

Abstract
The ability of cells to organize into multicellular structures in precise patterns requires that they “recognize” one another with high specificity. We discuss recent progress in understanding the molecular basis of cell-cell recognition, including unique phenomena associated with neuronal interactions. We describe structures of select adhesion receptor complexes and their assembly into larger intercellular junction structures and discuss emerging principles that relate cell-cell organization to the binding specificities and energetics of adhesion receptors. Armed with these insights, advances in protein design and gene editing should pave the way for breakthroughs toward understanding the molecular basis of cell patterning in vivo.
eTOC blurb – Honig
Differential adhesion is one of the fundamental mechanisms that allows cells to organize into tissues. In this Review, Honig & Shapiro review the molecular basis of cellular adhesion from structures to concepts.
II. Introduction
Cell-cell adhesion receptors play critical roles in the patterning of multicellular structures with their effects depending critically on the selectivity and adhesive strengths of interactions between receptors. A conceptual foundation for one type of patterning phenomenon involving the coalescence of cells based on differential interaction strengths – the differential adhesion hypothesis (DAH) – was developed many years ago (Foty and Steinberg, 2005). However, its impact was limited, in part because quantification of cellular interactions was crude and was not related directly to the properties of the adhesion receptors. It is only recently that interactions among adhesion receptors – often members of large multi-protein families – have been quantitatively assessed. In light of these developments, along with a deeper structural understanding of the assemblies formed by adhesion receptors engaged between cells, underlying principles have begun to emerge as to how protein-protein interactions mediate cell-cell recognition. We draw on our current understanding of the structure and function of select cell-cell adhesion receptors to illustrate how their properties relate to the effects they mediate in cell patterning. The review is by no means comprehensive. Rather, we have chosen topics so as to best highlight general principles.
Adhesion proteins exhibit precisely coded and conserved binding affinities and specificities that differ from one family member to another despite, in many cases, nearly identical sequences and structures. In numerous cases, clear relationships between the evolutionary design of adhesion protein families and biological function have become evident. These connections have been established by combining knowledge of protein structures, biophysical measurements, computational modeling and assessments of cell adhesive function through in vitro cell assays. What has been learned, both technically and conceptually, now opens the door to experiments designed to test specific ideas about how adhesion receptor interactions control cell patterning in vivo.
We consider the molecular basis of three main classes of cell patterning and organization phenomena: (1) The emergence of structure among populations of coalescent cells. This phenomenon involves the organization of multiple cell populations into distinct cell layers and other structures based, at least in part, on the strength of cell-cell interactions. (2) The targeted formation of highly specific interactions between two cells, such as the synaptic connections between neurons. In synapse formation a single axon, after it has been guided to sites where its targets are located, chooses an appropriate target from an array of possible choices (Sanes and Zipursky, 2020). (3) Neurite-neurite repulsion. In the nervous system, sister neurites (from the same neuron) avoid or repel one another, while neurites from different cells can freely interact (Kramer and Stent, 1985). This process, which requires the ability to distinguish “self” from “non-self” neurites, is also mediated by adhesion proteins (Zipursky and Sanes, 2010).
III. The assembly of multicellular structures
The differential adhesion hypothesis
Selective cell adhesion was first demonstrated by marine biologist H.V. Wilson who knew that cells of a sponge could be separated by forcing the sponge through a strainer (Wilson, 1907). The dissociated sponge cells, when left for a period of time, would spontaneously re-associate into a reformed sponge. Wilson reasoned that if he mixed the cells of a yellow sponge and a purple sponge, they would re-associate to form a yellow-and-purple sponge, thus forming a new hybrid animal. But this hoped-for hybrid failed to form. Rather, yellow cells associated with yellow cells, and purple with purple. Although Wilson had failed in creating a hybrid sponge, he had succeeded in demonstrating selective recognition between cells, and moreover had shown that some cells adhere homotypically: that is, they adhere specifically to other cells of the same type. Although a molecular basis for adhesive cell-cell interactions would not emerge for close to a century (Takeichi, 1988), Wilson’s experiments showed that selective adhesion provided a basis for the coalescence of genetically similar cells into tissue structures such as epithelia and cell layers.
The DAH, proposed by cell biologist Malcolm Steinberg, suggested a physical basis for the patterning of homotypic tissues ((Foty and Steinberg, 2005). The DAH relates the adhesive strengths of cells to cell sorting behavior. In essence, just as the properties of immiscible liquids (like oil and water) are defined by their intermolecular interactions, Steinberg and colleagues modeled the behavior of tissues based on the strengths of interactions between cells which were inferred primarily based on the surface tensions of cell-cell aggregates. The DAH makes distinct predictions about cell aggregation and tissue sorting behavior based on the principle that cells will organize so as to minimize their adhesive free energy. The DAH has been verified in many settings, for example largely accounting for the self-organization in vitro of amphibian embryonic cells in concentric layers and for co-aggregation behaviors of cell lines transfected with different adhesion receptors (Foty and Steinberg, 2005).
In the simplest case of a single adhesion receptor that undergoes trans (cell-to-cell) interactions, cell-cell adhesive strength – or avidity – will depend primarily on the expression level of the receptor and on its binding affinity (Katsamba et al., 2009). For example, two cell populations can have different avidities if they express different numbers of the same cell surface protein or similar numbers of different proteins that have different binding affinities. The patterns that are formed in in vitro cell aggregation experiments depend critically on whether the adhesion receptors interact homophilically, heterophilically or both. Figure 1 describes various idealized outcomes for two cell populations, each expressing a different adhesion protein. At one extreme (Figure 1A), if homophilic interactions are strong and heterophilic interactions are weak, separate non-interacting aggregates will form. At the other extreme (Figure 1B), if heterophilic interactions are stronger than homophilic interactions, a checkerboard pattern will form so as to maximize the number of heterotypic cell-cell contacts. Intermediate cases where all interactions are of comparable magnitude can lead to complex patterns depending on the actual affinities. Figure 1C corresponds to a case where homophilic interactions are somewhat stronger than heterophilic interactions, but the latter are also significant. Here, separate homotypic aggregates will form but they will adhere to one another driven by heterophilic interactions (Katsamba et al., 2009). Figure 1D corresponds to the case where heterophilic interactions are intermediate in strength between the two homophilic interactions. In this case a central homotypic core will form, surrounded by more weakly self-interacting cells that bind heterotypically to the central core.
Figure 1.
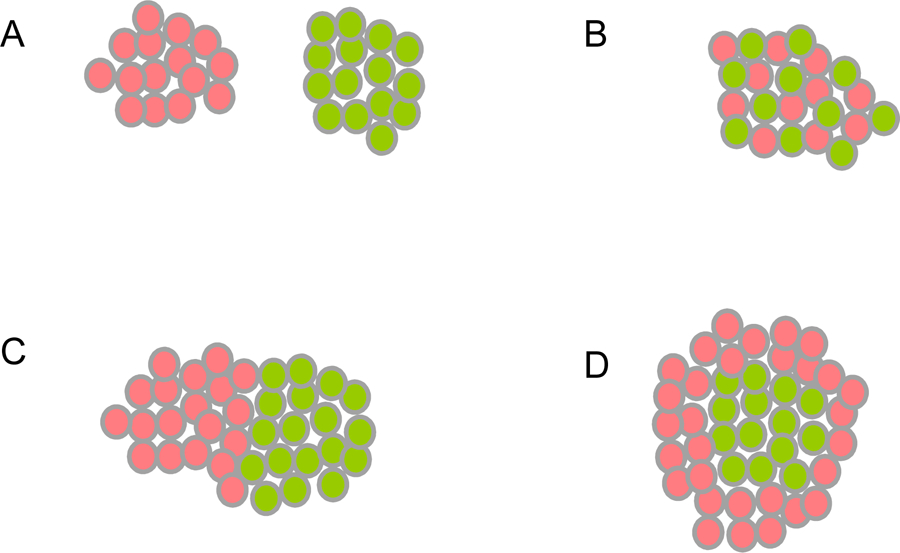
Cell Aggregation Outcomes from the Differential Adhesion Hypothesis (DAH).
Red and green spheres represent cells. In all cases red cells express one adhesion protein, and green cells express another. Homotypic cell-cell interactions are formed between red and red or green and green cells. Heterotypic cell-cell interactions are formed between red and green cells.
(A) When homophilic protein-protein interactions are strong and heterophilic interactions are weak, separate homotypic cell aggregates are formed.
(B) When heterophilic protein-protein interactions are strong, and homophilic interactions are weak, a heterotypic checkerboard pattern is formed.
(C) When homophilic protein-protein interactions are somewhat stronger than heterophilic interactions, two separate homotypic aggregates are formed and these adhere heterotypically.
(D) When heterophilic protein-protein interactions are intermediate in strength between the two homophilic interactions, a heterotypic core is formed by the cells expressing the adhesion protein with stronger homophilic affinity (green cells), which are surrounded by cells expressing the adhesion protein with weaker homophilic affinity (red cells).
A recent paper by Lim and coworkers offers striking examples of the application of the DAH, but in a way that is based on the binding affinities of individual proteins (Toda et al., 2018). Using a cellular system engineered to control expression levels of different type I cadherins, they were able to generate multiple patterns, all of which are consistent with expression levels and known heterophilic and homophilic affinities of the expressed cadherins (Vendome et al., 2014). These, and earlier cell aggregation experiments demonstrate a clear link between biophysical measurements on isolated molecules and the behavior of proteins on cell surfaces. In some cases, complex in vivo behavior can also be explained based on molecular affinities. For example, nectins exhibit strong heterophilic interactions (Harrison et al., 2012) and, as predicted (Figure 1B), helps generate a checkerboard pattern of individual cells in the inner ear (Togashi et al., 2011). In another example, the organization of axon bundles in the Drosophila visual system can be explained, and manipulated, entirely based on N-cadherin expression levels in individual cells (Schwabe et al., 2014).
Despite the overall success of the DAH, it was initially limited by its reliance on macroscopic variables such as surface tension. However, as more has become known about the properties of individual adhesion proteins, it has become possible to use microscopic molecular properties such as expression levels and binding affinities to describe cell-cell adhesive behavior (Katsamba et al., 2009). Implicit to the DAH is the assumption that adhesive strength between two cells is a property of entire cells with adhesion receptors spread uniformly over their surfaces. Assuming uniform distribution of adhesion receptors and no cis interactions, the adhesive strength will be proportional to the number of trans dimers formed, and to the binding free energy of each dimer. The number of dimers formed will in turn depend on the two-dimensional (2D) protein densities. It can be shown that under these conditions, the adhesive strength is given by - RTCiCjVAnln[KD(i,j)]/KD(i,j), where Ci and Cj are 3D concentrations of the interacting domains in cells i and j, V is the volume of the region between interacting cells that is accessible to these domains and allows a transformation between 2D and 3D affinities, An is Avogadro’s number and KD(i,j) is the dissociation constant for the dimerization of the receptors on cells i and j (Katsamba et al., 2009; Wu et al., 2010). An important consequence of this expression is that small differences in binding affinity between two different receptors will be magnified at the cellular level. For example, E-cadherin and N-cadherin have homophilic binding free energies of −5.3 and −6.5 kcal/mole, respectively and, hence, at the same expression levels, more N-cadherin trans dimers will be formed than E-cadherin trans dimers. Thus, not only will each N-cadherin dimer contribute more to adhesive energy than each E-cadherin dimer, there will be more of them, leading to an amplification of adhesion at the cellular level. Cell-cell adhesive strength therefore increases at a greater than linear rate with respect to the molecular binding affinities of the adhesion receptors.
Moreover, the DAH ignores essential features of cellular recognition, such as the localization of adhesion receptors to cell contact regions, the formation of junctions in cell-cell interfaces and the downstream signaling processes mediated by the trans binding of adhesion receptors. Different adhesion proteins may also localize to different regions of a cell surface, in which case the DAH no longer provides a reasonable model. Effects involving multiple adhesion proteins may be important for the development of complex cell patterns. A deeper understanding of cell-cell recognition requires that the structure and biophysical properties of adhesion receptors be integrated into a mechanistic description of cell-cell adhesion.
Structure, binding affinities, and specificities of cell-cell adhesion proteins
Many cell adhesion proteins evolved from two related protein folds: cadherin domains (Figure 2) and immunoglobulin-like (Ig) domains (Figure 3). As can be seen in both figures, very different regions of the protein surface are used to mediate trans complex formation, even when the interacting domains share a common fold. As illustrated in Figure 2, the classical cadherins (type I and type II) (Figure 2B,C, D) bind in trans through a parallel interaction mediated through a strand-swap mechanism involving extracellular cadherin domain 1 (EC1) (Brasch et al., 2018; Harrison et al., 2011; Shapiro et al., 1995); T-cadherin (Figure 2E) also dimerizes in a parallel orientation, but the interface comprises the linker region between EC1 and EC2 (Ciatto et al., 2010); clustered (Figure 2A) (Goodman et al., 2016a; Nicoludis et al., 2015; Rubinstein et al., 2015) and non-clustered protocadherins form an antiparallel interface comprised of EC1-EC4 (Cooper et al., 2016; Harrison et al., 2020; Modak and Sotomayor, 2019). Ig domain proteins also exploit multiple interfaces (Figure 3). Nectins (Figure 3A and B) (Harrison et al., 2012; Narita et al., 2011), DIPs and Dprs (Figure 3C) (Carrillo et al., 2015; Cosmanescu et al., 2018), Sygs (Ozkan) and SynCAMs (Figure 3D) (Dong et al., 2006; Peng et al., 2010) interact through their Ig1 domains in a very similar handshake-like orientation; Sidekicks (Figure 3F) (Goodman et al., 2016c), Axonin-1 (Figure 3G) (Freigang et al., 2000), Hemolin (He et al., 2009), and other Ig-like molecules form a horseshoe-like paddle domain consisting of the four N-terminal Ig domains which form distinct trans interfaces in a variety of orientations; Dscams (Figure 3E), which make use of the N-terminal paddle in addition to other interface elements, fold into an S-shaped structure that enables the formation of three distinct antiparallel interfaces (Sawaya et al., 2008).
Figure 2.
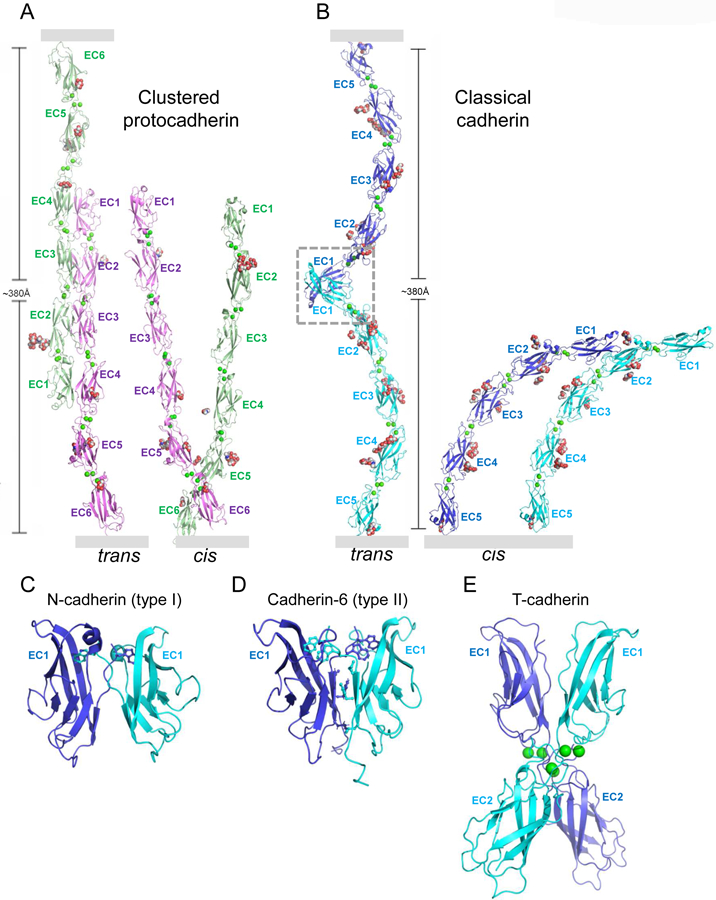
Cis and trans interactions of cadherin superfamily adhesion proteins.
(A) Clustered protocadherin trans dimer (left) and cis dimer (right) from Pcdh γB4 (Brasch et al., 2019). Membrane positions indicated by gray rectangles.
(B) Type I classical cadherin trans dimer (left) with EC1-interface region boxed, and cis dimer (right) from N-cadherin (Harrison et al., 2011). Membrane positions indicated by gray rectangles.
(C-E) Close-up views of trans interfaces from (C) type I N-cadherin (Harrison et al., 2011), (D) type II cadherin-6 (Brasch et al., 2018), with stick representation shown for the swapping tryptophan anchor residues. (E) T-cadherin (Ciatto et al., 2010). Proteins are shown in ribbon representation with key interfacial residues shown as sticks, Ca2+ ions as green spheres, and N-linked glycan as red, white, and blue spheres.
Figure 3.
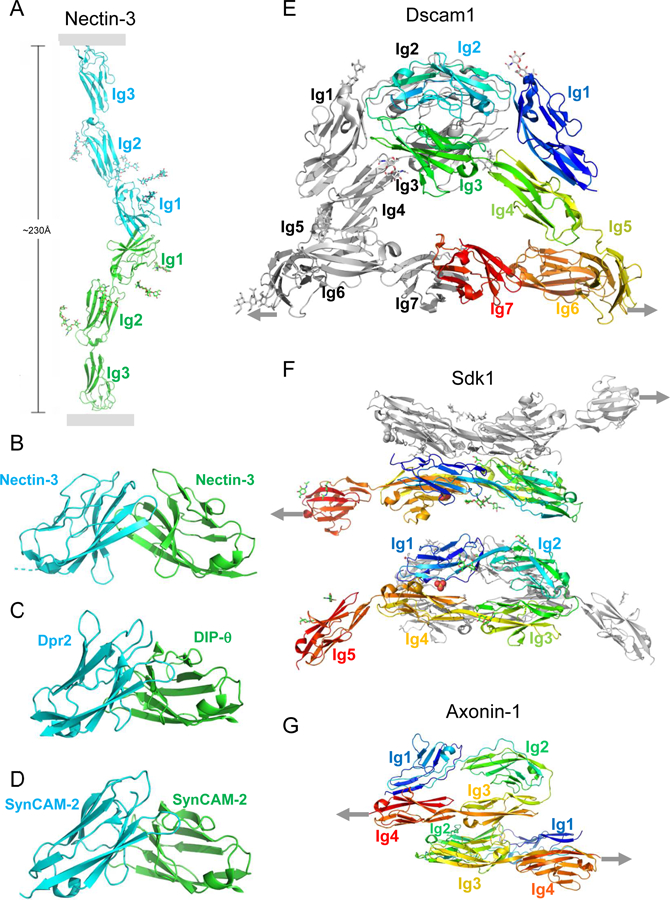
Trans interactions of immunoglobulin superfamily adhesion proteins.
(A) Nectin trans dimer from nectin-3 (Harrison et al., 2012). Membrane positions indicated by gray rectangles. Glycans are shown in stick representation.
(B-D) Close-up views of trans interfaces from (B) nectin-3:nectin-3 (Harrison et al., 2012); (C) Dpr2:DIP-θ (Cosmanescu et al., 2018), and (D) SynCAM2:SynCAM2 (Fogel et al., 2010).
(E) Dscam1 trans dimer interactions (Sawaya et al., 2008).
(F) Sidekick 1 trans dimer interactions (Goodman et al., 2016c).
(G) Axonin 1 trans dimer interactions (Freigang et al., 2000). (E-G) Linkages to the membrane are indicated by gray arrows.
In addition to trans interactions, which are obviously an essential feature of all cell-cell adhesion receptors, there are numerous examples of cis (same cell surface) interactions as well (see Figure 1 for cadherin and protocadherin cis interactions). These also exploit multiple sites on the protein surface, and are often involved in the formation of oligomeric or polymeric assemblies (see discussion below).
The multiple interaction modes observed for adhesion receptors enables the formation of recognition complexes that use numerous regions of the protein surface to encode specificity for different types of cell-cell interactions. Most of the interfaces that are formed display what appear to be highly controlled affinities and specificities that are essential for their biological function. The interplay between structure, affinity and specificity is central to the understanding of the relationship between protein-protein and cell-cell interactions, and is a recurrent theme in the discussion of individual protein families that follows.
Type I “classical” cadherins -
Cadherins comprise a large superfamily of calcium-dependent cell surface receptors that are found in a wide array of species ranging from unicellular animals with multi-cellular life stages to mammals (Takeichi, 2018). In vertebrates, there are two classical cadherin subfamilies – the closely related type I and type II cadherins. Type I cadherins, including vertebrate E- and N-cadherins, are broadly expressed in epithelia with each cell in general expressing only a single type. Type II cadherins are typically expressed in more fine-grained patterns, often with many subtypes co-expressed in the same cell, and play specialized roles – particularly in the nervous system (Basu et al., 2017). Both type I and type II “classical” cadherins – named thusly due to their early characterization – interact with the actin cytoskeleton indirectly through a complex involving α- and β-catenins (Gumbiner and McCrea, 1993; Pappas and Rimm, 2006). The α-catenin interaction has been used as a criterion to classify cadherins as members of the classical subfamily in other organisms including invertebrates (Hulpiau and van Roy, 2009) and single-celled organisms (Abedin and King, 2008). Notably, extracellular regions of the classical cadherins of invertebrates differ substantially from their vertebrate counterparts (Jin et al., 2012).
The vertebrate classical cadherins share a common ectodomain structure (Figure 2) with five tandem EC domains, with stereotyped binding sites for three Ca2+ ions situated between each pair of domains (Shapiro et al., 1995). Ca2+ binding rigidifies the ectodomain in a curved shape such that the long axes of the N- and C-terminal EC domains have orientations that differ by up to 90°. This enables their interaction in trans between cells through the strand-swap mechanism mentioned above, mediated by the N-terminal strand and anchored by a conserved Trp 2 residue (Figure 2B and C).
The five type I classical cadherins conserved in vertebrate genomes (E, N, R, P and M) exhibit trans-homophilic affinities ranging from about 10–20 µM for the closely related N- and R-cadherins to about 100 µM for E-cadherin (Katsamba et al., 2009; Vendome et al., 2014). Cadherin subtype affinities appear to be evolutionarily conserved and arise from small changes in sequence that might be expected to have little effect on affinities since the relevant amino acid substitutions are often conservative. The heterophilic N-/E- binding affinity lies between the two homophilic values and, indeed, mixed aggregates of cells containing N- and E-cadherin form patterns, as predicted from the DAH, similar to that depicted in Figure 1D (Katsamba et al., 2009). It is important to emphasize that combinations of homophilic and heterophilic interactions at the molecular level can lead to distinct cell patterning outcomes, as depicted in the examples shown in Figure 1. In cases where homophilic molecular interactions dominate, homophilic cell-cell interactions will form, but the separate aggregates will stick together through the weaker heterophilic molecular interactions, as in Figures 1C and 1D. These patterns appear to be mirrored in development; for example, the separation of the neural tube from the ectoderm (Hatta and Takeichi, 1986), driven in part by the separation of N- and E-cadherin expressing cells, is reminiscent of the pattern shown in Figure 1D.
The energetic factors that determine type I cadherin binding affinities have been analyzed in great detail (Vendome et al., 2014). Cadherin mutants were designed based on computational analysis and tested experimentally, showing KDs could be increased or decreased by up to two orders of magnitude. These mutants provide a powerful tool to study the role of absolute and relative binding affinities in controlling a wide range of cell patterning phenomena.
In mature tissues, type I cadherins often concentrate into adherens junctions – intercellular assemblies that have characteristic structural features when observed by electron microscopy (Farquhar and Palade, 1963). Biophysical studies have revealed that the ectodomains of type I cadherins spontaneously assemble – between membrane surfaces and in protein crystals – to form a two-dimensional lattice-like structure mediated by cis and trans interactions (Harrison et al., 2011; Taveau et al., 2008) (see Figure 6A). Critically, lattices of the same character have been observed for all type I cadherins investigated, and depend on the type I cadherin-conserved cis interaction. Remarkably, this interaction is too weak to be detected in solution, but appears to be enhanced in the constrained 2D environment between adherent membranes (Wu et al., 2011).
Figure 6.
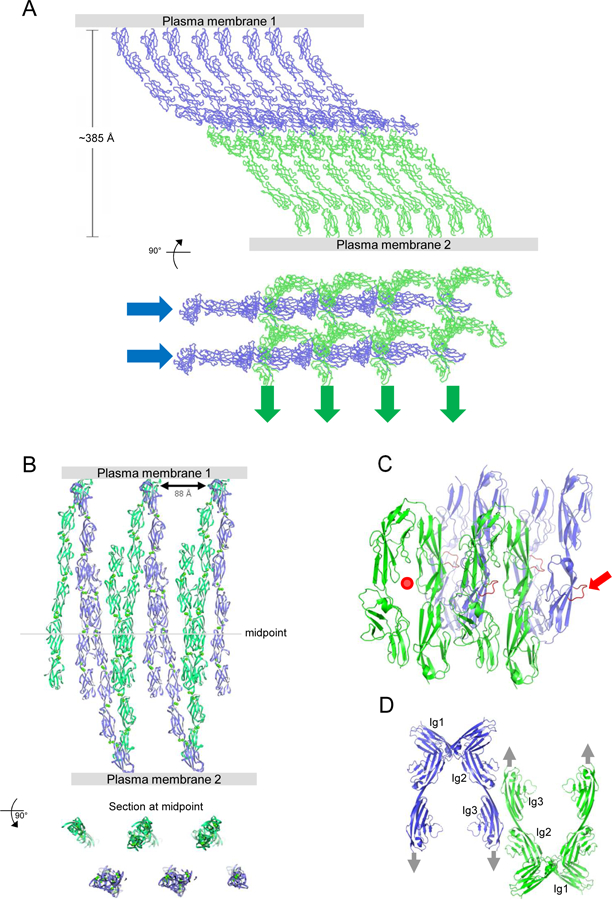
Intercellular Assemblies formed by Select Adhesion Proteins.
(A) Crystal lattice of N-cadherin, shown in two orthogonal views, is composed of cadherin ectodomain polymers formed by cis interactions (as in Fig. 2E) connected across the interacting membranes by trans interactions. Cis interacting linear arrays are indicated by blue and green arrows in the lower panel. A similar structure is seen between adherent membranes by EM (Harrison et al., 2011).
(B) Crystal lattice of Pcdh γB4, shown in two orthogonal views, is composed of discrete Pcdh ectodomain dimers formed by cis interactions connected by trans interactions to form a linear chain. Similar structures are seen between adherent membranes by EM, where multiple chains pack against one another to form two-dimensional assemblies (Brasch et al., 2019).
(C) Assembly observed in axonin-1 crystals. Each four-domain Ig paddle includes a protruding loop (red), which inserts into the paddle interface of a mate from the opposite membrane. In the figure one paddle interface is empty (red dot), and one protruding loop is available for continued growth of the assembly (red arrow).
(D) Assembly observed in NCAM domain 1–3 crystals. Cis dimers are formed between Ig1 domains, with trans dimers formed by Ig domains 2 and 3. In combination, these interfaces define a linear zipper (Soroka et al., 2003). Gray arrows indicate the direction to the membrane in which each molecule is embedded.
Type II classical cadherins -
There are twelve members of the type II cadherin subfamily (Cadherin 6 – 12, 18, 19, 20, 22, 24) and these are widely expressed – particularly in the CNS –often with multiple type II cadherins co-expressed in the same cells demarcating distinct tissue subregions (Price et al., 2002). Type II cadherins have been shown to function in neuronal targeting (Duan et al., 2014), as well as the sorting of motor neuron cell bodies into distinct compartments (Price et al., 2002) – “motor pools” – a process which is largely consistent with expectations based on their heterophilic and homophilic binding affinities (Brasch et al., 2018).
Type II ectodomain structures are nearly identical to those of type I cadherins, but the N-terminal α-strand that mediates their strand-swapped interfaces is anchored by two tryptophan residues (Trp 2 and Trp 4), rather than by the single Trp 2 residue of type I cadherins (Brasch et al., 2018; Patel et al., 2006). This makes type I and type II interfaces incompatible, allowing type I and type II adhesion systems to function independently. Homophilic and heterophilic trans interactions between type II family members tend to be stronger since their binding interfaces are larger and more hydrophobic. Type II cadherins exhibit strong heterophilic interactions among subfamily members as demonstrated in cell aggregation studies (Shimoyama et al., 2000), as well as surface plasmon resonance (SPR) experiments with purified ectodomains (Brasch et al., 2018). Based on their heterophilic binding specificities, they can be divided into three distinct “specificity groups” (cadherin 8, 11, and 24; cadherins 6, 9, and10; and cadherins 7, 12, 18, 20, and 22), which correspond to the branches on which they are situated in the type II cadherin phylogenetic tree (Brasch et al., 2018). The correspondence between SPR measurements and the specificities observed in cell aggregation experiments provide yet another example of the close connection between molecular affinities and cell-cell recognition.
Structure-based analysis of sequence patterns within specificity groups enabled the identification of interfacial residues that facilitate complex formation between group members, but which interfere with the binding of members of different groups. Such “negative constraints” are essential for the evolutionary design of specificity and will be discussed further below. Site-directed mutations at these positions have been used to convert the binding specificity of members of one group to that of another (Brasch et al., 2018). Figure 4 shows how type II cadherins accumulate in cell-cell contact regions based on their binding affinities. Wild-type proteins localize so as to interact homophilically and heterophilically with other group members, but do not accumulate in interfaces with cells expressing members of other groups. This behavior can be changed in predictable ways using mutant proteins with altered specificities. Figure 4 also illustrates how cells can form both homotypic and heterotypic “junctions” (used in this context only to indicate the accumulation or proteins in cell-cell contact regions). It is clear that far more complex behavior will be observed when cells express multiple proteins with differing affinities.
Figure 4.
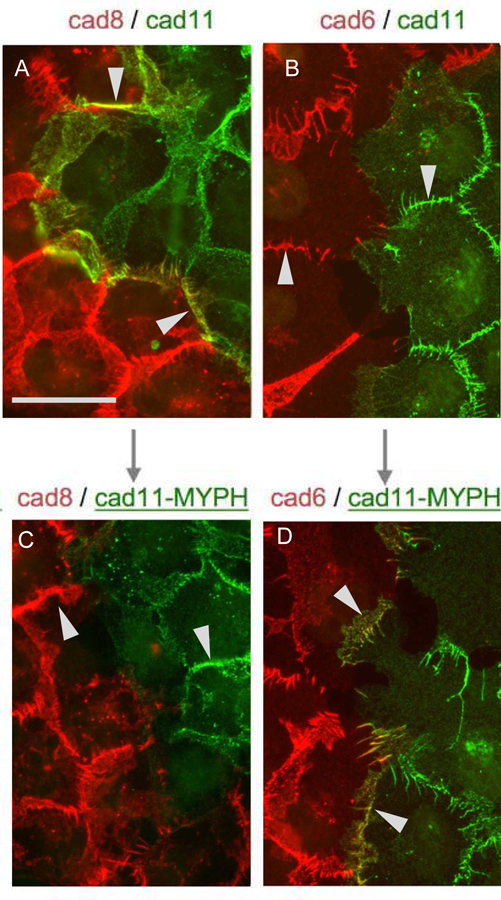
Type II Cadherin Localization at Homotypic and Heterotypic Contact Sites.
Cells transfected with type II cadherins tagged with either cherry (red) or dendra (green) were visualized by fluorescence microscopy (Brasch et al, 2018). Cadherins-8 and −11 are in the same specificity group, with strong heterophilic affinity for one another; cadherin-6 is in a different specificity group, and does not interact with cadherins −8 or −11. Each of the type II cadherins shows strong homophilic interactions.
(A) Cells expressing wild-type cadherin-8 (red) and wild-type cadherin-11 (green) concentrate at both homotypic and heterotypic junctions consistent with both strong homophilic interactions and the strong heterophilic interactions between cadherins-8 and −11. Arrows indicate heterotypic junctions. Scale bar 50 µm.
(B) Cells expressing wild-type cadherin-6 (red) and wild-type cadherin-11 (green) concentrate only at homotypic junctions (indicated by arrows) due to the lack of heterophilic interactions between cadherins-6 and −11.
(C) Cells expressing wild-type cadherin-8 (red) and a mutant cadherin-11 (green) with trans specificity converted to that of the cadherin-6 specificity group with mutations to MYPH at positions 3, 20, 89, and 97, so that it no longer interacts heterophilically with cadherin-8. Only homophilic junctions are observed.
(D) Cells expressing wild-type cadherin-6 (red) and the same mutant cadherin-11 (green) now form heterotypic junctions.
Nectins –
The nectins are a small family of immunoglobulin-like cell surface proteins which are conserved in vertebrates and function in cell-cell adhesion. Their ectodomains are composed of three immunoglobulin-like domains, with an N-terminal variable-like domain (Ig1) followed by two constant-like domains, a single transmembrane region and a cytoplasmic domain that binds to the actin cytoskeleton through the cytoplasmic adaptor protein afadin (Ogita et al., 2010). Cell adhesion by nectins is mediated by trans dimers formed exclusively through the Ig1 domain, with the α-sheets formed by the GFCC’C” strands of each molecule packing against one another in a “handshake” manner (Harrison et al., 2012). This is a mode of interaction common to several families of immunoglobulin-like adhesion proteins (Figure 3A–D). A highly related family of proteins, referred to as nectin-like proteins 1–5 (Necl-1–Necl-5), shares a similar domain organization, bind in trans through a similar interface, but do not bind to cytoplasmic afadin (Liu et al., 2019). Necl-1 through Necl-4 (but not the more divergent Necl-5) are referred to as synaptic cell adhesion molecules or SynCAMs (Frei and Stoeckli, 2017). Nectins bind in trans both homophilically and heterophilically, but their strongest interactions are heterophilic (Harrison et al., 2012). Nectin-1 and nectin-3 exhibit the strongest pairwise interaction; these two adhesion proteins are expressed in the auditory epithelium of the cochlea, where sensory hair cells and supporting cells are arranged in a checkerboard pattern (Togashi et al., 2011). Sensory hair cells express nectin-1, while supporting cells express nectin-3. Mouse knockout studies show that disruption of nectin-1/nectin-3 heterophilic adhesion leads to disruption of the checkerboard pattern in agreement with predictions of the DAH (Figure 1B).
“Non-clustered” δ–protocadherins –
The non-clustered δ–protocadherins – called “non-clustered” because their genes are widely distributed as opposed to the clustered protocadherins, which are encoded in a large gene cluster (see below) – comprise nine typical members in human and mouse (Hulpiau and van Roy, 2009). These are divided into two subfamilies, δ1 and δ2, with four and five members, respectively. δ1-family protocadherins contain seven EC repeats, while δ2-family protocadherins contain six.
δ–protocadherins are expressed in the nervous system in spatially and temporally regulated overlapping expression patterns (Kim et al., 2007), and like type II cadherins are often expressed with multiple subtypes in each cell. Cell aggregation studies with multiply transfected cells suggest that δ–protocadherin cell adhesive specificity could be derived from combinatorial expression (Bisogni et al., 2018). Each δ–protocadherin so far studied can mediate homophilic adhesion, and can also mediate heterophilic interactions within each subfamily (Harrison et al., 2020)
The best understood of the δ–protocadherins is the δ–2-family member Pcdh19. The crystal structure of a Pcdh19 trans dimer reveals binding through anti-parallel association between EC1- EC4 regions of partner molecules (Cooper et al., 2016), similar to the trans binding of clustered protocadherins (see below). Mutation of Pcdh19 underlies the X-linked Pcdh19 “girls clustering epilepsy” (Pcdh19-GCE), and the defects found in this disease yield important clues as to δ- protocadherin function. In a transgenic mouse model, due to the X-linked inheritance of Pcdh19, cells from female mice express Pcdh19 from either one of the two X-chromosome alleles such that individual neurons of heterozygous females express exclusively either wild-type or inactive mutant forms (Pederick et al., 2016). This mosaic expression leads to abnormal sorting in which neural progenitor cells expressing wild-type Pcdh19 separate from those expressing the inactive mutant to form inappropriate homotypic aggregates (Pederick et al., 2018). This developmental defect showed that Pcdh19 homophilic adhesion can substantially impact cell localization in the brain, and provides another example of in vivo cell behavior consistent with the DAH.
III. Cell patterning through pairwise interactions of adherent cells - Synaptic Partner Choice in the Nervous System
Cell patterning of populations of coalescent cells based on the strengths of their cell-cell interactions, as described by the DAH, is based on stereotyped interactions among the sub-populations and the drive toward adhesive-energetic equilibrium. However, some of the most specific cell-cell interactions occur between only two cells, the most highly studied examples being synaptic connections between neurons. In synapse formation, a single axon chooses an appropriate target from an array of choices [see the extensive discussion of this topic in the review by Sanes and Zipursky (2020)]. To achieve such specificity, the incoming axon presents on its surface adhesion proteins that bind cognate partners presented on the surface of the presumptive synaptic partner cell. Recognition between these molecules then leads to an adhesive interaction that may activate localized processes to promote synapse formation. In only a few cases have the relevant adhesion proteins been characterized both in vivo in terms of targeting, and in vitro in terms of structure and biophysics. We describe two such examples here: vertebrate sidekick proteins and invertebrate DIPs and Dprs.
Sidekicks –
In a series of studies, Sanes and coworkers showed that four immunoglobulin superfamily adhesion proteins — Dscam, DscamL, and sidekicks Sdk1 and Sdk2, all homophilic adhesion proteins — strongly bias layer-specific targeting in the inner plexiform layer (IPL) neuropil in chick retina [Yamagata and Sanes, 2008; and see accompanying review by Sanes and Zipursky]. Overall, their experiments demonstrate that sidekicks are both necessary and sufficient to determine the laminar choices of retinal neurons.
Sdk1 and Sdk2 each include an extracellular region with 6 immunoglobulin domains followed by 13 fibronectin type III (FNIII) domains, a single transmembrane helix, and a relatively short intracellular domain terminating in a PDZ binding motif (Yamagata and Sanes, 2008). Crystal structures of cell-adhesive homodimers of Sdk1 and Sdk2 revealed the four N-terminal immunoglobulin domains form a horseshoe-like conformation, similar to many other immunoglobulin-like adhesion proteins (Goodman et al., 2016c). While this horseshoe conformation is in common with other adhesion proteins, their antiparallel back-to- back mode of interaction is unique and generates large Ig1:Ig2 and Ig3:Ig4 interfaces (Figure 3E). Site-directed mutagenesis showed this crystallographically identified interface to be required for cell adhesion. Remarkably, due to a high degree of sequence conservation, the Sdk1:Sdk1 and Sdk2:Sdk2 interfaces are nearly identical (Goodman et al., 2016c). SPR characterization showed that, while homophilic binding is indeed favored, there is significant off-target heterophilic binding between Sdk1 and Sdk2. Since these molecules have strong homophilic interactions, their heterophilic KDs cannot be easily quantified (Katsamba et al., 2009). However, heterophilic SPR responses appear to be about one-third of homophilic responses.
Sdk1-expressing neurons that synapse in the S4 sublayer of the retina must first migrate through the S2 lamina where Sdk2 is expressed (Yamagata and Sanes, 2008). Despite the significant heterophilic interaction observed between Sdk1 and Sdk2, Sdk1-expressing neurons do not arborize or form synapses in the S2 layer where Sdk2 is concentrated. It is possible that the affinity of the heterophilic Sdk1:Sdk2 interaction is too weak to control these neuronal events, whereas the stronger homophilic Sdk1:Sdk1 or Sdk2:Sdk2 interactions are sufficient. However, this possibility is yet to be tested experimentally in vivo. Experiments with designed Sdks with altered affinities could address this issue.
DIPs and Dprs –
DIPs and Dprs are closely related families of immunoglobulin-like adhesion proteins with 11 and 21 members, respectively in Drosophila melanogaster (Carrillo et al., 2015; Ozkan et al., 2013; Tan et al., 2015). RNA sequencing of Drosophila visual system neurons revealed DIPs and Dprs to have remarkable cell type-specific expression (Tan et al., 2015). One or a few DIPs are expressed per neuron, whereas many neurons express multiple Dprs (Cosmanescu et al., 2018), and members of each family show layer-specific expression patterns. A qualitative “interactome”, mapping DIP/Dpr interactions revealed that each DIP binds to a subset of Dprs and vice versa (Ozkan et al., 2013). This work was later expanded through a comprehensive set of SPR experiments, which quantified DIP/Dpr binding affinities, providing KDs for each interaction, and identifying a subset of DIPs that can also engage in homophilic interactions (Cosmanescu et al., 2018). Based on measured binding preferences, phylogenetic profiles and homology models of complexes derived from available crystal structures, DIPs and Dprs were organized into seven distinct specificity groups such that, DIPs and Dprs in one group bind strongly to other proteins within the group, but bind weakly or not at all to DIPs in other groups (Figure 5) (Sergeeva et al., 2020).
Figure 5.
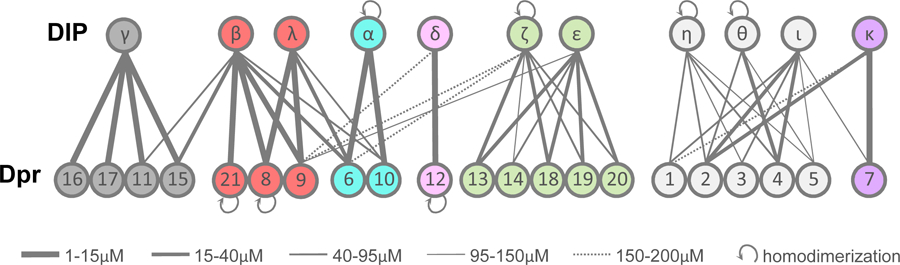
DIP/Dpr Quantitative Interactome.
The interactome is divided into seven specificity groups defined based on heterophilic interactions.
Each grey line connects interacting DIP/Dpr pairs with KDs as indicated by the line width. Self-pointing arrows for Dprs 21, 8 and 12 and DIPs α, ζ, η and θ indicate homodimerization.
Zipursky and co-workers studied DIPα and its binding partners Dpr6 and Dpr10 in flies, and demonstrated that DIPα-Dpr6/Dpr10 interactions promote synapse formation between Dpr6/10- and DIPα-expressing neurons (Xu et al., 2018) see Sanes/Zipursky review). DIPα, Dpr6, and Dpr10 are each concentrated in the M3 layer of the developing medulla neuropil. The DIPα-expressing Dm4 and Dm12 neurons extend axons and arborize in the developing M3 layer, and form synapses with Dpr6/Dpr10-expressing L3 neurons. These observations are consistent with the idea that Dprs expressed in one neuron may promote synapse formation with neurons expressing the cognate DIP. Indeed, both loss-of-function and gain-of-function studies indicate a central role for specific DIP/Dpr interactions in the generation of specific neuronal connections. For example, DIPα-Dpr10 interactions have also been shown to be critical for terminal branching patterns after motor neuron axons reach the vicinity of their muscle targets in Drosophila (Ashley et al., 2018; Venkatasubramanian et al., 2019). Similarly, expression of the cognate DIPγ-Dpr11 pair is critical in patterning interactions between yellow R7 photoreceptors, which express Dpr11, and Dm8 amacrine neurons, which express DIPγ (Menon et al., 2019). Overall, this correlation between the binding specificity of neuronal surface proteins and synaptic specificity is reminiscent of findings by Garcia and colleagues, which showed that, in C. elegans, specification of proper synapse formation is correlated with heterophilic SYG1/SYG2 affinity (Ozkan et al., 2014).
Whether only the strongest interactions, such as DIPα-Dpr6/Dpr10 and DIPγ-Dpr11 have functional importance, or whether there are also roles for the weaker interactions depicted in Figure 5 remains unclear. In addition, homophilic DIP/DIP and Dpr/Dpr interactions have been reported (Cheng et al., 2019a; Cheng et al., 2019b; Cosmanescu et al., 2018), and a mutant deficient in homophilic DIPα/DIPα interactions has been shown to result in an increase in the number of Dm4 synapses (Xu et al., 2018). Functional studies of the roles of DIP/Dpr interactions are still in their earliest stages, with many unanswered questions still to be explored. Are their observed neuronal and synaptic functions based mainly on adhesive actions, or do still-unknown signaling pathways activated by DIP/Dpr trans interactions play a decisive role? If adhesion is indeed the main factor, how do intermolecular affinities, the number of molecules interacting between neurons, or adhesive forces come into play? New types of experiments will need to be undertaken to begin to answer such questions.
IV. Neuron-neuron repulsion in the nervous system and neuronal “barcoding”
The phenomenon of cell-cell repulsion has been described in many systems and it is an integral component of axon guidance mechanisms (Tessier-Lavigne and Goodman, 1996). Axon guidance receptors such as the ephrin, semaphorin, robo, and netrin families can mediate both attractive and repulsive signals and it has become clear that they function in a modular fashion [see e.g., (Bashaw and Goodman, 1999)]. The extracellular domains of these transmembrane receptors are responsible for binding specificity, while cytoplasmic regions mediate signaling processes downstream from the initial recognition event,, which are thought to lead to cytoskeletal changes that result either in adhesion or repulsion. This “repulsion” has been proposed to arise either from the down-regulation of adhesion (Garrett et al., 2018), or from motility away from the site of contact mediated by the cytoskeleton (Sundararajan et al., 2019).
One special case is self-repulsion, where neurites from the same neuron avoid one another, while neurites from different neurons do not. This assures that sister neurites arborize extensively, while those from different neurons can interdigitate to occupy the same field and, when appropriate, form synapses. In an early study of the leech nervous system, Kramer and Stent noted that the phenomenon of self-repulsion required a mechanism whereby neurites can distinguish self from non-self; this would require that neurites possess a cell surface identity specific to each neuron (Kramer and Stent, 1985). Thus, “barcoding” mechanisms must exist to ensure that developing neurites that encounter one another express a different repertoire of surface proteins, and the diversity of such repertoires must be sufficient such that no two neurons encountering one another present the same repertoire by chance (Sanes and Zipursky, 2020; Zipursky and Grueber, 2013). In Drosophila, this neuronal self-identity is defined by the expression of single-cell-specific subsets of Dscam1 isoforms generated by stochastic alternative RNA splicing (Zipursky and Grueber, 2013). In vertebrates, by contrast, neuronal self-identity is provided by the expression of distinct subsets of clustered protocadherins (cPcdhs) [(Schreiner and Weiner, 2010; Thu et al., 2014) and see review by Sanes and Zipursky], whose genes are found in three tandemly arranged clusters –Pcdhα, Pcdhβ, and Pcdhγ (Wu and Maniatis, 1999 ) – and whose expression in each cell is determined by stochastic promoter choice (Canzio et al., 2019).
The molecular logic underlying neuronal barcoding in both invertebrates and vertebrates has become increasingly clear. Both Dscams (Wojtowicz et al., 2007) and cPcdhs (Schreiner and Weiner, 2010; Thu et al., 2014) appear to have strict homophilic recognition specificity, with heterophilic interactions between different family members weak or absent, even when closely related in sequence (see also below). Since all neurites from a single neuron present the same set of cPcdhs or Dscams on their surfaces, when sister neurites encounter one another their identical protein repertoires will initially engage in adhesive homophilic trans interactions. This engagement is thought to activate still poorly understood signaling events, dependent on cytoplasmic regions, to generate repulsion.
The unique challenge faced by these protein families is non-self recognition; that is, when two neurites from different cells encounter one another they should not incorrectly recognize one another as self. This would be simply accomplished if the isoform composition of the two cells was completely different because in this case, since these proteins are strictly homophilic, no binding would occur. But the probability of overlap in the identity of expressed isoforms in different neurons is non-negligible, even for the case of Dscams for which 19,008 distinct extracellular isoforms are produced. For example, Hattori et al. (2009) showed using Monte-Carlo simulations that with 30 isoforms randomly selected per neuron from a pool of 20,000, the probability that two neurons express at least one common isoform is about 4% (Hattori et al., 2008). They further showed that if one common isoform was sufficient to trigger repulsion, then only three neurons could reliably acquire unique identities. This number is clearly too small, then implying that neurons must be able to tolerate some fraction of common isoforms without triggering repulsion. Hattori et al. demonstrated that about a 20% “tolerance” for common isoforms would be sufficient, given a pool of thousands of isoforms, to provide tens to thousands of neurons with identities that are functionally unique (Hattori et al., 2008).
Since the number of cPcdh isoforms expressed per vertebrate neuron is about 50 – 60, the probability that two neurons will express a large fraction of common isoforms is much higher than for Dscam1 isoforms. Thus, cPcdhs must exploit a mechanism other than massive isoform diversity to mediate the barcoding of vertebrate neurons. As will be discussed, the mechanism involves reducing the tolerance to overlapping isoforms to the extent that even when all isoforms but one between two interacting neurons are identical. That is, even a single mismatch is sufficient to ensure that two neurites will not recognize one another as self (Rubinstein et al., 2015).
Generation of unique neuronal identities in invertebrates: Dscams –
Ectodomains of Dscam1 proteins consist of 8 Ig domains that fold into an S-shaped structure that enables the formation of a symmetric dimer such that Ig2, Ig3 and Ig7 in one protomer each interact with another copy of the same domain on the other protomer (Sawaya et al., 2008) (Figure 3E). Each Ig2:Ig2, Ig3:Ig3 and Ig7:Ig7 pair forms an antiparallel interface. A truncated Dscam1 protein containing only the N-terminal horseshoe structure that includes the Ig2:Ig2 and Ig3:Ig3 interfaces behaves as a monomer (Meijers et al., 2007), suggesting the requirement that all three interfaces must form to support binding. Consistent with this idea, high-throughput binding experiments with Dscam1 protein isoforms showed that matches in all three specificity domains were required for binding in the vast majority of cases (Wojtowicz et al., 2007). That is, all three domains must be perfectly matched for a dimer to form. This feature is essential if heterophilic trans dimerization is to be avoided. Specifically, if for example, Ig2 and Ig3 were identical in two protomers and could dimerize on their own, then binding could occur even if the two Ig7 domains were different and hence would not contribute to functional diversity. Thus, it is essential that individual Dscam1 domains bind weakly, but that together they form a stable complex.
While it has been firmly established that highly specific trans dimerization of multiple Dscam1 isoforms provides the molecular basis of neuronal self-identity in insects, it is not yet clear how trans binding activates intracellular signaling processes that drive neurite-neurite repulsion. It has been suggested that Dscam1 dimers have a different conformation in the Ig5-Ig8 regions than the S-shaped monomers and that this conformational change is transmitted to the cytoplasm (Sawaya et al., 2008). Another possibility involves the formation of clusters of Dscam1 dimers, perhaps driven by cis interactions. A third possibility invokes cis interactions with the ectodomains of other transmembrane receptors, but at present there is little evidence pointing to any of these mechanisms.
Generation of unique neuronal identities in vertebrates: Clustered protocadherins –
The ectodomains of clustered Pcdhs contain six EC domains and form trans dimers through a four-domain interface formed by EC1-EC4 regions, which bind in a head-to-tail orientation such that EC1 interacts with EC4 and EC2 interacts with EC3 (Goodman et al., 2016a; Goodman et al., 2016b; Nicoludis et al., 2015; Rubinstein et al., 2015) (Figure 2A, left panel). Thus, despite their evolutionary relationship to classical cadherins (Figure 2B), they interact in a very different way: the classical cadherins bend into a banana-shaped structure that enables their EC1 domains to form a parallel interface (Shapiro et al., 1995; Harrison et al., 2011) while the cPcdhs are much straighter and can only interact in trans in an antiparallel orientation (Figure 2A, left panel). As is the case for Dscams, all four EC domains are required for trans dimerization – smaller ectodomain fragments behave as monomers (Rubinstein et al., 2015; Goodman et al., 2016b). The rationale is identical to that discussed above for Dscams: if for example the EC2:EC3 dimer was stable on its own then the EC1:EC4 interface could not be effectively exploited to mediate homophilic specificity, which is an essential feature of both Dscams and cPcdhs. cPcdhs also form cis dimers mediated by EC5 and EC6 (Figure 2A, right panel) (Goodman et al., 2017). As opposed to trans binding which is strictly homophilic, cis interactions are thought to be largely promiscuous (Goodman et al., 2017; Schreiner and Weiner, 2010; Thu et al., 2014), amplifying cPcdh diversity through the formation of many distinct cis dimers formed from different monomeric subunits on neuronal surfaces.
The evidence for homophilic cPcdh trans dimerization is largely based on cell aggregation assays which reveal that homotypic cell aggregates are formed for essentially every cPcdh isoform, but heterotypic aggregates are never observed (Schreiner and Weiner, 2010; Thu et al., 2014). Cell assays have also revealed a remarkable feature of cPcdhs: for cells expressing multiple isoforms, all isoforms must be identical (“matched”) for two cells to adhere. Even when four out of five expressed isoforms are identical and one is different, no heterotypic aggregation is observed (Thu et al., 2014). It appears then that one mismatched isoform is able to “interfere” with the trans binding of four matched isoforms – a surprising phenomenon and, to our knowledge, one that has not been described for other systems. This interference phenomenon is at the core of the molecular mechanism of cPcdh-mediated barcoding in vertebrate neurons.
As shown in Figure 6B, cPcdhs form linear zipper-like structures, which are observed both in crystals and between the membranes of adherent liposomes (Brasch et al., 2019). Such a zipper depends on the formation of a continuous array of protomers assembled through alternating cis and trans interactions. This structure suggests a chain termination model whereby even a single trans mismatch would terminate the growth of the linear array (Brasch et al., 2019; Rubinstein et al., 2015). When identical isoforms are present, chain length is limited by the copy numbers of cPcdh isoforms expressed per cell, whereas in the presence of even a single mismatch, only truncated assemblies will be formed. Monte Carlo simulations show that this chain termination model yields an essentially binary signal with step-function-like behavior (Rubinstein et al., 2015). Underlying this mechanism is the notion that assembly size plays a crucial role in signaling such that large assemblies would transduce an intracellular signal initiating repulsion while the signal from small mismatched assemblies would remain below a critical threshold. This mechanism also depends on de-adhesion to enable separation of the two neurites, and consistent with this idea that cPcdhs are proteolysed by presenilins and matrix metalloproteases, which could play roles in this process (Buchanan et al., 2010).
It can be shown that the probability of two cells expressing 15 identical cPcdh isoforms stochastically chosen from a pool of 58, with a tolerance of zero as implied by the chain termination mechanism, is actually lower than the probability of two cells expressing 15 Dscam isoforms from a pool of 19,000 with a tolerance of 20% (Rubinstein et al., 2015). Thus, the chain termination model effectively accounts for the ability of a relatively small number of cPcdhs to provide a unique identity to a vast number of vertebrate neurons.
V. How specificity and affinity are encoded on protein surfaces
Over the course of evolution, gene duplications followed by sequence divergence have generated numerous protein families whose homologous members have distinct binding specificities. Families of cell-cell adhesion proteins offer many examples where seemingly small changes in sequence generate new protein-protein interaction specificities. As new family members are generated they must lose specificity for an existing partner as they gain specificity for another. In general, a trade-off between affinity and specificity is to be expected in protein families with only limited sequence divergence. For example, if two family members, say ‘a’ and ‘b’, have a strong binding affinity, then it is a challenge to generate a new family member, ‘c’, that is similar in sequence to ‘b’, but interacts very weakly with ‘a’. If on the other hand the affinity between ‘a’ and ‘b’ is low, then a small number of mutations in ‘b’ are more likely to ablate binding. Thus, the higher the specificity requirements, the greater the constraints on absolute affinities.
Simple principles can be inferred from interactions of adhesion proteins with single-domain interfaces. In the case of type II cadherins, family members sort into three specificity groups; the molecular basis of this specificity is determined by conserved residues that provide stabilizing interactions within a specificity group, but negative constraints on interactions between groups (Figure 7A). Nectin specificity is determined by a number of positions in the interface but, in particular, exploit a simple electrostatic “trick” to favor heterophilic over homophilic interactions (Harrison et al., 2012). Critically each nectin positions a single charged residue at the interface center, near the 2-fold axis (Figure 7B). Relative trans-binding specificity is largely controlled by interactions of these charged residues. If two different nectins have oppositely charged residues located at this position, they will interact favorably in a heterodimer. In contrast, nectins with like charges at this position will incur electrostatic repulsion and bind poorly. Nectin homodimers necessarily place two like-charged residues close to one another, thus weakening homophilic binding, and explaining the overall preference of heterophilic binding among nectins.
Figure 7.

Interaction Interface Specificity in Select Adhesion Proteins.
(A) Specificity determinants (magenta) of the cadherein-6 and cadherin-11 specificity groups shown as sticks on superposed EC1 domain homodimer structures from each group. Trp2 and Trp4, the residues that anchor the swapped strand, are shown for orientation. The boxed region shows the non-swapped hydrophobic interface region unique to type II cadherins.
(B) Nectin interfaces shown as top views, upper panels, and bottom views, lower panels. Conserved interfacial residues are labeled in the top views. Bottom views highlight a central charged residue (Glu in nectin-1 and Lys in nectin-3), which primarily underlies heterophilic preference. Homodimer structures are shown, although they form more weakly than heterophilic complexes.
(C) Specificity-determining palindromic β-strand interfaces from Dscam1 Ig2 domains, for Ig2 splice forms 1 and 44. Hydrogen-bonding patterns differ in each, providing a molecular basis for homophilic specificity.
DIPs and Dprs exhibit more complex specificity patterns (Carrillo et al., 2015; Cosmanescu et al., 2018; Cheng et al., 2019a; Sergeeva et al., 2020). Based on the interactome shown in Figure 5, there are a total of forty-nine possible DIP/Dpr subfamily pairings, which can be subdivided into seven ‘affinity groups’. Although interactions are all based on a common core, there are forty-two sets of negative constraints that preclude incorrect pairing (Sergeeva et al., 2020). The physical origins of these constraints include: replacing a charge in an ion pair with one of opposite sign leading to Coulombic repulsion; replacing one member of an ion pair with a neutral group, which has the effect of burying an unsatisfied charge; mutations in the hydrophobic core to larger amino acids that create steric clashes or to smaller ones that create cavities that lead to packing defects and to weaker hydrophobic contributions to binding. Of course, the same “trick” cannot be reused at the same location for 42 different subgroup pairs. This is why 38 of 66 interfacial residues, covering almost the entire 1900 Å2 interface, are utilized to generate negative constraints.
Invertebrate Dscams would appear to present the most remarkable example of negative constraints since most of the 19,008 isoforms exhibit no detectable heterophilic binding. At the structural level, each of the three interfaces must encode sufficient variability to distinguish 12, 48 and 33 isoforms for Ig2, Ig3 and Ig7 respectively, utilizing ~550–1300 Å2 buried surface area per domain (Sawaya et al., 2008). These sub-interfaces each involve antiparallel interactions between one or more β-strands (Figure 7C). Although cPcdh:cPcdh trans interfaces bury approximately 4000–5000 Å2, only half of the two-fold symmetric interface is available for the generation of diversity since the EC1:EC4 and EC2:EC3 interfaces are duplicated (Goodman et al., 2016a; Goodman et al., 2016b). cPcdhs thus succeed in discriminating among 58 isoforms using ~2000– 2500 Å2 of unique buried surface area. Described in terms of the homophilic specificity encoded per Å2 interfacial surface area, cPcdhs are similar to Dscams (58 specificities created using 2000– 2500 Å2 versus 12, 48 and 33 isoforms created using ~1000 Å2 for each). It is the combinatorial nature of the overall Dscam1 interface that makes it possible to encode 19,008 functionally distinct isoforms, but its individual domain:domain interfaces are not designed more efficiently than those of cPcdhs.
VII. Ordered molecular assemblies in cell-cell interfaces
The existence and biological roles of micron-scale liquid-liquid phase separation within cells have become topics of widespread interest (Banani et al., 2017). Liquid-like phases appear in the 3D environment of the cytoplasm where proteins can translate and rotate and where the phase separation can be described as a liquid-liquid transition. In contrast, cell surface receptors diffuse in the 2D environment of the cell membrane. These proteins, if they undergo trans interactions, will accumulate in cell-cell interfaces, a process that can be viewed as a 2D liquid-liquid phase transition. In some cases, crystal-like ordered assemblies are formed in a process which can be roughly described as a 2D liquid-solid phase transition. These terms are used here for purposes of analogy with cytoplasmic phenomena, but analyzing the underlying mechanisms in molecular terms can provide more insight.
Trans interactions alone will result in the enrichment of adhesion proteins in cell-cell contact regions with, at the steady state, the binding energy of complex formation balancing the entropic penalty associated with depletion of receptors on the entire cell surface. This process can be thought of as a “diffusion trap” which implies that proteins that form trans bonds diffuse into the interface and are trapped there (Wu et al., 2010). However, unless complexes form with an unusually large barrier to dissociation, the phenomenon is thermodynamically, not kinetically, driven. The accumulation of different proteins in contact regions can generate complex structures, as for example, the bulls eye pattern characteristic of the immunological synapse (Kaizuka et al., 2007). The immunological synapse consists of multiple proteins that partition into separate regions depending in part on the size of the receptors (Aricescu et al., 2007), but there is no evidence for the formation of ordered crystal-like protein assemblies in this system.
In contrast, there are numerous examples where adhesion receptors assemble into ordered, linear oligomeric arrays – commonly referred to as zippers (Aricescu and Jones, 2007; Schwartz et al., 2001). In principle zipper assembly can be driven by cis interactions alone, but in most cases the zipper involves both cis and trans interactions (Figure 6). Examples include axonin-1 (Freigang et al., 2000; Kunz et al., 2002), NCAM (Soroka et al., 2003), hemolin – the Drosophila L1 adhesion protein (Su et al., 1998), and the ephrin/ephrin receptor complex (Himanen et al., 2010; Seiradake et al., 2010). As opposed to the linear zippers formed by these proteins, type I cadherins and the closely related desmosomal cadherins form two-dimensional lattices (Al-Amoudi et al., 2011; He et al., 2003). Three type I cadherins (N-, E- and C-cadherin) form nearly identical crystal lattices formed by stereotyped cis and trans interactions; these correspond closely to structures formed between the adherent membranes of cadherin-coated liposomes (Figure 6A) (Boggon et al., 2002; Harrison et al., 2011), which appear similar to adherens junctions observed by electron microscopy (Harrison et al., 2011). Mutations that weaken the trans interaction ablate cell-cell adhesion, while mutations that weaken the cis interaction disrupt the ordered lattice and destabilize adherens junctions (Harrison et al., 2011; Indra et al., 2018).
The ordered structures formed by cell-cell adhesion proteins are in some ways a special case of phase separation phenomena on membrane surfaces (Banjade and Rosen, 2014). In these cases oligomerization is facilitated by a concentration build-up in cell-cell interfaces that is a natural consequence of the diffusion trap mechanism. In addition, cis interactions are enhanced in the 2D environment of a membrane since complex formation involves the loss of fewer translational and rotational degrees of freedom than in a 3D environment where proteins are not as constrained (Wu et al., 2011). Thus, it is perhaps not surprising that phase transitions are favored on membrane surfaces.
What is the role of ordered inter-cellular structures? One possibility is that they simply provide a mechanism to build up a large concentration of receptors which are required for downstream signaling. Alternatively, the regular arrangement of junctional adhesion receptors could produce ordered cytoplasmic structures recognized by intracellular factors to activate downstream signaling – analogous to the dimerization of receptor tyrosine kinases (Schlessinger, 2014). Another possibility is that the existence of a rigid structure offers a platform for the coupling of forces with the cytoskeleton and for the transmission of force between neighboring cells. Indeed in the case of adherens junctions, there is a clear coupling between the stability of cadherin ectodomain clusters and that of tensile actin bundles to which they are linked via β- and α-catenin interactions (Indra et al., 2020). Overall however, it is fair to say that the detailed mechanism of information transfer between cell-cell molecular recognition events and cytoskeletal phenomena is still poorly understood.
VIII. Concluding remarks
Perhaps the most important theme of this review is the role of molecular affinities in cell-cell recognition. In vitro experiments clearly establish a relationship between affinities, expression levels and cell aggregation phenomena and there are numerous examples where the DAH – redefined in terms of molecular properties – accounts, at least in part, for the organization of multicellular structures. Moreover, the barcoding of vertebrate and invertebrate neurons requires the existence of protein families comprised of exclusively homophilic members, thus offer an extreme example of the importance of affinities where they provide a strictly binary yes/no signal as to whether two proteins bind. However, the more general case is represented by classical cadherins, or by DIPs and Dprs where binding is both homophilic and heterophilic, where affinities vary in a limited range and do not generate a binary signal. But do affinities and expression levels constitute the entire story, or even its most important element?
Questions such as these can now begin to be addressed with the use of designed affinity and specificity mutants. There are now numerous examples where protein structure has been used as a basis to swap specificity between isoforms and/or to vary homophilic and heterophilic affinities so as to make them stronger or weaker (see for example Ozkan et al., 2014). Introducing such mutants into model organisms and organoid systems has the potential to significantly expand our understanding of the molecular underpinnings of cell-cell recognition. Ultimately, one can conceive of rewiring neuronal connections or, for example, modifying organ structure based on the expression of adhesion proteins with altered properties.
The focus in this review has been on the role of individual proteins and has for the most part ignored what might occur when multiple proteins with different affinities are expressed in a single cell [See e.g. (Loza et al., 2017)]. Experiments with multiple clustered Pcdhs revealed the interference phenomenon discussed above, but this is clearly a special case. As to more general cases, for example, what happens when one or more type I and type II cadherins, which don’t bind in trans to one another, are expressed in the same cell? Do they cluster in the same interface or do they diffuse to different contact zones, which would then naturally generate more complex shapes than have been seen so far in vitro? Such situations go beyond the confines of the DAH since their essential feature is a mosaic rather than a uniform protein surface, with different adhesion proteins localized to different regions. Simulations of such events could refine our understanding and help develop new insights, especially when combined with cell aggregation experiments of the type described above from the Lim lab (Toda et al., 2018), or in vivo approaches yet to be undertaken.
We are entering an era where cryo-EM techniques, super resolution microcopy and CRISPR technologies offer breathtaking new approaches in multiple fields. When combined with possibilities offered by our current understanding of the structural and energetic basis of molecular interactions in cell-cell recognition, and the ability to alter these interactions through protein design, there is every reason to expect that major breakthroughs will continue to emerge in the coming years.
Acknowledgements
We thank Phini Katsamba and Alina Sergeeva for their careful reading and helpful comments on the manuscript, and all of our lab members for their contributions to the underlying work described here. We are grateful to Josh Sanes and Larry Zipursky for their careful reading of the manuscript and for helpful suggestions. This work was supported by National Science Foundation grant MCB-1914542 and NIH grants MH11481707, AR073846, MH11481707, and DC016960.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abedin M, and King N (2008). The premetazoan ancestry of cadherins. Science 319, 946–948. [DOI] [PubMed] [Google Scholar]
- Al-Amoudi A, Castano-Diez D, Devos DP, Russell RB, Johnson GT, and Frangakis AS (2011). The three-dimensional molecular structure of the desmosomal plaque. Proceedings of the National Academy of Sciences of the United States of America 108, 6480–6485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aricescu AR, and Jones EY (2007). Immunoglobulin superfamily cell adhesion molecules: zippers and signals. Curr Opin Cell Biol 19, 543–550. [DOI] [PubMed] [Google Scholar]
- Aricescu AR, Siebold C, Choudhuri K, Chang VT, Lu W, Davis SJ, van der Merwe PA, and Jones EY (2007). Structure of a tyrosine phosphatase adhesive interaction reveals a spacer-clamp mechanism. Science 317, 1217–1220. [DOI] [PubMed] [Google Scholar]
- Ashley J, Sorrentino V, Nagarkar-Jaiswal S, Tan L, Xu S, Xiao Q, Zinn K, and Carrillo RA (2019). Transsynaptic interactions between IgSF proteins DIP-α and Dpr10 are required for motor neuron targeting specificity in Drosophila. eLife 8, e42690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banani SF, Lee HO, Hyman AA, and Rosen MK (2017). Biomolecular condensates: organizers of cellular biochemistry. Nat Rev Mol Cell Biol 18, 285–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banjade S, and Rosen MK (2014). Phase transitions of multivalent proteins can promote clustering of membrane receptors. eLife 3, e04123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bashaw GJ, and Goodman CS (1999). Chimeric axon guidance receptors: the cytoplasmic domains of slit and netrin receptors specify attraction versus repulsion. Cell 97, 917–926. [DOI] [PubMed] [Google Scholar]
- Basu R, Duan X, Taylor MR, Martin EA, Muralidhar S, Wang Y, Gangi-Wellman L, Das SC, Yamagata M, West PJ, et al. (2017). Heterophilic Type II Cadherins Are Required for High-Magnitude Synaptic Potentiation in the Hippocampus. Neuron 96, 160–176 e168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisogni AJ, Ghazanfar S, Williams EO, Marsh HM, Yang JY, and Lin DM (2018). Tuning of delta-protocadherin adhesion through combinatorial diversity. eLife 7, e41050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boggon TJ, Murray J, Chappuis-Flament S, Wong E, Gumbiner BM, and Shapiro L (2002). C-cadherin ectodomain structure and implications for cell adhesion mechanisms. Science 296, 1308–1313. [DOI] [PubMed] [Google Scholar]
- Brasch J, Goodman KM, Noble AJ, Rapp M, Mannepalli S, Bahna F, Dandey VP, Bepler T, Berger B, Maniatis T, et al. (2019). Visualization of clustered protocadherin neuronal self-recognition complexes. Nature, 569, 280–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brasch J, Katsamba PS, Harrison OJ, Ahlsen G, Troyanovsky RB, Indra I, Kaczynska A, Kaeser B, Troyanovsky S, Honig B, et al. (2018). Homophilic and Heterophilic Interactions of Type II Cadherins Identify Specificity Groups Underlying Cell-Adhesive Behavior. Cell reports 23, 1840–1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchanan SM, Schalm SS, and Maniatis T (2010). Proteolytic processing of protocadherin proteins requires endocytosis. Proceedings of the National Academy of Sciences of the United States of America 107, 17774–17779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canzio D, Nwakeze CL, Horta A, Rajkumar SM, Coffey EL, Duffy EE, Duffie R, Monahan K, O’Keeffe S, Simon MD, et al. (2019). Antisense lncRNA Transcription Mediates DNA Demethylation to Drive Stochastic Protocadherin alpha Promoter Choice. Cell 177, 639–653 e615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrillo RA, Ozkan E, Menon KP, Nagarkar-Jaiswal S, Lee PT, Jeon M, Birnbaum ME, Bellen HJ, Garcia KC, and Zinn K (2015). Control of Synaptic Connectivity by a Network of Drosophila IgSF Cell Surface Proteins. Cell 163, 1770–1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CP (2006). Theoretical And Experimental Insights Into Specificity Determinants And Molecular Mechanisms Of Cadherin Mediated Adhesion In Biochemistry & Molecular Biophysics (New York: Columbia University; ), pp. 120. [Google Scholar]
- Cheng S, Ashley J, Kurleto JD, Lobb-Rabe M, Park YJ, Carrillo RA, and Ozkan E (2019a). Molecular basis of synaptic specificity by immunoglobulin superfamily receptors in Drosophila. eLife 8, e41028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciatto C, Bahna F, Zampieri N, VanSteenhouse HC, Katsamba PS, Ahlsen G, Harrison OJ, Brasch J, Jin X, Posy S, et al. (2010). T-cadherin structures reveal a novel adhesive binding mechanism. Nat Struct Mol Biol 17, 339–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper SR, Jontes JD, and Sotomayor M (2016). Structural determinants of adhesion by Protocadherin-19 and implications for its role in epilepsy. eLife 5, e18529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosmanescu F, Katsamba P, Sergeev A, Ahlsen G, Patel SD, Brewer JJ, Tan L, Xu S, Xiao Q, Nagarkar-Jaiswal S, et al. (2018). Neuron sub-type specific expression, interaction affinities, and specificity determinants of DIP/Dpr cell recognition proteins. Neuron 100, 1385–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong X, Xu F, Gong Y, Gao J, Lin P, Chen T, Peng Y, Qiang B, Yuan J, Peng X, et al. (2006). Crystal structure of the V domain of human Nectin-like molecule-1/Syncam3/Tsll1/Igsf4b, a neural tissue-specific immunoglobulin-like cell-cell adhesion molecule. The Journal of biological chemistry 281, 10610–10617. [DOI] [PubMed] [Google Scholar]
- Duan X, Krishnaswamy A, De la Huerta I, and Sanes JR (2014). Type II cadherins guide assembly of a direction-selective retinal circuit. Cell 158, 793–807. [DOI] [PubMed] [Google Scholar]
- Farquhar MG, and Palade GE (1963). Junctional complexes in various epithelia. The Journal of cell biology 17, 375–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogel AI, Li Y, Giza J, Wang Q, Lam TT, Modis Y, and Biederer T (2010). N-glycosylation at the SynCAM (synaptic cell adhesion molecule) immunoglobulin interface modulates synaptic adhesion. The Journal of biological chemistry 285, 34864–34874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foty RA, and Steinberg MS (2005). The differential adhesion hypothesis: a direct evaluation. Dev Biol 278, 255–263. [DOI] [PubMed] [Google Scholar]
- Frei JA, and Stoeckli ET (2017). SynCAMs - From axon guidance to neurodevelopmental disorders. Mol Cell Neurosci 81, 41–48. [DOI] [PubMed] [Google Scholar]
- Freigang J, Proba K, Leder L, Diederichs K, Sonderegger P, and Welte W (2000). The crystal structure of the ligand binding module of axonin-1/TAG-1 suggests a zipper mechanism for neural cell adhesion. Cell 101, 425–433. [DOI] [PubMed] [Google Scholar]
- Garrett AM, Khalil A, Walton DO, and Burgess RW (2018). DSCAM promotes self-avoidance in the developing mouse retina by masking the functions of cadherin superfamily members. Proceedings of the National Academy of Sciences of the United States of America 115, E10216–E10224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman KM, Rubinstein R, Dan H, Bahna F, Mannepalli S, Ahlsen G, Aye Thu C, Sampogna RV, Maniatis T, Honig B, et al. (2017). Protocadherin cis-dimer architecture and recognition unit diversity. Proceedings of the National Academy of Sciences of the United States of America 114, E9829–E9837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman KM, Rubinstein R, Thu CA, Bahna F, Mannepalli S, Ahlsen G, Rittenhouse C, Maniatis T, Honig B, and Shapiro L (2016a). Structural Basis of Diverse Homophilic Recognition by Clustered alpha- and beta-Protocadherins. Neuron 90, 709–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman KM, Rubinstein R, Thu CA, Mannepalli S, Bahna F, Ahlsen G, Rittenhouse C, Maniatis T, Honig B, and Shapiro L (2016b). gamma-Protocadherin structural diversity and functional implications. eLife 5, e19058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman KM, Yamagata M, Jin X, Mannepalli S, Katsamba PS, Ahlsen G, Sergeeva AP, Honig B, Sanes JR, and Shapiro L (2016c). Molecular basis of sidekick-mediated cell-cell adhesion and specificity. eLife 5, e19058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumbiner BM, and McCrea PD (1993). Catenins as mediators of the cytoplasmic functions of cadherins. J Cell Sci Suppl 17, 155–158. [DOI] [PubMed] [Google Scholar]
- Harrison OJ, Brasch J, Katsamba P, Ahlen SP, Noble AJ, H., D., Sampogna RV, Potter CS, Carragher B, Honig B, et al. (2020). Family-wide structural and biophysical analysis of binding interactions among non-clustered delta-protocadherins. Cell reports, 30,2655–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison OJ, Jin X, Hong S, Bahna F, Ahlsen G, Brasch J, Wu Y, Vendome J, Felsovalyi K, Hampton CM, et al. (2011). The extracellular architecture of adherens junctions revealed by crystal structures of type I cadherins. Structure 19, 244–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison OJ, Vendome J, Brasch J, Jin X, Hong S, Katsamba PS, Ahlsen G, Troyanovsky RB, Troyanovsky SM, Honig B, et al. (2012). Nectin ectodomain structures reveal a canonical adhesive interface. Nat Struct Mol Biol 19, 906–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatta K, and Takeichi M (1986). Expression of N-cadherin adhesion molecules associated with early morphogenetic events in chick development. Nature 320, 447–449. [DOI] [PubMed] [Google Scholar]
- Hattori D, Millard SS, Wojtowicz WM, and Zipursky SL (2008). Dscam-mediated cell recognition regulates neural circuit formation. Annu Rev Cell Dev Biol 24, 597–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He W, Cowin P, and Stokes DL (2003). Untangling desmosomal knots with electron tomography. Science 302, 109–113. [DOI] [PubMed] [Google Scholar]
- He Y, Jensen GJ, and Bjorkman PJ (2009). Cryo-electron tomography of homophilic adhesion mediated by the neural cell adhesion molecule L1. Structure 17, 460–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Himanen JP, Yermekbayeva L, Janes PW, Walker JR, Xu K, Atapattu L, Rajashankar KR, Mensinga A, Lackmann M, Nikolov DB, et al. (2010). Architecture of Eph receptor clusters. Proceedings of the National Academy of Sciences of the United States of America 107, 10860–10865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulpiau P, and van Roy F (2009). Molecular evolution of the cadherin superfamily. Int J Biochem Cell Biol 41, 349–369. [DOI] [PubMed] [Google Scholar]
- Indra I, Choi J, Chen CS, Troyanovsky RB, Shapiro L, Honig B, and Troyanovsky SM (2018). Spatial and temporal organization of cadherin in punctate adherens junctions. Proceedings of the National Academy of Sciences of the United States of America 115, E4406–E4415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Indra I, Troyanovsky R, Shapiro L, Honig B, and Troyanovsky S (2020). Sensing actin dynamics through adherens junction. Cell reports,30, 2820–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin X, Walker MA, Felsövályi K, Vendome J, Bahna F, Mannepalli S, Cosmanescu F, Ahlsen G, Honig B, and Shapiro L (2012). Crystal structures of Drosophila N-cadherin ectodomain regions reveal a widely used class of Ca2+-free interdomain linkers. Proceedings of the National Academy of Sciences 109, E127–E134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaizuka Y, Douglass AD, Varma R, Dustin ML, and Vale RD (2007). Mechanisms for segregating T cell receptor and adhesion molecules during immunological synapse formation in Jurkat T cells. Proceedings of the National Academy of Sciences of the United States of America 104, 20296–20301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsamba P, Carroll K, Ahlsen G, Bahna F, Vendome J, Posy S, Rajebhosale M, Price S, Jessell TM, Ben-Shaul A, et al. (2009). Linking molecular affinity and cellular specificity in cadherin-mediated adhesion. Proceedings of the National Academy of Sciences of the United States of America 106, 11594–11599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SY, Chung HS, Sun W, and Kim H (2007). Spatiotemporal expression pattern of non-clustered protocadherin family members in the developing rat brain. Neuroscience 147, 996–1021. [DOI] [PubMed] [Google Scholar]
- Kramer AP, and Stent GS (1985). Developmental arborization of sensory neurons in the leech Haementeria ghilianii. II. Experimentally induced variations in the branching pattern. J Neurosci 5, 768–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunz B, Lierheimer R, Rader C, Spirig M, Ziegler U, and Sonderegger P (2002). Axonin-1/TAG-1 mediates cell-cell adhesion by a cis-assisted trans-interaction. The Journal of biological chemistry 277, 4551–4557. [DOI] [PubMed] [Google Scholar]
- Liu X, An T, Li D, Fan Z, Xiang P, Li C, Ju W, Li J, Hu G, Qin B, et al. (2019). Structure of the heterophilic interaction between the nectin-like 4 and nectin-like 1 molecules. Proceedings of the National Academy of Sciences of the United States of America 116, 2068–2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loza O, Heemskerk I, Gordon-Bar N, Amir-Zilberstein L, Jung Y, and Sprinzak D (2017). A synthetic planar cell polarity system reveals localized feedback on Fat4-Ds1 complexes. eLife 6, e24820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijers R, Puettmann-Holgado R, Skiniotis G, Liu JH, Walz T, Wang JH, and Schmucker D (2007). Structural basis of Dscam isoform specificity. Nature 449, 487–491. [DOI] [PubMed] [Google Scholar]
- Menon KP, Kulkarni V, Takemura SY, Anaya M, and Zinn K (2019). Interactions between Dpr11 and DIP-gamma control selection of amacrine neurons in Drosophila color vision circuits. eLife 8, e48935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modak D, and Sotomayor M (2019). Identification of an adhesive interface for the non-clustered delta1 protocadherin-1 involved in respiratory diseases. Communications biology 2, 354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narita H, Yamamoto Y, Suzuki M, Miyazaki N, Yoshida A, Kawai K, Iwasaki K, Nakagawa A, Takai Y, and Sakisaka T (2011). Crystal Structure of the cis-Dimer of Nectin-1: implications for the architecture of cell-cell junctions. The Journal of biological chemistry 286, 12659–12669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoludis JM, Lau SY, Scharfe CPI, Marks DS, Weihofen WA, and Gaudet R (2015). Structure and Sequence Analyses of Clustered Protocadherins Reveal Antiparallel Interactions that Mediate Homophilic Specificity. Structure 23, 2087–2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogita H, Rikitake Y, Miyoshi J, and Takai Y (2010). Cell adhesion molecules nectins and associating proteins: Implications for physiology and pathology. Proceedings of the Japan Academy Series B, Physical and biological sciences 86, 621–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozkan E, Carrillo RA, Eastman CL, Weiszmann R, Waghray D, Johnson KG, Zinn K, Celniker SE, and Garcia KC (2013). An extracellular interactome of immunoglobulin and LRR proteins reveals receptor-ligand networks. Cell 154, 228–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pappas DJ, and Rimm DL (2006). Direct interaction of the C-terminal domain of alpha-catenin and F-actin is necessary for stabilized cell-cell adhesion. Cell Commun Adhes 13, 151–170. [DOI] [PubMed] [Google Scholar]
- Patel SD, Ciatto C, Chen CP, Bahna F, Rajebhosale M, Arkus N, Schieren I, Jessell TM, Honig B, Price SR, et al. (2006). Type II cadherin ectodomain structures: implications for classical cadherin specificity. Cell 124, 1255–1268. [DOI] [PubMed] [Google Scholar]
- Pederick DT, Homan CC, Jaehne EJ, Piltz SG, Haines BP, Baune BT, Jolly LA, Hughes JN, Gecz J, and Thomas PQ (2016). Pcdh19 Loss-of-Function Increases Neuronal Migration In Vitro but is Dispensable for Brain Development in Mice. Sci Rep 6, 26765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pederick DT, Richards KL, Piltz SG, Kumar R, Mincheva-Tasheva S, Mandelstam SA, Dale RC, Scheffer IE, Gecz J, Petrou S, et al. (2018). Abnormal Cell Sorting Underlies the Unique X-Linked Inheritance of PCDH19 Epilepsy. Neuron 97, 59–66 e55. [DOI] [PubMed] [Google Scholar]
- Peng H, Ruan Z, Long F, Simpson JH, and Myers EW (2010). V3D enables real-time 3D visualization and quantitative analysis of large-scale biological image data sets. Nat Biotechnol 28, 348–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price SR, De Marco Garcia NV, Ranscht B, and Jessell TM (2002). Regulation of motor neuron pool sorting by differential expression of type II cadherins. Cell 109, 205–216. [DOI] [PubMed] [Google Scholar]
- Rubinstein R, Thu CA, Goodman KM, Wolcott HN, Bahna F, Mannepalli S, Ahlsen G, Chevee M, Halim A, Clausen H, et al. (2015). Molecular logic of neuronal self-recognition through protocadherin domain interactions. Cell 163, 629–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanes JS, and Zipursky SL (2020). Recognition Molecules, Synaptic Specificity and the Assembly of Neural Circuits. Cell In Press.
- Sawaya MR, Wojtowicz WM, Andre I, Qian B, Wu W, Baker D, Eisenberg D, and Zipursky SL (2008). A double S shape provides the structural basis for the extraordinary binding specificity of Dscam isoforms. Cell 134, 1007–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlessinger J (2014). Receptor tyrosine kinases: legacy of the first two decades. Cold Spring Harb Perspect Biol 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiner D, and Weiner JA (2010). Combinatorial homophilic interaction between gamma-protocadherin multimers greatly expands the molecular diversity of cell adhesion. Proceedings of the National Academy of Sciences of the United States of America 107, 14893–14898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwabe T, Borycz JA, Meinertzhagen IA, and Clandinin TR (2014). Differential adhesion determines the organization of synaptic fascicles in the Drosophila visual system. Curr Biol 24, 1304–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz JC, Zhang X, Fedorov AA, Nathenson SG, and Almo SC (2001). Structural basis for co-stimulation by the human CTLA-4/B7–2 complex. Nature 410, 604–608. [DOI] [PubMed] [Google Scholar]
- Seiradake E, Harlos K, Sutton G, Aricescu AR, and Jones EY (2010). An extracellular steric seeding mechanism for Eph-ephrin signaling platform assembly. Nature Structural & Molecular Biology 17, 398–U327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sergeeva A, Katsamba P, Cosmanescu F, Brewer JJ, Ahlsen G, Mannepalli S, Shapiro L, and Honig B (2020). DIP/Dpr interactions and the evolutionary design of specificity in protein families. bioRxiv, 10.1101/2020.01.13.899120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro L, Fannon AM, Kwong PD, Thompson A, Lehmann MS, Grubel G, Legrand JF, Als-Nielsen J, Colman DR, and Hendrickson WA (1995). Structural basis of cell-cell adhesion by cadherins. Nature 374, 327–337. [DOI] [PubMed] [Google Scholar]
- Shimoyama Y, Tsujimoto G, Kitajima M, and Natori M (2000). Identification of three human type-II classic cadherins and frequent heterophilic interactions between different subclasses of type-II classic cadherins. Biochem J 349, 159–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soroka V, Kolkova K, Kastrup JS, Diederichs K, Breed J, Kiselyov VV, Poulsen FM, Larsen IK, Welte W, Berezin V, et al. (2003). Structure and interactions of NCAM Ig1–2-3 suggest a novel zipper mechanism for homophilic adhesion. Structure 11, 1291–1301. [DOI] [PubMed] [Google Scholar]
- Su XD, Gastinel LN, Vaughn DE, Faye I, Poon P, and Bjorkman PJ (1998). Crystal structure of hemolin: a horseshoe shape with implications for homophilic adhesion. Science 281, 991–995. [DOI] [PubMed] [Google Scholar]
- Sundararajan L, Smith CJ, Watson JD, Millis BA, Tyska MJ, and Miller DM 3rd (2019). Actin assembly and non-muscle myosin activity drive dendrite retraction in an UNC-6/Netrin dependent self-avoidance response. PLoS Genet 15, e1008228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeichi M (1988). The cadherins: cell-cell adhesion molecules controlling animal morphogenesis. Development 102, 639–655. [DOI] [PubMed] [Google Scholar]
- Takeichi M (2018). Historical review of the discovery of cadherin, in memory of Tokindo Okada. Dev Growth Differ 60, 3–13. [DOI] [PubMed] [Google Scholar]
- Tan L, Zhang KX, Pecot MY, Nagarkar-Jaiswal S, Lee PT, Takemura SY, McEwen JM, Nern A, Xu S, Tadros W, et al. (2015). Ig Superfamily Ligand and Receptor Pairs Expressed in Synaptic Partners in Drosophila. Cell 163, 1756–1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taveau JC, Dubois M, Le Bihan O, Trepout S, Almagro S, Hewat E, Durmort C, Heyraud S, Gulino-Debrac D, and Lambert O (2008). Structure of artificial and natural VE-cadherin-based adherens junctions. Biochem Soc Trans 36, 189–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tessier-Lavigne M, and Goodman CS (1996). The molecular biology of axon guidance. Science 274, 1123–1133. [DOI] [PubMed] [Google Scholar]
- Thu CA, Chen WV, Rubinstein R, Chevee M, Wolcott HN, Felsovalyi KO, Tapia JC, Shapiro L, Honig B, and Maniatis T (2014). Single-cell identity generated by combinatorial homophilic interactions between alpha, beta, and gamma protocadherins. Cell 158, 1045–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toda S, Blauch LR, Tang SKY, Morsut L, and Lim WA (2018). Programming self-organizing multicellular structures with synthetic cell-cell signaling. Science 361, 156–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Togashi H, Kominami K, Waseda M, Komura H, Miyoshi J, Takeichi M, and Takai Y (2011). Nectins establish a checkerboard-like cellular pattern in the auditory epithelium. Science 333, 1144–1147. [DOI] [PubMed] [Google Scholar]
- Vendome J, Felsovalyi K, Song H, Yang Z, Jin X, Brasch J, Harrison OJ, Ahlsen G, Bahna F, Kaczynska A, et al. (2014). Structural and energetic determinants of adhesive binding specificity in type I cadherins. Proceedings of the National Academy of Sciences of the United States of America 111, E4175–4184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatasubramanian L, Guo Z, Xu S, Tan L, Xiao Q, Nagarkar-Jaiswal S, and Mann RS (2019). Stereotyped terminal axon branching of leg motor neurons mediated by IgSF proteins DIP-alpha and Dpr10. eLife 8, e42692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson HV (1907). On some phenomena of coalescence and regeneration in sponges. J Exp Zool 5, 245–258. [Google Scholar]
- Wojtowicz WM, Wu W, Andre I, Qian B, Baker D, and Zipursky SL (2007). A vast repertoire of Dscam binding specificities arises from modular interactions of variable Ig domains. Cell 130, 1134–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Q, and Maniatis T (1999). A striking organization of a large family of human neural cadherin-like cell adhesion genes. Cell 97, 779–790. [DOI] [PubMed] [Google Scholar]
- Wu Y, Jin X, Harrison O, Shapiro L, Honig BH, and Ben-Shaul A (2010). Cooperativity between trans and cis interactions in cadherin-mediated junction formation. Proceedings of the National Academy of Sciences of the United States of America 107, 17592–17597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Vendome J, Shapiro L, Ben-Shaul A, and Honig B (2011). Transforming binding affinities from three dimensions to two with application to cadherin clustering. Nature 475, 510–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S, Xiao Q, Cosmanescu F, Sergeev A, Yoo J, Lin Y, Katsamba P, Ahlsen G, Kaufman J, Linaval N, et al. (2018). DIP-α, Dpr6/10 interaction, layer-specific circuit, cellular interaction, visual system, circuit assembly. Neuron 100, 1369–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagata M, and Sanes JR (2008). Dscam and Sidekick proteins direct lamina-specific synaptic connections in vertebrate retina. Nature 451, 465–469. [DOI] [PubMed] [Google Scholar]
- Zipursky SL, and Grueber WB (2013). The molecular basis of self-avoidance. Annu Rev Neurosci 36, 547–568. [DOI] [PubMed] [Google Scholar]
- Zipursky SL, and Sanes JR (2010). Chemoaffinity revisited: dscams, protocadherins, and neural circuit assembly. Cell 143, 343–353. [DOI] [PubMed] [Google Scholar]