

Abstract
Introduction:
The myofibroblast is a gastrointestinal stromal cell that is a target of tumor necrosis factor-alpha (TNF-α), a pro-inflammatory cytokine strongly implicated in colitis-associated cancer. Crosstalk between TNF-α and other pro-inflammatory mediators amplify inflammatory signaling but the mechanism is unknown. Angiogenin (ANG) is a 14-kDa angiogenesis protein that is regulated in patients with inflammatory bowel disease. However, the role of ANG on inflammatory mediator crosstalk in the myofibroblast is unknown.
Methods:
The human colonic myofibroblast cell line 18Co, as well as primary mouse and human colonic myofibroblasts, were exposed to TNF-α (10 ng/ml) and bradykinin (BK, 100 nM). ANG was quantified by ELISA. The expression of cyclo-oxygenase-2 (COX-2) and phosphorylation of PKD was assessed by Western Blot.
Results:
Primary mouse and human colonic myofibroblasts exposed to TNF-α/BK led to enhanced PKD phosphorylation and synergistic COX-2 expression. 18Co cells secrete high levels of ANG (24h, 265 ± 5 pg/ml). The monoclonal antibody 26-2F, which neutralizes ANG, inhibited TNF-α/BK-mediated PKD phosphorylation and synergistic COX-2 expression in primary human myofibroblasts. Likewise, in primary mouse myofibroblasts that do not express ANG (ANG-KO), TNF-α/BK failed to enhance PKD phosphorylation and COX-2 expression.
Conclusions:
TNF-α/BK enhance PKD phosphorylation and COX-2 expression in primary mouse and human colonic myofibroblasts. Angiogenin is produced by the myofibroblast, and inhibition of ANG signaling, either by its absence (ANG-KO) or by pharmacologic inhibition, blocks enhanced PKD phosphorylation and synergistic COX-2 expression induced by TNF-α/BK. ANG mediates crosstalk signaling between TNF-α/BK in the regulation of stroma-derived COX-2 and may be a novel therapeutic target for the management of colitis-associated cancer.
Keywords: Myofibroblast, Colitis-associated cancer, Angiogenin, Protein kinase D, COX-2
1. Introduction
Chronic inflammatory bowel disease (IBD) predisposes to colorectal cancer, occurring in up to 18% of patients [1]. Colitis-associated cancer is characterized by rapid progression and high mortality compared to sporadic forms [2,3], and the risk increases with the extent, duration, and severity of colitis. While the association between chronic inflammation and cancer is well established [3,4], the precise mechanism(s) leading to neoplasia, and the contribution of specific cell populations on this process, remain unclear.
The myofibroblast is a gastrointestinal stroma cell that regulates epithelial proliferation [5,6], mucosal repair [5], and fibrosis [7,8] through paracrine signaling, and has been implicated in colitis-associated cancer [9–12]. The myofibroblast is the predominant nonmalignant stromal cell of the tumor microenvironment [13,14] and a major reservoir of stroma-derived cyclo-oxygenase-2 (COX-2) [15,16]. COX-2, encoded by the PTGS2 gene, is an early response enzyme that is inducibly expressed by inflammatory mediators [17,18], leading to the production of prostaglandins that not only participate in the GI response to colitis [5] but also predispose to cancer [12,15,17,19].
The myofibroblast is a target of inflammatory mediators like TNF-α [9,18,20], a potent pro-inflammatory cytokine that plays a key role in both IBD as well as colitis-associated cancer [9–11,21]. In the myofibroblast cell line 18Co, we have previously shown that TNF-α enhances the physiologic responses of the myofibroblast to G protein-coupled receptor (GPCR) agonist signaling, leading to a synergistic upregulation of PTGS2 (COX-2) mRNA, COX-2 protein, PTGES (mPGES-1) mRNA, and PGE2 [18,20]. This involved the enhanced activation of protein kinase D (PKD), a ubiquitous serine-threonine kinase known to participate in biological responses to inflammation. While TNF-α did not independently activate PKD [18], TNF-α augmented GPCR agonist-mediated PKD signaling [18,20]. Furthermore, inhibition of PKD with pharmacologic PKD inhibitors, as well as with PRKD1 (PKD) siRNA, completely blocked the synergistic expression of PTGS2 (COX-2) mRNA and COX-2 protein expression [18,20], illustrating the important role that PKD plays in this process.
The goal of our present study was to further elucidate the signaling interactions between TNF-α and GPCR’s within the myofibroblast, and to confirm that our findings were not specific to a myofibroblast cell line. Here we report for the first time that angiogenin, an angiogenesis protein with growth and survival properties, is a required element of this pro-inflammatory mediator crosstalk and may be an important link connecting the processes of inflammation and carcinogenesis.
2. Materials and methods
Animal experiments were approved by the Institutional Animal Care and Use Committee of Tufts Medical Center. The animal facility is accredited by the AALAC.
2.1. Cell culture
18Co cells (CRL-1459) were purchased from American Type Culture Collection (Rockville, MD). 18Co cells mimic colonic myofibroblasts structurally and functionally [22]. 18Co cells, along with primary mouse and human myofibroblasts, were maintained at 37 °C in DMEM supplemented with 10%FBS in a humidified atmosphere containing 10%CO2–90% air. Cells were plated in 35-mm dishes and grown in DMEM containing 10%FBS for 5–7days until confluent and used from passages 8–14.
2.2. Isolation of primary mouse myofibroblasts
Primary mouse myofibroblasts were isolated from C57BL/6 mice (Jackson Laboratory, Bar Harbor, ME) as previously described [23]. Briefly, the colon of male/female 8–10-week-old C57BL/6 mice was washed with ice cold sterile PBS and incubated in HBSS containing 5 mM EDTA in a shaking air bath, de-epithelializing the tissue. The tissue was incubated in RPMI-5 [RPMI with 5%FCS, 10 mM HEPES, 2 mM L-glutamine, 1 mM sodium pyruvate, 100U/ml Pen-Strep] containing 10U dispase (GIBCO-Invitrogen, Carlsbad, CA) and 2000U of collagenase D (Roche Diagnostics, Indianapolis, IN) in a shaking 37 °C air bath for 60min. The tissue was pelleted and resuspended with ACK lysis buffer (150 mM NH4Cl, 10 mM KHCO3, 0.1 mM Na2EDTA, pH = 7.2–7.4), filter sterilized (0.2-mm filter), then re-pelleted and re-suspended in RPMI-5, and passed through a 70 μM mesh strainer. After a 3h incubation, non-adherent cells were washed away leaving adherent cells consisting of epithelial cells, macrophages and myofibroblasts. After several days, only cells with a myofibroblast-like phenotype remain viable. We have previously confirmed that the isolated cells stain positive for α-SMA and vimentin and are negative for desmin [23]. Primary myofibroblasts were grown in cell culture and used 1–2 weeks after initial isolation.
2.3. Isolation of primary human myofibroblasts
A protocol to obtain human tissue from surgical patients was approved by our institutional review board. Human colon tissue immediately taken from surgically resected colon was washed with ice cold sterile PBS and shaken five times for 15min in HBSS containing 5 mM EDTA, which de-epithelialized the tissue. The tissue was incubated in 20 ml of RPMI-5 [RPMI with 5%FCS, 10 mM HEPES, 2 mM L-glutamine, 1 mM sodium pyruvate, 100U/ml Pen-Strep] containing 10.5 mg of Dispase (GIBCO-Invitrogen, Carlsbad, CA) and 7.2 mg of collagenase D (Roche Diagnostics, Indianapolis, IN) for 2h in a shaking 37 °C incubator. The digested tissue was treated with ACK lysis buffer for 5min, then passed through a 70-μM cell strainer into 100-mm dishes in RPMI-5. After a 3h incubation, the nonadherent cells were washed away, leaving adherent cells consisting mainly of macrophages and myofibroblasts. After several days the macrophages died, leaving cells with a myofibroblast phenotype (α-SMA and vimentin positive). Primary colonic myofibroblast cultures were used for experiments up to passage 4.
2.4. SDS-PAGE and immunoblotting
Cell lysis was performed using Triton buffer (50 mM Tris, pH 7.5, 1 mM EDTA, 1 mM EGTA, 1 mM Na3VO4, 5 mM sodium pyrophosphate, 10 mM sodium glycerophosphate, 1% Triton X-100, 50 mM NaF plus 1% Calbiochem Protease Inhibitor Cocktail) and lysates were assayed for protein using the Bradford protein assay, then diluted with 5x Laemmli loading buffer for SDS-PAGE. Equal amounts of protein were loaded in 4–20% Tris/glycine gels and electrophoresed for 120 min at 130 V. The gel was blotted onto a PVDF membrane by electrophoretic transfer at 25 V overnight. The membrane was washed, blocked with 5% milk, and probed with primary antibodies. Secondary antibodies conjugated to horse-radish peroxidase (Pierce, Rockford, IL) and a chemiluminescent substrate (SuperSignal, Pierce, Rockford, IL) were used to visualize immunoreactive bands.
2.5. ELISA
Angiogenin was quantified from the supernatant of serum-starved, confluent 18Co cells. The collected supernatant was centrifuged at 5,000g for 5min to remove cell debris. Absorbance readings were set between 405 and 420 nm on a spectrophotometer.
3. Materials and reagents
HBSS, EDTA, Dispase and RPMI-1640 were purchased from Thermo Fisher Scientific (Waltham MA). DMEM, FBS, penicillin G potassium, streptomycin, fungizone, and glutamine were purchased from Invitrogen (Carlsbad, CA). TNF-α was purchased from R&D Systems (Minneapolis, MN). Bradykinin and α-SMA antibody were purchased from Sigma-Aldrich (St. Louis, MO). COX-2 antibody was purchased from Cell Signaling Technology (Beverly, MA). C527 was provided by the Hu laboratory. The phospho-PKD polyclonal antibodies pSer916 was purchased from (Millipore, Bill-erica, MA). Antibody to GAPDH was purchased from Santa Cruz (Dallas, TX).
4. Results
TNF-α enhances bradykinin-mediated PKD phosphorylation and leads to synergistic COX-2 expression in primary mouse and human colonic myofibroblasts.
While the pro-inflammatory cytokine TNF-α does not independently activate PKD [18], we have previously demonstrated in the myofibroblast cell line 18Co that TNF-α augments GPCR-mediated PKD signaling [18,20], leading to a synergistic upregulation of COX-2 protein expression. To demonstrate that this finding was not cell line-specific, experiments were verified using primary myofibroblasts isolated from both mouse (Fig. 1) and human (Fig. 2) colon tissue using a well-established technique [24]. In Fig. 1, primary mouse myofibroblasts were exposed to bradykinin (100 nM) and TNF-α (10 ng/ml), either alone or in combination, for 4h. Phosphorylation of PKD at Ser916 and COX-2 protein expression were analyzed by Western blot. Consistent with previously reported data [18,20], TNF-α did not independently lead to phosphorylation of PKD but did result in a modest increase in COX-2 protein expression. Exposure to bradykinin alone led to phosphorylation of PKD that was evident at 1h, with a steady decline over 4h, followed by a modest increase in COX-2 expression at 2h and 4h. Confirming our prior data, simultaneous exposure of primary mouse myofibroblasts to both TNF-α and bradykinin led to enhanced PKD phosphorylation that was statistically significant at 1h and 2h (Fig. 1B), with a corresponding increase in COX-2 protein expression at 1h, 2h, and 4h (Fig. 1C).
Fig. 1.
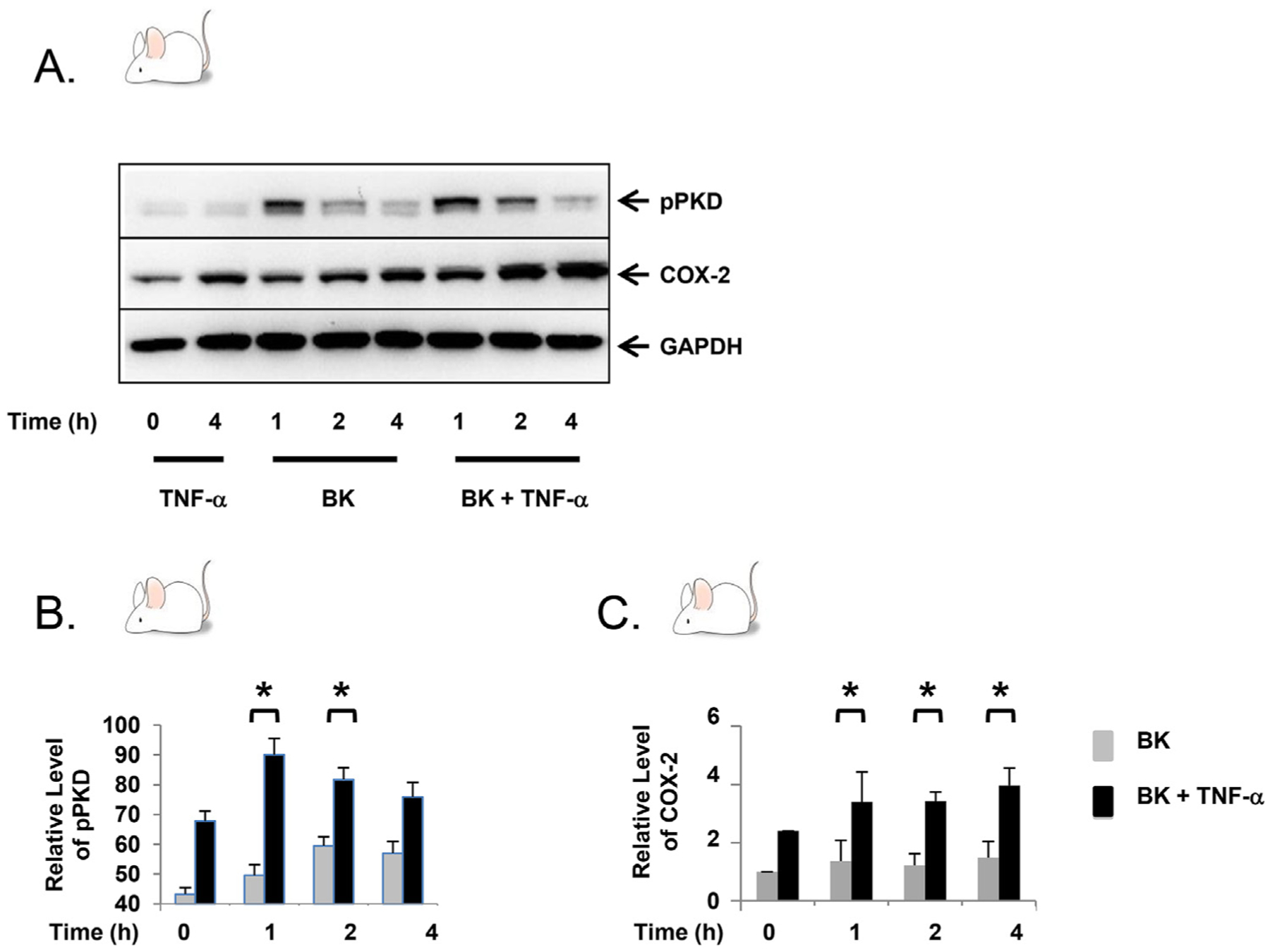
TNF-α enhances bradykinin-mediated PKD phosphorylation and leads to synergistic COX-2 expression in primary mouse myofibroblasts. A. Confluent primary mouse myofibroblasts were equilibrated in serum-free media for 30min, then exposed to TNF-α (10 ng/ml) ± BK (100 nM) for up to 4h. B–C. The results shown are the mean ± S.E. n ≥ 3, expressed as a relative expression level of pPKD and COX-2. * denotes p < 0.05.
Fig. 2.
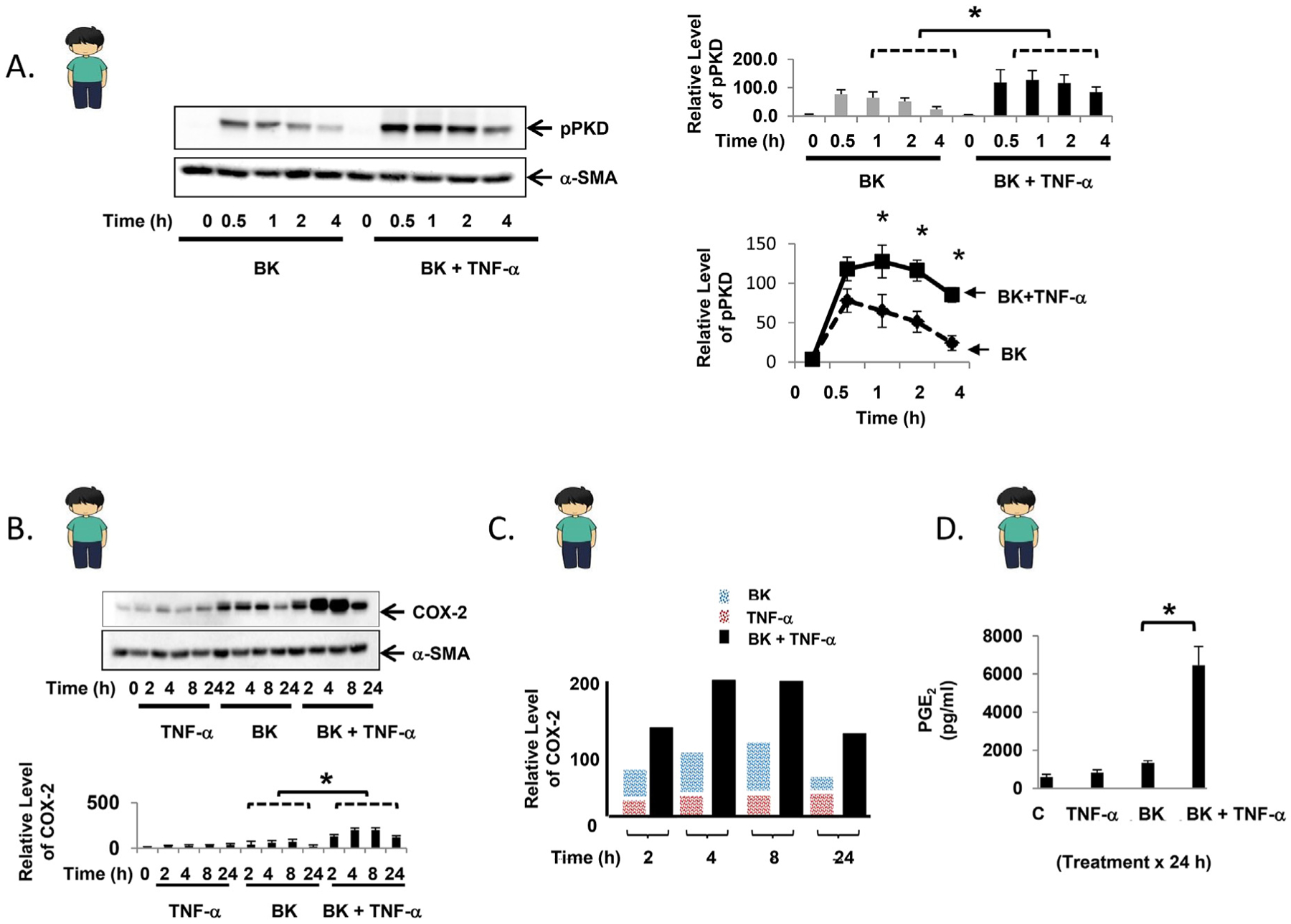
TNF-α enhances bradykinin-mediated PKD phosphorylation and leads to synergistic COX-2 expression in primary human myofibroblasts. A. Confluent primary human myofibroblasts were equilibrated in serum-free media for 30min and exposed to BK (100 nM) ± TNF-α (10 ng/ml) for up to 4h. Results are shown graphically as the mean ± S.E. n ≥ 3, expressed as the relative expression level of pPKD. B. Confluent primary human myofibroblasts were equilibrated in serum-free media for 30min and exposed to TNF-α (10 ng/ml) ± BK (100 nM) for up to 24h. The results are shown graphically as the mean ± S.E. n ≥ 3, expressed as the relative expression level of COX-2. C. Graphical representation of COX-2 protein expression induced by BK alone (blue bar), TNF-α alone (red bar), or the combination (black bar) over 24h. D. Following exposure of confluent primary human myofibroblasts to TNF-α (10 ng/ml) ± BK (100 nM) for 24h, PGE2 concentration was quantified from cell culture supernatant by ELISA. Control: 598.0 ± 131.1 pg/ml, TNF-α: 841.7 ± 135.4 pg/ml, BK: 1340.3 ± 120.8 pg/ml, TNF-α + BK: 6462.2 ± 989.4 pg/ml * denotes p < 0.05. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
Experiments were also performed using primary myofibroblasts that were isolated from surgically resected human colon tissue. TNF-α (10 ng/ml) alone did not activate PKD in primary human myofibroblasts (data not shown), while bradykinin (100 nM) led to phosphorylation of PKD that was evident at 0.5h, with a steady decline over 4h (Fig. 2A). Consistent with the responses seen in the myofibroblast cell line 18Co and in primary mouse myofibroblasts, TNF-α augmented bradykinin-mediated PKD phosphorylation, with statistically significant differences seen at 1h, 2h, and 4h compared to primary myofibroblasts exposed to bradykinin alone (Fig. 2A). The effect of TNF-α and bradykinin, alone and in combination, on COX-2 expression in primary human myofibroblasts was analyzed for up to 24h (Fig. 2B). Exposure to TNF-α alone led to a modest increase in COX-2 protein over 24h. Exposure of primary human myofibroblasts to bradykinin led to a more pronounced increase in COX-2 protein expression, with the highest levels occurring at 2 and 4h followed by a decrease in protein expression at 8h and 24h. However, exposure to both TNF-α and bradykinin led to a synergistic increase in COX-2 protein expression (Fig. 2C) that was evident at 2h and most pronounced at 4h and 8h. The synergistic increase in COX-2 expression was sustained at 24h, with a 2-old increase in protein expression at each time point compared to the additive response (Fig. 2C). At 24h, the corresponding synergistic increase in PGE2 production was even more pronounced (Fig. 2D), quantified by ELISA.
The Myofibroblast is a Source of Angiogenin.
Angiogenin is a 14-kDa protein that is a member of the pancreatic ribonuclease superfamily initially isolated from the media of the human colon cancer cell line HT-29 as the first tumor-derived angiogenesis protein [25]. Human patients with active IBD have elevated serum levels of angiogenin [26,27], suggesting that angiogenin may play a role in the pathophysiology or counter-regulatory response to colitis. In an effort to identify cell populations that generate angiogenin, we found that the myofibroblast cell line 18Co is a robust source (Fig. 3A). Confluent 18Co cells were placed in serum-free media and angiogenin concentration was analyzed by ELISA. 18Co cells secrete high levels of angiogenin (265 ± 5 pg/ml) in serum-free media after 24h, a finding consistent with recently reported data that angiogenin is secreted by human colon cancer-associated myofibroblasts [28].
Fig. 3.
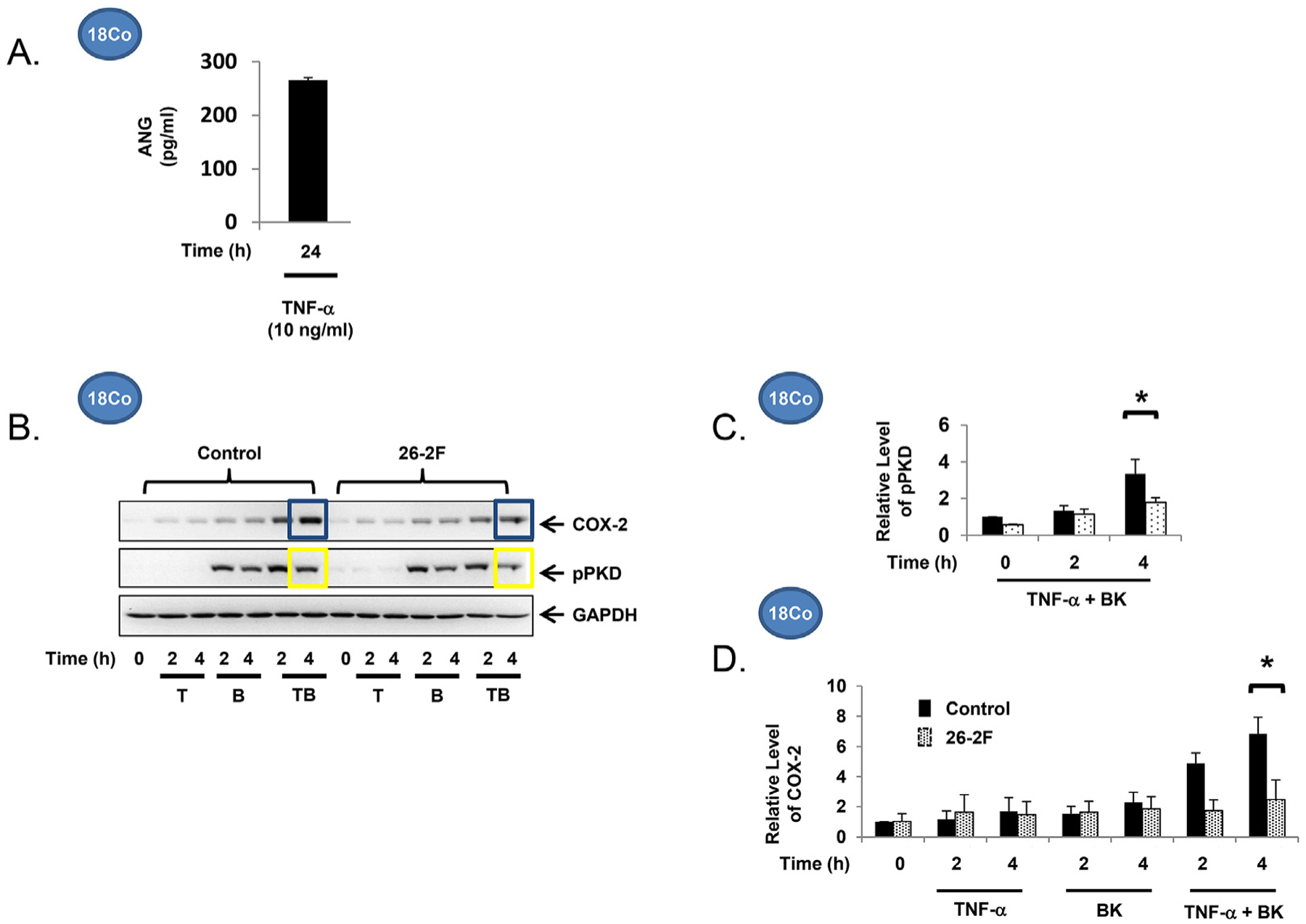
The myofibroblast is a source of angiogenin (265 ± 5 pg/ml in serum-free media after 24 h), quantified by ELISA. n ≥ 3. B. Confluent 18Co cells were equilibrated in serum-free media for 30min, then exposed to TNF-α (10 ng/ml) ± BK (100 nM) in the presence or absence of 26-2F, a neutralizing ANG antibody, for up to 4h. C. The relative levels of pPKD and COX-2 levels (D) following exposure to TNF-α (10 ng/ml) ± BK (100 nM) are shown graphically, expressed as the mean ± S.E. n ≥ 3.
Angiogenin Regulates PKD activation and COX-2 Expression induced by TNF-α and Bradykinin in the Colonic Myofibroblast.
Having demonstrated that the myofibroblast produces angiogenin, this raises the possibility that myofibroblast function may be regulated through angiogenin signaling. To explore whether angiogenin is involved in TNF-α/GPCR-induced, PKD-mediated COX-2 expression, we utilized 26-2F, a murine monoclonal antibody which is an IgG1kappa with an IC binding affinity of 1.6 nM that neutralizes human ANG [29]. While pre-treatment of 18Co cells with 26-2F did not regulate PKD activation or COX-2 expression induced by either TNF-α or bradykinin alone (Figs. 3B), 26-2F inhibited the enhanced PKD phosphorylation (Fig. 3C) as well as the synergistic upregulation of COX-2 (Fig. 3D) induced by their combination. These findings were verified using myofibroblasts isolated from angiogenin knockout (ANG-KO) C57BL/6 mice, a whole-body homozygous knockout strain. ANG-KO C57BL/6 mice are phenotypically normal and do not express angiogenin anywhere, including the gastrointestinal tract. Myofibroblasts isolated from the colon of ANG-KO C57BL/6 mice were grown in cell culture. PKD activation and COX-2 expression was analyzed following exposure to TNF-α (10 ng/ml) and bradykinin (100 nM). Consistent with the findings in 18Co cells following 26-2F treatment, ANG-KO myofibroblasts failed to demonstrate enhanced PKD activation or synergistic COX-2 expression at both 1h and 4h (Fig. 4).
Fig. 4.
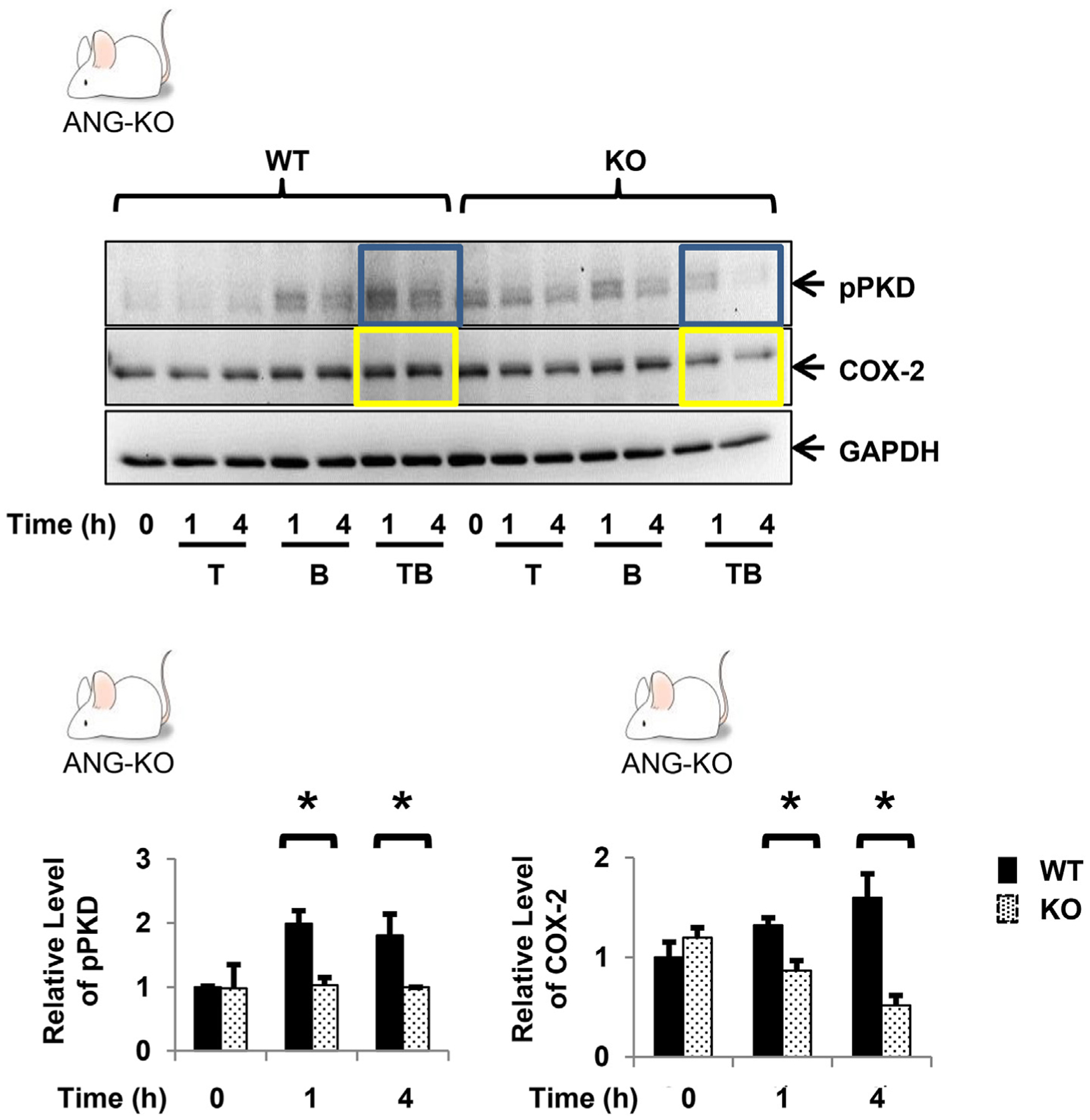
Confluent primary myofibroblasts from WT and ANG-KO mice were equilibrated in serum-free media for 30min and exposed to TNF-α (10 ng/ml) ± BK (100 nM) for up to 4h. The relative levels of pPKD and COX-2 following exposure to TNF-α (10 ng/ml) and BK (100 nM) are shown graphically, expressed as the mean ± S.E. n ≥ 3. * denotes p < 0.05.
5. Discussion
Dynamic GI epithelial-stromal cell interactions regulate the development of inflammation-associated cancer. Understanding the stromal contribution to carcinogenesis may provide new therapeutic avenues that do not currently exist, since traditional treatments focus on the primary tumor. The myofibroblast has been gaining considerable attention as a potential therapeutic target because of its role as a major source of COX-2 in the gastrointestinal tract. In the present study, we show that angiogenin, a multifunctional ribonuclease that is dynamically regulated in colitis and colorectal cancer, is a required element of the activity of pro-inflammatory mediators to enhance GPCR-induced COX-2 expression signaling within the myofibroblast.
Considerable evidence supports a role of COX-2 in the pathophysiology of colorectal cancer [19]. COX-2 is induced in the stroma of patients with IBD [15,16,30], and has been implicated in all stages of colorectal cancer development, from adenoma formation [31] to tumor progression [32,33]. Selective and non-selective COX-2 inhibitors have demonstrated clinical benefit as a therapeutic agent in the prevention and treatment of colorectal cancer in both general [34,35] and high-risk populations [36]. In this context, COX-2 is an attractive molecular target of anti-cancer therapy. However, GI and cardiovascular side effects have limited their use [37,38], and COX-2 inhibitors are generally contraindicated in patients with IBD for this reason [4,39,40]. As an alternative to systemic COX-2 inhibition, regional inhibition of COX-2 and its by-products by targeting the stroma may be more effective and less toxic. Local prostaglandin release by GI stromal cells have important regulatory functions that rely as much on close cellular proximity as concentration [5,41]. Moreover, several studies provide experimental evidence that stroma-directed therapies are feasible [42,43]. Within this context, important questions remain regarding the relative contribution of myofibroblast-derived COX-2 on the development of colitis-associated cancer, and whether its selective inhibition can be developed as an anti-cancer strategy that avoids systemic toxicity.
In conclusion, angiogenin regulates crosstalk signaling between TNF-α and GPCR’s to enhance COX-2 expression via PKD in the myofibroblast. This is supported by experiments utilizing neutralizing monoclonal antibodies of angiogenin, as well as ANG-KO myofibroblasts. Taken together, these findings suggest that myofibroblast-derived angiogenin may play an important role as a paracrine mediator of cell-cell crosstalk in the setting of colitis-associated cancer. These findings may serve as a foundation for new therapeutic approaches that target angiogenin signaling within stroma.
Funding
The authors have no disclosures.
Footnotes
Declaration of competing interest
The authors have no conflicts of interest.
References
- [1].Eaden JA, Abrams KR, Mayberry JF, The risk of colorectal cancer in ulcerative colitis: a meta-analysis, Gut 48 (2001) 526–535, 10.1136/gut.48.4.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Terzi c J, Grivennikov S, Karin E, Karin M, Inflammation and colon cancer, Gastroenterology 138 (2010), 10.1053/j.gastro.2010.01.058. [DOI] [PubMed] [Google Scholar]
- [3].Watanabe T, Konishi T, Kishimoto J, Kotake K, Muto T, Sugihara K, Japanese Society for Cancer of the Colon and Rectum, Ulcerative colitis-associated colorectal cancer shows a poorer survival than sporadic colorectal cancer: a nationwide Japanese study, Inflamm. Bowel Dis. 17 (2011) 802–808, 10.1002/ibd.21365. [DOI] [PubMed] [Google Scholar]
- [4].Bansal P, Sonnenberg A, Risk factors of colorectal cancer in inflammatory bowel disease, Am. J. Gastroenterol 91 (1996) 44–48. http://www.ncbi.nlm.nih.gov/pubmed/8561142. (Accessed 4 October 2019). [PubMed] [Google Scholar]
- [5].Brown SL, Riehl TE, Walker MR, Geske MJ, Doherty JM, Stenson WF, Stappenbeck TS, Myd88-dependent positioning of Ptgs2-expressing stromal cells maintains colonic epithelial proliferation during injury, J. Clin. Invest 117 (2007) 258e269, 10.1172/JCI29159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Uribe G, Villeger R, Bressollier P, Dillard RN, Worthley DL, Wang TC, Powell DW, Urdaci MC, V Pinchuk I, Lactobacillus rhamnosus GG increases cyclooxygenase-2 expression and prostaglandin E2 secretion in colonic myofibroblasts via a MyD88-dependent mechanism during homeostasis, Cell Microbiol. 20 (2018), e12871, 10.1111/cmi.12871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Bansal R, Nakagawa S, Yazdani S, van Baarlen J, Venkatesh A, Koh AP, Song WM, Goossens N, Watanabe H, Beasley MB, Powell CA, Storm G, Kaminski N, van Goor H, Friedman SL, Hoshida Y, Prakash J, Integrin alpha 11 in the regulation of the myofibroblast phenotype: implications for fibrotic diseases, Exp. Mol. Med 49 (2017), 10.1038/emm.2017.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Powell DW, Mifflin RC, Valentich JD, Crowe SE, Saada JI, West AB, Myofibroblasts. I. Paracrine cells important in health and disease, Am. J. Physiol. Physiol 277 (1999) C1–C19, 10.1152/ajpcell.1999.277.1.C1. [DOI] [PubMed] [Google Scholar]
- [9].Armaka M, Apostolaki M, Jacques P, Kontoyiannis DL, Elewaut D, Kollias G, Mesenchymal cell targeting by TNF as a common pathogenic principle in chronic inflammatory joint and intestinal diseases, J. Exp. Med 205 (2008) 331–337, 10.1084/jem.20070906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Burstein E, Fearon ER, Colitis and cancer: a tale of inflammatory cells and their cytokines, J. Clin. Invest 118 (2008) 464–467, 10.1172/JCI34831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Wilson JAP, Tumor necrosis factor α and colitis-associated colon cancer, N. Engl. J. Med 358 (2008) 2733–2734, 10.1056/NEJMcibr0803116. [DOI] [PubMed] [Google Scholar]
- [12].Elmashad N, Ziada D, Hasby E, el motaleb Mohamed A, Immunohisto-chemical expression of proinflammatory enzyme COX-2 and p53 in ulcerative colitis and its associated dysplasia and colorectal carcinoma, J. Microsc. Ultrastruct 4 (2016) 195, 10.1016/j.jmau.2016.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Kaler P, Owusu BY, Augenlicht L, Klampfer L, The role of STAT1 for crosstalk between fibroblasts and colon cancer cells, Front. Oncol 4 (2014), 10.3389/fonc.2014.00088. APR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Owusu BY, Vaid M, Kaler P, Klampfer L, Prognostic and predictive significance of stromal fibroblasts and macrophages in colon cancer, Biomarkers Canc. 7s1 (2015), 10.4137/bic.s25247.BIC.S25247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Shattuck-Brandt RL, Varilek GW, Radhika A, Yang F, Washington MK, DuBois RN, Cyclooxygenase 2 expression is increased in the stroma of colon carcinomas from IL-10(−/−) mice, Gastroenterology 118 (2000) 337–345, 10.1016/s0016-5085(00)70216-2. [DOI] [PubMed] [Google Scholar]
- [16].Konstantinopoulos PA, Vandoros GP, V Karamouzis M, Gkermpesi M, Sotiropoulou-Bonikou G, Papavassiliou AG, EGF-R is expressed and AP-1 and NF-kappaB are activated in stromal myofibroblasts surrounding colon adenocarcinomas paralleling expression of COX-2 and VEGF, Cell. Oncol 29 (2007) 477–482. http://www.ncbi.nlm.nih.gov/pubmed/18032824. (Accessed 4 October 2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Ma X, Aoki T, Tsuruyama T, Narumiya S, Definition of prostaglandin E2-EP2 signals in the colon tumor microenvironment that amplify inflammation and tumor growth, Canc. Res 75 (2015) 2822e2832, 10.1158/0008-5472.CAN-15-0125. [DOI] [PubMed] [Google Scholar]
- [18].Yoo J, Chung C, Slice L, Sinnett-Smith J, Rozengurt E, Protein kinase D mediates synergistic expression of COX-2 induced by TNF-α and bradykinin in human colonic myofibroblasts, Am. J. Physiol. Cell Physiol 297 (2009), 10.1152/ajpcell.00184.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Prescott SM, Is cyclooxygenase-2 the alpha and the omega in cancer? J. Clin. Invest 105 (2000) 1511e1513, 10.1172/JCI10241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Perez CER, Nie W, Sinnett-Smith J, Rozengurt E, Yoo J, TNF-α potentiates lysophosphatidic acid-induced COX-2 expression via PKD in human colonic myofibroblasts, Am. J. Physiol. Gastrointest. Liver Physiol 300 (2011), 10.1152/ajpgi.00381.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Popivanova BK, Kitamura K, Wu Y, Kondo T, Kagaya T, Kaneko S, Oshima M, Fujii C, Mukaida N, Blocking TNF-α in mice reduces colorectal carcinogenesis associated with chronic colitis, J. Clin. Invest 118 (2008) 560–570, 10.1172/JCI32453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Valentich JD, Popov V, Saada JI, Powell DW, Phenotypic characterization of an intestinal subepithelial myofibroblast cell line, Am. J. Physiol. Cell Physiol (1997), 10.1152/ajpcell.1997.272.5.c1513. [DOI] [PubMed] [Google Scholar]
- [23].Khalil HA, Lei NY, Nie W, Lewis MS, Stelzner MG, Martín MG, Dunn JCY, Yoo J, Primary myofibroblasts maintain short-term viability following sub-mucosal injection in syngeneic, immune-competent mice utilizing murine colonoscopy, PloS One 10 (2015), e0127258, 10.1371/journal.pone.0127258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Yoo J, Perez CER, Nie W, Edwards RA, Sinnett-Smith J, Rozengurt E, TNF-α induces upregulation of EGFR expression and signaling in human colonic myofibroblasts, Am. J. Physiol. Gastrointest. Liver Physiol 302 (2012), 10.1152/ajpgi.00522.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Barcena C, Stefanovic M, Tutusaus A, Martinez-Nieto GA, Martinez L, García-Ruiz C, De Mingo A, Caballeria J, Fernandez-Checa JC, Marí M, Morales A, Angiogenin secretion from hepatoma cells activates hepatic stellate cells to amplify a self-sustained cycle promoting liver cancer, Sci. Rep 5 (2015), 10.1038/srep07916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Koutroubakis IE, Xidakis C, Karmiris K, Sfiridaki A, Kandidaki E, Kouroumalis EA, Serum angiogenin in inflammatory bowel disease, Dig. Dis. Sci 49 (2004) 1758–1762, 10.1007/s10620-004-9565-4. [DOI] [PubMed] [Google Scholar]
- [27].Oikonomou KA, Kapsoritakis AN, Kapsoritaki AI, Manolakis AC, Tiaka EK, Tsiopoulos FD, Tsiompanidis IA, Potamianos SP, Angiogenin, angiopoietin-1, angiopoietin-2, and endostatin serum levels in inflammatory bowel disease, Inflamm. Bowel Dis 17 (2011) 963–970, 10.1002/ibd.21410. [DOI] [PubMed] [Google Scholar]
- [28].Drebert Z, MacAskill M, Doughty-Shenton D, De Bosscher K, Bracke M, Hadoke PWF, Beck IM, Colon cancer-derived myofibroblasts increase endothelial cell migration by glucocorticoid-sensitive secretion of a pro-migratory factor, Vasc. Pharmacol 89 (2017) 19–30, 10.1016/j.vph.2016.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Fett JW, Olson KA, Rybak SM, A monoclonal antibody to human angiogenin. Inhibition of ribonucleolytic and angiogenic activities and localization of the antigenic epitope, Biochemistry 33 (1994) 5421–5427, 10.1021/bi00184a010. [DOI] [PubMed] [Google Scholar]
- [30].Adegboyega PA, Ololade O, Saada J, Mifflin R, Di Mari JF, Powell DW, Subepithelial myofibroblasts express cyclooxygenase-2 in colorectal tubular adenomas, Clin. Canc. Res 10 (2004) 5870–5879, 10.1158/1078-0432.CCR-0431-03. [DOI] [PubMed] [Google Scholar]
- [31].Sandler RS, Halabi S, Baron JA, Budinger S, Paskett E, Keresztes R, Petrelli N, Pipas JM, Karp DD, Loprinzi CL, Steinbach G, Schilsky R, A randomized trial of aspirin to prevent colorectal adenomas in patients with previous colorectal cancer, N. Engl. J. Med 348 (2003) 883–890, 10.1056/NEJMoa021633. [DOI] [PubMed] [Google Scholar]
- [32].Ng K, Meyerhardt JA, Chan AT, Sato K, Chan JA, Niedzwiecki D, Saltz LB, Mayer RJ, Benson AB, Schaefer PL, Whittom R, Hantel A, Goldberg RM, Venook AP, Ogino S, Giovannucci EL, Fuchs CS, Aspirin and COX-2 inhibitor use in patients with stage III colon cancer, J. Natl. Cancer Inst 107 (2015), 10.1093/jnci/dju345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Chan AT, Ogino S, Fuchs CS, Aspirin use and survival after diagnosis of colorectal cancer, J. Am. Med. Assoc 302 (2009) 649–658, 10.1001/jama.2009.1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Rothwell PM, Wilson M, Elwin CE, Norrving B, Algra A, Warlow CP, Meade TW, Long-term effect of aspirin on colorectal cancer incidence and mortality: 20-year follow-up of five randomised trials, Lancet 376 (2010) 1741–1750, 10.1016/S0140-6736(10)61543-7. [DOI] [PubMed] [Google Scholar]
- [35].Chan AT, Giovannucci EL, Primary prevention of colorectal cancer, Gastroenterology 138 (2010) 2029–2043, 10.1053/j.gastro.2010.01.057, e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Steinbach G, Lynch PM, Phillips RK, Wallace MH, Hawk E, Gordon GB, Wakabayashi N, Saunders B, Shen Y, Fujimura T, Su LK, Levin B, Godio L, Patterson S, Rodriguez-Bigas MA, Jester SL, King KL, Schumacher M, Abbruzzese J, DuBois RN, Hittelman WN, Zimmerman S, Sherman JW, Kelloff G, The effect of celecoxib, a cyclooxygenase-2 inhibitor, in familial adenomatous polyposis, N. Engl. J. Med 342 (2000), 10.1056/NEJM200006293422603, 1946–52. [DOI] [PubMed] [Google Scholar]
- [37].Grosser T, Fries S, FitzGerald GA, Biological basis for the cardiovascular consequences of COX-2 inhibition: therapeutic challenges and opportunities, J. Clin. Invest 116 (2006) 4–15, 10.1172/JCI27291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Lanas A, Baron JA, Sandler RS, Horgan K, Bolognese J, Oxenius B, Quan H, Watson D, Cook TJ, Schoen R, Burke C, Loftus S, Niv Y, Ridell R, Morton D, Bresalier R, Peptic ulcer and bleeding events associated with rofecoxib in a 3-year colorectal adenoma chemoprevention trial, Gastroenterology 132 (2007) 490–497, 10.1053/j.gastro.2006.11.012. [DOI] [PubMed] [Google Scholar]
- [39].Kefalakes H, Stylianides TJ, Amanakis G, Kolios G, Exacerbation of inflammatory bowel diseases associated with the use of nonsteroidal anti-inflammatory drugs: myth or reality? Eur. J. Clin. Pharmacol 65 (2009) 963–970, 10.1007/s00228-009-0719-3. [DOI] [PubMed] [Google Scholar]
- [40].Velayos FS, Terdiman JP, Walsh JM, Effect of 5-aminosalicylate use on colorectal cancer and dysplasia risk: a systematic review and metaanalysis of observational studies, Am. J. Gastroenterol 100 (2005) 1345–1353, 10.1111/j.1572-0241.2005.41442.x. [DOI] [PubMed] [Google Scholar]
- [41].Li H-J, Reinhardt F, Herschman HR, Weinberg RA, Cancer-stimulated mesenchymal stem cells create a carcinoma stem cell niche via prostaglandin E2 signaling, Canc. Discov 2 (2012) 840–855, 10.1158/2159-8290.CD-12-0101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Knoop K, Schwenk N, Schmohl K, Müller A, Zach C, Cyran C, Carlsen J, Böning G, Bartenstein P, Göke B, Wagner E, Nelson PJ, Spitzweg C, Mesenchymal stem cell-mediated, tumor stroma-targeted radioiodine therapy of metastatic colon cancer using the sodium iodide symporter as theranostic gene, J. Nucl. Med 56 (2015) 600e606, 10.2967/jnumed.114.146662. [DOI] [PubMed] [Google Scholar]
- [43].Shinagawa K, Kitadai Y, Tanaka M, Sumida T, Onoyama M, Ohnishi M, Ohara E, Higashi Y, Tanaka S, Yasui W, Chayama K, Stroma-directed imatinib therapy impairs the tumor-promoting effect of bone marrow-derived mesenchymal stem cells in an orthotopic transplantation model of colon cancer, Int. J. Canc 132 (2012) 813–823, 10.1002/ijc.27735. [DOI] [PubMed] [Google Scholar]