

Abstract
Telomerase is a ribonuceloprotein complex responsible for maintaining telomeres and protecting chromosomal integrity. The human telomerase reverse transcriptase (hTERT) is expressed in ~90% of cancer cells where it confers the capacity for limitless proliferation. Along with its established role in telomere lengthening, telomerase also serves non-canonical extra-telomeric roles in oncogenic signaling, resistance to apoptosis, and enhanced DNA damage response. We report a new class of natural product-inspired covalent inhibitors of telomerase that target the catalytic active site.
Graphical Abstract
The ends of chromosomes in human cells are capped by telomeres, 5- to 10-kb tandem arrays of 5’-TTAGGG-3’ sequence1 that, with the cognate six-protein shelterin complex,2 protect chromosome ends against recognition as DNA damage. As telomeres progressively shorten with each cell division cycle, one or more may reach a critical length. The resulting DNA damage response blocks further proliferation and promotes apoptosis or cellular senescence.3 Stem cells, embryonic cells, and germ cells overcome this proliferation limit by expressing telomerase, a ribonucleoprotein (RNP) complex that counteracts telomere shortening by synthesizing TTAGGG repeats at chromosome 3’ ends.4 The human telomerase core is composed of an RNA-dependent DNA polymerase, the human telomerase reverse transcriptase (hTERT),5 and the human telomerase RNA (hTR)6 which serves as the template for telomere repeat synthesis. Telomerase binds to the 3’ single-strand telomeric overhang, aligning with the complementary sequence found on hTR, priming the DNA for nucleotide addition catalyzed by hTERT (Fig. 1A). Following nucleotide addition the RNA template translocates by six nucleotides relative to the catalytic center to promote an additional round of nucleotide addition. While hTR is broadly expressed, hTERT is normally silenced in somatic cells, although it is detectable in certain proliferating cells, stem cells and germ cells.7 Reactivation of hTERT is observed in ~90% of human malignancies and has been directly linked to cancer cell immortality, making hTERT a compelling target for inhibition.8 In addition to telomere maintenance, genetic studies of hTERT have identified multiple extra-telomeric activities that may promote malignancy including deregulation of oncogenic signaling pathways,9 resistance to apoptosis10, enhanced DNA repair11 and promotion of telomere protective complexes.12
Fig. 1:

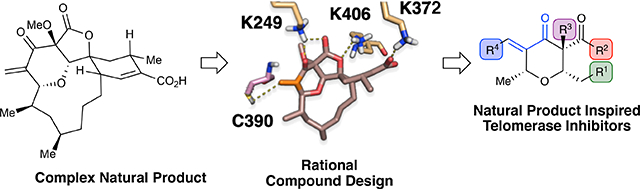
A) Telomere extension by telomerase. B) Natural products postulated to inhibit telomerase activity through a covalent mechanism. C) Workflow for rational design of chrolactomycin inspired chemical probes.
In recent decades, diverse strategies have been pursued to target telomerase in cancer,13 including the non-competitive small-molecule inhibitor BIBR1532,14 antisense oligonucleotide hTR binders such as imetelstat,15 and G-quadruplex stabilizers designed to block telomerase from extending telomeres.16 Moreover, nature has provided numerous unique scaffolds that have been proposed to inhibit telomerase activity17 such as epigallocatechin gallate,18 helenalin19 and costunolide20 (Fig. 1B). While nature serves as valuable information for the design of therapeutics and chemical probes, their implementation as tools to study biological processes is often limited by their isolation or synthesis.21 Overall, these agents have contributed significantly to our understanding of telomerase and telomere biology, yet fail to provide a means to covalently and irreversibly block telomerase activity, a unique inhibitory mechanism that may provide further insight into the biological functions of telomerase. Despite concerns of off-target reactivity, small-molecule covalent inhibitors can have clear advantages over non-covalent therapeutics.22 Their irreversible inhibition decreases dependence on affinity or drug levels and can raise the barrier to drug resistance.23 The clinical success of ibrutinib,24 a first-generation covalent inhibitor of Bruton’s tyrosine kinase, has led to multiple second generation investigational agents with increased specificity, establishing the value of combining rational compound design and proteomic validation to accelerate development of targeted covalent inhibitors. Therefore, our goal was to design and develop natural product-inspired, synthetically accessible covalent inhibitors of telomerase as new tools to study the telomeric functions of human telomerase (Fig. 1C).
RESULTS AND DISCUSSION
Among the diverse natural products that have been examined as candidate telomerase inhibitors, chrolactomycin (1), a tetrahydropyran macrolide antibiotic isolated from Streptomyces sp. 569N-3, was reported to display an IC50 of 0.5 μM in in vitro telomerase assays.25 Notably, chrolactomycin’s activity was hypothesized to be due to conjugation of the exomethylene group to an undetermined nucleophile. Efforts toward de novo synthesis of the closely related macrolide okilactomycin have been successful, but chrolactomycin has not been accessed by synthesis to date and the length of these routes leaves structure-activity studies essentially unexplored.26 Drawing on our laboratory’s previous work on similar tetrahydropyran macrolides,27 we applied rational compound design to chrolactomycin to lead to synthetically tractable, small-molecule analogues. Our goal in these efforts was to preserve the covalent mechanism for hTERT inhibition, providing a novel chemical path toward tool development targeting telomerase.
To validate a covalent mechanism for telomerase inhibition by chrolactomycin, we examined effects on the well-studied TERT protein from Tribolium castaneum, tcTERT. Proteolysis and tandem mass spectrometry of purified tcTERT treated with 1 μM chrolactomycin in vitro yielded a single modified peptide, HPQDEIPYCGK (Fig. 2A, see the Supporting Information). Notably, inclusion of costunolide and helenalin, two other natural products postulated to inhibit telomerase activity through a covalent mechanism, showed no adduct formation with any tcTERT peptides, indicating that their telomerase inhibitory mechanism is likely not due to covalent interactions with the reverse transcriptase. Based on the tcTERT crystal structure, the modified residue, C390 (C931 in hTERT), lies within the putative active site.28 Lacking a high resolution structure for hTERT to model chrolactomycin binding,29 tcTERT was utilized for our in silico studies. Docking of chrolactomycin in the active site of tcTERT was examined using the Schrödinger suite (Glide module). By placing the terminal carbon atom of the α, β-enone of chrolactomycin in close proximity (3.3 Å) to the C390 sulfur atom in the active site, several non-bonding interactions (K249, K406, K372) could be observed with potential to stabilize binding and facilitate delivery of the electrophilic warhead (Fig. 2B). This docking exercise revealed few, if any contacts, between tcTERT and the chrolactomycin macrocycle, which was solvent exposed and played a minimal role in the computed binding energy. Additionally, our computational studies identified that the macrocycle likely serves to rigidify chrolactomycin to orient the exocyclic methylene proximal to C390 for covalent addition.
Fig. 2:

A) Chrolactomycin inhibits telomerase by a covalent mechanism B) Cheminformatics model for binding of chrolactomycin. C) Synthetic sequence for modular access to chrologs
Drawing on these insights, we designed simplified chrolactomycin analogues (dubbed “chrologs”) that might maintain probe-target interactions but could be obtained via an efficient and modular synthetic sequence. Chrologs were designed using the Pipeline Pilot module of the Accelrys software package,30 and compounds were selected based on favorable computed drug-likeness.31 We scored probes using in silico docking with the Glide module32 and CovDock33 of the Schrödinger suite, where compound prioritization was assigned based on a computed binding energy between C390 and the exocyclic methylene. To commence, bicyclic dioxinones were obtained by a one-pot vinylogous Mukaiyama aldol reaction between aldehydes and silyl dioxinone dienolates using BF3·OEt2 at −78 °C, followed by warming up the reaction mixture to 0 °C and charging of a different aldehyde and additional BF3·OEt2.34 Dioxinone opening and subsequent trapping of the acyl ketene intermediate by an alcohol nucleophile afforded the β-ketoester. Lastly, the target structures were accessed by α-methylation and then a modified Mannich-type methylenation (Fig. 2C). Notably, these simplified analogues could be accessed in 4 steps vs. >25 steps required for the syntheses of okilactomycin.
In developing a preliminary structural activity relationship (SAR) for the chrologs, we divided the structure of the pyran into four main components: (1) C–2 substituents (carboxylic acid of chrolactomycin); (2) C–6 substituents (chrolactomycin macrolide loop); (3) C–3 substituents (fused γ-lactone of chrolactomycin) and (4) inclusion of the exocyclic methylene group. We carried out systematic modifications of each structural unit and evaluated the effects on telomerase activity as measured by a PCR-based telomerase activity assay (TRAP assay).8
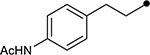
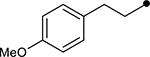
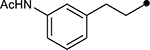
First, the effects of C–2 substitution were investigated (Table 1). The inclusion of a hydrogen-bonding group was found to be crucial, where a para N-acetylphenethyl group showed loss of telomerase activity (1.5 μM), compared to phenethyl and isobutyl groups (>100 μM). Exchange of the para N-acetylphenethyl group for para-fluorophenethyl, para-trifluoromethylphenethyl, para-methoxyphenethyl and all meta-substituted compounds showed reduced potency. Subsequently, we evaluated the role that C–6 substitution had on telomerase activity (Table 1). Surprisingly there was minimal steric tolerance at this position, where substitution of the C–6 methyl group for more sterically encumbered groups such as isobutyl and phenethyl showed a drop in activity. This loss of activity is likely due to the larger alkyl groups at C–6 providing a greater hydrophobic surface that destabilizes compound binding to TERT prior to covalent modification. The smaller C–6 methyl group provide minimal hydrophobic interactions, allowing for the predisposition of the exocyclic methylene proximal to C390 to facilitate C–S bond formation. Additionally, this trend in biological activity is reinforced by computed binding energies.
Table 1:
Structure-Activity Relationships of C–2 and C–6 Groups
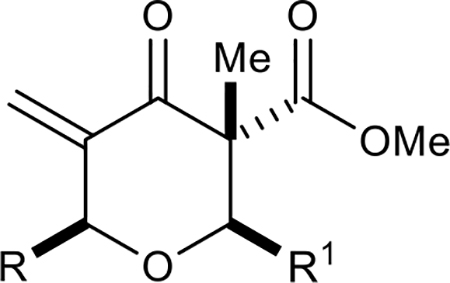 | ||||
|---|---|---|---|---|
| Compound | R | R1 | IC50 (μM) | ΔG Binding (kcal/mol) |
| 4a | 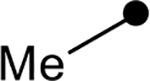 |
 |
1.5 | −31.9 |
| 4b | 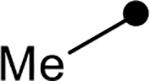 |
>100 | N/A | |
| 4c | 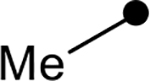 |
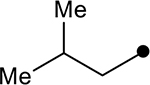 |
>100 | N/A |
| 4d | 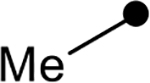 |
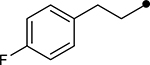 |
42 | −27.5 |
| 4e | 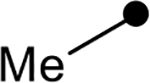 |
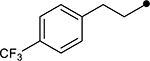 |
55 | −22.8 |
| 4f | 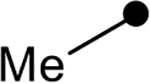 |
 |
62 | −21.1 |
| 4g | 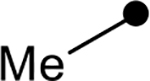 |
 |
11 | −28.6 |
| 4h | 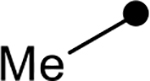 |
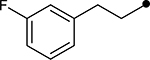 |
>100 | N/A |
| 4i | 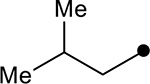 |
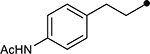 |
45 | −27.7 |
| 4j | 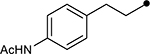 |
>100 | N/A | |
Next, we investigated the role of the β-ketoester in telomerase activity (Table 2). More sterically encumbered esters such as isopropyl, methylcyclopropyl, benzyl, and n-butyl proved deleterious to biological activity. Inclusion of H-bonding elements (methoxyethane) pendant on the alcohol nucleophile showed no demonstrable increases in activity. Unfortunately, attempts to access β-ketoamide derivatives were unsuccessful as these substrates were prone to decomposition during installation of the exocyclic methylene group. Introduction of the α-methoxy group either through direct methoxylation or hydroxylation-alkylation strategies was unfruitful, providing only enone structures. To identify if potential H-bonding due to the α-methoxy group in chrolactomycin was beneficial for biological activity, we conducted a direct comparison between chrolactomycin and okilactomycin, where the only structural difference is the exchange of the α-methoxy group for a α-methyl. This change showed that the α-methoxy group minutely impacted telomerase inhibition (0.5 μM to 2.1 μM), leading us to be content with the installation of alkyl groups at the C–3 position. We identified that a α-methyl group at C–3 provided the highest level of activity. Lastly, removal of the exocyclic methylene showed a dramatic loss of activity, indicating a covalent warhead remained critical for telomerase inhibition.
Table 2:
Structure-Activity Relationships of C–3 Group
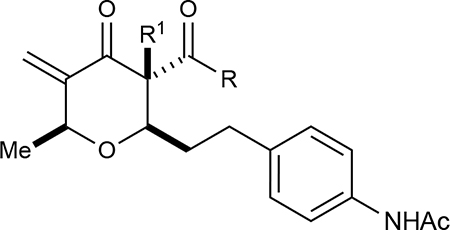 | ||||
|---|---|---|---|---|
| Compound | R | R1 | IC50 (μM) | ΔG Binding (kcal/mol) |
| 4a | 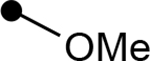 |
Me | 1.5 | −31.1 |
| 4k | 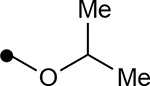 |
Me | 45 | −25.9 |
| 4l | 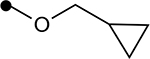 |
Me | 75 | −18.0 |
| 4m | Me | 35 | −27.9 | |
| 4n | Me | >100 | N/A | |
| 4o | Me | 85 | −16.5 | |
| 4p | Me | 25 | −23.1 | |
| 4q | 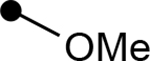 |
Et | 15 | −25.6 |
| 4r |  |
iPr | 62 | −22.1 |
While our synthetic sequence allowed for efficient access to racemic analogues, attempts to access enantioenriched chrologs via the β-hydroxy dioxinones through a variety of vinylogous Mukaiyama aldol approaches were unsuccessful.35 Fortunately, the utilization of a ketoreductase (KRED) mediated asymmetric reduction from the corresponding β-keto dioxinone was able to afford the desired β-hydroxy dioxinone in near quantitative yield and enantioselectivity (see Supporting Information).36 Utilizing this enantiopure β-hydroxy dioxinone, the enantiopure para N-acetylphenethyl analogue was accessed in 4 steps and showed an IC50 of 0.9 μM, a near 2-fold potency increase relative to the racemic analogue. Notably, the opposite antipode was also evaluated in the TRAP assay, providing an IC50 of 4.5 μM. With a first generation of chrologs synthesized and evaluated, we compared the computed binding energy to the observed biological activity, hoping to support our docking model and inform further compound design. The computed binding energy and IC50 values were plotted against each other and were found to be in good agreement (R2 = 0.81), strongly supporting our computational docking model.
With the necessary enantioenriched β-hydroxy dioxinone in hand, we subsequently re-evaluated our cheminformatics model to accommodate for this stereochemical information. Compounds were designed as previously mentioned using Pipeline Pilot.30 We then selected ~330 compounds that had favorable computed drug-likeness.31 We scored probes using in silico docking with the Glide module32 and CovDock33 of the Schrödinger suite to select and synthesize ~150 compounds, focusing on C–2 modification. SAR analysis was focused on increasing H-bonding interactions by targeting structure classes poised for late-stage diversification through amide bond formations and [3+2] cycloadditions to facilitate rapid library development (Fig. 3A). The goal in this 2nd generation library synthesis was to obtain a chrolactomycin analogue with an IC50 < 100 nM.
Fig. 3:

A) Workflow for probe synthesis. B) Optimized analogue NU-1 and des-exomethylene inactive analogue NU-2. C) Cheminformatics model for binding of NU-1. Telomerase inhibition of NU-1 as measured by TRAP assay in telomerase-positive D) cell lysates and E) cells in culture
Briefly, a series of simple amines were investigated as amide bond coupling partners with carboxylic acid pyranones 5 to obtain amide analogues 6 (see SI). Notably, analogues that had electron withdrawing groups on the aryl ring showed increased activity relative to unsubstituted analogues, indicating that there may be some H-bonding involved at that position. To attempt to take further advantage of H-bonding at the C–2, analogues that contained either a piperazine or 4-aminopiperidine linker were designed. We were pleased to find that this series of compounds showed higher activity in comparison to the simple amide analogues, indicating that the initial presumption that larger groups at this C–2 position that could participate in additional H-bonding was worth pursuing. Subsequently, we utilized alkyne containing pyranone 7 to start evaluating triazole analogues 8, where we were pleased to find that triazole analogues displayed higher activity in comparison to the amide analogues (see SI). Additionally, the same trend regarding analogues that had electron withdrawing groups on the aryl ring showing increased activity was observed (see Supporting Information for all relevant docking experiments). Optimization led to lead compound NU-1 that displayed an IC50 = 90 nM in TRAP assays in MCF7 cell lysate in vitro and an IC50 = 270 nM in MCF7 cells in culture (Fig. 3B). Additionally, we were pleased to find that NU-1 had the highest binding energy of all compounds synthesized and evaluated in the TRAP assay (–36.5 kcal/mol), further reinforcing our computational design approach (Fig. 3C). Notably, the electron withdrawing groups present on the aromatic ring of NU-1 showed the capability to pick up H-bonding interactions with K372 and K189 found in the TERT active site, lending additional evidence to our observation that substituents on the aromatic ring play a noticeable role in binding energy and telomerase inhibition.
These results were confirmed in four other telomerase-positive human cancer cell lysates and whole cells; A549 non-small cell lung cancer, ACHN renal carcinoma, MDA-MB-231 triple negative breast cancer and HeLa cervical cancer (Fig. 3D–E). To evaluate the role that the exocyclic methylene warhead plays in biological activity, we also synthesized the des-exocyclic methylene analogue NU-2. Notably, NU-2 displayed no appreciable inhibition of telomerase activity, indicating a covalent mechanism remained critical for telomerase inhibition.
NU-1 displayed satisfactory performance in plasma and microsomal stability assays, and showed favorable lipophilicity and CYP450 inhibition profiles (see the Supporting Information). To assess cysteine reactivity, we utilized the glutathione mimic N-acetyl cysteine (NAC) and measured the rate of chemical reactivity of NU-1 by HPLC.37 Briefly, 1 μM NU-1 was incubated with 5 mM of NAC (where both are at a concentration to best mimic cellular assay conditions) and the half-life was calculated by fitting to a pseudo-first-order kinetic rate equation relative to natural log transformed percent remaining NU-1. We were pleased to find that NU-1 demonstrated moderate thiol reactivity (t1/2 = 42 min) that is comparable to previously established guidelines for suitable reactivity windows for targeted covalent inhibitors.38
To evaluate if NU-1 was reactive with other potential cellular nucleophiles, NU-1 was incubated with N-acetyl serine and N-acetyl lysine, and the rate of chemical reactivity was measured by HPLC. No amino acid adducts were found, indicating that NU-1 is specific for thiol nucleophiles. Moreover, incubation of NU-1 with glycine showed no ketone-Schiff base formation. When NU-1 was incubated with tcTERT, proteolysis and mass spectrometry yielded a single modified peptide HPQDEIPYCGK, localizing the site of modification to the active site C390 and confirming a shared mechanism of action with chrolactomycin (see the Supporting Information).
To further evaluate NU-1, inhibition of telomerase activity was assessed by direct (non-PCR) primer extension assay. Human telomerase purified from telomerase-overexpressing HEK293T cells39 was treated with 1, NU-1 or NU-2. NU-1 and 1 inhibited telomere repeat synthesis in the low-micromolar range, while NU-2 displayed no inhibition (Fig. 4A), verifying the critical role of the enone warhead. Incubation of purified telomerase with 100 μM NU-1 displayed irreversible inhibition kinetics with a t1/2 of ~15 min at 23 °C, with complete inhibition observed after ~6 h (Fig. 4B). We then assessed inhibition of endogenous telomerase in MCF-7 and A549 cells after culture in the presence of 10 μM NU-1 or NU-2 control for 24 h. NU-1 completely eliminated endogenous telomerase activity, whereas NU-2 was indistinguishable from the DMSO (vehicle) control (Fig. 4C). The difference between the telomerase inhibition observed for the TRAP assay (IC50 = 500 nM for 1 and IC50 = 90 nM for NU-1) and the primer extension assay (1 ~ 10x more active than NU-1) could potentially be due to artifacts that are impacting the PCR amplification step of the TRAP assay such as impurities in the cell lysate40 or that either NU-1 or 1 inhibit Taq polymerase at differing levels. Notably, even a modest impact on Taq polymerase activity would have greatly augmented effects on the assay due to PCR amplification. This contrasts with direct primer extension assay, which is carried out with purified telomerase enzyme and the specific reason we employed this method. In addition, the determination of true “potency” as traditionally defined is difficult using the primer extension assay because the system is not at equilibrium; only when all activity is killed is the system at equilibrium (i.e., every molecule of hTERT is covalently inhibited).
Fig. 4:

Direct telomerase activity assays: reaction of 5’-biotin-CTAGACCTGTCATCA(TTAGGG)3 (S) with human telomerase and dTTP, dATP, and α−32P-dGTP. (A) Incubation of purified telomerase with varying concentrations of chrolactomycin 1, NU-1, or control compound NU-2; DMSO is the vehicle control. Asterisk indicates a 32Plabelled recovery/loading control DNA. (B) Time course of irreversible inhibition of purified telomerase by NU-1. (C) Endogenous telomerase activity from A549 or MCF7 cells after 24 h treatment with DMSO (vehicle control), NU-1, or NU-2.
To evaluate the impacts of telomerase inhibition on cell proliferation, MCF7 cells were cultured in media containing 1, NU-1 or NU-2. Both exomethylene-bearing compounds conferred similar dose-responsive loss of viability (IC50 of 21 μM and 27 μM respectively), while NU-2 had no effect. A similar pattern was observed in A549, ACHN, MDA-MB-231, and HeLa telomerase-positive cells. By contrast, like DMSO control or 2, treating the telomerase-negative cancer cell lines Saos-2 or VA-1341 with up to 100 μM 1 or NU-1 did not affect viability (Fig. 5A). These results are in-line with previous observations demonstrating that acute hTERT or hTR knockdown and telomerase inhibition results in a decrease in cell proliferation and viability independent of telomere length and integrity.42
Fig. 5:

A) Cell viability measurement of NU-1 in telomerase positive and negative cell lines. B) Different time points for cell viability-washout experiments. C) Different time points for TRAP-washout experiments. D) Washout experiments with non-covalent telomerase inhibitors
Consistent with a covalent mechanism, washing the treated cells to remove free NU-1 and then incubation in fresh culture media did not restore viability or telomerase activity even after 24 h (Fig. 5B–C). Comparing NU-1 to the reversible telomerase inhibitors BIBR1532 (IC50 ~1 μM) or MST-312 (IC50 ~1 μM) in MCF7 cells revealed qualitatively similar effects on viability, though NU-1 displayed the lowest apparent IC50.However, when treated MCF7 cells were washed and allowed to recover for 4 h, telomerase activity was restored in cells treated with BIBR1532 or MST-312 (Fig. 5D) but remained blocked in cells treated with NU-1.
To assess the full extent of protein labeling in telomerase positive cells (and thereby potential off-target reactivity that may complicate biological analysis), we utilized a competitive gel imaging technique employed in competitive activity-based protein profiling (ABPP)43, where compounds are assayed for their ability to block fluorescent probe labeling. A key advantage of competitive ABPP is that it allows for the evaluation of potency and selectivity of inhibitors without modulating the core structure of the parent compound.
Briefly, the MCF-7 proteome was dose-response treated with NU-1 or chrolactomycin (10 nM to 100 μM) for 1 hr, followed by treatment with a thiol reactive fluorescein-iodoacetamide (10 μM, 1 hr). The reactions were then quenched, separated by SDS-PAGE, and fluorescein-labeled proteins were detected by in-gel fluorescence scanning. Notably, we observed minimal loss of protein banding and streaking, where loss of protein banding is indicative of NU-1 and chrolactomycin having off-target reactivity and labeling proteins prior to fluoescein-iodoacetamide labeling (Fig. 6A–C).
Fig. 6:

A) Workflow for competitive gel analysis of NU-1/chrolactomycin with 5-iodoacetamido-fluorescein. Gel image of B) NU-1 and C) chrolactomycin
CONCLUSIONS
A new and effective class of telomerase inhibitors has been developed inspired by the natural product chrolactomycin, a covalent inhibitor of human telomerase reverse transcriptase (hTERT) catalytic activity. Rational compound design resulted in the lead compound NU-1, a simplified analogue accessible in four synthetic steps that binds to hTERT and directs an exomethylene group to react with an active-site cysteine, leading to irreversible enzyme inhibition. Despite concerns about potential reactivity with cellular nucleophiles, NU-1 had negligible effects on telomerase-negative cancer cells, and has shown to have comparable thiol reactivity to known targeted covalent inhibitors. This study provides proof-of-principle for sensitive, covalent chemical probes to dissect telomerase functions in cells and lead compounds to validate hTERT as a target to enhance conventional genotoxic cancer therapies.
METHODS
General Information
Chrolactomycin was isolated from Actinomycete Actinospica (NAICONS Laboratory, Milan Italy). The telomerase inhibitors BIBR1532, MST-312, costunolide and helenalin were purchased from Sigma Aldrich and used without further purification. All cell lines used were purchased from ATCC (MCF-7, MDA-MB-231, A549, HeLa, ACHN, Saos-2, VA-13) and tested for mycoplasma every six months. All cell lines were cultured according to the manufacturer’s instructions at 37 °C in 5 % CO2.
In-silico studies
Utilizing the ligand based enumeration techniques implemented in Pipeline pilot platform44 for library generation and considering a truncated version of chrolactomycin as the reference ligand ~10,000 drug-like structures had been generated. These structures had been filtered out using the PAINS filiters45 and 330 structures were selected for covalent docking studies. The CovDock46 (covalent docking) module available in Schrodinger suite has five distinct steps involved in the docking process of the ligand and the protein of interest. The steps involved are i) non-covalent docking; ii) receptor sampling; iii) covalent bond formation; iv) refinement; and v) apparent affinity scoring. Furthermore, CovDock also has two sub-modules such as CovDock-Screen and CovDock-Thorough which is coupled with the binding energy computation using MM-GBSA47 (Molecular Mechanical Generalized Born Surface Area Continuum Solvent Approach).
Ligand preparation
All the 330 filtered structures were subjected to ligand preparation panel available in Schrodinger at pH = 7.4±1 to obtain best initial geometry for all the structures. The ligands van der Waals radii were scaled to 0.8 Å with partial atomic charges < 0.15 esu.
Grid generation
The docking engine Glide available in Schrodinger has been built upon grid based techniques. In order to generate a grid box, we considered the apo TERT structure available in protein database with the accession code 3DU5. Before generating the grid box, the protein structure was processed through the protein preparation module in order to correct irrelevant side chains, added missing atoms, eliminated partial occupant rotamers, fixed the undesired orientation of Asn, Gln and His residues, and finally replaced the “b” values by the optimized potential for liquid simulations (OPLS3) charges.48 Since this was an apo structure, we applied the SiteMap algorithm49 to identify putative small molecule ligand binding pocket. After identifying the ligand binding site a grid box of 10×10×10 Å was constructed which contained the reactive C390 residue along with few critical residues such as K189, K249, K372, and K406.
CovDock
The very first step in CovDock was to define the type of chemical reaction that would occur in between the reactive residue and the ligand. We selected Michael Addition as the chemical reaction and the engine identified the atom in the electrophile of the ligand set where the reaction had to occur. Then the reactive residue C390 was selected and CovDock-Screen engine was used to screen the ligand set. This screening resulted in 200 hits and we then carried out CovDock-Thorough to these hits in order to compute the binding energy using MMGBSA. Based on the binding energy and the docked poses 150 structures were selected for synthetic effort.
Protease Digestion and Mass Spectrometry Analysis
tcTERT (50 μg, ~600 pmol) was treated with chrolactomycin, NU-1, helenalin, costunolide or DMSO for 1 hr at 37 °C. The reaction was diluted with ammonium bicarbonate buffer (pH 8.0), then reduced for 30 min at 56 °C with 10 mM DTT. After cooling to 23 °C, the protein was alkylated with 25 mM iodoacetamide for 30 min at 23 °C in the dark, followed by trypsin digestion (2.5 μg, Promega) overnight at 37 °C. Digested peptides (~2 pmol) were injected onto a self-packed precolumn (4 cm POROS10R2) and eluted into the mass spectrometer (LTQ Orbitrap Velos, Thermo Fisher). Peptides were subjected to MS2 by CAD (electron multiplier detection, relative collision energy 35%, q = 0.25).
Telomeric Repeat Amplification Protocol Assay
The telomeric repeat amplification protocol (TRAP) assay was performed using the TRAPeze RT Telomerase Detection Kit (EMD Millipore) and iTaq DNA polymerase (Bio-Rad) according to the manufacturer’s protocol.
Cell Lysate TRAP Analysis
MCF-7, HeLa, ACHN, MDA-MB231 and A549 cells were cultured in DMEM media supplemented with 10% Fetal Bovine Serum (FBS), 1% pen/strep and with 5% CO2 and 37 °C. Cells were plated in 2 x T75 flasks and cultured for 3 days. Flasks were rinsed twice with PBS and treated with 0.05% trypsin-EDTA (Thermo Fisher) to detach cells. The cell suspension was washed twice in PBS and the cell pellet was resuspended in 200 μl of 1X CHAPS Lysis Buffer (10 mM Tris-HCl, pH 7.5; 1 mM MgCl2; 1 mM EGTA; 0.1 mM benzamidine; 5 mM β-mercaptoethanol; 0.5% CHAPS; 10% glycerol)/106 cells, and incubated on ice for 30 min. Cell extracts were transferred to microcentrifuge tubes, 200 μl/tube, and centrifuged at 12,000 x g for 20 min at 4 °C to remove cell debris. Stock solutions of compounds were made in DMSO with a concentration of 100 mM and a 6-fold dilution was performed in DMSO in a 96-well plate to produce compound solutions of 100, 10, 1, 0.1, 0.01, and 0.001 mM (1000X of the final concentration). The 1000X stock solutions of the compound samples were transferred to a 96-well plate using the Echo 550 (Labcyte) and then diluted with the cell extracts to yield final compound concentrations of 100, 10, 1, 0.1, 0.01, and 0.001 μM. The plate was covered and shaken at room temperature for 30 min. For TRAPeze reactions, a “master mix” was prepared according to the TRAPeze protocol, containing 5 μl of the TRAPeze reaction mix; 0.4 μl Taq polymerase; 17.6 μl PCR grade, nuclease-free water. The master mix and treated cell extracts were transferred using the Mosquito Crystal Liquid Handling System (TTP Labtech) into separate wells in a white, 384-well RT-PCR plate. Each well contained 1.2 μl of the master mix and 1.2 μl of the treated cell extract to give a final volume of 2.4 μl, with two duplicates for each sample. The RT-PCR plate was then sealed and centrifuged for 5 min. A sample containing only CHAPS Lysis Buffer was included as negative control. The TSR8 standards were included to generate a standard curve. Samples were amplified in a CFW384 RT-PCR (BioRad) detection system with PCR parameters of 30 min at 30 °C for 1 cycle, 2 min at 95 °C for 1 cycle, and then 45 cycles of 15 s at 94 °C, 60 s at 59 °C, and 10 s at 45 °C. Real-time fluorescent data was obtained during the 10 s at 45 °C step. The number of cycles at which the fluorescence level reached a threshold (Ct) was averaged between the duplicates for each sample. Using a calibration curve obtained from the TSR8 standards, a linear equation was obtained to convert the Ct values into arbitrary telomerase activity units, which were plotted as a function of compound concentration to construct dose-response curves and calculate IC50.
Cultured Cells TRAP Analysis
MCF-7, HeLa, ACHN, MDA-MB231 and A549 cells were cultured in DMEM media supplemented with 10% Fetal Bovine Serum (FBS), 1% pen/strep and with 5% CO2 and 37 °C. Cells were plated in 96 well plates and allowed to attach for 24 h. Media was removed, and fresh media containing various concentrations of compound was added, and the plate was incubated for 24 h. Media was removed, the cells were washed 2 times with PBS, and the cells were lysed in 20 μl of 1X CHAPS Lysis Buffer (10 mM Tris-HCl, pH 7.5; 1 mM MgCl2; 1 mM EGTA; 0.1 mM benzamidine; 5 mM β-mercaptoethanol; 0.5% CHAPS; 10% glycerol). The cell extract was then centrifuged at 12,000 x g for 20 min at 4 °C to remove cell debris. Note: “fresh” media constitutes media supplemented with 10% Fetal Bovine Serum (FBS) and 1% pen/strep that has not been previously used for cell culture (e.g., not conditioned media). TRAP analysis was conducted as above.
Compound Washout TRAP analysis
MCF-7 cells were plated in 96 well plates and allowed to attach for 24 h. Media was removed, and fresh media containing various concentrations of compound was added, and the plate was incubated for 24 h. Media was removed, cells were washed 2 times with PBS, and fresh media without compound was added. Cells were incubated at 37 °C for an additional “X” hours. Media was removed, the cells were washed 2X with PBS, and the cells were lysed in 20 μl of 1X CHAPS Lysis Buffer (10 mM Tris-HCl, pH 7.5; 1 mM MgCl2; 1 mM EGTA; 0.1 mM benzamidine; 5 mM β-mercaptoethanol; 0.5% CHAPS; 10% glycerol). The cell extract was then centrifuged at 12,000g for 20 min at 4 °C to remove cell debris. Note: “fresh” media constitutes media supplemented with 10% Fetal Bovine Serum (FBS) and 1% pen/strep that has not been previously used for cell culture (e.g., not conditioned media”). TRAP analysis was conducted as above.
Cell Viability Measurements
MCF-7, HeLa, MDA-MB231 and A549 cells were cultured in DMEM media supplemented with 10% Fetal Bovine Serum (FBS), 1% pen/strep and with 5% CO2 and 37 °C. Cells were plated in 96 well plates and allowed to attach for 24 h. Media was removed, and fresh media containing various concentrations of compound was added, and the plate was incubated for 24 h. The plate was removed from the incubator and allowed to equilibrate to room temperature for 30 min. 50 μl of freshly prepared CellTiter-Glo (Promega) reagent was added, and the plate was mixed on an orbital shaker for 10 min to induce cell lysis and stabilize luminescent signal. The luminescence was recorded on a PerkinElmer Enspire multimode plate reader.
Compound Washout Cell Viability Measurements
MCF-7 cells were plated in 96 well plates and allowed to attach for 24 h. Media was removed, and fresh media containing various concentrations of compound was added, and the plate was incubated for 24 h. Media was removed, cells were washed 2 times with PBS, and fresh media without compound was added. Cells were incubated at 37 °C for an additional “X” h and analyzed as above. Note: “fresh” media constitutes media supplemented with 10% Fetal Bovine Serum (FBS) and 1% pen/strep that has not been previously used for cell culture (e.g., not conditioned media).
Primer-Extension Telomerase Activity Assays
Telomerase activity assays were performed as previously described.39b Experiments in Figures 4A and 4B utilized telomerase immunopurified with the hTERT antibody as described in the reference above; telomerase stock solution was at ~10 nM in “telomerase buffer”: 50 mM HEPES-KOH (pH 8); 300 mM KCl; 2 mM MgCl2; 10% v/v glycerol; 0.1% v/v Triton X-100; 1 mM DTT.
For Figure 4A, extension reactions were performed in a total volume of 100 μL. NU-1 or NU-2 were prepared in DMSO at 20× final concentration (2000 μM, 200 μM, or 20 μM). 5 μL of DMSO or the respective compound/DMSO dilution was placed in a tube, followed by 45 μL of a solution containing: 2.2 mM MgCl2; 1.1 mM DTT; 2.2 mM dTTP, 2.2 mM dATP, 22 μM dGTP, ~5 μCi α−32P-dGTP (Perkin Elmer), and the DNA substrate 5’-biotin-CTAGACCTGTCATCA(TTAGGG)3-3’ (2.2 μM). Reactions were initiated by addition of 50 μL telomerase solution, providing final concentrations of: ~5 nM telomerase; 25 mM HEPES-KOH; 150 mM KCl; 2 mM MgCl2; 0.05% v/v Triton X-100; 5% v/v glycerol; 1 mM DTT; 5% v/v DMSO; 1 mM dATP/dTTP; 10 μM dGTP. Reactions were incubated at 37 °C for 8 h, then quenched with addition of 400 μL STOP solution: 10 mM Tris-HCl (pH 7.5); 2 M NaCl; 1 mM EDTA; ~5,000 cpm 5’-α−32P-(CTAGACCTGTCATCA)2-biotin-3’ (recovery/loading control). Solutions were incubated with 30 μL Dynabeads M-280 Streptavidin suspension (Thermo-Fisher, suspension used as is) with rotation overnight. Beads were recovered by Dyna-magnet and washed twice with 500 μL [10 mM Tris-HCl (pH 7.5); 2 M NaCl; 1 mM EDTA]; followed by 300 μL [10 mM Tris-HCl (pH 7.5); 1 mM EDTA]. Beads were suspended in 30 μL [9:1 v/v Formamide/TBE:D-biotin solution (5 mM in TE)]. Samples were denatured at 80 °C for 10 min, and the product solutions recovered on the Dyna-magnet. Products (6 μL) were electrophoresed over a 10% denaturing acrylamide/8 M urea gel at 75 Watts until bromophenol blue was ~3/4 down the gel (~50 min).
For Figure 4B, two 500 μL binding reactions were prepared, each containing a 25 μL aliquot of either NU-1 or NU-2 (2 mM stock in DMSO = 100 μM final concentration) and ~10 nM telomerase in telomerase buffer. Immediately upon mixing (t = 0), and at t = 10, 20, 40, 60, 120, 240, and 360 min, 50 μL of each reaction was combined with 50 μL extension mix [2 mM MgCl2, 1 mM DTT, 2 mM dTTP, 2 mM dATP, 20 μM dGTP, ~5 μCi α−32P-dGTP (Perkin Elmer), and the DNA substrate 5’-biotinCTAGACCTGTCATCA(TTAGGG)3-3’ (2 μM)]. All reactions were incubated at 37 °C overnight, and then processed as described above.
For Figure 4C, the control or treated cells were prepared and collected as pellets of 5 × 106 cells each at NW and pellets were shipped to CMRI on dry ice. Frozen cell pellets were thawed on wet ice and suspended in 1 mL telomerase buffer supplemented with 1 mM PMSF. Endogenous telomerase was immunopurified and assayed as described.
CYP450 Inhibition Assay
Assay was conducted using the commercially available Vivid CYP1A2 Screening Kit (Thermo Fisher) according to the manufacturer’s protocol. α-napthoflavone and DMSO were used as a positive and negative control respectively based on the manufacturer’s protocol.
Non-Cell-Based Assay for Drug Transport (PAMPA assay)
Assay was conducted using the commercially available parallel artificial membrane permeation assay (PAMPA, Millipore Sigma) according to the manufacturer’s protocol.
Liver Microsomal Stability Assay
Assay was conducted using the Corning UltraPool HLM 150 Mixed Gender microsomal kit (Corning Life Sciences) according to the manufacturer’s protocol.
N-Acetyl Cysteine, N-Acetyl Lysine and N-Acetyl Serine Kinetic Studies
Assay was conducted as described with minor modifications.37 Briefly, a 1 mM stock solution of NU-1 with internal standard (phenacetin) in DMSO was added to a 50 mM solution of either N-acetyl cysteine, N-acetyl lysine or N-acetyl serine in 67 mM phosphate buffer (pH 7.4), as well as added to a solution of 67 mM phosphate buffer (pH 7.4) control. Samples were taken every 2 min for 2 h and quenched immediately with MeOH and centrifuged upon sampling. Samples were placed on a Waters Acquity UPLC-MS system and measured for the ratio of NU-1 to internal standard intensity to calculate the amount of NU-1 present at each time point relative to the amount of NU-1 at the first injection of the compound in buffer. Assay was conducted in triplicate.
Schiff-Base Measurement
A 1 mM stock solution of NU-1 in DMSO-d6 was added to a solution of 5 mM solution of glycine in D2O in an NMR tube. The mixture was analyzed by 1H NMR over 48 hours to analyze formation of Schiff-bases with the ketone of NU-1.
Kinetic Solubility Measurements of NU-1
Solubility readings were taken on a Synergy HTX plate reader using a 384 well plate format. Total volume of solution 50 μl with a final DMSO concentration at 1 % v/v in PBS. Solubility curve was established using concentration ranges from 1–500 μM that were prepared by serial dilution of NU-1 (50 mM in DMSO). Measurements were made in triplicate.
Competitive Gel Image Analysis
MCF-7 cells were cultured in DMEM media supplemented with 10% Fetal Bovine Serum (FBS), 1% pen/strep and with 5% CO2 and 37 °C. Cells were plated cultured for 3 days. Flasks were rinsed twice with PBS and treated with 0.05% trypsin-EDTA (Thermo Fisher) to detach cells. The cell suspension was washed twice in PBS and the cell pellet was resuspended in 200 μl of M-PER extraction reagent (Thermo Fisher)/106 cells, and incubated for 30 min. Cell extracts were transferred to microcentrifuge tubes, 200 μl/tube, and centrifuged at 12,000 x g for 20 min to remove cell debris. The corresponding lysate was then further diluted to a total protein concentration of 2 mg/mL as measured by Nanodrop (Thermo Fisher). 48 μL of MCF-7 lysate was added to twelve microcentrifuge tubes (six per compound), followed by treatment of cell lysate with 1 μL solution of NU-1/chrolactomycin (equating to doses ranging from 10 nM to 100 μM). The samples were incubated for 1 hr, followed by addition of 1 μL of 5-iodoacetamido-fluorescein to a final concentration of 10 μM. The samples were again incubated for 1 hr, followed by quenching the reaction with 15 μL NuPAGE LDS Sample Buffer (4x). The samples were heated in a water bath at 70 °C for 10 minutes, followed by loading 30 μL of the samples onto a 10% NuPAGE Bis-Tris gel with 20 μL of a 2:1 mixture of PageRuler Prestained protein ladder (Thermo Fisher). SDS-page was ran for 30 min at constant voltage (250 V) using a Mini Gel Tank (Thermo Fisher) and a PowerEase 300W (Thermo Fisher) power supply. Upon completion, the gels were removed from their cases, rinsed in DI water for 5 minutes, and imaged using a IBright FL1000 imager (Thermo Fisher).
Supplementary Material
ACKNOWLEDGMENT
R.C.B. was supported in part by the Chicago Cancer Baseball Charities at the Lurie Cancer Center of Northwestern University and the NIGMS Chemical-Biology Interface T32 Training Grant GM105538. Y.L. and S.J.K. were supported by NIH R01 CA217182. S.B.C acknowledges support from the Ernest & Piroska Major Foundation and Kids Cancer Alliance (Australia). The authors thank Dr. Morianna Iorio at NAICONS Laboratories, Milan Italy, for providing Actinomycete Actinospica for chrolactomycin isolation and Dr. Emmanuel Skordalakes at the Wistar Institute, Philadelphia PA, for generously providing tcTERT.
Footnotes
Competing Interests
A provisional composition of matter patent for NU-1 and related analogues has been filed. The authors declare no other competing financial interests.
ASSOCIATED CONTENT
The Supporting Information is available free of charge on the ACS Publications website.
REFERENCES
- (1).Moyzis RK, Buckingham JM, Cram LS, Dani M, Deaven LL, Jones MD, Meyne J, Ratliff RL, and Wu JR (1988) A highly conserved repetitive DNA sequence, (TTAGGG)n, present at the telomeres of human chromosomes, Proc Natl Acad Sci U S A 85, 6622–6626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).a de Lange T (2005) Shelterin: the protein complex that shapes and safeguards human telomeres, Genes Dev 19, 2100–2110; [DOI] [PubMed] [Google Scholar]; b de Lange T (2018) Shelterin-Mediated Telomere Protection, Annu. Rev. Genet. 52, 223–247. [DOI] [PubMed] [Google Scholar]
- (3).a Hayflick L, and Moorhead PS (1961) The serial cultivation of human diploid cell strains, Exp. Cell. Res. 25, 585–621; [DOI] [PubMed] [Google Scholar]; b Hayflick L (1965) The limited in vitro lifetime of human diploid cell strains, Exp. Cell. Res 37, 614–636; [DOI] [PubMed] [Google Scholar]; c Harley CB, Futcher AB, and Greider CW (1990) Telomeres Shorten during Aging of Human Fibroblasts, Nature 345, 458–460. [DOI] [PubMed] [Google Scholar]
- (4).a Greider CW, and Blackburn EH (1985) Identification of a specific telomere terminal transferase activity in tetrahymena extracts, Cell 43, 405–413; [DOI] [PubMed] [Google Scholar]; b Greider CW, and Blackburn EH (1987) The telomere terminal transferase of tetrahymena is a ribonucleoprotein enzyme with two kinds of primer specificity, Cell 51, 887–898; [DOI] [PubMed] [Google Scholar]; c Greider CW, and Blackburn EH (1989) A Telomeric Sequence in the Rna of Tetrahymena Telomerase Required for Telomere Repeat Synthesis, Nature 337, 331–337. [DOI] [PubMed] [Google Scholar]
- (5).a Lingner J, Hughes TR, Shevchenko A, Mann M, Lundblad V, and Cech TR (1997) Reverse Transcriptase Motifs in the Catalytic Subunit of Telomerase, Science 276, 561–567; [DOI] [PubMed] [Google Scholar]; b Nakamura TM, Morin GB, Chapman KB, Weinrich SL, Andrews WH, Lingner J, Harley CB, and Cech TR (1997) Telomerase catalytic subunit homologs from fission yeast and human, Science 277, 955–959. [DOI] [PubMed] [Google Scholar]
- (6).a Feng J, Funk WD, Wang SS, Weinrich SL, Avilion AA, Chiu CP, Adams RR, Chang E, Allsopp RC, Yu J, and al e. (1995) The RNA component of human telomerase, Science 269, 1236–1241; [DOI] [PubMed] [Google Scholar]; b Weinrich SL, Pruzan R, Ma LB, Ouellette M, Tesmer VM, Holt SE, Bodnar AG, Lichtsteiner S, Kim NW, Trager JB, Taylor RD, Carlos R, Andrews WH, Wright WE, Shay JW, Harley CB, and Morin GB (1997) Reconstitution of human telomerase with the template RNA component hTR and the catalytic protein subunit hTRT, Nat. Genet. 17, 498–502; [DOI] [PubMed] [Google Scholar]; c Beattie TL, Zhou W, Robinson MO, and Harrington L (1998) Reconstitution of human telomerase activity in vitro, Curr. Biol. 8, 177–180. [DOI] [PubMed] [Google Scholar]
- (7).Wright WE, Piatyszek MA, Rainey WE, Byrd W, and Shay JW (1996) Telomerase activity in human germline and embryonic tissues and cells, Dev Genet 18, 173–179. [DOI] [PubMed] [Google Scholar]
- (8).Kim NW, Piatyszek MA, Prowse KR, Harley CB, West MD, Ho PLC, Coviello GM, Wright WE, Weinrich SL, and Shay JW (1994) Specific Association of Human Telomerase Activity with Immortal Cells and Cancer, Science 266, 2011–2015. [DOI] [PubMed] [Google Scholar]
- (9).a Park J-I, Venteicher AS, Hong JY, Choi J, Jun S, Shkreli M, Chang W, Meng Z, Cheung P, Ji H, McLaughlin M, Veenstra TD, Nusse R, McCrea PD, and Artandi SE (2009) Telomerase modulates Wnt signalling by association with target gene chromatin, Nature 460, 66–72; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Ghosh A, Saginc G, Leow SC, Khattar E, Shin EM, Yan TD, Wong M, Zhang Z, Li G, Sung W-K, Zhou J, Chng WJ, Li S, Liu E, and Tergaonkar V (2012) Telomerase directly regulates NF-κB-dependent transcription, Nat. Cell Biol. 14, 1270. [DOI] [PubMed] [Google Scholar]
- (10).a Holt SE, Glinsky VV, Ivanova AB, and Glinsky GV (1999) Resistance to apoptosis in human cells conferred by telomerase function and telomere stability, Mol. Carcinog. 25, 241–248; [PubMed] [Google Scholar]; b Bermudez Y, Erasso D, Johnson NC, Alfonso MY, Lowell NE, and Kruk PA (2006) Telomerase confers resistance to caspase-mediated apoptosis, Clin Interv Aging 1, 155–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).a Sharma GG, Gupta A, Wang H, Scherthan H, Dhar S, Gandhi V, Iliakis G, Shay JW, Young CSH, and Pandita TK (2003) hTERT associates with human telomeres and enhances genomic stability and DNA repair, Oncogene 22, 131–146; [DOI] [PubMed] [Google Scholar]; b Masutomi K, Possemato R, Wong JMY, Currier JL, Tothova Z, Manola JB, Ganesan S, Lansdorp PM, Collins K, and Hahn WC (2005) The telomerase reverse transcriptase regulates chromatin state and DNA damage responses, Proc. Natl. Acad. Sci. U.S.A. 102, 8222–8227 [DOI] [PMC free article] [PubMed] [Google Scholar]; c Doksani Y, and de Lange T (2014) The role of double-strand break repair pathways at functional and dysfunctional telomeres, Cold Spring Harb Perspect Biol 6, a016576–a016576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Perera ON, Sobinoff AP, Teber ET, Harman A, Maritz MF, Yang SF, Pickett HA, Cesare AJ, Arthur JW, MacKenzie KL, and Bryan TM (2019) Telomerase promotes formation of a telomere protective complex in cancer cells, Sci. Adv [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).a Rezler EM, Bearss DJ, and Hurley LH (2002) Telomeres and telomerases as drug targets, Curr. Opin. Pharmacol. 2, 415–423; [DOI] [PubMed] [Google Scholar]; b Neidle S, and Parkinson G (2002) Telomere maintenance as a target for anticancer drug discovery, Nat. Rev. Drug Discov. 1, 383–393; [DOI] [PubMed] [Google Scholar]; c Ouellette MM, Wright WE, and Shay JW (2011) Targeting telomerase-expressing cancer cells, J. Cell. Mol. Med. 15, 1433–1442; [DOI] [PMC free article] [PubMed] [Google Scholar]; d Sekaran V, Soares J, and Jarstfer MB (2014) Telomere maintenance as a target for drug discovery, J. Med. Chem. 57, 521–538 [DOI] [PubMed] [Google Scholar]; e Arndt GM, and MacKenzie KL (2016) New prospects for targeting telomerase beyond the telomere, Nat. Rev. Canc 16, 508. [DOI] [PubMed] [Google Scholar]
- (14).a Damm K, Hemmann U, Garin-Chesa P, Hauel N, Kauffmann I, Priepke H, Niestroj C, Daiber C, Enenkel B, Guilliard B, Lauritsch I, Müller E, Pascolo E, Sauter G, Pantic M, Martens UM, Wenz C, Lingner J, Kraut N, Rettig WJ, and Schnapp A (2001) A highly selective telomerase inhibitor limiting human cancer cell proliferation, The EMBO Journal 20, 6958–6968; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Pascolo E, Wenz C, Lingner J, Hauel N, Priepke H, Kauffmann I, Garin-Chesa P, Rettig WJ, Damm K, and Schnapp A (2002) Mechanism of Human Telomerase Inhibition by BIBR1532, a Synthetic, Non-nucleosidic Drug Candidate, J. Biol. Chem. 277, 15566–15572; [DOI] [PubMed] [Google Scholar]; c El-Daly H, Kull M, Zimmermann S, Pantic M, Waller CF, and Martens UM (2005) Selective cytotoxicity and telomere damage in leukemia cells using the telomerase inhibitor BIBR1532, Blood 105, 1742–1749; [DOI] [PubMed] [Google Scholar]; d Mueller S, Hartmann U, Mayer F, Balabanov S, Hartmann JT, Brummendorf TH, and Bokemeyer C. (2007) Targeting telomerase activity by BIBR1532 as a therapeutic approach in germ cell tumors, Invest New Drug 25, 519–524. [DOI] [PubMed] [Google Scholar]
- (15).Roth A, Harley CB, and Baerlocher GM (2010) Imetelstat (GRN163L) - Telomerase-Based Cancer Therapy, Recent Results Canc 184, 221–234. [DOI] [PubMed] [Google Scholar]
- (16).a Kim M-Y, Vankayalapati H, Shin-ya K, Wierzba K, and Hurley LH (2002) Telomestatin, a Potent Telomerase Inhibitor That Interacts Quite Specifically with the Human Telomeric Intramolecular G-Quadruplex, J. Am. Chem. Soc. 124, 2098–2099; [DOI] [PubMed] [Google Scholar]; b Burger AM, Dai F, Schultes CM, Reszka AP, Moore MJ, Double JA, and Neidle S (2005) The G-Quadruplex-Interactive Molecule BRACO-19 Inhibits Tumor Growth, Consistent with Telomere Targeting and Interference with Telomerase Function, Cancer Res. 65, 1489; [DOI] [PubMed] [Google Scholar]; c De Cian A, Cristofari G, Reichenbach P, De Lemos E, Monchaud D, Teulade-Fichou MP, Shin-Ya K, Lacroix L, Lingner J, and Mergny JL (2007) Reevaluation of telomerase inhibition by quadruplex ligands and their mechanisms of action, Proc. Natl. Acad. Sci. U S A 104, 17347–17352; [DOI] [PMC free article] [PubMed] [Google Scholar]; d Neidle S (2010) Human telomeric G-quadruplex: The current status of telomeric G-quadruplexes as therapeutic targets in human cancer, FEBS Journal 277, 1118–1125; [DOI] [PubMed] [Google Scholar]; e Li Q., Xiang JF, Zhang H, and Tang YL (2012) Searching Drug-Like Anti-cancer Compound(s) Based on G-Quadruplex Ligands, Curr. Pharm. Desig 18, 1973–1983. [DOI] [PubMed] [Google Scholar]
- (17).Ganesan K, and Xu B (2018) Telomerase Inhibitors from Natural Products and Their Anticancer Potential, Int. J. Mol. Sci. 19, 1–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).a Naasani I, Seimiya H, and Tsuruo T (1998) Telomerase inhibition, telomere shortening, and senescence of cancer cells by tea catechins, Biochem. Biophys. Res. Commun. 249, 391–396; [DOI] [PubMed] [Google Scholar]; b Seimiya H, Oh-hara T, Suzuki T, Naasani I, Shimazaki T, Tsuchiya K, and Tsuruo T (2002) Telomere shortening and growth inhibition of human cancer cells by novel synthetic telomerase inhibitors MST-312, MST-295, and MST-1991, Mol Cancer Ther 1, 657–665. [PubMed] [Google Scholar]
- (19).Huang P-R, Yeh Y-M, and Wang T-CV (2005) Potent inhibition of human telomerase by helenalin, Cancer Lett. 227, 169–174. [DOI] [PubMed] [Google Scholar]
- (20).Choi S-H, Im E, Kang HK, Lee J-H, Kwak H-S, Bae Y-T, Park H-J, and Kim ND (2005) Inhibitory effects of costunolide on the telomerase activity in human breast carcinoma cells, Cancer Lett. 227, 153–162. [DOI] [PubMed] [Google Scholar]
- (21).a Harvey AL (2008) Natural products in drug discovery, Drug Discov. Today 13, 894–901; [DOI] [PubMed] [Google Scholar]; b Harvey AL, Edrada-Ebel R, and Quinn RJ (2015) The re-emergence of natural products for drug discovery in the genomics era, Nat. Rev. Drug Discov 14, 111; [DOI] [PubMed] [Google Scholar]; c Thomford EN, Senthebane AD, Rowe A, Munro D, Seele P, Maroyi A, and Dzobo K (2018) Natural Products for Drug Discovery in the 21st Century: Innovations for Novel Drug Discovery, Int. J. Mol. Sci 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).a Johnson DS, Weerapana E, and Cravatt BF (2010) Strategies for discovering and derisking covalent, irreversible enzyme inhibitors, Future Med. Chem. 2, 949–964; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Jackson PA, Widen JC, Harki DA, and Brummond KM (2017) Covalent Modifiers: A Chemical Perspective on the Reactivity of α,β-Unsaturated Carbonyls with Thiols via Hetero-Michael Addition Reactions, J. Med. Chem. 60, 839–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Singh J, Petter RC, Baillie TA, and Whitty A (2011) The resurgence of covalent drugs, Nat. Rev. Drug Discov. 10, 307–317. [DOI] [PubMed] [Google Scholar]
- (24).Byrd JC, Furman RR, Coutre SE, Flinn IW, Burger JA, Blum KA, Grant B, Sharman JP, Coleman M, Wierda WG, Jones JA, Zhao W, Heerema NA, Johnson AJ, Sukbuntherng J, Chang BY, Clow F, Hedrick E, Buggy JJ, James DF, and O’Brien S (2013) Targeting BTK with Ibrutinib in Relapsed Chronic Lymphocytic Leukemia, N. Eng. J. Med 369, 32–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).a Nakai R, Kakita S, Asai A, Cniba S, Akinaga S, Mizukami T, and Yamashita Y (2001) Chrolactomycin, a Novel Antitumor Antibiotic Produced by Streptomyces sp., J. Antibiot (Tokyo) 54, 836–839; [DOI] [PubMed] [Google Scholar]; b Nakai R, Ishida H, Asai A, Ogawa H, Yamamoto Y, Kawasaki H, Akinaga S, Mizukami T, and Yamashita Y (2006) Telomerase inhibitors identified by a forward chemical genetics approach using a yeast strain with shortened telomere length, Chem. Biol. 13, 183–190; [DOI] [PubMed] [Google Scholar]; c Iorio M, Maffioli SI, Gaspari E, Rossi R, Mauri P, Sosio M, and Donadio S (2012) Chrolactomycins from the actinomycete actinospica, J. Nat. Prod. 75, 1991–1993. [DOI] [PubMed] [Google Scholar]
- (26).a Tenenbaum JM, Morris WJ, Custar DW, and Scheidt KA 2011) Synthesis of (−) - Okilactomycin by a Prins - Type Fragment - Assembly Strategy, Angew. Chem. Int. Ed. 50, 5892–5895; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Smith AB, Basu K, and Bosanac T (2007) Total Synthesis of (−)-Okilactomycin, J. Am. Chem. Soc. 129, 14872–14874. [DOI] [PubMed] [Google Scholar]
- (27).a Custar DW, Zabawa TP, and Scheidt KA (2008) Total Synthesis and Structural Revision of the Marine Macrolide Neopeltolide, J. Am. Chem. Soc. 130, 804–805; [DOI] [PubMed] [Google Scholar]; b Custar DW, Zabawa TP, Hines J, Crews CM, and Scheidt KA (2009) Total Synthesis and Structure-Activity Investigation of the Marine Natural Product Neopeltolide, J. Am. Chem. Soc. 131, 12406–12414; [DOI] [PMC free article] [PubMed] [Google Scholar]; c Crane EA, and Scheidt KA (2010) Prins-Type Macrocyclizations as an Efficient Ring-Closing Strategy in Natural Product Synthesis, Angew Chem Int Edit 49, 8316–8326; [DOI] [PMC free article] [PubMed] [Google Scholar]; d Crane EA, Zabawa TP, Farmer RL, and Scheidt KA (2011) Enantioselective Synthesis of (−) - Exiguolide by Iterative Stereoselective Dioxinone - Directed Prins Cyclizations, Angew. Chem. Int. Ed. 50, 9112–9115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).a Gillis AJ, Schuller AP, and Skordalakes E (2008) Structure of the Tribolium castaneum telomerase catalytic subunit TERT, Nature 455, 633–636 [DOI] [PubMed] [Google Scholar]; b Mitchell M, Gillis A, Futahashi M, Fujiwara H, and Skordalakes E (2010) Structural basis for telomerase catalytic subunit TERT binding to RNA template and telomeric DNA, Nat. Struct. Mol. Biol. 17, 513. [DOI] [PubMed] [Google Scholar]
- (29).Nguyen THD, Tam J, Wu RA, Greber BJ, Toso D, Nogales E, and Collins K (2018) Cryo-EM structure of substrate-bound human telomerase holoenzyme, Nature 557, 190–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).a Hassan M, Brown RD, Varma-O’Brien S, and Rogers D (2006) Cheminformatics analysis and learning in a data pipelining environment, Mol. Div. 10, 283–299; [DOI] [PubMed] [Google Scholar]; b Hu Y, Lounkine E, and Bajorath J (2009) Improving the Search Performance of Extended Connectivity Fingerprints through Activity-Oriented Feature Filtering and Application of a Bit-Density-Dependent Similarity Function, ChemMedChem 4, 540–548; [DOI] [PubMed] [Google Scholar]; c Warr WA (2012) Scientific workflow systems: Pipeline Pilot and KNIME, J. Comput. Aided Mol. Des. 26, 801–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Ghose AK, Viswanadhan VN, and Wendoloski JJ (1999) A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases, J. Comb. Chem. 1, 55–68. [DOI] [PubMed] [Google Scholar]
- (32).a Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE, Francis P, and Shenkin PS (2004) Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy, J. Med. Chem. 47, 1739–1749; [DOI] [PubMed] [Google Scholar]; b Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT, and Banks JL (2004) Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening, J. Med. Chem. 47, 1750–1759; [DOI] [PubMed] [Google Scholar]; c Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Sanschagrin PC, and Mainz DT (2006) Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein–Ligand Complexes, J. Med. Chem. 49, 6177–6196. [DOI] [PubMed] [Google Scholar]
- (33).a Toledo Warshaviak D, Golan G, Borrelli KW, Zhu K, and Kalid O (2014) Structure-Based Virtual Screening Approach for Discovery of Covalently Bound Ligands, J. Chem. Inf. Mod. 54, 1941–1950; [DOI] [PubMed] [Google Scholar]; b Zhu K, Borrelli KW, Greenwood JR, Day T, Abel R, Farid RS, and Harder E (2014) Docking Covalent Inhibitors: A Parameter Free Approach To Pose Prediction and Scoring, J. Chem. Inf. Mod. 54, 1932–1940. [DOI] [PubMed] [Google Scholar]
- (34).Morris WJ, Custar DW, and Scheidt KA (2005) Stereoselective Synthesis of Tetrahydropyran-4-ones from Dioxinones Catalyzed by Scandium(III) Triflate, Org. Lett. 7, 1113–1116. [DOI] [PubMed] [Google Scholar]
- (35).a Singer RA, and Carreira EM (1995) Catalytic, Enantioselective Dienolate Additions to Aldehydes: Preparation of Optically Active Acetoacetate Aldol Adducts, J. Am. Chem. Soc. 117, 12360–12361; [Google Scholar]; b Krüger J, and Carreira EM (1998) Apparent Catalytic Generation of Chiral Metal Enolates: Enantioselective Dienolate Additions to Aldehydes Mediated by Tol-BINAP·Cu(II) Fluoride Complexes, J. Am. Chem. Soc. 120, 837–838; [Google Scholar]; c Denmark SE, and Beutner GL (2003) Lewis Base Activation of Lewis Acids. Vinylogous Aldol Reactions, J. Am. Chem. Soc. 125, 7800–7801; [DOI] [PubMed] [Google Scholar]; d De Rosa M, Acocella MR, Villano R, Soriente A, and Scettri A (2003) A convenient catalytic procedure for the highly enantioselective aldol condensation of O-silyldienolates, Tetrahedron: Asymmetry 14, 2499–2502 [Google Scholar]; e Gondi VB, Gravel M, and Rawal VH (2005) Hydrogen Bond Catalyzed Enantioselective Vinylogous Mukaiyama Aldol Reaction, Org. Lett 7, 5657–5660; [DOI] [PMC free article] [PubMed] [Google Scholar]; f Denmark SE, Heemstra JR, and Beutner GL (2005) Catalytic, Enantioselective, Vinylogous Aldol Reactions, Angew. Chem. Int. Ed. 44, 4682–4698. [DOI] [PubMed] [Google Scholar]
- (36).Betori RC, Miller ER, and Scheidt KA (2017) A Biocatalytic Route to Highly Enantioenriched β-Hydroxydioxinones, Adv. Synth. Catal. 359, 1131–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).a Kathman SG, Xu Z, and Statsyuk AV (2014) A Fragment-Based Method to Discover Irreversible Covalent Inhibitors of Cysteine Proteases, J. Med. Chem. 57, 4969–4974; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Flanagan ME, Abramite JA, Anderson DP, Aulabaugh A, Dahal UP, Gilbert AM, Li C, Montgomery J, Oppenheimer SR, Ryder T, Schuff BP, Uccello DP, Walker GS, Wu Y, Brown MF, Chen JM, Hayward MM, Noe MC, Obach RS, Philippe L, Shanmugasundaram V, Shapiro MJ, Starr J, Stroh J, and Che Y (2014) Chemical and Computational Methods for the Characterization of Covalent Reactive Groups for the Prospective Design of Irreversible Inhibitors, J. Med. Chem. 57, 10072–10079; [DOI] [PubMed] [Google Scholar]; c Cee VJ, Volak LP, Chen Y, Bartberger MD, Tegley C, Arvedson T, McCarter J, Tasker AS, and Fotsch C (2015) Systematic Study of the Glutathione (GSH) Reactivity of N-Arylacrylamides: 1. Effects of Aryl Substitution, J. Med. Chem. 58, 9171–9178; [DOI] [PubMed] [Google Scholar]; d Palkowitz MD, Tan B, Hu H, Roth K, and Bauer RA (2017) Synthesis of Diverse N-Acryloyl Azetidines and Evaluation of Their Enhanced Thiol Reactivities, Org. Lett. 19, 2270–2273. [DOI] [PubMed] [Google Scholar]
- (38).a Ward RA, Anderton MJ, Ashton S, Bethel PA, Box M, Butterworth S, Colclough N, Chorley CG, Chuaqui C, Cross DAE, Dakin LA, Debreczeni JÉ, Eberlein C, Finlay MRV, Hill GB, Grist M, Klinowska TCM, Lane C, Martin S, Orme JP, Smith P, Wang F, and Waring MJ (2013) Structure- and Reactivity-Based Development of Covalent Inhibitors of the Activating and Gatekeeper Mutant Forms of the Epidermal Growth sFactor Receptor (EGFR), J. Med. Chem. 56, 7025–7048; [DOI] [PubMed] [Google Scholar]; b Wilson AJ, Kerns JK, Callahan JF, and Moody CJ (2013) Keap Calm, and Carry on Covalently, J. Med. Chem. 56, 7463–7476; [DOI] [PubMed] [Google Scholar]; c Ding Y, Li D, Ding C, Wang P, Liu Z, Wold EA, Ye N, Chen H, White MA, Shen Q, and Zhou J (2018) Regio- and Stereospecific Synthesis of Oridonin D-Ring Aziridinated Analogues for the Treatment of Triple-Negative Breast Cancer via Mediated Irreversible Covalent Warheads, J. Med. Chem. 61, 2737–2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).a Cohen SB, and Reddel RR (2008) A sensitive direct human telomerase activity assay, Nat. Met. 5, 355; [DOI] [PubMed] [Google Scholar]; b Tomlinson CG, Sasaki N, Jurczyluk J, Bryan TM, and Cohen SB (2017) Quantitative assays for measuring human telomerase activity and DNA binding properties, Methods 114, 85–95. [DOI] [PubMed] [Google Scholar]
- (40).a Wright WE, Shay JW, and Piatyszek MA (1995) Modifications of a telomeric repeat amplification protocol (TRAP) result in increased reliability, linearity and sensitivity, Nucleic Acids Res. 23, 3794–3795; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Wu Y-Y, Hruszkewycz AM, Delgado RM, Yang A, Vortmeyer AO, Moon Y-W, Weil RJ, Zhuang Z, and Remaley AT (2000) Limitations on the quantitative determination of telomerase activity by the electrophoretic and ELISA based TRAP assays, Clin. Chim. Acta 293, 199–212. [DOI] [PubMed] [Google Scholar]
- (41).Bryan TM, Englezou A, Gupta J, Bacchetti S, and Reddel RR (1995) Telomere elongation in immortal human cells without detectable telomerase activity, EMBO J. 14, 4240–4248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).a Li S, Rosenberg JE, Donjacour AA, Botchkina IL, Hom YK, Cunha GR, and Blackburn EH (2004) Rapid Inhibition of Cancer Cell Growth Induced by Lentiviral Delivery and Expression of Mutant-Template Telomerase RNA and Anti-telomerase Short-Interfering RNA, Cancer Res. 64, 4833; [DOI] [PubMed] [Google Scholar]; b Zhao P, Wang C, Fu Z, You Y, Cheng Y, Lu X, Lu A, Liu N, Pu P, Kang C, Salford LG, and Fan X (2007) Lentiviral vector mediated siRNA knock-down of hTERT results in diminished capacity in invasiveness and in vivo growth of human glioma cells in a telomere length-independent manner, Int. J. Oncol. 31, 361–368; [PubMed] [Google Scholar]; c Shen Y, Zhang Y-W, Zhang Z-X, Miao Z-H, and Ding J (2008) hTERT-targeted RNA interference inhibits tumorigenicity and motility of HCT116 cells, Canc. Biol. Ther. 7, 228–236; [DOI] [PubMed] [Google Scholar]; d Mender I, Gryaznov S, Dikmen ZG, Wright WE, and Shay JW (2015) Induction of Telomere Dysfunction Mediated by the Telomerase Substrate Precursor 6-Thio-2′-Deoxyguanosine, Cancer Disc. 5, 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).a Leung D, Hardouin C, Boger DL, and Cravatt BF (2003) Discovering potent and selective reversible inhibitors of enzymes in complex proteomes, Nat. Biotechnol. 21, 687–691; [DOI] [PubMed] [Google Scholar]; b Chiang KP, Niessen S, Saghatelian A, and Cravatt BF (2006) An Enzyme that Regulates Ether Lipid Signaling Pathways in Cancer Annotated by Multidimensional Profiling, Chem. Biol. 13, 1041–1050; [DOI] [PubMed] [Google Scholar]; c Ahn K, Johnson DS, Fitzgerald LR, Liimatta M, Arendse A, Stevenson T, Lund ET, Nugent RA, Nomanbhoy TK, Alexander JP, and Cravatt BF (2007) Novel Mechanistic Class of Fatty Acid Amide Hydrolase Inhibitors with Remarkable Selectivity, Biochemistry 46, 13019–13030; [DOI] [PubMed] [Google Scholar]; d Long JZ, Li W, Booker L, Burston JJ, Kinsey SG, Schlosburg JE, Pavón FJ, Serrano AM, Selley DE, Parsons LH, Lichtman AH, and Cravatt BF (2009) Selective blockade of 2-arachidonoylglycerol hydrolysis produces cannabinoid behavioral effects, Nat. Chem. Biol. 5, 37–44; [DOI] [PMC free article] [PubMed] [Google Scholar]; e Long JZ, Nomura DK, Vann RE, Walentiny DM, Booker L, Jin X, Burston JJ, Sim-Selley LJ, Lichtman AH, Wiley JL, and Cravatt BF (2009) Dual blockade of FAAH and MAGL identifies behavioral processes regulated by endocannabinoid crosstalk in vivo, Proc. Natl. Acad. Sci. U.S.A. 106, 20270; [DOI] [PMC free article] [PubMed] [Google Scholar]; f Bachovchin DA, Ji T, Li W, Simon GM, Blankman JL, Adibekian A, Hoover H, Niessen S, and Cravatt BF (2010) Superfamily-wide portrait of serine hydrolase inhibition achieved by library-versus-library screening, Proc. Natl. Acad. Sci. U.S.A. 107, 20941; [DOI] [PMC free article] [PubMed] [Google Scholar]; g Nomura DK, and Casida JE (2011) Activity-Based Protein Profiling of Organophosphorus and Thiocarbamate Pesticides Reveals Multiple Serine Hydrolase Targets in Mouse Brain, J. Agric. Food. Chem. 59, 2808–2815; [DOI] [PMC free article] [PubMed] [Google Scholar]; h Zuhl AM, Mohr JT, Bachovchin DA, Niessen S, Hsu K-L, Berlin JM, Dochnahl M, López-Alberca MP, Fu GC, and Cravatt BF (2012) Competitive Activity-Based Protein Profiling Identifies Aza-β-Lactams as a Versatile Chemotype for Serine Hydrolase Inhibition, J. Am. Chem. Soc. 134, 5068–5071; [DOI] [PMC free article] [PubMed] [Google Scholar]; i Kaschani F, Nickel S, Pandey B, Cravatt BF, Kaiser M, and van der Hoorn RAL (2012) Selective inhibition of plant serine hydrolases by agrochesmicals revealed by competitive ABPP, Biorg. Med. Chem. 20, 597–600; [DOI] [PMC free article] [PubMed] [Google Scholar]; j Camara K, Kamat SS, Lasota CC, Cravatt BF, and Howell AR (2015) Combining cross-metathesis and activity-based protein profiling: New β-lactone motifs for targeting serine hydrolases, Biorg. Med. Chem. Lett. 25, 317–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Gilad Y, Nadassy K, and Senderowitz H (2015) A reliable computational workflow for the selection of optimal screening libraries, J Cheminform 7, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Baell JB, and Holloway GA (2010) New Substructure Filters for Removal of Pan Assay Interference Compounds (PAINS) from Screening Libraries and for Their Exclusion in Bioassays, J. Med. Chem. 53, 2719–2740. [DOI] [PubMed] [Google Scholar]
- (46).Zhu K, Borrelli KW, Greenwood JR, Day T, Abel R, Farid RS, and Harder E (2014) Docking covalent inhibitors: a parameter free approach to pose prediction and scoring, J Chem Inf Model 54, 1932–1940. [DOI] [PubMed] [Google Scholar]
- (47).Kuhn B, Kollman PA, and Stahl M (2004) Prediction of pKa shifts in proteins using a combination of molecular mechanical and continuum solvent calculations, J. Comput. Chem. 25, 1865–1872. [DOI] [PubMed] [Google Scholar]
- (48).Harder E, Damm W, Maple J, Wu C, Reboul M, Xiang JY, Wang L, Lupyan D, Dahlgren MK, Knight JL, Kaus JW, Cerutti DS, Krilov G, Jorgensen WL, Abel R, and Friesner RA (2016) OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins, J. Chem. Theory Comput. 12, 281–296. [DOI] [PubMed] [Google Scholar]
- (49).Halgren T (2007) New method for fast and accurate binding-site identification and analysis, Chem Biol Drug Des 69, 146–148. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.