

Abstract
Rationale:
Barth syndrome (BTHS) is an X-linked cardiac and skeletal myopathy caused by mutation of the gene Tafazzin (TAZ). Currently there is no targeted treatment for BTHS. Lack of a proper genetic animal model that recapitulates the features of BTHS has hindered understanding of disease pathogenesis and therapeutic development.
Objective:
We characterized murine germline (TAZ-KO) and cardiac specific (TAZ-CKO) Taz knockout models and tested the efficacy of AAV-mediated TAZ gene replacement therapy.
Methods and Results:
TAZ-KO caused embryonic and neonatal lethality, impaired growth, dilated cardiomyopathy, and skeletal myopathy. TAZ-KO mice that survived the neonatal period developed progressive, severe cardiac dysfunction and fibrosis. Cardiomyocyte specific inactivation of floxed Taz in CMs using Myh6-Cre caused progressive dilated cardiomyopathy without fetal or perinatal loss. Using both constitutive and conditional knockout models, we tested the efficacy and durability of Taz replacement by AAV gene therapy. Neonatal AAV-TAZ rescued neonatal death, cardiac dysfunction, and fibrosis in TAZ-KO mice, and both prevented and reversed established cardiac dysfunction in TAZ-KO and TAZ-CKO models. However, both neonatal and adult therapies required high CM transduction (~70%) for durable efficacy.
Conclusions:
TAZ-KO and TAZ-CKO mice recapitulate many of the key clinical features of BTHS. AAV-mediated gene replacement is efficacious when a sufficient fraction of CMs are transduced.
Keywords: Barth Syndrome, Tafazzin, gene therapy, cardiomyopathy, mitochondria, animal disease model, Animal Models of Human Disease, Myocardial Biology
Graphical Abstract
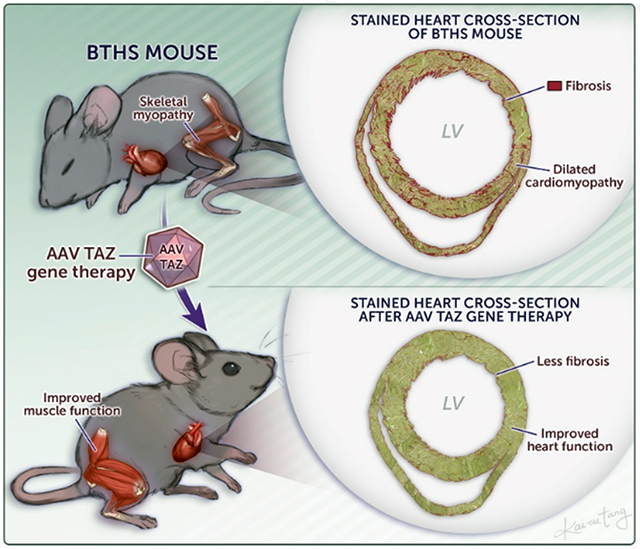
INTRODUCTION
Barth Syndrome (BTHS) is an X-linked, potentially lethal genetic disease that affects about 1 in 0.3 to 0.4 million live births1. Hallmarks of BTHS are cardiomyopathy, skeletal myopathy, neutropenia, growth delay, poor feeding, and organic aciduria, with cardiac disease and neutropenia being the leading causes of BTHS-related mortality1,2. Over 70% of BTHS patients develop cardiomyopathy in their first year, and 14% of BTHS patients require heart transplantation1. The skeletal myopathy results in life-altering, debilitating fatigue that severely limits activities3.
Mutation of the gene Tafazzin (TAZ) causes BTHS4. TAZ is a mitochondrial protein associated with the mitochondrial inner membrane. TAZ is required for the normal biogenesis of cardiolipin (CL)5, the signature phospholipid of mitochondria. CL is synthesized in nascent form with four non-specific acyl chains and undergoes TAZ-dependent remodeling, in which the acyl chains acquire a characteristic fatty acid composition, e.g. tetralinoleoyl cardiolipin in striated muscle6. The characteristic fatty acid composition of mature CL promotes its association with proteins in the inner mitochondrial membrane, facilitating the formation of mitochondrial super complexes7,8. Protein binding protects CL from degradation to monolysocardiolipin (MLCL), which lacks one of CL’s four fatty acid residues9. TAZ mutation impairs CL remodeling and protein binding, resulting in reduced mature CL and elevated MLCL10,11. This change in CL composition impairs the normal function of enzymes housed within the inner mitochondrial membrane, resulting in impaired electron transport chain function8, increased production of reactive oxygen species (ROS), and inefficient ATP synthesis12.
Several experimental models of BTHS have been reported12–16, including a doxycycline-induced short hairpin RNA TAZ knockdown mouse in which high dose doxycycline leads to 80-90% TAZ protein depletion15,16. However, important limitations of this model are residual TAZ expression, relatively mild cardiac involvement, high inter-animal variability, and the need for continuous, high dose doxycycline treatment, which itself impacts mitochondrial function17 and metalloprotease activity18. The lack of a TAZ knockout mouse that recapitulates the cardinal features of the human condition has hindered BTHS research.
Currently there are no targeted therapies for BTHS. Patients are treated with supportive medical management for cardiomyopathy (standard heart failure medications; transplantation) and neutropenia (GM-CSF). One attractive strategy is AAV gene therapy to replace mutant TAZ. TAZ gene replacement normalized function of BTHS human iPSC-CMs12, and AAV-TAZ partially normalized cardiac and skeletal muscle function in the TAZ knockdown model19. While promising, this proof-of-concept study had several important caveats that limit extrapolation to clinical translation. First, the study was based on the TAZ knockdown model, which has residual TAZ expression and relatively mild disease. Second, the study did not evaluate dose-response, a key issue given failure of a recent clinical cardiac AAV gene therapy trial likely as a result of insufficient dosing20. Third, the study did not evaluate the ability of AAV-TAZ to reverse established cardiac disease. Fourth, the study did not evaluate the durability of the therapeutic response.
Here we characterize the phenotype of mice with germline or cardiac-specific TAZ knockout. Germline TAZ knockout caused substantial fetal and perinatal demise that was likely due to skeletal muscle weakness. Survivors developed progressive cardiomyopathy and cardiac fibrosis, as did mice with cardiac-specific TAZ knockout. AAV-TAZ rescued neonatal demise, prevented cardiac dysfunction, and reversed established heart disease. However, therapeutic efficacy and durability were dependent on transduction of at least 70% cardiomyocytes (CMs). These results establish a platform for testing of BTHS therapies and demonstrates the efficacy of TAZ gene therapy when administered at a sufficient dose.
METHODS
The authors declare that all supporting data are available within the article and its online supplementary files. The AAV constructs can be obtained through Addgene. The mouse model can be obtained by contacting the authors.
Please see the Supplemental Materials for Expanded Materials and Methods. Major resources from this study are summarized in the Major Resources Table of the Online Supplemental Materials.
Animal.
Mice harboring the Tazfl and TazΔ allele were described previously21. Mice were genotyped by PCR using the primers indicated in Online Table I.
AAV Vectors.
AAV-GFP, AAV-Luciferase, and AAV-hTAZ contained a CAG promoter driving GFP, luciferase or codon optimized human full-length Tafazzin cDNA (hTAZ). AAV was administered by subcutaneous injection (age < P10), and to older mice by retro-orbital injection. High and medium AAV doses were 2x1010 and 1x1010 vg/g, respectively.
RNA in situ hybridization.
Transcripts of hTAZ and Actn2 were visualized using RNAscope® Multiplex Fluorescent Reagent Kit and probes from Advanced Cell Diagnostics.
Behavior studies
Open field assay of spontaneous activity was performed before and after mild treadmill exercise. Mice were also evaluated by treadmill exercise to exhaustion.22
Statistics.
Data are presented as mean ± standard deviation. Statistical testing was performed using GraphPad Prism 8 as described in each figure legend.
RESULTS
Global deletion of Tafazzin in mice caused embryonic and neonatal lethality.
The TazΔ allele was generated by treating Tazfl sperm with Cre (Fig. 1A)21. Since male mice lacking Taz are sterile23, heterozygous females (Taz+/Δ) were used to generate TazΔ/Y constitutive knockout (TAZ-KO) and littermate control (Taz+/Y) offspring. The mutant and wild-type alleles were easily distinguished by PCR genotyping (Fig. 1B). Immunoblotting using a capillary western system confirmed TAZ deletion in TAZ-KO (Fig. 1C). TAZ-KO mice were born at below the expected Mendelian ratio (Fig. 1D), although this was not statistically significant due to the relatively small sample size. Most liveborn TAZ-KO died in the neonatal period, so that only ~20% of live born mice survived to term (Fig. 1E). TAZ-KO neonates had mild to moderate ventricular systolic dysfunction (Fig. 1F).
Figure 1. Characterization of TAZ-KO mice.
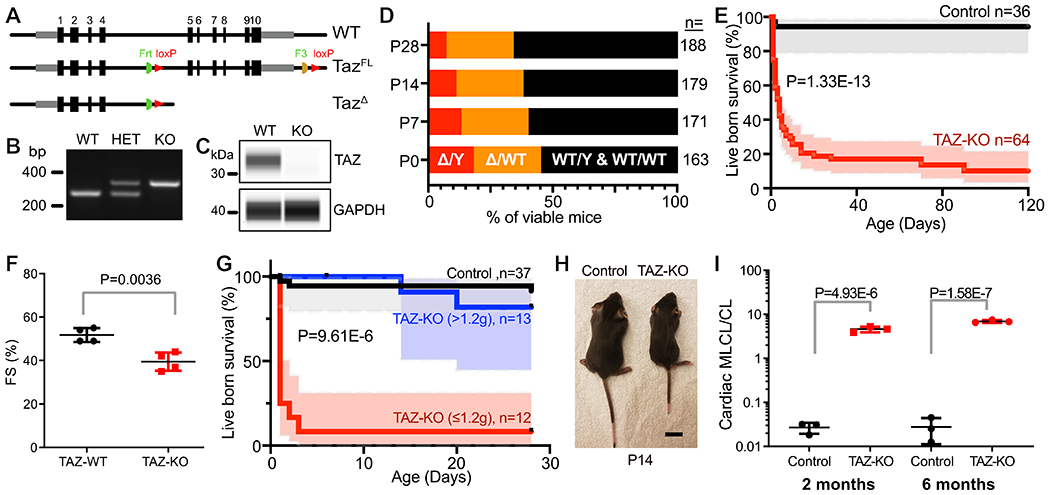
A. Schematic of TAZ wild-type, floxed, and deleted alleles. B. PCR genotyping. C. Loss of TAZ protein in TAZ-KO. Capillary immunoblotting of cardiac tissue from TAZ-KO and wild-type mice. D. Survival of TAZ-KO mice. TAZ-KO (Δ/Y) mice were present at below the expected Mendalian ratio from birth. E. Survival curve for live born control and TAZ-KO mice. Shading, 95% confidence interval. F. Cardiac contraction evaluated by echocardiography at P1. FS, fractional shortening. G. Survival curve for live born mice, with TAZ-KO mice classified by body weight at P1. Shading, 95% confidence interval. Statistical significance between TAZ-KO groups is shown. H. Photograph of TAZ-KO and control littermates at P14. Bar = 1 cm. I. Cardiac MLCL/CL ratio at 2 months and 6 months, as measured by mass spectrometry. Statistical tests: E,G: Mantel-Cox; F,I: t-test.
Like BTHS patients1,2, TAZ-KO newborn mice exhibited growth retardation and poor feeding. They also had reduced movement, hunchback, and dropping forelimbs, signs of neuromuscular weakness24 (Online Movie I and Online Fig. I-A). A milk spot was rarely observed in TAZ-KO neonates, and body weight declined between P1 and P2 in TAZ-KO whereas it increased in control littermates (Online Fig. I-B). Neonatal survival was related to birth weight, as TAZ-KO mice with body weight greater than 1.2 g at postnatal day 1 (P1) survived better than those with lower body weight (Fig. 1G). TAZ-KO mice who survived the newborn period had lower body weight and reduced body length throughout life (Fig. 1H). TAZ is required for CL remodeling, and BTHS patients have abnormal CL profiles with elevated MLCL:CL ratio10,25. Mass spectrometry of lipids isolated from TAZ-KO hearts demonstrated that the MLCL:CL ratio was markedly elevated compared to controls (Fig. 1I and Online Fig. I-C).
Together, these data demonstrate that Taz is essential for embryonic and neonatal development and survival, and indicate that TAZ-KO mice recapitulate many of the clinical and biochemical hallmarks of BTHS patients.
Cardiac phenotype of TAZ-KO mice.
Although most TAZ-KO mice did not survive the neonatal period, through intensive breeding we obtained sufficient TAZ-KO mice for analysis of adult phenotypes. In these TAZ-KO survivors, which largely had initial body weight > 1.2g at P1, the left ventricle was dilated and thin-walled compared to control, consistent with a dilated cardiomyopathy phenotype (Fig. 2A). Although our neonatal studies showed cardiac dysfunction (Fig. 1F), at 4-8 weeks-of-age TAZ-KO survivors had relatively normal cardiac function (Fig. 2B). This difference likely reflects survival bias. In the TAZ-KO survivor cohort, cardiac dysfunction and dilation typically manifested after 8 weeks-of-age and became progressively more severe (Fig. 2B–C). Adult TAZ-KO hearts had significantly increased fibrosis (Fig 2D–E). We confirmed that human BTHS hearts also have considerable cardiac fibrosis (Online Fig. II). Because cardiac fibrosis often occurs in the setting of increased CM death, we measured CM apoptosis in TAZ-KO and littermate control hearts. TUNEL staining demonstrated significantly increased CM apoptosis in TAZ-KO (Fig 2F–G).
Figure 2. Cardiac phenotype of TAZ-KO mice.
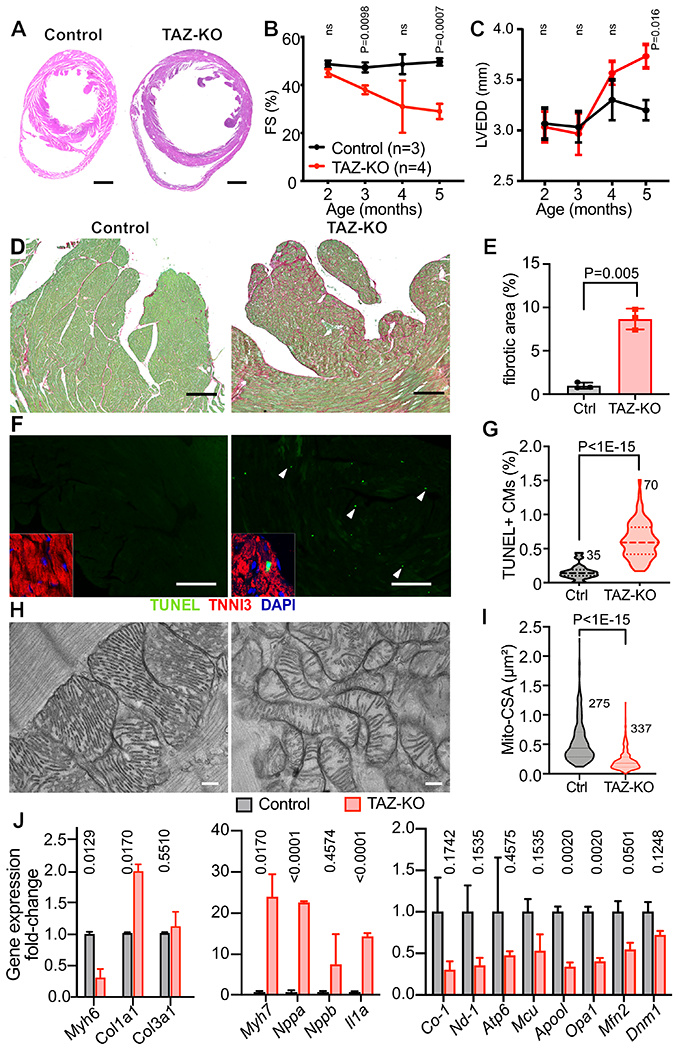
A. H&E stained sections demonstrating thin-walled, dilated TAZ-KO LV at 6 months-of-age. Bar = 1 mm. B-C. Echocardiography of TAZ-KO and control mice. FS, fractional shortening. LVEDD, LV end diastolic diameter. D-E. TAZ-KO cardiac fibrosis. Heart sections were stained with fast green/sirius red. Fibrotic area (red) was quantified as a fraction of myocardial area (green). Bar = 200 μm. n=3. F-G. CM apoptosis, measured by TUNEL labeling. Apoptotic cells were TUNEL-positive (arrowheads). Insets show TUNEL+ signal overlapping with a cardiac marker TNNI3 and DAPI. Bar = 200 μm. H. EM of cardiac mitochondria. Bar = 200 nm. I. Quantification of mitochondrial cross-sectional area (CSA). n=3 per group. J. Evaluation of markers of heart failure, fibrosis, and genes critical for mitochondrial functions and morphology by qRTPCR. n=3. B, C: Repeated measures two way ANOVA followed by Holm-Sidak multiple testing correction. E: t-test. G, I: Mann-Whitney test. J, Multiple t-tests with Holm-Sidak correction.
We used electron microscopy to evaluate the ultrastructure of TAZ-KO cardiomyocytes and mitochondria. TAZ-KO sarcomere Z-lines had normal morphology, but the A bands and M lines were poorly delineated, indicating abnormal sarcomere structure (Online Fig. III-A). TAZ-KO mitochondria displayed a drastic reduction in internal complexity (Fig 2H) and disorganized cristae, in contrast to the neatly organized and densely packed cristae of control mitochondria. Compared to controls, TAZ-KO mitochondria were smaller (Fig. 2I), and as a result there was a greater number of mitochondria per unit area (Online Fig. III-B). The spatial organization of mitochondria was also impaired by TAZ deficiency: whereas control mitochondria were aligned adjacent to sarcomeres, TAZ-KO mitochondria formed poorly organized clusters (Online Fig. III-C).
We analyzed TAZ-KO and control ventricular RNA for expression of genes related to cardiac stress, inflammation, fibrosis, and mitochondrial function (Fig. 2J). Consistent with myocardial stress, in TAZ-KO Myh6 was downregulated, and Myh7 and Nppa were markedly upregulated. The inflammatory cytokine Il1a was also strongly upregulated. Collagen type I (Col1a1) was significantly upregulated whereas the expression of collagen type III (Col3a1) was unchanged. Several nuclear mitochondrial transcripts were significantly downregulated (Apool, Opa1) or had a tendency to downregulation (Mfn2). Levels of mitochondrially encoded transcripts Co-1, Nd-1, and Atp6 were lower in TAZ-KO, although these comparisons did not reach statistical significance after multiple testing correction.
Non-cardiac phenotypes of TAZ-KO mice.
Skeletal muscle weakness/fatigability is a cardinal feature of BTHS3. We further characterized the skeletal muscle phenotype of TAZ-KO mice. TAZ-KO muscle fibers had smaller caliber and were more fibrotic than controls (Online Fig. IV A–F). As with cardiac mitochondria, skeletal muscle mitochondria were small and had severely compromised cristae (Online Fig. IV-G–J).
Next we measured skeletal muscle performance (Online Fig. IV-K). To evaluate endurance, we ran TAZ-KO and WT mice on a treadmill and measured the time to exhaustion (see Methods). On average, six month old TAZ-KO mice ran 110 seconds, whereas wild-type mice did not show exhaustion by the end of the test (840 seconds; P<0.05; Online Fig. IV-L). BTHS patients experience severe fatigue after sub-maximal exertion. To assess this phenotype in mice, animals were placed individually in open field chambers equipped to measure movement, before and after exercise. TAZ-KO mice had normal baseline activity prior to exercise (Online Fig. IV-M–N), but after exercise TAZ-KO had significantly voluntary movement (Online Fig. IV-O–P).
Neutropenia is another clinical feature of BTHS patients. The number of circulating neutrophils in 6-month-old TAZ-KO was significantly lower than WT (Online Fig. V). However, the value for both groups was within the normal range for mice (0.1–2.4K/μL; Online Fig. V). Neutropenia in BTHS patients can be sporadic, so further studies of are required to fully evaluate neutropenia in TAZ-KO mice.
Cardiomyocyte-restricted TAZ deletion.
To assess the contribution of Taz inactivation in cardiomyocytes to the TAZ-KO phenotype, we performed cardiomyocyte-specific TAZ inactivation using Myh6-Cre (Fig. 3A). Cardiomyocyte-specific Taz mutant mice (TAZ-CKO; Tazfl/Y; Myh6-Cre) were compared to littermate controls (Ctrl; Taz+/Y; Myh6-Cre). We validated the loss of TAZ protein by capillary immunoblotting (Fig. 3B) and abnormal cardiolipin remodeling by mass spectrometry (Fig. 3C).
Figure 3. Phenotype of CM-restricted TAZ-KO mice (TAZ-CKO).
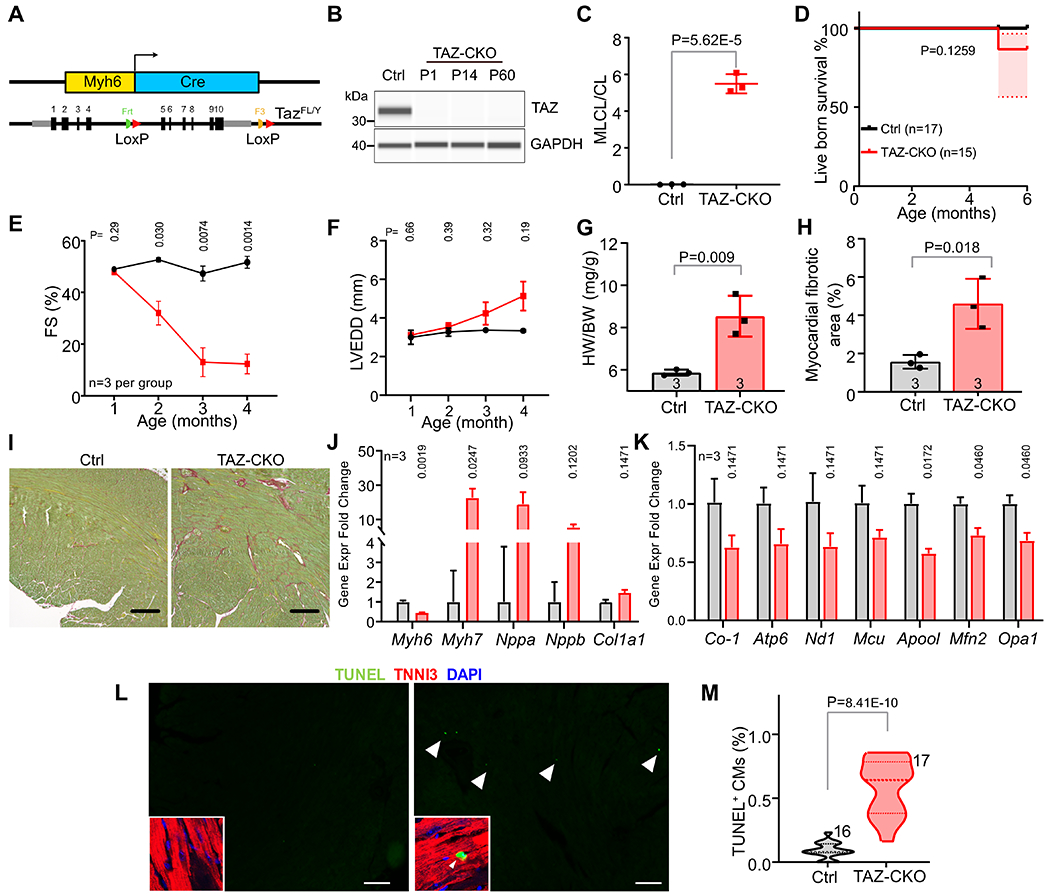
A. Schematic of cardiomyocyte-specific deletion by Myh6-Cre. B. TAZ protein expression bu capillary immunoblotting at P1, P14 and 2 months-of-age. C. MLCL/CL ratio from 2 month-old Ctrl and TAZ-CKO hearts. D. Normal survival of TAZ-CKO mice. E-F. Cardiac contraction (fractional shortening (FS)) and LV end diastolic diameter (LVEDD) evaluated by echocardiography. G. Heart weight to body weight ratio at 6 months. H-I. Ctrl and TAZ-CKO heart section at 6 months were stained with sirius red/fast green. Bars=200μm. Percentage of fibrotic area (red) in the myocardium (green) is shown in H. J. Expression of cardiac stress markers by qRT-PCR. Numbers above bars indicate p values. n=3. K. qRT-PCR measurement of mitochondria-related gene expression. n=3. L. Apoptotic CMs in the myocardium, which are marked by double-labeling by TNNI3 (red) and TUNEL-positive (green), shown in insets. Arrowheads point to selected examples. Bar = 200 μm. M. Quantification of apoptotic CMs. Numbers indicate sections examined from 3 different hearts per genotype. C, G, H: t-test. D: Mantel-Cox test. E, F: Repeated measures two way ANOVA followed by Holm-Sidak multiple testing correction. J, K: Multiple t-tests with Holm-Sidak multiple testing correction. M: Mann-Whitney U test.
TAZ-CKO mice were born at the expected Mendelian ratio with body weight comparable to controls. The mice survived normally to adulthood (Fig. 3D) and during 6 months of observation. TAZ-CKO had normal LV size and function at 1 month (Fig. 3E–F). LV function progressively declined on subsequent monthly echocardiograms (Fig. 3E), accompanied by an increase in LV chamber size (Fig. 3F). At 6 months, heart weight normalized to body weight was elevated (Fig. 3G). Histological evaluation demonstrated myocardial fibrosis and CM apoptosis in TAZ-CKO (Fig. 3H–I, L–M). Consistent with reduced cardiac function in TAZ-CKO hearts, cardiac stress markers Myh6, Myh7, and Nppa were significantly dysregulated., while nuclear encoded mitochondrial genes ApooI, Mfn2, and Opa1 were significantly downregulated (Fig. 3J–K).
Collectively, the TAZ-CKO model does not develop the fetal and perinatal loss observed in the whole body TAZ-KO, perhaps due to fetal or perinatal requirement of TAZ in non-cardiomyocytes, e.g. skeletal muscle. On the other hand, TAZ inactivation in CMs was sufficient to reproduce the progressive dilated cardiomyopathy and cardiac fibrosis observed in adult TAZ-KO mice.
AAV-hTAZ rescue of TAZ-KO neonatal lethality.
AAV gene therapy is an attractive strategy to treat Barth syndrome. We generated AAV9 in which full length, codon-optimized human TAZ was expressed from the potent and widely expressed CAG promoter (AAV-hTAZ; Fig. 4A). Both AAV9 and CAG are components of the FDA-approved gene therapy Zolgensma. AAV carrying the coding sequence of luciferase (AAV-Ctrl) was used as the control virus. First, we studied the impact of AAV-hTAZ on the demise of low body weight (< 1.2 g at P1) TAZ-KO mice in the first week of life (Fig. 4B). At birth, TAZ-KO pups were weighed and genotyped. At P1, low body weight TAZ-KO or control littermates were treated with either AAV-hTAZ or AAV-Ctrl via subcutaneous injection at a dose sufficient to transduced 65% of cardiac and 60% of skeletal muscle cells, as measured at P7 by RNA in situ hybridization using a probe specific to the codon-optimized hTAZ transcript (Fig. 4C) or by administration of a similar dose of AAV-GFP (Online Fig. VI-A). The primary endpoint was survival to P28. Secondary endpoints were cardiac function, as assessed by echocardiography monthly for 4 months and cardiac fibrosis at 4 months. Conversion of the single stranded AAV genome to a double-stranded episome is a rate-limiting step of AAV transduction26, and this process can be expedited by self-complementary AAV27 (scAAV; Online Fig. VI-A). Because the rapid neonatal loss of TAZ-KO mice made transduction kinetics a potential concern, we also included scAAV-hTAZ (Fig. 4A) in the study. We estimated the level of AAV-mediated hTAZ expression by qRTPCR. Because hTAZ and mTaz nucleotide sequences differ, qPCR primers did not amplify both transcripts with the same efficiency. We circumvented this problem by using standard curves (Online Fig. VI-B) to compare relative expression AAV-expressed hTAZ and endogenous mTAZ (Online Fig. VI-C; see Detailed Methods). AAV-hTAZ and scAAV-hTAZ drove equivalent levels of hTAZ cardiac expression when evaluated at 4 months after administration (Online Fig. VI-C), which we estimated to be 10-fold higher than endogenous mTAZ. Both AAV-hTAZ and scAAV-hTAZ partially corrected MLCL:CL to a similar degree (Online Fig. VI-D).
Figure 4. Effect of AAV-mediated TAZ replacement therapy on neonatal survival of TAZ-KO mice.
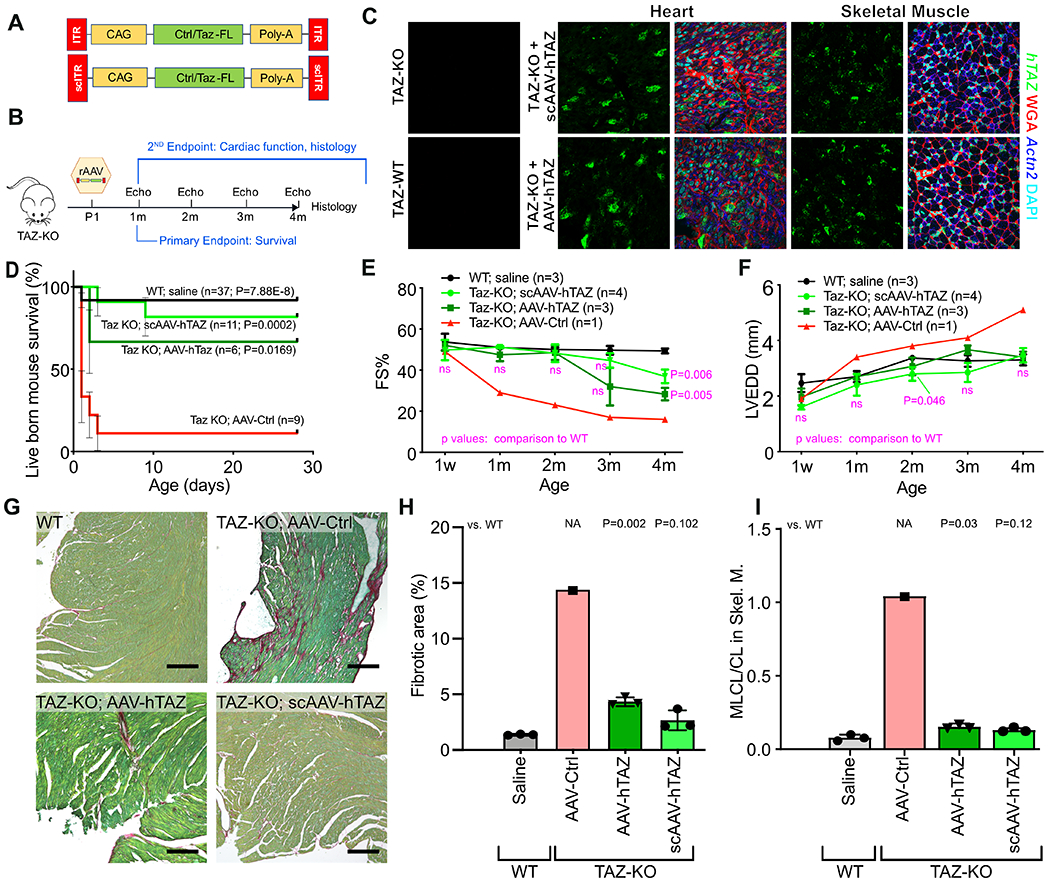
A. Schematic of AAV-hTAZ, and scAAV-hTAZ. Luciferase-expressing AAV was used as control virus (AAV-Ctrl). B. Experimental design. Neonatal TAZ-KO mice were treated at P1. Survival to weaning (P28) was the primary endpoint, and echocardiography and histological parameters were secondary endpoints. C. Viral transduction of cardiac and skeletal muscle was evaluated 7 days after AAV injection by in situ hybridization using probes specific to hTAZ (green puncta) and cardiomyocyte marker Actn2 (blue). D. Survival curve of mice after treatment with AAV at P1. Bars, standard error. P-values, comparison to TAZ-KO treated with AAV-Ctrl. Using Bonferonni correction for 3 comparisons, the significance threshold is p=0.0167. E-F. Serial echocardiography. Treated mice were not distinguishable from WT until 4 months, when the treatment groups showed reduced systolic function. G. Myocardial sections stained by fast green/sirius red. Bar=200 μm. H. Quantification of fibrosis. I. MLCL/CL in P7 skeletal muscle. D: Mantel-Cox. E, F: Repeated measures two way ANOVA followed by Tukey’s post hoc test. H, I: one-way ANOVA with Tukey post-hoc testing.
Both AAV-hTAZ and scAAV-hTAZ dramatically enhanced neonatal survival compared to AAV-Ctrl (Fig. 4D). Survival was comparable between scAAV-hTAZ and AAV-hTAZ.
We used echocardiography to monitor the heart function of rescued mice to 4 months-of-age. Due to the high frequency of neonatal demise among low birth weight TAZ-KO, we were only able to analyze one AAV-Ctrl treated low body weight TAZ-KO mouse, and this mouse exhibited progressive dilated cardiomyopathy that was more severe than observed in unselected TAZ-KO mice (which were almost all in the high body weight category; Fig. 1G and Fig. 2B). Both AAV-hTAZ and scAAV-hTAZ treated mice were protected against cardiac dysfunction for 2 months (Fig. 4E and Online Data I). At later time points, both hTAZ-treated groups began to show declining cardiac function, which became significantly depressed at 4 months-of-age. However, neither treatment group developed consistent LV dilatation during the 4 month study period (Fig. 4F).
At 4 months-of-age, we evaluated cardiac fibrosis. Control-treated TAZ-KO heart had extensive fibrosis whereas both hTAZ-treated groups had substantially reduced fibrosis (Fig. 4G–H), with the level of fibrosis in scAAV-hTAZ treated hearts comparable to WT. Histological analysis suggested that declining function at later time points was possibly due to insufficient CM transduction: whereas the dose used transduced 65% CMs at P7, at 21 days and 90 days after the initial treatment only 24% cardiac cells retain transgene expression (Online Fig. VI-E). A similar reduction of AAV-transduced cells was also observed in skeletal muscle (Online Fig. VI-E). This decrease in transduced cells was likely due to perinatal cardiomyocyte and skeletal muscle proliferation.28,29
TAZ-CKO mice survived normally but TAZ-KO mice did not, suggesting that the mortality in TAZ-KO mice is due to its TAZ deficiency in non-cardiomyocytes. Therefore TAZ expression in organs besides the heart is likely to be responsible for AAV-hTAZ rescue of TAZ-KO neonatal death. Given the skeletal muscle weakness of the TAZ-KO mice, we hypothesized that the skeletal muscle weakness and failure to compete with stronger littermates for nutrition are important contributors to neonatal death, and that AAV-hTAZ rescues by improving cardiolipin metabolism in skeletal muscle. To test this hypothesis, we constructed two additional scAAV vectors. scAAV2i8 cTNT-hTAZ used the cardiac specific cTNT promoter and the AAV2i8 capsid, which efficiently transduces heart and skeletal muscle, but detargets liver30. scAAV2i8 MHCK7-hTAZ used the MHCK7 striated muscle promoter31 and the AAV2i8 capsid. Using GFP as a reporter, we validated that the former expresses selectively in the heart, whereas the latter expresses selectively in heart and skeletal muscle (Online Fig. VI-F,G). scAAV2i8 cTNT-hTAZ did not significantly improve survival of TAZ-KO mice, whereas scAAV2i8 MHCK7-hTAZ had comparable effect to scAAV9 CAG-hTAZ (Online Fig. VI-G). Furthermore, scAAV2i8 MHCK7-hTAZ corrected the skeletal muscle CL profile at P7 (Fig. 4I). Together, these data demonstrate that hTAZ gene replacement, using either single-stranded or self-complementary AAV, efficiently prevents neonatal death by TAZ replacement in skeletal muscle. AAV-hTAZ protects cardiac function of TAZ-KO mice for at least 3 months, with relatively low cardiomyocyte transduction perhaps accounting for limited therapeutic durability.
AAV-hTAZ prevention of cardiomyopathy in TAZ-CKO mice.
Because the TAZ-CKO model circumvents difficulties with survival in the TAZ-KO model, it is more convenient for assessing therapeutic efficacy and dose response on the cardiomyopathic phenotype. Given that AAV-hTAZ and scAAV-hTAZ had similar efficacy, we focused on AAV-hTAZ. AAV expressing luciferase was again used as the control virus (AAV-Ctrl). We treated TAZ-CKO mice with AAV-hTAZ or AAV-Ctrl at P20 by intravascular (retro-orbital) injection, prior to the onset of cardiac dysfunction, to determine if gene therapy prevents the development of cardiomyopathy (Fig. 5A). Cardiac function was examined monthly to 4 months of age, when hearts were analyzed for histological endpoints. To evaluate dose-response, we tested medium and high doses of AAV, which transduced ~33% and >70% of cardiomyocytes, respectively (Online Fig. VII-A). CM transduction efficiency was similar between 21 and 90 days, indicating that the viral genome was stable in CMs during this period (Online Fig. VII-A).
Figure 5. AAV-hTAZ prevented development of cardiac dysfunction in TAZ-CKO in a dose dependent manner.
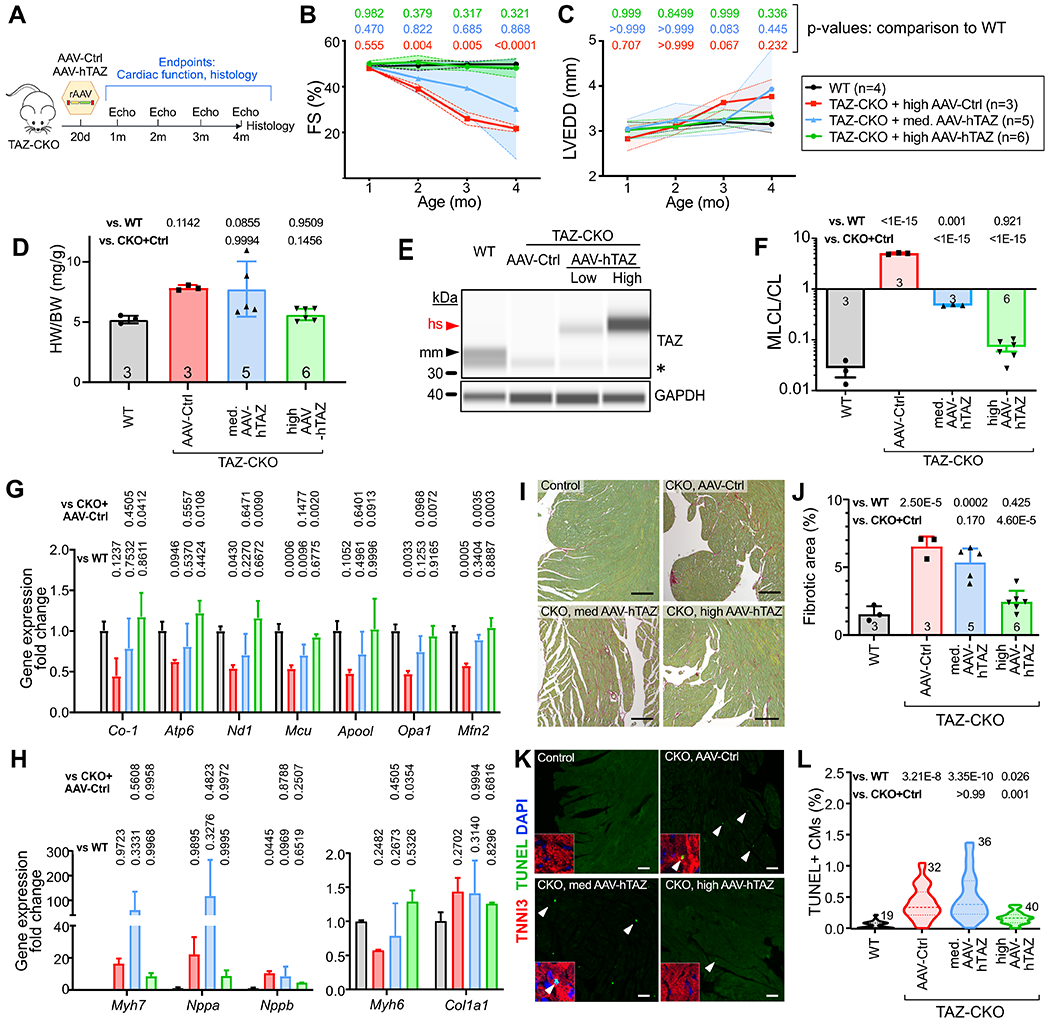
A. Experimental outline. B-C. Echocardiography of TAZ-CKO mice. FS, fractional shortening. LVEDD, left ventricular end diastolic diameter. Shading, standard deviation. Full statistical comparisons are in Online Data I. D. Cardiac hypertrophy, shown by the heart weight vs. body weight ratio, was examined 3 months after treatment. E. Capillary immunoblotting of TAZ in heart extracts. AAV-hTAZ delivered human (hs) TAZ has higher molecular weight than murine (mm) TAZ. * marks a non-specific band. F. Cardiac MLCL/CL measured by mass spectrometry. G-H. Transcriptional correction of genes critical for mitochondrial function and heart failure program. n=4 (TAZ-CKO+med. AAV-hTAZ) or 3 (others). P values are indicated above bars. I-J. Cardiac fibrosis measured by sirius red/fast green staining of cardiac samples at 4 months of age. Quantification is shown in J. Bar=200 μm. K-L. Cardiac apoptosis measured by TUNEL staining. Percentage of TUNEL-positive CMs was quantified in L. Insets, magnified CM nuclei. Arrowheads, example TUNEL+ CMs. Bar=200 μm. Numbers indicate sections analyzed, from at least 3 different hearts per group. B,C: Repeated two way ANOVA followed by Tukey’s post-hoc test. D, F, G, H, J: One way ANOVA test followed by Tukey’s post-hoc test. L: Kruskal Wallis with Dunn’s multiple comparison test.
In AAV-Ctrl treated TAZ-CKO mice, cardiac function became abnormal at 2 months and progressively declined at 3 and 4 months (Fig. 5B–C and Online Data I). High dose AAV-hTAZ provided consistent and durable protection against cardiac dysfunction. In contrast, medium dose AAV-hTAZ resulted in variable results, with a subset of mice exhibiting little improvement in cardiac function compared to control treatment, and other mice exhibiting normalization of cardiac function.
At 4 months of age, hearts were collected for molecular and histological analysis. Cardiac hypertrophy seen in TAZ-CKO mice was consistently reduced by high dose AAV-hTAZ but the response was variable in the medium dose group (Fig. 5D). This result did not reach statistical significance in ANOVA (p=0.146 with Tukey’s post-hoc test) due to the large variance in the medium dose group (p=0.0001 when medium dose group was excluded). TAZ expression was measured by capillary western blotting, in which endogenous murine TAZ and hTAZ can be distinguished by virtue of a primate-specific exon. This assay confirmed loss of endogenous mTAZ in TAZ-CKO hearts and dose-dependent expression of hTAZ by AAV-hTAZ (Fig. 5E). AAV-hTAZ normalized the MLCL/CL ratio in a dose-dependent manner, with the medium dose reducing it to an intermediate level, and the high dose making it comparable to control mice (Fig. 5F). High dose AAV-hTAZ treatment restored expression of genes important for mitochondrial function (Mcu, Opa1, Mfn2, Co-1, Atp6, Nd1) and normalized Myh6 expression (Fig. 5G–H). There was variable response in the medium dose group, with the subset of non-responsive mice continuing to have elevation of Myh7, Nppa, and Nppb (Fig. 5H). As a result, changes in the expression of these genes did not reach statistical significance by ANOVA. High dose AAV-hTAZ reduced cardiac fibrosis (Fig. 5I–J) and apoptosis (Fig. 5K–L) to normal levels, whereas medium dose AAV-hTAZ showed partial improvement.
We conclude that with sufficient cardiomyocyte transduction, AAV-hTAZ treatment successfully prevents the development of cardiomyopathy.
AAV-hTAZ reversal of established cardiac dysfunction.
Many patients with BTHS will likely have established cardiac dysfunction by the time that gene therapy is considered. To test the efficacy and dose-response of AAV-hTAZ in a more clinically relevant context, we performed serial echocardiograms on TAZ-CKO mice, and enrolled mice when their shortening fraction was below 40% (typically around 2 months of age). We then randomized mice to treatment with high dose or medium dose AAV-hTAZ, or high dose AAV-Ctrl, where the high and medium doses were calibrated to transduced ~70% and ~33% CM, respectively (Online Fig. VII-A). After treatment, mice were followed by echocardiography monthly for 3 months. Hearts were then collected and subjected to molecular and histological studies.
Monthly echocardiography demonstrated progressive deterioration of heart function with AAV-Ctrl treatment (Fig. 6B and Online Data I). In contrast, high dose AAV-hTAZ reverted cardiac function to normal levels, whereas the medium dose stabilized cardiac dysfunction (Fig. 6B). Both doses of AAV-hTAZ prevented LV dilatation and cardiac hypertrophy (Fig. 6C–D). Capillary western blotting of heart extracts confirmed loss of mTAZ in AAV-Ctrl treated TAZ-CKO mice, and dose-dependent replacement with full length human TAZ by AAV-hTAZ (Fig. 6E). Cardiolipin analysis confirmed dose-dependent correction of the MLCL/CL ratio by AAV-hTAZ (Fig. 6F). Expression of genes critical for mitochondrial function and morphology and cardiac stress markers were normalized by high dose AAV-hTAZ (Fig. 6G–H). High dose AAV-hTAZ reduced but did not completely normalize cardiac fibrosis and cardiomyocyte apoptosis (Fig. 6I–L), whereas the medium dose mildly reduced the cell death but did not significantly improve the overall fibrosis. Together, our results indicate that AAV-hTAZ administered at a sufficient dose reverses mild cardiomyopathy in TAZ-CKO hearts.
Figure 6. AAV-hTAZ reversal of established cardiac dysfunction in TAZ-CKO mice.
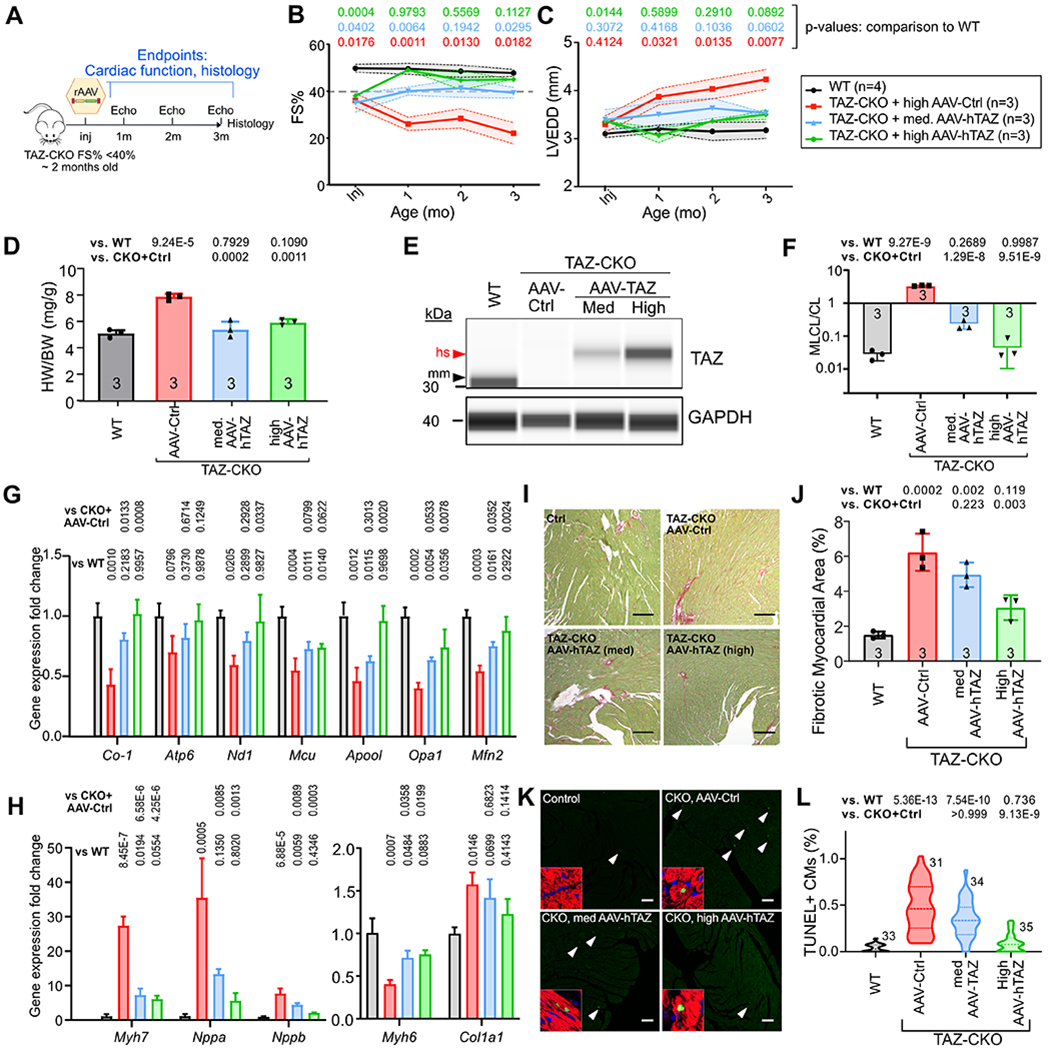
A. Experimental outline. TAZ-CKO mice with established heart dysfunction (FS<40%, ~ 2-month-old) were treated with medium or high doses of AAV, which were calibrated to transduce ~33% or ~70% CMs. B-C. Echocardiographic measurement of LV systolic function (B) and end diastolic diameter (C). Shading indicates standard deviation. Full statistical comparisons are provided in Online Data I. D. Cardiac hypertrophy, shown by the ratio of heart weight vs. bodyweight, was examined 3 months after treatment. E. Capillary immunoblotting of TAZ in heart extracts. AAV-hTAZ delivered human (hs) TAZ is longer than murine (mm) TAZ. F. Cardiac cardiolipin composition measured by mass spectrometry. G-H. Transcriptional correction of genes critical for mitochondrial function and heart failure program. n=3 per group. P values are indicated above bars. I-J. Cardiac fibrosis measured by sirius red/ fast green staining of cardiac samples at 4 months of age. Quantification is shown in J. Bar=200 μm. K-L. Cardiac apoptosis measured by TUNEL staining. Apoptotic CMs were identified with TNNI3 shown in insets. Percentage of TUNEL-positive CMs was quantified in L. Insects show TUNEL signal overlapping with TNNI3 and DAPI to identify CMs. Bar= 200 μm. Numbers indicate sections analyzed, from at least 3 different hearts per group. B, C: Repeated two way ANOVA followed by Tukey’s multiple comparison test. D, F, G, H, J: One way ANOVA test followed by Tukey’s multiple comparison test. L: Kruskal-Wallis with Dunn’s multiple comparisons test.
Because global TAZ knockout in TAZ-KO mice likely involves multiple organ systems, which may modify the effectiveness of AAV-hTAZ gene therapy for cardiomyopathy, we next asked if AAV-hTAZ reverses established cardiomyopathy in the germline TAZ-KO model (Fig. 7A). Although most TAZ-KO mice die before weaning, through intensive breeding we collected enough TAZ-KO mice to study. In initial experiments, we established that TAZ-KO mice had mild LV dysfunction (FS% < 40%), hypertrophy and mild histological abnormalities (Fig. 7B and Online Fig. VIII) at 3 months-old. We performed a therapeutic trial to reverse established cardiac dysfunction. 3-month-old TAZ-KO mice with FS% < 40% were enrolled and received AAV-hTAZ or AAV-Ctrl (encoding luciferase) at a dose calibrated to transduce ~70% CMs. The transgene remained stable in the heart for at least 90 days after injection (Online Fig. VII-B top panel). Mice underwent monthly echocardiography for 3 months, and then hearts were removed for further analysis.
Figure 7. AAV-TAZ improves cardiac function in TAZ-KO with established cardiomyopathy.
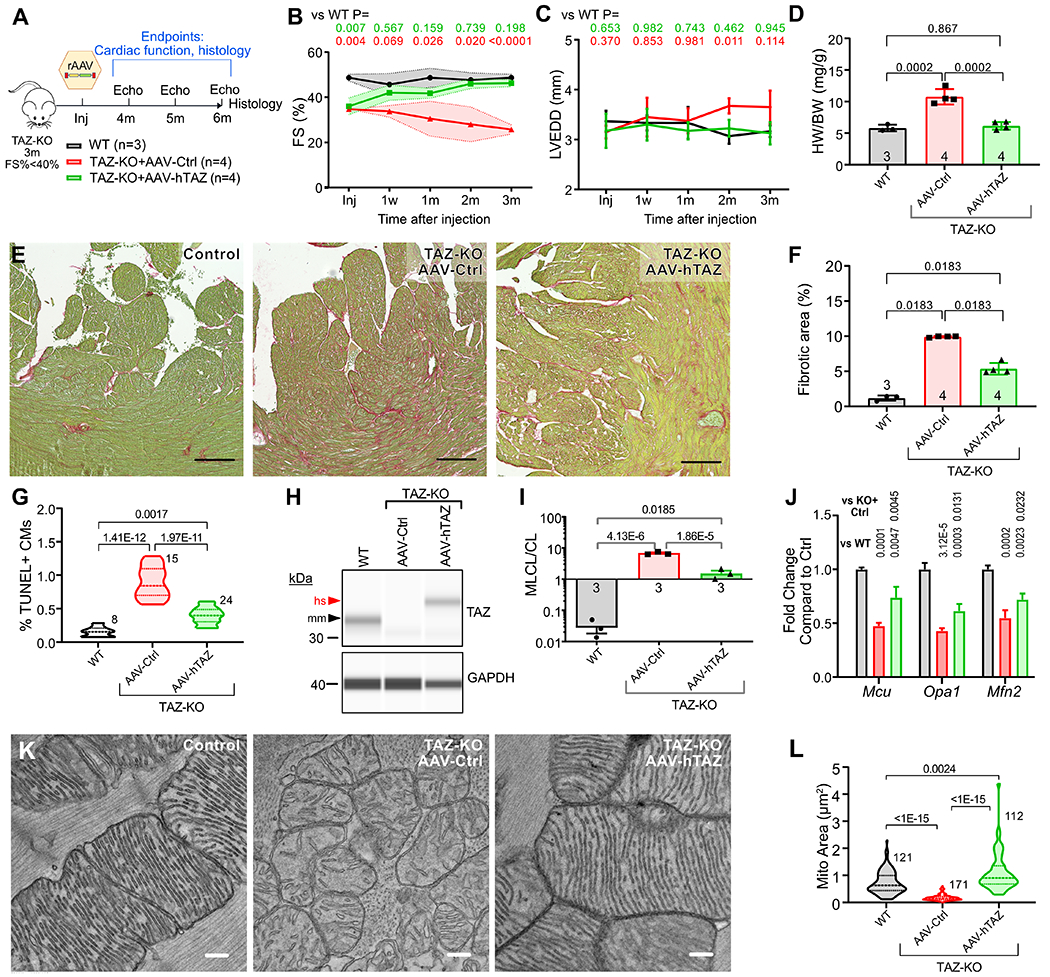
A. Experimental plan. TAZ-KO mice with FS<40% at ~3 months of age were treated with no agent (control), AAV-Ctrl, or AAV-hTAZ at a high dose (~70% CM transduction). Mice were followed for 3 months by echocardiography and then hearts underwent histological and molecular studies. Samples sizes for B-D are indicated. B-C. LV systolic function and LV end diastolic diameter measured by echocardiography. Shaded areas indicate standard deviation. Numbers at top indicate p-value of comparison to WT. Full statistical comparison is provided in Online Data I. D. Heart weight to body weight ratio. E-F. Histological sections stained with fast green and picrosirus red. Bar=200 μm. Percentage of myocardial tissue area that stained red is quantified in F. G. Percentage of TUNEL+ CMs. Numbers indicate sections analyzed, from at least 3 hearts per group. H. Capillary immunoblot of heart protein extracts probed with antibody to TAZ or GAPDH. mm, murine TAZ. hs, human TAZ. I. MLCL/CL was measured by mass spectrometry. n=3 per group. J. Cardiac RNA was analyzed by qRTPCR. Results were normalized to Gapdh. Numbers indicate p-values. n=3. K. EM showing mitochondrial morphology. Bar=200 nm. L. Quantification of mitochondrial cross-sectional area. Numbers indicate mitochondria measured, from at least 3 different hearts. B,C, Repeated measures two way ANOVA followed by Tukey’s post-hoc test. D, F, G, H, I, J: One way ANOVA with Tukey post-hoc test. L: Kruskal-Wallis with Dunn’s multi-test correction.
AAV-hTAZ treated mice had progressive improvement in heart function, and by 3 months after treatment FS% was not significantly different from control mice (Fig. 7B and Online Data I). The LV was not dilated at the start of the trial and tended to become more dilated over time in AAV-Ctrl but not in AAV-hTAZ (Online Fig. VIII-A and Fig. 7C). AAV-hTAZ prevented cardiac hypertrophy, as assessed by the heart weight to body weight ratio (Fig. 7D). On histological sections, AAV-hTAZ reduced myocardial fibrosis compared to AAV-Ctrl, although the extent of fibrosis remained elevated compared to control mice (Fig. 7E–F). AAV-hTAZ likewise reduced CM apoptosis compared to AAV-Ctrl, although the frequency of apoptotic CMs remained elevated compared to controls (Fig. 7G).
By qRT-PCR and capillary western blotting, AAV-hTAZ restored TAZ to at least control levels (Fig. 7H and Online Fig. IX-A) and markedly improved the cardiolipin profile (Fig. 7I). However, MLCL/CL remained elevated after AAV-hTAZ compared to the control genotype, consistent with transduction of most but not all cardiac cells.
Finally, we evaluated mitochondrial ultrastructure and gene expression. By qRT-PCR with Gapdh normalization, AAV-hTAZ partially normalized expression of mitochondrial genes (Fig. 7J and Online Fig. IX-B). AAV-hTAZ improved mitochondrial morphology, increased the density and regularity of mitochondrial cristae (Fig. 7K), and normalized mitochondrial cross-sectional area (Fig. 7L). AAV-hTAZ normalized the spatial distribution of mitochondria, with mitochondria aligning normally along sarcomeres (Online Fig. IX-C).
Overall, AAV-hTAZ reversed established cardiomyopathy, reduced cardiac fibrosis and cardiomyocyte apoptosis, and improved mitochondrial morphology and gene expression in the germline TAZ-KO model.
Effect of AAV-hTAZ on Skeletal Muscle Phenotype.
Although high dose AAV9 described above efficiently transduced the heart and improved cardiac function, when administered to adult mice it did not comparably transduce skeletal muscle cells (27% in quadriceps) (Online Fig. VII-B lower panel and Online Fig. X-A). Viral genome was further gradually lost from skeletal muscle over time, as the transduction efficiency was 17% at 60 days and 11% at 90 days after AAV administration (Online Fig. VII-B and 10B). Viral genome dilution in skeletal muscle was seen in other gene therapy trials targeting skeletal muscle28,29.
AAV-hTAZ increased expression of hTAZ transcripts in the quadriceps (Online Fig. X-C), although expression was low compared to heart (Online Fig. VI-C). The MLCL/CL ratio did not significantly improve, in keeping with the low fraction of transduced cells (Online Fig. X-D). Nevertheless, AAV-hTAZ improved mitochondrial gene expression in skeletal muscle (Online Fig. X-E). By EM, mitochondria in AAV-hTAZ treated skeletal muscle had significantly improved cross-sectional area (Online Fig. X-F–H). TAZ-KO muscle fiber cross sectional area was greater AAV-hTAZ compared to AAV-Ctrl (Online Fig. X-I), but remained less than WT. Functionally, AAV-hTAZ showed a tendency to improve endurance of TAZ-KO compared to AAV-Ctrl (Online Fig. X-J), but the difference did not reach statistical significance (p=0.06).
Together, our results indicate the dose of AAV-hTAZ sufficient to correct cardiomyopathy had a mild but measurable salutary effect on skeletal muscle function in TAZ-KO mice. These results suggest that the dose sufficient for improving heart function might be insufficient for correcting skeletal muscle defects, potentially due to the more efficient and durable transduction of cardiac compared to skeletal muscle by AAV9.
DISCUSSION
Constitutive and cardiac specific TAZ knockout models recapitulate multiple features of BTHS, including: fetal and perinatal demise; poor feeding; growth retardation; progressive cardiac dysfunction; cardiac fibrosis; skeletal muscle weakness and hypoplasia; low neutrophil count; and impaired CL remodeling. Our detailed characterization of the natural history of these models sets the stage for using them to evaluate the efficacy of potential BTHS therapies. Compared to the doxycycline-induced short hairpin RNA knockdown model reported previously15,16, this model has clear advantages, including greater similarity to BTHS patients, the lack of residual TAZ, far less inter-individual variation, and freedom from high dose doxycycline, which itself can affect mitochondrial function17 and metalloprotease activity18.
A notable finding from this model is that cardiac fibrosis and cardiomyocyte apoptosis are important features of the BTHS-related cardiomyopathy. We validated that human BTHS hearts that require transplantation also exhibited marked cardiac fibrosis, in both infants and adolescents. This is consistent with clinical findings in which heart failure symptoms can be more severe than would be expected by the degree of systolic dysfunction, suggestive of diastolic dysfunction. Since cardiolipin interaction with cytochrome C regulates a key apoptotic trigger32, a molecular pathway may link TAZ deficiency to CM apoptosis. Cell autonomous predisposition of TAZ deficient CMs to apoptosis has important implications for gene therapy, since non-transduced CMs would continue to be at risk for death.
We used the newly characterized models to evaluate the efficacy and dose-response of AAV-TAZ gene therapy for BTHS. A recent study used the shRNA TAZ knockdown mouse model to provide proof-of-concept that AAV-TAZ gene therapy might be effective for BTHS19. Here we answered several questions crucial for clinical translation that were not addressed by the prior study. We showed that gene replacement therapy could be successful in a model without residual TAZ expression. We demonstrated that high level cardiomyocyte transduction successfully maintained contractile function and prevented cardiac fibrosis and cardiomyocyte apoptosis. Moreover, high dose AAV-TAZ was able to reverse mild, established cardiac dysfunction. Through dose-response studies, we showed that high level cardiomyocyte transduction is required for durable efficacy. This is a critical consideration for clinical translation -- whereas transduction of over 90% CMs is readily achievable with AAV9 in rodents, achieving sufficiently high CM transduction in humans or other large animals may be more challenging and require improved vectors or methods to permit repeated vector dosing.
Consistent with prior reports on the TAZ knockdown mouse model16,33 and human BTHS patients34, our TAZ-KO model showed morphological and functional abnormalities in the skeletal muscles. AAV-hTAZ partially rescued these phenotypes when administered to adult mice, despite low transduction efficiency and diminishing fraction of TAZ+ skeletal myocytes over time. Further investigation of skeletal muscle responses to AAV-TAZ gene therapy will require use of conditional and inducible skeletal muscle deficient models, to circumvent limitations imposed by the high loss of TAZ-KO neonates. Such studies of the skeletal muscle abnormalities and their responses to therapy are beyond the scope of this study, but they are important to consider in translating AAV-based gene therapy to treating BTHS patients.
We were surprised to find that neonatal demise of TAZ-KO mice was due to skeletal muscle rather than cardiac insufficiency. BTHS infants are hypotonic and often have feeding issues, although due to supportive care this is a not significant cause of mortality in human patients. Survival is an easily scored phenotype which will be useful to characterize BTHS therapies in the TAZ-KO model.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What Is Known?
Barth Syndrome (BTHS) is a rare and potentially fatal disease caused by loss-of-function mutation of Tafazzin (TAZ), which is critical for mitochondrial function.
Cardiomyopathy and skeletal muscle fatigue are major phenotypes of BTHS.
Adeno-associated virus (AAV)-mediated TAZ replacement were previously shown to improve cardiac and skeletal muscle function in mice with incomplete TAZ depletion.
What New Information Does This Article Contribute?
Detailed characterization of a new mouse model with complete TAZ ablation shows that the model recapitulates major cardiac and skeletomuscular phenotypes of BTHS patients.
Myocardial fibrosis and cardiomyocyte apoptosis are important components of the myopathic process in BTHS mice.
AAV-mediated TAZ replacement prevents the onset of cardiomyopathy and restores contractile function in BTHS mice with pre-existing cardiac insufficiency, but durablility and efficacy depend on transduction of a high percentage of cardiomyocytes.
Understanding pathogenic mechanisms of BTHS and testing of potential therapies has been hindered by the lack of a robust animal model. Previous in vivo experiments relied on a doxycycline-inducible knock-down system, which suffered from high inter-animal variability and incomplete Taz depletion. Here we characterized the first Taz germline and conditional knock-out mouse models, with a focus on the cardiac and skeletal muscular phenotypes. In the C57BL6/J background, germline Taz knockout mice displayed rapid neonatal lethality. Survivors, or cardiac specific knockout, resulted in progressive cardiomyopathy with histological evidence of cardiomyocyte apoptosis and myocardial fibrosis, and marked exercise intolerance. Using AAV to direct TAZ expression, we showed that TAZ gene replacement rescued neonatal survival, prevented the development of cardiomyopathy, and even restored cardiac function in mice with established cardiomyopathy. Moreover, we demonstrate that transduction of a large fraction of cardiomyocytes is required for durable efficacy of gene therapy. Our results suggest that AAV-TAZ is an attractive strategy to treat BTHS cardiomyopathy, provided that sufficient cardiac transduction can be achieved.
ACKNOWLEDGEMENTS
The authors thank Nick Andrews, PhD, from the Boston Children’s Behavioral Neurosciences Core for expert advice with animal behavioral studies and Brian Polizzotti’s lab for assistance with cardiolipin extraction.
SOURCES OF FUNDING
WTP was funded by NIH (R01HL128694), the Barth Syndrome Foundation, and by charitable donations from Edwin August Boger, Jr. Fund and the Boston Children’s Hospital Department of Cardiology. MS was supported by NIH (R01 GM115593).
Nonstandard Abbreviations and Acronyms:
- AAV
adeno-associated virus
- AAV-GFP
AAV9 expressing green fluorescent protein
- AAV-hTAZ
AAV9 expressing human full length tafazzin
- BTHS
Barth syndrome
- CAG
CMV early enhancer - chicken beta-actin promoter
- CM
cardiomyocyte
- CL
cardiolipin
- FS
fractional shortening
- LVEDD
left ventricle end diastolic diameter
- MLCL
monolysocardiolipin
- scAAV
self complementary AAV
- TAZ-KO
germline TAZ knockout mice
- TAZ-CKO
cardiomyocyte-specific TAZ knockout mice
Footnotes
CONFLICTS OF INTEREST
WTP is a member of the Medical and Scientific Advisory Board of the Barth Syndrome Foundation. WTP is a consultant for AveXis.
REFERENCES
- 1.Clarke SLN, Bowron A, Gonzalez IL, Groves SJ, Newbury-Ecob R, Clayton N, Martin RP, Tsai-Goodman B, Garratt V, Ashworth M, Bowen VM, McCurdy KR, Damin MK, Spencer CT, Toth MJ, Kelley RI, Steward CG. Barth syndrome. Orphanet J Rare Dis. 2013;8:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roberts AE, Nixon C, Steward CG, Gauvreau K, Maisenbacher M, Fletcher M, Geva J, Byrne BJ, Spencer CT. The Barth Syndrome Registry: distinguishing disease characteristics and growth data from a longitudinal study. Am J Med Genet A. 2012;158A:2726–2732. [DOI] [PubMed] [Google Scholar]
- 3.Spencer CT, Byrne BJ, Bryant RM, Margossian R, Maisenbacher M, Breitenger P, Benni PB, Redfearn S, Marcus E, Cade WT. Impaired cardiac reserve and severely diminished skeletal muscle O2 utilization mediate exercise intolerance in Barth syndrome. Am J Physiol Heart Circ Physiol. 2011;301:H2122–9. [DOI] [PubMed] [Google Scholar]
- 4.Bione S, D’Adamo P, Maestrini E, Gedeon AK, Bolhuis PA, Toniolo D. A novel X-linked gene, G4.5. is responsible for Barth syndrome. Nat Genet. 1996;12:385–389. [DOI] [PubMed] [Google Scholar]
- 5.Schlame M, Greenberg ML. Biosynthesis, remodeling and turnover of mitochondrial cardiolipin. Biochim Biophys Acta Mol Cell Biol Lipids. 2017;1862:3–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mejia EM, Nguyen H, Hatch GM. Mammalian cardiolipin biosynthesis. Chem Phys Lipids. 2014;179:11–16. [DOI] [PubMed] [Google Scholar]
- 7.Zhang M, Mileykovskaya E, Dowhan W. Gluing the respiratory chain together. Cardiolipin is required for supercomplex formation in the inner mitochondrial membrane. J Biol Chem. 2002;277:43553–43556. [DOI] [PubMed] [Google Scholar]
- 8.Pfeiffer K, Gohil V, Stuart RA, Hunte C, Brandt U, Greenberg ML, Schägger H. Cardiolipin stabilizes respiratory chain supercomplexes. J Biol Chem. 2003;278:52873–52880. [DOI] [PubMed] [Google Scholar]
- 9.Xu Y, Phoon CKL, Berno B, D’Souza K, Hoedt E, Zhang G, Neubert TA, Epand RM, Ren M, Schlame M. Loss of protein association causes cardiolipin degradation in Barth syndrome. Nat Chem Biol. 2016;12:641–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schlame M, Kelley RI, Feigenbaum A, Towbin JA, Heerdt PM, Schieble T, Wanders RJ, DiMauro S, Blanck TJ. Phospholipid abnormalities in children with Barth syndrome. J Am Coll Cardiol. 2003;42:1994–1999. [DOI] [PubMed] [Google Scholar]
- 11.Vreken P, Valianpour F, Nijtmans LG, Grivell LA, Plecko B, Wanders RJ, Barth PG. Defective remodeling of cardiolipin and phosphatidylglycerol in Barth syndrome. Biochem Biophys Res Commun. 2000;279:378–382. [DOI] [PubMed] [Google Scholar]
- 12.Wang G, McCain ML, Yang L, He A, Pasqualini FS, Agarwal A, Yuan H, Jiang D, Zhang D, Zangi L, Geva J, Roberts AE, Ma Q, Ding J, Chen J, Wang D-Z, Li K, Wang J, Wanders RJA, Kulik W, Vaz FM, Laflamme MA, Murry CE, Chien KR, Kelley RI, Church GM, Parker KK, Pu WT. Modeling the mitochondrial cardiomyopathy of Barth syndrome with induced pluripotent stem cell and heart-on-chip technologies. Nat Med. 2014;20:616–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gu Z, Valianpour F, Chen S, Vaz FM, Hakkaart GA, Wanders RJA, Greenberg ML. Aberrant cardiolipin metabolism in the yeast taz1 mutant: a model for Barth syndrome. Mol Microbiol. 2004;51:149–158. [DOI] [PubMed] [Google Scholar]
- 14.Xu Y, Condell M, Plesken H, Edelman-Novemsky I, Ma J, Ren M, Schlame M. A Drosophila model of Barth syndrome. Proc Natl Acad Sci U S A. 2006;103:11584–11588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Phoon CKL, Acehan D, Schlame M, Stokes DL, Edelman-Novemsky I, Yu D, Xu Y, Viswanathan N, Ren M. Tafazzin knockdown in mice leads to a developmental cardiomyopathy with early diastolic dysfunction preceding myocardial noncompaction. J Am Heart Assoc [Internet]. 2012;1 Available from: 10.1161/JAHA.111.000455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Soustek MS, Falk DJ, Mah CS, Toth MJ, Schlame M, Lewin AS, Byrne BJ. Characterization of a transgenic short hairpin RNA-induced murine model of Tafazzin deficiency. Hum Gene Ther. 2011;22:865–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moullan N, Mouchiroud L, Wang X, Ryu D, Williams EG, Mottis A, Jovaisaite V, Frochaux MV, Quiros PM, Deplancke B, Houtkooper RH, Auwerx J. Tetracyclines Disturb Mitochondrial Function across Eukaryotic Models: A Call for Caution in Biomedical Research. Cell Rep. 2015;10:1681–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Golub LM, Lee HM, Ryan ME, Giannobile WV, Payne J, Sorsa T. Tetracyclines inhibit connective tissue breakdown by multiple non-antimicrobial mechanisms. Adv Dent Res. 1998;12:12–26. [DOI] [PubMed] [Google Scholar]
- 19.Suzuki-Hatano S, Saha M, Rizzo SA, Witko RL, Gosiker BJ, Ramanathan M, Soustek MS, Jones MD, Kang PB, Byrne BJ, Cade WT, Pacak CA. AAV-Mediated TAZ Gene Replacement Restores Mitochondrial and Cardioskeletal Function in Barth Syndrome. Hum Gene Ther [Internet]. 2018;Available from: 10.1089/hum.2018.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Greenberg B, Butler J, Felker GM, Ponikowski P, Voors AA, Desai AS, Barnard D, Bouchard A, Jaski B, Lyon AR, Pogoda JM, Rudy JJ, Zsebo KM. Calcium upregulation by percutaneous administration of gene therapy in patients with cardiac disease (CUPID 2): a randomised, multinational, double-blind, placebo-controlled, phase 2b trial. Lancet. 2016;387:1178–1186. [DOI] [PubMed] [Google Scholar]
- 21.Ren M, Xu Y, Erdjument-Bromage H, Donelian A, Phoon CKL, Terada N, Strathdee D, Neubert TA, Schlame M. Extramitochondrial cardiolipin suggests a novel function of mitochondria in spermatogenesis. J Cell Biol. 2019;218:1491–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Castro B, Kuang S. Evaluation of Muscle Performance in Mice by Treadmill Exhaustion Test and Whole-limb Grip Strength Assay. Bio Protoc [Internet]. 2017;7 Available from: 10.21769/BioProtoc.2237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cadalbert LC, Ghaffar FN, Stevenson D, Bryson S, Vaz FM, Gottlieb E, Strathdee D. Mouse Tafazzin Is Required for Male Germ Cell Meiosis and Spermatogenesis. PLoS One. 2015;10:e0131066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Turgeon B, Meloche S. Interpreting neonatal lethal phenotypes in mouse mutants: insights into gene function and human diseases. Physiol Rev. 2009;89:1–26. [DOI] [PubMed] [Google Scholar]
- 25.Kulik W, van Lenthe H, Stet FS, Houtkooper RH, Kemp H, Stone JE, Steward CG, Wanders RJ, Vaz FM. Bloodspot assay using HPLC-tandem mass spectrometry for detection of Barth syndrome. Clin Chem. 2008;54:371–378. [DOI] [PubMed] [Google Scholar]
- 26.Ferrari FK, Samulski T, Shenk T, Samulski RJ. Second-strand synthesis is a rate-limiting step for efficient transduction by recombinant adeno-associated virus vectors. J Virol. 1996;70:3227–3234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McCarty DM. Self-complementary AAV vectors; advances and applications. Mol Ther. 2008;16:1648–1656. [DOI] [PubMed] [Google Scholar]
- 28.Le Hir M, Goyenvalle A, Peccate C, Précigout G, Davies KE, Voit T, Garcia L, Lorain S. AAV genome loss from dystrophic mouse muscles during AAV-U7 snRNA-mediated exon-skipping therapy. Mol Ther. 2013;21:1551–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vulin A, Barthélémy I, Goyenvalle A, Thibaud J-L, Beley C, Griffith G, Benchaouir R, le Hir M, Unterfinger Y, Lorain S, Dreyfus P, Voit T, Carlier P, Blot S, Garcia L. Muscle function recovery in golden retriever muscular dystrophy after AAV1-U7 exon skipping. Mol Ther. 2012;20:2120–2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Asokan A, Conway JC, Phillips JL, Li C, Hegge J, Sinnott R, Yadav S, DiPrimio N, Nam H-J, Agbandje-McKenna M, McPhee S, Wolff J, Samulski RJ. Reengineering a receptor footprint of adeno-associated virus enables selective and systemic gene transfer to muscle. Nat Biotechnol. 2010;28:79–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Salva MZ, Himeda CL, Tai PW, Nishiuchi E, Gregorevic P, Allen JM, Finn EE, Nguyen QG, Blankinship MJ, Meuse L, Chamberlain JS, Hauschka SD. Design of tissue-specific regulatory cassettes for high-level rAAV-mediated expression in skeletal and cardiac muscle. Mol Ther. 2007;15:320–329. [DOI] [PubMed] [Google Scholar]
- 32.Kagan VE, Tyurin VA, Jiang J, Tyurina YY, Ritov VB, Amoscato AA, Osipov AN, Belikova NA, Kapralov AA, Kini V, Vlasova II, Zhao Q, Zou M, Di P, Svistunenko DA, Kurnikov IV, Borisenko GG. Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat Chem Biol. 2005;1:223–232. [DOI] [PubMed] [Google Scholar]
- 33.Powers C, Huang Y, Strauss A, Khuchua Z. Diminished Exercise Capacity and Mitochondrial bc1 Complex Deficiency in Tafazzin-Knockdown Mice. Front Physiol 2013;4:74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hornby B, McClellan R, Buckley L, Carson K, Gooding T, Vernon HJ. Functional exercise capacity, strength, balance and motion reaction time in Barth syndrome. Orphanet J Rare Dis. 2019;14:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Agah R, Frenkel PA, French BA, Michael LH, Overbeek PA, Schneider MD. Gene recombination in postmitotic cells. Targeted expression of Cre recombinase provokes cardiac-restricted, site-specific rearrangement in adult ventricular muscle in vivo. J Clin Invest. 1997;100:169–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guo Y, Jardin BD, Zhou P, Sethi I, Akerberg BN, Toepfer CN, Ai Y, Li Y, Ma Q, Guatimosim S, Hu Y, Varuzhanyan G, VanDusen NJ, Zhang D, Chan DC, Yuan G-C, Seidman CE, Seidman JG, Pu WT. Hierarchical and stage-specific regulation of murine cardiomyocyte maturation by serum response factor. Nat Commun 2018;9:3837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sun G, Yang K, Zhao Z, Guan S, Han X, Gross RW. Matrix-assisted laser desorption/ionization time-of-flight mass spectrometric analysis of cellular glycerophospholipids enabled by multiplexed solvent dependent analyte-matrix interactions. Anal Chem. 2008;80:7576–7585. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.