 complex. The path starts with interactions with Y154, N155, I435, W158 and F438 (blue) on the surface and ends at the binding site lined by L374, L379, N413, R417 (highly conserved among all LOXs) and I421 (orange) in the catalytic site. Catalytic residues (H373, H378, H553) and I676 are highlighted in green sticks. NO• is colored in red (oxygen atom) and blue (nitrogen atom) spheres. Pink sphere represents the Fe2+ ion.
High selectivity and specificity of 15-LOX for AA-PE oxidation is gained upon the interaction of the enzyme with a scaffold protein, PEBP1 9, 36. The binding interface between 15-LOX and PEBP1 overlaps with, and therefore obstructs, one of the entrances (entrance 2 in Fig. 6) into the oxygen channel through which NO• migrates to reach the catalytic site, as also noted earlier 40. Thus, only one of the two entrances, near Y154, N155, W158, I421, I435 and F438 on 15-LOX-2 surface, is presumably utilized by NO• (and O2) for transport to the immediate proximity of the catalytic Fe. Additional simulations of 15-LOX-2/PEBP1 complex dynamics using the previously generated structural model for the complex 9 confirmed the inaccessibility of entrance 2 and the competing NO•/O2 binding, and exposed the important roles of L374, L379, N413, R417 and I421 in mediating NO•/O2 binding. Moreover, the presence of PEBP1 induces an increase in stability at the binding site, which causes persistent interactions with the first binding NO• (through entrance 1 exclusively) and consequently a decrease in NO• turnover due to the extended occupancy of the binding site.
NO• readily interacts with LOO• in a chemical radical/radical addition reaction 41, 42. Notably, the common reaction intermediate of the lipoxygenase (LOX)-catalyzed di-oxygenation of polyunsaturated fatty acids (PUFA) and free radical PUFA oxidation is the peroxyl radical (LOO•). In fact, the inhibitory effect of NO• on the LOX-catalyzed peroxidation of PUFA has been considered by O’Donnell et al., 15. Thus, the inhibitory effect of NO• cannot be diagnostic of and usable for distinguishing between the enzymatic (LOX-catalyzed) vs non-enzymatic random chemical lipid peroxidation process. In this work, we established that selective peroxidation reaction of AA-PE catalyzed by a complex of 15LOX with a scaffold protein, PEBP1, and leading to the formation of the pro-ferroptotic signal 15-HpETE-PE 9, 36 is effectively suppressed by iNOS/NO• in M1-polarized macrophages and microglia (but not in M2 alternatively activated cells that do not express significant amounts of iNOS). Notably, KD of iNOS in M1 macrophages markedly increased their vulnerability to ferroptosis whereas NO• donors conferred resistance to ferroptosis in M2 RAW264.7 macrophages (Fig. 1a). Finally, exogenously added 15LOX from P. aeruginosa (pLoxA) was effective in inducing ferroptosis during co-incubation with target HBE cells and this ferroptotic effect was prevented by NO• donors (Fig. 3e and Supplementary figure 4e). By using high-resolution LC-MS we were able to detect nitroxygenated intermediates of this reaction in M1 RAW264.7 macrophages (Fig. 4e). While 15-HpETE-PE is a good predictive biomarker of ferroptosis, it is possible that its decomposition products – oxidatively-truncated highly electrophilic PE derivatives – may be essential secondary mediators attacking the nucleophilic sites on target proteins to form the adducts representing the proximate ferroptosis executioners. The chemical/biochemical nature of this secondary process has not been deciphered yet. This 15-HpETE-PE decomposition reaction may occur non-enzymatically via the formation of free radical intermediates. Therefore, we tested whether the nitroxygenated species of these intermediates could be formed and detected such nitroxygenated derivatives in activated M1 (but not M2) RAW264.7 macrophages. The common opinion in the field is that 15LOX is the initiator of the selective and specific peroxidation of AA-PE 23, 37 whereas chemical free radical cleavage of the 15-HpETE-PE product may include a non-enzymatic component. Given this highly complex chemistry/biochemistry of lipid peroxidation in ferroptosis, likely including enzymatic and non-enzymatic stages – significant future efforts will be necessary to fully characterize this process.
NO• has been suggested to act a pathophysiological modulator of cell proliferation, cell cycle arrest, and apoptosis 43. However, NO• can exert opposite effects under diverse conditions. Relative low concentrations of NO• favor cell proliferation and antiapoptotic responses and higher levels of NO• favor pathways inducing cell cycle arrest, suppression of mitochondrial respiration, senescence, or apoptosis 43, 44. The role of macrophage derived NO• in tumor microenvironment is also controversial. Although high level of iNOS/NO• expression is associated with pro-inflammatory M1 macrophages that also express high level of IFN-γ, macrophages with high NO• production exert potent immune suppressive effect, promote tumor progression and metastasis 45. Our study demonstrates, for the first time, the specific role of NO• in protecting cell from ferroptosis, thus providing an important insight into the biology of macrophage regulation under pathological conditions. Similarly, in acute brain injury, iNOS/NO• has been shown to exert both beneficial and detrimental effects in terms of neurocognitive recovery 26. Here we show that iNOS/NO• expressing M1 macrophages and microglia exert high resistance to ferroptosis in contrast to a higher sensitivity of the M2-phenotype of microglial cells. Previous studies showed that M1 microglia and infiltrating macrophages can produce high levels of pro-inflammatory and cytotoxic mediators that hinder CNS repair, while the M2 phenotype generates neuroprotective factors and orchestrate neuro-restorative processes that are beneficial for neurological recovery after TBI 27. While we and others 1, 9, 25, 46 have shown that neuronal ferroptosis is important in mediating secondary injury after TBI, the contribution of ferroptotic death in other cell types to ferroptosis remains known. Given strong pro-inflammatory effects of ferroptotically dying cells, this information is important for designing optimized therapies for TBI.
Overall, iNOS may be a potent regulator of ferroptotic cell death acting upstream of GPX4 – thus substantially broadening the ability to keep ferroptosis under control. Our demonstration of the iNOS/NO• regulation of ferroptosis in vivo points to many biologically important scenarios where the regulatory role of NO• against ferroptosis may find significant clinical applications in a variety of acute and chronic degenerative conditions with essential functions of professional phagocytes. Our previous work has identified necroptotic death triggered by P. aeruginosa pLoxA as a contributing pathogenic factor in cystic fibrosis and lower respiratory infections 21. A recent work has implicated macrophage ferroptosis as a major mechanism of necrosis in Mycobacterium tuberculosis infection and as a target for host-directed therapy of tuberculosis 47. This may also relate to cancer where death of immunosuppressive M2-like macrophages in the tumor microenvironment may be a desirable outcome in the complex immuno-therapy of cancer 33. In a number of neurological diseases, the precise timing for the activation of microglia and macrophages into pro- or anti-inflammatory states determines the overall outcome. Given strong pro-inflammatory effects of ferroptotically dying cells 48, the regulation of ferroptosis by iNOS/NO• may be important in controlling the inflammatory and immune responses. Interestingly, during inflammation, uptake of apoptotic cells is confined to a population of 15-LOX-expressing, alternatively activated resident macrophages, which blocks uptake of ACs into freshly recruited inflammatory monocytes in a 15-LOX-dependent manner thus maintaining the self-tolerance 49.
Online methods
Animals
Wild type (WT) and iNOS (NOS2) KO C57B6 mice were obtained from Jackson Laboratories (Bar Harbor). The male WT and KO mice, 8~12 weeks of age, were used in all experiments, and newborn pups (P0-2) of the WT were used for in vitro primary microglial cultures studies. These studies were carried out in strict accordance with the recommendation in the guide for the Care and Use of Laboratory Animals of the National Institute of Health. All efforts were made to minimize animal suffering. All animals were housed under specific pathogen-free conditions at the Animal Research Facility, University of Pittsburgh. All animal experiments were approved by the University of Pittsburgh Institutional Animal Care and Use Committee (IACUC).
TBI model and MRI contract agent administration
C57BL/6J (WT) mice and iNOS KO were given free access to food and water and were housed in laminar flow racks in a temperature-controlled room with 12 h day/night cycles. A previously described mouse model of controlled cortical impact (CCI) was used 1. Anesthesia was induced in male WT and iNOS KO mice (8-10 weeks old, weighing 25-30g) with 4% isoflurane in O2. A femoral venous catheter was surgically inserted for administration of micron-sized paramagnetic iron oxide (MPIO, Bangs Laboratories) particles to label brain macrophages/microglia in situ as described previously 2. MPIO particles are 0.9-μm superparamagnetic styrene-divinyl benzene inert polymer microspheres that contain a magnetite core and a fluorescein-5-isothiocyanate dye (Dragon Green fluorescent probe) contained within the cross-linked polymer sphere. Mice were then positioned in a stereotaxic frame, spontaneously breathing 1-2% isoflurane in 70% Nitrous Oxide (N2O)/30%O2. A temperature probe was inserted through a burr hole into the left frontal cortex. A craniotomy was made over the left parietal cortex. Once a brain temperature of 37°C±0.5°C was reached, it was maintained at that level for 5 min before a vertically directed CCI was delivered, using a 3-mm flat-tipped impounder with a depth of 1.4mm and a velocity of 6±0.2m/s. After injury, the bone flap was replaced, sealed with cement, and the scalp incision was closed, and the anesthetic was discontinued. MPIO particles were administered at a dose of 4.5mg Fe/kg body weight via femoral catheter after CCI. This dose was chosen based on a previous study 3. After the injury, mice were observed for 30min in 30% supplemental O2 and then returned to their cages. At 72h after CCI, mice underwent MPIO MRI imaging as described below. At 96h after CCI, mice were re-anesthetized with 4% isoflurane in O2 via nose cone, perfused transcardially with ice-cold saline and brains were quickly removed, divided into ipsilateral and contralateral hemispheres for flow cytometry analysis.
In vivo MRI acquisition and analysis
The mice were placed into a clear plexiglass anesthesia induction box that allows unimpeded visual monitoring of the animals. Anesthesia was induced with 3% isoflurane in O2 and maintained using 1-2% isoflurane in O2 with monitoring of respiration and rectal temperature (SA Instruments). In vivo brain MRI was carried out on a Bruker BioSpec 70/30 USR spectrometer (Bruker BioSpin MRI) operating at 7-Tesla field strength, equipped with an actively shielded gradient system and a quadrature radio-frequency volume coil with an inner-diameter of 35 mm. Multi-planar T2-weighted anatomical imaging was acquired with Rapid Imaging with Refocused Echoes (RARE) pulse sequence with the following parameters: field of view (FOV) = 2 cm, matrix = 256 X 256, slice thickness = 0.6 mm, in-plane resolution = 78 μm X 78 μm, RARE factor = 8, effective echo time (TE) = 48 msec, repetition time (TR) = 2585.3 msec, flip angle (FA) = 180⁰. Multi-planar T2*-weighted images were acquired with the Fast Low Angle Shot (FLASH) pulse sequence with the following parameters: FOV = 2 cm, matrix = 256 X 256, slice thickness = 0.6 mm, in-plane resolution = 78 μm X 78 μm, TE = 10 ms, TR = 300 msec, FA = 30⁰. The MRI data were exported to DICOM format and analyzed by two blinded independent observers using the open source ITK-SNAP (http://www.itksnap.org) brain segmentation software. The volumes of the MPIO-labeled glial infiltration foci, edema, contusion, and total cerebrum are quantitated by manual segmentation of each region of interest. MPIO-labeled glial infiltration foci were defined as areas with hypointensity on the T2*-weighted FLASH images, whereas edema was defined as regions of hyperintensity on the T2-weighted RARE images. The segmented regional volumes in mutants and wild types were compared using paired T-test.
Zymosan peritonitis RSL3/IKE-ferroptotic models
WT mice were injected with zymosan A (from Saccharomyces cerevisiae, Sigma-Aldrich) at 100mg/kg, i.p. since the lower dose of zymosan have no significant effect on rectal temperature 4. In the experiments the mice received saline or RSL3 (40mg/kg) or IKE (imidazole ketone erastin, (50mg/kg) with /without DETA NONOate (120mg/kg), i.p. injection at 72 h after zymosan administration, and the peritoneal exudates were collected 5 at 5 h after the RSL3 and/or DETA NONOate administration.
Macrophage and microglial isolation and flow cytometry analysis
The macrophages/microglial cells were isolated from mouse brains and peritoneal exudates as described previously 5. Briefly, ipsilateral and contralateral cortical and hippocampal was rapidly micro-dissected in HBSS using a Tenbroeck homogenizer with three gentle strokes. The crude homogenate was passed through 70 μM and 40 μM cell strainer to yield a single cell suspension in Hanks Buffered Salt Solution (HBSS, Invitrogen). MPIO+ and CD11b+ cells from the single cell suspension were isolated by MPIO (positive selection), myelin Removal Beads II (Miltenyi Biotec, negative selection), and CD11b MicroBeads (Miltenyi Biotec, positive selection) using magnetic separation (Easy Sep, StemCell-Technologies, Vancouver, Canada). For peritoneal macrophages, CD11b MicroBeads isolation was performed using magnetic separation. Fc receptor-mediated non-specific interaction was blocked using anti-mouse CD16/CD32 antibody (Cat# 553140, BD Bioscience). Fluorescent staining of CD11b+ single macrophages and/or microglia was performed using the following antibodies: anti-mouse CD11b-PE (Clone: REA592; Cat# 130-113-806, 2 μl/106 cells in 100 μl, Miltenyi Biotec), anti-mouse CD86-APC (PO3.3, Cat# 130-102-558, 10 μl/106 cells in 100 μl, Miltenyi Biotec), anti-mouse CD45-PerCP (30F11, Cat# 130-102-469, 10 μl/106 cells in 100 μl, Miltenyi Biotec), anti-mouse Alexa Fluor® 700 CD206 (C068C2, Cat# 141733, 2 μl/106 cells in 100 μl, BioLegend, Inc.). Zombie UV™ Fixable Viability Kit (Cat#423107Biolegend) was used for assessing live vs. dead status of cells. Macrophages/microglia (M1 or M2) were analyzed using FACS Canto II (BD Bioscience) flow cytometer, and data were analyzed using FlowJo Software (FlowJo. LLC). The gating strategy is presented in Supplementary Fig. 9.
Primary microglial cell culture
Primary microglial cells were cultured using a standard protocol as described previously 6. Briefly, primary microglial cell cultures were prepared from postnatal day 0-2 mouse pups. Brains were removed and immediately placed in ice-cold Ca2+- and Mg2+-free HBSS (Invitrogen) containing 10μg/ml gentamicin (Sigma-Adrich). Cortical hemispheres were dissected and meninges, blood vessels as well as white matter were removed. Tissue was cut into small pieces and transferred to pre-warmed HBSS containing 0.17% trypsin (37°C), chopped thoroughly and incubated for 15-20 min at 37°C. Supernatant was removed and the remaining trypsin was neutralized by addition of Dulbecco’s Modified Eagle’s Medium (DMEM) (Invitrogen) supplemented with 10% fetal bovine serum (FBS) and gentamicin (10 μg/ml) in mice. The cell suspension was then filtered using first a 100 μm cell strainer followed by a 40 μm cell strainer (BD Biosciences), and then were seeded onto T75 flask coated with 0.0005% poly-L-lysine in PBS at a ratio of 3-4 pups per T75 cell culture flask. After 24 h, the T75 flask was carefully tapped to dislodge sedimentary cell debris, and medium was exchanged (20 ml/flask). Cultures were maintained at 37°C in a humidified atmosphere of 5% CO2 and allowed to mature in vitro for 7-14 days before use. Microglial cells were harvested from confluent astrocyte monolayers at 14 days after the initial seeding by a combination of tapping the side of the culture flask and gently vortexing for ~1-min. The supernatant containing detached microglial cells was collected and centrifuged at 150 g for 7 min at room temperature. The microglial cells harvested were plated into plates containing conditioned media. The purity of the microglial cells was >95% as determined by Iba1 staining.
Bone marrow transplantation in mice
Bone marrow (BM) cells were isolated from 6-8 weeks old wild-type and iNOS (NOS2) KO C57BL6 mice (Jackson Lab). Recipient mice were lethally irradiated with a split dose 900Rad (with 5 h interval). 1.5 x 106 BM donor’s cells were injected i.v. and mice were observed for 12 weeks. After that time 5x105 Lewis Lung Carcinoma (LLC) tumor cells were injected s.c. Tumors were isolated 3 weeks later and evaluated. Tumors were weighed and cut into small pieces and digested with a Tumor Dissociation Kit according to the manufacture’s protocol (Miltenyi Biotec). Subsequently, the tumors were passed through a 70-μm cell strainer and were subjected to red-blood-cell lysis using ammonium-chloride-potassium (ACK) buffer. Cells were then washed with cold CSB and spun down, the pellet was resuspended in cold CSB and cells were counted using Trypan blue (VWR). Staining was performed for 15min at 4 °C in dark and all centrifugation was done at 1,500 r.p.m. at 4°C for 5min, unless recommended otherwise by the manufacturer. Usually up to 2x106 cells were incubated with Fc-block (BD Biosciences; clone 2.4G2; Cat. # 553142) in 50μl of CBS, then were washed in CBS and spun down before cell-surface staining with additional antibodies. After the final incubation, cells were washed in CBS, spun down and resuspended in 500μl of CBS before acquisition. Cells were run on Celesta flow cytometer (BD Biosciences) and data were analyzed by FlowJo Software (FlowJo. LLC).
Cell culture
RAW 264.7 and RAW 264.7 gamma NO(−) macrophages, the murine lung epithelial cell line MLE-12 and the mouse microglial cell line EOC 20 were obtained from the American Type Culture Collection (ATCC). RAW 264.7 and RAW 264.7 gamma NO(−) macrophages were cultured at 37°C and 5% CO2 in DMEM or RPMI (ATCC) supplemented with 10% heat-inactivated fetal calf serum (FCS) (Sigma-Aldrich), 50 U/ml penicillin-streptomycin (ThermoFisher Scientific). EOC 20 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Sigma), supplemented with 10 % heat-inactivated fetal calf serum (FCS), antibiotics, and recombinant colony stimulating factor (M-CSF, 10ng/ml, Miltenyi Biotec, mb130-094-129) as described previously 7. MLE-12 cells were cultured at 37°C and 5% CO2 in DMEM/F-12 (1:1) medium supplemented with 0.005 mg/ml insulin, 0.01 mg/ml transferrin, 30 nM sodium selenite (Life Technologies),10 nM hydrocortisone, 10 nM β-estradiol (Sigma-Aldrich), 2 mM L-glutamine, 10 mM HEPES, and 10% FBS (Life Technologies). Human bronchial epithelial cell (HBE) (originally established by Dr. Dieter Gruenert) was cultured as described previously 8. Briefly, cells were maintained in MEM (ThermoFisher) supplemented with 10% FCS (Gibco), 50 U/ml penicillin-streptomycin (ThermoFisher), plasmocin (InvivoGen), and l-glutamine (ThermoFisher).
Bacterial strains
Pseudomonas aeruginosa hyper-biofilm mutant (ΔwspF) strain used in this study was obtained from the P. aeruginosa transposon mutant library, University of Washington (Seattle, Washington, USA) as described previously.
Abiotic biofilm and supernatant collection
P. aeruginosa ΔwspF strain grown overnight in LB at 37°C, 220 rpm was diluted in MEM medium (without phenol red) to an OD600 of 0.05 and plated in 96-well vinyl microtiter plates (100 μl per well) (Costar). Plates were grown at 37°C without agitation for 24 h. Supernatant was collected by centrifugation at 3,000 g for 8 minutes and then frozen before further use in cell death assay.
Polarization of cells into M1/M2 states
RAW 264.7 macrophages were polarized by incubation in DMEM with 10% FBS, 50 U ml−1 penicillin-streptomycin containing interferon-γ (IFN-γ) (100 ng/ml) for M1 state or IL-4 (20 ng/ml) for M2 state for 48 h. RAW 264.7 gamma NO(−) cells were activated with IFN-γ alone (100 ng/ml) or IFN-γ (100 ng/ml) plus LPS (10 ng/ml) or with LPS only (10 ng/ml) for 48 h and then used for experiments. M1 polarization was attained by incubating EOC 20 or primary microglial cells in media supplemented with 10% fetal bovine serum and penicillin–streptomycin, followed by LPS (10 ng/ml, Sigma, L3012) plus INF-γ (100 ng/ml, Miltenyi Biotec, mb130-105-785) treatment for 48 h. M2 polarization was obtained by incubating EOC 20 or primary microglial cells with IL-4 (20 ng/ml, PeproTech, 214-14) for 48 h. Measurements performed using the CellInsight™ High Content Screening (HCS) Platform demonstrated that 24h after activation into M1 or M2 state, the number of M1 macrophages was ~1.2 times lower than the amounts M0 and M2 macrophages. In 48 h, the amount of M1 macrophages was 1.7 and 1.3 times lower than amounts of M0 and M2 macrophages, respectively (Supplementary Fig. 10). Based on these data, in experiments for assessment of ferroptosis in M0, M1 and M2 cells we initially seeded proportionally smaller amounts of M0 and M2 macrophages to obtain equal numbers of cells at the time of pro-ferroptotic treatment with RSL3. The results of these experiments were similar to those obtained before when the same amounts of cell were seeded into wells from the very beginning of the experiment.
Isolation of bone marrow derived macrophages
Cells were isolated from bones of mice as described in 9. Briefly, bones were washed “inside” by putting needle into bone. Obtained cell suspension was homogenized by putting several times through 18G needle, collected to tube and centrifuged at 4°C for 5 min and 1200 rpm. Cells were resuspended in fresh DMEM supplemented with 20 ng/ml M-CSF, plated in Petri dishes and cultured in 37°C and 5% CO2. On day 7 media was replaced by DMEM containing INF-γ (10ng/ml), LPS (100 ng/ml), M-CSF (20 ng/ml) (for stimulation into M1 state), or IL-13 (10ng/ml), IL-4 (10 ng/ml), M-CSF (20 ng/ml) (for stimulation into M2 state). To determine macrophage phenotype and intracellular expression of 15LOX and GPx4 molecules, cells were stained with the following anti-mouse antibodies: PerCp/Cy5.5-labeled CD11b (#101228, BioLegend); APC-labeled F4/80 (#123115, BioLegend); FITC-labeled CD206 (#141703, BioLegend); AF 488-labeled CD86 (#105017, BioLegend); anti-iNOS AF 488 conjugated antibody from (#53-5920-80, Invitrogen); anti-15LO2 AF647 conjugated (#sc-271290, Santa Cruz) and anti-GPx4 AF 488 conjugated (#sc-166120, Santa Cruz).
siRNA knockdown experiments
RAW 264.7 macrophages.
RAW 264.7 macrophages were transfected with a mix of two Dicer-substrate short interfering RNAs (DsiRNAs) against 15-LOX-2 (mm.Ri.Alox15.13.1 and mm.Ri.Alox15.13.2) or iNOS (mm.Ri.Nos2.13.1. and mm.Ri.Nos2.13.2) or Gpx4 (mm.Ri.Gpx4.13.1. and mm.Ri.Gpx4.13.2) or with control scrambled DsiRNA (51-01-14-04) (Integrated DNA Technology) using Lipofectamine 3000 (Life Technology) according to manufacturer’s instructions. The transfection mixture was removed after 12 h, cells were counted and seeded in 24 well plate or 6-cm dishes, activated for 48 h and then treated with RSL3 for cell death experiments or collected for western blotting.
Sequences of siRNA used for transfection of RAW 264.7 macrophages:
mm.Ri.Alox15.13.1
5’-rUrGrGrCrArArGrUrCrArUrGrArArUrCrGrGrUrArCrGrUGG-3’
5’-rCrCrArCrGrUrArCrCrGrArUrUrCrArUrGrArCrUrUrGrCrCrArGrA-3’
mm.Ri.Alox15.13.2
5’-rCrGrArGrGrArCrUrCrCrUrGrGrArUrArUrUrGrArCrArCTT-3’
5’-rArArGrUrGrUrCrArArUrArUrCrCrArGrGrArGrUrCrCrUrCrGrCrU-3’
mm.Ri.Nos2.13.1
5’-rUrCrUrGrArArGrArArArUrCrUrCrUrGrUrUrCrArUrGrCTT-3’
5’-rArArGrCrArUrGrArArCrArGrArGrArUrUrUrCrUrUrCrArGrArGrU-3’
mm.Ri.Nos2.13.2
5’-rArUrCrArUrGrArArGrArUrArUrCrUrUrCrGrGrUrGrCrAGT-3’
5’-rArCrUrGrCrArCrCrGrArArGrArUrArUrCrUrUrCrArUrGrArUrArA-3’
GPX4 (mm.Ri.GPX4.13.1)
5’-rArArCrUrUrUrArCrCrArArGrUrUrUrCrUrCrArUrUrGrAT A-3’
5’-rUrArUrCrArArUrGrArGrArArArCrUrUrGrGrUrArArArGrUrUrCrC-3’
GPX4 (mm.Ri.GPX4.13.1)
5’-rGrArGrCrCrArGrGrArArGrUrArArUrCrArArGrArArArUCA-3’
5’-rUrGrArUrUrUrCrUrUrGrArUrUrArCrUrUrCrCrUrGrGrCrUrCrCrU-3’
Control scrambled DsiRNA
5’-rCrUrU rCrCrU rCrUrC rUrUrU rCrUrC rUrCrC rCrUrU rGrUGA
5’-rUrCrA rCrArA rGrGrG rArGrA rGrArA rArGrA rGrArG rGrArA rGrGrA
All sequences are provided in 5′ → 3′ orientation
Microglia cells.
A pool of three target-specific 19-25 nt siRNAs designed to knock down iNOS and 15-LOX-2 expression in the EOC 20 cells was utilized. Control siRNA (sc-36869) and iNOS- (NOS2 siRNA (m): sc-36092) and 15-LOX-2-siRNA (15-LOX-2 siRNA (m): sc-45627) were designed and synthesized by Santa Cruz Biotechnology, Inc. Transfection of the control-siRNA and the target-siRNA was performed using Lipofectamine RNAi Max (Invitrogen) following the manufacturer’s instructions. EOC 20 cells were seeded onto 6 well-plates at 1~2x105 cells /1 ml /well for incubations up 7 h, then the transfection mixture was removed and cells were switched to DMEM supplemented with 10% FBS for 24 h. Knock down efficiency was assessed by western blot analysis using the following antibodies: anti-iNOS (ab178945, Abcam, 1:500), anti-15-LOX-2 (sc-67143, Santa Cruz, 1:500), horseradish peroxidase (HRP)-conjugated anti-rabbit IgG H&L (Sigma, A0545, 1:2000) secondary antibody, and anti-β-actin-peroxidase antibody (ThermoFisher, Cat # A3854,1:5000). To assess the effect of KD on ferroptosis cells were plated in 24-well plate (15000 - 20000 cells per well), incubated with RSL3 and cell death was assessed by measuring released LDH activity.
Sequences of siRNA used for transfection of EOC 20:
15-LO2 siRNA (m) is a pool of 3 different siRNA duplexes:
1) sc-45627A:
5’-CCAACAUCCUCAAUGGAAAtt
5’-UUUCCAUUGAGGAUGUUGGtt
2) sc-45627B:
5’-CUGGGAAGUUGAUAGACAAtt
5’-UUGUCUAUCAACUUCCCAGtt
3) sc-45627C:
5’-CUACCUCACUUCUUCUCAAtt
5’-UUGAGAAGAAGUGAGGUAGtt
NOS2 siRNA (m) is a pool of 3 different siRNA duplexes:
1) sc-36092A:
5’-GAAGCUGUAACAAAGGAAAtt
5’-UUUCCUUUGUUACAGCUUCtt
2) sc-36092B:
5’-CCAGAAACGUUAUCAUGAAtt
5’-UUCAUGAUAACGUUUCUGGtt
3) sc-36092C:
5’-GUACUAUUGUGGACUACUAtt
5’-UAGUAGUCCACAAUAGUACtt
All sequences are provided in 5′ → 3′ orientation
Overexpression of iNOS and 15-LOX
RAW 264.7 cells were first differentiated into M2 or M1 sates by treating with IL4 or IFN-γ and then transfected with vector only (pCMV6) or iNOS (pCMV6-iNOS) (Ori Gene, MR211704) or 15-LOX-2 (pCMV6-15-LOX-2) (Ori Gene, MR226258) for 12 h. Cells were then seeded in 24 well plate for cell death assay and in 6 cm dish for western blotting.
Ferroptosis assay
RAW 264.7 macrophages were plated in 24-well plate (15000 – 20000 cells per well). After 24 h, cells in fresh media were treated with RSL3 (500 nM) for 5 h in the absence or in the presence of ferrostatin-1 (400 nM). For evaluation of ferroptotic cell death, M1 and M2 microglia were incubated with RSL3 (500 nM ~1 μM), ML162 (2 μM), IKE (2 μM) or erastin (20 μM) ± Fer-1 (400~800 nM), or ± DPTA NONOate (25 μM), ± SNAP (25 μM), ± DETA NONOate (25 μM), ± L-NIL (10 μM), or ± 1400W (2 μM) for 5~20 h. Cell death was determined by measuring released lactate dehydrogenase (LDH) activity. LDH activity was quantified using the CytoTox-ONE™ Cyto-toxicity Detection Kit (Promega) according to the manufacturer’s instructions. Cell death in control cells was lower than 8%. HBE cells were treated with P. aeruginosa supernatant (Sup.) alone or with NO-donors SNAP (25 and 50 μM), added every 6 h or DETA NONOate (25 and 50 μM) for 20 h. Cell death was estimated by PI-staining by flow cytometry as described previously 8.
Ferroptosis induced by exogenous 15-HpETE-PE
RAW 264.7 cells were pretreated with RSL3 (150 nM) for 3 h and then incubated with 15-HpETE-PE (1 μM) with or without DPTA NONOate (25 μM) for further 3 h. Cell death was monitored by LDH release assay.
Co-culture experiments
MLE cells were plated in 6-well plate (105 cells per well). After 16 h, cells were transferred into fresh media containing 2 μM of Cell Tracker Green for 45 min. After changing the medium M1 activated RAW 264.7 macrophages (2x105 cells per well) were added and in 1 h cells were treated with RSL3 (500 nM) for 5 h in the absence or in the presence of Fer-1 (400 nM). Cells were trypsinized, centrifuged at 700 g for 6 min, resuspended in PBS containing PI for 5 min on ice, and then monitored by flow cytometry using FACS Canto (BD Bioscience) flow cytometer.
Western blot analysis
Total cell lysates were prepared after washing cells with PBS and adding cell lysis buffer (25 mM Tris-HCl, pH 7.5, 150 mM NaCl, and 1% SDS) or RIPA buffer (ThermoFisher) containing freshly added protease-phosphatase cocktail inhibitor mix (ThermoFisher). Samples were sonicated to break down DNA and total protein amount was estimated by BCA protein assay (ThermoFisher). Samples were diluted in 2X-6X Laemmli buffer before loading in 8-16% Tris-glycine gradient gels (Life Technologies) and proteins transferred to nitrocellulose (Biorad) or polyvinylidene difluoride (PVDF) membranes. Blocking was performed by Super Block T20 blocking buffer (ThermoFisher, 37536) or by PBST (0.05% tween-200 containing 5% skim milk). Target proteins were immunodetected using anti-GPX4 (ab125066, Abcam,1:500), anti-15-LOX-2 (sc-67143, Santa Cruz, 1:500), anti-ACSL4 (sc-365230, sc-134507, Santa Cruz, 1:500), anti-iNOS (Abcam, ab3523, ab178945), anti-LPCAT3 (goat polyclonal, Santa Cruz, sc-161831), anti-transferrin receptor (H68.4, Invitrogen) and anti-βactin (1:1000, A2228, A3854 Sigma-Aldrich) antibodies following overnight incubation at 4°C. Membranes were washed 5 times with TBST or PBST (0.05% Tween) and then incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies-rabbit anti-mouse (A9044), goat anti-rabbit (ab150074, Abcam, A0545, Sigma) and rabbit anti-goat (Cat # A5420, Sigma-Aldrich, Cat # ab150129, Abcam) for 1h at RT. Membrane were developed with SuperSignal™ West Pico Chemiluminescent Substrate (ThermoFisher) SuperSignal west femto, Thermo 34095 or GE Health Care) using image systems Amersham Imager 600 (GE Health Care, Life Sciences) and Chemi Doc XRSt (Bio-Rad). Quantification of the bands was performed using ImageJ software from NIH (imagej.nih.gov/ij/).
LC/MS analysis of phospholipids
MS analysis of PLs was performed on an OrbitrapTM FusionTM LumosTM mass spectrometer (ThermoFisher). Phospholipids were separated on a normal phase column (Luna 3 μm Silica (2) 100Å, 150 x 2.0 mm, (Phenomenex)) at a flow rate of 0.2 ml/min on a Dionex Ultimate 3000 HPLC system. The column was maintained at 35 °C. The analysis was performed using gradient solvents (A and B) containing 10 mM ammonium acetate. Solvent A contained propanol:hexane:water (285:215:5, v/v/v) and solvent B contained propanol:hexane:water (285:215:40, v/v/v). All solvents were LC/MS grade. The column was eluted for 0-23 min with a linear gradient from 10 % to 32 % B; 23-32 min using a linear gradient of 32-65% B; 32-35 min with a linear gradient of 65-100 % B; 35-62 min held at 100% B; 62-64 min with a linear gradient from 100 % to 10 % B followed by and equilibration from 64 to 80 min at 10 % B. The instrument was operated with electrospray ionization probe in negative polarity mode. Analysis of LC/MS data was performed using software package Compound Discoverer™ (ThermoFisher) with an in-house generated analysis workflow and oxidized phospholipid database. Lipids were further filtered by retention time and confirmed by fragmentation mass spectrum. The raw data are available at the following link https://data.mendeley.com/datasets/t4jyhhf3gw/draft?a=76c6ac56-4305-463e-bebc-b004c2000edb
Identification of RSL3 using LC-MS/MS.
RSL3 were identified and quantified using an LC-MS/MS method. RSL3 was extracted from cells using a chloroform-methanol extraction procedure. Extracted RSL3 were concentrated and subjected to LC-MS/MS analysis. Briefly, a normalized amount of samples were injected into a normal phase liquid chromatography column connected to mass-spectrometer. The chromatography and mass spectrometry conditions were maintained identical to the previously described phospholipid analysis method. RSL3 was identified based on the exact mass and confirmed through fragmentation analysis. Quantification of RSL3 was performed based on the calibration values obtained from calibration curves prepared using varying concentration of RSL3.
Fluorescence microscopy
Live cell imaging and quantification
Cells were seeded on 35mm glass bottom dishes (MatTek Corporation) and incubated with Liperfluo (10 μM) (Dojindo Molecular Technology, Inc.) or diaminorhodamine-4M (DAR-4AM) (5 μM) for 30 minutes at 37 °C. Cells were washed with Phosphate-Buffered Saline (PBS), the media replaced and the dish inserted into a closed, thermo-controlled (37ºC) stage top incubator (Tokai Hit Co., Shizuoka-ken, Japan) atop the motorized stage of an inverted Nikon TiE fluorescent microscope (Nikon Inc.) equipped with a 60X oil immersion optic (Nikon, CFI PlanFluor, NA 1.43) and NIS Elements Software. Liperfluo or DAR-4AM was excited using a Lumencor diode-pumped light engine (SpectraX, Lumencor Inc.) and detected using an ORCA-Flash 4.0 sCMOS camera (HAMAMATSU Corporation) and excitation and emission filters from Chroma Technology Corp. For time-lapse experiments, data was collected every 5 minutes for 3 hours, on approximately 10-20 cells per stage position, with 10-15 stage positions in each of 3 separate experiments per condition. Data were analyzed using NIS Elements (Nikon Inc).
Fluorescence microscopy analysis of microglial cells
M1 and M2 (106 cells) were seeded onto glass coverslips in a 12-well plates. At indicated time points cells were fixed by 4% paraformaldehyde solution. 0.25% Triton X-100 (10 min at RT) was used for permeabilization before blocking with normal goat serum 10% (50062Z, ThermoFisher). Samples were incubated with mouse monoclonal anti-iNOS antibody (Cat # ab178945, Abcam), at 4 °C overnight. iNOS expression was visualized using a fluorophore-labeled secondary antibody, Alexa Fluor 555 goat anti-mouse IgG (H + L, ab150074, Abcam). Nuclear DNA was stained using mounting medium with DAPI (Vector, H-1500). Samples were analyzed by using a Nikon microscope, and DS-Ri1 camera was used for image acquisition through software NIS-Elements.
Mathematical modeling
The network model was built based on the data presented in this manuscript and our previous model 10. Supplementary figure 6A illustrates the reaction scheme (all acronyms used are listed in Table S1). Specifically, enzymes ASCL4 and LPCAT3 catalyze the formation of AA-PE. The conversion from AA-PE to HOO-AA-PE can be catalyzed by 15-LOX with non-heme iron; and the 15-LOX-generated HOO-AA-PE can be cleaved to yield oxidatively truncated species, denoted as PEOxTr. The accumulation of HOO-AA-PE and PEOxTr leads to ferroptotic cell death. OOH-AA-PE is reduced into OH-AA-PE by GPX4, while GSH is converted into GSSG. iNOS and NO• donors induce the production of NO•, which reduces the catalytic non-heme iron of 15-LOX to non-productive ferrous state, and reacts with lipid radicals including HOO-AA-PE and PEOxTr leading to the formation of nitroxygenated lipid products.
The network kinetics was modeled as a system of 16 ordinary differential equations (ODEs) (Table S2). The reaction rates for synthesis, degradation, association, and inhibition were modeled using mass action kinetics. The catalytic reactions were described using Michaelis-Menten kinetics 10. We defined a score indicating cell death signal strength as a linear combination of the concentrations of OOH-AA-PE and PEOxTr:
where S0 is the basal level of cell death, α and β are coefficients. We then use the integration of Sdeath over time to predict cell death level. The ODE system was analyzed using the BioNetGen tool 11. Model calibration and sensitivity analysis were performed with statistical model checking-based methods 12.
Molecular dynamics simulations
Human 15-LOX-2 (PDB code: 4nre ) and 15-LOX-2-PEBP1 complex (4nre, 1beh ) have been simulated in the presence of randomly distributed NO• and O2 molecules (five of each). Four runs (Systems 1-4) of 150 ns with different spatial distributions of NO• and O2 molecules were performed for each structure (15-LOX-2 and its complex with PEBP1) using the NAMD MD simulation software with CHARMM27 force fields and 2 fs time steps 13. The proteins were solvated with TIP3P water at physiological salt concentrations. Docking simulations with the Gramm-X 14 were used to generate structural models for 15-LOX-2-PEBP1 complex, as reported previously 15. CHARMM force field parameters for NO•, O2 molecules and covalently bonded Fe2+ were created based on bonded O2, HEME group (default CHARMM force field parameters) and using Gaussian package 16. Prior to productive runs, we adopted following protocol: 0.2 ns of water equilibration, 10,000 steps of minimization, 0.35 ns of heating from 0 to 300 K, and 0.15 ns equilibration of the whole system. Simulations were performed with a cutoff of 12 Å for non-bonded interactions and Langevin piston algorithm to maintain the temperature at 300 K and the pressure at 1 atm. We used VMD for visualization and the ProDy API 17 for trajectory analysis.
Our code used for the analysis of the MD simulations of NO• interactions with 15LOX has been made available in two formats (i) Jupyter Notebook and (ii) HTML at the following link: https://onedrive.live.com/?authkey=%21AFEVmP5sOP1km0s&id=4960CA1B5C7F3FD%219568&cid=04960CA1B5C7F3FD.
For the mathematical modeling of the network of the major ferroptotic interactions and regulatory pathways involving NO• reactions we provided the following files: inos.bngl: the mathematical model file in BioNetGen language, inos_ode.h and inos_pathway.m the c and matlab code for parameter estimation using our SMC tool (now the tool is called SMOKE). These files are uploaded together with the source code of SMC tool to github at the following link: https://github.com/liubing1020/smoke
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis
The data in figure legends are presented as means ± s.d. values from at least three experiments unless otherwise specified. In general, the exact value of sample size (n) is presented in figure legends and reflects either the number of experimental replications with cells or biochemical model systems or the number of animals used in in vivo experiments. Statistical analyses were performed by unpaired Student’s t-tests unless otherwise specified. The statistical significance of differences was set at p < 0.05.
Data and Code availability
The raw data are available at the following link https://data.mendeley.com/datasets/t4jyhhf3gw/draft?a=76c6ac56-4305-463e-bebc-b004c2000edb
Code used for the analysis of the MD simulations of NO• interactions with 15LOX has been made available in two formats (i) Jupyter Notebook and (ii) HTML at the following link: https://onedrive.live.com/?authkey=%21AFEVmP5sOP1km0s&id=4960CA1B5C7F3FD%219568&cid=04960CA1B5C7F3FD.
Supplementary Material
Acknowledgements
This work was supported by NIH (HL114453-06, U19AI068021, CA165065-06, NS076511, NS061817, P41GM103712) and by Russian academic excellence project “5-100”.
Footnotes
DECLARATION OF INTERESTS
The authors have declared that no conflict of interest exists.
References
- 1.Stockwell BR et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 171, 273–285 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dixon SJ et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Friedmann Angeli JP et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nature cell biology 16, 1180–1191 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stoyanovsky DA et al. Iron catalysis of lipid peroxidation in ferroptosis: Regulated enzymatic or random free radical reaction? Free Radic Biol Med (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gugiu BG et al. Identification of oxidatively truncated ethanolamine phospholipids in retina and their generation from polyunsaturated phosphatidylethanolamines. Chem Res Toxicol 19, 262–271 (2006). [DOI] [PubMed] [Google Scholar]
- 6.Pizzimenti S et al. Interaction of aldehydes derived from lipid peroxidation and membrane proteins. Front Physiol 4, 242 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hoff HF, O’Neil J, Wu Z, Hoppe G & Salomon RL Phospholipid hydroxyalkenals: biological and chemical properties of specific oxidized lipids present in atherosclerotic lesions. Arterioscler Thromb Vasc Biol 23, 275–282 (2003). [DOI] [PubMed] [Google Scholar]
- 8.Doll S et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol 13, 91–98 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wenzel SE et al. PEBP1 Wardens Ferroptosis by Enabling Lipoxygenase Generation of Lipid Death Signals. Cell 171, 628–641 e626 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kagan VE et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nature chemical biology 13, 81 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Conrad M et al. Regulation of lipid peroxidation and ferroptosis in diverse species. Genes Dev 32, 602–619 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Matsushita M et al. T cell lipid peroxidation induces ferroptosis and prevents immunity to infection. J Exp Med 212, 555–568 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Feng H & Stockwell BR Unsolved mysteries: How does lipid peroxidation cause ferroptosis? PLoS Biol 16, e2006203 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Murray PJ Macrophage Polarization. Annu Rev Physiol 79, 541–566 (2017). [DOI] [PubMed] [Google Scholar]
- 15.O’Donnell VB et al. 15-Lipoxygenase catalytically consumes nitric oxide and impairs activation of guanylate cyclase. J Biol Chem 274, 20083–20091 (1999). [DOI] [PubMed] [Google Scholar]
- 16.Lorsbach RB, Murphy WJ, Lowenstein CJ, Snyder SH & Russell SW Expression of the nitric oxide synthase gene in mouse macrophages activated for tumor cell killing. Molecular basis for the synergy between interferon-gamma and lipopolysaccharide. J Biol Chem 268, 1908–1913 (1993). [PubMed] [Google Scholar]
- 17.Gao M, Monian P, Quadri N, Ramasamy R & Jiang X Glutaminolysis and Transferrin Regulate Ferroptosis. Mol Cell 59, 298–308 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Corna G et al. Polarization dictates iron handling by inflammatory and alternatively activated macrophages. Haematologica 95, 1814–1822 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang Y et al. Imidazole Ketone Erastin Induces Ferroptosis and Slows Tumor Growth in a Mouse Lymphoma Model. Cell Chem Biol 26, 623–633 e629 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Borland C et al. Permeability and diffusivity of nitric oxide in human plasma and red cells. Nitric Oxide 78, 51–59 (2018). [DOI] [PubMed] [Google Scholar]
- 21.Dar HH et al. Pseudomonas aeruginosa utilizes host polyunsaturated phosphatidylethanolamines to trigger theft-ferroptosis in bronchial epithelium. J Clin Invest 128, 4639–4653 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yamanaka K et al. A novel fluorescent probe with high sensitivity and selective detection of lipid hydroperoxides in cells. RSC Advances 2, 7894–7900 (2012). [Google Scholar]
- 23.Shah R, Shchepinov MS & Pratt DA Resolving the Role of Lipoxygenases in the Initiation and Execution of Ferroptosis. ACS Cent Sci 4, 387–396 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Monroe LL et al. Zymosan-Induced Peritonitis: Effects on Cardiac Function, Temperature Regulation, Translocation of Bacteria, and Role of Dectin-1. Shock 46, 723–730 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kenny EM et al. Ferroptosis Contributes to Neuronal Death and Functional Outcome After Traumatic Brain Injury. Crit Care Med (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bayir H et al. Enhanced oxidative stress in iNOS-deficient mice after traumatic brain injury: support for a neuroprotective role of iNOS. J Cereb Blood Flow Metab 25, 673–684 (2005). [DOI] [PubMed] [Google Scholar]
- 27.Loane DJ & Kumar A Microglia in the TBI brain: The good, the bad, and the dysregulated. Exp Neurol 275 Pt 3, 316–327 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bayir H et al. Neuronal NOS-mediated nitration and inactivation of manganese superoxide dismutase in brain after experimental and human brain injury. J Neurochem 101, 168–181 (2007). [DOI] [PubMed] [Google Scholar]
- 29.Foley LM et al. Magnetic resonance imaging assessment of macrophage accumulation in mouse brain after experimental traumatic brain injury. J Neurotrauma 26, 1509–1519 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brune B et al. Redox control of inflammation in macrophages. Antioxid Redox Signal 19, 595–637 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ivanov I, Kuhn H & Heydeck D Structural and functional biology of arachidonic acid 15-lipoxygenase-1 (ALOX15). Gene 573, 1–32 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lawrence T & Natoli G Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat Rev Immunol 11, 750–761 (2011). [DOI] [PubMed] [Google Scholar]
- 33.Mantovani A, Marchesi F, Malesci A, Laghi L & Allavena P Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol 14, 399–416 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Murray PJ et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity 41, 14–20 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kanazawa M, Ninomiya I, Hatakeyama M, Takahashi T & Shimohata T Microglia and Monocytes/Macrophages Polarization Reveal Novel Therapeutic Mechanism against Stroke. Int J Mol Sci. 18, 2135 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Anthonymuthu TS et al. Empowerment of 15-lipoxygenase catalytic competence in selective oxidation of membrane ETE-PE to ferroptotic death signals, HpETE-PE. J Am Chem Soc (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zilka O et al. On the Mechanism of Cytoprotection by Ferrostatin-1 and Liproxstatin-1 and the Role of Lipid Peroxidation in Ferroptotic Cell Death. ACS Cent Sci 3, 232–243 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cao JY & Dixon SJ Mechanisms of ferroptosis. Cell Mol Life Sci 73, 2195–2209 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rubbo H et al. Nitric oxide inhibition of lipoxygenase-dependent liposome and low-density lipoprotein oxidation: termination of radical chain propagation reactions and formation of nitrogen-containing oxidized lipid derivatives. Arch Biochem Biophys 324, 15–25 (1995). [DOI] [PubMed] [Google Scholar]
- 40.Saam J, Ivanov I, Walther M, Holzhutter HG & Kuhn H Molecular dioxygen enters the active site of 12/15-lipoxygenase via dynamic oxygen access channels. Proc Natl Acad Sci U S A 104, 13319–13324 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rubbo H et al. Nitric oxide regulation of superoxide and peroxynitrite-dependent lipid peroxidation. Formation of novel nitrogen-containing oxidized lipid derivatives. J Biol Chem 269, 26066–26075 (1994). [PubMed] [Google Scholar]
- 42.O’Donnell VB et al. Nitric oxide inhibition of lipid peroxidation: kinetics of reaction with lipid peroxyl radicals and comparison with alpha-tocopherol. Biochemistry 36, 15216–15223 (1997). [DOI] [PubMed] [Google Scholar]
- 43.Napoli C et al. Effects of nitric oxide on cell proliferation: novel insights. J Am Coll Cardiol 62, 89–95 (2013). [DOI] [PubMed] [Google Scholar]
- 44.Thomas DD et al. The chemical biology of nitric oxide: implications in cellular signaling. Free Radic Biol Med 45, 18–31 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Perrotta C et al. Nitric Oxide Generated by Tumor-Associated Macrophages Is Responsible for Cancer Resistance to Cisplatin and Correlated With Syntaxin 4 and Acid Sphingomyelinase Inhibition. Front Immunol 9, 1186 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xie BS et al. Inhibition of ferroptosis attenuates tissue damage and improves long-term outcomes after traumatic brain injury in mice. CNS Neurosci Ther 25, 465–475 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Amaral EP et al. A major role for ferroptosis in Mycobacterium tuberculosis-induced cell death and tissue necrosis. J Exp Med (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Martin-Sanchez D et al. Ferroptosis, but Not Necroptosis, Is Important in Nephrotoxic Folic Acid-Induced AKI. J Am Soc Nephrol 28, 218–229 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Uderhardt S et al. 12/15-lipoxygenase orchestrates the clearance of apoptotic cells and maintains immunologic tolerance. Immunity 36, 834–846 (2012). [DOI] [PubMed] [Google Scholar]
ONLINE METHODS REFERENCES
- 1.Bayir H et al. Enhanced oxidative stress in iNOS-deficient mice after traumatic brain injury: support for a neuroprotective role of iNOS. J Cereb Blood Flow Metab 25, 673–684 (2005). [DOI] [PubMed] [Google Scholar]
- 2.Foley LM et al. Magnetic resonance imaging assessment of macrophage accumulation in mouse brain after experimental traumatic brain injury. J Neurotrauma 26, 1509–1519 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wu YL et al. In situ labeling of immune cells with iron oxide particles: an approach to detect organ rejection by cellular MRI. Proc Natl Acad Sci U S A 103, 1852–1857 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Monroe LL et al. Zymosan-Induced Peritonitis: Effects on Cardiac Function, Temperature Regulation, Translocation of Bacteria, and Role of Dectin-1. Shock 46, 723–730 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ray A & Dittel BN Isolation of mouse peritoneal cavity cells. J Vis Exp (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lian H, Roy E & Zheng H Protocol for Primary Microglial Culture Preparation. Bio Protoc 6 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Elmore MR et al. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron 82, 380–397 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dar HH et al. Pseudomonas aeruginosa utilizes host polyunsaturated phosphatidylethanolamines to trigger theft-ferroptosis in bronchial epithelium. J Clin Invest 128, 4639–4653 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weischenfeldt J & Porse B Bone Marrow-Derived Macrophages (BMM): Isolation and Applications. CSH Protoc 2008, pdb.prot5080 (2008). [DOI] [PubMed] [Google Scholar]
- 10.Doll S et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol 13, 91–98 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Harris LA et al. BioNetGen 2.2: advances in rule-based modeling. Bioinformatics 32, 3366–3368 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu B, Gyori B & Thiagarajan P Statistical model checking based analysis of biological networks. Automated Reasoning for Systems Biology and Medicine in press, arXiv:1812.01091 (2019). [Google Scholar]
- 13.Phillips JC et al. Scalable molecular dynamics with NAMD. J Comput Chem 26, 1781–1802 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tovchigrechko A & Vakser IA GRAMM-X public web server for protein-protein docking. Nucleic Acids Res 34, W310–314 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wenzel SE et al. PEBP1 Wardens Ferroptosis by Enabling Lipoxygenase Generation of Lipid Death Signals. Cell 171, 628–641 e626 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frisch M et al. Gaussian 03, revision B. 05;. Gaussian. Inc., Pittsburgh, PA: 12478 (2003). [Google Scholar]
- 17.Bakan A et al. Evol and ProDy for bridging protein sequence evolution and structural dynamics. Bioinformatics 30, 2681–2683 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw data are available at the following link https://data.mendeley.com/datasets/t4jyhhf3gw/draft?a=76c6ac56-4305-463e-bebc-b004c2000edb
Code used for the analysis of the MD simulations of NO• interactions with 15LOX has been made available in two formats (i) Jupyter Notebook and (ii) HTML at the following link: https://onedrive.live.com/?authkey=%21AFEVmP5sOP1km0s&id=4960CA1B5C7F3FD%219568&cid=04960CA1B5C7F3FD.