

Abstract
Metabolic inactivation of 1,25(OH)2D3 requires molecular recognition between the mitochondrial enzyme cytochrome P450 24A1 (CYP24A1) and its cognate redox partner Adrenodoxin (Adx). Recent evidence supports a model of CYP24A1 function in which substrate binding and Adx recognition are structurally linked. However, the details of this allosteric connection are not clear. In this study, we utilize chemical cross-linking coupled to mass spectrometry, nuclear magnetic resonance (NMR) spectroscopy, and CYP24A1 functional assays to inform a working model of a CYP24A1-Adx complex. We report that differential cross-linking internal to CYP24A1 point toward an Adx-induced conformational change that perturbs the F and G helices, which are required for substrate binding. Moreover, the modeled complex suggests that a semi-conserved nonpolar interaction at the interface may influence CYP24A1 regioselectivity. Taken together, these findings contribute to our understanding of Adx recognition in a critical vitamin-D inactivating enzyme as well as provide broader insight regarding the variability inherent in CYP-Adx interactions.
Keywords: Cytochrome P450, Adrenodoxin, Vitamin-D, Chemical cross-linking, NMR, Allostery
GRAPHICAL ABSTRACT
INTRODUCTION
Membrane-associated cytochrome P450 enzymes (CYPs) play integral roles in the homeostatic maintenance of bioactive vitamin-D. In mitochondria, the enzyme CYP27B1 catalyzes 1α-hydroxylation of 25(OH)D3 to produce the active hormone, 1,25(OH)2D3.1 The multi-functional enzyme CYP24A1 then mediates deactivation of 1,25(OH)2D3 by carrying out carbon-23 or carbon-24 hydroxylation reactions.2–5 Subsequently, CYP24A1 also catalyzes four additional reactions that produce either water-soluble calcitroic acid (via the carbon-24 pathway) or 1,25(OH)2D3-26,23-lactone (via the carbon-23 pathway). The latter product is of particular interest for therapeutic reasons, as it is known to mediate bone resorption by acting as an antagonist of the vitamin-D receptor.6, 7
Variable site-specific hydroxylation of 1,25(OH)2D3 between species appears to be based in part on the nature of a single residue in the CYP24A1 active site; an alanine at residue 326 correlates with the carbon-24 hydroxylation reaction, whereas a glycine in the same position correlates with carbon-23 hydroxylation.8 While the identification of an active site residue that affects regioselectivity likely points toward reorientation of the substrate, it does not fully explain why some CYP24A1 isoforms that contain a glycine at position 326 are either complete carbon-24 hydroxylases (rat) or dual carbon-23/carbon-24 hydroxylases (human).
We have developed a working hypothesis that an external factor, in the form of molecular recognition of the cognate electron donor protein Adrenodoxin (Adx), also influences the regioselectivity of the reaction. This hypothesis is supported by two of our previous studies. First, we utilized protein NMR to demonstrate that CYP24A1 enzymes from species that are known to display distinctive substrate regioselectivity also rely on different secondary interactions to bind Adx.9 Subsequently, we also demonstrated that in a carbon-24 hydroxylase system (from rat), allosteric communication exists between the active site and the proximal Adx recognition surface of CYP24A1.10
A comprehensive understanding of the CYP24A1-Adx interface and how it influences CYP24A1 function (aside from electron delivery) would help address these unanswered questions. However, structural details of the CYP24A1-Adx redox complex, and of CYP-Adx protein complexes in general, remain unclear. Existing information is extrapolated from either of two sources; the crystal structure of CYP11A1 expressed as a fusion with Adx, in which orientation of Adx may be influenced by the presence of the protein linker,11 and from protein docking of CYP11B1 and CYP11B2 with a dimer of Adx.12 We should note that an ultracentrifugation study of full-length Adx indicated that in the reduced (productive) state, Adx occupies a monomeric form.13 Therefore, the structural details of a functional, unconstrained, and stoichiometric (1:1) complex of a mitochondrial CYP with Adx remain unclear.
In this study, we aim to understand molecular recognition in a functional 1:1 complex of rat CYP24A1 (a carbon-24 hydroxylase) with full-length rat Adx. We report the use of comparative chemical cross-linking using the zero-length cross-linker 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC) coupled to mass spectrometric analysis of the isolated 1:1 complex. We identify a pattern of inter protein cross-linked peptides that, when combined with site-directed mutagenesis and functional CYP24A1 assays, support the presence of a distinct mode of Adx recognition from those described previously. Moreover, we found that the pattern of intra-protein cross-linking in CYP24A1 is sensitive to the presence of Adx or to the combined presence of 1,25(OH)2D3 and Adx, consistent with induction of conformational changes in the CYP. These findings are discussed in the context of a working model of the CYP24A1-Adx complex.
MATERIALS AND METHODS
Protein Expression and Purification –
The gene encoding full-length rat Adx (GenBank accession number NP_05882.1) containing a C-terminal six histidine tag was custom-synthesized (Genscript) and cloned into a pET-15b expression vector (Novagen). Overexpression and purification of unlabeled and 15N labeled Adx were carried out as described previously.10 Purity was determined by a A415/A280 ratio greater than 0.8. Adx concentration was calculated using an extinction coefficient for the iron cofactor of 11 mM−1 cm−1. Point mutations of rat Adx were also generated by Genscript and were purified identically to the wild-type protein. The generation of recombinant rat CYP24A1 (GenBank accession number NP_963966.1) was also carried out as described previously.10 Briefly, the CYP24A1 gene was designed similar to the structural reference 14 with a S57D mutation and without the 32 residue leader sequence, and cloned into a pTrc vector (Novagen). Recombinantly expressed CYP24A1 was extracted from membrane using 1% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS), then bound to an Adx affinity column under low salt conditions (10 mM potassium phosphate, 20% glycerol, 0.5% CHAPS, 2 mM b-mercaptoethanol, pH 7.4) and eluted under high salt conditions (500 mM potassium phosphate, 300 mM NaCl, 20% glycerol, 0.5% CHAPS, 2 mM b-mercaptoethanol, pH 7.4). CYP24A1 purity was determined by a Rz ratio (A417/A280) greater than 1.0. The bovine adrenodoxin reductase used in reconstituted activity assays was also prepared as described by Sagara et al.15 Lastly, CYP11B1 and CYP11B2 enzymes, along with bovine Adx and bovine Adx L80K, were gifts from Dr. Richard Auchus at the University of Michigan.
Chemical Crosslinking –
Purified rat CYP24A1 was incubated with Adx under low-salt conditions (10mM potassium phosphate buffer, .025 % CHAPS, pH 7.4) and in the presence of the zero-length cross-linker EDC (2mM working concentration) (Thermo Scientific). Due to instability of isolated CYP24A1 at the low salt concentrations necessary to promote the electrostatic interaction with Adx, Adx was included prior to buffer exchange. 2-10 uM of CYP24A1 was combined with 40 uM Adx or mutants in a 20-ul reaction volume. After a 2-hour incubation period at 25°C, the reaction was terminated by addition of an equal volume of Laemmli loading dye (Bio-Rad). The samples were then resolved by gel electrophoresis and stained with Coomassie Blue. Substrate bound samples were prepared by step-wise additions of 1,25(OH)2D3 (ApexBio) in 5 nmole increments to 200 pmoles of CYP24A1 to reach a final substrate concentration of 40 μM.
Mass Spectrometry –
Gel bands containing the crosslinked product were excised and subjected to an in-gel digestion procedure. In brief, gel bands were first diced into 1 mm3 cubes, washed by water and de-stained by 1 ml 50% pH 8.4 Tris-formic acid (FA)/acetonitrile (ACN) overnight. De-stained gel cubes were completely dehydrated by ACN (1 ml for 10 mins, 3 times), and reduced in 500 ul 10 mM dithiothreitol (DTT) at 56 °C for 30 mins. Protein was then alkylated in 500 ul 25 mM iodoacetamide at 37 °C in darkness for 30 mins. Next, gel cubes were incubated with 300 ul 0.0125 ug/ul MS grade chymotrypsin (Pierce) for 30 min on ice. Chymotrypsin solution was then replaced by 200 uL pH 8.4 Tris-FA and gel cubes were incubated at 37 °C overnight for protein digestion. To note, a 10-min dehydration procedure by 1 ml ACN and air-drying were required between every two steps. Protein digestion was terminated by addition and incubation with 20 ul 5% FA for 15 mins, and liquid was transferred to a new tube. 500 ul 50% pH 8.4 Tris-FA/ACN and 500 ul ACN were then added sequentially to completely squeeze liquid out of the gel cubes. The liquid was combined in the same tube and dried by SpeedVac. Digested peptides were then re-suspended by 50 uL 0.05% trifluoroacetic acid in 2% ACN, centrifuged at 18,000 g, 4 °C for 30 mins, and transferred to LC vials for LC-MS analysis.
The LC-MS system consists of a Dionex Ultimate 3000 nano LC system, a Dionex Ultimate 3000 micro LC system with a WPS-3000 autosampler, and an Orbitrap Fusion Lumos mass spectrometer. A large-inner diameter (i.d.) trapping column (300-um i.d. x 5 mm) was implemented before the nano LC column (75-um i.d. x 65 cm, packed with 2.5-um Xselect CSH C18 material) for high-capacity and selective sample loading and delivery. For each sample, 4 ul of derived peptides were injected for LC-MS analysis. Mobile phases A and B were 0.1% FA in 2% ACN and 0.1% FA in 88% acetonitrile. The 180-min LC gradient profile was: 4% B for 3 mins, 4–11% B for 5 mins, 11–32% B for 117 mins, 32–50% B for 10 mins, 50–97% B for 5 mins, 97% B for 7 mins, and then equilibrated to 4% for 27 mins. The mass spectrometer was operated under data-dependent acquisition (DDA) mode with a maximal duty cycle of 3 s. MS1 spectra were acquired by Orbitrap under 120k resolution for ions within the m/z range of 400 – 1500. Automatic Gain Control and maximal injection time were set at 5E5 and 50 ms, and dynamic exclusion was also applied (45 s, ± 10 ppm). Precursor ions with a charge state of 2 – 7 were filtered by quadrupole based on intensity using a 1.2-Th wide window and fragmented by high-energy collisional dissociation (HCD) at a normalized energy of 30%. MS2 spectra were acquired by Orbitrap under 15k resolution. Detailed LC-MS setting and relevant information can be found in Shen et al.16
LC-MS files were matched against a contatenated protein sequence database which contains CYP24A1, Adx and Swiss-Prot Escherichia coli protein sequences, using the Sequest HT engine embedded in Proteome Discoverer 1.4 (Thermo Fisher Scientific). Search parameters include: 1) peptide length: 6 – 144; 2) maximal missed cleavages: 2; 3) mass tolerance: 10 ppm for precursors, 0.02 Da for fragments; 4) dynamic modifications: peptide N-terminal acetylation, methionine oxidation; 5) static modification: cysteine carbamidomethylation. The target-decoy searching approach was employed for false positive control. Search results were combined by Scaffold 4.8.4 (Proteome Software, Inc.) for peptide filtering, protein inference, false-discovery rate estimation, and peptide quantification. To calculate the peptide ratios between different samples, quantitative results were exported from Scaffold, and all peptides identified were divided into two groups, one with lysines and the other without lysines. The sum intensities of peptides without lysines were used as the internal standard in each sample, against which the intensities of each peptide were normalized. Ratios and p-values (by Student’s t-test) for lysine-containing peptides were calculated separately, and peptides with a ratio of < 0.5 and a p-value of < 0.05 were determined to be altered between the groups compared. To further identity potential crosslinking sites, pLink 2.3 was employed following the standard workflow and parameters.17 Results were exported and manually curated in Microsoft excel, and crosslinked peptide spectra were exported from pLabel 2.4.
NMR –
1H-15N HSQC data were acquired at 25°C on a Bruker Avance 800 MHz spectrometer using samples of uniformly labeled 15N-Adx and 15N-Adx containing the L80Q and L80K mutations. NMR buffers consisted of 50 mM potassium phosphate (pH 7.4), 50 mM NaCl and 5% D2O. The concentration of labeled protein was kept constant at 100 μM. Acquisition parameters consisted of 4 scans over 128 increments. Spectra were processed using nmrPipe18 and analyzed with NMRViewJ19. The 15N chemical shift assignments that were determined previously10 were used to analyze peak broadening in response to addition of 0.2 molar equivalents of CYP24A1. The addition of ligand-free CYP24A1 was also carried out as described previously.9
CYP24A1 Functional Assays –
Reconstituted CYP24A1 functional assays were carried out in triplicate 100-ul volumes and contained equal concentrations (1.5 μM) of rat CYP24A1, rat Adx, and bovine adrenodoxin reductase. 1,25(OH)2D3 was kept constant at 5 μM. Final assay conditions consisted of assay buffer (50 mM potassium phosphate) and trace amounts of CHAPS from addition of stock CYP24A1 (0.035 %). The reaction mixtures were first pre-incubated in assay buffer for 5 minutes at 37°C, then initiated by the addition of NADPH to a final concentration of 1 mM. After a further 4 minutes incubation at 37°C, the reactions were quenched by addition of two volumes of methanol. 25(OH)D3 was added as an internal standard to a final concentration of 1 μM. Samples were then subjected to centrifugation at 8,000 g for 30 minutes. 150 ul of the supernatant was removed and diluted with additional methanol to a final volume of 200 ul. Metabolites were resolved by liquid chromatography using an Agilent 1260 Infinity II liquid chromatography system with a Poroshell 120 EC-C18 column (4.6 mm X 250 mm) and using an isocratic mobile phase of 70% acetonitrile. Elution peaks were detected by monitoring absorbance at 264 nm. The remaining substrate for each sample was calculated based on a peak volume ratio of the internal standard. Fractional depletion was then calculated as a ratio of substrate concentration from control reactions that were quenched prior to addition of NADPH. Statistical significance of differences between WT and mutant Adx reactions was determined using a one-way ANOVA test and an ANOVA post-test (GraphPad), using a significance level of α of 0.05. Final values are represented as boxplots generated using Interactive Dotplot.20
Molecular Dynamics –
The rat CYP24A1 sequence was modeled onto the rat CYP24A1 X-ray crystal structure (PDB ID: 3K9V) using the MODELLER21 interface in UCSF Chimera.22 The model was checked using ModRefiner yielding a TM-Score of 0.9948 and RMSD of 0.568.23 This model was parameterized using the GROMOS 54A7 force field.24 The resulting topology files were modified to reflect the cysteinyl ligation to heme25, 26 and immersed in a simulated cubic water box 11.951 nm per side containing ~55000 waters and five Cl- ions to balance charge. The system was energy minimized by the method of steepest descent to remove Van der Waals contact between overlapping waters and the amino acids of the protein. Constant volume and temperature (NVT) equilibration was performed using V-rescale temperature coupling27 at 300 K for 100 ps followed by 1000 ps of constant pressure and temperature (NPT) equilibration using V-rescale temperature coupling and the Particle-Particle Particle-Mesh algorithm with analytical derivative (P3M-AD) pressure coupling28 at 300 K and periodic boundary conditions in all directions. During the NPT equilibration, the protein was position restrained to allow waters to fill cavities. An unrestrained production simulation of 50 ns was performed using V-rescale temperature coupling and P3M-AD pressure coupling at 300 K on the University of Connecticut High Performance Computing Center in Storrs. To approximate chemical crosslinking observed within CYP24A1 (Glu-151:Lys-459 and Glu-154:Lys-459), separate 25 ns pulling simulations were performed using an umbrella potential of 5000 kJ mol-1 nm-2 and 0.3 nm between one glutamate epsilon oxygen and the lysine zeta nitrogen in the pair of residues. In parallel, an unbiased 25 ns simulation without pulling was performed as a control.
In silico modeling and protein-protein docking –
Docking of the CYP24A1, Adx complex was performed using the HADDOCK 2.2 server29 and was carried out using the NMR informed homology model of rat Adx described previously,10 along with either chain A of the rat CYP24A1 (PDB ID 3K9Y)14 or the 25 ns dynamics simulation of CYP24A1 in which Glu-154 has been constrained with Lys-459 (CYP24A1154-459). The constraints used were based on cross-linking results (CYP24A1:Lys-77 – Adx:Glu-109; CYP24A1:Lys-382 – Adx:Glu-35), previous NMR titrations,10 substrate depletion assays in the current study which indicate involvement of Asp-76 at the interface (CYP24A1:Arg-465 – Adx:Asp-76), and the requirement for a favorable alignment of the iron centers as required for productive electron transfer (CYP24A1:Cys-462 – Adx:Thr-49). The docking results obtained as clusters were evaluated based on 1) favorable HADDOCK scores, and 2) protein orientation consistent with the empirically derived restraints. Analysis of the complex, visualization, and distance calculations between interacting residues were performed using UCSF Chimera22.
RESULTS
Analysis of CYP24A1-Adx inter-protein cross-linking.
Productive molecular recognition leading to electron transfer between CYP24A1 and Adx requires electrostatically mediated contact between the proximal surface of CYP24A1 and one or more of the short anionic helices that frame the [2Fe2S] cluster of Adx. We’ve previously reported that the formation of the complex, as measured by solution NMR, can be disrupted by substitution of numerous surface charges of Adx.9, 10 In particular, disruption occurs when the highly conserved a-helix 3, the putative CYP recognition site, is made less anionic. However, the corresponding surface contacts on CYP24A1 remain unknown. One obstacle toward addressing this question is the fact that current protocols to purify recombinant CYP24A1 require the use of an Adx affinity column,14, 30 thus surface mutations of CYP24A1 designed to interrogate recognition of Adx cannot currently be isolated.
In order to address this question, we expanded our previous application of the zero-length cross-linker EDC and analyzed the cross-linked product by SDS-PAGE and mass spectrometry in order to identify inter-protein contacts. EDC is an effective way of trapping CYP, redox partner complexes,12, 31 owing to its reactivity with both the carboxylic groups of anionic redox proteins and the primary amines on the proximal surface of CYPs. We observed that, upon resolving the cross-linked product by SDS-PAGE, the complex appeared as a significant band at approximately 70 kDa (Figure 1a), consistent in size with 1:1 stoichiometric binding of rat CYP24A1 (56 kDa) with full-length rat Adx (14.5 kDa). Since the reduction of Adx is known to promote the monomeric form of the protein,13 it follows that the 1:1 complex likely involves inter-protein contacts that are functionally relevant. To prevent any false positives resulting from non-specific cross-linking between multimers of CYP24A1, the resolved 1:1 complex was excised from the polyacrylamide gel prior to chymotrypsin digestion and analysis by LC-MS Orbitrap. Two inter-protein cross-linked products were identified (Figures 1b and 1c); Lys-177 of CYP24A1 was linked to Glu-109 of Adx, and Lys-382 of CYP24A1 to Glu-35 of Adx.
Figure 1.
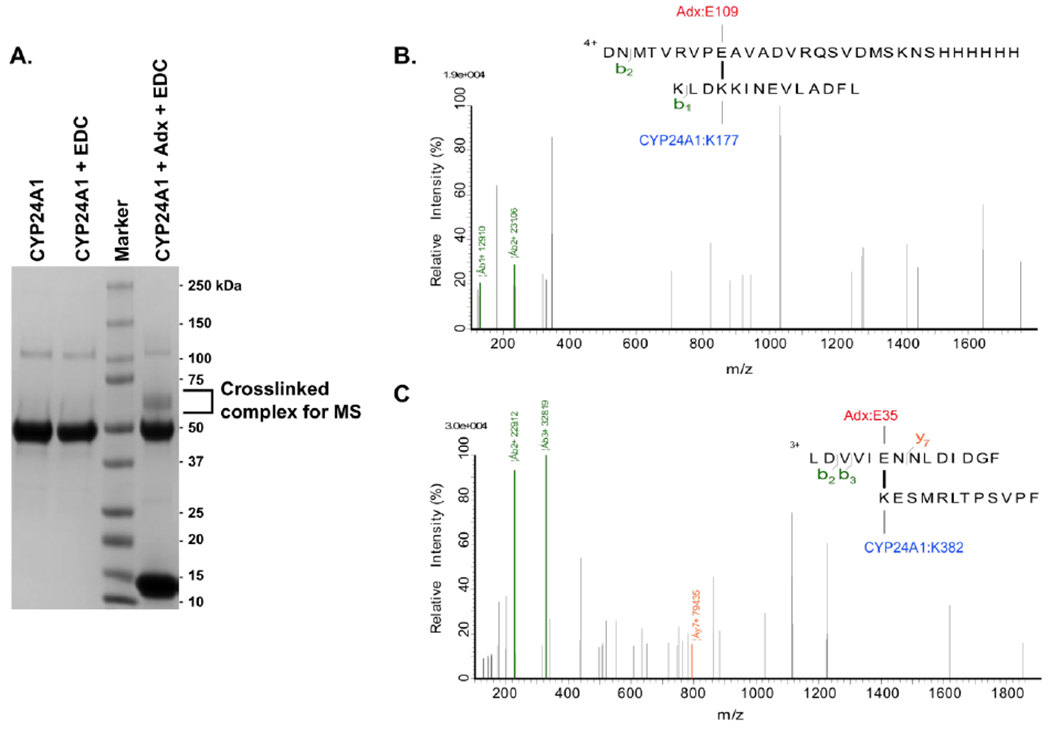
EDC and LC-MS identification of inter-protein cross-links between CYP24A1 and Adx. (A) SDS PAGE gel image of the purified cross-linked product of a 1:1 protein complex. (B) Cross-linked peptide products of Adx: Glu-109 linked to CYP24A1: Lys-177, and (C) ADX:Glu-35 linked to CYP24A1:Lys-382.
While Lys-177 may represent a peripheral binding site, Lys-382 is located at the center of the K-helix on the proximal (non-membrane) surface of CYP24A1. K-helix recognition of Adx has been reported previously for other CYPs.11, 12 In each of those studies, the K-helix residues were reported in contact with α-helix 3 of Adx. Interestingly, our results indicate a K-helix interaction with Glu-35 located on the first Adx α-helix, thus suggesting that CYP24A1 may recognize Adx in the 1:1 complex via a distinct binding mode from those described previously.
Intra-protein cross-linking of CYP24A1
In the course of analyzing the LC-MS peptides resulting from EDC cross-linking of the 1:1 redox complex, we also observed a pattern of internal CYP24A1 cross-linking (Table S1). All of the intra-linked peptides involve solvent-exposed carboxylic side chains and primary amines that appear to undergo some reactivity with EDC. This pattern included several cross-linked sites that are not positioned to form salt bridges in the CYP24A1 crystal structure.32 Since the products are from an isolated 1:1 complex, this raised some concerns that some of these products may result from denaturing of the CYP24A1 tertiary structure. However, there were also numerous cross-linked sites that correspond with native salt-bridges in CYP24A1. For example, the peptide containing Asp-105 forms a product with a peptide containing Lys-100. These side chains are closely associated as a salt-bridge on anti-parallel beta-strands in CYP24A1. Another example is a cross-linked site between Glu-213 and Lys-209, which are located one full turn apart and form a salt-bridge at the center of the E-helix.
In order to determine whether the presence of Adx effects internal cross-linking of CYP24A1, the peptides identified in the presence of Adx were compared with a control condition in which CYP24A1 alone was incubated with EDC for the same length of time. The CYP24A1-only reaction was then resolved by SDS-PAGE and the band excised for analysis by mass spec. Here we observed that several internal cross-linked sites occur in the presence and the absence of Adx. These include the two aforementioned salt-bridges in CYP24A1 as well as some of the non-native cross-linked products. However, we also noted the absence of several linked peptides in the vicinity of the CYP24A1 heme and the proximal surface that appear to be induced only when CYP24A1 is bound to Adx. Specifically, Glu-151 and Glu-154 at the N-terminal end of the C-helix are linked to Lys-459 of the proximal surface (Figure 2a). These cross-linked sites appear in three independent peptides when Adx is present, yet are not detected when Adx is withheld. In the CYP24A1 structure, the Glu-151 and Glu-154 side chains are located at distances of 11.5 and 17.2 Å away, respectively, from the side chain of Lys-459 (Figure 2b). Our interpretation of these results is that, while these side chains are not likely to interact in the CYP24A1 conformation represented in the crystal structure, binding of Adx promotes an alternative CYP24A1 conformation in which Glu-151 and Glu-154 are brought closer to Lys-459, thus increasing reactivity with EDC at these sites.
Figure 2.
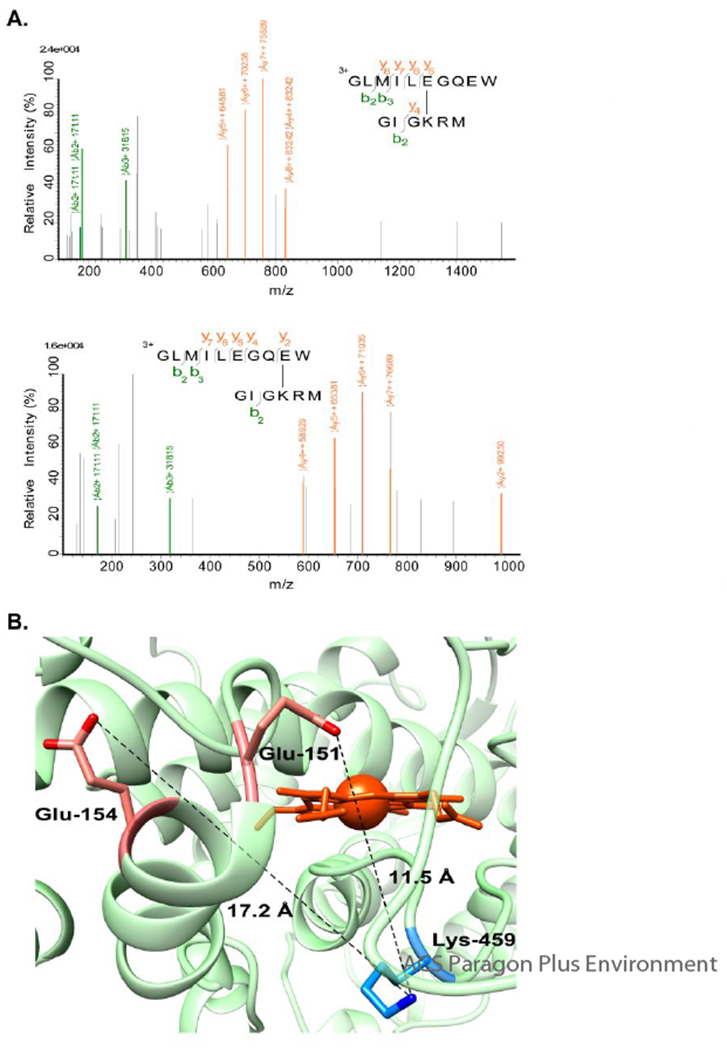
Intra-CYP24A1 EDC cross-linking induced by addition of Adx. (A) The CYP24A1 residue Lys-459 was identified covalently linked to residues Glu-151 and Glu-154. (B) Identified residues displayed in the crystal structure of CYP24A1 (PDB ID 3K9Y).
Next, we wanted to examine the effects of a conformational change in CYP24A1 that occurs in a manner that abides by the presence of a salt bridge between these residues. We carried out a set of 25 ns molecular dynamics simulations in which the CYP24A1 structure was allowed to adjust to energy minimized conformations either without bias or with the addition of either crosslinking derived salt bridge as constraints. The final modeled structures are designated as CYP24A1151-459, CYP24A1154-459 or CYP24A1unbiased. It should be noted that the available structures of rat CYP24A1 (PDB ID: 3K9V and 3K9Y) were crystallized in the presence of two molecules of CHAPS or CYMAL detergent positioned in what is likely the substrate access channel.32 As such, the beginning structures for the MD simulations represent a semi-open CYP24A1 structure in which the F-G helical cassette is oriented away from the distal side of the active site. In all simulations, the CYP24A1 structure adjusts into a closed conformation. Predictably, the constrained structures CYP24A1151-459 and CYP24A1154-459 also reflect a perturbation in the C-helix near Glu-151 and Glu-154. In contrast, the loop containing Lys-459 is largely unchanged, likely due to the covalent anchoring of this region by residue Cys-462, which is the axial coordination site for the heme cofactor.
Interestingly, in addition to local changes near the cross-linked sites, CYP24A1151-459 and CYP24A1154-459 also reflect long-distance changes in the global structure (Figure 3). These include repositioning of the H-helix, the I-helix, and more significantly, the F-G helical cassette. Figure 3a shows an overlay between CYP24A1unbiased and CYP24A1154-459. Significant changes occur in the F-helix, the short FG loop, and the beginning of the G helix, where the protein backbone in CYP24A1154-459 is pulled closer over the putative substrate access channel by approximately 3 Å. Another notable difference is the subtle repositioning of the M-helix on the proximal surface of CYP24A1. The M-helix contains two highly conserved arginine residues, Arg-465 and Arg-466, and has been reported previously as participating in recognition of Adx.11, 33–35 In this study, despite the absence of Adx in the simulation, this surface is repositioned farther toward the center of the active site cavity of CYP24A1154-459. In total, net differences result in a global compression of CYP24A1 toward a structure that is further closed than was observed in CYP24A1unbiased. An overlay of CYP24A1154-459 with the open structure (PDB 3K9Y) (Figure 3b) reflects the extent of the open-to-closed transition suggested by the data, including considerable reordering of the protein backbone and rearrangement of the central I-helix bordering the active site.
Figure 3.
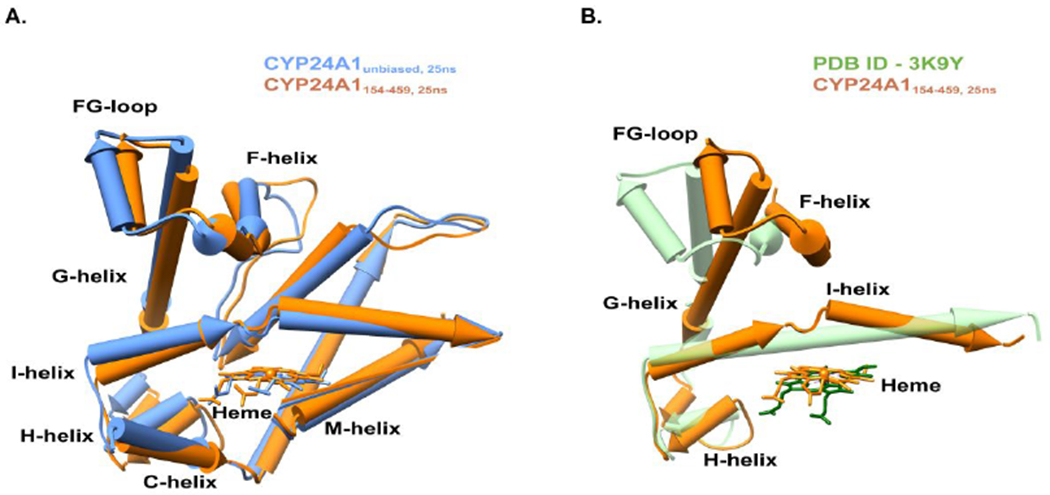
Comparison of CYP24A1 in the unbiased and constrained model after 25ns MD simulation. (A) The Cα-backbone RMSD for CYP24A1unbiased, 25ns vs CYP24A1154-459, 25ns is 2.5Å. An overlay of CYP24A1 without simulation (PDB ID −3K9Y) and in the constrained simulation (CYP24A1154-459, 25ns) is shown in panel (B), with a RMSD value of the Cα-backbone of 23.8Å. Reorganization of helices is notable for the I-helix, with a kink of 35° in the center, a 20° shift in the F-helix, and an 18° shift in the G-helix toward a closed conformation. For clarity, other helices were removed from the display.
1,25(OH)2D3 and Adx exert a coordinated change in CYP24A1 structure
The possibility of an Adx-induced change in CYP24A1 structure that leads to a closed conformation with a more restrictive active site environment, as suggested by the molecular dynamics simulations, can in theory link access or positioning of 1,25(OH)2D3 (substrate binding, step 1 in CYP catalysis) with electron delivery from Adx (step 2 in CYP catalysis). To further evaluate the presence of conformational coupling between these two events, we carried out an expanded analysis of EDC reactivity of CYP24A1 under substrate-bound and substrate-free conditions, as well as in the presence or absence of Adx. Gel bands containing EDC-treated CYP24A1 by itself or the 1:1 complex were isolated from three independent replicate reactions and analyzed by chymotrypsin digest and LC-MS. The ratios of peptides containing lysine were compared using the intensities of peptides without lysine as an internal standard for inter-sample normalization. Peptides with a ratio of < 0.5 and a p-value of < 0.05 were determined to exhibit significantly different levels. These data were then filtered with data from a reference condition in which 1,25(OH)2D3 had been withheld. While this process does not yield specific residue cross-links, peptides identified by this strategy are assessed as having been cross-linked by EDC in a manner dependent on the formation or disruption of native CYP24A1 salt bridges.
Here we observed that internal CYP24A1 cross-linking is not altered when CYP24A1 is bound to 1,25(OH)2D3, as no peptides were identified that were differentially quantified between substrate-free and substrate-bound samples (Figure 4a, left panel). However, analysis of the CYP24A1-Adx complex bound to 1,25(OH)2D3 resulted in the identification of four peptides (Figure 4b, summarized in Table S2) that were differentially modified when compared to the complex in the absence of substrate. 0ur interpretation is that these peptides represent sub-substructures that are further altered beyond the effects exerted by Adx alone. These peptides are mapped in Figure 4c. Some of the mapped regions form a near-contiguous surface on CYP24A1 that is encompassed by the EF loop, the N-terminal end of the F-helix, and the C-terminal end of the G-helix. This region is nearly contiguous with a region on the proximal surface consisting of the C-helix, D-helix bend. A fourth identified peptide maps onto the A-helix located opposite of the putative solvent access channel.
Figure 4.
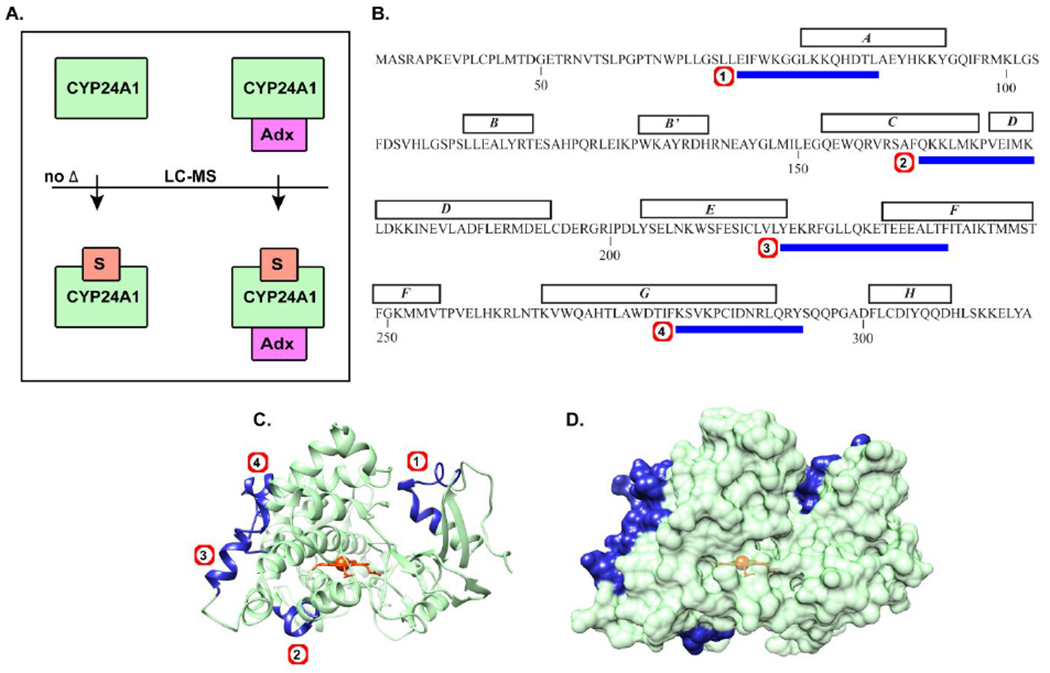
Adx and substrate exert a coordinated effect on CYP24A1 structure. (A) EDC reactivity of CYP24A1 was compared with and without substrate (left), where no difference was observed, and with or without substrate but in the presence of Adx (right), where four peptides (B) were differentially affected. Mapping of the peptides across the A, C-D, F, and G helices are shown in (C) and as a surface representation in (D).
Since the presence of 1,25(OH)2D3 alone did not induce any changes in this pattern, we conclude that the presence of substrate and Adx, when combined, promote further structural changes in these regions. Based on molecular dynamics simulations using specific cross-linked sites as constraints (Figure 3), we hypothesize that the transition represented by this experiment is actually an open-to-close change in the CYP24A1 structure. This theory is supported by a close comparison of these sites in the open structure (PDB 3K9V at 0 ns simulation) and the simulated closed structure (CYP24A1154-459 at 25 ns simulation), as shown in Supplemental Figure S1. In three of the peptides identified in this experiment, with the exception of the proximal C/D-helices, closing of CYP24A1 appears to require the breaking of a native salt bridge interaction, thus potentially impacting EDC reactivity at these sites. Therefore, identification of these specific peptides is in keeping with an open-to-close equilibrium that exhibits more “closed” conformations than when Adx is present alone.
Activity of CYP24A1 in response to surface mutations of Adx
Our efforts to use EDC cross-linking coupled to mass spectrometry resulted in fewer inter-protein contacts than expected (Figure 1). Notable for its absence in these data was any peptide representing α-helix 3 of Adx. The conserved anionic residues on α-helix 3, Asp-76 in particular, are generally acknowledged as necessary residues for electron transfer to CYP enzymes.12, 36, 37 Our previous NMR titration data also demonstrate the importance of a-helix 3 residues in the interaction with CYP24A1.9, 10 One possible reason for the absence of a-helix 3 peptides in these experiments is that in all samples, full coverage of CYP24A1 was not achieved. The peptide 455GIGKRMCIGRRL467 was routinely not detected. This sequence contains the heme coordination site, Cys-462, and two highly conserved arginines, Arg-465 and Arg-466, that are solvent-exposed and precede the M-helix. Clinical mutation of a residue corresponding to Arg-465 was found to correlate with loss of function in the related vitamin-D 1α-hydroxylase CYP27B1, most likely due to disruption of electron transfer.35 Therefore, we assess that this gap in CYP24A1 coverage correlates with the absence of Adx α-helix 3 peptides due to an inter-molecular interaction at these sites.
In light of these cross-linking results, and in order to confirm the anticipated role of Adx α-helix 3 in binding to CYP24A1, we carried out reconstituted CYP24A1 hydroxylation assays using a variety of charge-neutralizing mutations of Adx. These included the putative recognition site as well as additional non-conserved residues (Figure 5a). As expected, the mutant D76N of Adx resulted in significant disruption of CYP24A1 mediated depletion of 1,25(OH)2D3, thus supporting its role in electron transfer (Figure 5b). The combination of this mutation with D31N had a similar result. However, individual point mutations of α-helix 1, D31N and E35Q, also led to the statistically significant disruption of CYP24A1 function to a similar degree as the mutation of the a-helix 3 residue Asp-72, resulting in fraction depletion of the substrate of approximately half of that observed with WT Adx. In our assessment, this finding confirms at least a secondary role for α-helix 1 of Adx in binding CYP24A1. Two additional surface mutations of Adx, E47Q and E65Q, did not significantly disrupt CYP24A1 function.
Figure 5.
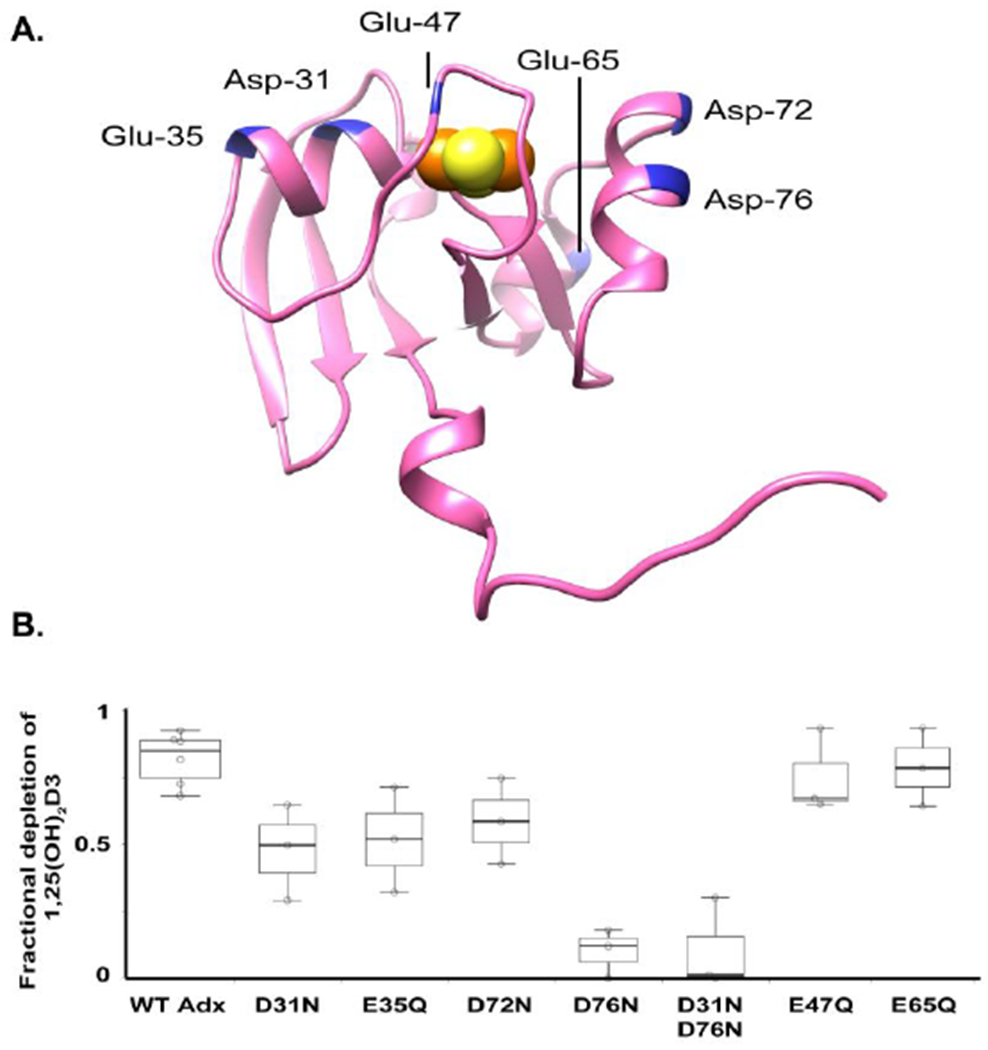
Effects of Adx mutations on CYP24A1-mediated depletion of 1,25(OH)2D3. (A) Distribution of anionic residues targeted for point mutations on rat Adx, including residues in the putative recognition site (Asp-72 and Asp-76), and residues on secondary surfaces (Asp-31 and Glu-35). (B) A plot of fractional depletion of 1,25(OH)2D3 by CYP24A1 for six single charge-neutralizing mutations and one double mutation.
Docking of the open and closed forms of CYP24A1 with Adx
Inter-protein crosslinking data points toward at least two contacts between CYP24A1 and Adx. These contacts suggest a general orientation of Adx on the proximal surface of CYP24A1 that is distinct from other descriptions of CYP-Adx complexes. For example, the involvement of residues on α-helix 1 of Adx is not evident from the co-structure of Adx with CYP11A1 or from modeling with the CYP11B enzymes.11, 12 However, our findings implicating this surface are supported by a previous observation that mutation of anionic side chains on α-helix 1 results in disruption of the complex as measured by NMR titrations10 as well as by a functional comparison of the enzyme activity in response to α-helix 1 mutations in this study (Figure 5).
To understand how the CYP24A1-Adx complex compares to other similar complexes in mitochondrial CYPs, we carried out a series of molecular docking experiments using the docking program HADDOCK 2.229 using both the open form of CYP24A1 (represented by PDB 3K9V) and the simulated closed form of the enzyme (CYP24A1151-459). Inter-molecular constraints were informed by the empirical data that include both of the inter-protein cross-linked side chains described earlier. An additional constraint was included based on the functional data in which mutation of Asp-76 of Adx led to significant disruption of the metabolism of 1,25(OH)2D3. Asp-76 was constrained in the docking experiment by an interaction with the surface residue Arg-465 of CYP24A1. This proximal surface residue is conserved in all mitochondrial P450 and its involvement at the interface has been reported previously.33–35 Moreover, the absence of any Adx a-helix 3 cross-linked peptides correlated with a gap in LC-MS coverage of CYP24A1 for a peptide containing Arg-465, which would be consistent with an interaction between these two sites. Lastly, in order to model a complex capable of electron transfer, a final constraint was added between Leu-50 of Adx, located adjacent to residues that coordinate the [2Fe2S] cluster, and Cys-462, which represents the axial heme coordination site of CYP24A1. The resulting docking solutions were evaluated based on 1) an orientation of Adx that adhered to the empirical data, and 2) energy minimization of the docked complex. The complexes selected for analysis had HADDOCK scores of −137.4 with open CYP24A1 and −111.0 with closed CYP24A1.
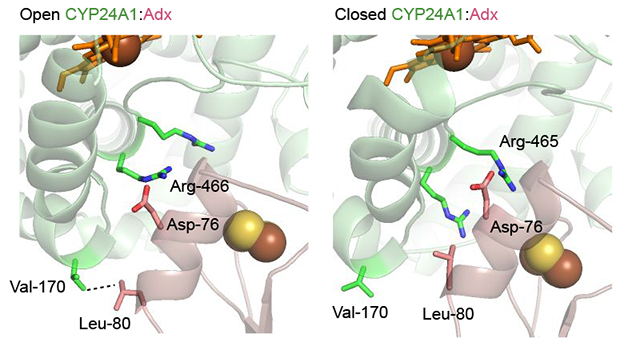
Close inspection of the complex interface modeled from the open form (Figures 6a and 6b) indicates that in this orientation, Asp-76 of Adx α-helix 3 is in position to interact with Arg-466 of CYP24A1 while Asp-72 is in position to interact with Arg-465 of CYP24A1. Notably, an arrangement of the complex that brings the iron centers within a distance suitable for electron transfer also forces the EDC derived cross-linked sites, (Adx:Glu-109 – CYP24Al:Lys-177) and (Adx:Glu-35 – CYP24Al:Lys-382) into distances of approximately 25 and 12 Å, respectively. This may point toward the involvement of these sites as electrostatic steering interactions instead of as primary recognition sites for electron transfer. This would be in keeping with the observation that mutation of α-helix 1 residues of Adx reduce the metabolism of 1,25(OH)2D3, but do not abolish it. Interestingly, we also observed that this orientation of Adx implicates a non-polar interaction between Leu-80 of Adx with Val-170 and Pro-169 of CYP24A1. Val-170 is located on the N-terminal side of the CYP24A1 C-helix, adjacent to the C-D helical bend. This same proximal region was also revealed by differential EDC cross-linking and LC-MS to be responsive to the presence of 1,25(OH)2D3 (Figure 4). A notable difference was observed for this hydrophobic interaction when compared to the complex modeled with the closed form of the enzyme (Figure 6c). Here Adx is repositioned at the binding site so that Asp-76 may be the remaining anionic surface charge (thus explaining its central role in functional assays), and that the non-polar contact is partially broken, with the distance between the Val-170 side chain and Leu-80 of Adx increasing by 1.5 Å. These models support differential Adx binding between the open and closed forms of CYP24A1, thus also possibly linking redox partner binding to conformational changes in the CYP.
Figure 6.
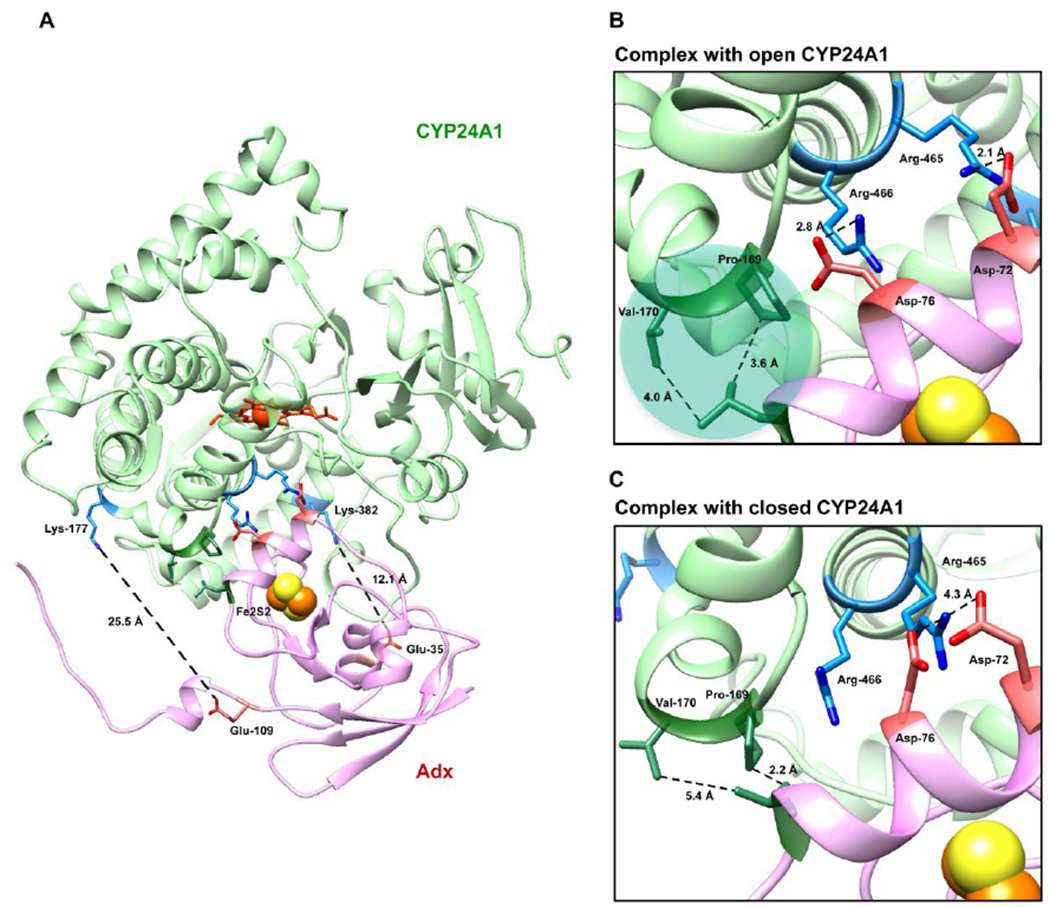
Docked model of the CYP24A1 and Adx complex (1:1) using HADDOCK 2.2. (A) An overview of the modeled complex with Adx (pink) oriented on the proximal surface of CYP24A1 (green). The two cross-linked LC-MS derived constraints (Adx:Glu-109 – CYP24A1:Lys-177 and Adx:Glu-35 – CYP24A1:Lys-382) provide a general docking orientation. (B) Details at the interface reflect the prominent role of Asp-76, which contributes key electrostatic interactions in both the open form as well as the closed form shown in (C). The nonpolar interaction between Val-170 of CYP24A1 and Leu-80 of Adx is partially broken when CYP24A1 is in the closed conformation.
Functional and structural analysis of Leu-80 mutations of Adx
The Adx residue Leu-80 has been investigated previously with respect to CYP11B1 and CYP11B2 function and was found to be essential.12 Our earlier NMR titrations showing differential peak broadening at this residue also indicate that Leu-80 is at least affected by the interaction with CYP24A1 as well.10 These previous findings correlate with identification in this study via molecular docking of a hydrophobic interaction between Leu-80 of Adx and the C-D helical bend of CYP24A1; a site also implicated in substrate-induced changes in CYP24A1 conformation (Figure 4). In order to explore the role of Leu-80 further, we generated substitutions of Leu-80 with lysine, as was done previously in the context of CYP11B enzyme function, as well as with glutamine in order to prevent possible electrostatic repulsion between lysine and the positively charged proximal surfaces of CYP24A1.
First, based on simple EDC cross-linking, L80K mutation of Adx produced a comparable cross-linked product to that of WT Adx (Figure 7a). We then wanted to compare this result with the previously reported ability of L80K to disrupt the interaction with CYP11B enzymes. The same reaction conditions were applied to detect cross-linking between human CYP11B1 and CYP11B2 with bovine Adx and L80K mutants. Here we observed that, while the L80K mutation of Adx caused considerable disruption of the interaction with CYP11B enzymes, matching the previously reported findings for these systems12, Leu-80 appears to play a different role in molecular recognition between rat CYP24A1 and rat Adx.
Figure 7.
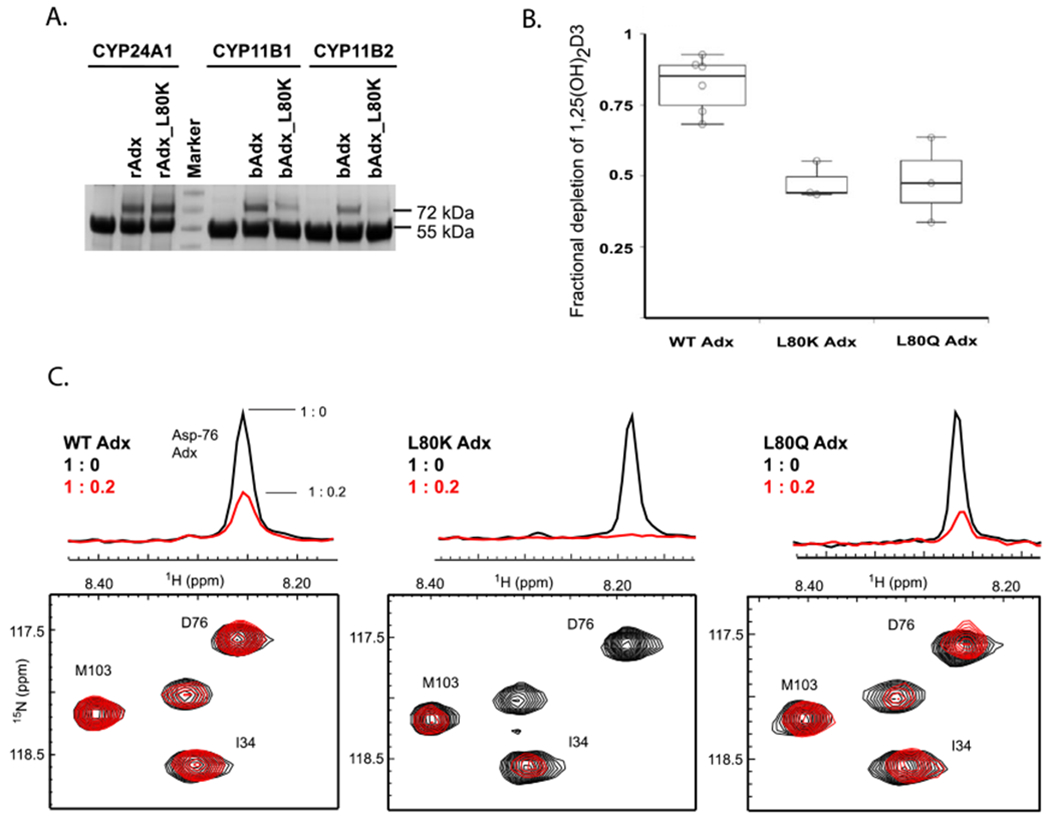
Evaluation of the Leu-80 mutations of Adx on CYP24A1 function. (A) The L80K mutation of Adx disrupts EDC cross-linking with CYP11B1 and CYP11B2 enzymes, yet does not disrupt cross-linking with CYP24A1. (B) Fractional depletion of 1,25(OH)2D3 in reconstituted functional assays (triplicate samples) is represented by the box plot, indicating the presence of a functional contribution by the Leu-80 residue. (C) Nevertheless, L80Q and L80K result in comparable (L80Q) or higher (L80K) extent of NMR peak broadening for the resonance corresponding to Asp-76.
Next, we tested L80K and L80Q mutant function using reconstituted CYP24A1 assays. We observed that the Adx mutants also appear able to support metabolism of 1,25(OH)2D3, albeit to a lesser extent than WT Adx (Figure 7b). Both of these results indicate that a distinctive mode of recognition occurs between CYP24A1 and Adx, since the L80K mutant failed to either cross-link or support the function of CYP11B enzymes,12 yet merely decrease the catalytic activity of CYP24A1.
Lastly, in order to evaluate the interaction between CYP24A1 and L80K and L80Q mutants of Adx (without contributions from a covalent linker), we acquired 15N-Adx and 15N-Adx mutant NMR HSQC spectra with and without ligand-free CYP24A1. Based on an overlay of the HSQC spectra, the mutations did not disrupt the global fold of Adx (Figure S2). In previous NMR binding assays,10 we observed considerable peak broadening of the NMR signal upon addition of 0.2 molar equivalents of CYP24A1, as expected for a protein-protein interaction that occurs in the intermediate chemical exchange timescale. In these binding assays, charge-neutralizing mutations of Adx were shown to reduce overall peak broadening, likely due to disruption of the CYP24A1-Adx complex. However, here we observed an unexpected increase in the extent of peak broadening for both L80 mutants in response to 0.2 molar equivalents of CYP24A1 (Figure 7c). Albeit an indirect measure of affinity, enhanced peak broadening is an indicator of a stronger interaction and was not expected in light of the reduced CYP24A1 function caused by the mutations. The most significant difference was observed for the L80K mutant when compared to peak broadening induced by the interaction of WT Adx with CYP24A1. The resonance for the key interacting residue, Asp-76, is broadened beyond detection when the complex forms with L80K Adx.
It should be noted that NMR binding assays likely encompass binding contributions from both monomeric and dimeric forms of Adx. However, in light of functional CYP24A1 assays and molecular docking experiments, our interpretation of these data is that substitution of a hydrophobic contact on the Adx surface may modulate the Adx binding mode in such a way that promotes tighter binding with CYP24A1, yet results in an orientation between the proteins that modestly disrupts CYP24A1 function. When compared to binding data from CYP11B enzymes, these data provide additional support for variable CYP-Adx interactions in the mitochondrial matrix.
DISCUSSION
Electron delivery to non-mitochondrial CYPs is somewhat variable and includes substrate-induced protein interactions with a requisite redox partner in the form of a CYP reductase or the modulatory protein and conditional electron donor, cytochrome b538–40 In contrast, electron delivery in mitochondrial CYPs appears, initially, to be more straightforward as it relies on a CYP interaction with a single ferredoxin protein. However, a close examination of reported binding data and crystallographic structures suggests a significant amount of variability in CYP-Adx interactions. For example, EDC cross-linking of Adx with the closely related enzymes CYP11B1 and CYP11B2 reveals a protein interface this is opposite of the orientation observed in the fused Adx-CYPllAl structure.11, 12
While it is possible that the orientation of Adx is constrained in the fused Adx-CYP11A1 structure, our own NMR titration data between Adx and CYP24A1 supports variability even between species.9 Specifically, CYP24A1 from opossum, human, and rat were all found to rely primarily on an interaction with the conserved α-helix 3 of Adx for complex formation. However, charge-neutralizing mutations at secondary sites of Adx were found to affect the complex in different ways, thus implying that CYP24A1 isoforms that have a distinct regioselective preference for the site of vitamin-D hydroxylation also form distinct complexes with Adx.
In the present study, we rely on a combination of EDC cross-linking, mass spectrometry, vitamin-D hydroxylation assays, protein NMR, and molecular dynamics in order to inform a working model of a functional CYP24A1-Adx complex. In order to avoid the introduction of artifacts resulting from combining redox partners from different species, recombinant proteins matching the amino acid sequence of CYP24A1 and Adx from rat (a carbon-24 hydroxylase system) were used. Furthermore, due to monomerization of Adx under reducing conditions, we focused our efforts on elucidating details of a stoichiometric 1:1 complex. Our expectation is that furthering our understanding of molecular recognition in a species-matched complex will provide mechanistic insight toward how Adx, directly and indirectly, influences vitamin-D metabolism.
A significant finding is that internal EDC cross-linking in CYP24A1 was differentially affected when Adx was present (Figure 2) and when Adx and 1,25(OH)2D3 were present together (Figure 4). Mapping of the peptides that were responsive to the presence of substrate creates a near-contiguous map connecting the FG helices on the distal side of the enzyme with the proximal surface of CYP24A1. In general, a substrate-induced effect in the FG helical region is not entirely unexpected, since repositioning of this sub-structure is known to be important for substrate binding. However, what is more intriguing is a substrate-induced effect of the FG helices that also correlates with long-range changes on the proximal surface at the C/D-helical bend, which is located adjacent to an inter-protein Adx binding site (Lys-177) that was identified earlier in this study. We have previously used the substrate analog 1α(OH)D3 to confirm that proper recognition of CYP24A1 substrate is important for productive binding of Adx.10 However, this feature does not appear to be unique to CYP24A1, as the addition of Adx has been shown to reposition 22-hydroxycholesterol in the active site of CYP11A1, thus allowing carbon monoxide to bind only when reduced Adx is present.41 Therefore, the peptides linking distinct regions in CYP24A1 likely offer important clues regarding how information is allosterically relayed between the proximal surface of the CYP to the distal side of the structure and in a way that may influence substrate binding and orientation.
Based on insight gained by the molecular docking experiments, we also evaluated the role of Leu-80 of Adx in CYP24A1 function. We observed unexpected enhancement in the degree of NMR peak broadening for both mutants, but for L80K in particular, indicating a stronger interaction with CYP24A1. One possible explanation for a tighter binding complex in response to L80K is that CYP24A1 residue Glu-171, located adjacent to the suspected interaction with Val-170, may substitute an electrostatic interaction with the lysine side chain of L80K. In contrast, a hypothetical reorientation of Adx so that Leu-80 is positioned opposite of the CD-helical bend would offer no candidate residues for a complimentary charge pairing interaction with L80K. In our assessment, this finding further supports a modeled orientation of Adx that brings Leu-80 in close contact with Val-170 of CYP24A1. Moreover, the fact that disruption of this nonpolar contact results in enhanced NMR peak broadening, implying a stronger interaction with CYP24A1, yet negatively affects the turnover of 1,25(OH)2D3, confirms that hydrophobic residues at the interface play a supplementary role in CYP24A1 function. Such a role is consistent with supporting allosteric communication between the F-G helices and the proximal surface of the CYP. We should note that Val-170 of CYP24A1 is not a conserved residue in mitochondrial CYPs. Human CYP27B1, for example contains a glutamine in the corresponding position and human CYP11A1 contains a glutamate. This may suggest a distinctive mode of Adx recognition that is unique to CYP24A1, as indicated by comparative cross-linking with CYP11B enzymes (Figure 7a). However, there are also indicators that Adx recognition by CYP24A1 is species-specific. Sequence analysis of additional CYP24A1 isoforms reveals an interesting correlation between the predicted regioselectivity of vitamin-D hydroxylation and semi-conservation of certain residues near the C/D helical bend (Figure 8a). Grouping of CYP24A1 sequences based on conservation of the active site residue 326, which consists of an alanine in carbon-23 hydroxylases and a glycine in carbon-24 hydroxylases,8 also reveals semi-conservation of residues at position 170. Specifically, CYP24A1 isoforms confirmed or predicted to be carbon-23 hydroxylases contain a threonine residue at this site, whereas isoforms confirmed or predicted to be carbon-24 hydroxylases contain either a non-polar residue or, in the case of the human enzyme which exhibits both regiospecific activities, a glycine at this same position. Additional residues in the C-helix that are largely semi-conserved along the same pattern include residues 153 and 164. On the rat CYP24A1 structure, these three residues are surface exposed with side chains that are generally oriented in the same direction (Figure 8b). A closer analysis of these residues is a focus of continuing studies in order to establish their link to regioselective positioning of vitamin-D substrate.
Figure 8.
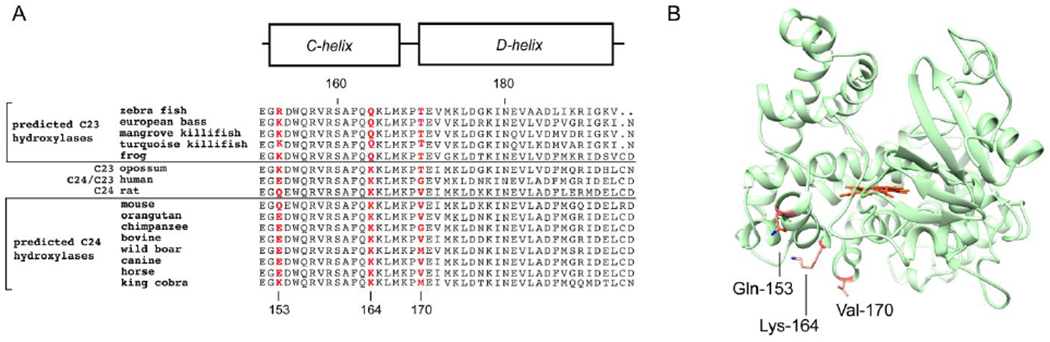
Multiple sequence alignment of confirmed and predicted carbon-23 and carbon-24 CYP24A1 hydroxylases. (A) Semi-conservation of residues at positions 153, 164, and 170 of the C-D helices correlate with species predicted to have a regioselective preference for 1,25(OH)2D3 hydroxylation. Panel (B) demonstrates the locations of these residues in rat CYP24A1, in which all of the semi-conserved side chains are surface exposed and located near the Adx binding surface.
CONCLUSIONS
The findings presented in this study represent evidence in support of conformational coupling in the CYP24A1 structure in response to both Adx and 1,25(OH)2D3. Specifically, Adx binding to CYP24A1 appears to populate a closed form of the enzyme that is reflected in the F and G helices on the distal side of the structure. Similarly, the combined presence of Adx with 1,25(OH)2D3 induces a change in the reactivity of proximal surface sub-structures to the salt-bridge sensitive cross-linker EDC, which is again consistent with an open-to-close change in the enzyme. This insight is combined with a modeled CYP24A1-Adx complex that leads us to a revised hypothesis in which conformational coupling is also important in determining site-specific hydroxylation of CYP24A1 substrate. This work not only provides insight into CYP24A1 function but also underscores the importance of understanding how variable redox partner binding affects mitochondrial CYP function.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to acknowledge the kind gift of CYP11B enzymes from the laboratory of Dr. Richard Auchus at the University of Michigan. NMR data were acquired at the NMR core facility at the SUNY College of Environmental Science and Forestry in Syracuse, NY. The molecular dynamics research presented in this paper was supported by the systems, services, and capabilities provided by the University of Connecticut High Performance Computing (HPC) facility on the Storrs campus.
FUNDING SOURCES
Funding for this research was provided by R00GM112862 (DFE) and R35GM133375 (DFE).
ABBREVIATIONS
- CYP
cytochrome P450
- Adx
Adrenodoxin
- NMR
Nuclear Magnetic Resonance
- EDC
1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride
- 1,25(OH)2D3
1,25-dihydroxyvitamin-D
Footnotes
ASSOCIATED CONTENT
Please find the file Supporting Information, which includes a listing of CYP24A1 peptides and peptide masses, peptide intensity ratios, an analysis of native salt-bridge interactions in CYP24A1, and full XH-15N HSQC NMR spectra for Leu-80 mutations of Adx.
REFERENCES
- [1].Jones G, Prosser DE, and Kaufmann M (2014) Cytochrome P450-mediated metabolism of vitamin D, J. Lipid Res 55, 13–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Axen E, Postlind H, Sjoberg H, and Wikvall K (1994) Liver mitochondrial cytochrome P450 CYP27 and recombinant-expressed human CYP27 catalyze 1 alpha-hydroxylation of 25-hydroxyvitamin D3, Proceedings of the National Academy of Sciences of the United States of America 91, 10014–10018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Chen KS, Prahl JM, and DeLuca HF (1993) Isolation and expression of human 1,25-dihydroxyvitamin D3 24-hydroxylase cDNA, Proceedings of the National Academy of Sciences of the United States of America 90, 4543–4547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Ohnuma N, and Norman AW (1982) Identification of a new C-23 oxidation pathway of metabolism for 1,25-dihydroxyvitamin D3 present in intestine and kidney, J. Biol. Chem 257, 8261–8271. [PubMed] [Google Scholar]
- [5].Sakaki T, Sawada N, Komai K, Shiozawa S, Yamada S, Yamamoto K, Ohyama Y, and Inouye K (2000) Dual metabolic pathway of 25-hydroxyvitamin D3 catalyzed by human CYP24, Eur. J. Biochem 267, 6158–6165. [DOI] [PubMed] [Google Scholar]
- [6].Ishizuka S, Kurihara N, Reddy SV, Cornish J, Cundy T, and Roodman GD (2005) (23S)-25-Dehydro-1{alpha}-hydroxyvitamin D3-26,23-lactone, a vitamin D receptor antagonist that inhibits osteoclast formation and bone resorption in bone marrow cultures from patients with Paget’s disease, Endocrinology 146, 2023–2030. [DOI] [PubMed] [Google Scholar]
- [7].Mizwicki MT, Bula CM, Mahinthichaichan P, Henry HL, Ishizuka S, and Norman AW (2009) On the mechanism underlying (23S)-25-dehydro-1alpha(OH)-vitamin D3-26,23-lactone antagonism of hVDRwt gene activation and its switch to a superagonist, J. Biol. Chem 284, 36292–36301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Prosser DE, Kaufmann M, O’Leary B, Byford V, and Jones G (2007) Single A326G mutation converts human CYP24A1 from 25-OH-D3-24-hydroxylase into -23-hydroxylase, generating 1alpha,25-(OH)2D3-26,23-lactone, Proceedings of the National Academy of Sciences of the United States of America 104, 12673–12678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Estrada DF (2018) The cytochrome P450 24A1 interaction with adrenodoxin relies on multiple recognition sites that vary among species, J. Biol. Chem 293, 4167–4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kumar A, and Estrada DF (2019) Specificity of the Redox Complex between Cytochrome P450 24A1 and Adrenodoxin Relies on Carbon-25 Hydroxylation of Vitamin-D Substrate, Drug Metab Dispos 47, 974–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Strushkevich N, MacKenzie F, Cherkesova T, Grabovec I, Usanov S, and Park HW (2011) Structural basis for pregnenolone biosynthesis by the mitochondrial monooxygenase system, Proceedings of the National Academy of Sciences of the United States of America 108, 10139–10143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Peng HM, and Auchus RJ (2017) Molecular Recognition in Mitochondrial Cytochromes P450 That Catalyze the Terminal Steps of Corticosteroid Biosynthesis, Biochemistry 56, 2282–2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Behlke J, Ristau O, Muller EC, Hannemann F, and Bernhardt R (2007) Self association of adrenodoxin studied by using analytical ultracentrifugation, Biophys. Chem 125, 159–165. [DOI] [PubMed] [Google Scholar]
- [14].Annalora AJ, Goodin DB, Hong WX, Zhang Q, Johnson EF, and Stout CD (2010) Crystal structure of CYP24A1, a mitochondrial cytochrome P450 involved in vitamin D metabolism, J. Mol. Biol 396, 441–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Sagara Y, Wada A, Takata Y, Waterman MR, Sekimizu K, and Horiuchi T (1993) Direct expression of adrenodoxin reductase in Escherichia coli and the functional characterization, Biol. Pharm. Bull 16, 627–630. [DOI] [PubMed] [Google Scholar]
- [16].Shen X, Shen S, Li J, Hu Q, Nie L, Tu C, Wang X, Orsburn B, Wang J, and Qu J (2017) An IonStar Experimental Strategy for MS1 Ion Current-Based Quantification Using Ultrahigh-Field Orbitrap: Reproducible, In-Depth, and Accurate Protein Measurement in Large Cohorts, J Proteome Res 16, 2445–2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Chen ZL, Meng JM, Cao Y, Yin JL, Fang RQ, Fan SB, Liu C, Zeng WF, Ding YH, Tan D, Wu L, Zhou WJ, Chi H, Sun RX, Dong MQ, and He SM (2019) A high-speed search engine pLink 2 with systematic evaluation for proteome-scale identification of cross-linked peptides, Nat Commun 10, 3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, and Bax A (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes, J. Biomol. NMR 6, 277–293. [DOI] [PubMed] [Google Scholar]
- [19].Johnson BA (2004) Using NMRView to visualize and analyze the NMR spectra of macromolecules, Methods in molecular biology 278, 313–352. [DOI] [PubMed] [Google Scholar]
- [20].Weissgerber TL, Savic M, Winham SJ, Stanisavljevic D, Garovic VD, and Milic NM (2017) Data visualization, bar naked: A free tool for creating interactive graphics, J. Biol. Chem 292, 20592–20598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Webb B, and Sali A (2016) Comparative Protein Structure Modeling Using MODELLER, Curr Protoc Protein Sci 86, 291–2937. [DOI] [PubMed] [Google Scholar]
- [22].Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004) UCSF Chimera--a visualization system for exploratory research and analysis, J Comput Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- [23].Xu D, and Zhang Y (2011) Improving the physical realism and structural accuracy of protein models by a two-step atomic-level energy minimization, Biophys. J 101, 2525–2534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Schmid N, Eichenberger AP, Choutko A, Riniker S, Winger M, Mark AE, and van Gunsteren WF (2011) Definition and testing of the GROMOS force-field versions 54A7 and 54B7, Eur. Biophys. J 40, 843–856. [DOI] [PubMed] [Google Scholar]
- [25].Oda A, Yamaotsu N, and Hirono S (2005) New AMBER force field parameters of heme iron for cytochrome P450s determined by quantum chemical calculations of simplified models, J Comput Chem 26, 818–826. [DOI] [PubMed] [Google Scholar]
- [26].Froelich JW, Hearshen DO, Halpert RD, and Patel S (1985) Nuclear magnetic resonance: current and future clinical applications, Henry Ford Hosp Med J 33, 122–127. [PubMed] [Google Scholar]
- [27].Bussi G, Donadio D, and Parrinello M (2007) Canonical sampling through velocity rescaling, J Chem Phys 126, 014101. [DOI] [PubMed] [Google Scholar]
- [28].Neelov A, and Holm C (2010) Interlaced P3M algorithm with analytical and ik-differentiation, J Chem Phys 132, 234103. [DOI] [PubMed] [Google Scholar]
- [29].van Zundert GCP, Rodrigues J, Trellet M, Schmitz C, Kastritis PL, Karaca E, Melquiond ASJ, van Dijk M, de Vries SJ, and Bonvin A (2016) The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes, J. Mol. Biol 428, 720–725. [DOI] [PubMed] [Google Scholar]
- [30].Hartfield KA, Stout CD, and Annalora AJ (2013) The novel purification and biochemical characterization of a reversible CYP24A1:adrenodoxin complex, J. Steroid Biochem. Mol. Biol 136, 47–53. [DOI] [PubMed] [Google Scholar]
- [31].Zhao C, Gao Q, Roberts AG, Shaffer SA, Doneanu CE, Xue S, Goodlett DR, Nelson SD, and Atkins WM (2012) Cross-linking mass spectrometry and mutagenesis confirm the functional importance of surface interactions between CYP3A4 and holo/apo cytochrome b(5), Biochemistry 51, 9488–9500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Annalora A, Bobrovnikova-Marjon E, Serda R, Lansing L, Chiu ML, Pastuszyn A, Iyer S, Marcus CB, and Omdahl JL (2004) Rat cytochrome P450C24 (CYP24A1) and the role of F249 in substrate binding and catalytic activity, Archives of biochemistry and biophysics 425, 133–146. [DOI] [PubMed] [Google Scholar]
- [33].Cui N, Xia W, Su H, Pang L, Jiang Y, Sun Y, Nie M, Xing X, Li M, Wang O, Yuan T, Chi Y, Hu Y, Liu H, Meng X, and Zhou X (2012) Novel mutations of CYP27B1 gene lead to reduced activity of 1alpha-hydroxylase in Chinese patients, Bone 51, 563–569. [DOI] [PubMed] [Google Scholar]
- [34].Urushino N, Yamamoto K, Kagawa N, Ikushiro S, Kamakura M, Yamada S, Kato S, Inouye K, and Sakaki T (2006) Interaction between mitochondrial CYP27B1 and adrenodoxin: role of arginine 458 of mouse CYP27B1, Biochemistry 45, 4405–4412. [DOI] [PubMed] [Google Scholar]
- [35].Zalewski A, Ma NS, Legeza B, Renthal N, Fluck CE, and Pandey AV (2016) Vitamin D-Dependent Rickets Type 1 Caused by Mutations in CYP27B1 Affecting Protein Interactions With Adrenodoxin, J. Clin. Endocrinol. Metab 101, 3409–3418. [DOI] [PubMed] [Google Scholar]
- [36].Coghlan VM, and Vickery LE (1991) Site-specific mutations in human ferredoxin that affect binding to ferredoxin reductase and cytochrome P450scc, J. Biol. Chem 266, 18606–18612. [PubMed] [Google Scholar]
- [37].Usanov SA, Graham SE, Lepesheva GI, Azeva TN, Strushkevich NV, Gilep AA, Estabrook RW, and Peterson JA (2002) Probing the interaction of bovine cytochrome P450scc (CYP11A1) with adrenodoxin: evaluating site-directed mutations by molecular modeling, Biochemistry 41, 8310–8320. [DOI] [PubMed] [Google Scholar]
- [38].Lampe JN (2017) Advances in the Understanding of Protein-Protein Interactions in Drug Metabolizing Enzymes through the Use of Biophysical Techniques, Front Pharmacol 8, 521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Peng HM, Im SC, Pearl NM, Turcu AF, Rege J, Waskell L, and Auchus RJ (2016) Cytochrome b5 Activates the 17,20-Lyase Activity of Human Cytochrome P450 17A1 by Increasing the Coupling of NADPH Consumption to Androgen Production, Biochemistry 55, 4356–4365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Scott EE, Wolf CR, Otyepka M, Humphreys SC, Reed JR, Henderson CJ, McLaughlin LA, Paloncyova M, Navratilova V, Berka K, Anzenbacher P, Dahal UP, Barnaba C, Brozik JA, Jones JP, Estrada DF, Laurence JS, Park JW, and Backes WL (2016) The Role of Protein-Protein and Protein-Membrane Interactions on P450 Function, Drug Metab Dispos 44, 576–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Mast N, Annalora AJ, Lodowski DT, Palczewski K, Stout CD, and Pikuleva IA (2011) Structural basis for three-step sequential catalysis by the cholesterol side chain cleavage enzyme CYP11A1, J. Biol. Chem 286, 5607–5613. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.