

Abstract
Introduction
Individuals with Down syndrome (DS) show enhanced amyloid beta (Aβ) deposition in the brain. A new positron emission tomography (PET) index of amyloid load (AβL) was recently developed as an alternative to standardized uptake value ratios (SUVrs) to quantify Aβ burden with high sensitivity for detecting and tracking Aβ change.1
Methods
AβL was calculated in a DS cohort (N = 169, mean age ± SD = 39.6 ± 8.7 years) using [C‐11]Pittsburgh compound B (PiB) PET imaging. DS‐specific PiB templates were created for Aβ carrying capacity (K) and non‐specific binding (NS).
Results
The highest values of Aβ carrying capacity were found in the striatum and precuneus. Longitudinal changes in AβL displayed less variability when compared to SUVrs.
Discussion
These results highlight the utility of AβL for characterizing Aβ deposition in DS. Rates of Aβ accumulation in DS were found to be similar to that observed in late‐onset Alzheimer's disease (AD; ≈3% to 4% per year), suggesting that AD progression in DS is of earlier onset but not accelerated.
Keywords: Alzheimer's disease, amyloid load, amyloid PET, Down syndrome, longitudinal, PiB
1. INTRODUCTION
Amyloid β (Aβ) plaques are a pathological feature of Alzheimer's disease (AD) and have been shown to precede symptoms of dementia by two decades. 2 Adults with Down syndrome (DS) are at increased risk for developing AD compared to the general population, with a sharp increase in prevalence after 50 years of age. 3 An early presence of brain Aβ is evident in DS beginning as early as in adolescence with severe cortical prominence by age 40, 4 , 5 which is several decades earlier than reported in late‐onset AD. 6 This stems from the triplication of chromosome 21, containing the gene encoding the production of the amyloid precursor protein and, the resulting increase in Aβ production. 7 , 8 Similar to autosomal dominant AD (ADAD), the mechanism for AD pathophysiology in DS is likely more heavily influenced by Aβ overproduction than the failure of Aβ clearance mechanisms postulated for late‐onset AD. 9 , 10 Although the biological mechanisms underlying the disease differ, the structure of Aβ plaques between DS and late‐onset AD are indistinguishable. 11 , 12
Use of positron emission tomography (PET) with Aβ‐targeting radioligands provides an in vivo assay of the spatial extent of Aβ deposition and can monitor Aβ progression during the course of AD. 13 An early study using [C‐11]Pittsburgh compound B (PiB) PET in non‐demented DS demonstrated the feasibility of conducting scans in this population and reported elevated PiB retention as early as age 38, with earliest retention in the striatum. 14 This striatum‐first pattern of Aβ deposition was previously observed in presenilin‐1 mutation carriers. 15 Cross‐sectional PET studies in DS with increased sample sizes revealed that positivity for Aβ was detectable as early as the late 30s, with cortical Aβ deposition showing consistency with the spatial pattern displayed in late‐onset AD. 16 , 17 , 18 , 19 , 20 , 21 , 22 , 23 , 24 , 25 , 26 , 27 In addition, a majority of DS individuals who were Aβ(+) displayed striatal Aβ retention, 16 , 22 highlighting the striatum as a target region for early detection in this population. 28 Longitudinal studies in non‐demented DS revealed that participants converting from Aβ(−) to Aβ(+) showed Aβ signal increases of ≈3% to 4% per year, with the striatum showing the earliest and most prominent change. 29 , 30 , 31
Understanding the trajectory of Aβ accumulation in DS is a critical step toward characterizing the similarities and differences with the trajectory in late‐onset AD. Accurate assessment for detecting these subtle changes in Aβ burden requires a quantitative metric that is representative of the concentration of Aβ protein. A common outcome measure for PET imaging of Aβ is the standardized uptake value ratio (SUVr), calculated as the quotient of the PET‐measured signal from a target region and off‐target region (eg, precuneus/cerebellum). For [C‐11]PiB, SUVr has been validated as an accurate proxy for a more precise metric of distribution volume ratio (DVR). 32 Both global and regional PiB SUVrs have been used to distinguish Aβ‐positivity in preclinical AD, 33 and demonstrated to detect increases in Aβ signal over time in DS. 29 , 30 Although SUVr is used routinely to track changes in Aβ burden for longitudinal studies, it can be prone to high variability during assessment of longitudinal Aβ change, 34 , 35 resulting in lower power to detect biological significance from the data. 1
The metric of amyloid load (AβL) was developed as a global (ie, whole brain) outcome measure to reduce variability by utilizing template‐based images of Aβ‐specific and non‐specific radioligand uptake. 1 Proper voxel‐based weighting of these images can then be used to provide a global estimate of AβL. Using model inputs of SUVr and canonical images of indexes for radioligand‐specific (K) and non‐specific (NS) binding, 36 the AβL index shows reduced longitudinal variability by measuring only the SUVr signal corresponding to specific Aβ binding. A benefit to this method is that it can be fully implemented as a template‐based approach. This allows for PET data processing without the need for magnetic resonance imaging (MRI) for spatial normalization, 37 , 38 , 39 which is advantageous for populations prone to significant motion artifacts that negatively affect segmentation and registration.
Highlights
The amyloid load index was calculated from Pittsburgh compound B (PiB) positron emission tomography (PET) images of adults with Down syndrome
Amyloid load displayed high sensitivity to detect longitudinal changes in amyloid
The rates of amyloid accumulation in Down syndrome and late‐onset Alzheimer's disease are similar
RESEARCH IN CONTEXT
Systematic review: Longitudinal positron emission tomography (PET) studies in adults with Down syndrome (DS) have revealed elevated levels of brain amyloid beta (Aβ) at younger ages compared to the general population. The amyloid load index (AβL) serves as a metric for quantifying and tracking changes in Aβ with high sensitivity. However, AβL was evaluated in research studies of late‐onset Alzheimer's disease (AD) and has not been characterized in other populations.
Interpretation: Our findings in DS confirm the high sensitivity of AβL to detect Aβ change reported from the Alzheimer's Disease Neuroimaging Initiative (ADNI) data. The longitudinal rates of AβL change between late‐onset AD and DS were similar, suggesting that AβL can be a standardized marker for drawing direct comparisons across populations.
Future directions: Future research should characterize AβL across different Aβ PET radioligands and populations. A direct comparison between AβL and other standardized methods of quantification (eg, Centiloids) should be explored to evaluate which metric is most sensitive to detect Aβ change.
The Alzheimer's Biomarker Consortium—Down Syndrome (ABC‐DS) is an ongoing study to characterize the natural history of AD‐related biomarkers in individuals with DS. Aβ PET scans, using [C‐11]PiB, have been acquired on participants from this study and reported in the literature using longitudinal data from legacy studies. 29 , 30 , 31 The overall aim of this work is to assess and compare longitudinal Aβ change using AβL and SUVr in a large DS population. The algorithm for generating AβL was implemented as a template‐based approach and modified for use in DS through inclusion of striatal Aβ in the model parameters.
2. METHODS
2.1. Participants
The cohort of participants with DS (N = 169; 39.6 ± 8.7 years) was initially recruited through a project studying the natural history of Aβ deposition in DS by the University of Wisconsin‐Madison Waisman Center and the University of Pittsburgh Medical Center, and then expanded to include a subsequent project of ABC‐DS, which also included the University of Cambridge Intellectual and Developmental Disabilities Research Group and Barrow Neurological Institute. Participant demographics are included in Table 1. Consent was obtained during enrollment into the study by the participant or legally designated caregiver. Trisomy of chromosome 21 was confirmed using genetic testing. Inclusion criteria included age >25 years and having receptive language ≥3 years. Exclusion criteria included having a prior diagnosis of dementia or a psychiatric condition that impaired cognitive functioning. Of the 169 participants, 68 completed two cycles of PET imaging and neuropsychological evaluation (mean ± SD = 2.9 ± 0.7 years apart), 50 completed three cycles (2.6 ± 0.8 years apart), and 14 completed four cycles (1.7 ± 0.3 years apart). Thus, 301 PET images were collected in total for this cohort. During the course of the study, 15 participants were classified having mild cognitive impairment (MCI)/AD, 134 were considered cognitively unimpaired, and a consensus on the cognitive status for the remaining 20 has yet to be determined.
TABLE 1.
Down syndrome participant demographics by sex, age, and cognitive status
| N | 169 |
|---|---|
| Male | 84 |
| Female | 85 |
| Age (mean ± SD) | 39.6 ± 8.7 years |
| MCI/AD consensus | 15 |
2.2. Imaging
T1‐weighted MRIs were acquired during each imaging cycle for all participants. A target dose of 15 mCi of [C‐11]PiB was injected intravenously, and PET scanning was performed from 50 to 70 minutes post‐injection (four 5‐minute frames). Individual PET frames were re‐aligned to correct for motion, summed to generate a static image, and spatially normalized to the Montreal Neurological Institute 152 space (MNI152 space) using Statistical Parametric Mapping 12 (SPM12) via a DS‐specific PiB PET template. 21 All PET images were cross‐checked with the MRI scans prior to spatial normalization. SUVr images were generated by voxel intensity normalization to cerebellar gray matter (formed by combining Automated Anatomical Labeling atlas (AAL) regions 91‐108, smoothing the mask with a 6 mm Gaussian kernel, and only keeping voxels with values ≥0.7). Global PiB was computed as the average SUVr from regions of interest (ROIs) defining the anterior cingulate, frontal cortex, parietal cortex, precuneus, temporal cortex, and striatum.
2.3. Generation of DS‐specific parametric maps for NS and K
The first step in calculating the AβL is to generate canonical images of the non‐specific binding (NS) and the carrying capacity (K) components of the [C‐11]PiB SUVr image for the DS population. The methods followed those outlined by Whittington et al. using [F‐18]florbetapir in the ADNI population, 36 with the modification to include the striatal region in the global PiB SUVr measure. Longitudinal rates of global PiB SUVr change were plotted with respect to the baseline measure, and the data were then fit by a restricted cubic spline function as described previously. 40 The spline fit was integrated with respect to time via the modified Euler method, and the resulting Aβ growth curve (Figure 1) was used to represent the change in global PiB SUVr over the time course of preclinical AD for the population. 40 The change in SUVr with respect to time was then modeled by the logistic growth function (Equation 1), 36
| (1) |
and the exponential growth rate (r) and time point of half‐maximal SUVr (T50) were estimated from the fit. The logistic growth model was applied to the SUVr images at the voxel level while keeping values for r and T50 fixed. Parametric maps of NS and K were generated through linearization of the model. 36 Because the striatum reveals early Aβ deposition in DS, this analysis was repeated while considering only striatal SUVr to observe how striatum‐specific values for r and T50 compared to the global estimates.
FIGURE 1.
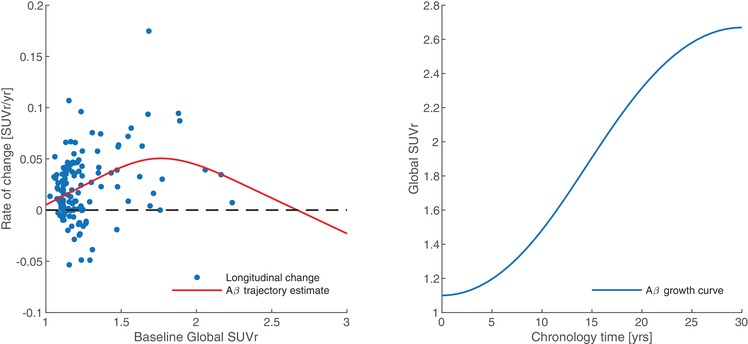
Longitudinal rate of PiB change relative to baseline global SUVr fit by a restricted cubic spline (left). Integral of the fit representing AD chronology with respect to global SUVr (right)
2.4. Calculation of AβL
AβL was determined from each subject's SUVr image in MNI152 space utilizing published methods. 1 AβL is calculated from the SUVr image, the population‐derived PiB template images of non‐specific binding (NS) and carrying capacity (K), using the functional equation:
| (2) |
where ns is the coefficient of non‐specific binding used to weight the component of non‐specific PiB signal (via the NS image) in the SUVr image. In its current form, Equation 2 represents an overdetermined system of linear equations, where AβL and ns can be solved simultaneously using the linear least‐squares method on the SUVr, NS, and K matrices. A single AβL and ns are estimated for each subject across all of the voxels (i) in the images. These parameters can then be used to generate an SUVrfit image for comparison with the measured SUVr. The residual difference between the SUVr and the SUVrfit was calculated to evaluate how well the model estimated the SUVr based on the input parameters.
2.5. Definition of AβL cutoff for Aβ(+)
Using sparse k‐means clustering with resampling, regional and global cutoffs for Aβ(+) were established for PiB SUVr data in a control population and applied to DS data. 30 , 33 The Aβ(+) cutoff for AβL was determined by performing a linear regression between global PiB SUVr and AβL. The resulting fit was used to convert the global SUVr cutoff into a value of AβL.
2.6. Comparison of longitudinal AβL and SUVr change
For participants with longitudinal data (n = 68), the percent change per year between imaging cycles for AβL was calculated. Similarly, percent change in global PiB SUVr per year was calculated for all participants using a DS‐specific atlas. 21 Mean rates of change were calculated and compared across Aβ(−) and Aβ(+) groups.
3. RESULTS
3.1. Parametric imaging of NS and K
The fit of the logistic growth model (Equation 1) to population‐level global SUVr yielded fixed global values for r (=0.21 years−1) and T50 (=14.8 years) as shown in Figure 1. When the growth model was applied to just striatal SUVr, the resulting parameters for r and T50 were identical to the global estimates. With these parameters for r and T50, the logistic growth model was applied at the voxel level for all participants, yielding the parametric images for NS and K (Figure 2). The highest values for carrying capacity were found in the striatum and precuneus (2.21 and 2.29 SUVr units, respectively). The image of NS was consistent with the known non‐specific binding of PiB to white matter.
FIGURE 2.
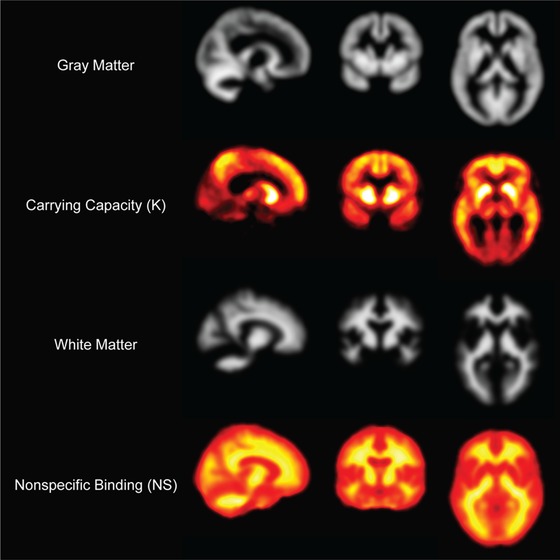
DS‐specific parametric images for carrying capacity (K) and non‐specific binding (NS) displayed as orthographic planes with corresponding gray and white matter maps
3.2. Quantification of AβL
AβL showed a positive linear correlation (Pearson's r = 0.97; P value: .00001) with global PiB SUVr (Figure 3). There was no observable dependence of ns (non‐specific binding coefficient) on SUVr (Pearson's r = −0.01; P value: .86). The quality of fit for SUVrfit was evaluated by assessing the percentage of voxels in the brain which satisfied |SUVr—SUVrfit| >0.3, as described previously. 1 Across 301 images, the mean percentage of voxels in the brain exceeding this threshold was 5.8% ± 4.2%, suggesting that the SUVr was well modeled by the algorithm. Of the poorly modeled voxels, 31.5% ± 6.95% resided in high K regions, 30.0% ± 6.48% resided in high NS regions (subcortical/cerebellar white matter), and 38.5% ± 9.20% resided in low K/NS regions (low PiB‐binding regions, cerebrospinal fluid [CSF] spaces). No correlation was observed between AβL and poorly modeled voxels in high K regions (Pearson's r = 0.018; P value: .76). However, a slight negative correlation was observed between AβL and high NS regions (Pearson's r = −0.26; P value: .00001) and a slight positive correlation between AβL and low K/NS regions (Pearson's r = 0.17; P value: .01). When compared with age at the time of imaging, AβL showed cross‐sectional increases with age at baseline as well as longitudinal increases over time within individuals that underwent multiple cycles of data collection (Figure 4).
FIGURE 3.
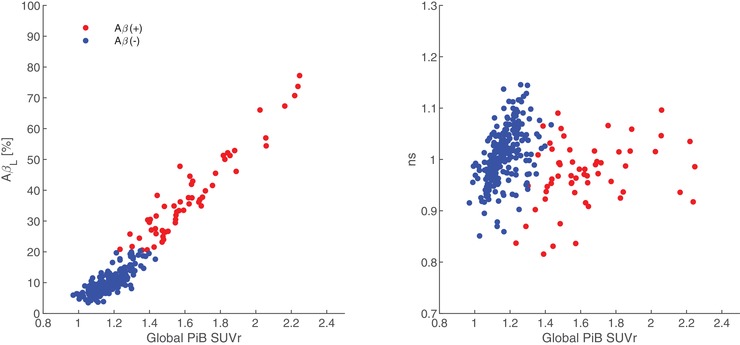
Comparison of AβL and non‐specific uptake (ns) to Global PiB SUVr. Participants were classified as Aβ(+) if their AβL exceeded the cutoff of 20.0%
FIGURE 4.
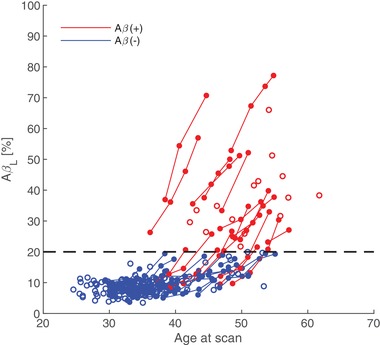
Dependence of AβL on age at the time of scan. Empty circles represent participants that have completed just one cycle of PiB imaging. The dashed line represents the cutoff for Aβ(+)
3.3. Classification of Aβ(+) based on AβL
The AβL cutoff for Aβ(+) determined through the linear regression of AβL and global PiB SUVr was calculated as 20.0%. Participants were classified as Aβ(+) if their AβL exceeded this threshold. 27 participants were classified as Aβ(+) at baseline while seven converted to Aβ(+) during the study duration compared to 35 at baseline and 15 converting using a previously established regional ROI‐based threshold (Table 2). Of our participants, 25% converted to Aβ(+) between ages 35 and 49, 47% between ages 50 and 59, and 100% above age 60, which is consistent with the reported clinical disease conversion rates of 23%, 55%, and 75%‐100% in DS for these respective age ranges. 41 For the ROI‐based threshold, the striatum was the first region to surpass the Aβ(+) threshold for all but one participant.
TABLE 2.
Number of participants converting to Aβ(+) at each imaging cycle classified with AβL and SUVr metrics
| Cycle 1 | Cycle 2 | Cycle 3 | Cycle 4 | |
|---|---|---|---|---|
| Aβ(+) Classification | n = 169 | n = 68 | n = 50 | n = 14 |
| AβL | 27 | 0 | 6 | 1 |
| ROI SUVr | 35 | 6 | 7 | 2 |
For the ROI‐based threshold, all but one participant converted to Aβ(+) in the striatum first.
3.4. Assessment of longitudinal Aβ change in DS
Across all cycles of longitudinal data, the distribution of rates of change for AβL and global SUVr are shown in Figure 5. The mean rate of AβL change per year (with 95% confidence intervals [Cis]) was 0.60% [0.40, 0.80] in the Aβ(−) group and 3.32% [2.72, 3.91] in the Aβ(+) group. The mean rate of global SUVr change was 1.24% [0.80, 1.68] in the Aβ(−) group and 3.23% [2.33, 4.14] in the Aβ(+) group. These results show that the longitudinal variability is lower for AβL change compared to SUVr in the Aβ(−) group. In addition, AβL shows similar increase to SUVr in the Aβ(+) group.
FIGURE 5.
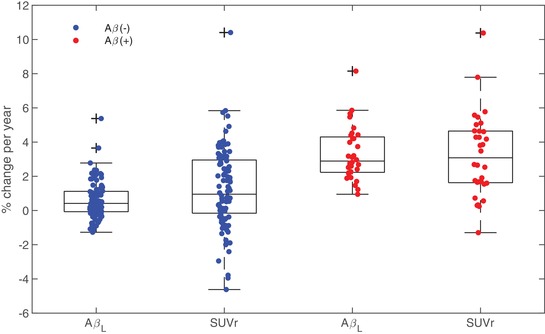
Distribution of longitudinal change in AβL and global PiB SUVr. Lower longitudinal variability is observed for AβL compared to SUVr for both Aβ(−) and Aβ(+) cases
4. DISCUSSION
With earlier age at onset and a higher risk of AD development compared to the general population, DS may serve as a powerful model to understand disease progression. Because individuals with DS are at very high risk for developing AD, there is motivation to prepare this population for involvement in clinical trials aimed at AD prevention, as in the ongoing Dominantly Inherited Alzheimer Network Trials Unit (DIAN‐TU) project. 42
The results presented above illustrate the utility of the AβL index for tracking Aβ burden in the DS population. Given inputs of typically measured SUVr and the canonical images of non‐specific binding and carrying capacity, a global AβL was calculated that shows close agreement with global PiB SUVr determined from a subset of brain regions. The canonical images also revealed the precuneus and striatum to have the highest capacity to carry Aβ in the DS brain (2.29 and 2.21 SUVr units, respectively), consistent with the pattern of Aβ deposition observed in ADAD. 15 , 43 The AβL model has not yet been evaluated in cases of ADAD, but would be insightful to compare the spread of Aβ in forms of AD influenced by Aβ overproduction. A direct comparison of carrying capacities between DS and ADAD with APP mutations may be useful to further classify the extent of Aβ deposition in these regions. High Aβ carrying capacities were observed in the parietal cortex (1.81) and anterior cingulate (1.68), while the frontal (1.35) and temporal cortices (1.02) had moderate carrying capacities. When the AβL index was characterized for the Alzheimer's Disease Neuroimaging Initiative (ADNI) population, the regions with the highest Aβ carrying capacities were the anterior cingulate and precuneus. 36 The parietal cortex also displayed high carrying capacities, whereas the striatum, frontal, and temporal cortices displayed moderate carrying capacities. 36 Compared to the ADNI data, our findings reveal that the spatial distribution of Aβ and carrying capacities are very similar between DS and late‐onset AD, with the exception of the striatum. This finding further highlights this region as a potential target of interest in the monitoring of the presence of early AD pathology in DS.
Estimating the trajectory of Aβ accumulation in DS at the population level using the logistic growth model yielded an uninhibited growth rate (r) of 0.21 years−1 and a time point of half‐maximal Aβ accumulation (T50) of 14.8 years based on the global PiB SUVr images. As derived from the ADNI dataset, global values of r and T50 were estimated as 0.20 years−1 and 14.9 years, respectively. 36 These findings indicate that even though the underlying mechanisms driving the production of Aβ and age at onset of PET‐detectable deposition may differ, the rates of Aβ accumulation are indistinguishable between these populations. Furthermore, the findings from the ADNI data revealed no spatial heterogeneity in r and T50 across brain regions, suggesting that an estimate of these parameters from a global composite ROI would be representative of the rate of Aβ accumulation in each of the individual regions. 36 Based on these findings, we chose to estimate r and T50 from a global composite ROI in the DS data rather than from each ROI individually. However, because early striatal Aβ accumulation is unique to DS, we applied the logistic growth model to the striatal SUVr from all DS participants to evaluate whether striatal r and T50 differed from the global estimates. The estimated striatal values of r and T50 were found to be identical to the global values, indicating that the only difference observed in the striatum is its higher carrying capacity, which leads to earlier detection by PET. One limitation to the estimate of the DS‐specific Aβ growth curve was that individuals in the late stages of Aβ progression were underrepresented, mainly because participants were enrolled in the study as non‐demented. As a result, there is higher uncertainty in the upper limit of the growth curve when estimated from the available population data (seen in Figure 1). To account for this, a sensitivity analysis was performed by varying the value of r within two standard deviations (SD) of the original estimate to encompass different limits of global Aβ carrying capacity, and then calculating values of AβL using these new parameters. We found that changing these limits had minimal influence (<2%) on the value of AβL, and when applied to the longitudinal data there was no change in variance between baseline and follow‐up scans, affirming that our original estimates of these parameters were suitable for use in DS. In addition, the lower and upper limits of the Aβ growth curve from our original estimate were similar to those observed when evaluated in a late‐onset AD study using [C‐11]PiB. 40
This work has demonstrated that AβL can be derived from static PET images acquired from short scanning periods and can be implemented as a template‐based approach without the need for an anatomical MRI, allowing feasibility of use in DS. Compared to conventional SUVr analysis, AβL results were similar to SUVr when evaluated at the cross‐sectional level but showed lower variability when evaluating longitudinal change in Aβ. However, for the purposes of determining Aβ(−) or Aβ(+) status, our results suggest that AβL is not as sensitive at classifying Aβ(+) cases compared to an ROI‐based SUVr threshold in DS. Because the AβL index is derived from all Aβ‐carrying voxels in the brain, it will have reduced sensitivity for detecting smaller focal increases in Aβ accumulation, such as those seen in the striatum. As the striatum contains fewer voxels than the cortical regions, the AβL will be more heavily influenced by the lower intensity cortical voxel values during the early stages of Aβ progression. If the objective is to identify the earliest evidence of regional Aβ accumulation, it may be more advantageous to focus on striatal SUVr, since this region was the first to show significant increase for all but one individual from our DS cohort. It is not fully understood why the striatum is the first to show elevated PiB binding in DS, but it is speculated that the signal observed is a result of large amounts of diffuse Aβ plaques in this region. 44 However, further histopathological investigation is needed to better understand this phenomenon. For the outlying case, Aβ(+) was reached globally with the highest signal in the precuneus and very low signal in the striatum that did not increase across longitudinal scans.
Preclinical AD studies have implemented use of a wide variety of Aβ PET radioligands, each with different binding characteristics and variability in quantification methods, making direct comparisons between studies challenging. Thus, there is motivation to standardize the methods of Aβ quantification from PET imaging to allow for a more practical interpretation of results in the clinical setting. One current method of standardizing Aβ PET quantification is implementation of the Centiloid scale, in which PET signal (units of SUVr) is linearly scaled to anchors determined in young controls (∼0 Centiloids) and “typical” AD patients (∼100 Centiloids). 45 , 46 The Centiloid methodology allows for a direct comparison between imaging studies regardless of the radioligand used, since uptake values of every radioligand can be converted to the same scale. Similar to the Centiloid scale, AβL would allow for direct comparisons between radioligands because the PET signal is scaled dependent on the Aβ burden relative to the total theoretical Aβ carrying capacity. The total Aβ carrying capacity can be derived for a population using any Aβ‐specific radioligand, thus a conversion factor between AβL for different PET radioligands would not be necessary. Because the Centiloid values are derived directly from SUVr data without any additional processing to remove the non‐specific signal, this method of quantification would suffer from the same longitudinal variability as observed with SUVr. Compared to Centiloids, AβL provides the advantage of being a standardized measure of Aβ burden that is less susceptible to non‐specific binding signal in the images, so quantification is indicative of only Aβ‐specific signal. Studies using multiple Aβ PET radioligands will be required to evaluate the differences in these two outcome measures.
5. CONCLUSION
Using a fully template‐based approach, AβL has been effectively modeled in DS from static [C‐11]PiB PET scans. Furthermore, this template‐based approach can make image acquisition and analysis in DS more feasible. At the cross‐sectional level, AβL showed consistency with SUVr in evaluating global Aβ burden. However, given its global nature, AβL was not as sensitive to classify Aβ(+) cases compared to a ROI‐based SUVr approach. In the longitudinal analysis, AβL showed reduced variability between scans compared to SUVr while displaying high sensitivity to detect Aβ change, suggesting AβL as a promising PET outcome measure for Aβ analysis.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
Supporting information
Supporting Information
ACKNOWLEDGMENTS
ABC‐DS: The Alzheimer's Biomarkers Consortium—Down Syndrome (ABC‐DS) project is a longitudinal study of cognition and blood‐based, genetic and imaging biomarkers of Alzheimer's disease. This study is funded by the National Institute on Aging (NIA) grants [U01AG051406, R01AG031110] and the National Institute for Child Health and Human Development (NICHD), grant [U54HD090256]. We thank the ABC‐DS study participants and the ABC‐DS research and support staff for their contributions to this study. We also thank Alex Whittington for insightful discussions on the implementation and use of the amyloid load index.
This manuscript has been reviewed by ABC‐DS investigators for scientific content and consistency of data interpretation with previous ABC‐DS study publications. We acknowledge the ABC‐DS study participants and the ABC‐DS research and support staff for their contributions to this study. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health (NIH).
Zammit MD, Laymon CM, Betthauser TJ, et al. Amyloid accumulation in Down syndrome measured with amyloid load. Alzheimer's Dement. 2020;12:e12020 10.1002/dad2.12020
REFERENCES
- 1. Whittington A, Gunn RN. Amyloid Load—a more sensitive biomarker for amyloid imaging. J Nucl Med. 2018;60(4):536‐540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Villemagne VL, Burnham S, Bourgeat P, et al. Amyloid β deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer's disease: a prospective cohort study. Lancet Neurol. 2013;12(4):357‐367. [DOI] [PubMed] [Google Scholar]
- 3. Schupf N. Genetic and host factors for dementia in Down's syndrome. Br J Psychiatry. 2002;180(5):405‐410. [DOI] [PubMed] [Google Scholar]
- 4. Mann DMA. Alzheimer's disease and Down's syndrome. Histopathology. 1988;13:125‐137. [DOI] [PubMed] [Google Scholar]
- 5. Wisniewski KE, Wisniewski HM, Wen GY. Occurrence of neuropathological changes and dementia of Alzheimer's disease in Down's syndrome. Ann Neurol. 1985;17:278‐282. [DOI] [PubMed] [Google Scholar]
- 6. Jack CR, Lowe VJ, Weigand SD, et al. Serial PIB and MRI in normal, mild cognitive impairment and Alzheimer's disease: implications for sequence of pathological events in Alzheimer's disease. Brain. 2009;132:1355‐1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Oyama F, Cairns NJ, Shimada H, Oyama R, Titani K, Ihara Y. Down's syndrome: Up‐regulation of β‐amyloid protein precursor and τ mRNAs and their defective coordination. J Neurochem. 1994;62:1062‐1066. [DOI] [PubMed] [Google Scholar]
- 8. Rumble B, Retallack R, Hilbich C, et al. Amyloid A4 protein and its precursor in down's syndrome and Alzheimer's disease. N Engl J Med. 1989;320:1446‐1452. [DOI] [PubMed] [Google Scholar]
- 9. Russo C, Saido TC, DeBusk LM, Tabaton M, Gambetti P, Teller JK. Heterogeneity of water‐soluble amyloid β‐peptide in Alzheimer's disease and Down's syndrome brains. FEBS Lett. 1997;409:411‐416. [DOI] [PubMed] [Google Scholar]
- 10. Teller JK, Russo C, Debusk LM, et al. Presence of soluble amyloid β–peptide precedes amyloid plaque formation in Down's syndrome. Nat Med. 1996;2:93‐95. [DOI] [PubMed] [Google Scholar]
- 11. Ellis WG, McCulloch JR, Corley CL. Presenile dementia in down's syndrome: Ultrastructural identity with alzheimer's disease. Neurology 1974;24:101‐106. [DOI] [PubMed] [Google Scholar]
- 12. Ohara PT. Electron microscopical study of the brain in down's syndrome. Brain. 1972;95:681‐684. [DOI] [PubMed] [Google Scholar]
- 13. Klunk WE, Engler H, Nordberg A, et al. Imaging brain amyloid in Alzheimer's disease with Pittsburgh Compound‐B. Ann Neurol. 2004;55:306‐319. [DOI] [PubMed] [Google Scholar]
- 14. Handen BL, Cohen AD, Channamalappa U, et al. Imaging brain amyloid in nondemented young adults with Down syndrome using Pittsburgh compound B. Alzheimers Dement. 2012;8:496‐501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Klunk WE, Price JC, Mathis CA, et al. Amyloid Deposition Begins in the Striatum of Presenilin‐1 Mutation Carriers from Two Unrelated Pedigrees. J Neurosci. 2007;27:6174‐6184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Annus T, Wilson LR, Hong YT, et al. The pattern of amyloid accumulation in the brains of adults with Down syndrome. Alzheimers Dement. 2016;12:538‐545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cole JH, Annus T, Wilson LR, et al. Brain‐predicted age in Down syndrome is associated with beta amyloid deposition and cognitive decline. Neurobiol Aging. 2017;56:41‐49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hartley SL, Handen BL, Devenny DA, et al. Cognitive functioning in relation to brain amyloid‐β in healthy adults with Down syndrome. Brain 2014;137:2556‐2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jennings D, Seibyl J, Sabbagh M, et al. Age dependence of brain β‐amyloid deposition in Down syndrome. Neurology 2015;84:500. [DOI] [PubMed] [Google Scholar]
- 20. Landt J, D'Abrera JC, Holland AJ, et al. Using Positron Emission Tomography and Carbon 11–Labeled Pittsburgh Compound B to Image Brain Fibrillar β‐Amyloid in Adults With Down Syndrome: Safety, Acceptability, and Feasibility. Arch Neurol. 2011;68:890‐896. [DOI] [PubMed] [Google Scholar]
- 21. Lao PJ, Handen BL, Betthauser TJ, et al. Imaging neurodegeneration in Down syndrome: brain templates for amyloid burden and tissue segmentation. Brain Imaging Behav. 2019;13(2):345‐353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lao PJ, Betthauser TJ, Hillmer AT, et al. The effects of normal aging on amyloid‐β deposition in nondemented adults with Down syndrome as imaged by carbon 11–labeled Pittsburgh compound B. Alzheimers Dement. 2016;12:380‐390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mak E, Bickerton A, Padilla C, et al. Longitudinal trajectories of amyloid deposition, cortical thickness, and tau in Down syndrome: A deep‐phenotyping case report. Alzheimers Dement Diagn Assess Dis Monit. 2019;11:654‐658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Matthews DC, Lukic AS, Andrews RD, et al. Dissociation of Down syndrome and Alzheimer's disease effects with imaging. Alzheimers Dement Transl Res Clin Interv. 2016;2:69‐81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rafii M, Wishnek H, Brewer J, et al. The down syndrome biomarker initiative (DSBI) pilot: proof of concept for deep phenotyping of Alzheimer's disease biomarkers in down syndrome. Front Behav Neurosci. 2015;9:239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rafii MS, Lukic AS, Andrews RD, et al. PET Imaging of Tau Pathology and Relationship to Amyloid, Longitudinal MRI, and Cognitive Change in Down Syndrome: Results from the Down Syndrome Biomarker Initiative (DSBI). J Alzheimers Dis. 2017;60:439‐450. [DOI] [PubMed] [Google Scholar]
- 27. Sabbagh MN, Chen K, Rogers J, et al. Florbetapir PET, FDG PET, and MRI in Down syndrome individuals with and without Alzheimer's dementia. Alzheimers Dement. 2015;11:994‐1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cohen AD, McDade E, Christian B, et al. Early striatal amyloid deposition distinguishes Down syndrome and autosomal dominant Alzheimer's disease from late‐onset amyloid deposition. Alzheimers Dement. 2018;14:743‐750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hartley SL, Handen BL, Devenny D, et al. Cognitive decline and brain amyloid‐β accumulation across 3 years in adults with Down syndrome. Neurobiol Aging. 2017;58:68‐76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lao PJ, Handen BL, Betthauser TJ, et al. Longitudinal changes in amyloid positron emission tomography and volumetric magnetic resonance imaging in the nondemented Down syndrome population. Alzheimers Dement Diagn Assess Dis Monit. 2017;9:1‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tudorascu DL, Minhas DS, Lao PJ, et al. The use of Centiloids for applying [11C]PiB classification cutoffs across region‐of‐interest delineation methods. Alzheimers Dement Diagn Assess Dis Monit. 2018;10:332‐339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lopresti BJ, Klunk WE, Mathis CA, et al. Simplified Quantification of Pittsburgh Compound B Amyloid Imaging PET Studies: A Comparative Analysis. J Nucl Med. 2005;46:1959‐1972. [PubMed] [Google Scholar]
- 33. Cohen AD, Mowrey W, Weissfeld LA, et al. Classification of amyloid‐positivity in controls: Comparison of visual read and quantitative approaches. NeuroImage. 2013;71:207‐215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Landau SM, Fero A, Baker SL, et al. Measurement of Longitudinal β‐Amyloid Change with 18F‐Florbetapir PET and Standardized Uptake Value Ratios. J Nucl Med. 2015;56:567‐574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tryputsen V, DiBernardo A, Samtani M, et al. Optimizing Regions‐of‐Interest Composites for Capturing Treatment Effects on Brain Amyloid in Clinical Trials. J Alzheimers Dis. 2015;43:809‐821. [DOI] [PubMed] [Google Scholar]
- 36. Whittington A, Sharp DJ, Gunn RN. Spatiotemporal Distribution of β‐Amyloid in Alzheimer Disease Is the Result of Heterogeneous Regional Carrying Capacities. J Nucl Med. 2018;59:822‐827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Della Rosa PA, Cerami C, Gallivanone F, et al. A Standardized [18F]‐FDG‐PET Template for Spatial Normalization in Statistical Parametric Mapping of Dementia. Neuroinformatics. 2014;12:575‐593. [DOI] [PubMed] [Google Scholar]
- 38. Friston KJ, Ashburner J, Frith CD, Poline J‐B, Heather JD, Frackowiak RSJ. Spatial registration and normalization of images. Hum Brain Mapp. 1995;3:165‐189. [Google Scholar]
- 39. Meyer JH, Gunn RN, Myers R, Grasby PM. Assessment of Spatial Normalization of PET Ligand Images Using Ligand‐Specific Templates. NeuroImage. 1999;9:545‐553. [DOI] [PubMed] [Google Scholar]
- 40. Jack CR, Wiste HJ, Lesnick TG, et al. Brain β‐amyloid load approaches a plateau. Neurology. 2013;80:890‐896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Strydom A, Coppus A, Blesa R, et al. Alzheimer's disease in Down syndrome: An overlooked population for prevention trials. Alzheimers Dement Transl Res Clin Interv. 2018;4:703‐713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Morris JC, Aisen PS, Bateman RJ, et al. Developing an international network for Alzheimer research: The Dominantly Inherited Alzheimer Network. Clin Investig. 2012;2:975‐984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Villemagne VL, Ataka S, Mizuno T, et al. High Striatal Amyloid β‐Peptide Deposition Across Different Autosomal Alzheimer Disease Mutation Types. Arch Neurol. 2009;66(12):1537‐1544. [DOI] [PubMed] [Google Scholar]
- 44. Abrahamson EE, Head E, Lott IT, et al. Neuropathological correlates of amyloid PET imaging in Down syndrome. Dev Neurobiol. 2019;79:750‐766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Klunk WE, Koeppe RA, Price JC, et al. The Centiloid Project: Standardizing quantitative amyloid plaque estimation by PET. Alzheimers Dement. 2015;11:1‐15.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Su Y, Flores S, Hornbeck RC, et al. Utilizing the Centiloid scale in cross‐sectional and longitudinal PiB PET studies. NeuroImage Clin. 2018;19:406‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information