

SUMMARY
Recent developments in controlled C–H functionalization transformations continue to inspire new retrosynthetic disconnections. One tactic in C–H functionalization is the intermolecular C–H insertion reaction of rhodium bound carbenes. These intermediates can undergo highly selective transformations through the modulation of the ligand framework of the rhodium catalyst. This work describes our continued efforts towards differentiating C–H bonds in the same molecule by judicious catalyst choice. Substituted cyclobutanes which exist as a mixture of interconverting conformers and possess neighboring C–H bonds within a highly strained framework are the targets herein for challenging the current suite of catalysts. While most C–H functionalization tactics focus on generating 1,2-disubstituted cyclobutanes via substrate-controlled directing group methods, the regiodivergent methods in this paper provide access to chiral 1,1-disubstituted and cis-1,3-disubstituted cyclobutanes simply by changing the catalyst identity, thus permitting entry to novel chemical space.
Keywords: Asymmetric, rhodium, cyclobutanes, diazo compound, C–H functionalization
eTOC Blurb
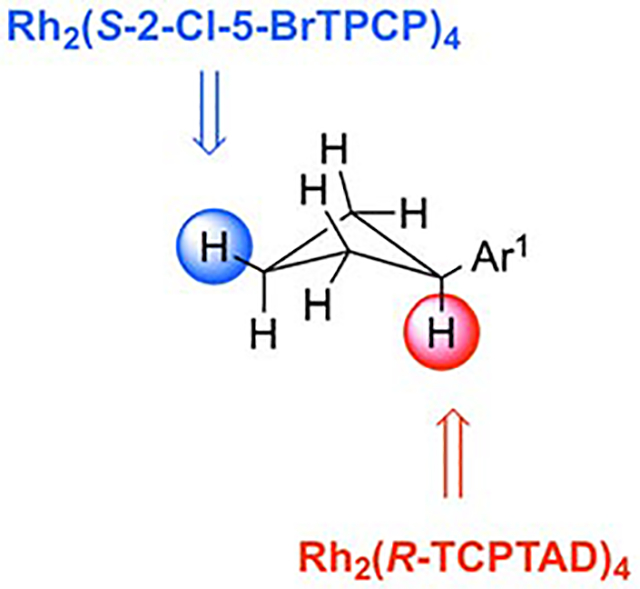
This study shows how to control site-selectivity in C-H functionalization by simply using the correct catalyst. Cyclobutanes were used as the scaffold to illustrate the impact of catalyst control because the core unit is incorporated into various structures of biomedical interest. The catalysts control whether the chemistry occurs at C1 or C3 of the cyclobutane.
Graphical Abstract
INTRODUCTION
Catalyst-controlled C–H functionalization is of considerable current interest.1,2 The ability to control which C–H bond in a particular compound is functionalized simply by selecting the right catalyst would offer exciting opportunities in organic synthesis. Considerable effort has been directed towards developing small catalysts3–5 and evolved enzymes6–8 that can achieve such selectivity without resorting to well-defined functional groups on substrates. We have been developing the dirhodium-catalyzed reactions of donor/acceptor carbenes as a robust system for site-selective C–H functionalization reactions. 9 The combination of both a donor and acceptor group on the carbene leads to an intermediate that is still sufficiently reactive to functionalize C–H bonds, and due to the modulating effect of the donor group, the system is prone to subtle catalyst control effects. In recent years, we have prepared a series of catalysts that are capable of site-selective functionalization of unactivated C–H bonds (Figure 1A).10 The catalysts adopt unique geometries that dictate the substrate trajectory towards the rhodium-bound carbene thus enabling single C–H bonds to be transformed. So far, catalysts have been designed to selectively functionalize either primary11, secondary12, or tertiary13 C–H bonds (Figure 1B). Further ligand development has resulted in catalysts that distinguish between benzylic and unactivated methylene C–H bonds,14 and most recently, we have introduced a catalyst capable of desymmetrizing substituted cyclohexane rings.15
Figure 1. Site-Selective C–H Functionalization by Carbenoid Insertion Reactions.

(A) Dirhodium tetracarboxylate catalysts possess structural diversity which enables functionalization of specific C–H bonds.
(B) Primary, secondary, and tertiary C–H bonds can be selectively functionalized by judicious catalyst choice.
(C) The work outlined herein showcases a regioselective C–H insertion reaction which permits access to new chemical space.
The work described in this manuscript details our efforts toward applying this growing suite of catalysts and our understanding of the behavior of C–H bonds for the selective functionalization of arylcyclobutanes, either at C1 or C3, depending on which catalyst is used (Figure 1C). Arylated cyclobutane rings occur frequently in both natural products and pharmaceuticals. Many of the natural products arise directly from photochemical [2+2] dimerization processes of simpler alkene building blocks, as in the truxillic and truxinic acids (from cinnamic acid)16 and sceptrin (from hymenidin).17 Beyond these dimeric compounds, arylcyclobutane scaffolds are also observed in monoterpenoids such as cannabicyclol,18 murrayamine M,19 rhodonoids A and B,20 and cochlearol B.21 Arylcyclobutanes are considered intriguing systems for drug discovery because they place functionality in a defined spacial orientation.,22 These compounds can exhibit wide-ranging bioactivity, as exemplified by the anti-clotting agent piperaborenine D,23 the glucocortecoid receptor binding (−)-endiandrin A,24 and the CYP3 inhibitor dipiperamide A.25 1,1-cis disubstituted arylcyclobutanes are represented in a number of modern drug candidates. Recently, researchers at Abbvie, inc. have reported on a potent and selective TRPV3 antagonist which is a 1,1-difunctionalized arylcyclobutane.26,27 In this study, the stereochemistry at the cyclobutane ring was shown to have a significant effect on the bioactivity of the analogs. In light of the significance of these substances, a novel method for preparing chiral 1,1-difunctionalized arylcyclobutanes and 1,3-cis-disubstituted arylcyclobutanes could be of significant value to the scientific community.
Considerable effort has been expended in generating elaborate cyclobutanes by means of C–H functionalization tactics which rely on the use of directing groups on the substrate to control regiochemistry.28–34 While these methods have provided elegant entries into complex natural products and access to enantioenriched cyclobutane scaffolds, the methods are restricted to, and primarily successful with, the formation of sp3-sp2 carbon-carbon bonds. Additionally, we are aware of only one example of a carbene-induced C–H functionalization of a cyclobutane where a sp3-sp3 C-C bond is formed, but this was an intramolecular example.35
Even though phenylcyclobutane is a small molecule, it represents an interesting challenge for C–H functionalization (Figure 2). The compound would be expected to exist as two interconverting conformers A and B, with conformer A being favored where the substituent occupies an equatorial position (when R = Br the equatorial isomer is favored by 1 kcal/mol36). From previous studies with donor/acceptor carbenes conducted on alkylcyclohexane15and acyclic alkanes11–13, we would not expect the methylene sites adjacent to the substituent to be sterically accessible. Therefore, we would expect carbene insertion to be a competition between C1 (substituted carbon) and C3 (distal) functionalization. We also know from studies on the C–H functionalization of cyclohexane that equatorial sites react about 140 times faster than reactions at axial sites.15 Hence, we would expect that reaction at C3 would occur at the equatorial site. If the reaction occurs at the major conformer A, the resulting cyclobutane would be cis disubstituted, whereas reaction of the minor conformer would give a trans disubstituted product. Reaction at C1 is electronically preferred due to the influence of the aryl ring but is sterically encumbered as a tertiary site. The minor conformer B would be expected to be the most reactive because the tertiary C–H bond is equatorial. However, it will not be possible to distinguish whether the tertiary C–H bond functionalization is occurring through A or B because each will generate the same C1 functionalized product. In order to achieve insertion at the C3 position, a sterically bulky catalyst like 2 or 3 would be expected to be required, whereas attack at C1 would be expected to occur with a less bulky catalyst like 4 or 5.
Figure 2. Analysis of Cyclobutane C–H Functionalization.

Selective intermolecular C–H functionalization of cyclobutanes poses significant challenges. Potential functionalization can occur at three carbon sites. On the basis of our understanding of donor/acceptor carbene-induced C-H functionalization, we would expect the equatorial C-H bonds at C1 and C3 to be prefered and distinguished by appropriate choice of catalyst.
RESULTS
This study began by surveying dirhodium tetracarboxylate catalysts for functionalizing readily available cyclobutane 6 (Figure 3).37 The reactions were conducted in refluxing methylene chloride with 2,2,2-trichloroethyl 2-(4-bromophenyl)-2-diazoacetate (7)38 as the carbene precursor. The use of 2,2,2-trihaloethyl ester groups is often advantageous in the dirhodium-catalyzed reactions of donor/acceptor carbenes because they can enhance enantioselectivity.39 Our analysis above of the current catalyst toolbox and our understanding of selective C–H bond transformations assisted which catalysts were selected for the initial reaction screen. The original prolinate catalyst, Rh2(S-DOSP)4,40 is considered to be a relatively uncrowded catalyst, and gave clean reaction at C1 to form 8 with no indication of the C3 product 9. However, it is well established this catalyst does not give high levels of enantioselectvity in dichloromethane,41 and this behavior was confirmed in this case, with 8 formed in only 32% ee. Similarly high site selectivities were observed with the phathlimido catalysts 4 and 5, but now the enantioselectivity was also very high, reaching 98% ee with 4. The bulky catalysts 2 and 3, designed for functionalization of primary or secondary C–H bonds,12,14 caused a dramatic change in selectivity, favoring the C3 product 8 (2:1 for 2 and 5:1 for 3). Catalyst, 1, designed for primary C–H functionalization,11 also preferred attack at C3 (3:1) but did not outperform the site selectivity exhibited by catalyst 3 and also generated a further undefined isomer of the C-H functionalization products. On the basis of these studies, Rh2(S-TCPTAD)4 (3) was selected as the optimum catalyst for C1 functionalization and Rh2(S-2-Cl-5-BrTPCP)4 (3) was selected for C3 functionalization.
Figure 3. Optimization of C–H Functionalization of Substituted Cyclobutanes.

The reaction conditions were the following: the diazo compound 6 (0.25 mmol) in 1.5 mL solvent was added over 3 h to a solution of the cyclobutane substrate 7 (0.75 mmol, 3.0 equiv.) and catalyst (1.0 mol %) in 3.0 mL dichloromethane at room temperature. The reaction was allowed to stir an additional 2 h. All yields are isolated yields (98% of unreacted 6 was recovered). The enantiomeric excess (ee) was determined by chiral HPLC analysis of the isolated product.
With these regiodivergent methods in place, the substrate scope was evaluated (Figure 5 and 6). The cyclobutane substrates were readily accessed by a 2-step protocol (Grignard addition to cyclobutanone followed by reduction). The functionalization of the benzylic site was first examined using Rh2(S-TCPTAD)4 (4) as catalyst (Figure 4). A variety of different arylcyclobutanes were used in this reaction along with a number of aryl diazoacetates to generate the C1 functionalized products 10-21. As the cyclobutane derivatives are the most valuable of the two reagents, these reactions were conducted with 2 equivalents of the diazo compound. Notable examples include the use of heteroaromatic groups in the formation of 18-21. Also notable is the functionalization of the cyclobutane ring in preference to the primary benzylic site as shown for 15; in this instance, Rh2(S-TCPTAD)4 exhibits complete selectivity for the more substituted benzylic site even though it is more sterically crowded. The absolute configuration of the tertiary substitution was verified by X-ray crystallographic analysis of 8, and the other examples were tentatively assigned by analogy.
Figure 5. Reaction Scope for C–H Functionalization of Arylcyclobutanes at C3.

The Rh2(S-2-Cl-5-BrTPCP)4 (3)-catalyzed reactions of aryldiazoacetates results in the selective C–H functionalization of the C3 secondary site of the arylcyclobutanes. The catalyst is sterically demanding and reacts at the sterically most accessible secondary site. ee (enantiomeric excess). 1The tertiary substituted product was obtained in 84% ee. 2The tertiary substituted product was obtained in 90% ee.
Figure 6. Competition studies for site-selective C-H functionalization.

The relative rates of functionalization of the cyclobutane C-H bonds versus the unstrained benzylic C-H bonds in isopropyl-, ethyl- and methylbenzene The tertiary benzylic site of cyclobutane (marked in red) is more reactive than the tertiary benzylic site is an unstrained system for both the Rh2(S-TCPTAD)4 and Rh2(S-2-Cl-5-BrTPCP)4 catalyzed reactions (1.7 times and >15 times more reactive, respectively). The electronically unactivated secondary site of the cyclobutane (marked in blue) is 2.8 times more reactive in the Rh2(S-2-Cl-5-BrTPCP)4-catalyzed reaction and >10 times more reactive in the Rh2(S-TCPTAD)4-catalyzed reaction.
Figure 4. Reaction Scope for C–H Functionalization of Arylcyclobutanes at C1.

The Rh2(S-TCPTAD)4 (4)-catalyzed reactions of aryldiazoacetates results in the selective C–H functionalization of the tertiary benzylic site of the arylcyclobutane. The catalyst is relatively uncrowded and reacts at the electronically preferred site. ee (enantiomeric excess).
We next investigated the selective functionalization of the distal site, C3, using Rh2(S-2-Cl5-BrTPCP)4 as catalyst (Figure 5). Despite our initial success, the development of the cis-1,3disubstitued cyclobutane scope would be more challenging because the C1 position is electronically activated. Additionally, the conformational interconversion of different substituted cyclobutanes may lead to varying levels of diastereoselectivity. In fact, a variety of different aryl substituted cyclobutanes and aryldiazoacetates were competent to form the C3 functionalized products 22-32, and the diastereoselectivity for distal functionalization was good for all the examples. Perhaps unsurprisingly, electron-deficient aryl rings on the cyclobutanes lowered the production of the tertiary side-product as seen for 23versus22, but importantly, Rh2(S-2-Cl-5-BrTPCP)4 was able to generate the desired cis-1,3-disubstituted cyclobutanes as the major product in all instances. The X-ray crystallographic analysis of 27 enabled the unambiguous assignment of the major stereoisomer as cis, and this was further verified by nOe analysis wherein the hydrogens on the same face of the cyclobutane were coupled. This validated the hypothesis that the equatorial C–H bonds in conformer A would be most susceptible to functionalization. This reaction was also amenable to heteroaryldiazoacetates and heteroarylcyclobutanes as illustrated in the formation of 29-32.
The successful C-H functionalization of the cyclobutanes is unexpected because the C-H bonds in a cyclobutane ring have greater s character and are stronger than C–H bonds contained in an unstrained system. Therefore, we conducted competition experiments of 4-tert-butylphenylcyclobutane with substrates containing unstrained primary, secondary and tertiary benzylic C–H bonds (Figure 6, see supporting information for details). The competition reactions with Rh2(S-TCPTAD)4 revealed that the tertiary benzylic C-H bond on the bicyclobutane is more reactive that the tertiary site in isopropyl benzene. In the reaction with the sterically crowded catalyst Rh2(S-2-Cl-5-BrTPCP)4-catalyzed reaction, the electronically unactivated secondary site of the cyclobutane is more reactive that the secondary benzylic site in ethyl benzene. These results indicate that the cyclobutane C–H bonds are more reactive compared to their unstrained counterparts, presumably because the C–H bonds would be more sterically exposed or possibly because of hyperconjugative effects in the strained ring.
DISCUSSION
C–H functionalization tactics for generating substituted cyclobutanes are dominated by the use of substrate control and directing groups which limits the accessibility of a variety of desirable substituted cyclobutanes. This report details an intermolecular C–H insertion approach to chiral substituted cyclobutanes which can generate both 1,1-disubstituted and 1,3-disubstituted cyclobutanes. The lessons we learned about specific substrate interactions with our growing catalyst toolbox assisted the rapid discovery of reaction conditions which were suitable for generating each set of substituted cyclobutanes. A less bulky dirhodium catalyst favors attack at the tertiary benzylic site, whereas a bulky catalyst favors attack at the sterically most accessible secondary site. These studies illustrate the potential of C–H functionalization strategies to rapidly access novel chiral scaffolds of potential pharmaceutical relevance.
EXPERIMENTAL PROCEDURES
General Procedure for the C–H Functionalization of Cyclobutanes
An oven-dried vial was equipped with magnetic stir bar and sealed with septa and cap. This was cooled under vacuum, and then flame-dried once. After cooling to room temperature, the vial was loaded with rhodium catalyst (0.5–1 mol %), the cyclobutane substrate (1 equiv), and anhydrous dichloromethane (2 mL dichloromethane / 1 mmol cyclobutane). The mixture was allowed to stir under argon (the mixture was heated if heat was applied) while the diazo compound was prepared. A solution of the diazo compound (2 equiv) was prepared by dissolving in the dichloromethane (3 mL dichloromethane / 0.25 mmol diazo compound) and then this mixture was added dropwise by syringe pump over 3 h. Upon completion of the addition, the reaction was stirred an additional 2–4 h. Residual solvent was removed under reduced pressure (if the reaction was heated, it was allowed to come to room temperature before removing residual solvent), and the crude product was purified by silica gel chromatography. The regisomeric ratio (rr) and the diastereomeric ratio (dr) were determined by NMR analysis of the crude reaction mixture. The enantioselectivity (ee, enantiomeric eccess) was determined by HPLC analysis of the material after flash chromatography. Images of the key spectral and analytical data of the compounds generated in this study are included in Figures S1–S138 in the Suplemental Information.
DATA AND SOFTWARE AVAILABILITY
Crystallographic data for compounds 8 and 25 have been submitted to the Cambridge Crystallographic Database under deposition numbers CCDC:1921680-1921681.
Supplementary Material
Bigger Picture.
Traditional synthetic strategies have viewed most C-H bonds as chemically inert and utilize functional groups for transformations. C–H Functionalization is an attractive alternative strategy for the synthesis of complex organic molecules because it leads to the possibility of rapidly accessing novel chemical space. To fully develop this alternative approach, it is necessary to identify ways for reacting at specific C–H bonds even when a number of similar C–H bonds may exist in a substrate molecule. It would be particularly beneficial if a collection of catalysts were available, each with a preference for reaction at a specific C–H bond. Over the last few years, we have developed such a collection of catalysts for C–H functionalization chemistry of rhodium bound carbenes. In this paper, we illustrate how these catalysts can be applied to the selective functionalization of cyclobutanes, leading to the formation of pharmaceutically relevant chiral building blocks.
Highlights.
Catalyst-controlled C-H functionalization
Synthesis of chiral cyclobutane derivatives
New strategy for the synthesis of pharmaceutically relevant compounds
ACKNOWLEDGMENTS
This work was supported by the NSF Center for Selective C-H Functionalization (CHE 1700982) and NIGMS of the NIH under award number F32GM130020 (Z.J.G.). Additional financial support was provided by AbbVie. Instrumentation used in this work was supported by the National Science Foundation (CHE1531620 and CHE1626172). We wish to thank the members of the NSF Center for C-H Functionalization (CHE 1700982) for helpful discussions regarding this work. We thank Dr. John Bacsa and Mr. Thomas Pickel at the Emory X-ray Crystallography Facility for the X-ray structural analysis.
Footnotes
DECLARATION OF INTERESTS
H.M.L.D. is a named inventor on a patent entitled ‘Dirhodium catalyst compositions and synthetic processes related thereto’ (US 8,974,428, issued March 10, 2015). The other authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information includes detailed experimental data and images of the key spectral and analytical data (Figures S1–S138).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES AND NOTES
- 1.He J; Wasa M; Chan KSL; Shao Q; Yu J-Q Palladium-catalyzed transformations of alkyl C–H bonds. Chem. Rev 117, 8754–8786 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hartwig JF; Larsen MA Undirected, homogeneous C–H bond functionalization: challenges and opportunities. ACS Cent. Sci 2, 281–292 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.White MC; Zhao JP, Aliphatic C-H oxidations for late-stage functionalization. J. Am. Chem. Soc 140, 13988–14009 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roizen JL; Harvey ME; Du Bois J, Metal-catalyzed nitrogen-atom transfer methods for the oxidation of aliphatic C-H Bonds. Acc. Chem. Res 45, 911–922, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nishikawa T; Ryu I, Site-selective C-H functionalization by decatungstate anion photocatalysis: Synergistic control by polar and steric effects expands the reaction scope. Acs Catalysis 8, 701–713(2018). [Google Scholar]
- 6.Zhang RK; Chen K; Huang X; Wohlschlager L; Renata H; Arnold FH Enzymatic assembly of carbon–carbon bonds via iron-catalysed sp3 C–H functionalization. Nature 565, 67–72 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dydio P; Key HM; Hayashi H; Clark DS; Hartwig JF Chemoselective, enzymatic C-H bond amination aatalyzed by a cytochrome P450 containing an Ir(Me)-PIX cofactor. J. Am. Chem. Soc 139, 1750–1753 (2017). [DOI] [PubMed] [Google Scholar]
- 8.Zwick CZ; Renata H Remote C–H hydroxylation by an α-ketoglutarate-dependent dioxygenase enables efficient chemoenzymatic synthesis of manzacidin C and proline analogs. J. Am. Chem. Soc 140, 1165–1169 (2018). [DOI] [PubMed] [Google Scholar]
- 9.Davies HML; Morton D Guiding principles for site selective and stereoselective intermolecular C-H functionalization by donor/acceptor rhodium carbenes. Chem. Soc. Rev 40, 1857–1869 (2011). [DOI] [PubMed] [Google Scholar]
- 10.Davies HML; Liao K Dirhodium tetracarboxylates as catalysts for selective intermolecular C–H functionalization, Nat. Rev. Chem 3, 347–360 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liao K; Negretti S; Musaev DG; Bacsa J; Davies HML Site-selective and stereoselective functionalization of unactivated C–H bonds. Nature, 533, 230–234 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Liao K; Pickel TC Boyarskikh V; Bacsa J; Musaev DG; Davies HML Site-selective and stereoselective functionalization of non-activated tertiary C-H bonds. Nature, 551, 609–613(2017). [DOI] [PubMed] [Google Scholar]
- 13.Liao K; Yang Y-F; Li Y; Sanders JN; Houk KN; Musaev DG; Davies HML Design of catalysts for site-selective and enantioselective functionalization of non-activated primary C–H bonds. Nature Chemistry, 10, 1048–1055 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu W; Ren Z; Bosse AT; Liao K; Goldstein EL; Bacsa J; Musaev DG; Stoltz BM; Davies HML Catalyst-controlled selective functionalization of unactivated C–H bonds in the presence of electronically activated C–H bonds. J. Am. Chem. Soc. 140, 12247–12255 (2018). [DOI] [PubMed] [Google Scholar]
- 15.Fu J; Ren Z; Bacsa J; Musaev DG; Davies HML Desymmetrization of cyclohexanes by site- and stereoselective C–H functionalization. Nature 564, 395–399 (2018). [DOI] [PubMed] [Google Scholar]
- 16.Krauze-Baranowska M Truxillic and truxinic acids-occurrence in plant kingdom. Acta poloniae pharmaceutica 59, p. 403–410 (2002). [PubMed] [Google Scholar]
- 17.Ma Z; Wang X; Wang X; Rodriguez RA; Moore CE; Gao S; Tan X; Ma Y; Rheingold AL; Baran PS; Chen C Asymmetric syntheses of sceptrin and massadine and evidence for biosynthetic enantiodivergence. Science 346, p. 219–224 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yeom H-S; Li H; Tang Y; Hsung RP Total Syntheses of Cannabicyclol, Clusiacyclol A and B, Iso-Eriobrucinol A and B, and Eriobrucinol. Org. Lett 15, p. 3130–3133 (2013). [DOI] [PubMed] [Google Scholar]
- 19.Gassner C; Hesse R; Schmidt AW; Knolker H-J Total synthesis of the cyclic monoterpenoid pyrano[3,2-a]carbazole alkaloids derived from 2-hydroxy-6methylcarbazole. Org. Biomol. Chem 12, p. 6490–6499 (2014). [DOI] [PubMed] [Google Scholar]
- 20.Liao H-B; Lei C; Gao L-X; Li J-Y; Li J; Hou A-J Two Enantiomeric Pairs of Meroterpenoids from Rhododendron capitatum. Org. Lett 17, p. 5040–5043 (2015). [DOI] [PubMed] [Google Scholar]
- 21.Dou M; Di L; Zhou L-L; Yan Y-M; Wang X-L; Zhou F-J; Yang Z-L; Li R-T; Hou F-F; Cheng Y-X Cochlearols A and B, polycyclic meroterpenoids from the fungus Ganoderma cochlear that have renoprotective activities. Org. Lett 16, p. 6064–6067 (2014). [DOI] [PubMed] [Google Scholar]
- 22.Wager TT; Pettersen BA; Schmidt AW; Spracklin DK; Mente S; Butler TW; Howard H Jr, Lettiere DJ; Rubitski DM; Wong DF; Nedza FM; Nelson FR; Rollema H; Raggon JW; Aubrecht J; Freeman JK; Marcek JM; Cianfrogna J; Cook KW; James LC; Chatman LA; Iredale PA; Banker MJ; Homiski ML; Munzner JB; Chandrasekaran RY Discovery of two clinical histamine H3 receptor antagonists:trans-N-ethyl-3-fluoro-3-[3-fluoro-4-(pyrrolidinylmethyl)-phenyl]cyclobutanecarboxamide (PF-03654746) and trans-3-fluoro-3-[3-fluoro-4-(pyrrolidin-1-ylmethyl)phenyl]-N-(2-methylpropyl)cyclobutanecarboxamide (PF-03654764). J. Med. Chem 54, p. 7602–7620 (2011). [DOI] [PubMed] [Google Scholar]
- 23.Tsai I-L; Lee F-P; Wu C-C; Duh C-Y; Ishikawa T; Chen J-J; Chen Y-C; Seki H; Chen I-S New cytotoxic cyclobutanoid amides, a new furanoid lignan and anti-platelet aggregation constituents from Piper arborescens. Planta. Med 71, 535–542 (2005). [DOI] [PubMed] [Google Scholar]
- 24.Davis RA; Carroll AR; Duffy S; Avery VM; Guymer GP; Forster PI; Quinn RJ Endiandrin A, a potent glucocorticoid receptor binder isolated from the Australian plant Endriandra anthropophagorum. J. Nat. Prod 70, 1118–1121 (2007). [DOI] [PubMed] [Google Scholar]
- 25.Tsukamoto S; Cha B-C; Ohta T Dipiperamides A, B, and C: bisalkaloids from the white pepper Piper nigrum inhibiting CYP3A4 activity. Tetrahedron, 58, 1667–1671 (2002). [Google Scholar]
- 26.Gomtsyan A; Schmidt RG; Bayburt EK; Gfesser GA; Voight EA; Daanen JF; Schmidt DL; Cowart MD; Liu H; Altenbach RJ; Kort ME; Clapham B; Cox PB; Shrestha A; Henry R; Whittern DN; Reilly RM; Puttfarcken PS; Brederson J-D; Song P; Li B; Huang SM; McDonald HA; Neelands TR; McGaraughty SP; Gauvin DM; Joshi SK; Banfor PN; Segreti JA; Shebley M; Faltynek CR; Dart MJ; Kym PR Synthesis and pharmacology of (pyridin-2-yl)methanol derivatives as novel and selective transient receptor potential vanilloid 3 antagonists. J. Med. Chem 59, 4926–4947 (2016). [DOI] [PubMed] [Google Scholar]
- 27.Voight EA; Daanen JF; Schmidt RG; Gomtsyan A; Dart MJ; Kym PR Stereoselective synthesis of a dipyridyl transient receptor potential vanilloid-3 (TRPV3) antagonist. J. Org. Chem 81, 12060–12064 (2016). [DOI] [PubMed] [Google Scholar]
- 28.Gutekunst WR; Baran PS Total synthesis and structural revision of the piperarborenines via sequential cyclobutane C–H arylation. J. Am. Chem. Soc 133, 19076–19079 (2011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Namyslo JC; Kaufmann DE The application of cyclobutane derivatives in organic synthesis. Chem. Rev 103, 1485–1537 (2003), [DOI] [PubMed] [Google Scholar]
- 30.Akula PS; Hong B-C; Lee G-H Catalyst- and substituent-controlled switching of chemoselectivity for the enantioselective synthesis of fully substituted cyclobutane derivatives via 2 + 2 annulation of vinylogous ketone enolates and nitroalkene. Org. Lett 20, 7835–7839 (2018). [DOI] [PubMed] [Google Scholar]
- 31.Chapman LM; Beck JC; Wu L; Reisman SE Enantioselective total synthesis of (+)-psiguadial B. J. Am. Chem. Soc 138, 9803–9806 (2016). [DOI] [PubMed] [Google Scholar]
- 32.Wu Q-F; Wang X-B; Shen P-X; Yu J-Q Enantioselective C–H arylation and vinylation of cyclobutyl carboxylic amides. ACS Catal. 8, 2577–2581 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Beck JC; Lacker CR; Chapman LM; Reisman SE A modular approach to prepare enantioenriched cyclobutanes: synthesis of (+)-rumphellaone A. Chem. Sci 10, 2315–2319 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ren R; Zhao H; Huan L; Zhu C Manganese-catalyzed oxidative azidation of cyclobutanols: regiospecific synthesis of alkyl azides by C-C bond cleavage. Angew. Chem. Int. Ed 54, 12692–12696 (2015). [DOI] [PubMed] [Google Scholar]
- 35.Rao VB, George CF, Wolff S, Agosta WC Synthetic and structural studies in the [4.4.4.5]fenestrane series. J. Am. Chem. Soc 107, 5732–5739 (1985). [Google Scholar]
- 36.Glendening ED; Halpern AM Ab initio study of cyclobutane: Molecular structure, ring-puckering potential, and origin of the inversion barrier. J. Phys. Chem. A 109, 635–642 (2005). [DOI] [PubMed] [Google Scholar]
- 37.Chen H; Hu L; Ji W; Yao L; Liao X Nickel-catalyzed decarboxylative alkylation of aryl iodides with anhydrides. ACS Catal. 8, 10479–10485 (2018). [Google Scholar]
- 38.Fu L Mighion JD; Voight EA; Davies HML. Synthesis of 2,2,2-trichloroethyl aryl- and vinyldiazoacetates by palladium-catalyzed cross-coupling,. Chem. Eur. J 23, 3272–3275 (2017). [DOI] [PubMed] [Google Scholar]
- 39.Guptill DM; Davies HML 2,2,2-Trichloroethyl aryldiazoacetates as robust reagents for the enantioselective C-H functionalization of methyl ethers. J. Am. Chem. Soc 136, 17718–17721 (2014). [DOI] [PubMed] [Google Scholar]
- 40.Davies HML; Bruzinski P; Hutcheson DK; Fall MJ Asymmetric cyclopropanations by rhodium(II) N-(arylsulfonyl)prolinate catalyzed decomposition of vinyldiazomethanes in the presence of alkenes. Practical enantioselective synthesis of the four stereoisomers of 2-phenylcyclopropane-1-amino acid, J. Am. Chem. Soc 118, 6897 (1996). [Google Scholar]
- 41.Davies HML Dirhodium tetra(N-arylsulfonylprolinates) as chiral catalysts for asymmetric transformations of vinyl- and aryldiazoacetates. Eur. J. Org. Chem 10, 2459–2469 (1999). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Crystallographic data for compounds 8 and 25 have been submitted to the Cambridge Crystallographic Database under deposition numbers CCDC:1921680-1921681.