

Abstract
Budesonide is a potential therapeutic option for the prevention of bronchopulmonary dysplasia in mechanically ventilated premature neonates. The dose and concentrations of budesonide that drive effective prophylaxis are unknown, due in part to the difficulty in obtaining serial blood samples from this fragile population. Of primary concern is the limited total blood volume available for collection for the purposes of a pharmacokinetic study. Dried blood spots (DBS), which require the collection of <200 µL whole blood to fill an entire card, are an attractive low-blood volume alternative to traditional venipuncture sampling. We describe a simple and sensitive method for determining budesonide concentrations in DBS using an ultra-high-performance liquid chromatography – tandem mass spectrometry assay. Budesonide was liberated from a single 6 mm punch using a basified methyl tert-butyl ether extraction procedure. The assay was determined to be accurate and precise in the dynamic range of 1 to 50 ng/mL. The validated assay was then successfully applied to DBS collected as part of a multi-center, dose-escalation study of budesonide administered in surfactant via intra-tracheal instillation to premature neonates between 23 and 28 weeks gestational age. These findings show that DBS are a useful technique for collecting pharmacokinetic samples in premature neonates and other pediatric populations.
Keywords: Budesonide, Dried blood spots, UHPLC-MS/MS, Premature neonates
1. Introduction
Bronchopulmonary dysplasia (BPD) is a chronic lung disease involving injury and inflammation of the immature lung that is associated with ongoing pulmonary illness, including asthma, neurodevelopmental complications, and mortality [1]. BPD occurs in a vast majority (>70%) of extremely low gestational age newborns (ELGAN) who require ventilator support in the immediate post-natal period [2,3]. Despite extensive research, few therapeutic options have demonstrated the ability to safely protect ELGAN from BPD. One option, prophylactic corticosteroid anti-inflammatory therapy was beneficial when administered systemically, however, this approach was associated with an increased risk of toxicity [4–7]. It is therefore of interest to assess whether corticosteroid administration that is non-systemic can be protective, while reducing the risk of toxicity.
One of the potential routes of administration that may avoid high systemic exposure of corticosteroids is intra-tracheal instillation. The glucocorticoid budesonide exhibits anti-inflammatory effects in the lung following intra-tracheal administration in a surfactant vehicle, making it a potential candidate for BPD prophylaxis in ELGAN [8,9]. For budesonide to be a successful prophylactic agent for BPD, its systemic penetration through the lung must be minimal in order to avoid toxicity. However, measuring systemic drug exposures in ELGAN is impeded by the limited blood volume available for collection. Dried blood spot (DBS) collection is a low blood volume technique that offers several advantages compared to traditional blood sampling techniques [10–13]. Furthermore, DBS have previously demonstrated utility for measuring systemic exposures and testing for inborn errors of metabolism in pediatric populations, including neonates [11,14–16]. This article describes the development and validation of an ultra-high-performance liquid chromatography – tandem mass spectrometry (UHPLC-MS/MS) method for measuring budesonide concentrations in DBS to assess the systemic exposure of budesonide following intra-tracheal instillation in ELGAN.
2. Methods
2.1. Chemicals and materials
The reference standards budesonide (molecular weight (MW) = 430.53, purity = 98%) and budesonide-D8 (MW = 438.58, purity = 95%) were purchased from Toronto Research Chemicals (Ontario, Canada). Reagents including methanol (Honeywell-Burdick & Jackson®, Muskegon, MI), ammonium hydroxide (NH4OH) (Fisher Scientific, Fairlawn, NJ), OmniSolv® methyl tert-butyl ether (MtBE) (MilliporeSigma, Burlington, MA), and ammonium bicarbonate (NH4HCO3) (Sigma-Aldrich, St. Louis, MO) were all analytical grade. Ultrapure water (UP H2O) was prepared in house from deionized water with a Milli-Q® Gradient A10 system (Millipore Corp., Bedford, MA). Sarstedt polypropylene test tubes (13 × 100 mm) and MicroLiter Autosampler Vials (300 µL) were obtained from VWR™ International (Radnor, PA). DBS collection materials, including Whatman® Protein Saver cards, humidity indicators, dessicant packs, and zippered bags were purchased through Fisher Scientific™ (Fairlawn, NJ).
2.2. Preparation of standards, internal standards, and quality controls
Stock solutions of both budesonide and budesonide-D8 were prepared in methanol to a concentration of 1.0 mg/mL. The stock solution of budesonide-D8 was first diluted to an intermediate concentration of 2 µg/mL in methanol, followed by another dilution to the working internal standard solution at a concentration of 2 ng/mL in methanol. The budesonide stock solutions used to prepare calibration standards and quality control (QC) samples were weighed and prepared separately. An intermediate budesonide stock solution at 2.5 ng/µL in methanol was made for both the standard and QC stocks, and used to prepare working standard and QC solutions. Working standard stocks ranged between 0.05 and 2.5 ng/µL, while working QC stocks ranged between 0.25 and 2.0 ng/µL. For standards, 2 mL of human whole blood (hematocrit = 41%, Biological Specialty Corp., Colmar, PA) with EDTA as an anti-coagulant was pipetted into a polypropylene tube and combined with 40 µL of the appropriate working standard stock. This resulted in calibration standard concentrations of 1.0, 2.5, 5.0, 10, 20, 25, 40, and 50 ng/mL in whole blood. For QCs, 5 mL of whole blood was pipetted into a polypropylene tube, followed by the addition of 100 µL of the appropriate working QC stock, resulting in whole blood concentrations of 5.0 (low QC), 15 (mid QC), and 40 (high QC) ng/mL. The fortified whole blood was then spotted (30 µL/spot) onto labeled DBS cards for each standard or QC solution, until the solution was exhausted, then dried for 2 h on the bench-top. DBS cards were stored in a zippered bag with a desiccant pack at −80 °C until assayed.
2.3. Extraction overview
For each sample type (standards, QCs, unknowns), a 6 mm Harris Uni-Core™ punch was taken from the center of the DBS and transferred to an appropriately labeled 13 × 100 mm polypropylene tube. After each punch, a separate punch of blank filter paper was obtained and discarded, and after every analytical batch the punching device was thoroughly rinsed with methanol. Both steps were conducted to limit the risk of carry-over within and between analytical batches. A 50 µL volume of the working internal standard was then added to every sample (including a single blank “zero” sample containing an unfortified DBS punch) other than the double blank, to which a 50 µL volume of methanol was added.
After the addition of internal standard, 500 µL of freshly-prepared 10% NH4OH followed by 2 mL of MtBE were added to the polypropylene tube, and vortexed well. The solution was then sonicated for 15 min in a water bath held at ambient temperature, to accelerate the rate at which drug was liberated from the DBS card paper. The mixture was then centrifuged (Beckman GPR Centrifuge, Beckman Coulter®, Indianapolis, IN) at 3200 rpm for 20 min to separate the aqueous and organic layers, and placed in a 80 °C freezer for 30 min to freeze the aqueous layer. The organic layer was then decanted into a fresh polypropylene tube. The organic layer was then dried under lab air (15 psi) in a 40 °C water bath (Zymark TurboVap® LV, Biotage, Uppsala, Sweden) for 18 min, followed by reconstitution with 100 µL of 50:50 methanol:5 mM NH4HCO3. The samples were then centrifuged at 3200 rpm for 5 min to collect the entire reconstitution volume at the bottom of the polypropylene test tube, and transferred from the test tube to a 300 µL polypropylene autosampler vial for analysis. A schematic of the sample extraction procedure is shown in Fig. 1.
Fig. 1.

Schematic describing the sample extraction procedure.
2.4. UHPLC-MS/MS instrumentation and conditions
Sample detection was accomplished with a Waters Micromass Quattro Premier XE® MS/MS coupled to a Waters Acquity® UPLC instrument (Waters Corp., Milford, MA). Data were captured using MassLynx v4.1 software. An Acquity® BEH C18 1.7 µm, 2.1 × 50 mm analytical column was used with a guard column for chromatographic separation. Separation was achieved using a gradient method combining 5 mM NH4HCO3 at pH 8 (A) and methanol (B) at a flow rate of 0.300 mL/min. For each sample, the instrument was equilibrated at 50% A:50% B (v/v) prior to injection and held at that proportion for the first 30 s after injection. At 30 s the gradient was linearly increased to 100% B until 3 min post injection. Mobile phase was subsequently held at 100% B for one minute, after which the composition was immediately returned to 50% A:50% B and maintained for 1 min to re-equilibrate. The column was heated to 45 °C throughout the injection, while the autosampler tray was maintained at 10 °C. A 50 µL sample loop was used, and 25 µL was injected for each sample in partial loop mode.
The MS/MS was operated in positive ionization mode. Selected Reaction Monitoring (SRM) transitions for budesonide and budesonide-D8 were 431.2->147.1 and 439.3->147.1, respectively. Capillary voltage, cone voltage, and collision energy were set at and desolvation temperatures were 100 °C and 400 °C, respectively. 3.5 kV, 20 V, and 30 eV respectively, for both compounds. The source The cone gas flow was set to 50 L/hr, while the desolvation gas flow was set to 800 L/hr.
3. Results
3.1. Method optimization
Three primary factors were considered when optimizing the extraction and UHPLC-MS/MS method, in order to achieve the greatest sensitivity possible. We first sought to determine the MS/MS and mobile phase conditions needed to optimize the response of budesonide in the MS/MS detector. Second, multiple extraction methods and reconstitution solvents were tested to determine which best liberated drug from the DBS punch and yielded the highest sensitivity. Finally, we tested whether borosilicate glass or polypropylene plastic tubes impacted assay sensitivity.
Optimization of UHPLC-MS/MS conditions included product ion selection and mobile phase changes. The initial MS/MS conditions used 15 eV collision energy and the product ions at m/z 323 for both the analyte and internal standard. Increasing the collision energy to 30 eV generated product ions at m/z 173 of comparable abundance to that at m/z 323, but yielded a more abundant product ion at m/z 147. Using the m/z 147 product ion was found to improve both sensitivity and selectivity when analyzing extracted samples, therefore was used for quantitation. Initially, the aqueous mobile phase component was 5 mM ammonium acetate, pH 5, and the organic component was methanol. Mobile phases containing acetonitrile reduced retention and sensitivity as compared to those with methanol. A comparison of aqueous mobile phases including 0.1% formic acid, 10 mM ammonium formate, pH 3.5, 5 mM ammonium acetate, pH 5, and 5 mM ammonium bicarbonate, pH 8, demonstrated that ammonium bicarbonate yielded the highest response from a pooled sample extract.
Three different extraction methods were investigated, including simple methanol sonication, a combined methanol/MtBE extraction, and finally, an MtBE only extraction. In the initial stages of method development, it was anticipated that using methanol to liberate drug from the DBS punch would yield the highest recovery, with acceptable sample cleanliness. However, initial extractions using this method resulted in frequent clogging of the injection valve of the autosampler, which we attributed to paper fibers from the DBS punch that may remain in the sample following extraction. Furthermore, this extraction method was associated with an interference peak that was unable to be baseline resolved from budesonide without compromising the peak shape of budesonide. We therefore evaluated an MtBE-based sample clean-up procedure, both with and without the use of methanol to liberate drug from the punch. While MtBE decreased absolute signal response in the MS/MS (due to ion suppression), it eliminated the interference peak, as well as the issue of autosampler clogging. Removing the interference peak greatly improved the signal-to-noise ratio for the samples, and eased peak integration by the data system, improving the achieved method sensitivity. The use of methanol did not yield increased signal, so was not further evaluated. Finally, during method development, we found that reconstitution of samples with a mixture of methanol and NH4HCO3 buffer demonstrated enhanced signal compared to the use of a methanol:water mixture, or water alone.
Lastly, we performed an experiment to determine whether budesonide adhered to either borosilicate glass or polypropylene plastic tubes during the extraction process. This experiment was conducted in large part due to the observation of nonlinearity in the dynamic range of the assay during method development, when borosilicate glass was used. To determine whether response was affected by the composition of the tube used during extraction, we extracted triplicate blank DBS punches with internal standard added in both borosilicate and polypropylene tubes. A two-fold higher response was observed in the polypropylene tubes compared with the response in the borosilicate tube. Furthermore, an extracted calibration curve using polypropylene tubes was found to be linear. Combined, this suggests that budesonide adhered to the borosilicate, thereby limiting assay sensitivity and introducing nonlinearity into the dynamic range of the assay.
3.2. Accuracy and precision
Method accuracy and precision were calculated from replicate analysis (n = 6) of each QC level (n = 3) as part of three separate analytical runs (n = 18 total replicates at each QC level). In addition, duplicate calibration curves were co-extracted and analyzed with each analytical batch. Acceptance criteria for both standards and QCs were ±15% for both accuracy (compared to nominal as % difference) and precision (determined as % coefficient of variation) at all concentrations but the lower limit of quantitation (LLOQ), where ±20% was allowed.
Standard curves were fit with a linear regression using 1/x2 weighting. The dynamic range of the assay was between 1 and 50 ng/mL. Standard curve precision was within 10.5% across the concentration range, while accuracy was within ±8.8% for all back-calculated standards (Table 1). Four (of 48 total) standard samples (one each of the 5.0, 10, 25, and 50 ng/mL standards) were removed from the calibration curve due to back-calculated results outside of the ±15% acceptance criteria. Three of these four samples occurred during the same analytical run. The slopes of the standard curves had a precision of 3.7%, and all standard curves had R2>0.9857 (Table 1). Across all analytical batches (validation and sample analysis), the 1 ng/mL LLOQ calibration standard had an accuracy of −0.2% and a precision of 7.0%. Additionally, a 1 ng/mL calibration standard was analyzed independently from the calibration curve to assess sensitivity at the start of every analytical batch (n = 8), and all were found to back-calculate within 11.2% of nominal. These data support the accuracy and reproducibility of the assay at the LLOQ.
Table 1.
Standard curve performance from 3 validation runs with duplicate standard curves (n = 6 replicates total). The standard curve was fit with a linear regression with 1/x2 weighting.
| Calibration Standards (1.0–50 ng/mL) | |
|---|---|
| Interassay Accuracy (%diff) | −8.8 to 7.4 |
| Interassay Precision (%CV) | 5.7 to 10.5 |
| Slope, Mean | 0.0828 |
| Slope, Precision (%CV) | 3.7 |
| R2, Mean | 0.9913 |
Inter-assay precision was within 8.2%, and inter-assay accuracy was within ±6.0% for all QC levels (Table 2). Similarly acceptable results were observed for inter-assay precision (<7.7%) using the analysis of variance (ANOVA) approach described by Wille et al [17]. Intra-assay precision and accuracy were within 9.4% and ±7.2%, respectively, across all analytical runs and QC levels (Table 2). Two (of the 54) QCs (one 15 and one 40 ng/mL QC) failed the ±15% acceptance criteria; both of these samples occurred during the same analytical batch.
Table 2.
Performance of each of the 3 QC levels analyzed in 6 replicates in each of 3 different validation runs (n = 18 replicates total).
| Low QC (5 ng/mL) | Mid QC (15 ng/mL) | High QC (40 ng/mL) | |
|---|---|---|---|
| Interassay Accuracy (%diff) | −6.0 | −3.9 | −3.3 |
| Interassay Precision (%CV) | 5.5 | 7.7 | 8.2 |
| Intraassay Accuracy (%diff) | −7.2 to −4.5 | −5.9 to −0.4 | −6.9 to 1.5 |
| Intraassay Precision (%CV) | 3.0 to 8.8 | 4.4 to 9.4 | 6.6 to 9.3 |
Example chromatograms of a double blank, LLOQ, and patient sample are shown in Fig. 2a–c.
Fig. 2.

Representative blank (A), LLOQ (1 ng/mL, B), and patient sample (10.7 ng/mL, C) chromatograms. The top row represents budesonide, while the bottom row shows chromatograms for the internal standard, budesonide-d8.
3.3. Selectivity and specificity
DBS selectivity was assessed by preparing DBS from 3 separate lots of blank whole blood. Instrument carryover was assessed by injecting a blank sample following the high standard. Crosstalk was assessed by injecting an extracted 50 ng/mL sample without internal standard, and an extracted sample containing only internal standard. There was no significant budesonide or internal standard signal in any of the blank lots of blood, supporting the specificity of the method. No carryover was observed, nor was there any crosstalk between the analyte and internal standard, supporting method selectivity.
3.4. Matrix effects and extraction recovery
An investigation to determine recovery (RE), matrix effects (ME), and process efficiency (PE) was conducted using the experiment published by Mastuzewski et al. [18]. Briefly, three sets of samples at three concentrations (QCs: 5, 15, and 40 ng/mL) were prepared in triplicate, and analyzed to determine peak response. Set 1 contained budesonide spiked into the reconstitution solvent, set 2 consisted of blank DBS punches that were extracted and then spiked with budesonide, while set 3 samples represented full extracts of the QCs. Set 2 and 3 used a single lot of whole blood. A 50 µL volume of internal standard was added to each set of samples. In set 1, budesonide and the internal standard were added to a tube then dried prior to adding the reconstitution solvent; in set 2, budesonide and the internal standard were added to the tube following evaporation of the MTBE, then dried prior to adding the reconstitution solvent; in set 3, internal standard was added as described in the extraction overview. ME was determined by comparing set 2 to set 1, RE was calculated by comparing set 3 and set 2, and PE was found by comparing set 3 and 1. Ion suppression was negligible for both budesonide (ME = 94.2%) and the internal standard (ME = 95.2%). These results, combined with the use of an isotopic internal standard, minimize the potential impact of the DBS matrix on quantitation. Budesonide (RE = 77.9%) and the internal standard (RE = 85.7%) were adequately recovered from the DBS punch extraction. Across all concentrations in the RE experiment, the precision was 9.8% for budesonide (IS = 3.4%), and RE was not correlated with concentration. Notably, the similarity in RE between budesonide (in the DBS punch) and the internal standard (not in the DBS punch) suggests that the efficiency of eluting budesonide from the DBS punch was high. Total process efficiency for budesonide (PE = 73.4%) and its internal standard (PE = 81.6%) demonstrated minimal loss of sensitivity during the course of analysis.
3.5. Correlation of DBS and plasma concentrations of budesonide
In order to relate concentrations obtained from the DBS matrix to those described in literature from plasma, we conducted an experiment to assess the correlation between concentrations quantified in the two matrices. A total of 8 samples were created in-house by spiking varying concentrations of budesonide into 1 mL of whole blood, similar to the preparation of DBS standards and QCs, followed by a 10 min incubation period at room temperature. These samples were then centrifugated to isolate plasma. Duplicate 50 µL aliquots of plasma were extracted using the same method described for DBS samples. Plasma sample concentrations were interpolated from a standard curve that was prepared directly in plasma (and confirmed with duplicate QCs at a low (5 ng/mL), medium (15 ng/mL), and high (40 ng/mL) concentration, all of which were within ±10.6% of the expected concentration), then correlated to the expected DBS concentration using a linear regression. The regression demonstrated that DBS and plasma concentrations were correlated (R2= 0.978), and that plasma concentrations were 12.5% higher than those determined in DBS, as determined by the slope of the regression line (Fig. 3).
Fig. 3.

Correlation between DBS and plasma concentrations of spiked samples.
3.6. Hematocrit
The impact of hematocrit on determining budesonide concentrations in DBS was tested by comparing lab-prepared samples with differing hematocrits. Blank whole blood (hematocrit = 48%) was obtained from an adult male and diluted with plasma to make whole blood with a hematocrit of 24% (500 µL whole blood with 500 µL plasma) and 14.4% (300 µL whole blood with 700 µL plasma). Additionally, a 1 mL plasma aliquot was isolated to represent a 0% hematocrit. The blank blood at each hematocrit was then fortified to represent the medium (15 ng/mL) and high (40 ng/mL) QC levels, and spotted onto the DBS cards. These samples were then compared to QC samples at the same concentration (hematocrit = 41%). Acceptance criteria for this experiment was a mean concentration within ±15% of the determined concentrations for the QC samples (a hematocrit of 41%).
Fig. 4 shows the results from this experiment for the medium (A) and high (B) QC samples. Notably, while there was a trend for decreased concentration in samples with lower hematocrits, only the medium QC sample at a hematocrit of 48% and the 0% hematocrit samples fell outside the acceptance criteria. Visual observation showed minimal difference of the blood spot diameter between the samples, except for the spot consisting of a 0% hematocrit, which had a much larger diameter than the other samples. The greater area on the DBS card for these plasma only samples is a likely reason behind this sample not passing the experiment’s acceptance criteria. The hematocrits observed in the ELGAN participating in the clinical study described below ranged between 21 and 46%, which, in combination with the results of this experiment, suggest that hematocrit did not have a significant impact on budesonide concentrations from clinical samples.
Fig. 4.
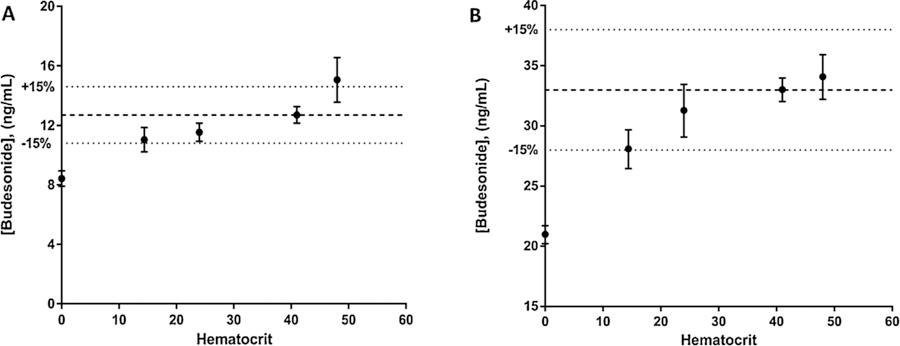
Effect of hematocrit on budesonide from DBS spiked at the medium (A) and high (B) QC concentrations. Data points represent mean (SD) of triplicate samples at each hematocrit value. Dashed line represents the mean value of the sample at 41% hematocrit (the hematocrit of standard and QC samples), while dotted lines represent the acceptance criteria for the experiment (±15% of the sample at 41% hematocrit).
3.7. Stability
The stability of budesonide both in stock solutions and in DBS were tested under a variety of conditions using both the low and high QCs. Budesonide was considered stable under the test condition if both precision and accuracy (as determined by % difference between the test sample and either the nominal or control sample) were within ±15%. Long term storage of methanolic stock solutions at −20 °C was tested by comparing stock solutions prepared a year apart. Long term DBS stability at 80 °C was tested by comparing QC samples that were stored for 3 months against freshly prepared samples. Finally, we tested the conditional stability of these samples after three freeze/thaw cycles (a thaw cycle consisted of 3 h at room temperatures) or 24 h storage at room temperature. The stability of budesonide in the reconstituted matrix was tested by comparing freshly extracted samples to extracted samples that were previously injected and then stored in the autosampler (4 °C) for 24 h.
Methanolic stock solutions stored for 1 year at −20 °C were stable (%diff = 1.4%), as were DBS stored at 80 °C for 3 months (%diff < ±15.0%). DBS subjected to freeze/thaw cycles were stable (%diff < 14.9%), however, those samples stored at room temperature for 24 h were not stable, with percent differences ranging between 13.2–29.1% lower than nominal. Finally, reconstituted samples were able to be reinjected 24 h later (%diff < ± 11.8%).
3.8. Clinical application
The clinical application of this analytical method was demonstrated using samples collected as part of a multi-center pilot study investigating the efficacy and safety of increasing doses of budesonide mixed into a surfactant vehicle for preventing BPD in mechanically ventilated premature neonates between 23 and 28 weeks gestational age (NCT 02907593, IND Number 128102). DBS samples were collected as part of an IRB-approved study from participants whose parents provided consent for their enrollment into the study. Budesonide was delivered via intra-tracheal instillation at doses of 0.025, 0.05 and 0.10 mg/kg given every 24 h, with 8 participants receiving each dose level. Blood was primarily collected via a central line into a micro-syringe, then spotted in volumes per spot, before being dried for between 1 and 4 h, followed by storage at 80 °C. Samples were collected 15 min, 1 h, and 4 h after the first dose, as well as immediately prior to doses 2, 3, 4, and 5. Mean (%CV) 1 h concentrations were 6.63 (51%), 6.59 (62%), and 12.6 (26%) ng/mL for the 0.025, 0.05, and 0.10 mg/kg doses, respectively (Fig. 5). When adjusted for the dose of budesonide, the observed concentrations are similar to those described in a previous study in preterm neonates conducted by Yeh et al. [9] Samples obtained at 24 h after dosing were predominantly below the 1 ng/mL limit of quantitation (n = 71 of 73 collected samples). Twenty of 24 patients had a 24 h post dose sample with a chromatogram that mirrored that of the double blank samples in the analytical batch, providing further evidence of the method’s selectivity.
Fig. 5.
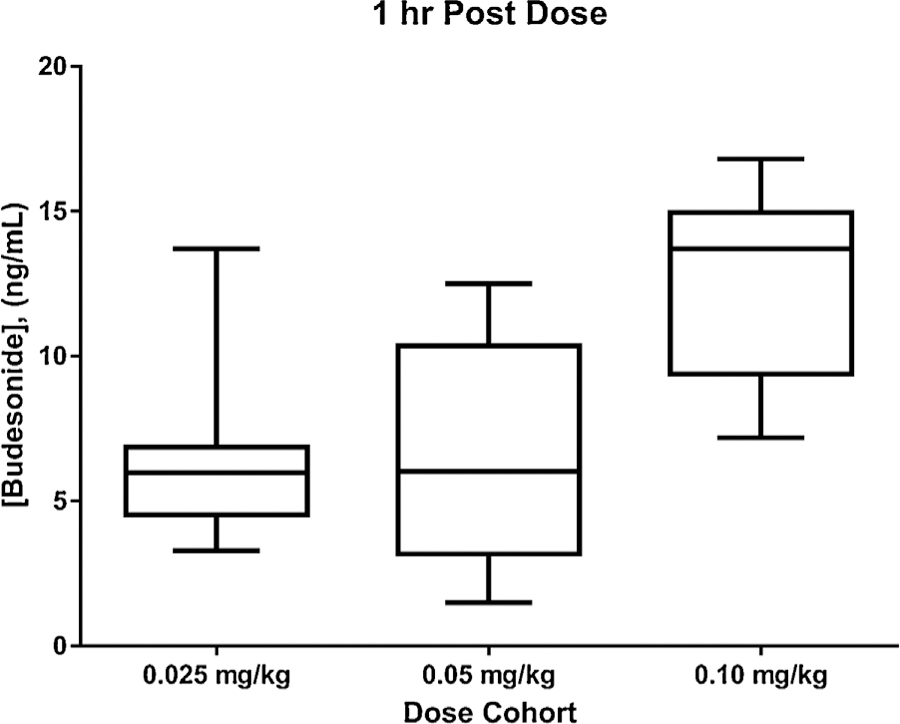
Box (mean, interquartile range) and whiskers (range) plot of 1 h post-dose budesonide concentrations in DBS, separated by dosing cohort (0.025, 0.05, and 0.10 mg/kg).
4. Discussion
The described method achieves several goals that were established at the initiation of method development, most importantly the attainment of a 1 ng/mL LLOQ from a low blood volume sample. The extraction was simple, and resulted in accurate and precise quantitation of budesonide in DBS. The method demonstrated that DBS are a viable technique for conducting PK studies in ELGAN and other pediatric populations. Method application yielded important data that describe the PK of budesonide in ELGAN following intratracheal instillation.
Previous descriptions of analytical methods measuring budesonide from human blood have primarily used large volumes of plasma to attain the requisite sensitivity. These descriptions include the use of 1 mL of plasma to achieve a LLOQ of 0.1 ng/mL [19], 0.35 mL plasma for a LLOQ of 0.05 ng/mL [20], and 0.5 mL plasma for a LLOQ of 0.0075 ng/mL [21], among others [22–24]. The whole blood volume needed to yield these plasma volumes is unattainably large for a clinical study collecting serial PK samples in ELGAN, necessitating the use of DBS in our study. One prior publication by Thomas et al. describes the use of a 20 µL DBS to attain a limit of detection of 0.25 ng/mL [25]. However, the method by Thomas et al. included a total of 26 target compounds, and was therefore not optimized for the extraction and determination of budesonide. Notably, we were able to apply our method to a study in ELGAN, showing the utility of DBS in this population, whereas the method by Thomas et al. utilized only fortified DBS.
DBS offer many advantages to traditional venipuncture sampling, especially in the setting of ELGAN and other pediatric populations. Notably, no parent refused consent to participate in the study due to the collection of DBS. Chief among these benefits is that DBS require collection of a substantially (> 5-fold) less blood than venipuncture sampling. The decrease in blood volume enables the collection of serial blood samples, which can be used to more accurately describe PK in this under-studied population. While venipuncture collection requires a needle stick to access blood, DBS can be collected via finger or heel-stick or a pre-existing central line, techniques which are less invasive and potentially more comfortable for the patient. Parents or other caregivers can be trained to collect DBS via finger or heel-stick, opening the possibility for in-home collection of samples. Once dried, DBS are safe to ship without biohazardous labeling, and, depending on drug stability, may be shipped at ambient conditions. Notably, the ambient temperature stability of budesonide in the DBS matrix may not be adequate for ambient shipping, based on the results from our stability experiments. These many advantages position DBS to be a useful matrix for collecting PK samples from ELGAN and other pediatric populations. We successfully collected DBS samples from ELGAN between the ages of 23 and 28 weeks gestation, supporting the utility of DBS for multi-center PK studies in this fragile population.
While DBS offer many advantages, there are some disadvantages compared to method development for traditional venipuncture plasma samples. Of primary concern is the uniformity with which blood spreads on the DBS filter paper following collection. Many factors, including spot volume, hematocrit, punch location, and the site of blood collection (finger-stick, heel-stick, venipuncture) may impact the extent to which blood wicks on the DBS, resulting in samples which are not uniform within a single DBS spot, the spots for an individual patient, or the entire study population. [12,26,27] The impact of these various factors can and should be assessed during method validation, and/or carefully controlled during the course of a clinical study. For the described assay, we chose to control these factors as best as possible throughout the conduct of the study. Spot volumes were to be equal (30 µL) across all patient samples, as well as the standards and QCs made in house. Hematocrit may impact the extent to which blood spreads on the DBS card, which would in turn lead to changes in the blood volume assayed within a single punch and/or extraction recovery. Importantly, the variability in hematocrit in the patients enrolled in the current study was relatively minimal (mean (%CV): 36 (14%)). This decreases the likelihood that the spread of blood on the DBS card was sufficiently different between study samples to impact assay results. In combination with experimental data suggesting that budesonide concentrations did not vary outside of acceptance criteria in hematocrits between 14 and 41%, (48% at the high QC) the impact of hematocrit in this study of ELGAN does not appear to be significant. Budesonide is expected to be highly bound to plasma protein [28,29], therefore, we do not anticipate the drug to concentrate into red blood cells, which is another mechanism by which hematocrit could impact quantitation in this study. The 6 mm diameter punch used covered the majority of DBS, meaning that only center punches could be used for this study. Finally, blood was primarily collected through a central line into a syringe, therefore the impact of collection site was not assessed for this validation.
The vast majority of PK literature uses concentration data determined from plasma. Therefore, a comparison between plasma and DBS concentrations of the drug of interest is needed to place results in the context of prior literature. Where possible, this is best done using blood collected from patients via venipuncture, from which DBS are spotted prior to plasma isolation. The use of patient samples for this assessment is the most accurate representation of how a drug is distributed within the blood milieu in vivo. However, we were unable to collect paired samples from the patients in our study, which meant that we had to rely upon laboratory prepared samples to determine the correlation between the two matrices. From this analysis, we found that plasma concentrations of budesonide are 12.5% higher than those determined in DBS. This finding is similar to previously published data, which suggests that the blood:plasma ratio of budesonide is . [30] Future in vivo studies are needed to confirm that this correlation represents the distribution of budesonide in circulating blood.
To summarize, the described method is an accurate, precise, and robust approach for measuring budesonide concentrations in ELGAN. The success of this approach supports the use of DBS for future studies involving ELGAN and other pediatric populations where blood volume limitations are an obstacle to collecting rich PK data.
Acknowledgements
The authors would like to thank the study participants and their families, as well as the Steroids and Surfactant in Extremely Low Gestational Age Infants (SASSIE) study team for their help in obtaining the samples used in this analysis. We would also like to acknowledge the contributions Dr. Chris Stockman made in designing the clinical study prior to his untimely passing. The work described in this manuscript was funded by the Thrasher Foundation, Thrasher Research Fund Award #12450 (C. McEvoy).
Funding source
The work described in this manuscript was funded by the Thrasher Foundation, Thrasher Research Fund Award #12450 (C. McEvoy). The Thrasher Foundation was not involved in study design, or in the collection, analysis, or interpretation of the data, or in the writing or submission of this manuscript.
Footnotes
Competing interests
The authors have no competing interests relevant to this publication.
References
- [1].McEvoy CT, Jain L, Schmidt B, Abman S, Bancalari E, Aschner JL, Bronchopulmonary dysplasia: NHLBI workshop on the primary prevention of chronic lung diseases, Ann. Am. Thorac. Soc 11 (Supplement 3) (2014) S146–S153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Ballard RA, Truog WE, Cnaan A, Martin RJ, Ballard PL, Merrill JD, Walsh MC, Durand DJ, Mayock DE, Eichenwald EC, Inhaled nitric oxide in preterm infants undergoing mechanical ventilation, N. Engl. J. Med 355 (4) (2006) 343–353. [DOI] [PubMed] [Google Scholar]
- [3].Laughon MM, Langer JC, Bose CL, Smith PB, Ambalavanan N, Kennedy KA, Stoll BJ, Buchter S, Laptook AR, Ehrenkranz RA, Prediction of bronchopulmonary dysplasia by postnatal age in extremely premature infants, Am. J. Respir. Crit. Care Med 183 (12) (2011) 1715–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Halliday HL, Ehrenkranz RA, Doyle LW, Early (< 8 days) postnatal corticosteroids for preventing chronic lung disease in preterm infants, Cochrane Database Syst. Rev 1 (1) (2009). [DOI] [PubMed] [Google Scholar]
- [5].Halliday HL, Ehrenkranz RA, Doyle LW, Moderately Early (7–14 Days) Postnatal Corticosteroids for Preventing Chronic Lung Disease in Preterm Infants, The Cochrane Library, 2003. [DOI] [PubMed]
- [6].Halliday HL, Ehrenkranz RA, Doyle LW, Late (& 7 days) postnatal corticosteroids for chronic lung disease in preterm infants, Cochrane Database Syst. Rev 1 (2009). [DOI] [PubMed] [Google Scholar]
- [7].Barrington KJ, The adverse neuro-developmental effects of postnatal steroids in the preterm infant: a systematic review of RCTs, BMC Pediatr 1 (1) (2001) 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Yeh TF, Chen CM, Wu SY, Husan Z, Li TC, Hsieh WS, Tsai CH, Lin HC, Intratracheal administration of budesonide/surfactant to prevent bronchopulmonary dysplasia, Am. J. Respir. Crit. Care Med 193 (1) (2016) 86–95. [DOI] [PubMed] [Google Scholar]
- [9].Yeh TF, Lin HC, Chang CH, Wu TS, Su BH, Li TC, Pyati S, Tsai CH, Early intratracheal instillation of budesonide using surfactant as a vehicle to prevent chronic lung disease in preterm infants: a pilot study, Pediatrics 121 (5) (2008) e1310–e1318. [DOI] [PubMed] [Google Scholar]
- [10].Wilhelm AJ, den Burger JCG, Swart EL, Therapeutic drug monitoring by dried blood spot: progress to date and future directions, Clin. Pharmacokinet 53 (11) (2014) 961–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Patel P, Mulla H, Tanna S, Pandya H, Facilitating pharmacokinetic studies in children: a new use of dried blood spots, Arch. Dis. Child 95 (6) (2010) 484–487. [DOI] [PubMed] [Google Scholar]
- [12].Spooner N, Lad R, Barfield M, Dried blood spots as a sample collection technique for the determination of pharmacokinetics in clinical studies: considerations for the validation of a quantitative bioanalytical method, Anal. Chem 81 (4) (2009) 1557–1563. [DOI] [PubMed] [Google Scholar]
- [13].Wagner M, Tonoli D, Varesio E, Hopfgartner G, The use of mass spectrometry to analyze dried blood spots, Mass Spectrom. Rev 35 (3) (2016) 361–438. [DOI] [PubMed] [Google Scholar]
- [14].Lawson G, Patel P, Mulla H, Tanna S, Dried blood spot sampling with LC-MS analysis for routine therapeutic caffeine monitoring in neonates, ISRN Chromatogr 2012 (2012). [Google Scholar]
- [15].Patel P, Tanna S, Mulla H, Kairamkonda V, Pandya H, Lawson G, Dexamethasone quantification in dried blood spot samples using LC–MS: the potential for application to neonatal pharmacokinetic studies, J. Chromatogr. B 878 (31) (2010) 3277–3282. [DOI] [PubMed] [Google Scholar]
- [16].Dietzen DJ, Bennett MJ, Lo SF, Grey VL, Jones PM, Dried blood spot reference intervals for steroids and amino acids in a neonatal cohort of the national children’s study, Clin. Chem 2016 (2016), 263434. [DOI] [PubMed] [Google Scholar]
- [17].Wille SM, Peters FT, Di Fazio V, Samyn NJA, Assurance Q, Practical aspects concerning validation and quality control for forensic and clinical bioanalytical quantitative methods, Accred. Qual. Assur 16 (6) (2011) 279. [Google Scholar]
- [18].Matuszewski BK, Constanzer ML, Chavez-Eng CM, Strategies for the assessment of matrix effect in quantitative bioanalytical methods based on HPLC MS/MS, Anal. Chem 75 (13) (2003) 3019–3030. [DOI] [PubMed] [Google Scholar]
- [19].Wang Y, Tang Y, Moellmann H, Hochhaus G, Simultaneous quantification of budesonide and its two metabolites, 6þ-hydroxybudesonide and 16a-hydroxyprednisolone, in human plasma by liquid chromatography negative electrospray ionization tandem mass spectrometry, Biomed. Chromatogr 17 (2–3) (2003) 158–164. [DOI] [PubMed] [Google Scholar]
- [20].Nilsson K, Andersson M, Beck O, Phospholipid removal combined with a semi-automated 96-well SPE application for determination of budesonide in human plasma with LC–MS/MS, J. Chromatogr. B 970 (2014) 31–35. [DOI] [PubMed] [Google Scholar]
- [21].do Carmo Borges NC, Astigarraga RB, Sverdloff CE, Borges BC, Paiva TR, Galvinas PR, Moreno RA, Budesonide quantification by HPLC coupled to atmospheric pressure photoionization (APPI) tandem mass spectrometry. Application to a comparative systemic bioavailability of two budesonide formulations in healthy volunteers, J. Chromatogr. B 879 (3–4) (2011) 236–242. [DOI] [PubMed] [Google Scholar]
- [22].Li YNB, Tattam B, Brown KF, Seale JP, Quantification of epimeric budesonide and fluticasone propionate in human plasma by liquid chromatography–atmospheric pressure chemical ionization tandem mass spectrometry, J. Chromatogr. B Biomed. Sci. Appl 761 (2) (2001) 177–185. [DOI] [PubMed] [Google Scholar]
- [23].Streel B, Cahay B, Klinkenberg R, Using total error concept for the validation of a liquid chromatography–tandem mass spectrometry method for the determination of budesonide epimers in human plasma, J. Chromatogr. B 877 (23) (2009) 2290–2300. [DOI] [PubMed] [Google Scholar]
- [24].Kronkvist K, Gustavsson M, Wendel A-K, Jaegfeldt H, Automated sample preparation for the determination of budesonide in plasma samples by liquid chromatography and tandem mass spectrometry, J. Chromatogr. A 823 (1–2) (1998) 401–409. [DOI] [PubMed] [Google Scholar]
- [25].Thomas A, Geyer H, Schänzer W, Crone C, Kellmann M, Moehring T, Thevis M, Sensitive determination of prohibited drugs in dried blood spots (DBS) for doping controls by means of a benchtop quadrupole/Orbitrap mass spectrometer, Anal. Bioanal. Chem 403 (5) (2012) 1279–1289. [DOI] [PubMed] [Google Scholar]
- [26].Denniff P, Spooner NJB, The effect of hematocrit on assay bias when using DBS samples for the quantitative bioanalysis of drugs, Bioanalysis 2 (8) (2010) 1385–1395. [DOI] [PubMed] [Google Scholar]
- [27].Li W, Tse FL, Dried blood spot sampling in combination with LC-MS/MS for quantitative analysis of small molecules, Biomed. Chromatogr 24 (1) (2010) 49–65. [DOI] [PubMed] [Google Scholar]
- [28].Taylor S, Harker A, Modification of the ultrafiltration technique to overcome solubility and non-specific binding challenges associated with the measurement of plasma protein binding of corticosteroids, J. Pharm. Biomed. Anal 41 (1) (2006) 299–303. [DOI] [PubMed] [Google Scholar]
- [29].Zhang F, Xue J, Shao J, Jia L, Compilation of 222 drugs’ plasma protein binding data and guidance for study designs, Drug Discov. Today 17 (9–10) (2012) 475–485. [DOI] [PubMed] [Google Scholar]
- [30].Szefler SJ, Pharmacodynamics and pharmacokinetics of budesonide: a new nebulized corticosteroid, J. Allergy Clin. Immunol 104 (4) (1999) S175–S183. [DOI] [PubMed] [Google Scholar]