

SUMMARY
While autophagy is thought to be an essential process in some cancer cells, it is unknown if or how such cancer cells can circumvent autophagy inhibition. To address this, we developed a CRISPR/ Cas9 assay with dynamic live-cell imaging to measure acute effects of knockout (KO) of autophagy genes compared to known essential and non-essential genes. In some cancer cells, autophagy is as essential for cancer cell growth as mRNA transcription or translation or DNA replication. However, even these highly autophagy-dependent cancer cells evolve to circumvent loss of autophagy by upregulating NRF2, which is necessary and sufficient for autophagy-dependent cells to circumvent ATG7 KO and maintain protein homeostasis. Importantly, however, this adaptation increases susceptibly to proteasome inhibitors. These studies identify a common mechanism of acquired resistance to autophagy inhibition and show that selection to avoid tumor cell dependency on autophagy creates new, potentially actionable cancer cell susceptibilities.
In Brief
An area of controversy is whether cancer cells are dependent upon autophagy. Towers et al. show that some cells are indeed autophagy dependent but can adapt to circumvent the autophagy mechanism. These findings uncover an adaptation mechanism for acquired resistance to autophagy inhibition that creates new cancer susceptibilities.
Graphical Abstract
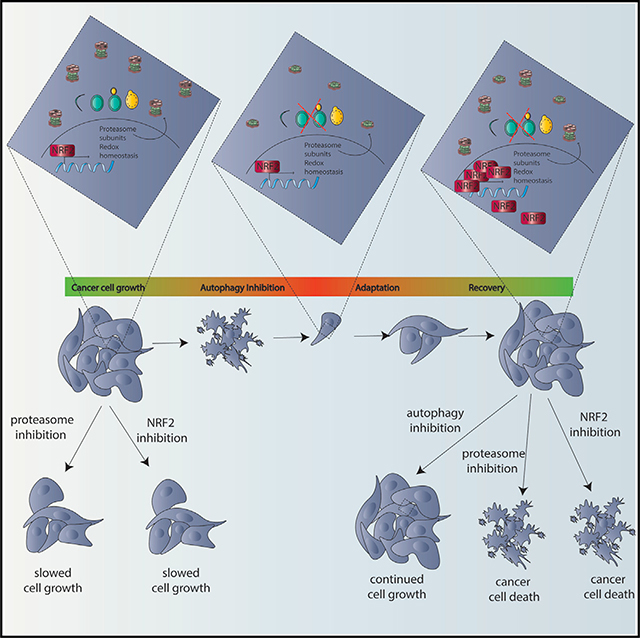
INTRODUCTION
Autophagy is a critical process by which cells degrade organelles, proteins, and other cell components via the lysosome. In established tumors, autophagy often promotes tumor growth and autophagy inhibition preferentially kills and inhibits growth of some cancers but not others. Many studies focus on tumor cells with RAS pathway mutations (Guo et al., 2011, 2013, 2016; Karsli-Uzunbas et al., 2014; Levy et al., 2014; Lock et al., 2011; Rao et al., 2014; Rosenfeldt et al., 2013; Strohecker et al., 2013; Yang et al., 2014, 2011), which may be particularly susceptible to autophagy inhibition, especially in combination with inhibitors of the RAF-MEK-ERK pathway (Levy et al., 2014; Mulcahy Levy et al., 2017; Bryant et al., 2019; Kinsey et al., 2019; Lee et al., 2019). However, autophagy dependence as determined by significant inhibition of growth and induction of apoptosis when autophagy regulators are genetically inactivated has also been reported in cancer cells without RAS pathway mutation (Maycotte et al., 2014). In animal models with established tumors, genetic deletion of critical ATG genes (specifically Atg5 or Atg7) alone is sufficient to decrease tumor growth and enhance overall survival (Guo et al., 2011, 2013; Karsli-Uzunbas et al., 2014; Rao et al., 2014; Rosenfeldt et al., 2013; Strohecker et al., 2013; Yang et al., 2014). Importantly, these effects are thought to be due to both tumor cell-autonomous and non-autonomous roles for autophagy that are essential for sustained growth of some tumors (Poillet-Perez et al., 2018; Yang et al., 2018). Such pre-clinical studies have prompted over 50 clinical trials targeting autophagy with lysosomal inhibitors, chloroquine (CQ), or hydroxychloroquine (HCQ) to block autophagy, usually in combination with other drugs (Levy et al., 2017; Towers and Thorburn, 2016). Improved clinical responses have been seen where autophagy inhibition increased response rates and overall survival (Rangwala et al., 2014; Rojas-Puentes et al., 2013) and reversed both in vitro resistance and clinically acquired resistance to other drugs (Levy et al., 2014; Mulcahy Levy et al., 2017; Ma et al., 2014). Together, these studies indicate that autophagy inhibition may be efficacious to treat some but probably not all cancers. However, clinical studies already demonstrate both inherent and acquired resistance to autophagy inhibition such that patients who initially display clinical benefit upon treatment with a lysosomal autophagy inhibitor, eventually experience tumor growth (Rangwala et al., 2014). These clinical observations suggest that even if cancer cells start off dependent on autophagy and responsive to autophagy inhibitors, they may be able to evolve mechanisms of resistance to autophagy inhibition.
To identify acute gene dependencies that might eventually get circumvented, we created an assay to detect effects on tumor cell growth immediately following gene targeting and employed it to test if a cell-autonomous dependency on autophagy can be circumvented. To this end, we developed a live-cell assay to monitor cell viability and growth in unselected cells in a mixed population within hours of CRISPR/Cas9 gene editing. This approach allowed us to measure how essential a gene of interest is compared to known non-essential and essential genes (Blomen et al., 2015; Hart et al., 2015; Wang et al., 2015). Specifically, genes required for 3 completely essential processes, DNA replication, gene transcription, and mRNA translation, were compared to 12 core autophagy genes (ATGs) needed for different steps in the autophagy process in 8 cancer cell lines on a rapid time scale. With this acute approach, we found that some cancer cells are highly autophagy dependent. But, even these cells can circumvent loss of a core autophagy protein and thus gain resistance to both genetic and pharmacological autophagy inhibition. The cells switched to become autophagy independent by upregulating the cyto-protective master-regulator NRF2 to maintain protein homeostasis. Importantly however, this creates new, potentially druggable vulnerabilities.
RESULTS
Quantitative Acute, Live-Cell Imaging CRISPR-RNP Assay to Quantify the Effects of KO of a Gene of Interest Compared with Known Essential Genes
To quantify acute autophagy dependencies in cancer cells and then monitor their ability to acquire mechanisms to circumvent such a requirement, we first created an acute, quantitative assay to compare the effects of gene knockout by CRISPR/Cas9. Nuclear mCherry and GFP were stably expressed in NCIH292 (H292) lung cancer, BT549 breast cancer, and HCT116 colon cancer lines. Double labeled cells were co-transfected with ribonucleic particles (RNPs) containing recombinant Cas9 protein and 4 in-vitro-transcribed guide RNAs (gRNAs): two that target a gene of interest and two that target GFP. Based on the assumption that cells competent to take up one RNP will take up all RNPs, knock out (KO), and loss of GFP was used as a surrogate for loss of another gene of interest. Quantitative live-cell imaging (with an Incucyte apparatus) was used to assess the effects on cell growth following loss of a gene of interest by measuring the cell count of GFP/mCherry+ cells over 7–14 days immediately following transfection (Figure 1A). All guide RNAs were empirically designed and tested for their ability to cut their target DNAs in vitro (Figure S1; Table S2). Incucyte results showed the assay is robust and quantitative in all cell lines tested and transfection of RNPs with guide RNAs targeting GFP (gGFP) resulted in a loss of GFP signal in >80% of the cells (Figures 1B and 1C). To test if loss of GFP can be used as a surrogate for loss of another gene, we transfected mCherry+/GFP+ cells with RNPs containing gRNAs targeting both GFP and mCherry. Flow cytometry analysis confirmed loss of both mCherry and GFP when gRNAs targeting both genes were co-transfected (Figures 1D–1F). Across multiple cell lines, the vast majority of cells either took up neither gRNA and remained double positive or took up both gRNAs and became double negative; very few cells lost only one fluorophore. These results indicate that when co-targeting RNPs with different gRNAs are transfected, loss of GFP can serve as an efficient, albeit not perfect, surrogate for inactivation of another gene. To allow comparison between cells with different co-transfection abilities, only cell lines where GFP loss was at least 90% efficient as a surrogate for loss of another, unrelated co-targeted gene (mCherry), were used for subsequent studies. To confirm loss of an endogenous gene of interest in the GFP population, gRNAs targeting GFP, and a known non-essential gene (PTEN or FOXO3a) were co-transfected into mCherry+/GFP+ cells. Western blotting showed a dramatic decrease in protein expression of non-essential genes in the GFP sorted fraction in pooled populations of cells (Figures 1G–1I). It is important to note that because of the expected false positive cells and the possibility of in-frame deletions that make a functional protein, complete KO in the GFP pooled cells of either mCherry or a known non-essential gene was not observed; however, in all cell lines used >90% of the co-targeted cells lost expression of the other gene of interest. Proof-ofprinciple experiments to test if this approach could reliably detect differences in growth rates following targeting of different genes were conducted in H292, BT549, and HCT116 mCherry+/ GFP+ cells using co-transfected gRNAs targeting GFP and genes that regulate three critical cellular processes—DNA replication, RNA transcription, and protein translation—by targeting proliferating cell nuclear antigen (PCNA), RNA Polymerase II subunit A (POLR2A), and eukaryotic elongation factor 2A (eEF2) (Hart et al., 2015; Wang et al., 2015). Live-cell quantification of GFP cells for 6–10 days immediately following transfection confirmed essentiality of these genes compared to loss of GFP alone or GFP in combination with known non-essential genes like the tumor suppressor, PTEN, or the pro-apoptotic transcription factor, FOXO3a (Figures 1J–1L). Five additional cancer cell lines were also studied (Figure S2). Each cell line confirmed expected behavior for known essential and non-essential genes (Figures S2K–S2O). These results indicate that the assay can accurately measure acute effects of KO of specific genes in unselected pooled populations of cells within hours of transfection and extending 1–2 weeks.
Figure 1. Design of a Quantitative Live-Cell Imaging CRISPR-RNP Assay to Identify Essential Genes.

(A) Schematic representation of the assay.
(B) Representative images and (C) Incucyte quantification of GFP+ cell count normalized to mCherry+ cell count in mCherry+/GFP+ H292, BT549, and HCT116 cells after transfection with gRNAs targeting GFP. Data are mean ± standard error of the mean (SEM) for technical replicates (N = 2–3) and graphs are representative of 3–6 individual experiments. Statistical analysis: two-way ANOVA and significance at the last time point is shown on the graph. Scale bars represent 200 mM.
(D–F) Flow cytometry was performed in mCherry+/GFP+ cells before and after transfection with gGFP or gGFP+ gmCherry. Data are representative of 2–3 experiments.
(G–I) Western blotting of the GFP sorted populations from mCherry+/GFP+ cells subject to RNP transfections with gRNAs targeting GFP and the indicated genes.
(J–L) Incucyte quantification of mCherry+/GFP cell count/mm2 after transfection with RNPs targeting GFP and indicated genes, data are mean ± SEM for technical replicates (N of 2–3). The graphs are representative of 3–6 experiments. Statistical analysis: two-way ANOVA and the significance at the last time point is shown. *p % 0.05, **p % 0.01, ***p % 0.001, ****p % 0.0001. See also Figures S1 and S2.
Live-Cell Imaging Identifies Autophagy-Dependent Cell Lines
To identify cancer cell lines that are particularly dependent on autophagy, the assay was employed using gRNAs that target core autophagy regulators. Previous studies testing autophagy dependence in cell lines or mice usually target just one or two ATGs. And, the manipulated genes were most often restricted to ATGs involved in the autophagy protein conjugation machinery such as ATG5 or ATG7. However, recent studies have highlighted autophagy-independent functions of ATG proteins (Cadwell and Debnath, 2018), suggesting that manipulation of only a handful of ATG genes may result in erroneous conclusions that autophagy is important for a given biological response when in fact an autophagy-independent function was critical. Therefore, we targeted 12 different ATGs required for different steps in the pathway. Cas9 RNPs containing confirmed gRNAs (Figure S1) that target ULK2, ATG13, RB1CC1 (FIP200), AMBRA1, PICK3C3 (VPS34), BECN1, ATG5, ATG12, ATG7, ATG3, STX17, and LAMP2 were compared to RNPs targeting known non-essential genes (PTEN or FOXO3a) and essential genes (PCNA, POLR2A, or eEF2). To allow quantitative comparison of effects of the different gRNAs across various cell lines, Incucyte data from each technical replicate (2–3 per experiment) was normalized to generate a CRISPR growth score (CGS) and technical replicates were then combined from 2–4 biological replicates. The area under the curve from the mCherry+/GFP growth curves for each gRNA was normalized to an essential and non-essential gene targeted for each cell line, such that the essential gene has a CGS of 0 and the non-essential gene has a CGS of 1 (Figure 2A). If a gene of interest scores as more essential than the positive control for essentiality, e.g., PCNA, the gene will receive a negative CGS score. The assay was first used in HCT116 colon cancer cells, a line that has previously been identified as autophagy independent with the ability to survive genetic loss of core autophagy genes (Eng et al., 2016; Maycotte et al., 2014). Growth curves from the quantified mCherry+/GFP cell counts when gRNAs targeting autophagy genes were co-transfected with gGFP resembled the growth curves generated from gRNAs targeting non-essential genes (Figure 2B; Videos S1 and S2) and these results were reflected in relatively high CGSs for each ATG (Figure 2C)—i.e., each gene behaved like a known non-essential gene. ULK2 was more essential in the HCT116 cells than other autophagy genes suggesting that ULK2 may have autophagy-independent functions that are essential for survival in these cells. There have been recent reports indicating autophagy-independent functions of the ULK kinases in a variety of different contexts including cellular trafficking (Joo et al., 2016), stress granule disassembly (Wang et al., 2019), as well as additional functions in neuronal development and function that are not phenocopied with manipulation of other upstream autophagy regulators like ATG13 or RB1CC1 (Wang and Kundu, 2017). This highlights the ability of this type of quantitative assay to identify potential autophagy-independent functions for ATGs. Taken together, the results from the panel of gRNAs targeting different autophagy regulators support the previous conclusion that autophagy is a non-essential process in the HCT116 cells. In contrast however, in H292 cells, the majority of autophagy genes scored very low CGS’s, some even below 0, indicating the genes scored as more essential by this assay than PCNA (Figures 2D and 2E; Videos S3 and S4). After the Incucyte data were collected, H292 cells were sorted by flow cytometry based on GFP expression and individual GFP clones screened for ATG KO (Figure S3). As expected, gRNAs that scored a low CGS like those targeting ATG5, FIP200, or ATG7 had very few clones that were null for protein expression and the majority of clones that grew up were false positives.
Figure 2. Identification of Autophagy-Dependent and -Independent Cells.

(A) Schematic depicting normalization to quantify gene essentiality. The area under the curve from the mCherry+/GFP growth curves for each gRNA was normalized to an essential and non-essential gene targeted for each cell line, such that the essential gene has a CGS of 0 and the non-essential gene has a CGS of 1.
(B and D) Incucyte quantification of mCherry+/GFP cell count after transfection with gRNAs targeting GFP and the indicated genes. Data are represented as mean ± standard deviation (SD) for technical replicates (N = 2–3) and the graphs are representative of 2–3 individual experiments. Statistical analysis: two-way ANOVA and the significance at the last time point is shown.
(C and E) The normalized CRISPR growth Score (CGS) was calculated for each autophagy-targeting gRNA based on the curves generated in (B or D) wherenormalized CGS for gPTEN is 1 and gPCNA/POLR2A is 0. Data are represented as mean ± SEM for experimental replicates (N = 2–3).
(F) Comparison of normalized CGS for all the autophagy genes in the panel of cell lines. Each data point represents the mean of 2–4 experimental replicates.
(G–L) The data graphed in (F) for each of the indicated cell lines. The data are represented as mean ± SEM for experimental replicates (N of 2–3). *p % 0.05, **p % 0.01, ***p % 0.001, **** p % 0.0001. See also Figures S3 and S4.
Six more cancer cell lines were tested including MCF7, MDAMB-468, and BT549 breast cancer cell lines, SJSA-1 (SJSA) and HT1080 sarcoma cells and the MAF-794 pediatric rhabdoid brain tumor cell line. Figure 2F shows a large variation in autophagy dependence across the 8 cell lines. Cell lines where 6 or more of the 12 autophagy genes scored a CGS (combined from multiple biological and technical replicates) of greater than or equal to 0.5 were classified as autophagy independent, lines where 3–5 autophagy genes scored a CGS R 0.5 were classified as intermediate, and lines were 2 or less autophagy genes scored a CGS R 0.5 were classified as autophagy dependent. Similar to the HCT116 cells, MCF7 breast cancer cells resulted in high CGSs for all the ATG genes and we classified MCF7 cells as an autophagy-independent cell line (Figures 2G and S4A). Again, this is consistent with previous studies where it was found that MCF7 cells are not autophagy dependent (Maycotte et al., 2014; Yang et al., 2011). SJSA and HT1080 cell lines were intermediate, with approximately half of the ATGs scoring CGS’s greater than 0.5 and the other genes scoring lower (Figures 2H, 2I, S4B, and S4C). Consistent with previous studies, 3 cell lines (BT549, MFA-794, and MDA-MB468) that had previously been shown to be highly dependent on autophagy following shRNA knockdown of ATG5 or ATG7 (Maycotte et al., 2014; Levy et al., 2014; Mulcahy Levy et al., 2017), all scored as autophagy-dependent with the majority of autophagy-targeting gRNAs scoring like essential genes with a CGS below 0.5 and many below 0 (Figures 2J, 2L, and S4D– S4F). These results indicate that cancer cell lines vary considerably in their autophagy dependency even in nutrient replete media with no additional exogenous stressors. Indeed, in this acute assay, some cell lines display a similar level of dependence on autophagy as they do for critical cellular functions such as DNA replication. For others the requirement for autophagy is minimal under these unstressed culture conditions and KO of ATGs has little effect on tumor cell survival and growth. And, targeting of ATGs affects subsequent cell growth similar to that seen upon CRISPR/Cas9 targeting of PTEN or even a completely irrelevant gene like GFP.
Autophagy-Dependent Cancer Cells Can Undergo Adaptation and Selection to Circumvent Complete Inactivation of an Autophagy Regulator
To test if highly autophagy-dependent cell lines could adapt to survive complete loss of autophagy core proteins, we repeated the acute-live cell CRISPR/Cas9 assay in H292 and BT549 cells with guide RNAs targeting ATG7. ATG7 was chosen because it is a core ATG that serves as an E1-like enzyme in protein conjugations required for autophagosome formation (Ichimura et al., 2000). It has also been commonly targeted in cancer cells both in vitro and in vivo. Consistent with the previous results, ATG7 resembled an essential gene for the first week in both BT549 and H292 cells (Figures 3A and 3B). Any surviving cells were allowed to remain in culture with continued nutrient replenishment. After several weeks, mCherry+/GFP cells grew enough to be sorted for clonal selection, then grown up and harvested for protein expression analysis. As expected, the majority of the subsequently selected clones were false positives that express ATG7 (Figures S3D and S3F). However, it was possible to isolate multiple clones that expressed no ATG7 (Figures 3C and 3D). Consistent with a lack of canonical autophagy, these clones accumulated the autophagy substrate, SQSTM1/p62, and had no conjugated ATG5–12. They also failed to display phosphatidylethanolamine-conjugated LC3-II even after induction of autophagy by amino acid starvation and lysosome block with CQ (Figures 3C, 3D, and S5A–S5D). Flow cytometry with a tandem LC3-mCherry-GFP (Gump and Thorburn, 2014) showed no amino acid starvation-induced autophagic flux in the absence of ATG7 (Figure S5C). Together, these results confirm that the ATG7-null clones derived from the originally autophagy-dependent H292 and BT549 cells were deficient for canonical autophagy. Surprisingly, live-cell imaging showed that weeks after their initial creation, the ATG7−/− clones had undergone adaptation so that they now grew identically to their wild-type counterparts in both nutrient-rich and -depleted conditions (Figures 3E–3H; Video S5). Moreover, ATG7−/− clones maintained growth curves similar to WT cells even under stress such as growth in galactose – forcing reliance on oxidative phosphorylation, or under hypoxic conditions—another stress known to induce autophagy (Figures S5E and S5F). To test their ability to grow under stresses found in tumors, WT and ATG7−/− clones derived from the autophagy-dependent H292 cell line were xenografted into mice and allowed to grow for 3 weeks. Tumors from ATG7−/− clones grew similarly to their matched WT cells (Figure S5G). These data suggest that after clonal selection following ATG KO, the autophagy-dependent cancer cells adapted so that they evolved a mechanism to circumvent loss of ATG7.
Figure 3. Autophagy-Dependent Cancer Cells Can Undergo Selection to Circumvent Inactivation of an Autophagy Regulator.

(A and B) Incucyte quantification of mCherry+/GFP cell count after transfection with gRNAs targeting GFP and the indicated genes. Data are represented as mean ± SEM for technical replicates (N = 2–3). Graphs are representative of 2–3 individual experiments. Statistical analysis: two-way ANOVA.
(C and D) Western blot analysis in clonal isolates of mCherry+/GFP cells weeks after treatment with gATG7. Data are representative of 2–3 experiments.
(E–H) Incucyte quantification of mCherry+ cell count of WT or ATG7−/− clones in (E and F) nutrient replete conditions or (G and H) starved conditions in EBSS. Data are represented as mean ± SEM for technical replicates (N = 2–3) and graphs are representative of 2–3 individual experiments. Statistical analysis: two-way ANOVA.
(I and J) Normalized CGS for WT or ATG7−/− clones calculated from Incucyte quantification of mCherry+/GFP cell count normalized to essential and non-essential genes after transfection with gRNAs targeting GFP and the indicated genes. Data are represented as mean ± SEM for technical replicates (N = 2–3) and graphs are representative of 2 individual experiments. Statistical analysis: two-way ANOVA. *p % 0.05, **p % 0.01, ***p % 0.001, **** p % 0.0001. Statistical significance indicated for the last time point for time course graphs. See also Figure S5.
The ATG7−/− and WT clones of both autophagy-dependent lines were tested again in the live-cell CRISPR assay to test if cells that circumvent loss of ATG7 had altered sensitivity to loss of the other genes in the autophagy panel. As expected, ATG7−/− clones were no longer sensitive to Cas9 RNPs containing gRNAs targeting ATG7. Interestingly, however, they were also less sensitive to loss of most of the other ATGs that scored as essential with a CGS of close to zero in the WT cells (Figures 3I and 3J). Interestingly, even the ULK2 CGS increased in the ATG7−/− clones in H292 cells, indicating that most of the ULK dependent growth effects are due to its canonical roles in the autophagy pathway. However, in the BT549 cells there was a more modest increase in the ULK2 CGS in the ATG7−/− cells. This suggests that some of the ULK2 mediated growth effects are autophagy independent in this cell line, similar to the HCT116 cells. Together, these data further support a context dependent role for autophagy independent functions of this kinase family. These results suggest that rather than specifically replacing ATG7’s E1 activity, the ATG7−/− clones circumvented the entire autophagy pathway. Consistent with this, the ATG7−/− clones were also much more resistant to CQ-induced apoptosis compared to their WT, autophagy-dependent, counterparts (Figures 4A–4E and S5H). To test resistance to autophagy inhibition in vivo, H292 WT and ATG7−/− clones were xenografted into mice and treated with vehicle or HCQ. Tumor growth was inhibited by HCQ in the WT tumors however, tumors derived from ATG7−/− clones showed no sensitivity to HCQ and trended toward increased growth compared to vehicle (Figure 4F). These studies indicate that even cancer cells that start off highly dependent on functional ATG genes can circumvent autophagy inhibition and become resistant to genetic and pharmacological inhibition of autophagy.
Figure 4. Newly Acquired Autophagy Independence Causes Resistance to Pharmacological Autophagy Inhibition.

(A) Incucyte mCherry+ cell count/mm2 in BT549 WT or ATG7−/− clones treated with CQ (50 mM ) normalized corresponding untreated wells. The data are represented as mean ± SEM for technical replicates (N = 3) and are representative of 4 individual experiments. Statistical analysis: two-way ANOVA.
(B) The Incucyte mCherry+ cell count/mm2 72 h after treatment with the indicated dose of CQ in BT549 WT or ATG7−/− clones normalized to corresponding untreated samples. Data are represented as mean ± SEM for technical replicates (N = 3) and are representative of 3 individual experiments. Statistical analysis: two-way ANOVA.
(C) Normalized Caspase3/7 CellEvent green count after treatment with CQ (50 mM) for BT549 WT and ATG7−/− clones. The data are represented as mean ± SEM for technical replicates (N = 3) and are representative of 2 individual experiments. Statistical analysis: two-way ANOVA.
(D) Representative Incucyte images of Caspase 3/7 CellEvent green 48 h after treatment with 100 mM CQ in BT549 WT and ATG7−/− clones. The scale bars represent 0–200 mM.
(E) Normalized Caspase3/7 CellEvent green count 72 h after treatment with CQ in BT549 WT and ATG7−/− clones. Data are represented as mean ± SEM for technical replicates (N = 3) and representative of 2 individual experiments. Statistical analysis: two-way ANOVA.
(F) Mean tumor volume as a percentage of pre-treatment baseline, following initiation of vehicle or HCQ-treatment in mice bearing H292 WT or ATG7−/− xenograft tumors. N = 5–7 mice per group. Data are represented as mean ± SEM. Statistical analysis: two-way ANOVA. *p % 0.05, **p % 0.01, *** p % 0.001, ****p % 0.0001, statistical significance indicated for the last time point for time course graphs. See also Figure S5.
We also tested if rescue of ATG7 expression in a naturally occurring ATG7−/− and autophagy deficient cell line, NCIH-1650, could confer any growth advantage. In support of our finding that tumor cells can circumvent loss of normally essential autophagy regulators and survive in their absence, rescue of ATG7 and canonical autophagy did not provide any growth advantage, even under nutrient starvation conditions (Figures S5I–S5L).
ATG7−/− Clones Have Defective Proteasomes
To better understand how autophagy-dependent cells circumvent autophagy inhibition, we investigated a critical cellular process that autophagy is known to regulate: proteasome-mediated protein turnover. Although it is established that basal autophagy turns over the proteasome (Marshall et al., 2015; Dikic, 2017), inhibition of autophagy with siRNAs or shRNAs decreases proteasome-mediated protein turnover (Korolchuk et al., 2009). To test if the ubiquitin-proteasome system (UPS) is altered in ATG7−/− clones of autophagy-dependent cells, proteasomal flux was measured using ubiquitin-primed GFP (Dantuma et al., 2000). BT549 ATG7−/− clones had decreased proteasome flux (Figure 5A). Three proteolytic activities (chymotrypsin-like, trypsin-like, and post-glutamyl peptide hydrolytic or caspase-like) contained within the 20S core contribute to the majority of protein degradation. An in vitro enzymatic assay revealed that the ATG7−/− clones had a significant decrease in function of all three proteolytic activities (Figure 5B). These data indicate that the ATG7−/− clones derived from autophagy-dependent cells have an impaired UPS and we hypothesized this could increase sensitivity to a proteasome inhibitor. Live-cell imaging showed ATG7−/− clones had reduced growth and increased caspase 3/7 activity after bortezomib treatment compared to WT cells (Figures 5C–5E). This increased sensitivity was specific to proteasome inhibitors as ATG7−/− clones were not more sensitive to DNA damaging drugs (Figures S6A and S6B).
Figure 5. ATG7−/− Clones Have Defective Proteasomes and Acquire Increased Sensitivity to Proteasome Inhibition.

BT549 WT and ATG7−/− clones (A) Left: Flow cytometry for GFP-ubiquitin expression after bortezomib treatment (50 nM, 24 h). Gated on 5% of the untreated cells and each sample shown is treated with bortezomib. Right: The data graphed represent the fold change (compared to WT) of bortezomib treated samples gated as GFP+ and are represented as the mean ± SEM for two individual experiments. Statistical analysis: two-tailed Student’s t test.
(B) in vitro protease assay. Data represented as mean ± SEM for biological replicates (N = 2–3). Statistical analysis: two-way ANOVA.
(C) Incucyte mCherry+ cell count/mm2 calculated 48 h after bortezomib treatment and normalized to TP0; fold change relative to vehicle is shown. Data represented as mean ± SEM for technical replicates (N = 3) and representative of 2 individual experiments. Statistical analysis: two-way ANOVA.
(D and E) Normalized Caspase3/7 CellEvent green count after treatment with (D) Bortezomib (50 nM) or (E) 6 days after treatment with indicated doses of bortezomib. The data are represented as mean ± SEM for technical replicates (N = 3), representative of 2 individual experiments. Statistical analysis: two-way ANOVA.
(F) Western blot analysis of BT549 WT and ATG7−/− clones, asterisks indicate the bands quantified in Figure S6E.
(G) qRT-PCR to measure mRNA levels of 26S proteasome subunits relative to 18S mRNA levels. Data are represented as mean ± SD for technical replicates(N = 2) representative of 3 individual experiments. Statistical analysis: one-way ANOVA. *p % 0.05, **p % 0.01, ***p % 0.001, ****p % 0.0001, statistical significance indicated for the last time point for time course graphs See also Figure S6.
A decrease in the UPS can lead to increased endoplasmic reticulum (ER) stress due to a buildup of damaged proteins (Obeng et al., 2006). Surprisingly, neither BT549 nor H292 ATG7−/− clones showed an increased ER stress response measured by IRE1a mediated splicing of X-box binding protein 1 (XBP1), or mRNA levels of ER stress response genes including C/EBP homologous protein (CHOP), immunoglobin binding protein (BiP/GRP78), ER-degradation-enhancing-a- mannosidase-like protein (EDEM), or activating transcription factor 4 (ATF4) (Figures S6C and S6D). Together with the lack of observed growth defects, this suggests that while not fully functional, the ATG7−/− clones still have enough proteasome function to maintain sufficient protein homeostasis to avoid excess ER stress. To better understand the mechanism of this regulation, we assayed protein levels of the 20S proteasome by western blot. Surprisingly, multiple subunits were increased (Figures 5F and S6E). These results were confirmed by quantitative real-time PCR to assess transcript levels of the 26S proteasome subunits, which showed that mRNA levels of the majority of the 14 subunits analyzed were increased in ATG7−/− clones derived from both the BT549s and H292 cell lines (Figures 5G and S6F). This suggests that the ATG7−/− clones compensate for loss of proteasome activity by increasing production of proteasome subunits. However, presumably because of lack of autophagic turnover of the proteasomes, those in the ATG7−/− cells are less effective at proteolysis than in WT cells.
Compensatory NRF2 Signaling Increases Proteasome Production
Nuclear factor erythroid 2-related factor 2 (NRF2) is a cytoprotective transcription factor that regulates proteins involved in multiple processes (Rojo de la Vega et al., 2018). Almost all of the proteasome mRNAs upregulated in the ATG7−/− clones are NRF2-regulated genes (Arlt et al., 2009; Kwak et al., 2003; Kapeta et al., 2010). ATG7−/− clones from both BT549 and H292 cell lines had a dramatic increase in NRF2 protein expression (Figures 6A and S7A). shRNA-mediated knockdown of NRF2 in the ATG7−/− clones reinstated 20S proteasome, both mRNA and protein, levels almost back to baseline indicating most of the compensatory increase in proteasome expression is mediated by NRF2 upregulation (Figures 6B, 6C, and S7D). NRF2 is regulated by the E3 ubiquitin ligase KEAP1, which targets it for proteasome degradation (Alam et al., 2003; Itoh et al., 2003). The autophagy cargo protein p62 binds to KEAP1 to prevent its binding to NRF2, resulting in stabilization and activation of NRF2 (Komatsu et al., 2010; Lau et al., 2010). To test if this mechanism is responsible for NRF2 upregulation in the ATG7−/− clones, shRNA-mediated knock down of p62 was conducted in the WT and ATG7−/− clones of the BT549 cells resulting in a loss of the upregulated NRF2 and 20S proteasome indicating a p62-dependent mechanism of regulation (Figures 6D and S7E). NRF2 is also an important anti-oxidant regulator and congruently, the ATG7−/− cells with elevated NRF2 expression showed an increase in glutathione (GSH) and a robust decrease in reactive oxygen species (ROS) (Figures S7B and S7C).
Figure 6. ATG7−/− Clones Upregulate NRF2.

(A–D) BT549 WT and ATG7−/− clones.
(A and B) Western blot analysis with shRNA-mediated KD of (B) NRF2; blots shown are representative of 2–5 experiments. Asterisks indicate the bands quantified in Figure S7D.
(C) qRT-PCR to measure mRNA levels of 26S proteasome subunits relative to 18S mRNA levels after shRNA-mediated KD of NRF2. Data are represented asmean ± SD for technical replicates (N = 2) representative of 2 individual experiments. Statistical analysis: one-way ANOVA.
(D) Western blot analysis after shRNA-mediated KD of p62. Dotted line indicates where unnecessary lanes were removed. Asterisks indicate the bands quantifiedin Figure S7E.
(E) Hierarchical clustered RNA-seq data displaying ATG7 status in cell lines and NRF2 gene signature activation. Statistical analysis: Two-sided Fisher’s exact test. *p % 0.05, **p % 0.01, *** p % 0.001, ****p % 0.0001. See also Figure S7.
We next asked if this mechanism to adapt to autophagy inhibition takes place in a biological context in which loss of an essential autophagy gene has naturally occurred. To this end, we analyzed over 1,000 cell lines from the Cancer Cell Line Encyclopedia (Barretina et al., 2012) and identified 11 cancer cell lines with a homozygous deletion of ATG7. Using the normalized RNA-seq data, we assessed the expression of two independently identified NRF2 gene signatures (Goldstein et al., 2016; Namani et al., 2018) and found that only 6% of cell lines that maintained ATG7 expression had a high NRF2 gene signature compared to 27%–36% of cell lines with a homozygous deletion of ATG7 (Fisher’s exact test: p = 0.004, p = 0.03, respectively) (Figure 6E). These results suggest that tumors that lost ATG7 while still growing in a patient have a higher propensity to upregulate NRF2 signaling and that a similar adaptation mechanism to the one we found during in vitro selection following CRISPR-mediated inactivation of ATG7 may also have happened during evolution of the tumors in patients from which these cell lines were derived.
Adaptation to Circumvent Loss of ATG7 Increases Cancer Cell Dependency on NRF2
To further investigate an increased NRF2 dependency after loss of ATG7, we tested if the ATG7−/− clones of BT549 and H292 cells depend on upregulation of NRF2 for survival. Live-cell imaging showed that shRNA knockdown of NRF2 in the ATG7−/− clones dramatically inhibited cell growth and increased caspase 3/7 activity in both BT549 and H292 cells (Figures 7A, 7B, and S7F). Strikingly, loss of NRF2 had a greater growth inhibitory effect (Figures 7A and S7F) and caused more caspase 3/7 activity (Figure 7B) in ATG7−/− clones compared to their WT counterparts. To test if elevated NRF2 is sufficient to decrease autophagy dependence, it was exogenously expressed in the autophagy-dependent WT BT549 cells or KEAP1 was knocked down with shRNA. In both cases, this increased NRF2 to similar levels as in the ATG7−/− clones (Figures 7C and 7E, insets) and led to decreased apoptosis after CQ treatment (Figures 7C–7F). These results indicate that upregulation of NRF2 is necessary and sufficient for autophagy-dependent cells to circumvent autophagy inhibition. Additionally, NRF2 knockdown further increased bortezomib sensitivity in the ATG7−/− clones (Figure 7G).
Figure 7. ATG7−/− Clones Are Dependent on NRF2 for Survival.

(A–G) Incucyte live cell imaging of (A and B) BT459 cells transduced with shNS or shNRF2. (A) KD of NRF2 induced effects on growth. Data are represented as mean ± SD of the fold change in Incucyte mCherry+ cell count/mm2 for shNRF2 over shNS for technical replicates (N = 3) representative of 2 individual experiments. Statistical analysis: two-way ANOVA.
(B) KD of NRF2 induced effects on apoptosis 8 days after transduction with indicated shRNAs measured as the fold change in CellEvent green count for shNRF2 relative to shNS. Data are represented as mean ± SD for technical replicates (N = 3) representative of 2 individual experiments. Statistical analysis: one-way ANOVA.
(C–G) Normalized Caspase3/7 Cell Event green in (C–F) parental BT549 mCherry-NLS cells with (C and D) stable overexpression of EV or NRF2-flag or (E and F) shNS or shRNAs targeting KEAP1 or (G) BT549 WT or ATG7−/− with KD of NRF2.
(C and E) Data represented as the mean fold change in treated over untreated cells ± SEM over time or (D and F) 3 days after treatment with indicated doses of CQ for technical replicates (N = 3) and representative of 2 individual experiments. Statistical analysis: two-way ANOVA. Inset: western blot of stable lines overexpressing NRF2.
(G) After transduction with shNS or shNRF2 and treatment with bortezomib (100 nM) at indicated time points. Data are represented as the mean ± SEM for technical replicates (N of 3) and the graphs shown are representative of 2 individual experiments. Statistical analysis: two-way ANOVA. *p % 0.05, **p % 0.01, ***p %0.01, ****p % 0.001, *****p % 0.0001, significance indicated for the last time point for time course graphs. See also Figures S6 and S7.
DISCUSSION
This is the first study to show that even when cancer cells start off highly dependent on autophagy, they can adapt to circumvent the autophagy pathway in its entirety by upregulating NRF2 signaling. Importantly, adaptation to survive complete genetic inactivation of an essential autophagy regulator, ATG7, also led to resistance to pharmacological autophagy inhibition and resistance to genetic KO of multiple other autophagy regulators. Previously, we and others have found some cancer cells are more dependent on autophagy than others even in the absence of additional exogenous stressors such as nutrient deprivation. In mice, loss of Atg5 or Atg7 can suppress tumors in several different KRAS- and BRAF-driven models of cancer (Yang et al., 2014, 2018; Xie et al., 2015; Strohecker et al., 2013; Rosenfeldt et al., 2013; Rao et al., 2014; Karsli-Uzunbas et al., 2014). In cell culture experiments, it has also been reported that some, but not all, cancer cells are highly dependent on autophagy (Yang et al., 2011; Maycotte et al., 2014; Levy et al., 2014; Degenhardt et al., 2006; Lock et al., 2011; Guo et al., 2011). These studies usually tested one or two autophagy regulators by shRNA knockdown followed by antibiotic selection to study the depleted cells and have been interpreted to mean that some cancers are autophagy-dependent.
However, a recent publication cast doubt on the conclusion that some cancer cells are dependent on autophagy when shRNAs targeting three autophagy regulators were not differentially selected in a genome-wide screen of a large panel of human cancer cell lines with and without RAS mutations (Eng et al., 2016). Moreover, in the same study, CRISPR-mediated knockout of ATG7 had no growth inhibitory effect in selected clones from several tumor cell lines that were expected to be autophagy-dependent. This study concluded that autophagy is dispensable for cancer cell growth, whether or not the cancer cell has RAS pathway mutations and has been used to argue that targeting autophagy is not a suitable therapeutic strategy in cancer.
Our work provides an explanation for this confusion. Using a rigorous and comprehensive assay for autophagy gene essentiality that quantitatively compares acute inactivation of a dozen autophagy regulator genes side by side with inactivation of genes required for other essential processes like DNA replication, our data show that some cancer cell lines are, in fact, extremely dependent on autophagy (Figures 1, 2, and 3). By directly assessing autophagy regulators rather than extracting data regarding ATG genes out of a full genome screen, the overall result (cancer cells vary greatly in their reliance on autophagy genes in the absence of added exogenous stressors) agrees with previous work from our own lab and several other laboratories where similar approaches using shRNAs and both inducible and constitutive knockouts of mouse ATG genes in tumors were performed. Most important, our work also tested whether selection can change the apparent autophagy dependence of tumor cells. We find that under sufficient selective pressure caused by complete inactivation of an essential autophagy regulator, a few surviving tumor cells can adapt and evolve so that autophagy’s critical functions are circumvented. Thus, after clonal selection, autophagy-dependent cell lines as defined by their reliance on each of a dozen different autophagy regulators can evolve to circumvent loss of autophagy (Figure 3). A similar selection process may have occurred in the previous study by Eng et al., 2016 thus explaining why they did not see evidence of ATG genes behaving like essential genes in any cells.
ATG7 deficient clones made from autophagy-dependent cell lines that circumvented autophagy inhibition grew similarly to their WT counterparts even under physiologically relevant stress and became resistant to CQ both in vitro and in vivo (Figures 3 and 4). Selection for resistance to ATG7 KO caused autophagy-dependent cells to upregulate the master-transcriptional regulator NRF2 via a p62-KEAP1-dependent mechanism (Figure 6). Importantly, we identified a common mechanism of acquired autophagy independence as every clone tested from two different autophagy-dependent cell lines showed an increased dependency on NRF2 for survival compared to their WT counterparts. This mechanism is linked to one of the core cellular functions of autophagy—regulation of protein homeostasis—such that elevated NRF2 signaling is needed to compensate for decreased proteasome turnover in the autophagy deficient clones (Figures 5, 6, and 7). NRF2 is also a master anti-oxidant regulator and accordingly the autophagy deficient clones showed decreased levels of ROS (Figure S7) suggesting that both NRF2 mediated proteasomal compensation and reduced ROS contribute to the adaptation and survival of the autophagy deficient cells.
NRF2 signaling has been linked to autophagy and the two cooperate in a regulatory loop with each other. Thus while NRF2 turnover is controlled by KEAP1 through p62 (Komatsu et al., 2010), several core autophagy proteins are direct transcriptional targets of NRF2 (Pajares et al., 2016). Our results show, for the first time, that autophagy deficient cells become addicted to NRF2 signaling (Figure 7). Conversely, NRF2 overexpression is sufficient to make autophagy-dependent cancer cells less sensitive to pharmacological autophagy inhibition (Figure 7). This implies the autophagy-NRF2 loop may be critical for determining autophagy dependence.
This mechanism of enhancing NRF2-dependent signaling makes the cells moresensitiveto depletion of NRF2and also leads to increased sensitivity to inhibitors of the proteasome (Figures 5, 6, and 7). Together our studies identified molecular mechanisms that lead to resistance to autophagy inhibition and also identified new, potentially targetable, susceptibilities that arise as a result of the selection to circumvent autophagy inhibition. These results also show the importance of developing intact animal models of acquired autophagy resistance, both pharmacological and genetic, to gain further insight into the ways that cancer cells adapt to loss of autophagy. We hypothesize that it will be possible to find both purely pharmacological mechanisms of resistance (Collins et al., 2018) and genetically driven alterations that confer resistance to autophagy inhibition. Importantly our work suggests that these mechanisms will include upregulation of NRF2 as discovered here, which appears to have also taken place in cell lines derived from tumors that presumably lost ATG7 expression during normal tumor evolution in a person. These findings suggest that clinical trials with autophagy inhibitors like CQ and HCQ should acknowledge the possibility of acquired resistance by collecting tumor samples before, during, and after treatment with autophagy inhibitors in order to detect if alterations in NRF2 that might increase susceptibilities to other interventions could be used to reduce the incidence of acquired or innate resistance to autophagy-targeted therapies.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Andrew Thorburn (andrew.thorburn@ucdenver.edu). This study did not generate new unique reagents other than guide RNAs and all sequences are provided in Table S1.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Lines
All Cell lines were maintained at 37C and 5 % CO2. HCT116 (male) cells were maintained in Dulbecco’s Modified Eagle Medium (DMEM) with 10 % fetal bovine serum (FBS). MCF7 (female) cells were maintained in Minimum Essential Media (MEM) with 10 % FBS and Insulin (0.01 mg/ml). BT549 (Female) cells were maintained in Roswell Park Memorial Institute medium (RPMI 1640) with 10 % FBS and Insulin (7.5 mg/ml). MDA-MB-468 (female) were maintained in Dulbecco’s Modified Eagle Medium/Nutrient Mixture F-12 with 10% FBS. NCIH292 (female), SJSA-1 (male), and NCIH1650 (male) cells were maintained in Roswell Park Memorial Institute medium (RPMI 1640) with 10 % FBS. MAF-794 were maintained in Optimem medium with 15% FBS. The HT1080 (male) cells were maintained in Minimum Essential Media (MEM) with Non-essential amino acids and sodium pyruvate. For experiments with galactose, RPMI 1640 media without glucose was supplemented with 10 mM galactose, 10% FBS, and 7.5 mg/ml insulin. NCIH-1650 cell lines with stable expression of ATG7 were made by transfecting PCDA3-EV or PCDNA3 containing human ATG7 isoform A, and then selected with hygromycin. All cell lines were maintained in penicillin streptomycin while in culture and periodically monitored for mycoplasma contamination. All cell lines were fingerprinted by short-tandem repeat profiling to confirm identity.
Animal Studies
All animal studies were performed in accordance with and approval by the Institutional Animal Care and Use Committee at Colorado State University. 6–8 week old female athymic nude (nu/nu) mice were purchased from The Jackson Laboratory, and housed in microisolator cages in the laboratory animal facility at Colorado State University. 2 × 106 H292 wild-type, or Atg7−/− cells were injected subcutaneously into the right dorsal flank of mice in 100 mL of serum free RPMI media. Tumor volume was measured daily in a blinded fashion with digital calipers using the formula short diameter2 × long diameter × 0.5. Once tumors reached 150 mm3, mice were randomized to vehicle control (0.9% saline) or hydroxychloroquine (HCQ) treatment groups, and treatment with 60 mg/kg HCQ, i.p. daily in 100 mL injection volume of 0.9% saline was initiated. Mice were weighed every other day for drug dosage calculations and sacrificed after 10% weight loss, or when tumor volume reached 1500 mm3.
METHOD DETAILS
CRISPR with RNPs
Each guide RNA (gRNA) was created by first running a nested PCR to generate a PCR template that contains the T7 sequence adjacent to a 20 base pair gRNA target sequence (designed using crispr.mit.edu) and a tracer RNA region according to previous reports (Liang et al., 2015). Specifically, an initial PCR reaction was used to amplify the tracer RNA from 20 ng of lentiCRISPRV2 plasmid using GTTTTAGAGCTAGAAATAGCAAG and AAAAGCACCGACTCGGTGCCAC oligonucleotides at 0.5 mM each and 0.5 ml of Phusion High Fidelity DNA Polymerase in a 50-ml reaction. Thermal cycles programed for 2 minutes at 98C for initial denaturation, followed by 35 cycles of 30 seconds at 98C for denaturation, 30 seconds at 55C as annealing, and 30 seconds at 72C for elongation and a final extension of 5 minutes at 72C. PCR products were examined by electrophoresis at 100–120 Volz for approximately 30 minutes in a 1% (w/v) agarose gel in 1x TAE buffer and the size was confirmed with a 1 KB DNA ladder. PCR products were purified with the Wizard SV Gel PCR clean up kit and eluted in 30 ml of RNAse/DNase free H2O. Subsequent nested PCRs were then amplified off of 17 ng of this product using a T7 FWD primer (TAATACGACTCACTATAG) and TrcRNA REV primer (AAAAGCACCGA CTCGGTGCCAC) each at 0.5 mM along with a Forward and Reverse primer corresponding to each specific gRNA sequence at 1.5 nM (see Oligos in Table S2) in a 50-ml reaction with 0.5 ml of Phusion High Fidelity DNA Polymerase. Thermal cycles programed for 2 minutes at 98C for initial denaturation, followed by 35 cycles of 30 seconds at 98C for denaturation, 30 seconds at 55C as annealing, and 30 seconds at 72C for elongation and a final extension of 5 minutes at 72C. PCR products were examined by electrophoresis at 100–120 Volz for approximately 30 minutes in a 1% (w/v) agarose gel in 1x TAE buffer and the size was confirmed with a 1 KB DNA ladder. PCR products were cleaned up with the Wizard SV Gel PCR clean up kit and eluted in 30 ml of RNase/DNase free H2O. 500 ng of each guide RNA template was then subject to In-vitro transcription with a MEGAshortscript T7 transcription kit according to manufactures instructions and incubated for 4 hrs at 36.5C followed by a 15-minute incubation with DNase treatment at 36.5C. The transcribed RNA was cleaned up with the MEGAclear transcription clean-up kit and eluted in 100 ml of elution buffer warmed to 98C. RNA concentration and purity was calculated with a nanodrop machine. RNA was then diluted to 10 ng/ml in RNase/DNase free H2O in single use aliquots and frozen at −80C. Five individual gRNAs were transcribed per gene and then tested for editing ability via an in-vitro Cas9 assay. Specifically, the region of DNA (300–600 base pairs) predicted to be cut was amplified from expression plasmids or cDNA via a standard PCR reaction. 60 ng of target PCRs were then incubated for 1 hr at 37 with bovine serum albumin (BSA), NEB Buffer #3, 100 ng of a single guide RNA and 150 ng of recombinant Cas9 protein from PNAbio. PCR products with and without Cas9 were examined by electrophoresis at 100–120 Volts for approximately 30–60 minutes in a 1% (w/v) agarose gel in 1x TAE buffer and the size was confirmed with a 1 KB DNA ladder. Based on their ability to edit in vitro and predicted off target binding 2 guide RNAs were chosen per gene to transfect into cells. Specifically, 200 ng of recombinant Cas9 (PNAbio) was incubated for 10 minutes with 5 ng of each guide RNA (two that target GFP and two that target the gene of interest). 7.4 ml of Opti-MEM reduced serum medium was then added to the eppendorf tubes along with 0.4 ml of Cas9 Plus Reagent (from the Lipofectamine CRISPR Max kit transfection kit) and allowed to incubate for another 10 minutes at room temperature. Simultaneously in separate eppendorf tubes 9.7 ml of Opti-MEM was incubated with 0.3 ml of CRISPRMAX reagent for 10 minutes at room temperature. The Cas9 plus incubation was then added to the CRISPRMAX incubation and allowed to incubate together for 15 minutes at room temperature. 20 ml of the transfection was added to each individual well of a 96 well plate where cells (mCherry+/GFP+) were seeded at 500–1000 cells per well in 100 ml in triplicate wells the previous day and allowed to incubate for 4.5 hours at 37C and 5% CO2. The media was then dumped off and 200 ml of full medium was added back to each well, and plates were monitored by Incucyte live cell imaging for 7–10 days with images taken every 2–4 hours. Medium was replaced as needed every 3–4 days. For each cell line gRNAs targeting essential and non-essential genes were tested in parallel in triplicate wells on each 96 well plate and used for normalization. Oligonucleotoides used to design guide RNAs are located in Table S2.
Incucyte Live Cell Imaging
Live cell imaging was performed with an Incucyte (dual color model 4459) at 4X magnification and images in the red and green channel were taken every 2–4 hours (for figure presentation, quantification ever 8–12 hours is shown). For mCherry cell count, mCherry+ cells/mm2 were masked (and optimized for each cell type). For caspase 3/7 activity: CellEvent Caspase 3/7 green reagent was added (2 mM) at the same time as drug and green events were masked (and optimized for each cell type). Green count/mm2 was then normalized to red count/mm2 to normalize for cell number at every time point. For quantification of GFP− cell count during the Live-cell CRISPR assay: the overlap mask (optimized for each cell type) was used to quantify the number of double positive cells. At each time point the overlap/mm2 count was subtracted from the red count/mm2 to quantify the GFP− cell count. To calculate normalized CRISPR growth scores (CGS): For each gene the area under the curve was calculated as described in Figure 3A using the trapezoidal function of GFP− cell count vs time for the average from duplicate or triplicate wells. The area under the curve for the essential gene was subtracted from the area of the curve of the non-essential gene and for each gene of interest. The subtracted value for each gene of interest was then divided by the subtracted value for the non-essential gene such that the essential gene ran in parallel would receive a CGS of 0 and the non-essential gene would have a CGS of 1. CGS values were then combined across experimental replicates.
Flow Cytometry
For the Measurement of GFP and/or mCherry Loss after RNP Transfection
500–1000 mCherry+/GFP+ cells were plated into 96 well plates and transfected as described above with gRNAs that target gGFP and/or gmCherry. 7–10 days after transfection, cells were trypsinized, pelleted by centrifugation, and resuspended in phenol-free medium. Flow cytometry was performed with a Gallios 561 (Beckman Coulter) using the 488 and 561 nM lasers for green and red fluorophore excitation, respectively. The appropriate side/forward scatter profile was used to exclude non-viable cells. The gates for GFP and mCherry positive cells were set based on unstained cells and Mcherry+/GFP+ for each cell line.
Measurement of Proteasomal Flux by Flow Cytometry
BT549 WT and ATG7−/− clones were made with stable expression of Ubiquitin (G76V) fused to the N-terminus of GFP (Dantuma et al., 2000). Each stable cell line was plated in duplicate wells; one of which was left untreated and the other treated with bortezomib (50 nM) for 16–24 hours. The two conditions for each stable line were trypsinized, pelleted by centrifugation, and resuspended in phenol-red free medium. Flow cytometry analysis was performed with a Gallios 561 (Beckman Coulter) using 488nm laser for green excitation. The appropriate side/forward scatter profile was used to exclude non-viable cells. The gates for each stable line were set such that 5% of the untreated cells were counted as GFP−. Proteasomal flux was then calculated as the increase in viable cells that shifted into the 5% gate after treatment with bortezomib where each cell line was re-gated based on the corresponding untreated sample.
Measurement of Autophagic Flux by Ratiometric Flow Cytometry
BT549 cells stably expressing mCherry-GFP-LC3 were used for flow cytometric analysis. Cells were either left untreated, or washed twice with PBS before receiving EBSS for 24 hrs, or treated with bafilomycin A1 (10 nM) for 24 hrs. Flow cytometry was performed with a Gallios 561 (Beckman Coulter) using 488 and 561 nM lasers for green and red fluorophore excitation, respectively. The appropriate side/forward scatter profile was used to exclude non-viable cells. Autophagic delivery of the tandem constructs to the lysosome quenches the GFP signal, therefore cells undergoing autophagy were defined as those expressing a high mCherry/GFP fluorescence ratio. The gate to define cells undergoing autophagy was set based on cells treated with bafilomycin A1, a condition that represents cells with little or no autophagic flux. The bottom of the gate for each set of flow cytometry experiments was therefore set at the rightward base of the bafilomycin A1-treated curve (24 hr at 10 mM) such that 5% of bafilomycin A1-treated cells were included in the gate.
Measurement of ROS with cellROX
Cells were treated with cellROX green (1 mM) for 1 hr, then trypsinized and harvested for flow cytometry analysis with a Gallios 561 (Beckman Coulter) using the 488nM laser for green fluorophore excitation. Forward and side scatter was used eliminate non-viable cells. The cellROX+ gate was set such that 10% of the WT cells were gated as positive and the percent of cells for each of the clones was analyzed based on this gate.
FACs Sorting
Mcherry+/GFP+ cells were subject to RNP transfections with gRNAs targeting GFP as well as a gene of interest as described above. 7–14 days later, the cells were trypsinized, pelleted by centrifugation, and resuspended in phenol-red free media. The appropriate side/forward scatter profile was used to exclude non-viable cells. The GFP− population was gated based on unstained cells (as shown in the figures), and sorted with a MoFlo XDP100 (Beckman Coulter). The sorted GFP- fraction was then plated and harvested 3–5 days later for western blot analysis of the GFP− pooled population.
Measurement of GSH
Glutathione (GSH) was measured in cell homogenates using a microtiter plate assay as previously described (Vandeputte et al., 1994). Briefly, samples were collected by scraping cells from a 60 mm tissue culture dish into 10 mM HCl and homogenates prepared by sonication of sample. A 50-ml aliquot was collected for protein determination and 62.5 ml of 6.5% (w/v) sulfosalicylic acid added to precipitate protein in the remaining 250 ml sample. The sample was then incubated on ice for 10 mins followed by centrifugation at 2000 RCF and the resulting supernatant collected. Twenty microliters of blank, standard or unknown were added to a 96-well plate followed by 20 ml of 143 mM phosphate buffer (pH 7.4) and 200 ml of assay mix (143-mM phosphate buffer (pH 7.4), 6.3 mM EDTA, 1 mM 5,5’-dithios-2-nitrobenzoic acid (DTNB) and 0.34 mM NADPH. The plate was incubated for 5 minutes at room temperature followed by the addition of 40 ml of glutathione reductase (8.5 IU/ml in phosphate buffer). The reaction was monitored for the change in absorbance at 405 nm for 5 minutes and GSH concentrations were calculated from a standard curve using known concentrations of GSH (3.125 – 50 mM).
Hypoxia Treatments
1,000 mCherry-NLS labeled cells were plated per well in 96 well plates. The following day the cells were switched to media buffered with HEPES (50 mM) and then placed in a humidified atmosphere at 37C, with 1% O2 and 5% CO2. Every 24 hrs the plate was removed from the incubator to scan in the Incucyte and mCherry+ cell counts were calculated over time.
Western Blotting
Whole cell lysate samples were harvested in stringent-RIPA buffer, with 1X Protease inhibitor cocktail added fresh before each use. Nuclear lysates were harvested by the following nuclear extraction protocol: Washed and pelleted cells were resuspended in cell lysis buffer (10 nM HEPES (pH 7.5), 10 mM KCL, 0.1 mM EDTA, 1 mM dithiothreitol (DTT), 0.5% Nonidet-40, and 1X 4-(2-aminoethyl) benzenesulfonyl fluoride hydrochloride (AEDSF), 1X protease inhibitor cocktail), allowed to swell on ice for 15–20 minutes with intermittent mixing. Tubes were vortexed to disrupt cell membranes and centrifuged at 12,000g at 4C for 10 minutes. After the cytoplasmic extract was removed, the pellets were washed thrice in cell lysis buffer, and resuspended in nuclear extraction buffer (20mM HEPES (pH 7.5), 400mM NaCl, 1mM EDTA, 1mM DTT, 1X AEBSF, 1X protease inhibitor cocktail) and incubated on ice for 30 minutes. Nuclear extracts were collected by centrifugation at 12,000g for 15 minutes. A Bradford assay was used to calculate protein concentrations relative to a bovine serum albumin (BSA) standard curve. Western blotting was performed using standard methods including protein separation on SDS-PAGE 1.5mm mini gels in running buffer at 100V for 2hrs, followed by transfer to PVDF membranes in transfer buffer using a semi-dry transfer apparatus at 15V for 70 minutes. Membranes were blocked in 5% milk for 1hr at room temperature with gentle rocking, washed twice in 1X TBST, and then incubated overnight at 4C with gentle rocking in primary antibodies. Membranes were then washed thrice in TBST and incubated for 1hr at room temperature with gently rocking in secondary antibodies followed by 3 more TBST washes. Membranes were developed with Immobilon Western chemiluminescent HRP substrate (Millipore) and analyzed on the OdysseyFc imaging system. Antibodies are listed in the Key Resources Table.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| NRF2 | Abcam | Cat# ab62352; RRID: AB_944418 |
| 20-s Proteasome | UBPBio | Y2010 |
| P62 | Novus | Cat# H00008878-M01; RRID: AB_548364 |
| LC3 | Novus | Cat# NB100-2220; RRID: AB_10003146 |
| ATG5 | Cell Signaling Technology | Cat# 9980S; RRID: AB_10829153 |
| ATG7 | Cell Signaling Technology | Cat# 8558; RRID: AB_10831194 |
| ATG12 | Cell Signaling Technology | Cat # 2010s; RRID: AB_2059086 |
| STX17 | Sigma-Aldrich | Cat# HPA001204; RRID: AB_1080118 |
| FIP200 | Novus | Cat# NBP1-31583; RRID: AB_2300812 |
| PTEN | Santa Cruz Biotechnology | Cat# sc-7974; RRID: AB_628187 |
| FOXO3a | Cell Signaling | Cat# 3938; RRID: AB_2106669 |
| β-Actin | Sigma-Aldrich | Cat# A5441; RRID: AB_476744 |
| β-Tubulin | Sigma-Aldrich | Cat# T5168; RRID: AB_477579 |
| Mouse-IgG | Cell Signaling Technology | Cat# 7076; RRID: AB_330924 |
| Rabbit-IgG | Cell Signaling Technology | Cat# 7074; RRID: AB_2099233 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Bafilomycin A1 | Sigma-Aldrich | B1792; CAS RN: 88899-55-2 |
| Chloroquine | MP-Biomedicals | 93919; CAS RN: 50-63-5 |
| Protease Inhibitor Cocktail | Roche | 11836153001 |
| Bortezomib | Selleck Chemicals | S1013 |
| Critical Commercial Assays | ||
| MEGAscript T7 Transcription Kit | Thermo Fisher | AM1354 |
| MEGclear Transcription Clean Up Kit | Thermo Fisher | AM1908 |
| Wizard SV Gel PCR clean up kit | Promega | A9282 |
| Lipofectamine CRISPR Max Cas9 transfection reagent | Thermo Fisher | CMAX00003 |
| RNeasy RNA isolation kit | Qiagen | 74104 |
| QuantiTect Reverse Transcription Kit | Qiagen | 205311 |
| SYBR Green CFX for QPCR Applied Biosystems | Applied Biosystems | 4472942 |
| Seahorse XF Mitochondrial Stress Test | Agilent | 103015–100 |
| Proteasome Activity Fluorometric Assay Kit II | UBPBio | J4120 |
| CellROX green flow cytometry assay kit | Thermo Fisher | C10492 |
| Experimental Models: Cell Lines | ||
| HCT116 | ATCC | NCI-DTP Cat# HCT-116; RRID: CVCL_0291 |
| MCF7 | ATCC | NCI-DTP Cat# MCF7; RRID: CVCL_0031 |
| HT1080 | ATCC | CLS Cat# 300216/p517_HT-1080; RRID: CVCL_0317 |
| NCIH292 | Kindly provided from Dr. Joaquin Espinosa | ECACC Cat# 91091815; RRID: CVCL_0455 |
| MAF-794 | Kindly provided from Dr. Jean Mulcahy-Levy | N/A |
| BT549 | ATCC | NCI-DTP Cat# BT-549; RRID: CVCL_1092 |
| SJSA-1 | Kindly provided from Dr. Joaquin Espinosa | ATCC Cat# CRL-2098; RRID: CVCL_1697 |
| NCIH-1650 | ATCC | ATCC Cat# CRL-5883; RRID: CVCL_1483 |
| Experimental Models: Organisms/Strains | ||
| Athymic Nude mice (nu/nu) | Jackson Laboratory | Stock number: 007850 |
| Oligonucleotides | ||
| Oligonucleotides for qRT-PCR primers: Table S1 | this study; Fitzwalter et al., 2018; Oslowski and Urano, 2011; Radhakrishnan et al., 2010; Vangala et al., 2014. | N/A |
| Oligonucleotides for guide RNA design: Table S2 | this study | N/A |
| Recombinant DNA | ||
| LentiCRISPR – ATG7 | (O’Prey et al., 2017) | N/A |
| PLJM1-GFP-3xNLSpuromycin | (Thorburn et al., 2017) | N/A |
| PLJM1-mCherry-3xNLS blasticidin | (Thorburn et al., 2017) | N/A |
| PCDNA3-EV | this paper | N/A |
| PCDNA3-hATG7 isoform A | this paper | N/A |
| PLKO.1 shRNA-Keap1 TRCN0000158081 | Sigma-Aldrich | N/A |
| PLKO.1 shRNA-Keap1 TRCN0000158076 | Sigma-Aldrich | N/A |
| PLKO.1-shRNA-NRF2/NFE2L2 TRCN0000007559 | Sigma-Aldrich | N/A |
| PLKO.1-shRNA-NRF2/NFE2L2 TRCN0000007558 | Sigma-Aldrich | N/A |
| PLKO.1-shRNA-p62/SQSTM1 TRCN0000007234 | Sigma-Aldrich | N/A |
| PLKO.1-shRNA-p62 TRCN0000007236 | Sigma-Aldrich | N/A |
| NC16 pCDNA3.1 FLAG NRF2 | (Camp et al., 2012) | Addgene plasmid #36971 |
| Ub-G76V-GFP | (Dantuma et al., 2000) | Addgene plasmid #11941 |
| Other | ||
| Phusion High Fidelity DNA Polymerase | New England Biolabs | M0530L |
| CellEvent Caspase3/7 Green Detection Reagent | Thermo Fisher Scientific | C10423 |
| TransIT LT1 Transfection Reagent | Mirus | MIR 2304 |
| Cas9 recombinant protein | PNAbio | CP01 |
Clonogenic Growth Assay
2,000 H1650 cells were plated into 12 well plates. The following day cells were washed and treated with the indicated percent of Media/EBSS. After 72 hours, the media was replaced with full media for another 7 days, with media changes every 3 days. Cells were then washed fixed and stained with crystal violet. To quantify the signal, the crystal violet was solubilized and the absorbance read at 590 nm.
qRT-PCR
Reverse Transciptase Quantitative Polymerase Chain Reaction was performed on an Applied Biosystems ViiA 7 by Life Technologies machine with an epMotion 5070 Eppendorf robot as follows: the RNA was isolated using a Qiagen RNeasy mini kit following the manufacturer’s instructions. Next, a reverse transcriptase reaction was performed using the Qiagen Quantitect RT kit according to manufacturer’s instructions. The resulting cDNA was then subject to quantitative PCR using SYBR green CFX master mix from Applied Biosystems. A standard curve was used from 0.04 ng to 50ng of cDNA. The relative quantities of cDNA in each sample were calculated relative to this standard curve and normalized to 18s rRNA as housekeep gene control. Primers are listed in the Table S1.
Protease Assays
The proteasome activity fluorometric assay kit II (UBPBio, catalog# J4120) was used to assess the chymotrypsin-like, trypsin-like, and caspase-like activities of the proteasome according to the manufacturer’s instructions. Cells were plated in 10 cm2 dishes (850,000cells/plate) and harvested the next day in 1 ml of cold lysis buffer (40 mM Tris pH 7.2, 50 mM NaCl, 2 mM b-mercaptoethanol (bME), 2 mM ATP, 5 mM MgCl2, 10% glycerol). After sonication, the lysates were centrifuged and the protein concentrations assessed by a Bradford’s assay compared to a BSA standard curve. 25 mg of protein was added to a black 96 well plate (Costar catalog#3631) in 50 ml in replicates of 3 for each sample. 50 ml of one of the three substrates, Suc-LLVY-AMC (chymotrypsin-like), Boc-LRR-AMC (Trypsin-like), and Z-LLE-AMC (caspase-like) (diluted down to 100 mM in 1X assay buffer) was added to each lysate containing well. The plate was read on a Synergy HT (BioTek) plate reader with excitation/emission filter sets at 360/40 nm and 460/40 nm, respectively. Readings were taken ever 1:30 minutes for a total of 30 minutes. The fluorometric arbitrary units were graphed over time and linear regression analysis performed in PRISM to calculate the slope for each condition.
Clustering Analysis
Normalized RNA-seq data was downloaded as transcripts per million from the Cancer Cell Line Encyclopedia. Cell lines with homozygous ATG7 deletions were identified using cBioPortal (Gao et al., 2013; Cerami et al., 2012). Expression was Z score normalized and selected to include only genes within two NRF2 gene signatures (Goldstein et al., 2016; Namani et al., 2018). Hierarchical clustering was performed using Euclidean distance with complete linkage. A heatmap displaying gene expression was generated using the Complex Heat map R package (v1.20.0) (PMID: 27207943). ATG7 and NRF2 classification was compared using a Fisher’s exact test.
QUANTIFICATION AND STATISTICAL ANALYSIS
All graphs and statistical analysis was performed with PRISM software. The statistical details for each experiment can be found in the corresponding figure legend. One-way analyses of variance (ANOVA), two-way analyses of variance, or unpaired Student’s t-tests were performed where indicated in figure legends using Prism/Graphpad. *p < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
DATA AND CODE AVAILABILITY
The datasets generated during this study are available through the Cancer Cell Line Encyclopedia available at https://portals.broadinstitute.org/ccle/about.
Supplementary Material
Highlights.
An acute CRISPR/Cas9 assay identifies autophagy-dependent cancer cell lines
Autophagy-dependent cells can undergo selection to circumvent loss of autophagy
Cancer cells acquire dependence on NRF2 signaling to maintain proteostasis
Adaptation to loss of autophagy increases sensitivity to proteasome inhibition
ACKNOWLEDGMENTS
This work was supported by NIH grants RO1CA150925 (to A.T.), RO1CA190170 (to A.T. and D.L.G.), 5 T32 CA 190216-2 (C.G.T), 1K01OD022982 (D.R.), the American Cancer Society Post-Doctoral fellowship AWD 183648 (C.G.T), and shared resources supported by the University of Colorado Cancer Center P30CA046934. We would like to thank Elma Kajtazovic, Darya Wodetzki, and Jackie Thorburn for help with experimentation.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.devcel.2019.07.010.
REFERENCES
- Alam J, Killeen E, Gong P, Naquin R, Hu B, Stewart D, Ingelfinger JR, and Nath KA (2003). Heme activates the heme oxygenase-1 gene in renal epithelial cells by stabilizing Nrf2. Am. J. Physiol. Renal Physiol 284, F743–F752. [DOI] [PubMed] [Google Scholar]
- Arlt A, Bauer I, Schafmayer C, Tepel J, Mu€erkö ster SS, Brosch M, Rö der C, Kalthoff H, Hampe J, Moyer MP, et al. (2009). Increased proteasome subunit protein expression and proteasome activity in colon cancer relate to an enhanced activation of nuclear factor E2-related factor 2 (Nrf2). Oncogene 28, 3983–3996. [DOI] [PubMed] [Google Scholar]
- Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehá r J, Kryukov GV, Sonkin D, et al. (2012). The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blomen VA, Má jek P, Jae LT, Bigenzahn JW, Nieuwenhuis J, Staring J, Sacco R, Van Diemen FR, Olk N, Stukalov A, et al. (2015). Gene essentiality and synthetic lethality in haploid human cells. Science 350, 1092–1096. [DOI] [PubMed] [Google Scholar]
- Bryant KL, Stalnecker CA, Zeitouni D, Klomp JE, Peng S, Tikunov AP, Gunda V, Pierobon M, Waters AM, George SD, et al. (2019). Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat. Med 25, 628–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadwell K, and Debnath J (2018). Beyond self-eating: the control of nonautophagic functions and signaling pathways by autophagy-related proteins. J. Cell Biol 217, 813–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camp ND, James RG, Dawson DW, Yan F, Davison JM, Houck SA, Tang X, Zheng N, Major MB, and Moon RT (2012). Wilms tumor gene on X chromosome (WTX) inhibits degradation of NRF2 protein through competitive binding to KEAP1 protein. J. Biol. Chem 287, 6539–6550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. (2012). The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins KP, Jackson KM, and Gustafson DL (2018). Hydroxychloroquine: a physiologically-based pharmacokinetic model in the context of cancerrelated autophagy modulation. J. Pharmacol. Exp. Ther 365, 447–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dantuma NP, Lindsten K, Glas R, Jellne M, and Masucci MG (2000). Short-lived green fluorescent proteins for quantifying ubiquitin/proteasomedependent proteolysis in living cells. Nat. Biotechnol 18, 538–543. [DOI] [PubMed] [Google Scholar]
- Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G, Mukherjee C, Shi Y, Gé linas C, Fan Y, et al. (2006). Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 10, 51–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dikic I (2017). Proteasomal and autophagic degradation systems. Annu. Rev. Biochem 86, 193–224. [DOI] [PubMed] [Google Scholar]
- Eng CH, Wang Z, Tkach D, Toral-Barza L, Ugwonali S, Liu S, Fitzgerald SL, George E, Frias E, Cochran N, et al. (2016). Macroautophagy is dispensable for growth of KRAS mutant tumors and chloroquine efficacy. Proc. Natl. Acad. Sci. USA 113, 182–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzwalter BE, Towers CG, Sullivan KD, Andrysik Z, Hoh M, Ludwig M, O’Prey J, Ryan KM, Espinosa JM, Morgan MJ, and Thorburn A (2018). Autophagy Inhibition Mediates Apoptosis Sensitization in Cancer Therapy by Relieving FOXO3a Turnover. Dev. Cell 44, 555–565 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. (2013). Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal 6, pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein LD, Lee J, Gnad F, Klijn C, Schaub A, Reeder J, Daemen A, Bakalarski CE, Holcomb T, Shames DS, et al. (2016). Recurrent loss of NFE2L2 Exon 2 is a mechanism for Nrf2 pathway activation in human cancers. Cell Rep. 16, 2605–2617. [DOI] [PubMed] [Google Scholar]
- Gump JM, and Thorburn A (2014). Sorting cells for basal and induced autophagic flux by quantitative ratiometric flow cytometry. Autophagy 10, 1327–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JY, Chen HY, Mathew R, Fan J, Strohecker AM, Karsli-Uzunbas G, Kamphorst JJ, Chen G, Lemons JM, Karantza V, et al. (2011). Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 25, 460–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JY, Karsli-Uzunbas G, Mathew R, Aisner SC, Kamphorst JJ, Strohecker AM, Chen G, Price S, Lu W, Teng X, et al. (2013). Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes Dev. 27, 1447–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JY, Teng X, Laddha SV, Ma S, Van Nostrand SC, Yang Y, Khor S, Chan CS, Rabinowitz JD, and White E (2016). Autophagy provides metabolic substrates to maintain energy charge and nucleotide pools in Ras-driven lung cancer cells. Genes Dev. 30, 1704–1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart T, Chandrashekhar M, Aregger M, Steinhart Z, Brown KR, Macleod G, Mis M, Zimmermann M, Fradet-Turcotte A, Sun S, et al. (2015). High-resolution CRISPR screens reveal fitness genes and genotypespecific cancer liabilities. Cell 163, 1515–1526. [DOI] [PubMed] [Google Scholar]
- Ichimura Y, Kirisako T, Takao T, Satomi Y, Shimonishi Y, Ishihara N, Mizushima N, Tanida I, Kominami E, Ohsumi M, et al. (2000). A ubiquitin-like system mediates protein lipidation. Nature 408, 488–492. [DOI] [PubMed] [Google Scholar]
- Itoh K, Wakabayashi N, Katoh Y, Ishii T, O’Connor T, and Yamamoto M (2003). Keap1 regulates both cytoplasmic-nuclear shuttling and degradation of Nrf2 in response to electrophiles. Genes Cells 8, 379–391. [DOI] [PubMed] [Google Scholar]
- Joo J, Wang B, Frankel E, Ge L, Xu L, Iyengar R, Li-Harms X, Wright C, Shaw T, Lindsten T, et al. (2016). The noncanonical role of ULK/ATG1 in ER-toGolgi trafficking is essential for cellular homeostasis. Mol. Cell 62, 491–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapeta S, Chondrogianni N, and Gonos ES (2010). Nuclear erythroid factor 2-mediated proteasome activation delays senescence in human fibroblasts. J. Biol. Chem 285, 8171–8184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karsli-Uzunbas G, Guo JY, Price S, Teng X, Laddha SV, Khor S, Kalaany NY, Jacks T, Chan CS, Rabinowitz JD, et al. (2014). Autophagy is required for glucose homeostasis and lung tumor maintenance. Cancer Discov. 4, 914–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinsey CG, Camolotto SA, Boespflug AM, Gullien KP, Foth M, Truong A, Schuman SS, Shea JE, Seipp MT, Yap JT, et al. (2019). Protective autophagy elicited by RAF>MEK>ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nat. Med 25, 620–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, Sou YS, Ueno I, Sakamoto A, Tong KI, et al. (2010). The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol 12, 213–223. [DOI] [PubMed] [Google Scholar]
- Korolchuk VI, Mansilla A, Menzies FM, and Rubinsztein DC (2009). Autophagy inhibition compromises degradation of ubiquitin-proteasome pathway substrates. Mol. Cell 33, 517–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak MK, Wakabayashi N, Itoh K, Motohashi H, Yamamoto M, and Kensler TW (2003). Modulation of gene expression by cancer chemopreventive dithiolethiones through the Keap1-Nrf2 pathway. Identification of novel gene clusters for cell survival. J. Biol. Chem 278, 8135–8145. [DOI] [PubMed] [Google Scholar]
- Lau A, Wang XJ, Zhao F, Villeneuve NF, Wu T, Jiang T, Sun Z, White E, and Zhang DD (2010). A noncanonical mechanism of Nrf2 activation by autophagy deficiency: direct interaction between Keap1 and p62. Mol. Cell. Biol 30, 3275–3285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CS, Lee LC, Yuan TL, Chakka S, Fellmann C, Lowe SW, Caplen NJ, Mccormick F, and Luo J (2019). MAP kinase and autophagy pathways cooperate to maintain RAS mutant cancer cell survival. Proc. Natl. Acad. Sci. USA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy JM, Thompson JC, Griesinger AM, Amani V, Donson AM, Birks DK, Morgan MJ, Mirsky DM, Handler MH, Foreman NK, et al. (2014). Autophagy inhibition improves chemosensitivity in BRAF(V600E) brain tumors. Cancer Discov. 4, 773–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy JMM, Towers CG, and Thorburn A (2017). Targeting autophagy in cancer. Nat. Rev. Cancer 17, 528–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang X, Potter J, Kumar S, Zou Y, Quintanilla R, Sridharan M, Carte J, Chen W, Roark N, Ranganathan S, et al. (2015). Rapid and highly efficient mammalian cell engineering via Cas9 protein transfection. J. Biotechnol 208, 44–53. [DOI] [PubMed] [Google Scholar]
- Lock R, Roy S, Kenific CM, Su JS, Salas E, Ronen SM, and Debnath J (2011). Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Mol. Biol. Cell 22, 165–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma XH, Piao SF, Dey S, Mcafee Q, Karakousis G, Villanueva J, Hart LS, Levi S, Hu J, Zhang G, et al. (2014). Targeting ER stress-induced autophagy overcomes BRAF inhibitor resistance in melanoma. J. Clin. Invest 124, 1406–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall RS, Li F, Gemperline DC, Book AJ, and Vierstra RD (2015). Autophagic degradation of the 26S proteasome is mediated by the dual ATG8/ubiquitin receptor RPN10 in Arabidopsis. Mol. Cell 58, 1053–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maycotte P, Gearheart CM, Barnard R, Aryal S, Mulcahy Levy JM, Fosmire SP, Hansen RJ, Morgan MJ, Porter CC, Gustafson DL, et al. (2014). STAT3-mediated autophagy dependence identifies subtypes of breast cancer where autophagy inhibition can be efficacious. Cancer Res.74, 2579–2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulcahy Levy JM, Zahedi S, Griesinger AM, Morin A, Davies KD, Aisner DL, Kleinschmidt-Demasters BK, Fitzwalter BE, Goodall ML, Thorburn J, et al. (2017). Autophagy inhibition overcomes multiple mechanisms of resistance to BRAF inhibition in brain tumors. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namani A, Matiur Rahaman M, Chen M, and Tang X (2018). Gene-expression signature regulated by the KEAP1-NRF2-CUL3 axis is associated with a poor prognosis in head and neck squamous cell cancer. BMC Cancer 18, 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obeng EA, Carlson LM, Gutman DM, Harrington WJ, Lee KP, and Boise LH (2006). Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood 107, 4907–4916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Prey J, Sakamaki J, Baudot AD, New M, Van Acker T, Tooze SA, Long JS, and Ryan KM (2017). Application of CRISPR/Cas9 to autophagy research. Methods Enzymol. 588, 79–108. [DOI] [PubMed] [Google Scholar]
- Oslowski CM, and Urano F (2011). Measuring ER stress and the unfolded protein response using mammalian tissue culture system. Methods Enzymol 490, 71–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pajares M, Jimé nez-Moreno N, Garcıá-Yagu€e ÁJ, Escoll M, De Ceballos ML, Van Leuven F, Rá bano A, Yamamoto M, Rojo AI, and Cuadrado A (2016). Transcription factor NFE2L2/NRF2 is a regulator of macroautophagy genes. Autophagy 12, 1902–1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poillet-Perez L, Xie X, Zhan L, Yang Y, Sharp DW, Hu ZS, Su X, Maganti A, Jiang C, Lu W, et al. (2018). Autophagy maintains tumour growth through circulating arginine. Nature 563, 569–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radhakrishnan SK, Lee CS, Young P, Beskow A, Chan JY, and Deshaies RJ (2010). Transcription factor Nrf1 mediates the proteasome recovery pathway after proteasome inhibition in mammalian cells. Mol. Cell 38, 17–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangwala R, Leone R, Chang YC, Fecher LA, Schuchter LM, Kramer A, Tan KS, Heitjan DF, Rodgers G, Gallagher M, et al. (2014). Phase I trial of hydroxychloroquine with dose-intense temozolomide in patients with advanced solid tumors and melanoma. Autophagy 10, 1369–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao S, Tortola L, Perlot T, Wirnsberger G, Novatchkova M, Nitsch R, Sykacek P, Frank L, Schramek D, Komnenovic V, et al. (2014). A dual role for autophagy in a murine model of lung cancer. Nat. Commun. 5, 3056. [DOI] [PubMed] [Google Scholar]
- Rojas-Puentes LL, Gonzalez-Pinedo M, Crismatt A, Ortega-Gomez A, Gamboa-Vignolle C, Nuñ ez-Gomez R, Dorantes-Gallareta Y, ArceSalinas C, and Arrieta O (2013). Phase II randomized, double-blind, placebo-controlled study of whole-brain irradiation with concomitant chloroquine for brain metastases. Radiat. Oncol 8, 209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojo de la Vega M, Chapman E, and Zhang DD (2018). NRF2 and the hallmarks of cancer. Cancer Cell 34, 21–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeldt MT, O’Prey J, Morton JP, Nixon C, Mackay G, Mrowinska A, Au A, Rai TS, Zheng L, Ridgway R, et al. (2013). p53 status determines the role of autophagy in pancreatic tumour development. Nature 504, 296–300. [DOI] [PubMed] [Google Scholar]
- Strohecker AM, Guo JY, Karsli-Uzunbas G, Price SM, Chen GJ, Mathew R, Mcmahon M, and White E (2013). Autophagy sustains mitochondrial glutamine metabolism and growth of BrafV600E-driven lung tumors. Cancer Discov. 3, 1272–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorburn J, Staskiewicz L, Goodall ML, Dimberg L, Frankel AE, Ford HL, and Thorburn A (2017). Non-cell-autonomous effects of autophagy inhibition in tumor cells promote growth of drug-resistant cells. Mol. Pharmacol 91, 58–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towers CG, and Thorburn A (2016). Therapeutic targeting of autophagy. EBioMedicine 14, 15–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandeputte C, Guizon I, Genestie-Denis I, Vannier B, and Lorenzon G (1994). A microtiter plate assay for total glutathione and glutathione disulfide contents in cultured/isolated cells: performance study of a new miniaturized protocol. Cell Biol. Toxicol 10, 415–421. [DOI] [PubMed] [Google Scholar]
- Vangala JR, Dudem S, Jain N, and Kalivendi SV (2014). Regulation of PSMB5 protein and beta subunits of mammalian proteasome by constitutively activated signal transducer and activator of transcription 3 (STAT3): potential role in bortezomib-mediated anticancer therapy. J. Biol. Chem 289, 12612–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, and Kundu M (2017). Canonical and noncanonical functions of ULK/Atg1. Curr. Opin. Cell Biol 45, 47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Maxwell BA, Joo JH, Gwon Y, Messing J, Mishra A, Shaw TI, Ward AL, Quan H, Sakurada SM, et al. (2019). ULK1 and ULK2 regulate stress granule disassembly through phosphorylation and activation of VCP/p97. Mol. Cell 74, 742–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Birsoy K, Hughes NW, Krupczak KM, Post Y, Wei JJ, Lander ES, and Sabatini DM (2015). Identification and characterization of essential genes in the human genome. Science 350, 1096–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie X, Koh JY, Price S, White E, and Mehnert JM (2015). Atg7 overcomes senescence and promotes growth of BrafV600E-driven melanoma. Cancer Discov. 5, 410–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang A, Herter-Sprie G, Zhang H, Lin EY, Biancur D, Wang X, Deng J, Hai J, Yang S, Wong KK, et al. (2018). Autophagy sustains pancreatic cancer growth through both cell autonomous and non-autonomous mechanisms. Cancer Discov. 8, 276–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang A, Rajeshkumar NV, Wang X, Yabuuchi S, Alexander BM, Chu GC, Von Hoff DD, Maitra A, and Kimmelman AC (2014). Autophagy is critical for pancreatic tumor growth and progression in tumors with p53 alterations. Cancer Discov. 4, 905–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H, Bause A, Li Y, Stommel JM, Dell’Antonio G, et al. (2011). Pancreatic cancers require autophagy for tumor growth. Genes Dev. 25, 717–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during this study are available through the Cancer Cell Line Encyclopedia available at https://portals.broadinstitute.org/ccle/about.