

Abstract
Porcine epidemic diarrhoea virus (PEDV) is an emerging and re‐emerging epizootic virus of swine that causes substantial economic losses to the pig industry in China and other countries. The variations in the virus, and its co‐infections with other enteric viruses, have contributed to the poor control of PEDV infection. In the current study, a broad epidemiological investigation of PEDV was carried out in 22 provinces or municipalities of China during 2015–2018. The enteric viruses causing co‐infection with PEDV and the genetic diversity of the PEDV S1 gene were also analysed. The results indicated that, of the 543 diarrhoea samples, 66.85% (363/543) were positive for PEDV, and co‐infection rates of PEDV with 13 enteric viruses ranged from 3.58% (13/363) to 81.55% (296/363). Among these enteric viruses, the signs of diarrhoea induced by PEDV were potentially associated with co‐infections with porcine enterovirus 9/10 (PEV) and torque teno sus virus 2 (TTSuV‐2) (p < .05). The 147 PEDV strains identified in our study belong to Chinese pandemic strains and exhibited genetic diversity. The virulence‐determining S1 proteins of PEDV pandemic strains were undergoing amino acid mutations, in which S58_S58insQGVN–N135dup–D158_I159del‐like mutations were common patterns (97.28%, 143/147). When compared with 2011–2014 PEDV strains, the amino acid mutations of PEDV pandemic strains were mainly located in the N‐terminal domain of S1 (S1‐NTD), and 21 novel mutations occurred in 2017 and 2018. Furthermore, protein homology modelling showed that the mutations in pattern of insertion and deletion mutations of the S1 protein of PEDV pandemic strains may have caused structural changes on the surface of the S1 protein. These data provide a better understanding of the co‐infection and genetic evolution of PEDV in China.
Keywords: co‐infection, mutation, PEDV, S1 gene
1. INTRODUCTION
Porcine epidemic diarrhoea virus (PEDV) is an enveloped, single‐stranded, positive‐sense RNA virus, belonging to the genus Alphacoronavirus. PEDV was identified as the causative agent of porcine epidemic diarrhoea (PED) in 1978 (Pensaert & de Bouck, 1978). This disease is characterized by acute watery diarrhoea, vomiting and dehydration, with high mortality that often reaches 100% in neonatal piglets. From 1984 to early 2010, PEDV circulated in the pig population in China, but there were no large‐scale outbreaks (Tamura, Stecher, Peterson, Filipski, & Kumar, 2013). At the end of 2010, a PEDV outbreak occurred in several pig‐producing provinces in southern China (Li et al., 2012). Since then, the disease has spread throughout other provinces of China and has led to enormous economic losses within the pork industry. Currently, PEDV infection is widespread in swine‐farming countries in Asia, Europe and North America (Lin, Saif, Marthaler, & Wang, 2016; Sun, Wang, Wei, Chen, & Feng, 2016; Wang et al., 2016). The emergence and re‐emergence of PEDV cause severe economic losses and pose significant public health concerns throughout the world.
In addition to PEDV, a large variety of porcine enteric pathogens has been found in diarrhoea samples from pigs, including porcine teschovirus (PTV), porcine transmissible gastroenteritis virus (TGEV), porcine sapelovirus (PSV), porcine enterovirus 9/10 (PEV), mammalian reovirus (MRV), porcine group A rotavirus (GARV), porcine astrovirus (PAstV), porcine torovirus (PToV), torque teno sus virus 2 (TTSuV‐2), porcine bocavirus (PBoV), porcine kobuvirus (PKV) and porcine deltacoronavirus (PDCoV); however, data have indicated that PEDV is the major cause of viral diarrhoeal disease in swine in China (Decaro et al., 2005; Song et al., 2015; Wang, Zhou, et al., 2014; Zell et al., 2000; Zhang, Tang, Yue, Ren, & Song, 2014; Zhang et al., 2013). Viral diarrhoeal diseases caused by a variety of porcine enteric pathogens, and co‐infection with multiple enteric pathogens, are very prevalent in piglets with diarrhoea (Chen et al., 2018; Zhang et al., 2014, 2013). In the case of PEDV, the variations in the virus and its co‐infections with multiple pathogens make it difficult to control the infection. However, limited data are available on co‐infections during PEDV infection in piglets with diarrhoea. Therefore, it is necessary to investigate the co‐infection of PEDV with other pathogens.
The S protein of coronaviruses is responsible for induction of neutralizing antibodies, specific receptor binding and cell membrane fusion (Li, 2015; Sun et al., 2008). Deletion and/or insertion mutations of the S protein are potentially associated with the pathogenicity and tissue tropism of coronaviruses (Lin et al., 2016). In December 2013, an S‐INDEL strain, OH851, with reduced virulence, was reported in the USA (Wang, Byrum, & Zhang, 2014). Subsequently, S‐INDEL strains similar to OH851 have been frequently reported in other countries (Yamamoto, Soma, Nakanishi, Yamaguchi, & Niinuma, 2015). In a recent study, Wang et al. (2016) reported that three PEDV strains, non‐S‐INDEL, ‘CV777’ S‐INDEL and ‘US’ S‐INDEL co‐circulated in the swine population in China (Wang et al., 2016). The S1 region of the S protein is under greater immune pressure than the S2 region (Aydin, Al‐Khooly, & Lee, 2014; Jarvis et al., 2016). Widespread epidemiological investigations have demonstrated that the virulence‐determining S1 gene of PEDV is undergoing rapid mutation. Additionally, sialic acid binding activity is located in the N‐terminal domain (NTD) of PEDV S1 protein (Li, Kuppeveld, He, Rottier, & Bosch, 2016); the S1‐NTD of Chinese PEDV pandemic/variant strains exhibits strong sialic acid binding activity when compared with the classic PEDV strain (Deng et al., 2016). Thus, patterns of insertion and deletion mutations of the S1 protein (S1‐IDMPs) are an important target for better understanding of the emergence and re‐emergence of PEDV in Asia, Europe and North America. Although genotyping of PEDV pandemic strains has been widely undertaken in the pig population in China (Chen et al., 2019; Wang et al., 2016; Wen et al., 2018), there is fairly limited information available on the S1‐IDMPs of PEDV pandemic strains.
In our study, to investigate the co‐infection of PEDV with other pathogens and the evolution of S1‐IDMPs of PEDV circulating in China, 543 intestinal samples from diarrhoeic piglets were collected from 2015 to 2018. All the samples were used to investigate the co‐infection of PEDV with other enteric pathogens, and the S1 genes obtained were subjected to analysis of S1‐IDMPs, phylogenetic analysis and homology modelling of the protein. Our aim was to provide a better understanding of the epidemiology of PEDV in China.
2. MATERIALS AND METHODS
2.1. Ethics statements
The study was approved by the Animal Experiments Committee of the Heilongjiang Bayi Agricultural University (registration protocol 201401002). The field study did not involve endangered or protected species. No specific permissions were required for the collection of samples because the samples were collected from public areas or nonprotected areas. All sampling and publication of the data were approved by the farm owners.
2.2. Sample collection
In this study, 543 samples of intestinal tissues from piglets (aged <10 days) with diarrhoea were collected from January 2015 to August 2018 in 22 provinces or municipalities of China including Anhui (n = 11), Beijing (n = 4), Fujian (n = 19), Gansu (n = 4), Guangdong (n = 22), Hebei (n = 34), Heilongjiang (n = 98), Henan (n = 18), Hubei (n = 43), Hunan (n = 9), Jiangsu (n = 32), Jiangxi (n = 19), Jilin (n = 30), Liaoning (n = 69), Shandong (n = 26), Shaanxi (n = 1), Shanxi (n = 27), Sichuan (n = 18), Shanghai (n = 33), Tianjin (n = 4), the Xinjiang Uygur Autonomous Region (n = 4) and the Inner Mongolia Autonomous Region (n = 18). All samples were stored at –80°C.
2.3. PCR detection of PEDV and other pathogens
The RNA extraction and cDNA synthesis were performed as described previously by Wang et al. (2016). The DNA extraction was carried out according to the protocol described by Qi et al. (2019). PEDV and 12 other enteric viruses, PTV, TGEV, PSV, PEV, MRV, GARV, PAstV, PToV, TTSuV‐2, PBoV, PKV and PDCoV, were detected by nested PCR as described in previous studies (Chu, Poon, Guan, & Peiris, 2008; Decaro et al., 2005; Elschner, Prudlo, Hotzel, Otto, & Sachse, 2002; Song et al., 2015; Van et al., 2016; Wang et al., 2016, 2009; Wang, Zhou, et al., 2014; Zell et al., 2000; Zheng et al., 2016) or as designed in this study (Table S1). The map of China was drawn with an online map‐generation website (http://pixelmap.amcharts.com/).
2.4. Sequencing and analysis of the S1 genes of PEDV
Amplification of the S1 gene of PEDV was carried out according to the protocol described by Wang et al. (2016). The amplified S1 genes were cloned into the vector pMD18‐T according to the manufacturer's instructions (TaKaRa Biotechnology Co., Ltd). Three positive clones from each sample were subjected to Sanger sequencing. All nucleotide sequences generated in our study were submitted to GenBank. Sequence analysis was carried out using the EditSeq tool in Lasergene DNASTAR™ 5.06 software (DNASTAR Inc). Multiple sequence alignments were carried out using the multiple sequence alignment tool of DNAMAN 6.0 software (Lynnon BioSoft). The sequence variation was described according to the nomenclature system (den Dunnen & Antonarakis, 2001). A divergence analysis of the S1 protein of PEDV was performed with WebLogo (http://weblogo.threeplusone.com/), online software for sequence logo generator (Crooks, Hon, Chandonia, & Brenner, 2004). The comparison of point mutations in the S1 protein of PEDV was made with GraphPad Prism® 8.0 (GraphPad Software, Inc).
2.5. Phylogenetic analysis
For the phylogenetic analysis, the S1 genes of PEDV strains were retrieved from GenBank (Table S2). These nucleotide sequences were used to generate a neighbour‐joining phylogenetic tree of the S1 gene using the ClustalX alignment tool in MEGA6.06 software (Tamura et al., 2013). A neighbour‐joining phylogenetic tree was built using the p‐distance model and 1,000 bootstrap replicates. The phylogenetic tree was annotated with the Interactive Tree Of Life (iTOL) software (http://itol.embl.de/), an online tool for the display and annotation of phylogenetic trees (Letunic & Bork, 2016).
2.6. Molecular modelling and analysis of the S1 protein of PEDV
The dominant S1 amino acid sequences from each year during 2015–2018 were aligned (named 2015_PEDV‐strain, 2016_PEDV‐strain, 2017_PEDV‐strain, 2018_PEDV‐strain, respectively) (Table S3) and then selected for investigation of the effect of S1‐IDMPs on the conformation of the S1 protein of PEDV. The predicted tertiary structures of the S1 region were modelled using the open‐source modelling server SWISS‐MODEL (https://swissmodel.expasy.org/) from the Swiss Institute of Bioinformatics (Biasini et al., 2014). The S1 monomer tertiary structures were prepared by using the spike protein of Human coronavirus NL63 (PDB ID: 5SZS) as a template. Illustrations and comparisons of these modelled tertiary structures were obtained using the python‐based molecular viewer PyMOL (The PyMOL Molecular Graphics System, Version 1.7.4 Schrödinger, LLC). In addition, the asparagine (N)‐linked glycosylation sites were predicted using the NetNGlyc 1.0 Server (http://www.cbs.dtu.dk/services/NetNGlyc/) (Gupta, Jung, & Brunak, 2004).
2.7. Statistical analysis
The correlation of PEDV infection with other pathogens (PTV, TGEV, PSV, PEV, MRV, GARV, PAstV, PToV, TTSuV‐2, PBoV, PKV, PDCoV and PCV‐3) was carried out in 2 × 2 contingency tables using the chi‐square (χ 2) test in IBM SPSS Statistics, version 22.0 (IBM® SPSS Inc). The significance level in all analyses was 5%, with a confidence interval of 95%. The difference in values was considered statistically significant or highly significant if the associated p value was <.05 or <.01, respectively. The results for PCV‐3 detection in these 543 diarrhoea samples have been published in our previous study (Qi et al., 2019).
3. RESULTS
3.1. Co‐infection of PEDV with multiple pathogens in diarrhoeic piglets
Of the 543 diarrhoea samples, PEDV was found in 66.85% (363/543), PKV in 81.22% (441/543), PBoV in 61.14% (332/543), PTV in 60.77% (330/543), PEV in 40.88% (222/543), TTSuV‐2 in 33.15% (180/543), GARV in 20.07% (109/543), PAstV in 16.39% (89/543), MRV in 11.23% (61/543), PToV in 9.21% (50/543), TGEV in 8.47% (46/543), PSV in 5.52% (30/543) and PDCoV in 4.79% (26/543) (Table S4; Figure 1a). Of the 543 diarrhoea samples, infection with PKV, PEDV, PBoV, PTV and PEV showed a wider geographical distribution than the other enteric viruses (Figure 1b). Among the 13 enteric viruses, 96.50% (524/543) of samples had at least two pathogens, 68.87% (374/543) of samples had three to five different pathogens and the mean number of pathogens per sample was 4.25 (Figure 1c). Of the 363 PEDV‐positive samples, co‐infection with PKV, PTV and PBoV reached 81.55% (296/363), 62.53% (227/363) and 55.65% (202/363), respectively (Figure 2a). Among the samples positive for the 13 enteric viruses, PEDV exhibited high co‐infection rates, ranging from 37.70% to 92.93% (Figure 2b). In addition, a statistical analysis showed that PEDV‐induced diarrhoea symptoms were potentially associated with co‐infection of PEV and TTSuV‐2 (p < .05) (Table 1).
Figure 1.
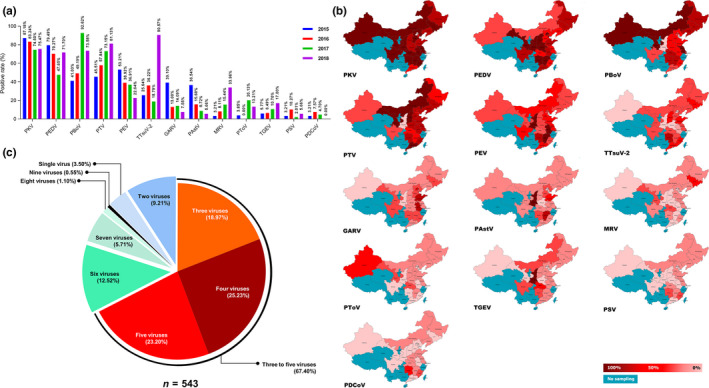
The positive rate and geographic distribution of the selected enteric viruses in the diarrhoea samples during 2015–2018. (a) The positive rate of the selected 13 enteric viruses in the 543 diarrhoea samples during 2015–2018. (b) The geographic distribution of the selected 13 enteric viruses in the 543 diarrhoea samples during 2015–2018. (c) The mix‐infection patterns of the selected 13 enteric viruses in the 543 diarrhoea samples during 2015–2018 [Colour figure can be viewed at wileyonlinelibrary.com]
Figure 2.
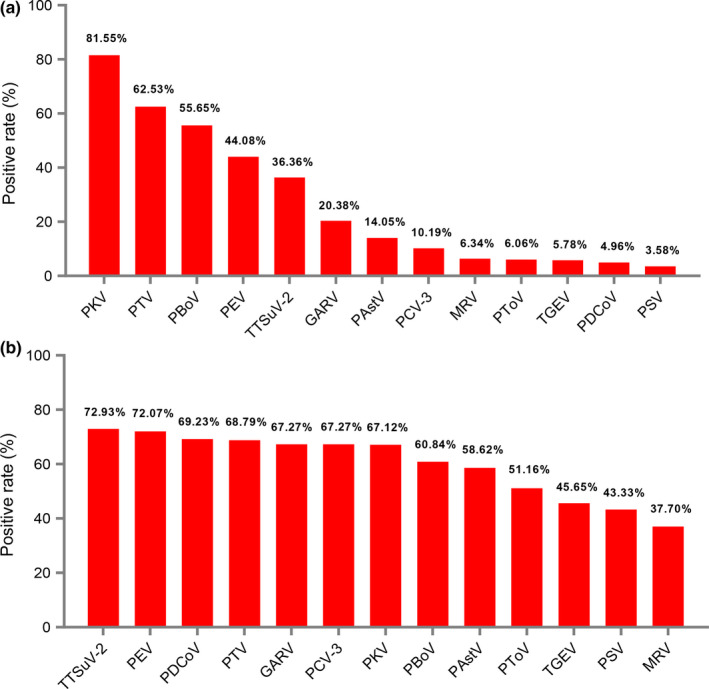
The co‐infection of PEDV with the selected enteric viruses in the diarrhoea samples during 2015–2018. (a) The co‐infection rate of the selected 13 enteric viruses in 363 PEDV‐positive samples. (b) The co‐infection rate of PEDV in the 13 enteric viruses positive samples [Colour figure can be viewed at wileyonlinelibrary.com]
Table 1.
The statistical analysis of correlations of PEDV with other pathogens
| P value | Odds ratio (OR) | 95% Confidence interval (95% CI) | |
|---|---|---|---|
| PKV | .782 | 1.066 | 0.677–1.680 |
| PBoV | .000 | 0.502 | 0.341–0.737 |
| PTV | .233 | 1.248 | 0.867–1.795 |
| PEV | .032 | 1.500 | 1.035–2.173 |
| TTSuV−2 | .033 | 1.528 | 1.033–2.260 |
| GARV | .797 | 1.061 | 0.677–1.662 |
| PAstV | .075 | 0.654 | 0.409–1.046 |
| MRV | .000 | 0.253 | 0.145–0.440 |
| PToV | .023 | 0.488 | 0.261–0.914 |
| TGEV | .001 | 0.381 | 0.207–0.701 |
| PSV | .005 | 0.356 | 0.169–0.751 |
| PDCoV | .871 | 0.934 | 0.408–2.138 |
| PCV−3 | .212 | 1.478 | 0.798–2.740 |
The PEDV‐induced diarrhea symptoms associated‐viruses (PEV, TTsuV‐2) are shown in Bold.
Abbreviations: CI, confidence interval; OR, odd ratio.
3.2. Sequencing and analysis of S1 genes of PEDV
In our study, a total of 147 S1 genes of PEDV were successfully sequenced from 2016–2018 PEDV‐positive samples (Table S5). The 147 S1 genes identified were 2,376 bp in length, except for three strains: JL/2016/47a (2,388 bp), JL/2016/47b (2,388 bp) and LN/DT/2016/510a (2,352 bp). Nucleotide identity of 95.5%–99.9% and amino acid (aa) identity of 94.2%–99.7% were revealed among the 147 S1 genes identified. The 147 PEDV strains exhibited 90.7%–91.8% nucleotide sequence homology and 89.3%–90.7% amino acid homology when compared with the PEDV CV777 strain (GenBank accession no. AF353511). The S1‐IDMPs of the 147 PEDV strains identified in our study were analysed using the S1 protein of the PEDV CV777 strain as the comparator. The results indicated that 143 S1 proteins exhibited S1‐IDMP S58_S58insQGVN–N135dup–D158_I159del, two S1 proteins from the same farm (JL/2016/47a and JL/2016/47b) showed S1‐IDMP S58_S58insQGVN–N135dup–D158_I159del–T380_V380insGQRS and one S1 protein (LN/DT/2016/510a) exhibited S1‐IDMP S58_S58insQGVN–N135dup–D158_I159del–N553_Y560del. From the same farm, the LN/DT/2016/510b strain exhibited a different S1‐IDMP S58_S58insQGVN–N135dup–D158_I159del when compared with the LN/DT/2016/510a strain. For PEDV, the sialic acid binding activity is located in the NTD (aa 9–433) of the S1 protein; the cellular receptor binding is located in the receptor‐binding domain (RBD, aa 501–629) of the S1 protein (Li et al., 2016). In addition, the S1‐IDMPs of 147 Chinese PEDV pandemic strains occurred in the NTD of the S1 protein; only the S1‐IDMP of the LN/DT/2016/510a strain, S58_S58insQGVN–N135dup–D158_I159del–N553_Y560del, was located in the NTD and RBD of the S1 protein (Figure 3a). These data indicate that the S1‐IDMP S58_S58insQGVN–N135dup–D158_I159del, accounting for 97.28% (143/147), was the pandemic insertion and deletion mutation pattern in the 147 PEDV strains identified.
Figure 3.

Sequence analysis of S1 proteins of the PEDV strains identified in our study. (a) Divergence analysis of S1 proteins of PEDV strains identified in our study during 2015–2018. (b) The comparison of amino acid mutations of S1 proteins of PEDV strains identified in our study during 2015–2018 with PEDV CV777 strain. (c) The comparison of amino acid mutation positions of S1 proteins of PEDV strains identified in our study during 2015–2018 with PEDV CV777 strain [Colour figure can be viewed at wileyonlinelibrary.com]
To explore further the evolution of S1 proteins of PEDV strains, the deduced amino acids of the 54 S1 genes from NCBI (2011–2014 = 24, 2015 = 29), and the 147 S1 genes identified in this study (2016 = 72, 2017 = 46, 2018 = 29) were aligned. The results showed that, when compared with the dominant S1 amino acid sequences of 2011–2014 PEDV strains (2011–2014_PEDV‐strain), the amino acid mutations were concentrated mainly in the S1‐NTD region (40.91%, 18/44) (Figure 3a). The mean number of amino acid mutations was 13.00 in 2015 PEDV strains, 17.11 in 2016 PEDV strains, 18.87 in 2017 PEDV strains and 16.17 in 2018 PEDV strains (Figure 3b). In addition, the probabilities (%) of amino acid mutation sites V13A, L17F, G68Y, G70R, N139D, Y182H, M214T, P229L, T235I, I287M, H301Q, S334A, I503T, S524A, K570N, G616V and P638S were increased from 2015 to 2018 PEDV strains (Figure 3c, red circle), mutation sites L14I, A193S, R195N, D223G, T379I, G432S, E640V, F674V, D683A, P770L and T781M occurred in 2017 PEDV strains, and mutation sites V77A, H110Y, A112S, I154T, S246L, D304G, A331V, Y610H, A612D and K637T occurred in 2018 PEDV strains.
3.3. Phylogenetic analysis
For the phylogenetic analysis, a phylogenetic tree was constructed based on the S1 genes of the 147 PEDV strains identified in this study and 131 reference PEDV strains from GenBank. The phylogenic tree was divided into groups GI and GII, and the GII group was composed of two subgroups (GIIa and GIIb). The 147 PEDV strains identified in our study had a close relationship with Chinese PEDV pandemic reference strains, but exhibited genetic diversity (Figure 4). Of the 147 PEDV strains identified in our study, 43 strains (29.25%, 43/147) were placed into the GIIa subgroup, and the remaining 104 strains (70.75%, 104/147) were classified into the GIIb subgroup. The phylogenic tree showed that 64.79% (46/71) of 2011–2015 Chinese PEDV strains, 50.62% (41/81) of 2016 Chinese PEDV strains and 8.86% (7/79) of 2017–2018 Chinese PEDV strains belonged to the GIIa subgroup; 19.72% (14/71) of 2011–2015 Chinese PEDV strains, 49.38% (40/81) of 2016 Chinese PEDV strains and 91.13% (72/79) of 2017–2018 Chinese PEDV strains belonged to the GIIb subgroup (Figure 4).
Figure 4.
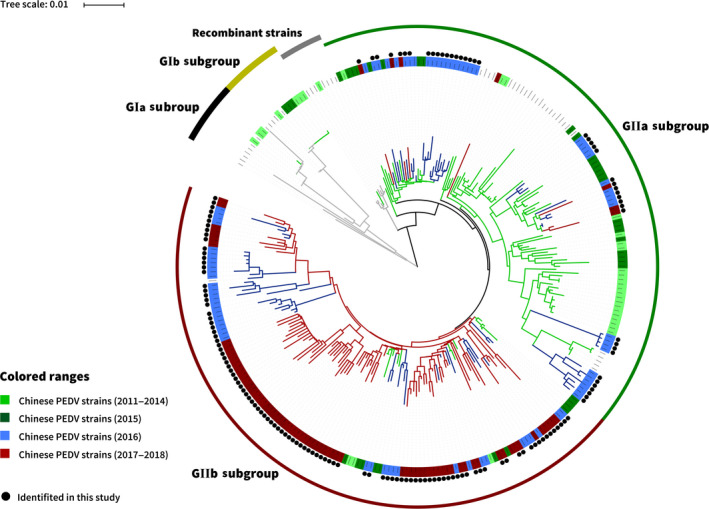
The S1‐based phylogenetic analysis of the PEDV strains identified in our study. Black circle diagram represents the 147 PEDV strains identified in this study [Colour figure can be viewed at wileyonlinelibrary.com]
3.4. Analysis of PEDV S1 protein modelling
Analysis of the S1 protein modelling indicated that the S1‐NTD of 2018_PEDV‐strain showed four major changes, in residues 55–64, 113–120, 130–142 and 157–165, when compared with the classical PEDV CV777 strain (Figure 5). When compared with 2015_PEDV‐strain, 2016_PEDV‐strain has an obvious structural change in residues 558–575, located in the S1‐RBD region (Figure 6a). Compared with 2016_PEDV‐strain, 2017_PEDV‐strain exhibited an obvious structural change in residues 130–143, located in the S1‐NTD region (Figure 6b). In addition, the predicted S1 structure of HLJ/2015/DP1‐1, which was identified in our previous study (Wang et al., 2016), has changed when compared with S1 modelling of 2015_PEDV‐strain. JL/2016/47a and LN/DT/2016/510a exhibited changes to varying degrees when compared with S1 modelling of 2016_PEDV‐strain, among which the deletion mutation N553VTNSYGY of the LN/DT/2016/510a strain was located in the RBD of the S1 protein of 2016_PEDV‐strain (Figure 7). In addition, the predicted N‐linked glycosylation sites of the S1 glycoprotein showed that HLJ/2015/DP1‐1 strain and LN/DT/2016/510a strain have lost one N‐linked glycosylation site at N381 and N556, predicted on the S1 of 2015_PEDV‐strain and 2016_PEDV‐strain, respectively (Figure 7a and c).
Figure 5.
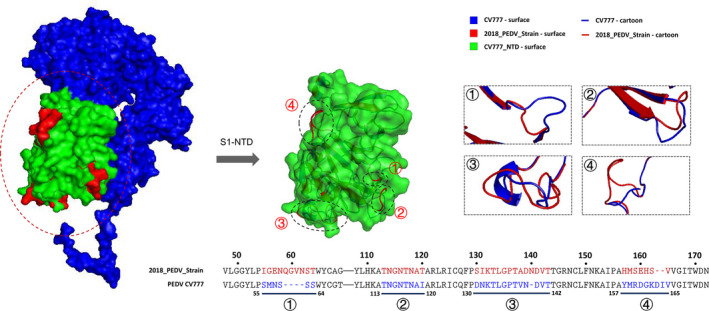
Comparative analysis of the predicted S1 protein modelling between PEDV CV777 strain and 2018_PEDV‐strain. The S1 protein modelling of PEDV CV777 strain was shown as surface and cartoon by blue. The S1 protein modelling of 2018_PEDV‐strian was shown as surface and cartoon by red. The mutant amino acid residues of S1 protein of 2018_PEDV‐strain were shown as surface by red. The S1‐NTD of PEDV CV777 strain was shown as surface by green [Colour figure can be viewed at wileyonlinelibrary.com]
Figure 6.
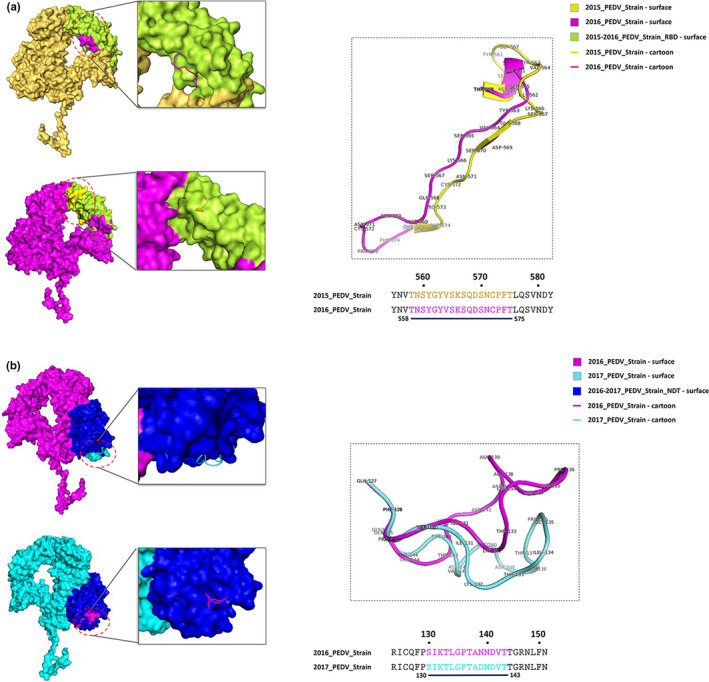
Comparative analysis of the predicted S1 protein modelling among 2015–2018 PEDV strains. (a) Comparative analysis of the predicted S1 protein modelling between 2015_PEDV‐strain and 2016_PEDV‐strain. (b) The compared S1 modelling of the 2016_PEDV‐strain and 2017_PEDV‐strain. The 2015_PEDV‐strain was shown as surface and cartoon by yellow; 2016_PEDV‐strain was shown as surface and cartoon by pink; 2017_PEDV‐strain was shown as surface and cartoon by cyan. The S1‐NDT of 2016‐2017_PEDV‐strain was shown as surface by blue. The S1‐RBD 2015‐2016_PEDV‐strain was shown as surface by limon [Colour figure can be viewed at wileyonlinelibrary.com]
Figure 7.
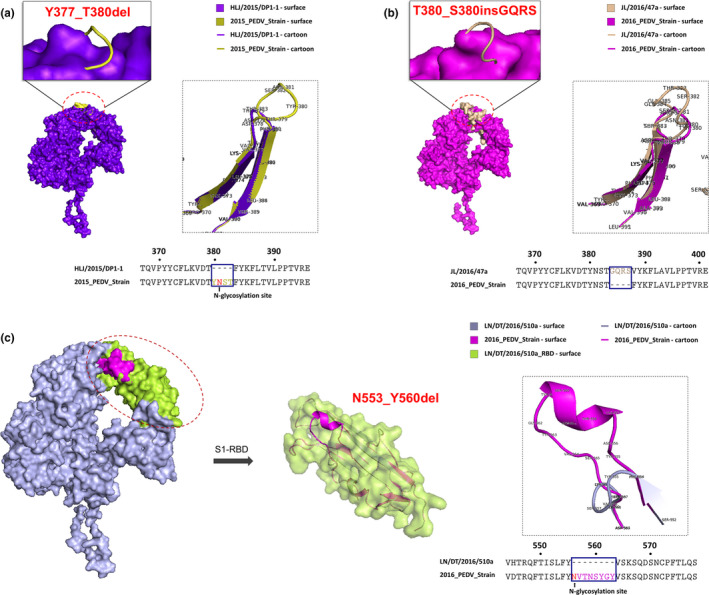
Comparative analysis of the predicted S1 protein modelling of PEDV HLJ/2015/DP1‐1, JL/2016/47a and LN/DT/2016/510a strains. (a) The compared S1 protein modelling between PEDV HLJ/2015/DP1‐1 strain and 2015_PEDV‐strain. (b) The compared S1 protein modelling between PEDV JL/2016/47a strain and 2016_PEDV‐strain. (c) The compared S1 protein modelling between PEDV LN/DT/2016/510a strain and 2016_PEDV‐strain. The HLJ/2015/DP‐1‐1 strain was shown as surface and cartoon by purpleblue. The 2015_PEDV‐strain was shown as cartoon by yellow. The JL/2016/47a strain was shown as surface and cartoon by wheat. The LN/DT/2016/510a strain was shown as surface and cartoon by lightblue. The 2016_PEDV‐strain was shown as surface and cartoon by pink. The S1‐RBD of LN/DT/2016/510a strain was shown as surface by limon [Colour figure can be viewed at wileyonlinelibrary.com]
4. DISCUSSION
PEDV has become the most important intestinal pathogen in swine in China (Wang et al., 2016), and a high prevalence of infection with PEDV, 66.9% (363/543), was detected in piglets with diarrhoea in our study. Many studies of the mechanism of PEDV infection and effective vaccines have been published. However, co‐infections with multiple pathogens make it difficult to understand and control PEDV infections. In order to better understand the prevalence of the co‐infection of PEDV with other pathogens in China, a broad epidemiological investigation of PEDV and 13 other enteric viruses was carried out in this study. Co‐infections of PEDV with other enteric pathogens have been reported in previous studies. Zhang et al. (2014) reported that the co‐infection of PEDV and PBoV was more prevalent in diarrhoea samples than non‐diarrhoea samples (Zhang et al., 2014). Chen et al. (2018) reported that, among 217 PEDV‐positive samples, 27% (58/217) of samples had PEDV infection alone whereas the remaining 73% (159/217) of samples exhibited two to nine pathogens (Chen et al., 2018). In our study, a high percentage of co‐infection among piglet diarrhoea samples was identified. Among the 543 diarrhoea samples, the mean number of pathogens per sample was 4.25, and 96.5% of samples were co‐infected with at least two different viruses, 67.4% of samples were co‐infected with three to five different pathogens and, notably, co‐infection with nine different pathogens was identified in three samples. According to this survey, single infection with PEDV occurred in only 0.92% of samples (5 out of 543), while most of the samples involved co‐infections.
Nantel‐Fortier, Lachapelle, Letellier, L'Homme, and Brassard (2019) reported that piglets with diarrhoea shed more Kobuvirus than healthy individuals during the late‐nursing stage (6–21 days old), and that piglets shed more Kobuvirus during the post‐weaning stage (on nursery farms) than during any of the other life stages. In our study, positive rates of PKV in the 543 diarrhoea samples ranged from 74.50% to 83.24% during 2015–2018, and PKV showed a high co‐infection rate in PEDV‐positive samples (81.55%). These data suggest that PKV has a potential role in PEDV‐induced diarrhoea symptoms. TTSuV‐2 is frequently detected in pigs affected with porcine post‐weaning multisystemic wasting syndrome (PMWS) (Wang et al., 2009), and PEV infections remain asymptomatic or show mild pathogenicity in pigs (Zell et al., 2000). Both TTSuV‐2 and PEV exhibited a high prevalence in diarrhoeic piglets in this study, and when compared with PEDV‐negative samples, a highly significant association of PEV (p = .033) and TTSuV‐2 (p = .032) infection with PEDV‐positive samples was observed. Although these data suggest that co‐infections of PEDV with PEV and TTSuV‐2 may result in the aggravation of clinical signs of diarrhoea in piglets, the evidence is limited, and further studies will need to be undertaken.
In the current study, the data indicate that ongoing variation of the S1 gene is occurring in the PEDV strains circulating in China, which is the one of most important risk factors for the failure of classic PEDV vaccines and the changes in pathogenesis and tissue tropism. Direct evidence has been reported that insertion and deletion mutations of the S protein change the pathogenicity and tissue tropism of PEDV (Deng et al., 2016; Lin et al., 2016). In this study, we summarized the S1‐IDMPs of the 147 PEDV strains identified using the S1 protein of prototype PEDV CV777 strain as the comparator. Among these Chinese PEDV pandemic strains, the S58_S58insQGVN–N135dup–D158_I159del or S1‐IDMPs similar to S58_S58insQGVN–N135dup–D158_I159del were the dominant insertion and deletion mutation pattern. Meanwhile, the S1‐IDMPs of nearly all of the 147 strains occurred in the NTD of the S1 protein of PEDV; the S1‐IDMP of only the LN/DT/2016/510a strain was located in both the NTD and RBD of the S1 protein. The evidence from S1‐IDMPs indicated that, although ongoing variation has been occurring in the S1 genes of Chinese PEDV pandemic strains in the past three years, the S1‐IDMPs of PEDV show genetic stability in the pig population in China.
The sialic acid binding activity of the S protein facilitates infection by PEDV and other coronaviruses (Deng et al., 2016; Li et al., 2016; Schwegmann‐Wessels et al., 2011). In our study, the pandemic S1‐IDMP S58_S58insQGVN–N135dup–D158_I159del of Chinese PEDV variant strains was also located in the NTD of the S1 protein, where it is associated with the sialic acid binding activity. Similar to that reported by Chen et al. (2019, the homology modelling of the S protein showed that, when compared with PEDV CV777, the 2018 PEDV strains exhibited four major structural changes at residues 55–64, 113–120, 130–142 and 157–165, which all reside on the surface of the S1‐NTD region. In addition, the amino acid mutations of the S1 protein are mainly located in the region of S1‐NTD. These data suggest that the S1‐IDMP S58_S58insQGVN–N135dup–D158_I159del, which is located in the S1‐NTD, may enhance the cellular entry of Chinese PEDV pandemic strains via alteration of the sialic acid binding activity.
The S1‐IDMP of the LN/DT/2016/510a strain identified in our study is a unique and novel S1‐IDMP because it is located not only in the NTD but also in the RBD; the S1‐IDMPs of the JL/2016/47 (a, b) strain and the HLJ2015/DP1‐1 strain identified in our previous study (Wang et al., 2016) were also novel because they exhibit insertion and deletion mutations at four sites of the S1‐NTD. The homology modelling of the S protein suggested that these S1‐IDMPs, occurring in the LN/DT/2016/510a, JL/2016/47 (a, b) and HLJ2015/DP1‐1 strains, had a potential influence on the structure of the S protein of PEDV. In addition, HLJ/2015/DP1‐1 and LN/DT/2016/510a have lost one N‐linked glycosylation site on the S1 protein. Previous study has revealed that the N‐linked glycosylation sites on the S protein of severe acute respiratory syndrome coronavirus (SARS‐CoV) are associated with virus replication and zoonotic transmission (Han, Lohani, & Cho, 2007; Zheng et al., 2018). These data suggest that the change in the N‐linked glycosylation site of HLJ/2015/DP1‐1 and LN/DT/2016/510a may play a potential role in viral infection.
Wen et al. (2018) reported that the S1 regions of spike sequences became genetically more diverse with time (Wen et al., 2018). In our study, when compared with 2011–2014 PEDV strains, an increasing number of amino acid mutations of the S1 gene was found among 2015–2018 PEDV strains, and 11 and 10 novel mutations occurred in 2017 and 2018, respectively. Moreover, analysis of the S protein modelling indicated that 2016 and 2017 PEDV strains exhibited an obvious potential structural change when compared with 2015 and 2016 PEDV strains, respectively. These data demonstrate that the emergence of novel mutations may be associated with self‐variation of PEDV, as with other RNA viruses, in response to the prolonged immune pressure from vaccination or infection.
In conclusion, our findings have provided evidence that PEDV exhibits the co‐infection with 13 enteric viruses, and the clinical signs of diarrhoea induced by PEDV are potentially associated with co‐infection with PEV and TTSuV‐2. The PEDV strains identified in our study belonged to Chinese pandemic strains and exhibited genetic diversity. The S1‐IDMP of the PEDV strains identified in our study was undergoing amino acid mutations, among which the S58_S58insQGVN–N135dup–D158_I159del‐like mutations were common patterns, and the novel patterns S58_S58insQGVN–N135dup–D158_I159del–T380_V380insGQRS and S58_S58insQGVN–N135dup–D158_I159del–N553_Y560del were identified in our study. Moreover, when compared with PEDV CV777, 10 novel mutation positions of the S1 gene were found in our study. These results could be of significance in understanding the co‐infection and genetic evolution of PEDV, and in the development of new strategies to control the disease.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ETHICAL APPROVAL
The study was approved by the Animal Experiments Committee of the Heilongjiang Bayi Agricultural University (registration protocol 201401002). All sampling and publication of the data were approved by the farm owners.
Supporting information
ACKNOWLEDGMENTS
The study was supported by the National Natural Science Foundation of China (grant no. 31873011), the Outstanding Youth Science Foundation of Heilongjiang province (grant no. JC2017007), the National Key Research and Development Program of China (grant no. 2017YFD0501604‐5), the Heilongjiang Bayi Agricultural University Support Program for San Heng San Zong (TDJH201804) and the Graduate innovative research projects in Heilongjiang Bayi Agricultural University (YJSCX2018‐Z02/YJSCX2017‐Z02).
Su M, Li C, Qi S, et al. A molecular epidemiological investigation of PEDV in China: Characterization of co‐infection and genetic diversity of S1‐based genes. Transbound Emerg Dis. 2020;67:1129–1140. 10.1111/tbed.13439
Su and Li are contributed equally to this work.
REFERENCES
- Aydin, H. , Al‐Khooly, D. , & Lee, J. E. (2014). Influence of hydrophobic and electrostatic residues on SARS‐coronavirus S2 protein stability: Insights into mechanisms of general viral fusion and inhibitor design. Protein Science, 23(5), 603–617. 10.1002/pro.2442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biasini, M. , Bienert, S. , Waterhouse, A. , Arnold, K. , Studer, G. , Schmidt, T. , Schwede, T. (2014). SWISS‐MODEL: Modeling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Research, 42, W252–W258. 10.1007/978-1-4939-8736-8_17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, P. , Wang, K. , Hou, Y. , Li, H. , Li, X. , Yu, L. , … Zhou, Y. (2019). Genetic evolution analysis and pathogenicity assessment of porcine epidemic diarrhea virus strains circulating in part of China during 2011–2017. Infection, Genetics and Evolution, 69, 153–165. 10.1016/j.meegid.2019.01.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Q. , Wang, L. , Zheng, Y. , Zhang, J. , Guo, B. , Yoon, K. J. , Li, G. (2018). Metagenomic analysis of the RNA fraction of the fecal virome indicates high diversity in pigs infected by porcine endemic diarrhea virus in the United States. Virology Journal, 15(1), 95. 10.1186/s12985-018-1001-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu, D. K. , Poon, L. L. , Guan, Y. , & Peiris, J. S. (2008). Novel astroviruses in insectivorous bats. Journal of Virology, 82, 9107–9114. 10.1128/JVI.00857-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooks, G. E. , Hon, G. , Chandonia, J. M. , & Brenner, S. E. (2004). WebLogo: A sequence logo generator. Genome Research, 14, 1188–1190. 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decaro, N. , Campolo, M. , Desario, C. , Ricci, D. , Camero, M. , Lorusso, E. , … Buonavoglia, C. (2005). Virological and molecular characterization of a mammalian orthoreovirus type 3 strain isolated from a dog in Italy. Veterinary Microbiology, 109(1‐2), 19–27. 10.1016/j.vetmic.2005.05.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Dunnen, J. T. , & Antonarakis, S. E. (2001). Nomenclature for the description of human sequence variations. Human Genetics, 109(1), 121–124. 10.1007/s004390100505 [DOI] [PubMed] [Google Scholar]
- Deng, F. , Ye, G. , Liu, Q. , Navid, M. T. , Zhong, X. , Li, Y. , Peng, G. (2016). Identification and comparison of receptor binding characteristics of the spike protein of two porcine epidemic diarrhea virus strains. Viruses, 8, 55. 10.3390/v80300550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elschner, M. , Prudlo, J. , Hotzel, H. , Otto, P. , & Sachse, K. (2002). Nested reverse transcriptase‐polymerase chain reaction for the detection of group A rotaviruses. Journal of Veterinary Medicine Series B, 49(2), 77–81. 10.1046/j.1439-0450.2002.00510.x [DOI] [PubMed] [Google Scholar]
- Gupta, R. , Jung, E. , & Brunak, S. (2004). NetNGlyc 1.0 Server. Center for Biological Sequence Analysis, Technical University of Denmark available from: http://www.cbs.dtudk/services/NetNGlyc [Google Scholar]
- Han, D. P. , Lohani, M. , & Cho, M. W. (2007). Specific asparagine‐linked glycosylation sites are critical for DC‐SIGN‐ and L‐SIGN‐mediated severe acute respiratory syndrome coronavirus entry. Journal of Virology, 81(21), 12029–12039. 10.1128/JVI.00315-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvis, M. C. , Lam, H. C. , Zhang, Y. , Wang, L. , Hesse, R. A. , Hause, B. M. , … Marthaler, D. (2016). Genomic and evolutionary inferences between American and global strains of porcine epidemic diarrhea virus. Preventive Veterinary Medicine, 123, 175–184. 10.1016/j.prevetmed.2015.10.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic, I. , & Bork, P. (2016). Interactive tree of life (iTOL) v3: An online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Research, 44(W1), W242–W245. 10.1093/nar/gkw290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, F. (2015). Receptor recognition mechanisms of coronaviruses: A decade of structural studies. Journal of Virology, 89(4), 1954–1964. 10.1128/JVI.02615-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, W. , Li, H. , Liu, Y. , Pan, Y. , Deng, F. , Song, Y. , He, Q. (2012). New variants of porcine epidemic diarrhea virus, China, 2011. Emerging Infectious Diseases, 18(8), 1350–1353. 10.3201/eid1808.120002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, W. , van Kuppeveld, F. J. , He, Q. , Rottier, P. J. , & Bosch, B. J. (2016). Cellular entry of the porcine epidemic diarrhea virus. Virus Reseach, 226, 117–127. 10.1016/j.virusres.2016.05.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, C. M. , Saif, L. J. , Marthaler, D. , & Wang, Q. (2016). Evolution, antigenicity and pathogenicity of global porcine epidemic diarrhea virus strains. Virus Reseach, 226, 20–39. 10.1016/j.virusres.2016.05.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nantel‐Fortier, N. , Lachapelle, V. , Letellier, A. , L'Homme, Y. , & Brassard, J. (2019). Kobuvirus shedding dynamics in a swine production system and their association with diarrhea. Veterinary Microbiology, 235, 319–326. 10.1016/j.vetmic.2019.07.023 [DOI] [PubMed] [Google Scholar]
- Pensaert, M. B. , & de Bouck, P. (1978). A new coronavirus‐like particle associated with diarrhea in swine. Archives of Virology, 58, 243–247. 10.1007/BF01317606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi, S. , Su, M. , Guo, D. , Li, C. , Wei, S. , Feng, L. I. , & Sun, D. (2019). Molecular detection and phylogenetic analysis of porcine circovirus type 3 in 21 Provinces of China during 2015–2017. Transboundary and Emerging Diseases, 66(2), 1004–1015. 10.1111/tbed.13125 [DOI] [PubMed] [Google Scholar]
- Schwegmann‐Wessels, C. , Bauer, S. , Winter, C. , Enjuanes, L. , Laude, H. , & Herrler, G. (2011). The sialic acid binding activity of the S protein facilitates infection by porcine transmissible gastroenteritis coronavirus. Virology Journal, 8(1), 10.1186/1743-422X-8-435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, D. , Zhou, X. , Peng, Q. , Chen, Y. , Zhang, F. , Huang, T. , … Tang, Y. (2015). Newly emerged porcine deltacoronavirus associated with diarrhoea in swine in China: Identification, prevalence and full‐length genome sequence analysis. Transboundary and Emerging Diseases, 62, 575–580. 10.1111/tbed.12399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, D. , Feng, L. , Shi, H. , Chen, J. , Cui, X. , Chen, H. , & Tong, G. (2008). Identification of two novel B cell epitopes on porcine epidemic diarrhea virus spike protein. Veterinary Microbiology, 131, 73–81. 10.1016/j.vetmic.2008.02.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, D. , Wang, X. , Wei, S. , Chen, J. , & Feng, L. (2016). Epidemiology and vaccine of porcine epidemic diarrhea virus in China: A mini‐review. Journal of Veterinary Medical Science, 78, 355–363. 10.1292/jvms.15-0446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura, K. , Stecher, G. , Peterson, D. , Filipski, A. , & Kumar, S. (2013). MEGA6: Molecular evolutionary genetics analysis version 6.0. Molecular Biology and Evolution, 30, 2725–2729. 10.1093/molbev/mst197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van, D. N. , Anh, P. H. , Van, C. N. , Hoa, N. T. , Carrique‐Mas, J. , Hien, V. B. , … Simmonds, P. (2016). Large‐scale screening and characterization of enteroviruses and kobuviruses infecting pigs in Vietnam. Journal of General Virology, 97(2), 378–388. 10.1099/jgv.0.000366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, L. , Byrum, B. , & Zhang, Y. (2014). New variant of porcine epidemic diarrhea virus, United States, 2014. Emerging Infectious Diseases, 20(5), 917–919. 10.3201/eid2005.140195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, E. , Guo, D. , Li, C. , Wei, S. , Wang, Z. , Liu, Q. , … Sun, D. (2016). Molecular characterization of the ORF3 and S1 genes of porcine epidemic diarrhea virus non S‐INDEL strains in seven regions of China, 2015. PLoS ONE, 11(8), e0160561. 10.1371/journal.pone.0160561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, M. M. , Zhou, Y. J. , Chen, Z. Y. , Yu, H. , Li, G. X. , Yan, L. P. , … Tong, G. Z. (2009). Identification and survey of Torque teno virus in pigs in China. Chinese Journal of Veterinary Science, 31, 751–755. [Google Scholar]
- Wang, Y. , Zhou, L. , Zhou, Y. C. , Cai, Y. H. , Zhu, L. , Xu, Z. W. , & Guo, W. Z. (2014). Establishment and application of a nest RT‐PCR method for detection of porcine torovirus. Chinese Journal of Veterinary Science, 7, 1039–1042. [Google Scholar]
- Wen, Z. , Li, J. , Zhang, Y. , Zhou, Q. , Gong, L. , Xue, C. , Cao, Y. (2018). Genetic epidemiology of porcine epidemic diarrhoea virus circulating in China in 2012‐2017 based on spike gene. Transboundary and Emerging Diseases, 65(3), 883–889. 10.1111/tbed.12825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto, R. , Soma, J. , Nakanishi, M. , Yamaguchi, R. , & Niinuma, S. (2015). Isolation and experimental inoculation of an S INDEL strain of porcine epidemic diarrhea virus in Japan. Research in Veterinary Science, 103, 103–106. 10.1016/j.rvsc.2015.09.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zell, R. , Krumbholz, A. , Henke, A. , Birch‐Hirschfeld, E. , Stelzner, A. , Doherty, M. , & Wurm, R. (2000). Detection of porcine enteroviruses by nRT‐PCR: Differentiation of CPE groups I‐III with specific primer sets. Journal of Virological Methods, 88, 205–218. 10.1016/S0166-0934(00)00189-0 [DOI] [PubMed] [Google Scholar]
- Zhang, B. , Tang, C. , Yue, H. , Ren, Y. , & Song, Z. (2014). Viral metagenomics analysis demonstrates the diversity of viral flora in piglet diarrhoeic faeces in China. Journal of General Virology, 95(Pt_7), 1603–1611. 10.1099/vir.0.063743-0 [DOI] [PubMed] [Google Scholar]
- Zhang, Q. , Hu, R. , Tang, X. , Wu, C. , He, Q. , Zhao, Z. , Wu, B. (2013). Occurrence and investigation of enteric viral infections in pigs with diarrhea in China. Archives of Virology, 158(8), 1631–1636. 10.1007/s00705-013-1659-x [DOI] [PubMed] [Google Scholar]
- Zheng, J. , Yamada, Y. , Fung, T. S. , Huang, M. , Chia, R. , & Liu, D. X. (2018). Identification of N‐linked glycosylation sites in the spike protein and their functional impact on the replication and infectivity of coronavirus infectious bronchitis virus in cell culture. Virology, 513, 65–74. 10.1016/j.virol.2017.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng, X. , Liu, G. , Opriessnig, T. , Wang, Z. , Yang, Z. , & Jiang, Y. (2016). Development and validation of a multiplex conventional PCR assay for simultaneous detection and grouping of porcine bocaviruses. Journal of Virological Methods, 236, 164–169. 10.1016/j.jviromet.2016.06.014 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials