

Abstract
Monoclonal antibody (mAb)–based blocking of the immune checkpoints involving the CTLA4-B7 and PD1-PDL1 inhibitory axes enhance T cell–based adaptive immune responses in cancer patients. We show here that antitumor responses by natural killer (NK) cells can be enhanced by a checkpoint-blocking mAb, 14-25-9, that we developed against proliferating cell nuclear antigen (PCNA). PCNA is expressed on the surface of cancer cells and acts as an inhibitory ligand for the NK cell receptor, NKp44-isoform1. We tested for cytoplasmic and membrane-associated PCNA by FACS- and ImageStream-based staining of cell lines and immunohistochemistry of human cancer FFPE tissues. The mAb 14-25-9 inhibited binding of chimeric NKp44 receptor to PCNA and mostly stained the cytoplasm and membrane of tumor cells, whereas commercial antibody (clone PC10) stained nuclear PCNA. NK functions were measured using ELISA-based IFNγ secretion assays and FACS-based killing assays. The NK92-NKp44-1 cell line and primary human NK cells showed increased IFNγ release upon co-incubation with mAb 14-25-9 and various solid tumor cell lines and leukemias. Treatment with 14-25-9 also increased NK cytotoxic activity. In vivo efficacy was evaluated on patient-derived xenografts (PDX)-bearing NSG mice. In PDX-bearing mice, intravenous administration of mAb 14-25-9 increased degranulation (CD107a expression) of intratumorally-injected patient-autologous or allogeneic NK cells as well as inhibited tumor growth when treated long term. Our study describes a mAb against the NKp44-PCNA innate immune–checkpoint that can enhance NK cell antitumor activity both in vitro and in vivo.
Keywords: Immune checkpoint (IC), Monoclonal antibody (mAb), NKp44, PCNA, PDX
Introduction
Immune checkpoints (ICs) regulate the extent and degree of physiological immune responses in peripheral tissues to maintain self-tolerance (1,2). Several studies have demonstrated that tumors harness IC pathways to gain immune resistance (1). Thus, suppression of innate and adaptive immunity emerged as a hallmark of cancer in the review by Weinberg and Hanahan (3). Immune checkpoints involving the CTLA4-B7 and the PD1-PDL1 inhibitory axes have been therapeutically disabled with blocking antibodies to reactivate tumor-responsive cytotoxic T cells, thereby leading to clinical responses in patients (4–6).
Natural Killer (NK) cells contribute to the cancer immunome within the tumor microenvironment (7,8). In a healthy individual, NK cell activity is regulated by a balance between its inhibitory and activating receptors (9,10). Primary NK inhibitory receptors bind with class-I human leukocyte antigen (HLA-I) molecules and signal through immunoreceptor tyrosine-based inhibitory motif (ITIM) domains. For NK-based innate ICs, those receptors are the first candidates to explore. Anti-KIR antibodies enhance anti-B-cell lymphoma NK activity as mono/combination therapy (11) and augment NK-mediated killing of human AML cells (12).Ab-based blocking of CD94/NKG2A improved NK cell dysfunction in CLL (13,14).
Binding of activating receptors with specific ligands initiates signaling through immunoreceptor tyrosine-based activation motifs (ITAM), which are found on cytoplasmic domains of receptor-associated adaptor molecules(9,15,16). Crucial tumor-reactive activating receptors include NKG2D and natural cytotoxicity receptors (NCRs), namely NKp30, NKp46, and NKp44 (10,15,16). NKp44 receptor is a transmembrane glycoprotein with one IgG type V-like extracellular domain, it is encoded by the NCR2 gene and there is no homolog in the mouse genome (17–19). NKp44 is expressed when NK cells are active and shows cytotoxic activity against both tumor and virus infected cells (19,20). Heparan sulphate and several viral proteins function as NKp44 ligands (21–23). A truncated isoform of MLL5 intracellular protein is also an NKp44 ligand (24). The soluble PDGF-DD is a primary ligand for NKp44 (25). ITAM-mediated activation signaling by NKp44 receptor is mediated through its association with DAP12 (26). NKp44 plays a dual role, both an activating/ inhibitory receptor. The inhibitory function of NKp44 is regulated by an ITIM motif in the receptor cytoplasmic domain (27–29). Engagement with the specific ligand called proliferating cell nuclear antigen (PCNA) triggers the NKp44/ITIM inhibitory axis (30,31). In healthy replicating cells, PCNA is expressed in the nucleus and helps in DNA replication and repair (32,33). However, PCNA is overexpressed in most cancers and associated with cancer virulence (34–37). Cancer cells have cytoplasmic and cell surface PCNA (30,31,38). Among the three mRNA splice variants of NKp44, NKp44 isoform 1 (NKp44-1) contains the cytoplasmic ITIM and the other two (NKp44–2 and −3) lack the ITIM. High mRNA expression of NKp44-1 in AML patients correlates with poor survival (39). Cell surface PCNA expressed by cancer cells exploits the NKp44-1/ITIM inhibitory axis as an IC to escape NK cell–mediated immune attack.
These findings led us to develop a specific monoclonal antibody (mAb), 14-25-9, that recognizes NKp44-PCNA IC. We show that mAb 14-25-9 can recognize membrane-associated PCNA on the surface of several cancer cell lines. It can also inhibit NKp44 binding with PCNA in ELISA. When applied to a co-culture of NK92-NKp44-1 and tumor cell lines, 14-25-9 can enhance NK cells function in terms of IFNγ secretion and cytotoxicity. The 14-25-9 antibody can also enhance in vivo cytotoxic function of NK92-NKp44-1 cells, as well as patient autologous models of NK cells. Systemic treatment with antibody and human NK cells inhibits growth of patient-derived xenografts (PDX) in vivo.
Materials and Methods
Recombinant human PCNA production
The pET-28 and pMAL-c2x vectors were used to produce soluble human His-tagged and MBP-tagged PCNA respectively in Rosetta™ 2 (DE3) cells. Plasmids containing the cDNA or coding sequence of PCNA were transformed into Rosetta™ 2 cells via heat shock, and grown on LB agar plates with kanamycin and chloramphenicol selection. Large scale synthesis of PCNA was then done as described previously (40). Purification of His-tagged PCNA was done using a Gravity-flow Column with “HisPur™ Ni-NTA Resin” Bead kit (Thermo-Scientific). [Binding Capacity ≤ 60mg of a 28kDa 6xHis-tagged protein from a bacterial source per milliliter of settled resin] according to the supplied kit protocol. Purification of MBP-tagged PCNA was done using Amylose resin beads (Catalog #E8035S) (40).
Generation and screening of hybridoma
For the production of membrane PCNA-specific monoclonal antibody 14-25-9, five 8-week old C57BL/6 mice were first immunized by IP injections of 50μg/mouse of human His-PCNA in CFA. Then at days 14 and 34, two boosting immunizations of 50μg/mouse of Human His-PCNA in IFA were done. At day 43, serum titer of PCNA polyclonal antibodies from all five mice were checked by ELISA. Two mice with the highest titer of polyclonal antibodies were chosen and boosted with 50μg/mouse Human His-PCNA without adjuvant. At day 53, the mice were euthanized, spleens were removed under aseptic conditions, and the splenocytes were fused with mouse Sp2/0 myeloma cells using standard fusion methods. After fusion, the cells were plated in 96-well plates to densities of 1 to 2 surviving hybridoma clones/well (1920 wells total) and selected with HAT (SIGMA, H0262). The cells were supplemented with complete 20% DMEM with all standard additives. Following 14 days of HAT selection, hybridoma supernatants were screened for inhibition of chimeric NKp44 receptor binding with PCNA in ELISA and tumor cell surface PCNA recognition in FACS (described in the following section). Positive hybridomas were selected, subcloned to ensure monoclonality, and PCNA interaction was confirmed. The 14-25-9 hybridoma was cultured in HT (SIGMA, H0137) selection medium and further in BIO-CHO/BIO-GROW medium for production and purification of antibody. Antibody purification was performed using a protein-G column and FPLC system (GE Healthcare).
ELISA assays
A 96-well ELISA plate (Costar) was coated overnight at 4°C with 0.1μM of the recombinant human PCNA (His and MBP tagged) and 0.1μM of His-DJ1 (Human, negative control, a kind gift from Dr. Dan Levy, BGU, Israel) in PBS. After washing, the plate was blocked with 2.5% skimmed milk in PBST (PBS+0.05% TWEEN-20 [Sigma, P1379]) for 1h at 37°C followed by washing and incubation with 2.5μg/ml of 14-25-9 and purified mouse IgG1κ isotype control (BioLegend, 401402). Detection was done using mouse IgG HRP-linked whole Ab (from Sheep) (GE Healthcare, NA931) (1:1000 final dilutions). Following washing, TMB (DAKO, S1599) was added. Optical density (O.D.) was measured at 650 nm (Thermo Electron Multiskan Spectrum).
In another setup, ELISA plates were coated overnight at 4°C with 0.1μM of the recombinant human PCNA (MBP tagged); blocking was done as above. After blocking the plates were washed and incubated with 10 to 0.65μg/ml of 14-25-9 and PC10, 2.5μg/ml of mouse IgG1 and 0.25% skimmed milk in PBST as control for 1h at 37°C and then NKp44-Ig (2 μg/ml, final concentration) was added without washing for 1h at 37°C. NKp44-Ig binding to PCNA was detected using peroxidase AffiniPure goat anti-human IgG (H+L) (Jackson ImmunoResearch Laboratories, Inc.) (1:1000 final dilution). Following washing, TMB (100μl/well DAKO, S1599) was added. O.D. was read at 650 nm (Thermo Electron Multiskan Spectrum).
Western blotting, membrane protein isolation and immunoprecipitation
A549, DU145, HeLa cells were individually lysed with 50mM HEPES (pH 7.4), 100 mM NaCl, 0.1% CHAPS, 1 mM DTT, 0.1 mM EDTA, and protease inhibitor cocktail (Calbiochem) containing buffer to extract the total cellular protein. Protein concentration was determined by Bradford assay (BIO-RAD, Protein Assay, 500–0006). 30μg of total cellular protein from each cell line and 10μg of recombinant human PCNAs (His- and MBP- tagged) and human DJ-1 (His- tagged) were electrophoresed on 10% SDS–polyacrylamide gels followed by their transfer onto nitrocellulose membranes. The membranes were blocked with 10% skimmed milk in TBST [Tris pH 7.4 0.02(M), NaCl 0.15(M), and 0.1% TWEEN-20] for 2h at room temperature, followed by overnight incubation at 4°C with 1μg/ml of 14-25-9 and PC10 (for cell lysates) and 14-25-9 (for recombinant PCNA) diluted in 1% BSA in TBST. Blots were then washed with TBST followed by incubation with peroxidase AffiniPure rabbit anti-mouse IgG (H+L) (Jackson Immunoresearch Laboratories, Inc.) at a dilution of 1:5000 in TBST. SuperSignalWest Pico chemiluminescent substrate (Thermo Scientific™) was used to develop the blots, and the images were taken using XRS+ imaging system (Bio-Rad). Membrane protein was extracted from 1×108 K562 cells using a plasma membrane protein extraction kit (Abcam, ab65400, Cambridge, United Kingdom) according to the manufacturer’s instructions. Immunoprecipitation was then performed with protein G-Sepharose beads (GE Healthcare) pre-coated with 14-25-9 (5μg) and IgG1 (5μg) and incubated overnight at 4°C, gently rocking with 50μg of membrane protein lysate. Elutes were then electrophoresed on 10% SDS–polyacrylamide gels and Coomassie staining was done. Total cellular protein was also isolated from NK92-NKp44-1 cell line, electrophoresed, transferred on nitrocellulose membrane, and immune blotted with antibody against total NFκβ and phospho-NFκβ (Santa Cruz Biotechnology, CA, USA). Western blot quantification was performed by measuring band intensity with ImageJ freeware (National Institutes of Health, Bethesda, MD, USA) (31,41).
Kinetic analysis of the antibody by surface plasmon resonance
A ProteOn™ XPR36 Protein Interaction Array System (Bio-Rad) was used to measure the affinity of the monoclonal antibody 14-25-9 to His-tagged recombinant PCNA. For the assay, a HTG sensor chip (#1765031) and ProteOn Manager Version 3.1.0.6 (Bio-Rad Laboratories) were employed. After activation of the chip using EDC/S-NHS amine coupling procedure, the ligand immobilization process was performed with His-tagged recombinant PCNA and His-tagged IL2 as control, at a flow rate of 30μl/min in different flow cells. Different analyte (14-25-9) concentrations (0–100nM) were injected at a flow rate of 40μl/min, with regeneration of the surface using 50mM NaOH after each analyte. Data was analyzed using the bivalent binding model (40).
Cell lines and cell cultures
Cell lines used in this study were as follows: A549, human lung adenocarcinoma (ATCC, CCL-185); DU145, human prostate carcinoma (ATCC, HTB-81); HeLa, human cervical adenocarcinoma (ATCC, CCL-2); HEK293T, SV40 large T Ag-transfected HEK293 cells (ATCC, CRL-11268); CAL-33, human tongue squamous cell carcinoma (RRID:CVCL_1108);K562, human chronic myelogenous leukemia (ATCC CCL-243) and 721.221Cw6, stable 721.221 (EBV-transformed human B-cell lymphoma) transfectants expressing HLA-C molecules. Cells were cultured in either DMEM or RPMI-1640 (Gibco, Life Technologies) medium supplemented with 10% FBS and 1% penicillin/streptomycin. The NK-92 (human NK cell leukemia) cell line (ATCC CRL-2407), was transduced by retrovirus to highly express the canonical sequence (splice variant 1) of NKp44 (designated as NK92-NKp44-1) (39,42), as well as with N-terminus GFP-tagged NKp44 splice variants 1, matching the following cDNA sequences: NKp44-1, NM_004828.3 (39) (NK92-NKp44-1-GFP). NK cell lines were then cultured as previously described (39,43). EL4 cells stably expressing human PCNA, named EL4-PCNA clone 1, 15, 25, 27 and 32, were kindly provided by Drs. Marco Colonna and Alexander Barrow, Washington University, USA (25). EL4 cells and EL4-PCNA clones were cultured in RPMI-1640 (Gibco, Life Technologies) medium supplemented with 10% FBS and 1% penicillin/streptomycin. Cell lines were tested yearly for mycoplasma contamination using Hy-mycoplasma Detection Kit (Hylabs, Cat No. KI50341). Cell lines were not authenticated in the past year; yet, we frequently thaw and use cells from freezing stocks generated upon the introduction of the cells to the laboratory. All cell lines employed in this study were negative for mycoplasma.
Flow cytometry for surface PCNA detection
All adherent cells were detached with Versene 1:5000(1X) (Gibco, Life Technologies), washed in complete 10% FBS containing DMEM, and incubated in a 37°C, 5% CO2 incubator with fresh medium for 2h. After washing with PBS, cells were counted, seeded in 96-well U-bottom plate (50,000 cells/well) and incubated on ice with 14-25-9 and PC10 (2μg/ml) for 1h 30min. The samples were then washed and stained with Allophycocyanin (APC) AffiniPureF(ab’)₂ fragment goat anti-mouse IgG (Jackson ImmunoResearch Laboratories, Inc). 7-AAD (BioLegend, 420404) viability staining solution was used to distinguish between live and dead cells. All suspension cells were washed with PBS and stained as mentioned above. EL4-PCNA clones were further gated for GFP+ cells since PCNA-transfected cells express GFP (25).
Xenograft tumors of patient 1, 2, and 3 were excised and digested using gentle MACS Octo Dissociator with Heaters (MiltenyiBiotec). After digestion, cells were washed twice with HBSS (Sigma, H6648) and seeded in 96-well U-bottom plates and stained as described above. Flow cytometry was done on a Gallios instrument (Beckman Coulter) and FACS data was analyzed only from live cells using FlowJo software (Tree Star, Inc.).
Imaging flow cytometry, immunocytochemistry and immunohistochemistry
For intracellular staining, HeLa cells were fixed, permeabilized and stained using Fixation buffer (cat. 420801, Biolegend) and Permeabilization Wash buffer (cat. 421002, Biolegend) according to manufacturer’s specifications. Cells were then incubated with 2μg/ml of 14-25-9, PC10 and mouse IgG1 on ice for 2h and 30min. After washing with Transcription Factor 1X Perm Buffer, cells were incubated with secondary antibody Alexa Fluor® 488 AffiniPure goat anti-mouse IgG (H+L) (Jackson Immunoresearch Laboratories, Inc) for 1h on ice. After washing the cells, 7AAD was added to stain the nucleus. Imaging flow cytometry was then performed on ImageStream Mark II high resolution microscopy in flow system (Amnis) using 60× magnification for HeLa cells. All data were analyzed using the ImageStream Data Analysis and Exploration Software (IDEAS) (Amnis). Nuclear and non-nuclear staining was then quantified using “Nuclear Localization and Similarity Feature” application (IDEAS; Amnis) (42).
HeLa cells were seeded in eight-chamber cover glass μ-slides (Ibidi) and cultured overnight. Cells were then fixed with 4% paraformaldehyde for 10min followed by washing with PBS and permeabilization with 0.1% Triton X-100 (Sigma, T8532) for 10min. Cells were then washed and blocked at room temperature for 30min in blocking solution (10% FCS; 0.01% Triton X-100). Primary antibody incubation was carried out with 2μg/ml of 14-25-9, PC10 and mouse IgG1 diluted in 1% FCS in 0.01% Triton-X at room temperature for 2h. After washing, staining was done with allophycocyanin (APC)-conjugated anti-mouse IgG; Nuclear staining was done using DAPI (BioLegend, 422801) (1:5000 final dilutions in PBS) for 20min. All images were acquired on a FluoView FV1000 confocal system using a 40× UPLSAPO objective (Olympus). Nuclear or cytoplasmic fluorescence was quantified using the ImageJ (Fiji) software package. The following formula was used to calculate the corrected total cell fluorescence (CTCF); CTCF = Integrated Density - (area of selected cell X mean fluorescence of background readings).
Sections for immunohistochemistry analysis were taken from FFPE blocks of five patients from the WINTHER cohort (44). For immunohistochemical staining, tissue sections were first deparaffinised by two rinses of 10min each in xylene and 100% ethanol. Next, slides were rinsed in 70% and 50% ethanol for 5min, washed in ultrapure water, and subjected to antigen retrieval at 95°C for 30min using antigen unmasking solution, Citrate Buffer (cat. 005000, ThermoFisher Scientific). Sections were then washed and blocked with blocking buffer (5% FCS in 1X PBS with 0.03% TritonX-100) for 2h at room temperature. After blocking, sections were washed and incubated overnight at 4°C with 4μg/ml of PC10, 14-25-9 and mouse IgG1 diluted in 1X PBS containing 1% FCS. Sections were then washed and APC-conjugated anti-mouse IgG was added as secondary antibody for 1h and 30 min. Nuclear staining was then done using DAPI (BioLegend, 422801) (1: 5000 final dilutions in PBS) for 20min.
Small interfering RNA and NKp44-derived peptide-based downregulation of PCNA
Small interfering RNA (siRNA) against human PCNA and control siRNA (Santa Cruz Biotechnology, sc-29440, sc-36869) were used to silence PCNA expression in HeLa cells. 50 pmols of hPCNA siRNA and control siRNA were transfected using Lipofectamine 2000 (Invitrogen, CA, USA) according to the manufacturer’s protocol. After approximately 48h of transfection, cells were stained and analyzed by FACS for surface PCNA with 14-25-9 (31,41).
For NKp44-derived peptide-based down regulation of PCNA, the following peptides were used: R11-NLS-pep8 and R11-NLS-pep8 short. Briefly, 2×105 HeLa cells were seeded in 6-well plates and incubated in sets of three at 37°C and 5% CO2. Upon reaching 80% confluency, they were treated with 2μM of either R11-NLS-pep8 or R11-NLS-pep8 short peptide diluted in 5% FBS-containing DMEM culture medium for 24h. Cells were stained and analyzed by FACS for surface PCNA with 14-25-9 (40).
Isolation and culture of primary human NK cells
Primary human NK cells were isolated from the peripheral blood of healthy donors using the RosetteSep Human NK Cell Enrichment Cocktail (StemCell Technologies) negative isolation kit according to the manufacturer’s protocol and cultured as previously described (43). The purity of CD3−CD56+ NK cells was >95%. Peripheral blood donations from healthy volunteer donors were carried out with their informed consent and with prior approval from the Ben-Gurion University of the Negev Institutional Review Board.
Cell stimulation and IFNγ assays
For NK cell IFNγ secretion assays, target adherent cells were first detached with 1X Versene (Gibco, 15040–033), washed in complete 10% FBS containing DMEM and incubated in 37°C 5% CO2 incubator with fresh medium for 2h, whereas suspension cells were used directly. Cells were then counted and 1.5×105 cells/well were incubated with 2, 5 and 10 μg/ml (for primary NK cells) and 10, 20 and 30μg/ml (for NK92-NKp44-1 cell line) of 14-25-9 and 5 and 20μg/ml of mouse IgG1 respectively for 1h on ice. 5× 104 cells/well of primary NK or NK92-NKp44-1 cell lines were then co-cultured with those target cells in 96-well U-bottom plates (NUNC) pre-coated with 1μg/mL concentration of anti-NKp30 (R&D systems, C34554) for 18h at 37°C 5% CO2 incubator. IFNγ concentrations in the supernatants were then assayed by standard ELISA assay (ELISA MAX, Biolegend).
NK cell cytotoxicity assays
Target cells were first labeled with CFSE (Invitrogen, C34554) according to the manufacturer’s recommendations (for EL4-PCNA clone1, gating was done on GFP+ cells and E: T ratio used were 1:1, 2.5:1, 5:1). NK92-NKp44-1 effector cells were then counted and 0.5×105 cells or its multiples/well were pre-incubated on ice for 1hr with 10, 20 and 30 μg/ml of 14-25-9 or 20 μg/ml of mouse IgG1. Then, effector and target cells were co-incubated (in the presence of the 14-25-9 or IgG1) for 6h in 96-well U-bottom plates (NUNC) pre-coated with 1μg/ml of anti-NKp30 (R&D systems, C34554). Then, cells were washed with PBS and labeled with PI (Santa Cruz Biotechnology, SC-3541). Quantification of cytotoxicity was made in accordance with the previously published method (42) and based on the following gating strategy: CFSE−/GFP− (excludes NK cells) then CFSE+ PI− or CFSE+ PI+ target cells were subjected to Live/Dead analysis.
Mouse strains
Six- to eight-week-old NSG (NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ) mice were purchased from Jackson Laboratory (#005557). Maintenance and breeding of all mice used in this study were done in the local animal care facility, approved by the Institutional Animal Care and Use Committee (IACUC) of Ben-Gurion University of the Negev (BGU). Revision and approval of all experimental procedures were done by the Institutional Animal Care and Use Committee (IACUC) of Ben-Gurion University of the Negev (BGU’s IACUC) according to specified protocols that aim to ensure animal welfare and reduce suffering (permit:IL80-12-2015).
Ethics statement:
The numbers of the Ethics Committee approvals are 0372-15-SOR, 0421-16-SOR, and 0103-17-SOR. The approved protocol allowed us to obtain fresh biopsy specimens from surgeries of HNSCC patients, on the basis of signed written informed consent from all patients.
In vivo mouse model
Freshly obtained tumor samples from HNSCC patients were received from Soroka Medical Center, Beer Sheva, Israel. Within 2–3 hours of receiving the samples, they were implanted subcutaneously in NSG mice to establish the PDXs. Once the size of the PDXs reached around 200 mm3, the mice were randomly allocated to two groups (n=4). Both groups were injected with 2×106 NK92-NKp44-1-GFP cells intratumorally. The control group received PBS, the treatment group received 10mg/kg body weight 14-25-9 intravenously. Mice were sacrificed 6h post antibody administration and tumors were excised and digested using gentleMACS Octo Dissociator with Heaters (Miltenyi Biotec). Cells were then washed twice with HBSS (Sigma, H6648) and seeded in 96-well U-bottom plates, stained with Brilliant Violet-conjugated human CD107a mAb (BioLegend) (1:300 final dilutions) for 1h on ice. The samples were then washed and stained with 7AAD. 50,000 cell events were acquired and CD107a expression was detected from GFP+ NK92-NKp44-1-GFP cells, as described elsewhere (see Flow Cytometry). For the experiments with patient autologous NK cells, after 3 weeks of culture, 2×106 autologous CD56+NKp44+ NK cells were injected intratumorally and the experiment was done in the same way as mentioned for the NK-92 cells above. After tumor digestion, cells were stained with Brilliant Violet-conjugated human CD107a mAb (BioLegend) (1:300 final dilutions) and PE-conjugated human CD16 mAb (BioLegend) (1:300 final dilutions). CD107a expression was detected from CD16+ NK cells.
Efficacy study in xenograft mouse model
To study the effect of 14-25-9 on tumor growth, we employed PDX models from two HNSCC patients. Mice were randomly allocated to three groups (n=5). On day 0, mice were given 250cGy total body irradiation by x-ray (45). On day 1, mice from two groups were infused IV with 5 million NK92-NKp44-1-GFP cells. Vehicle group received 15mg/Kg of mouse IgG1 (BioXcell, USA, cat noBE0083) and treatment group received 15mg/Kg of 14-25-9 IV on day 1 and every other day for 10 days. Both groups also received 10μg/mouse human recombinant IL2 (modified and lab produced) IP in three rounds- on day 1, 3 and 5. The third group received only the IL2 on the same schedule. Tumor volumes were measured every other day using digital calipers. At the end of the experiment on day 10, tumor volumes were measured, mice were sacrificed, and tumors were excised for further immunohistochemical analysis.
Statistics
Graphics and statistical analysis were performed using GraphPad Prism software. Statistical analysis of the data was performed using t test and ANOVA (with p-values of *<0.05, **<0.01 or ***<0.001, ****P<0.0001 as indicated on the figures).
Results
Anti-PCNA 14-25-9 stains tumor cell membrane and inhibits binding of NKp44 to PCNA
We previously reported that interaction of NK cell-expressed NKp44 isoform 1 (NKp44-1) with PCNA expressed on the membrane of tumor cells inhibits NK cell function (39). To block the NKp44-1-PCNA IC, we generated PCNA mAb capable of both staining the tumor cell surface and inhibiting NKp44 binding to PCNA. We screened supernatants from 384 PCNA+ colonies for staining of HEK293T cell membrane and for inhibiting NKp44-Ig binding to MBP-hPCNA. Only one hybridoma, 14-25-9, passed both tests. Purified 14-25-9 mAb bound with the recombinant hPCNA specifically as shown by ELISA and Western blot assays (Fig.1A, B). 14-25-9 mAb bound to recombinant human PCNA with moderate affinity (KD = 3.54 × 10–8 M) (Fig.1C).
FIGURE 1. Binding Specificity of 14-25-9 mAb with recombinant and endogenous hPCNA and inhibition of NKp44-Ig binding.
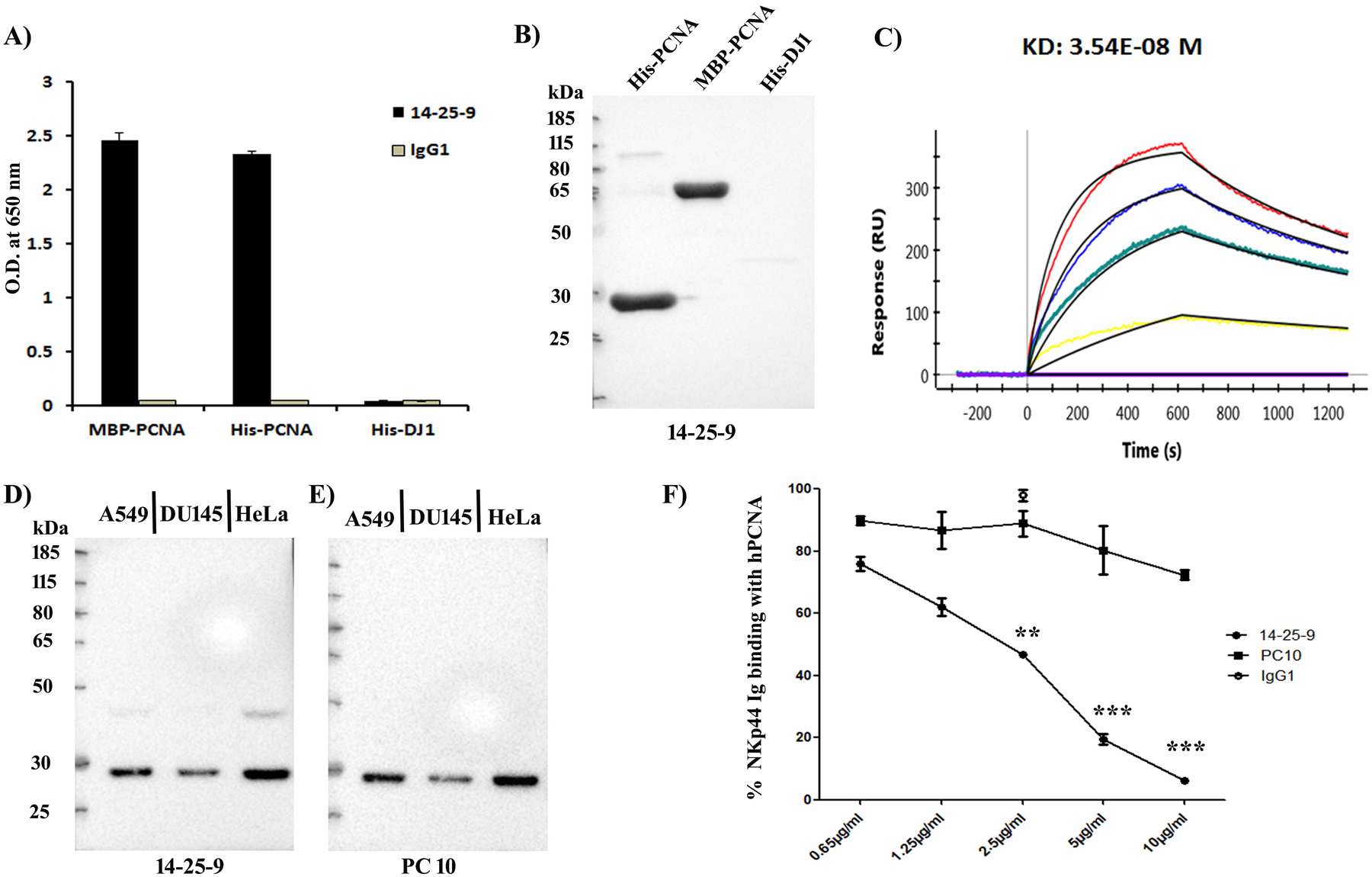
(A) ELISA demonstrating the binding specificity of 14-25-9 mAb with plate-bound recombinant MBP- and His-tagged hPCNA. His-DJ1 (negative control) and mIgG1 (isotype control). (B) Western blot image demonstrating specific binding of 14-25-9 with His-hPCNA (30kDa) and MBP-hPCNA (71.5kDa) and no binding with His-DJ1. (C) ProteOn array showing the affinity of 14-25-9 at a concentration range of 0–100nM to His-hPCNA. Concentrations represented with lines are, 0nM (Violet, base-line), 6.25nM (Yellow), 25nM (Green), 50nM (Blue), and 100nM (Red). Analysis: bivalent binding model. (D-E) Western blot images showing recognition of endogenous PCNA. Data are representative of three independent experiments. (F) ELISA demonstrating inhibition of NKp44-Ig binding with plate-bound MBP-hPCNA by titrated 14-25-9 and PC10. Percentages of NKp44-Ig binding, were calculated against no-antibody incubation value. Data represents mean±SD of two independent experiments in n=3 biological replicates for each dose. One-way ANOVA was performed among the groups; comparison was done between PC10 and 14-25-9 groups. * p<0.05, **p<0.01, ***p<0.001.
PC10 is a common human PCNA mAb employed both in PCNA research and in the clinic for the assessment of human cancer virulence (34). Cell lysates of three human cancer cell lines were tested for the binding of 14-25-9 and PC10. Both mAbs showed similar binding to cellular PCNA as assayed by Western blotting (Fig.1D & E, respectively). We observed that PC10 was not able to inhibit chimeric NKp44 binding to plate bound PCNA, whereas14-25-9 significantly inhibited NKp44 binding to PCNA at concentrations between 2.5–10μg/ml (Fig.1F). Mouse IgG1was employed as control, since 14-25-9 was determined to be an IgG1 isotype. The second selection criterion for which 14-25-9 was chosen among 384 PCNA+ colonies was its ability to bind cell membranes of tumor cells. 14-25-9, but not PC10, positively stained the cell membrane of several human cancer cell lines both from solid or leukemic source (Fig.2A). The HLA class I– leukemia line721–221 did not bind 14-25-9, whereas HLA-Cw6 transfected 721.221 (721.221-Cw6) did (Fig.2A). This is in accordance with the observations that HLA-Cw6 is interacting with PCNA (33) and that membrane-associated PCNA is transported to the membrane with HLA class I (30). The PC10 mAb did not stain the cell surface membrane of these cell lines (Fig.2A). Human and murine PCNA are different only by 5 of 261 AA and PC10 also recognizes both human and murine nuclear PCNA (33). Yet, 14–25.9, but not PC10, could positively stain the cell membrane of mouse cancer cell lines (e.g. 4T1, Fig.2A). We then assayed whether 14-25-9 can stain the cell membrane of human primary non-transformed cells. 14-25-9 and PC10 were not able to positively stain the cell membranes of human primary foreskin fibroblasts, embryonic stem cells or NK cells (Fig.2A). Primary human cancer cells were obtained after digestion of human xenograft tumors of head and neck squamous cell carcinoma (HNSCC) cancer patients, (patient 1 [Pat1], patient 2 [Pat2], and patient 3 [Pat3]), and stained with 14-25-9 and PC10. Again, 14-25-9, but not PC10, stained the cell surface of these cells positively (Fig.2B). We then sought to assess 14-25-9 staining pattern on a cell line that was transduced to overexpress human PCNA. EL4 cells and five overexpressing human PCNA-transfected clones were previously reported to show negative membrane staining with PC10 mAb (25). We repeated the staining with PC10 and with 14-25-9. EL4 cells showed no staining on the cell surface either with PC10 or with 14-25-9 (Fig.2C). Yet, all five hPCNA-overexpressing EL4 clones showed strong staining on the surface by 14-25-9. As reported before (25), PC10 staining remained negative for all of the EL4-PCNA clones (Fig.2D, E).
FIGURE 2. Endogenous and overexpressed cell surface PCNA recognition by 14-25-9 and non-specificity towards human primary cells.
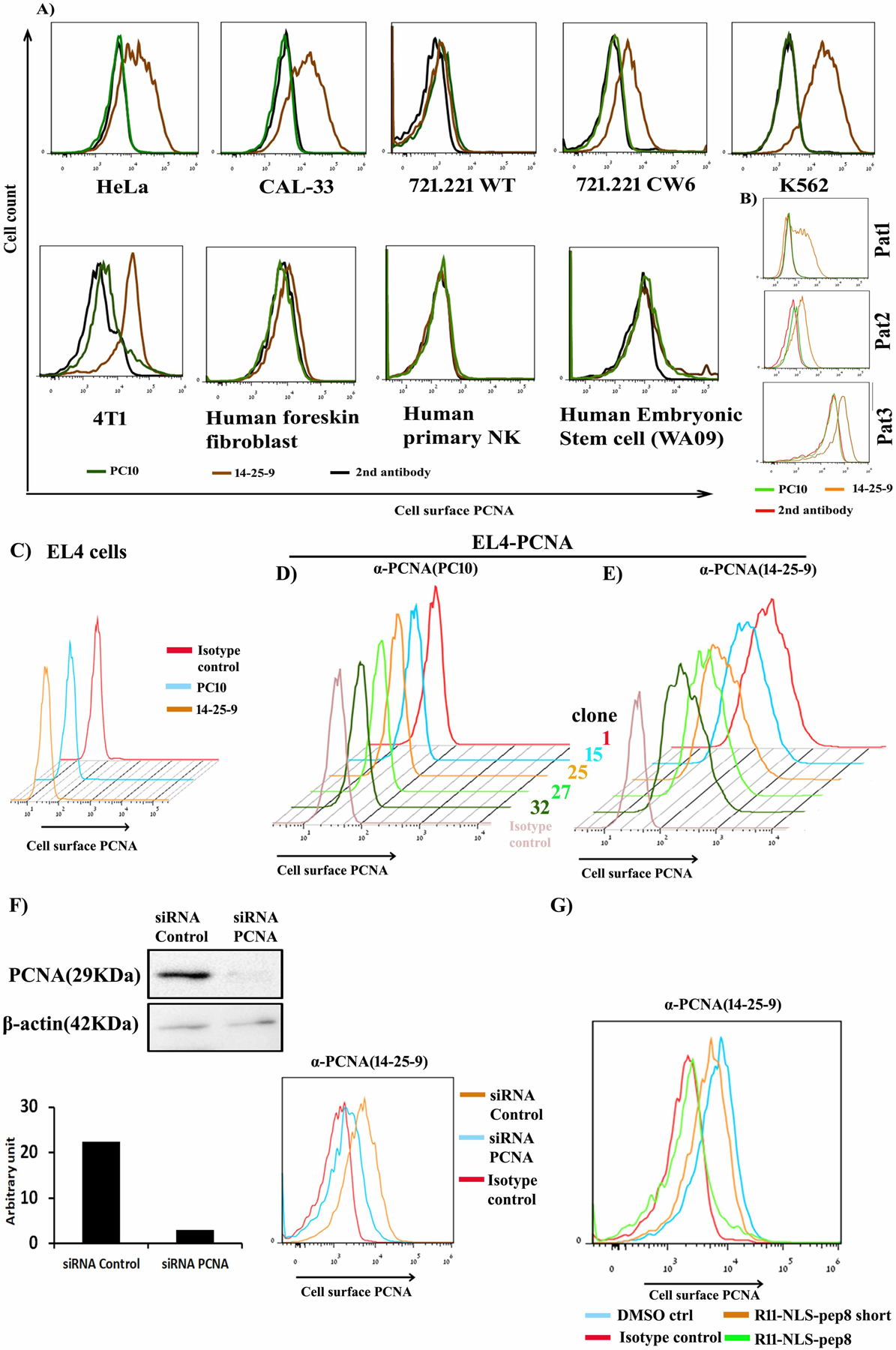
(A) Staining data showing cell surface PCNA staining by 14-25-9 (brown) and no staining by PC10 (green) compared to secondary anti-mouse IgG alone (black) for cancer cell lines and primary human cells: foreskin fibroblast, NK and human embryonic stem cell (WA09). (B) Primary cancer cells derived from PDX from HNSCC cancer patients (Pat1–3) were stained with 14-25-9 (orange), PC10 (green) and secondary anti-mIgG alone (red). (C) EL4 cells were stained with 14-25-9 (orange), PC10 (sky blue) and mIgG1 (red). (D-E) Human PCNA-transfected EL4 clones (clone 1, 15, 25, 27 and 32, depicted according to their histogram color) were stained with PC10 (D) and 14-25-9 (E). (F) siRNA based intracellular PCNA downregulation was done in HeLa cells to confirm PCNA staining by 14-25-9. (F) SiRNA study: western blot image representing downregulation of PCNA in HeLa by siRNA and quantitation (bar-histogram); FACS histogram: cell-surface staining by 14-25-9 in the same siRNA-based PCNA-silenced HeLa cells. (G) Cell-surface staining by 14-25-9 in HeLa cells treated with R11-NLS-pep8. DMSO and R11-NLS-pep8 short served as controls. Data are representative of three independent experiments.
We further asked (i) if manipulation of intracellular PCNA affects the expression of PCNA on the membrane of tumor cells and (ii) whether the PCNA moiety recognized by 14-25-9 on the cell membrane represents fragmented or full-length PCNA. To address the first question, we used siRNA-based and peptide-based downregulation of cellular PCNA. Downregulation of the endogenous PCNA within HeLa cells using PCNA-specific siRNA, but not control siRNA, reduced both endogenous PCNA (Fig.2F; the full blot is shown in Supplementary Fig.S1A) and surface staining of the HeLa cells by 14-25-9 (Fig.2F). We previously identified a NKp44-derived peptide named pep8, which can bind with PCNA, and when conjugated with cell penetrating peptide, induces tumor cell death by blocking PCNA function (40). Incubation of HeLa cells with a non-lethal dose (below IC50) of R11-NLS-pep-8 (2μM) reduced cell surface PCNA staining by 14-25-9, whereas control peptide R11-NLS-pep-8 short showed almost no reduction in staining (Fig.2G).
We also employed a plasma membrane protein extraction kit for isolation of plasma membrane protein from cultured K562 cells, and immuno-precipitated using 14-25-9. Coomassie staining showed the precipitation of PCNA from plasma membrane protein by 14-25-9 (Supplementary Fig.S1B).
14-25-9 stains cytoplasmic and membrane PCNA whereas PC10 stains nuclear PCNA.
We further analyzed the intracellular staining pattern of PCNA within cancer cell lines by fixing and permeabilizing the cells and analyzing staining results with the ImageStream flow cytometer and with a confocal microscope. Using the ImageStream, we observed that PC10 mainly recognized the nuclear PCNA whereas 14-25-9 mainly recognized non-nuclear PCNA (Fig.3A–B). Employing “Nuclear Localization and Similarity Feature” application software, we determined that 4.81% of HeLa cells were stained in the non-nuclear region by PC10 whereas 14-25-9 stained 62.7% in non-nuclear regions (Fig.3C–D). To quantify the same phenomenon on adherent cells, HeLa cells were fixed, permeabilized, and stained employing IFC; images were taken on a confocal microscope. 14-25-9 stained mainly cytoplasmic/membrane PCNA, whereas PC10 stained nuclear PCNA (Fig.3E). Using Fiji-ImageJ software, we showed that total nuclear fluorescence was significantly higher in the case of PC10 compared to 14-25-9; inversely, 14-25-9 stained mainly cytoplasm/membrane areas significantly higher as compared to PC10 (Fig.3F). A similar distribution was observed also in biopsies obtained from human cancer patients: head and neck, oral cavity, tongue and oral cavity, kidney and breast cancers. Representative immunofluorescence (IF) staining results of formalin fixed paraffin embedded tissue (FFPE) sections from tongue and oral cavity were shown in Fig.4. MAb 14-25-9 stained primarily cytoplasmic/membrane-associated PCNA (Fig.4A, middle panel), whereas PC10 stained mainly nuclear PCNA (Fig.4A, upper panel) and control IgG1 showed no staining (Fig.4A, lower panel). Figs. 4B–D show representative magnified images from PC10, 14-25-9 and IgG1 staining, respectively to elucidate the nuclear staining by PC10 and cytoplasmic/membrane-associated staining by 14-25-9. Fluorescence intensity was calculated for PC10 and 14-25-9 for these patients’ samples using the Fiji-ImageJ software. Average total nuclear staining in tumor sections was significantly higher for PC10 compared to 14-25-9 (Fig.4E). Inversely for cytoplasmic/nuclear staining: 14-25-9 stained significantly better than PC10 (Fig.4F). Note that sections of matched normal tissues for the same 5 patients were also stained with PC10 and 14-25-9. Significantly less intense PC10 nuclear staining was observed for matched normal tissue sections as compared to tumor sections (Fig.4E); this result is in accordance with >30 years’ experience of staining clinical samples with PC10. The lower nuclear staining of tumor cells with 14-25-9 was even lower in matched normal sections (Fig.4E). Cytoplasmic and membrane staining of sections from matched normal tissues with 14-25-9 showed little to no staining (Fig.4F).
FIGURE 3. Recognition of non-nuclear and nuclear PCNA of cancer cell line by 14-25-9 and PC10 antibodies.
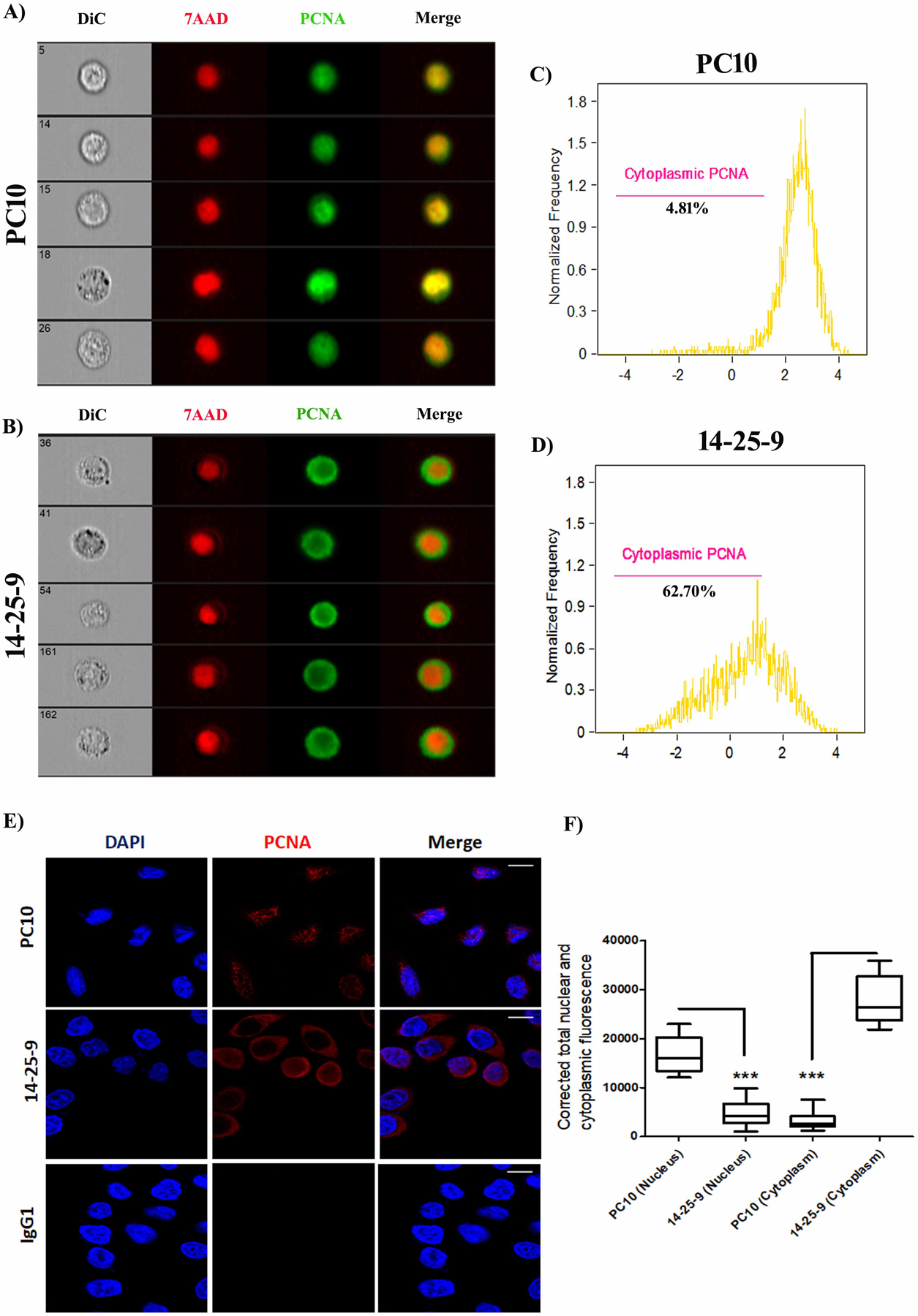
ImageStream (A-D) and confocal (E-F) analysis of fixed and permeabilized HeLa cells: (A) PC10 (A) and 14-25-9 (B) staining (Alexafluor 488) co-stained with 7AAD. Images are representative of three independent experiments. (C-D) Quantification of the ImageStream results; histograms represent percentage values for cytoplasmic staining of PC10 (C) and 14-25-9 (D). (E) PC10 (top panels), 14-25-9 (middle) and mIgG1 (bottom) staining (APC) co-stained with DAPI. Images are representative of three independent experiments. (F) The corrected total nuclear and cytoplasmic fluorescence values for images of (E) are presented for PC10 and 14-25-9. * p < 0.05, ANOVA.
FIGURE 4. MAb 14-25-9 can recognize nonnuclear PCNA of human cancer biopsy samples.
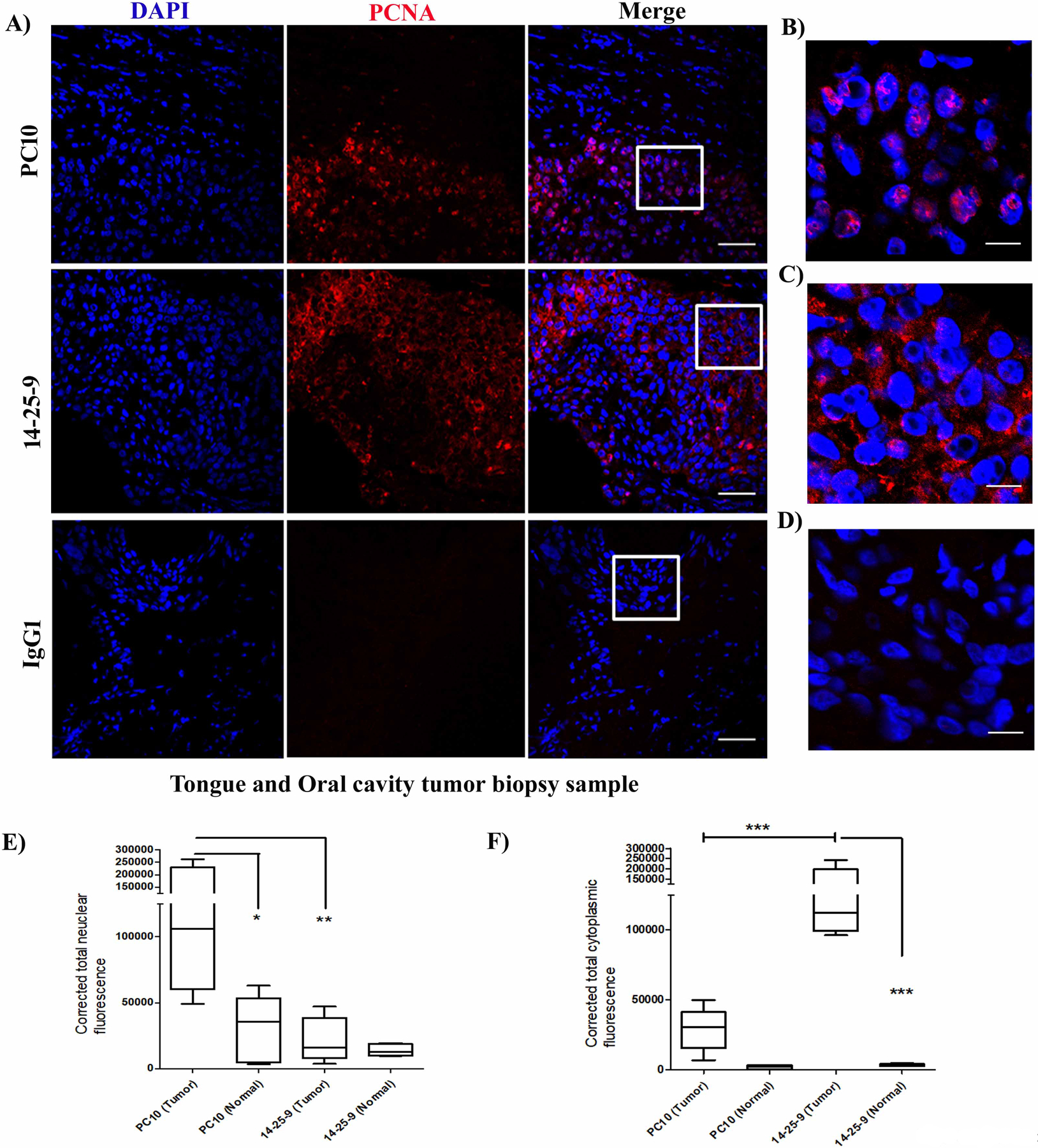
Fluorescence immunohistochemistry was performed on FFPE human cancer biopsy samples. (A) Representative confocal images showing PC10 (top panels), 14-25-9 (middle) and mIgG1 (bottom) staining (APC) co-stained with DAPI. Higher magnification images of regions marked in “merge” panels are shown for PC10 (B) 14-25-9 (C) and mIgG1 (D). Calculated nuclear (E) and cytoplasm/membrane (F) fluorescence values for both tumor and matched-normal tissues of all five patients are presented for PC10 and 14-25-9. * p<0.05, **p<0.01, ***p<0.001.
14-25-9 enhanced antitumor function of cultured and primary NK cells
The effect of 14-25-9 as an IC inhibitor was studied in vitro by culturing NK and tumor cells in the presence of 14-25-9 and checking for IFNγ secretion and target cell lysis. We employed effector NKp44–92-1 cells and IL2-cultured primary human NK cells that express NKp44 constitutively; as targets, we studied five different tumor cell lines. A dose dependent increase in the secretion of IFNγ was seen when primary human NK cells were incubated with mAb 14-25-9 in the presence of the tumor cells. In the presence of 10μg/ml 14-25-9, IFNγ secretion increased by 4.1, 2, 2, 1.6, and 1.3 fold for 721.221-Cw6, K562, A549, HeLa, and DU145, respectively (Fig.5A–E). IFNγ quantities in absolute value for 721.221-Cw6 with pNK are represented in Fig.5F. When NK92-NKp44-1 were used as effector cells, the effect of 14-25-9 was apparent in higher mAb doses. In the presence of 30μg/ml of 14-25-9, IFNγ secretion increased by 2.7, 1.8, 1.7, 1.8, and 2-fold for 721.221-Cw6, K562 A549, HeLa, and DU145, respectively (Fig.5G–K). IFNγ quantity in absolute value for 721.221-Cw6 with NKp44–92-1 cells is represented in Fig.5L. The increase in IFNγ secretion following 14-25-9 treatment was significantly higher compared to treatment with mIgG1 for all tested doses for NK92-NKp44-1, and for most tested doses for primary human NK cells (Fig.5A–L). Following treatment with 14-25-9, no significant increase in IFNγ secretion by NK92-NKp44–3 was observed when interacting with EL4 and EL4-PCNA clone-1 targets (Supplementary Fig.S2A–B); confirming the specificity of 14-25-9 to blocking of the interaction of membrane PCNA and NKp44-1. To confirm the specificity of 14-25-9 for cancer cell surface PCNA, we applied the antibody on human primary NK cells without target cells; there was no significant increase of IFNγ secretion by these NK cells (Supplementary Fig.S2C).
FIGURE 5. Effect of mAb 14-25-9 on IFNγ secretion and cytotoxic activity of NK92-NKp44-1cell line and human primary NK cells.
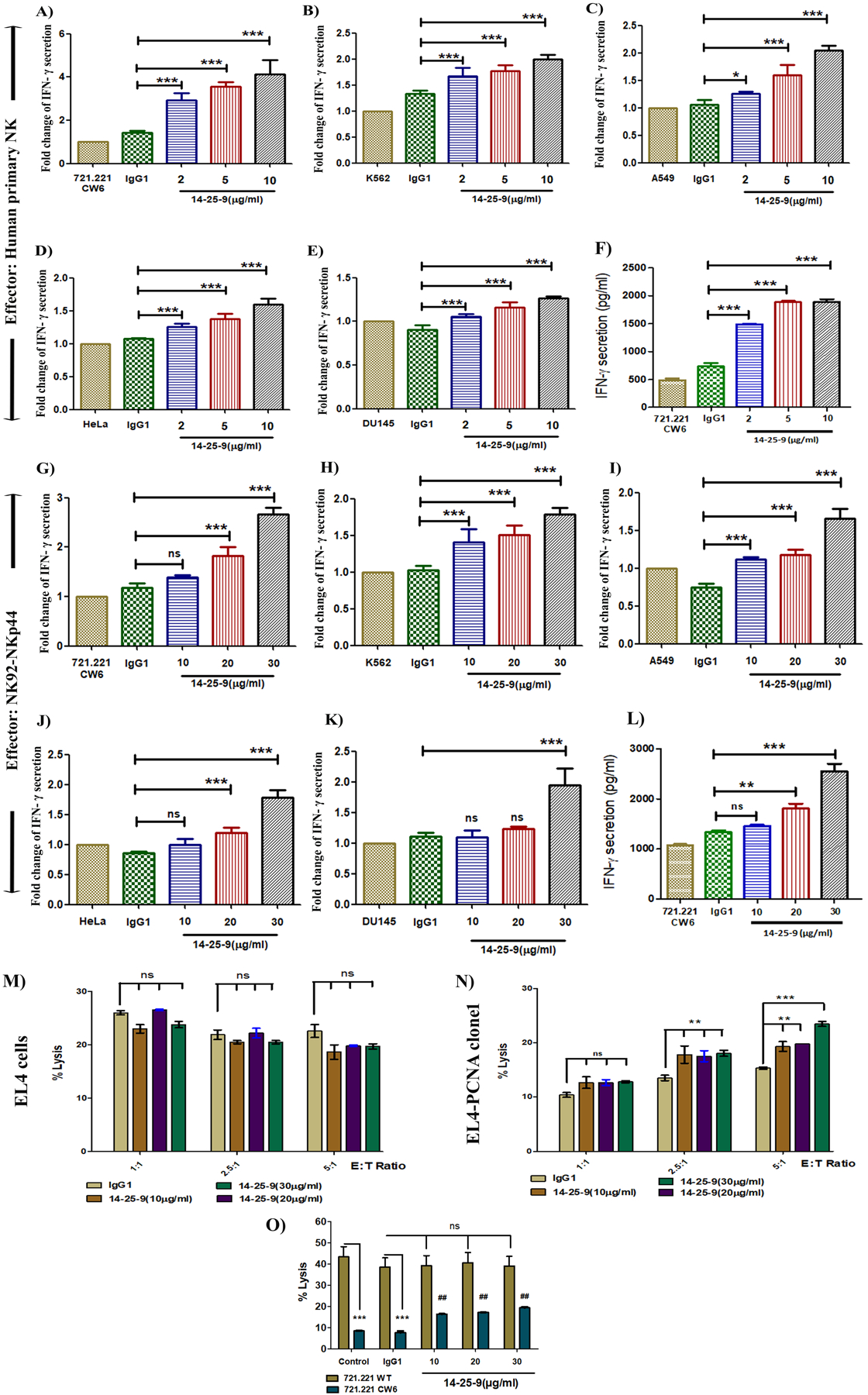
Interferon-γ ELISA results showing impact of 14-25-9 on primary human NK and NK92-NKp44-1 cells, while interacting with cancer cell lines. NK cells were co-incubated overnight with different cancer cell lines in the presence of titrated 14-25-9, 5μg/ml of mIgG1 and PBS as controls. IFNγ concentration (pg/ml) was assayed by ELISA. Fold change values of IFNγ with respect to control PBS were determined for (A) 721.221-Cw6 (B) K562 (C) A549 (D) HeLa and (E) Du145. (F) Representative bar graph showing absolute IFNγ concentration (pg/ml) for 721.221-Cw6 cells when interacting with pNK cells. NK92-NKp44-1 cells were co-incubated overnight with different cancer cell lines in presence of titrated 14-25-9, 20μg/ml of mIgG1 and PBS as controls. IFNγ concentration (pg/ml) was assayed by ELISA. Fold change values of IFNγ in respect to control PBS were determined for (G) 721.221-Cw6 (H) K562 (I) A549 (J) HeLa and (K) Du145. (L) Representative bar graph showing absolute IFNγ concentration (pg/ml) for 721.221-Cw6 cells when interacting with NK92-NKp44-1cells. Data represents mean±SD of two to three independent experiments in n = 3 biological replicates for each treatment. One-way ANOVA was performed among the groups, comparison was done between mIgG1 and 14-25-9 treatment groups. *p<0.05, **p<0.01, ***p<0.001, ns=not significant. To determine the effect of 14-25-9 on cytotoxic activity of NK92-NKp44-1 cells, they were incubated with EL4 cells (M) and hPCNA overexpressing EL4 clone1 cells (N) (E: T= 1:1, 2.5:1, 5:1) for 6h in presence of titrated 14-25-9 and 20μg/ml of mouse IgG1 as control. (O) 721.221 and 721.221-Cw6 cells were incubated with NK92-NKp44-1 cells (E: T= 1:1) for 6h in presence of titrated 14-25-9 and 20μg/ml of mIgG1 as control. Specific cytotoxicity was assayed by CFSE/GFP and PI-based FACS analysis. Two-way ANOVA and Bonferroni post tests were performed among the groups. Comparison was done among IgG1 and different treatment groups for EL4 and EL4-PCNA clone1. ‘ns’ represent no significant increase in cytotoxicity with treatment of 14-25-9 and **p<0.01, ***p<0.001 represent significant increase in cytotoxicity. For 721.221 versus 721.221-Cw6, comparisons were done both from control and mIgG1. Significant decrease in killing of 721.221-Cw6 compared to 721.221 control and treated with mouse IgG1 (***p<0.001). No significant increase (ns) is observed in killing of 721.221 treated with three doses of 14-25-9. Significant increase is observed in killing of 721.221-Cw6 cells treated with three doses of 14-25-9 compared to 721.221-Cw6 cells treated with mIgG1 ## (p<0.01). The results are from three experiments.
To determine the effect of 14-25-9 on the cytotoxic activity of NK92-NKp44-1cells, we assessed a pair of target cells that are negative and positive for cell surface staining with 14-25-9; e.g. negative EL4 and positive EL4-PCNA clone 1 in three different E:T ratios, and negative 721.221 and positive 721.221-Cw6 (Fig.5M–N–O). These target cells were incubated with NK cells in the presence of 14-25-9 or mIgG1. Target-specific cytotoxicity was assayed by CFSE- and PI-based FACS analysis. Note that EL4-PCNA clone 1 was lysed to a lesser extent than EL4 cells without mAb treatment or in the presence of mIgG1, as explained by the overexpression of membrane-associated PCNA by EL4-PCNA clone 1. Indeed, incubation with different doses of 14-25-9 enhanced the lysis of EL4-PCNA clone 1 to the equivalent of target EL4 cells (Fig.5M–N). Similar observations were shown for 721.221 and 721–221-Cw6, although lysis of 721.221-Cw6 did not reach that of 721.221 (Fig.5O), perhaps due to the presence of Cw6 in addition to the presence of membrane-associated PCNA that was blocked by 14-25-9. Therefore, we have demonstrated that the presence of the PCNA monoclonal antibody 14-25-9 can block the NKp44/PCNA-associated IC to enhance NK cell antitumor functions in-vitro, thus acting as an IC inhibitor.
Treatment of PDX mice with 14-25-9 enhances function of intratumorally injected NK cells
As NKp44 is not expressed in mouse, we employed immune deficient NSG: patient-derived HNSCC tumor tissue, grown as xenografts (PDX) following implantation in immune deficient NSG mice, were used as a tumor model. Freshly obtained tumor tissues from two different HNSCC patients (Pat1 and Pat2) were implanted into one mouse each to establish the PDX tumors. After subsequent passage, PDX bearing mice were distributed to two groups (n=4) for each patient. Initially, to determine the impact of 14-25-9 mAb mediated inhibition of NKp44-PCNA IC in vivo, GFP+NK92-NKp44-1-GFP effector cells were used. After 2 weeks, when tumor volume in both groups was around 200 mm3, 2×106 GFP+NK92-NKp44-1-GFP cells were injected intratumorally. The treatment group was subsequently injected intravenously with 10mg/kg of mAb 14-25-9 and the control group with PBS (Fig.6A). After 6h, tumors were excised and digested to generate single cell suspensions. Cells were then stained for human CD107a. When gated upon GFP+ NK cells, it was observed that mean CD107a expression values for the 14-25-9-treated groups were significantly higher for both Pat1 and Pat2 than for the respective control groups (Fig.6B–C).
FIGURE 6. Systemic treatment with 14-25-9 augments antitumor cytotoxic activity of allogeneic and autologous NK cells and halts tumor growth.
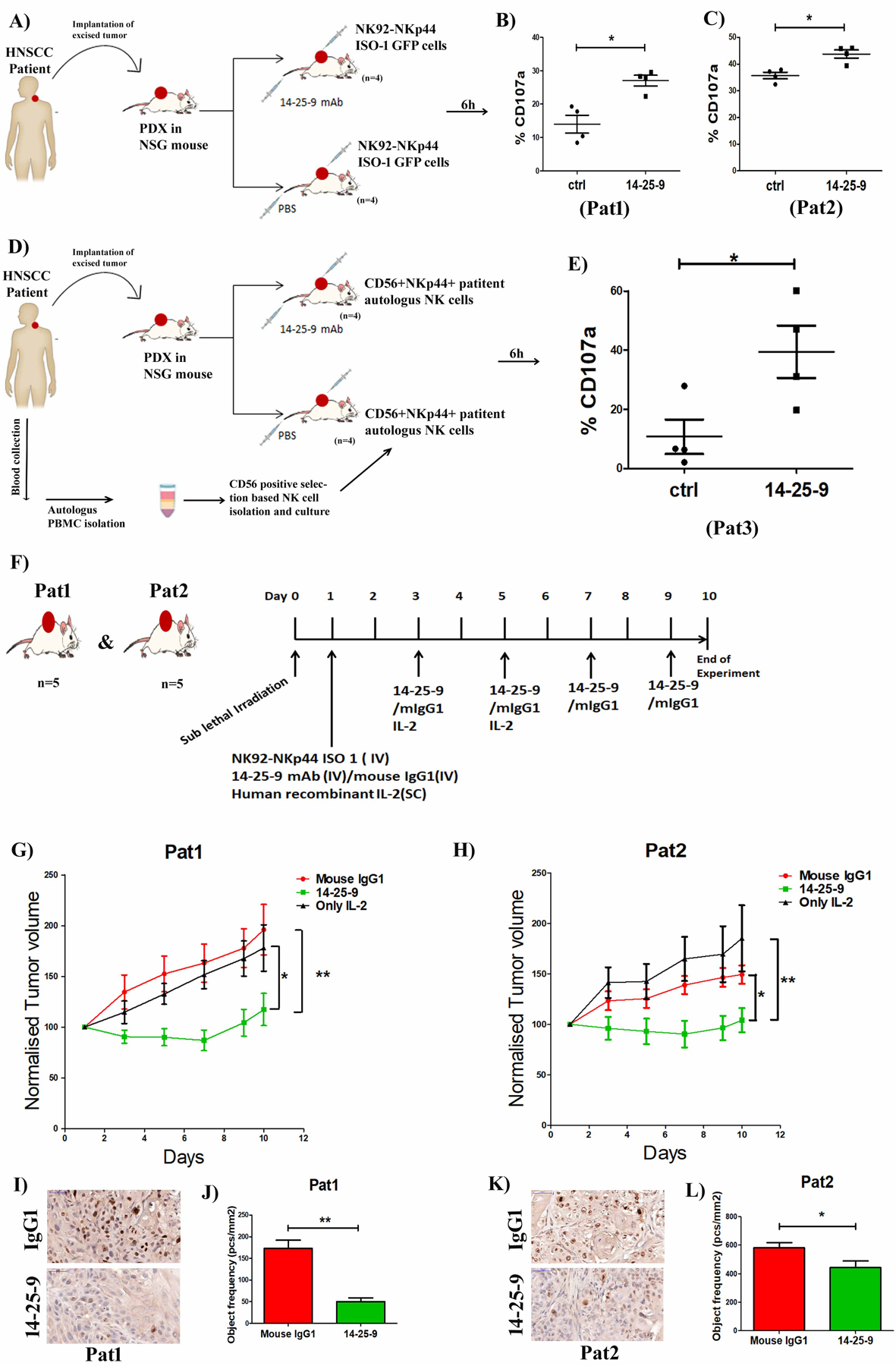
(A) Schematic diagram demonstrating engraftment and propagation of freshly obtained patient tumor samples in NSG mice and intratumoral injection of NK92-NKp44-1-GFP cells and subsequent intravenous administration of 14-25-9 (10mg/Kg) and control PBS. After 6h tumors from both control and 14-25-9 treated groups (n=4) were digested, washed and single cell suspension was stained for CD107a. Gating was set on GFP+ cells and percentages of CD107a+ cells were analyzed by flow cytometry. (B-C) Comparisons of CD107a+ cell percentage in each animal between control and 14-25-9 treated groups of Pat1 and Pat2, respectively. (D) Schematic diagram demonstrating engraftment and propagation of freshly obtained patient 3 (Pat3) tumor samples in NSG mice and CD56+ selection-based isolation and culture of autologous NK cells for three weeks and then intratumoral injection of autologous NK cells and subsequent intravenous administration of 14-25-9 (10mg/Kg) and control PBS. (E) After 6h, tumors from both control and 14-25-9 treated groups (n=4) were digested, washed and single cell suspensions were stained for CD107a and CD16. Gating was set upon CD16+ NK cells and percentages of CD107a+ cells were analyzed by flow cytometry. Graph representing mean ± SEM. * p < 0.05, Student’s t test. (F) Schematic diagram representing treatment schedule for PDXs of Pat1 and Pat2 for 10 days. (G) Tumor growth curve of Pat1 representing normalized tumor volume of mIgG1, 14-25-9 and only IL2 treated groups, over 10 days. (H) Tumor growth curve of Pat2 representing normalized tumor volume of mIgG1, 14-25-9 and only IL2 treated groups, over 10 days. One Way ANOVA was performed among the groups to calculate significance at day 10, *p<0.05, **p<0.01. (I-K) Representative DAB stained images (40X) of Pat1 and Pat2 PDX tumor respectively showing difference in Ki67 expression between mouse IgG1 and 14-25-9 treated groups at the end of experiment (day10). (J-L) Bar graphs showing quantification of Ki67 staining from PDX tumors (n=5) of Pat1 and Pat2 respectively at the end of experiment (day 10). * p < 0.05, **p<0.01Student’s t test.
We then sought to determine the impact of 14-25-9 mAb on patient autologous NK cell responses in our HNSCC PDXs. Freshly obtained tumor tissue from the third HNSCC patient (Pat3) was implanted in one mouse and subsequently passaged to generate the required number of PDX-bearing mice. Autologous NK cells were isolated from Pat3 and cultured in vitro with 300 IU/ml of human IL2 for 3 weeks (described in Materials & Methods). FACS staining data showed that Pat3 autologous NK cells were positive for CD56 (containing both CD56dim and CD56bright populations) and 56.4% of the cells expressed NKp44 after 3 weeks in culture (Supplementary Fig.S3A). When the tumor volume in both the groups was around 200mm3, 2×106 CD56+ NKp44+ NK cells were injected intratumorally. The treatment group was subsequently injected intravenously with 10mg/kg mAb 14-25-9 and the control group with PBS (Fig.6D). After 6h, the tumors were excised and digested to generate single cell suspensions. Cells were then stained for human CD107a and human CD16. When gated upon CD16+ NK cells, the mean CD107a expression value in the 14-25-9-treated group was significantly higher than the respective control group (Fig.6E).
Systemic injection of NK cells and 14-25-9 inhibits tumor growth in PDX-bearing mice
To validate the in vivo efficacy of 14-25-9, we used the HNSCC PDX models derived from Pat1 and Pat2. On day 0, PDX-bearing NSG mice were divided into three groups (n=5, keeping similar average of PDX size among groups) and mice were given sub-lethal x-ray irradiation (250cGy) to suppress residual NSG-immune system (45). On day 1, 5×106 NK92-NKp44-1-GFP cells were infused IV into two groups while the 3rd group received no NK cells. To sustain human NK cell viability, groups were treated with SC injection of human IL2 (10μg/mouse/dose) in three doses on day 1, 3 and 5 (45), including the group without inoculated NK cells (as control for IL2 effect). For the two NK-inoculated groups, one group received 15mg/Kg of 14-25-9 mAb IV on day 1 and then every other day for 10 days, while the 2nd group received 15mg/Kg mouse IgG1 (Fig.6F). Tumor volumes were measured on day 1 and then every other day until day 10. For Pat1, with the treatment of 14-25-9, tumor growth was slowed compared to both no NK (*, p<0.01) and NK+IgG1 (**, p<0.05) at day 10 (Fig 6G). Similar results were obtained for Pat2 (Fig 6H). Note that upon IL2 removal, some tumor re-growth was observed in the 14-25-9 treated group reflecting the IL2 dependence of functional NK even with the presence of 14-25-9.
On day 10, tumors were excised from treatment and vehicle groups and stained for Ki67. For Pat1 there was a significant reduction in Ki67 expression in the PDXs of 14-25-9 treated group compared to mIgG1 (Fig.6I–J). For Pat2, PDXs in the 14-25-9 treated group manifested significantly reduced Ki67 staining as compared to mIgG1 group (Fig.6K–L). Higher magnification of Ki67 stained images of representative tumors are shown in Supplementary Fig.S3B. These results correlate with the higher 14-25-9 staining of Pat1-PDX-derived tumor cells as compared to Pat2 (Fig.2B).
Discussion
Many cancer types harness ICs to present immune resistance (1,2,27). Monoclonal antibody-based blocking of ICs demonstrated success in treatment of several types of cancers (46–48). Cancer-associated ICs involving the immune receptors CTLA4 and PD1 have been documented, particularly for tumor-specific T cells. ICs that are more associated with NK cells have been also reported, e.g. KIR-HLA-C, NKG2A-HLAE and NKp44-membrane-associated PCNA (14,29,31). NK cells display rapid and potent immunity against metastatic or hematological cancers (7,8). With the success of immune-checkpoint inhibitors primarily targeting T cells, efforts are now being undertaken to exploit NK cell antitumor properties (39,40,49). Our group and others have reported that PCNA expressed on the surface of cancer cells can interact with NKp44 to inhibit NK cell antitumor function and thus act as an immune-checkpoint. We also reported that it is the NKp44-isoform 1 that mediates this checkpoint and not isoforms 2 and 3 of NKp44 (30,31,39); A blocker for this IC has been unavailable. Immune-checkpoints, shaped by ligand-receptor interactions, can be modulated by antibodies targeting either receptor or ligand. In the present study, we have developed a mAb against human PCNA, named 14-25-9, that can recognize cell surface PCNA on the surface of solid and leukemic cancer cell lines as well as tumor cells from PDXs and blocks the NKp44-PCNA IC.
PCNA is overexpressed in proliferative cells of all cancer types. Cancer virulence is associated with PCNA overexpression (35–37). In both standard and pathological laboratory settings, mAb PC10 is used to detect PCNA (34). We observed that our anti-PCNA, 14-25-9, recognizes total cellular PCNA in Western blots from several cancer cell lines, similar to PC10. 14-25-9 also recognized PCNA on the cancer cell surface membrane, which PC10 was unable to do. EL4 cells transfected to overexpress human PCNA, which are not stained by PC10 (25), show membrane staining with 14-25-9. PCNA is expressed mainly in the nucleus of healthy replicating cells and has a role in DNA replication and repair (32). PCNA also exists as a cytoplasmic and a cell surface protein in tumor cells (38,50,51). Witko-Sarsat et al. reported that relocation of PCNA from the nucleus into the cytoplasm occurs in mature neutrophils and that cytoplasmic PCNA plays an anti-apoptotic role via interacting with procaspases (52). Bouayad et al. also described a nuclear export signal in the PCNA sequence, which is exposed only when PCNA is monomeric. Nuclear export of PCNA involves chromosome region maintenance 1 exportin (53). Ohayon et al. reported that cytoplasmic PCNA enhances glycolytic metabolism by building a protein scaffold in AML blast cells and thus increasing their survival (51). In all of these studies, cytoplasmic PCNA was detected by polyclonal PCNA antibody. Here we identified one PCNA mAb (14–25–9) that can recognize both non-nuclear and membrane-associated PCNA of several cancer cell types in cell lines and cancer biopsies. We further showed that it is PCNA that 14-25-9 recognizes on the membrane. 14-25-9 can inhibit chimeric NKp44 receptor binding with plate bound human PCNA in a dose-dependent manner; PC10 failed to do so. We then demonstrated that 14-25-9 can overcome IC inhibitory potential by enhancing antitumor responses by NK cells both in vitro and in vivo.
A cytokine environment enriched with IL2, IL15 and IL12 and with NCRs interacting with tumor ligands stimulates mature NK cells (CD56dimCD16+) to produce IFNγ to suppress cancer cells (54). In a variety of tumor cell lines, IFNγ suppresses tumor angiogenesis and induces TRAIL- and FasL-mediated cellular susceptibility to apoptosis (55). Our hypothesis that mAb 14-25-9 could function as an IC blocker was validated, as we observed that NK92-NKp44-1 cells had increased IFNγ secretion when co-incubated with several solid and leukemic cancer cell lines in the presence of different doses of 14-25-9 mAb. We also observed this phenomenon in IL2 cultured human primary NK cells, which predominantly express the NKp44-1 receptor isoform (39). Among the three splice variants of NKp44 receptor (i.e. NKp44-1, NKp44–2, and NKp44–3), only NKp44-1 carries the ITIM-like sequence that transduces inhibitory signaling within NK cells upon interaction with cell surface PCNA on cancer cells (29). This effect is consistent with our observation of increased IFNγ secretion by primary NK cells as well as NK92-NKp44-1 cells in the presence of 14-25-9 mAb through blockade of inhibitory signaling. When tested with NK92-NKp44–3 effector cells and EL4 or EL4-PCNA clone-1 target cells, no increase in IFNγ secretion was observed following treatment with 14-25-9, confirming the specificity of the 14-25-9 antibody for blocking the interaction between membrane PCNA and NKp44-1. Nuclear factor-κB (NF-κB) is a transcription factor that plays a role in receptor-mediated NK cell effector functions (56). It has been reported that patient NK cells deficient for NF-κB essential modulator (NEMO) and inhibitor of κB (IκB) kinase β (IKKβ), exhibit impairment in IFNγ production and cytotoxic function. We observed that 14-25-9 mAb was capable of increasing phosphorylation of NF-κB within NK92-NKp44-1 cells when exposed to HeLa cells for only 30min. Although 14-25-9 mAb did not increase NK cell cytotoxicity towards other adherent cell lines, some cytotoxicity was observed against EL4-PCNA clone1 and 721.221-Cw6 cell lines by NK92-NKp44-1cells compared to only EL4 and 721.221 cells. These results correlate with the negative cell surface staining of EL4 and 721.221 cells by 14-25-9. In the case of EL4-PCNA clone1, the transfection-mediated overexpression of human PCNA in EL4 mediated the expression of membrane PCNA. In the case of 721.221-Cw6, PCNA binds to HLA-Cw6 and HLA class I is associated with expression of membrane PCNA, thus 721–221-Cw6 is positive for membrane PCNA as compared to 721.221 (30, 33). Since NKp44 is not expressed in mouse (39), we used PDX-bearing NSG mice to confirm that mAb 14-25-9 injected in the circulation can enhance in vivo antitumor activity of intratumorally-injected NK92-NKp44-1-GFP cells, as well as patient autologous NK cells, as measured in terms of CD107a expression. Furthermore, systemic treatment of PDX-bearing mice with 14-25-9, in the presence of NK92-NKp44-1 cells prevented the growth of the PDX, confirming the role of this NK-innate checkpoint as well as the blocking capacity of 14-25-9.
To recapitulate, we have produced an IC blocking monoclonal antibody against human PCNA, named 14-25-9, that can recognize membrane-associated PCNA of cancer cells and can block interaction of the inhibitory NKp44 receptor with PCNA. 14-25-9 can enhance the functional activity of both human primary NK cells and the NK-92 cell line overexpressing NKp44-Iso 1. 14-25-9 can halt tumor growth in PDX bearing NSG mice. In future, combining this monoclonal antibody with other IC blocker antibodies or targeted therapeutics could be a promising in treatment of cancer.
Supplementary Material
Acknowledgements:
We want to acknowledge Drs. Marco Colonna and Alexander Barrow, Washington University, USA for providing us the EL4-PCNA clones. We give special thanks to Dr. Alon Zilka for technical assistance in ProteOnarray, Dr. Uzi Hadad for technical assistance in ImageStream and Tatiana Rabinsky for providing valuable technical assistance to the undertaking of the research summarized in this article. This work was supported by the Israel Science Foundation grant 1188/16 (A. Porgador), the US/Israel Binational Science Foundation grant 2015344 (A. Porgador), the Israeli Ministry of Science and Technology/ German Cancer Research Center program Grant CA172 (A. Porgador), the Israeli Ministry of Science and Technology grant 54180 (A. Porgador), Israel Science Foundation grant 1700/16 (M. Elkabets), and NCI Comprehensive Cancer Center Support Grant CA06927 (FCCC). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. We would like to acknowledge Kreitman School of Advanced Graduate Studies (Ben-Gurion University of the Negev, Israel) for supporting with Ph.D. and short term post-doctoral scholarship to K. Kundu.
Funding-
This work was supported by the Israel Science Foundation grant 1188/16 (A. Porgador), the US/Israel Binational Science Foundation grant 2015344 (A. Porgador), the Israeli Ministry of Science and Technology/ German Cancer Research Center program Grant CA172 (A. Porgador), the Israeli Ministry of Science and Technology grant 54180 (A. Porgador), Israel Science Foundation grant 1700/16 (M. Elkabets), NCI Comprehensive Cancer Center Support Grant CA06927 (FCCC). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Disclosure of Potential Conflicts of Interest: No potential conflicts of interest were disclosed.
References-
- 1.Marin-Acevedo JA, Dholaria B, Soyano AE, Knutson KL, Chumsri S, Lou Y. Next generation of immune checkpoint therapy in cancer: new developments and challenges. Journal of Hematology & Oncology 2018;11(1):39 doi 10.1186/s13045-018-0582-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nature reviews Cancer 2012;12(4):252–64 doi 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hanahan D, Weinberg Robert A. Hallmarks of Cancer: The Next Generation. Cell 2011;144(5):646–74 doi 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 4.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. New England Journal of Medicine 2010;363(8):711–23 doi 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Peggs KS, Quezada SA, Chambers CA, Korman AJ, Allison JP. Blockade of CTLA-4 on both effector and regulatory T cell compartments contributes to the antitumor activity of anti–CTLA-4 antibodies. The Journal of Experimental Medicine 2009;206(8):1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, et al. Nivolumab plus Ipilimumab in Advanced Melanoma. New England Journal of Medicine 2013;369(2):122–33 doi 10.1056/NEJMoa1302369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Langers I, Renoux VM, Thiry M, Delvenne P, Jacobs N. Natural killer cells: role in local tumor growth and metastasis. Biologics : Targets & Therapy 2012;6:73–82 doi 10.2147/BTT.S23976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tarazona R, Duran E, Solana R. Natural Killer Cell Recognition of Melanoma: New Clues for a More Effective Immunotherapy. Frontiers in immunology 2016;6(649) doi 10.3389/fimmu.2015.00649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lanier LL. Up on the tightrope: natural killer cell activation and inhibition. Nature Immunology 2008;9:495 doi 10.1038/ni158110.1038/ni1581https://www.nature.com/articles/ni1581#supplementary-informationhttps://www.nature.com/articles/ni1581#supplementary-information . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moretta A, Biassoni R, Bottino C, Mingari MC, Moretta L. Natural cytotoxicity receptors that trigger human NK-cell-mediated cytolysis. Immunology Today 2000;21(5):228–34 doi 10.1016/S0167-5699(00)01596-6. [DOI] [PubMed] [Google Scholar]
- 11.Kohrt HE, Thielens A, Marabelle A, Sagiv-Barfi I, Sola C, Chanuc F, et al. Anti-KIR antibody enhancement of anti-lymphoma activity of natural killer cells as monotherapy and in combination with anti-CD20 antibodies. Blood 2014;123(5):678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Romagné F, André P, Spee P, Zahn S, Anfossi N, Gauthier L, et al. Preclinical characterization of 1–7F9, a novel human anti–KIR receptor therapeutic antibody that augments natural killer–mediated killing of tumor cells. Blood 2009;114(13):2667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McWilliams EM, Mele JM, Cheney C, Timmerman EA, Fiazuddin F, Strattan EJ, et al. Therapeutic CD94/NKG2A blockade improves natural killer cell dysfunction in chronic lymphocytic leukemia. OncoImmunology 2016;5(10):e1226720 doi 10.1080/2162402X.2016.1226720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Andre P, Denis C, Soulas C, Bourbon-Caillet C, Lopez J, Arnoux T, et al. Anti-NKG2A mAb Is a Checkpoint Inhibitor that Promotes Anti-tumor Immunity by Unleashing Both T and NK Cells. Cell 2018. doi 10.1016/j.cell.2018.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koch J, Steinle A, Watzl C, Mandelboim O. Activating natural cytotoxicity receptors of natural killer cells in cancer and infection. Trends in Immunology 2013;34(4):182–91 doi 10.1016/j.it.2013.01.003. [DOI] [PubMed] [Google Scholar]
- 16.Kruse Philip H, Matta J, Ugolini S, Vivier E. Natural cytotoxicity receptors and their ligands. Immunology and Cell Biology 2013;92(3):221–9 doi 10.1038/icb.2013.98. [DOI] [PubMed] [Google Scholar]
- 17.Allcock Richard JN, Barrow Alexander D, Forbes S, Beck S, Trowsdale J. The human TREM gene cluster at 6p21.1 encodes both activating and inhibitory single IgV domain receptors and includes NKp44. European Journal of Immunology 2003;33(2):567–77 doi 10.1002/immu.200310033. [DOI] [PubMed] [Google Scholar]
- 18.De Maria A, Ugolotti E, Rutjens E, Mazza S, Radic L, Faravelli A, et al. NKp44 expression, phylogenesis and function in non-human primate NK cells. International Immunology 2009;21(3):245–55 doi 10.1093/intimm/dxn144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vitale M, Bottino C, Sivori S, Sanseverino L, Castriconi R, Marcenaro E, et al. NKp44, a Novel Triggering Surface Molecule Specifically Expressed by Activated Natural Killer Cells, Is Involved in Non–Major Histocompatibility Complex–restricted Tumor Cell Lysis. The Journal of Experimental Medicine 1998;187(12):2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hershkovitz O, Jivov S, Bloushtain N, Zilka A, Landau G, Bar-Ilan A, et al. Characterization of the Recognition of Tumor Cells by the Natural Cytotoxicity Receptor, NKp44. Biochemistry 2007;46(25):7426–36 doi 10.1021/bi7000455. [DOI] [PubMed] [Google Scholar]
- 21.Brusilovsky M, Radinsky O, Cohen L, Yossef R, Shemesh A, Braiman A, et al. Regulation of natural cytotoxicity receptors by heparan sulfate proteoglycans in ‐cis: A lesson from NKp44. European Journal of Immunology 2014;45(4):1180–91 doi 10.1002/eji.201445177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hershkovitz O, Rosental B, Rosenberg LA, Navarro-Sanchez ME, Jivov S, Zilka A, et al. NKp44 Receptor Mediates Interaction of the Envelope Glycoproteins from the West Nile and Dengue Viruses with NK Cells. The Journal of Immunology 2009;183(4):2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ho JW, Hershkovitz O, Peiris M, Zilka A, Bar-Ilan A, Nal B, et al. H5-Type Influenza Virus Hemagglutinin Is Functionally Recognized by the Natural Killer-Activating Receptor NKp44. Journal of Virology 2008;82(4):2028–32 doi 10.1128/JVI.02065-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rajagopalan S, Long EO. Found: a cellular activating ligand for NKp44. Blood 2013;122(17):2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barrow AD, Edeling MA, Trifonov V, Luo J, Goyal P, Bohl B, et al. Natural Killer Cells Control Tumor Growth by Sensing a Growth Factor. Cell 2018;172(3):534–48.e19 doi 10.1016/j.cell.2017.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Campbell KS, Yusa S-i, Kikuchi-Maki A, Catina TL. NKp44 Triggers NK Cell Activation through DAP12 Association That Is Not Influenced by a Putative Cytoplasmic Inhibitory Sequence. The Journal of Immunology 2004;172(2):899. [DOI] [PubMed] [Google Scholar]
- 27.Cantoni C, Bottino C, Vitale M, Pessino A, Augugliaro R, Malaspina A, et al. NKp44, A Triggering Receptor Involved in Tumor Cell Lysis by Activated Human Natural Killer Cells, Is a Novel Member of the Immunoglobulin Superfamily. The Journal of Experimental Medicine 1999;189(5):787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cantoni C, Ponassi M, Biassoni R, Conte R, Spallarossa A, Moretta A, et al. The Three-Dimensional Structure of the Human NK Cell Receptor NKp44, a Triggering Partner in Natural Cytotoxicity. Structure 2003;11(6):725–34 doi 10.1016/S0969-2126(03)00095-9. [DOI] [PubMed] [Google Scholar]
- 29.Shemesh A, Brusilovsky M, Kundu K, Ottolenghi A, Campbell KS, Porgador A. Splice variants of human natural cytotoxicity receptors: novel innate immune checkpoints. Cancer Immunology, Immunotherapy 2017. doi 10.1007/s00262-017-2104-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Horton NC, Mathew SO, Mathew PA. Novel interaction between proliferating cell nuclear antigen and HLA I on the surface of tumor cells inhibits NK cell function through NKp44. PloS one 2013;8(3):e59552 doi 10.1371/journal.pone.0059552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rosental B, Brusilovsky M, Hadad U, Oz D, Appel MY, Afergan F, et al. Proliferating cell nuclear antigen is a novel inhibitory ligand for the natural cytotoxicity receptor NKp44. Journal of immunology (Baltimore, Md : 1950) 2011;187(11):5693–702 doi 10.4049/jimmunol.1102267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kelman Z PCNA: structure, functions and interactions. Oncogene 1997;14:629 doi 10.1038/sj.onc.1200886. [DOI] [PubMed] [Google Scholar]
- 33.Naryzhny SN. Proliferating cell nuclear antigen: a proteomics view. Cellular and Molecular Life Sciences 2008;65(23):3789–808 doi 10.1007/s00018-008-8305-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bologna-Molina R, Mosqueda-Taylor A, Molina-Frechero N, Mori-Estevez AD, Sánchez-Acuña G. Comparison of the value of PCNA and Ki-67 as markers of cell proliferation in ameloblastic tumor. Medicina Oral, Patología Oral y Cirugía Bucal 2013;18(2):e174–e9 doi 10.4317/medoral.18573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li N, Deng W, Ma J, Wei B, Guo K, Shen W, et al. Prognostic evaluation of Nanog, Oct4, Sox2, PCNA, Ki67 and E-cadherin expression in gastric cancer. Medical Oncology 2014;32(1):433 doi 10.1007/s12032-014-0433-6. [DOI] [PubMed] [Google Scholar]
- 36.Woods AL, Hall PA, Shepherd NA, Hanby AM, Waseem NH, Lane DP, et al. The assessment of proliferating cell nuclear antigen (PCNA) immunostaining in primary gastrointestinal lymphomas and its relationship to histological grade, S + G2 + M phase fraction (flow cytometric analysis) and prognosis. Histopathology 1991;19(1):21–8 doi 10.1111/j.1365-2559.1991.tb00890.x. [DOI] [PubMed] [Google Scholar]
- 37.Yin S, Li Z, Huang J, Miao Z, Zhang J, Lu C, et al. Prognostic value and clinicopathological significance of proliferating cell nuclear antigen expression in gastric cancer: a systematic review and meta-analysis. OncoTargets and therapy 2017;10:319–27 doi 10.2147/OTT.S126551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.NS N, Hoyun L Proliferating cell nuclear antigen in the cytoplasm interacts with components of glycolysis and cancer. FEBS Letters 2010;584(20):4292–8 doi doi: 10.1016/j.febslet.2010.09.021. [DOI] [PubMed] [Google Scholar]
- 39.Shemesh A, Brusilovsky M, Hadad U, Teltsh O, Edri A, Rubin E, et al. Survival in acute myeloid leukemia is associated with NKp44 splice variants. Oncotarget 2016. doi 10.18632/oncotarget.8782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shemesh A, Kundu K, Peleg R, Yossef R, Kaplanov I, Ghosh S, et al. NKp44-Derived Peptide Binds Proliferating Cell Nuclear Antigen and Mediates Tumor Cell Death. Frontiers in immunology 2018;9(1114) doi 10.3389/fimmu.2018.01114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kundu K, Dutta K, Nazmi A, Basu A. Japanese encephalitis virus infection modulates the expression of suppressors of cytokine signaling (SOCS) in macrophages: Implications for the hosts’ innate immune response. Cellular Immunology 2013;285(1):100–10 doi 10.1016/j.cellimm.2013.09.005. [DOI] [PubMed] [Google Scholar]
- 42.Michael B, Olga R, Limor C, Rami Y, Avishai S, Alex B, et al. Regulation of natural cytotoxicity receptors by heparan sulfate proteoglycans in ‐cis: A lesson from NKp44. European Journal of Immunology 2015;45(4):1180–91 doi doi: 10.1002/eji.201445177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shemer-Avni Y, Kundu K, Shemesh A, Brusilovsky M, Yossef R, Meshesha M, et al. Expression of NKp46 Splice Variants in Nasal Lavage Following Respiratory Viral Infection: Domain 1-Negative Isoforms Predominate and Manifest Higher Activity. Frontiers in immunology 2017;8(161) doi 10.3389/fimmu.2017.00161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rodon J, Soria JC, Berger R, Batist G, Tsimberidou A, Bresson C, et al. Challenges in initiating and conducting personalized cancer therapy trials: perspectives from WINTHER, a Worldwide Innovative Network (WIN) Consortium trial. Annals of Oncology 2015;26(8):1791–8 doi 10.1093/annonc/mdv191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Miller JS, Rooney CM, Curtsinger J, McElmurry R, McCullar V, Verneris MR, et al. Expansion and homing of adoptively transferred human natural killer cells in immunodeficient mice varies with product preparation and in vivo cytokine administration: implications for clinical therapy. Biology of blood and marrow transplantation : journal of the American Society for Blood and Marrow Transplantation 2014;20(8):1252–7 doi 10.1016/j.bbmt.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sharma P, Allison JP. The future of immune checkpoint therapy. Science 2015;348(6230):56 doi 10.1126/science.aaa8172. [DOI] [PubMed] [Google Scholar]
- 47.Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science 2015;348(6230):69 doi 10.1126/science.aaa4971. [DOI] [PubMed] [Google Scholar]
- 48.Okazaki T, Honjo T. PD-1 and PD-1 ligands: from discovery to clinical application. International Immunology 2007;19(7):813–24 doi 10.1093/intimm/dxm057. [DOI] [PubMed] [Google Scholar]
- 49.Huang BY, Zhan YP, Zong WJ, Yu CJ, Li JF, Qu YM, et al. The PD-1/B7-H1 pathway modulates the natural killer cells versus mouse glioma stem cells. PloS one 2015;10(8):e0134715 doi 10.1371/journal.pone.0134715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.De Chiara A, Pederzoli-Ribeil M, Burgel P-R, Danel C, Witko-Sarsat V. Targeting cytosolic proliferating cell nuclear antigen in neutrophil-dominated inflammation. Frontiers in immunology 2012;3(311) doi 10.3389/fimmu.2012.00311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ohayon D, De Chiara A, Chapuis N, Candalh C, Mocek J, Ribeil J-A, et al. Cytoplasmic proliferating cell nuclear antigen connects glycolysis and cell survival in acute myeloid leukemia. Scientific Reports 2016;6:35561 doi 10.1038/srep3556110.1038/srep35561https://www.nature.com/articles/srep35561#supplementary-informationhttps://www.nature.com/articles/srep35561#supplementary-information . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Witko-Sarsat V, Mocek J, Bouayad D, Tamassia N, Ribeil J-A, Candalh C, et al. Proliferating cell nuclear antigen acts as a cytoplasmic platform controlling human neutrophil survival. The Journal of Experimental Medicine 2010;207(12):2631–45 doi 10.1084/jem.20092241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bouayad D, Pederzoli-Ribeil M, Mocek J, Candalh C, Arlet J-B, Hermine O, et al. Nuclear-to-cytoplasmic Relocalization of the Proliferating Cell Nuclear Antigen (PCNA) during Differentiation Involves a Chromosome Region Maintenance 1 (CRM1)-dependent Export and Is a Prerequisite for PCNA Antiapoptotic Activity in Mature Neutrophils. Journal of Biological Chemistry 2012;287(40):33812–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Arase H, Arase N, Saito T. Interferon gamma production by natural killer (NK) cells and NK1.1+ T cells upon NKR-P1 cross-linking. The Journal of Experimental Medicine 1996;183(5):2391–6 doi 10.1084/jem.183.5.2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kelly JM, Takeda K, Darcy PK, Yagita H, Smyth MJ. A Role for IFN-γ in Primary and Secondary Immunity Generated by NK Cell-Sensitive Tumor-Expressing CD80 In Vivo. The Journal of Immunology 2002;168(9):4472. [DOI] [PubMed] [Google Scholar]
- 56.Kwon H-J, Choi G-E, Ryu S, Kwon SJ, Kim SC, Booth C, et al. Stepwise phosphorylation of p65 promotes NF-κB activation and NK cell responses during target cell recognition. Nature Communications 2016;7:11686 doi 10.1038/ncomms1168610.1038/ncomms11686https://www.nature.com/articles/ncomms11686#supplementary-informationhttps://www.nature.com/articles/ncomms11686#supplementary-information . [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.