

Abstract
Passive immunotherapy, i.e., the administration of exogenous antibodies that recognize a specific target antigen, has gained significant momentum as a potential treatment strategy for several central nervous system (CNS) disorders, including Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, and brain cancer, among others. Advances in antibody engineering to create therapeutic antibody fragments or antibody conjugates have introduced new strategies that may also be applied to treat CNS disorders. However, drug delivery to the CNS for antibodies and other macromolecules has thus far proven challenging, due in large part to the blood-brain barrier and blood-cerebrospinal fluid barriers that greatly restrict transport of peripherally administered molecules from the systemic circulation into the CNS. Here, we summarize the various passive immunotherapy approaches under study for the treatment of CNS disorders, with a primary focus on disease-specific and target site-specific challenges to drug delivery and new, cutting edge methods.
Graphical Abstract
INTRODUCTION
Antibodies are a class of serum glycoproteins called immunoglobulins (Igs) that facilitate the adaptive humoral immune response in vertebrates 1. Immunoglobulin G (IgG; ~150 kDa) is the most abundant serum isotype and consists of a crystallizable fragment (Fc; ~ 50 kDa) that binds to Fc receptors and elicits immune effector functions 1 and two antigen-binding fragments (Fab; ~ 50 kDa each), both of which contain a variable region capable of recognizing a highly specific target antigen (Figure.1). The administration of antibodies to target disease-specific antigens, also referred to as ‘passive immunotherapy’, has steadily gained momentum since César Milstein and Georges Köhler’s seminal discovery demonstrating the production of monoclonal antibodies using hybridomas 2. Indeed, antibody-based therapeutics have emerged as one of the fastest growing class of drugs 3 due to their high target specificity and capacity to be customized 4. Therapeutic antibodies can be full-length antibodies (e.g., IgG), which have a long half-life due to Fc binding to the Brambell receptor/ neonatal Fc receptor (FcRn) 5 and elicit effector functions by interacting with Fcγ receptors 6–9, or antibody fragments such as Fab or single domain antibodies (sdAbs) which are useful when long half-lives and effector functions are not needed 10. Additionally, antibody fragments are smaller and may penetrate physiological barriers better, as well as recognize more inaccessible antigen epitopes 10. The ability to engineer antibody fusion proteins, bispecific antibodies, and antibody-drug conjugates has further expanded the use of therapeutic antibodies 4,10,11.
Figure 1.

Summary of IgG, Fab, and sdAb structure and sizes. (A) Full length IgG is a Y shaped molecule made up of four polypeptide chains – two heavy chains (red) and two light chains (grey) that are linked by disulfide bonds. Each polypeptide chain has constant domains (C) and variable domains (V). There are two Fab arms, each containing an antigen-binding site made up of the variable domains of the heavy and light chains, which can recognize antigens with high specificity. The crystallizable fragment or Fc arm can interact with Fc receptors. (B) Camelids, sharks and other cartilaginous fish (Chondrichthyes) produce a unique IgG molecule consisting of heavy chains alone. A camelid IgG molecule is depicted here. A single heavy chain variable domain is also referred to as a single domain antibody or nanobody. Unlike the antibody variable domains in other species, camelid and cartilaginous fish variable domains do not aggregate when isolated and retain their antigen binding capacity; this has generated interest in their use as therapeutics when a smaller size and no Fc interactions are desired 10.
The massive burden placed on the healthcare system due to the increasing incidence of central nervous system (CNS) disorders and the paucity of disease-modifying drugs for these disorders underscores the need for better therapies 12. Antibodies have many promising applications in the treatment CNS disorders; they may elicit disease-modifying effects for neurodegenerative diseases by interfering with the aggregation of abnormal proteins and aiding their clearance, or they may have cytotoxic effects on tumor cells and be used in the treatment of brain cancers. However, therapeutic antibodies are large proteins, making their delivery to the CNS difficult due to the restrictive properties of the blood-brain barrier (BBB) 13 and blood-cerebrospinal fluid barriers (BCSFBs) 14,15. In this review, we discuss the application of antibody-based therapeutics for the treatment of several CNS disorders in the context of disease-specific pathology as well as strategies for their successful delivery to the brain and spinal cord.
BRAIN CANCER
There are several types of cancers that occur within the CNS and they may be classified based on their site of origin (primary or metastatic), the cell type they are derived from (e.g., astrocyte, neuron, meningeal cell, etc.), their level of malignancy, and the CNS region they affect. Primary and metastatic brain tumors may have adverse effects due to several reasons: increased mass causing a rise in intracranial pressure 16, physical encroachment on normal brain areas, and necrosis in tumors which may cause inflammation and cognitive decline due to neuronal cell death. Brain metastases from peripheral cancers are the most common type of intracranial tumors and typically arise from non-small cell lung cancer, breast cancer, or melanoma 17. Brain metastases are associated with a poor (8%) 2-year survival rate 18 and a median survival time of 4–12 months 19, with few treatment options thus far 17. Primary brain cancers originate from abnormal cells within the brain. The most common type of primary brain cancer is the glioma, which as the name suggests originates from glial cells. Gliomas cause the second highest level of morbidity in individuals under 15 and the fourth highest level of morbidity in individuals between 35 and 54 20 and account for over 60% of primary neoplasms 21. The prognosis for the most malignant form referred to as glioblastoma multiforme (GBM) continues to be poor, with most patients dying within a year of the initial diagnosis 20. The median survival time for GBM patients following diagnosis is 14.6 months and the 5-year survival rate is 9.8 % 22–24. Gliomas and brain metastases are typically diagnosed by neuroimaging in patients who present with symptoms such as chronic headaches, onset of seizures, nausea and vomiting, neurological deficits, and signs of increased intracranial pressure 18,25. Despite advances in new cancer therapeutics, the typical standard of care consists of surgical resection (when possible) followed by a combination of radio and chemotherapy with temozolomide (TMZ), which has limited benefit 23.
Passive immunotherapies for brain cancers.
Passive immunotherapies have emerged as a promising class of therapeutics for the treatment of brain cancers and can overcome several challenges specific to this pathology. First, there is significant heterogeneity in brain tumors observed across individuals, between different cells within a tumor, as well as at different stages of tumor growth 26,27. This heterogeneity underlies the need for therapies that are combinatorial and can be tailored to a specific antigen profile at different stages of brain cancer in a patient and across patients. Passive immunotherapy lends itself well to this purpose since antibody-based therapeutics have the ability to be highly selective in recognizing tumor-specific or relevant anti-tumor immunomodulatory antigens that can be targeted to either directly inhibit tumor growth or selectively target a cytotoxic payload of chemo or radiotherapy to kill tumor cells. Another challenge in the treatment of brain cancers is their aggressive growth. For example, gliomas often cannot be fully surgically resected due to their infiltrative and diffuse spread 28. Surgical resection is also far more challenging in the case of many pediatric glioma patients since the tumors are often in non-hemispheric regions such as the brainstem 29. Additionally, it is often challenging to strike a balance between the efficacy, pharmacokinetic characteristics, and safety profile for small molecule therapeutics, putting them at a disadvantage compared to highly specific and potent antibody-based therapies 30. Overall, passive immunotherapies have many potential advantages for the treatment of brain cancers. To facilitate our discussion of antibodies investigated as potential therapies for brain cancer, we will describe them in the context of five categories based on their targets and modes of action: (i) anti-angiogenic antibodies, (ii) checkpoint inhibitors, (iii) lymphocyte target, (iv) antibody drug conjugates, and (v) metastatic brain tumor target.
Anti-angiogenic antibodies:
The strategy to use anti-angiogenic agents as anti-cancer therapies was founded based on the correlation between pathological angiogenesis and tumorigenesis, first established by Judah Folkman over 40 years ago 31,32; it is summarized schematically in Figure 2. Folkman’s findings spurred the eventual isolation of the pro-angiogenic vascular endothelial growth factor (VEGF) 33–35. VEGF165 or VEGFA is the most physiologically relevant isoform and may get cleaved by plasmin or matrix metalloproteinase-9 (MMP-9) to release bioactive fragments that promote angiogenesis. In January 1997, Genentech filed an Investigational New Drug application and initiated clinical trials for bevacizumab (commercial name – Avastin) – a humanized monoclonal recombinant antibody that binds to all VEGFA isoforms and their bioactive fragments with high affinity and specificity, inhibiting their interaction with VEGFRs, and thus suppressing VEGF signaling 30. The US Food and Drug Administration (FDA) approved the use of bevacizumab first for the treatment of colorectal cancer in 2004 and later expanded the range of approved oncology indications to include the treatment of lung, breast, brain, cervical, and ovarian cancer over the next decade in keeping with new clinical trial data. Since GBM is associated with significant necrosis and high VEGF mRNA expression within clusters of necrotic tumor cells 36, it was hoped that ‘anti-angiogenesis’ therapies might offer a powerful treatment strategy for gliomas, which demonstrate the highest degree of angiogenesis of all human neoplasms 37,38.
Figure 2. Passive immunotherapy strategies for brain cancer using anti-angiogenic antibodies.

VEGFA binding to VEGFR2 triggers an increase in paracellular permeability, downregulation of tight junctional proteins, and the promotion of angiogenesis. Anti-VEGFA antibodies can bind to VEGFA and prevent angiogenesis thus inhibiting tumor growth and survival. Adapted from: 51. Abbreviations: VEGF – vascular endothelial growth factor; VEGFR – VEGF receptor.
Initial Phase 2 clinical studies investigating systemically administered bevacizumab monotherapy or combinatorial therapies for recurrent glioblastoma demonstrated a reduced radiological contrast enhancement and increase in progression free survival (PFS) with bevacizumab 39–43. The FDA subsequently provided accelerated approval for systemically administered bevacizumab as a monotherapy to treat patients with recurrent GBMs that had progressed following initial treatment with chemotherapy and radiation 44. However, the benefit of systemically administered bevacizumab for the treatment of recurrent glioblastoma as a monotherapy or in combination with radiotherapies and chemotherapies remains controversial 39. Results from initial phase 2 clinical studies 39–43 must be interpreted with care and have several caveats such as small sample sizes, insufficient controls, instances of poor correlation between radiological contrast enhancement and anti-tumor effects, and no significant indication of increased overall survival 39–43. The more recent bevacizumab and lomustine for recurrent GBM (BELOB) clinical trial was a randomized controlled multicenter phase 2 study that included three treatment arms receiving bevacizumab monotherapy, lomustine monotherapy, or bevacizumab in combination with lomustine. By including a treatment group that did not receive bevacizumab the BELOB trial provided the first objective phase 2 clinical assessment of bevacizumab monotherapy versus chemotherapy alone or in combination with bevacizumab 39,45. The primary outcome of overall survival at 9 months was lowest in the group receiving bevacizumab alone and did not justify further clinical study for systemically administered bevacizumab monotherapy for recurrent GBM. Additionally, initial clinical investigation in a randomized controlled trial also demonstrated that systemically administered bevacizumab provided no benefit for newly diagnosed glioblastoma 46. The poor clinical outcomes of systemically administered bevacizumab for GBM may be attributed in some part to insufficient delivery to the brain target site. The elevated production of VEGF by tumor cells 47 and the occurrence of VEGF/VEGFRs on both luminal and abluminal sides of tumor vasculature underscores the importance of successful delivery of anti-angiogenic therapies to the brain tumor and migrating tumor cells by overcoming or circumventing the blood-tumor barrier (BTB) and BBB 48 Furthermore, many of the adverse side-effects of bevacizumab treatment, e.g., hypertension, fatigue, headache, hemorrhage, and thromboembolic events 49, may in fact be a consequence of off-target anti-angiogenic effects at non-tumor sites 50. Thus, drug delivery strategies that minimize exposure to non-tumor sites will prove beneficial.
Checkpoint inhibitors:
To ensure specific targeting of abnormal or pathogenic entities versus normal host tissue, the immune system relies on the recognition of molecular checkpoints to make go/no-go decisions. Cancer cells have the ability to modulate these molecular checkpoints and thus escape attack from the immune system. Therefore, checkpoint inhibitors that may be antibodies or small molecules have emerged as a promising strategy to prevent checkpoint modulation by cancer cells and thus improve anti-tumor immune responses.
For example, although cancer cells often express antigens that can be recognized by T cells of the host immune system, they often escape T cell mediated elimination. This is because the appropriate priming and accomplishment of T cell effector functions requires not only the engagement of the T cell receptor (TCR) by antigen peptides presented on the major histocompatibility complex (MHC) of antigen presenting cells (APCs) and tumor cells, but also activation of additional co-stimulatory signals and suppression of inhibitory signals (immune check points) expressed by APCs and tumor cells (Figure. 3) 52. TCR engagement without the support of co-stimulatory signals results in a suppressed T cell immune responsive state referred to as ‘anergy’ 52. Both co-stimulatory and inhibitory signals that influence T cell response occur in peripheral lymphoid organs as well as in the tumor microenvironment. Augmenting co-stimulatory signals and blocking inhibitory signals to increase anti-tumor T cell activity has thus emerged as a viable strategy for cancer therapy 52.
Figure 3. T cell immune response and immune checkpoints in brain cancer.

T cells may recognize tumor antigen peptides presented via MHC class I/II molecules on tumor cells or antigen presenting cells (APCs) via the TCR, resulting in a weak immune stimulatory signal. Interaction between the TCR and tumor antigen peptide/MHC complex can only activate the T cell in the presence of other co-stimulatory immune signaling. However, tumor cells and APCs in the tumor microenvironment express high levels of programmed cell death-ligand-1 (PD-L1), a ligand for the programmed cell death - (PD-1) receptor expressed by T cells, which inhibits T cell activation. APCs presenting the tumor antigen peptide/MHC complex may migrate to the cervical lymph nodes where T cells recognizing the tumor antigen may be activated and directed to the tumor. In addition to the TCR-tumor antigen/MHC interaction, the T cell must receive co-stimulatory signals in order to be activated. This co-stimulatory signal is typically received when the classification determinant 28 (CD28) receptor on T cells interacts with the B7 ligand expressed by APCs. However, regulatory T cells (Treg cells) express high amounts of cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) – a receptor that mimics CD28 and has an even higher affinity for the B7 ligand. Thus, CTLA-4-B7 interaction can compete with the CD28-B7 interaction, resulting in the lack of appropriate co-stimulatory signaling to activate tumor antigen recognizing T cells. Adapted from: 53–55. Abbreviations: PD-1 – programmed cell death protein-1; PD-L1 – programmed cell death protein ligand-1; CTLA-4 – cytotoxic T-lymphocyte-associated protein 4; Treg – regulatory T cells; CD28 – classification determinant 28.
Checkpoint inhibitor immunotherapies for cancer currently include six FDA approved IgG antibodies that target CTLA-4 (ipilimumab), PD-1 (pembrolizumab, nivolumab), or PD-L1 (atezolimumab, avelumab, and durvalumab). Ipilimumab (commercial name – Yervoy; Bristol-Myers Squibb; approved in 2011) was the first checkpoint inhibitor immunotherapy approved by the FDA for the treatment of melanoma. Pembrolizumab (commercial name – Keytruda; Merck), nivolumab (commercial name – Opdivo; Bristol-Myers Squibb) were approved by the FDA in 2014 for advanced melanoma. Atezolimumab (commercial name – Tecentriq; Genentech) was approved by the FDA in 2016 for urothelial carcinoma and metastatic lung cancer. Avelumab (commercial name – Bavencio; Merck, Pfizer, & Eli Lilly) was approved in 2017 for urothelial carcinoma and metastatic merkel cell carcinoma). Durvalumab (commercial name – Imfinzi; Medimmune/Astrazeneca) was approved in 2017 for advanced bladder cancer and in 2018 for advanced non-small cell lung cancer.
The CD-28 receptor on T cells and its B7–1/B7–2 ligands expressed by APCs constitute an important co-stimulatory pathway that can increase anti-tumor T cell activity. Conversely, CTLA-4 (cytotoxic T lymphocyte antigen-4)56, an inducible CD-28 homologue expressed by T cells, binds to B7–1/B7–2 ligands with a higher affinity than CD-28 57 and initiates an inhibitory response that can suppress anti-tumor T cell activity 52. CTLA-4 expression has been shown to be upregulated on anti-tumor T cells and in particular on an immunosuppressive T cell population called regulatory T cells or Tregs. Thus, blocking CTLA-4 with antibodies such as ipilimumab offers a potentially promising strategy to allow immune recognition of cancer cells (Figure. 4). Initial clinical investigation in a small cohort of glioblastoma patients testing ipilimumab in combination with bevacizumab showed that the combination was well tolerated and was associated with positive radiographic responses over a 3 month period 58, possibly warranting further clinical examination.
Figure 4. Passive immunotherapy strategies for brain cancer using immune checkpoint inhibitory antibodies.

Interactions between T cells, antigen presenting cells (APCs), and tumor cells that inhibit appropriate activation of T cell cytotoxic immune responses may be modulated via passive immunotherapy. For example, anti-PD-1 antibodies can bind to the PD-1 receptor that is expressed by T cells and disrupt PD-1’s interaction with its ligand PD-L1, which is highly expressed on tumor cells and APCs in the tumor microenvironment. Alternatively, anti-PD-L1 antibodies can neutralize the PD-L1 ligand’s ability to bind to PD-1. Anti-CTLA-4 antibodies may be used to block the interaction between the CTLA-4 receptor on Treg cells and the B7 ligand on tumor cells and APCs; this would subsequently allow B7 interaction with the CD28 receptor on T cells, which provides a stimulatory signal for T cell activation. Adapted from: 53–55. Abbreviations: PD-1 – programmed cell death protein-1; PD-L1 – programmed cell death protein ligand-1; CTLA-4 – cytotoxic T-lymphocyte-associated protein 4; Treg – regulatory T cells; CD28 – classification determinant 28.
The expression of a receptor called PD-1 (programmed cell death-1) on activated T cells 59 and its ligand PD-L1 (programmed cell death ligand-1) 60 on APCs constitutes an important inhibitory pathway that under normal physiological conditions plays an important role in preventing autoimmunity. However, the high expression of PD-L1 on several tumor cell types results in the PD-1/PD-L1 inhibitory pathway preventing an appropriate anti-tumor T cell response 61. PD-L1 is highly expressed by GBM tumor cells, in particular at the tumor periphery, resulting in the formation of a “molecular shield” between the tumor boundary and host anti-tumor T cells 62 and is a promising target for passive immunotherapy (Figure. 4). Nivolumab and pembrolizumab are anti-PD-1 monoclonal antibodies that are currently being clinically investigated for the treatment of primary and metastatic brain cancers as monotherapies and in combination with radiation therapies, chemotherapies, or other immunotherapies 54,62–64. It remains to be seen whether CNS access of antibodies acting as checkpoint inhibitors is needed and, if so, whether such access is sufficient to alter the course of primary as well as metastatic brain cancers. As checkpoint inhibitors have been expected to primarily act in the periphery on T cells, it has been suggested that CNS access may not be needed for effects 17; however, the observation that brain metastases continue to occur with systemic application of these newer therapies and that extracranial responses are generally superior to intracranial responses suggests that CNS delivery may in fact be needed for more robust responses 17. Indeed, clinical trials are under way in which checkpoint inhibitor immunotherapies are administered both systemically and intrathecally (e.g., nivolumab; 65).
Lymphocyte target:
Lymphocytes are not typically present in the central compartment (cerebrospinal fluid (CSF) and CNS tissue) in large numbers except in disease conditions (e.g., multiple sclerosis); however, T cells commonly perform a CNS immune surveillance function even in healthy individuals 66,67. Although lymphocyte numbers in the CSF are very low under normal physiological conditions, recirculating lymphocytes have been shown to migrate into the CSF at levels similar to those observed in subcutaneous lymph 68. Primary CNS lymphoma (PCNSL) is a rare form of non-Hodgkin lymphoma that occurs in the brain, leptomeninges, or eyes 69,70. Median survival of PCNSL patients is 13 months with a 5 year survival rate less than 5 % 71. Immunodeficiency is a major risk factor for PCNSL, with a high rate of incidence observed in patients infected with the human immunodeficiency virus (HIV) or organ transplant recipients 70,72,73. PCNSL is thought to typically involve the malignant transformation of B cells within the brain microenvironment, although the precise biological details are still lacking 69. Malignant lymphocytes from the periphery extravasate at the level of arterioles and venules to first enter and spread along enlarged perivascular spaces, and eventually move into the CNS parenchyma as the outer boundary of the perivascular space is compromised 74. Once malignant lymphocytes enter the CNS they are not easily eradicated since the CNS is a relatively immune-privileged site. The adhesion molecule CD44 and its ligands likely play an important role in the extravasation of malignant lymphocytes; high CD44 expression is observed within PCNSL lesions in the white matter 74. Due to their diffuse progression, surgical resection is not a useful strategy for CNS lymphomas. High dose methotrexate (HD-MTX) chemotherapy for newly diagnosed PCNSL and whole brain radiotherapy (WBRT) for recurring PCNSL are the current standard of care 75. Poor penetration of methotrexate through the BBB due to the presence of efflux transporters (methotrexate is a substrate for many such transporters, including p-glycoprotein and breast cancer resistance protein 76) and toxicity associated with high doses of methotrexate pose additional challenges for this treatment strategy 77. Passive immunotherapy approaches targeting abnormal lymphocytes in PCNSLs are currently being explored (Figure. 5). For example, systemic administration of rituximab, a chimeric murine monoclonal antibody that recognizes the B cell specific cell surface antigen CD20 78, has been reported to elicit radiographic responses in 4 out of 12 patients in a small clinical study, and these may be synergistic when delivered in combination with chemotherapy 79. Osmotic disruption of the BBB in combination with intra-arterial methotrexate has also been demonstrated to improve patient outcomes by improving chemotherapeutic delivery to the CNS 71,80.
Figure 5. Passive immunotherapy strategies for antibodies recognizing lymphocyte antigens.

Antibodies that recognize molecules expressed by malignant infiltrating lymphocytes may be used to treat certain CNS lymphomas. Adapted from: 74,78.
Antibody drug conjugates:
Antibody drug conjugates (ADCs) are targeted antibodies linked to anti-tumor cytotoxic moieties and have been successfully used in the treatment of peripheral solid tumors 81. ADCs may allow tumor specific targeting of radio and chemotherapies, while reducing off-target side effects. However, the benefit of ADCs may be lost over chronic application if the expression of the targeted tumor antigen gets downregulated 25. Most passive immunotherapies with naked (unconjugated) antibodies for brain cancers have thus far demonstrated limited success in improving overall survival (e.g., bevacizumab in GBM). There are two possible reasons for these disappointing outcomes – (i) unconjugated mAbs are not eliciting sufficient pharmacological efficacy at their target site, possibly due to downregulation of target antigens or other tumor compensatory mechanisms, and (ii) antibodies are not being delivered effectively to the target sites due to challenges posed by CNS barriers such as the BBB and the BCSFBs. Using ADCs as a therapy for brain cancers is a potential way to navigate the first pharmacological challenge since they provide an additional benefit of delivering an effective cytotoxic payload (Figure. 6). Several radioimmuno-conjugates are being investigated in clinical trials for the treatment of brain cancer. For example, 188Re-nimotuzumab, a beta-emitting radioisotope of rhenium linked to an anti-epithelial growth factor receptor (EGFR) antibody is being investigated for the treatment of gliomas overexpressing EGFR 82. 211At-ch81C6, an alpha-emitting radioisotope of astatine linked to an anti-tenascin antibody and 131I-BC2/BC4, a beta and gamma emitting radioisotope of iodine linked to an anti-tenascin antibody are being investigated for the treatment of GBM 83. Tenascin C is an extracellular matrix protein whose expression is controlled by Notch signaling; in GBM tumor cells, aberrant notch signaling results in over-expression of Tenascin C resulting in increased tumor cell migration which aids the invasiveness of GBM tumors 84,85. Bacterial toxins conjugated to proteins such as transferrin and interleukin-13 are being investigated in the treatment of high-grade gliomas. A similar strategy with bacterial toxins conjugated to targeted antibodies might also serve as a related promising strategy. These toxins include molecules such as the diphtheria toxin and the Pseudomonas aeruginosa exotoxin A, among others 81. ABT-414 (Abbvie) – anti-EGFR antibody conjugated to the cytotoxin monomethyl auristatin F (MMAF) – an anti-mitotic agent that inhibits cell division – is currently under clinical evaluation for newly diagnosed GBM with EGFR amplification 86. AMG-595 (Amgen), an anti-EGFR antibody conjugated to the cytotoxin maytansinoid emtansine (DM1), is currently under clinical evaluation for newly diagnosed GBM with EGFR amplification; DM1 binds to the ends of microtubules and thereby destabilizes the cytoskeleton of tumor cells 87.
Figure 6. Passive immunotherapy strategies for brain cancer using antibody drug conjugates (ADCs).

ADCs combine the ability of antibodies to recognize specific antigens overexpressed by tumor cells (e.g., EGFR, IL-13R, or IL-4R) and the ability to deliver a cytotoxic payload that can lead to tumor cell death or arrest tumor growth. Typically an ADC has 3 main components – an antibody that can recognize a tumor antigen, a linker, and a cytotoxic payload. The cytotoxic payloads may be radioisotopes (e.g., I131) that can cause DNA damage within the tumor cell, bacterial immunotoxins (e.g., diphtheria toxin) that may interfere with microtubule assembly or protein translation, or anti-tumor chemotherapeutic drugs (e.g., MMAF). Since ADCs can deliver a cytotoxic payload to the tumor target with high specificity they minimize off-target effects. Adapted from: 81. Abbreviations: EGFR – epidermal growth factor receptor; IL – interleukin; I131 – iodine radioisotope; MMAF – monomethyl auristatin F
Metastatic brain tumor target:
Primary tumors in the periphery can metastasize to the brain 17,88. Metastatic brain cancers are as much as ten times more common than primary brain cancers, with brain metastases from lung (~50%), breast (~15–25%) and melanoma (~5–20%) being the most common 17,88,89. Brain cancer metastases are often non-angiogenic tumors, i.e., the metastatic cancer cells co-opt the existing brain vasculature, which may make anti-angiogenic therapies less effective in treating these tumors 90. Systemic treatment with trastuzumab, an anti-human epidermal growth factor receptor 2 (HER2) antibody (commercial name – Herceptin; Genentech/Roche) has been used to effectively treat extracranial breast cancer that overexpresses HER2 91 (Figure. 7). However, systemic trastuzumab treatment also has a significant correlation to increased incidence of brain metastasis 92 and this correlation is most likely the consequence of trastuzumab not being effectively delivered to the metastatic brain tumors across the BTB and BBB 93. Intrathecal administration of trastuzumab in breast cancer patients with leptomeningeal carcinomatosis has shown some promise warranting further investigation in a larger study 94,95. Similar considerations hold true of passive immunotherapies for the treatment of other types of brain metastases as such as non-small cell lung cancer (treatment – nivolumab; anti-PD1 antibody; commercial name – Opdivo; Bristol-Myers Squibb) 96, and melanoma (treatment – ipilimumab; anti-CTLA-4 antibody; commercial name – Yervoy; Bristol-Myers Squibb) 97,98.
Figure 7. Passive immunotherapy strategy to treat breast cancer brain metastases using anti-HER2 antibodies.

HER2 overexpressing breast cancer brain metastases may be treated with anti-HER2 antibodies. Anti-HER2 passive immunotherapy may have several effects. First, HER2 homo or hetero dimerization that drives downstream signaling that promotes tumor cell survival may be disrupted using anti-HER2 antibodies. Second, the extracellular domain of HER2 is typically shed in tumor cells, leaving behind a phosphorylated P95 that is membrane bound and can drive downstream signaling promoting tumor cell growth and survival; anti-HER2 antibodies can bind to the HER2 extracellular domain and prevent its cleavage. Third, anti-HER2 antibodies may bind to HER2 expressed on tumor cell surfaces and initiate an Fc-mediated immune effector function that targets tumor cells. Fourth and finally, anti-HER2 antibodies may bind to HER2 and cause its internalization by endocytosis, resulting in HER2 degradation. Adapted from: 91. Abbreviations: HER2 – human epidermal growth factor receptor 2.
Current strategies and challenges in delivering passive immunotherapies to brain tumors.
Delivering passive immunotherapies to treat brain cancers is difficult. Both systemic and central delivery approaches used clinically face unique challenges in the treatment of brain cancers, emphasizing the need for new approaches and strategies for tumor drug delivery. Targeting passive immunotherapies to brain tumors via systemic delivery suffers the inherent drawback of having a large fraction of the administered dose being potentially lost to the rest of the body and is heavily dependent on the capacity of antibodies to not only cross the BTB but also areas of normal BBB that tumor cells may be hidden behind. Hydrophilic macromolecules like antibodies are thought to cross the walls of peripheral microvessels typically via passive movement across fenestrations and interendothelial clefts or via active receptor-mediated transcytosis 99. In order of increasing permeability, brain tumor microvasculature may include non-fenestrated, continuous capillaries, which closely resemble those observed in normal brain tissue, fenestrated continuous capillaries, and fenestrated capillaries with interendothelial gaps as large as 1 μm 100,101; importantly, BTB permeability in animal models of brain cancer has been shown to exhibit marked heterogeneity ranging from minimal to marked permeability that is not easily predictable 102. Passive movement of large biologics like antibodies may only occur appreciably across capillaries with open fenestrations, large interendothelial gaps, or via transcytosis 103,104. To harness the potential of receptor mediated transcytosis across the walls of tumor microvessels, bispecific antibodies that recognize both a transcytosis receptor at the BTB and an anti-tumor antigen within the brain tumor may be used 105. However, transcytosis receptors at the BTB may also be expressed elsewhere within the body, which increases the possibility of off-target side effects 106,107. Therapeutic antibodies designed to exploit receptor-mediated transcytosis at the BTB for transport into the tumor may also face the challenge of having to compete with the endogenous ligand of the receptor 100. Typically, microvessel permeability within the tumor core is high and drops sharply at the tumor margins 108. However, cancer cells may reside in the tumor periphery and remain protected by the BBB, facilitating the possibility of tumor spread or recurrence. Overall, the permeability of microvessels within brain tumors and surrounding brain varies considerably depending on the type of tumor and the location of the microvessels 100, making systemic delivery of passive immunotherapies to brain tumors a complex task. Strategies such as transiently disrupting the BTB to enhance systemic drug delivery to brain tumors by systemic infusion of hyperosmolar mannitol appear to have some benefit 71,80. However, permeability of the BBB in normal brain tissue may be relatively more affected than the BTB by systemic osmotic approaches 109, resulting in neurotoxic sequelae in healthy tissue. Distribution of antibody-based therapeutics within solid tumors has often been found to be heterogeneous and sites of antibody accumulation often do not correlate with sites of high antigen expression 110. This phenomenon of problematic and uneven distribution of systemically administered antibody-based therapeutics within tumors has been attributed to the high interstitial pressure that builds within tumors due to the increased angiogenesis and vascular hydraulic conductivity in tumors 111, although other factors may also be at play. The more or less uniformly high interstitial pressure within tumors and sharp drop in pressure at the tumor periphery may result in systemically administered macromolecules like antibody-based therapeutics to accumulate close to blood vessels (points of entry) and the tumor periphery, with little delivery occurring to the rest of the tumor 112,113.
Strategies involving the direct administration of antitumor drugs into the CNS have emerged to overcome some of the challenges faced by systemic delivery. Methods such as convection-enhanced delivery (CED) 114 or injection/infusion of drugs directly into cavities following surgical tumor resection, can deliver passive immunotherapies directly to the brain while bypassing the BTB, the BBB, and the BCSFBs. However, in addition to being highly invasive, such strategies are likely to be practically restricted to local drug delivery due to the transport limitations associated with the brain extracellular spaces where long range distribution is limited by diffusion (224, 225); diffusive transport in brain extracellular spaces is size-dependent and will be particularly limited for large macromolecules like antibodies (Wolak 2015). While this transport limitation may be desirable to ‘target’ drugs to a small area of a brain tumor, cancer cells within the tumor periphery may still be beyond reach. Direct injection or infusion (including the aforementioned CED) into brain tumors (intratumoral, intracystic, and intralesional) or surrounding tissue has been utilized clinically to deliver a variety of antibody therapeutics 65. Examples of passive immunotherapies administered via CED to treat brain cancers include: 131I-chTNT-1/B (commercial name – Cotara; Peregrine Pharmaceuticals/Avid bioservices) – an ADC consisting of an iodine radioisotope conjugated to an anti-DNA-histone H1 complex monoclonal antibody 115; 123I- or 131I-labeled 81C6 (commercial name – Neurodiab; Bradmer Pharmaceuticals) – an ADC consisting of an iodine radioisotope conjugated to an anti-tenascin monoclonal antibody 116; 131I-8H9 (commercial name – Burtomab; Y-mAbs Therapeutics) – an ADC consisting of an iodine radioisotope conjugated to a murine anti-human B7-H3 monoclonal antibody 117,118; D2C7-IT – an ADC consisting of a Pseudomonas exotoxin (PE38KDEL) conjugated to a single chain variable fragment of an anti-EGFRwt/EGFRvIII monoclonal antibody 119; and Me1–14 F(ab’)2 – a F(ab’)2 antibody fragment of the anti-proteoglycan chondroitin sulfate-associated protein murine monoclonal antibody Me1–14) 120. Clinical trials infusing antibodies into a surgically created resection cavity have also been conducted with ADCs (e.g., 123I- or 131I-labeled 81C6 121).
Other methods of delivery that circumvent the BTB, BBB, and BCSFBs are intrathecal and intracerebroventricular (ICV) administration into CSF; these routes may provide more global delivery of antibody-based therapeutics within the CNS due to their capacity to access low-resistance pathways such as perivascular spaces surrounding leptomeningeal and cerebral blood vessels that potentially allow rapid distribution throughout the brain and exchange between the interstitial fluid and CSF 122. Numerous clinical trials have been or are currently being conducted for treatment of CNS cancer using CSF-administered antibodies. Intrathecal/ICV rituximab - an anti-CD20 monoclonal antibody (commercial name – MabThera/Rituxan; Genentech/Roche) -has been administered to treat CNS lymphoma 123. Intrathecal/ICV 131I-3F8 - a radiolabeled anti-GD2 ganglioside monoclonal antibody - has been used to treat primary and metastatic leptomeningeal or brain tumors, including a trial for medulloblastoma 118. Intrathecal/ICV administration of two anti-HER2 antibodies has been investigated for the treatment of leptomeningeal metastases associated with HER2+ breast cancer - trastuzumab (commercial name – Herceptin; Genentech/Roche) monotherapy or in combination with pertuzumab (commercial name – Perjeta; Genentech/Roche) 123,124. Intrathecal/ICV 131I-8H9 has been given Breakthrough Therapy Designation by the FDA for the treatment of neuroblastoma 118. 123I- or 131I-labeled 81C6, Me1–14 F(ab’)2, and LMB-7 (or B3(Fv)-PE38, a single-chain variable fragment of the murine B3 anti-Lewis Y-related carbohydrate monoclonal antibody conjugated to the a portion of the Pseudomonas exotoxin PE38) 125 have also been administered into the CSF for primary or metastatic brain cancer and leptomeningeal cancer. Finally, other non-conventional routes of administration (e.g., intranasal delivery) are also being actively investigated to target therapies to brain tumors 126. The intranasal route for drug delivery is thought to achieve some degree of CNS targeting by accessing pathways associated with the olfactory and trigeminal nerve systems in the nasal mucosae that allow brain entry at the level of the olfactory bulbs and brainstem, respectively 127. Intranasal delivery in particular may prove to be relevant for the treatment of brainstem gliomas, which are not particularly amenable to surgical resection or invasive drug delivery methods 128.
ALZHEIMER’S DISEASE
Alzheimer’s disease (AD) and related dementia are estimated to affect more than 47 million patients worldwide 129, with more than 5.7 million patients in the United States as of 2018 130. These numbers are likely to double by 2050, partly due to the rise of a more susceptible ageing demographic (Alzheimer’s association, 130. The clinical definition of AD has evolved over the last three decades from a cognitive syndrome 131 to a multi-faceted gamut of pathological changes that gradually lead to cognitive impairment over decades 132. Among AD patients, cognitive impairment often manifests as one or more progressively declining core domains (memory, executive function, language, visuospatial perception, and intellect) 133,134. AD can be difficult to diagnose since symptoms for AD may overlap with a variety of other neurological conditions, including (but not limited to) vascular dementia, dementia with Lewy bodies, frontotemporal dementia and cardiovascular disease 133. AD pathophysiology is typically characterized by simultaneous accumulation of two abnormal proteins and their aggregates – beta-amyloid and hyperphosphorylated tau 134.
Passive immunotherapies for Alzheimer’s disease.
Beta-amyloid and hyperphosphorylated tau occur in several different forms and stages of aggregation during the progression of AD pathology providing a wide range of targets for therapies. For example, amyloid β-peptide (Aβ) occurs as a heterogeneous mixture of monomeric peptides from the sequential cleavage of amyloid precursor protein (APP) by several different enzymes. Aβ peptides are typically in the range of 38–43 amino acids although other isoforms are also generated 135,136. APP cleavage by α-secretase or β-secretase generates amino-terminal fragments and carboxy-terminal fragments; the amino-terminal fragments are called secreted APP (sAPP) α or β respectively and the carboxy-terminal fragments (CTFs) are called CTF83 and CTF99 respectively 136. γ-secretase cleavage of CTF83 and CTF99 results in the generation of p3 and Aβ peptides respectively and the amino-terminal APP intracellular domain (AICD) 136. In the amyloidogenic pathway APP is primarily cleaved by β-secretase (beta-site APP-cleaving enzyme 1, or BACE1, in the brain) and γ-secretase resulting in the production of pathogenic Aβ isoforms 136–138. In the non-amyloidogenic pathway, observed in healthy individuals, APP is primarily cleaved by α-secretase and γ-secretase 136. α-secretase cleavage is thought to prevent Aβ formation since the α-secretase cleavage site occurs within the Aβ sequence 136. In AD pathology the less amyloidogenic Aβ40 is the predominant species present around the cerebral vasculature while the more amyloidogenic Aβ42 is the earliest and most abundant isoform within the parenchyma 132,139. N-terminally truncated forms of Aβ40/42 may also form very harmful pyroglutamate Aβ isomers (pGlu-Aβ(3–40/42)) following cyclization of the N-terminal glutamate residue 140. Increased production or the lack of efficient clearance of Aβ spurs CNS buildup and aggregation as multiple Aβ units fuse together to form toxic, soluble oligomers 141,142. These soluble oligomers further act as seeds for aggregation of insoluble, fibrillar species of beta-amyloid 143,144 leading to accumulation within the brain parenchyma as well as abnormal deposition around the smooth muscle layer of cerebral arteries, referred to as cerebral amyloid angiopathy (CAA) 132; both of these processes (and others) ultimately are responsible for the neurodegeneration observed in AD 132,135. While the focus typically has been on targeting insoluble fibrillar oligomers or ‘plaques’, recent evidence suggests that soluble oligomers may drive the levels of other Aβ aggregates; so therapeutic strategies engaging oligomers might be a promising approach moving forward 135,145. Likewise, abnormal hyperphosphorylated tau protein aggregates to form several different types of pathologic conformations such as paired helical filaments (PHFs), pre-neurofibrillary tangles (pre-NFTs) and eventually neurofibrillary tangles (NFTs) that disrupt the neuronal cytoskeleton and lead to cell death 146. Passive immunotherapy lends itself well to the task of targeting these different Aβ and hyperphosphorylated tau antigen profiles over the course of AD progression and has therefore emerged as a promising course of treatment 147,148. The fact that there are currently no disease-modifying therapies approved for AD 149 further emphasizes the need to investigate AD therapies.
In this review we will discuss antibodies as potential therapies for AD (Figure. 8) in the context of four broad categories based on their antigen/target: (i) anti-beta amyloid antibodies, (ii) anti-tau antibodies, (iii) anti-BACE 1antibodies, (iv) anti-apolipoprotein E (APOE) antibodies, and (v) anti-inflammatory antibodies.
Figure 8.

Passive immunotherapy strategies to treat Alzheimer’s disease (AD). Some of the major hallmarks of pathology in AD include: (i) excess production of amyloid β-peptide (Aβ) fragments catalyzed by the beta-site APP-cleaving enzyme 1 (BACE1) and γ-secretase enzyme complex cleavage of the amyloid precursor protein (APP); (ii) accumulation and aggregation of hyperphosphorylated tau within neurons leading to cell death and cell-to-cell transmission of extracellular tau; and (iii) accumulation and aggregation of Aβ within the brain parenchyma (Aβ42) and the perivascular compartments of cerebral arteries (Aβ40). Passive immunotherapy may be used to target these different features of AD pathology. (A) Anti-BACE1 antibodies can be used to block the BACE1 cleavage of APP and thus minimize abnormal and excess production of Aβ fragments. (B) Anti-tau antibodies that target hyperphosphorylated tau can be used to block intracellular tau aggregation (likely using intrabodies 169) and prevent the extracellular cell-to-cell transmission of pathologic tau (conventional antibodies 163,164). (C) Anti-Aβ42 antibodies can be used to target Aβ42 in the brain parenchyma and halt or reverse disease pathology by aiding microglia mediated Aβ42 clearance via Fc interactions, binding to monomers and oligomers and preventing their aggregation, and resolving plaques via serine protease activity. Anti-Aβ40 antibodies may be used to target Aβ40 accumulation in the perivascular compartment of cerebral arteries (also referred to as cerebral amyloid angiopathy or CAA) in a similar manner. Adapted from: 136,183–185.
Anti-beta amyloid antibodies:
Aβ was the first antigen target investigated for potential AD passive immunotherapies based on two independent studies that showed a reduction in Aβ levels in the brain via different mechanisms following chronic systemic administration of two different monoclonal anti-Aβ antibodies (3D6 and m266) 150,151. It was proposed that 3D6 demonstrated reduced plaque burden by engaging various forms of Aβ within the brain parenchyma while also mediating Aβ clearance via cell-mediated immune mechanisms (Fc-receptor-mediated phagocytosis) 150. In contrast, m266 was believed to primarily act by sequestering Aβ in the peripheral compartment, shifting the equilibrium between the CNS and the peripheral Aβ pools towards a greater accumulation in the periphery 151. In 2006, humanized versions of these ‘first-generation’ antibodies were eventually tested for clinical efficacy in phase 2 studies under the labels bapineuzumab (3D6; Janssen/Pfizer) and solanezumab (m266; Eli Lilly), respectively 152. Bapineuzumab clinical trials for AD were discontinued in 2013 for their inability to meet clinical endpoints 153. Systemic administration of solanezumab – a humanized monoclonal antibody that recognizes soluble Aβ – also failed to show significant improvement in primary cognitive outcomes in two phase 3 clinical trials (EXPEDITION 1 and EXPEDITION 2) in patients diagnosed with mild-to-moderate AD 154. Disappointing outcomes of the bapineuzumab and solanezumab clinical trials may potentially be attributed to many possible factors: (i) initiation of treatment too late in the disease process 155; (ii) the possibility that targeting Aβ alone may be insufficient to alter disease progression in some cases 155; and (iii) insufficient central delivery of systemically applied antibodies to the appropriate target sites 156. Further investigation into the possibility of therapeutic effects at an earlier stage of AD was spurred by secondary analysis of the EXPEDITION 1 and 2 trial data which showed that solanezumab treatment resulted in lesser cognitive and functional decline than placebo among trial participants diagnosed with mild AD 157. However, investigation of systemic solanezumab passive immunotherapy in a third Phase 3 trial specifically for mild AD (EXPEDITION 3; 400 mg solanezumab or placebo administered intravenously every 4 weeks for 76 weeks) also recently failed to show any significant effect on cognitive outcomes 158. Higher doses of solanezumab are currently being investigated in prodromal populations at risk for AD in two major clinical studies: (i) the Dominantly Inherited Alzheimer Network (DIAN) clinical trial investigating solanezumab as a preventative treatment in individuals at risk for early onset AD due to a dominantly inherited genetic mutation 159 and (ii) the A4 trial investigating solanezumab as a preventative treatment in older individuals at risk for AD due to amyloid plaque build-up but who do not yet show any cognitive impairment 160. The results of these later trials may ultimately better inform on solanezumab efficacy and its limitations.
Other anti-Aβ passive immunotherapies for which (i) clinical investigation has been discontinued due to failure to meet clinical endpoints and/or (ii) clinical study outcomes have not been fully reported include: ponezumab (Rinat Neuroscience/Pfizer), an anti-Aβ monoclonal antibody that specifically binds to the Aβ40 fragment that accumulates in the walls of blood vessels as part of the CAA process; GSK933776 (GlaxoSmithKline), an anti-Aβ monoclonal antibody that binds with higher affinity to Aβ monomers and has a modified Fc region that reduces effector-mediated functions to minimize the risk of side effects such cerebral edema or microhaemorrhages that are detected as amyloid-related imaging abnormalities (ARIA) 161; AAB-003 (Pfizer/Janssen), a modified version of bapineuzumab that has a modified Fc region that reduces effector-mediated functions to minimize the risk of ARIA 162; SAR228810 (Sanofi), an anti-Aβ monoclonal antibody that binds with higher affinity to Aβ protofibrils than Aβ oligomers or monomers and has reduced effector-mediated function to minimize the risk of ARIA; and MEDI1814 (AstraZeneca and Eli Lilly), an anti-Aβ monoclonal antibody that binds with high affinity to Aβ42.
Numerous anti-Aβ passive immunotherapy trials are still under clinical investigation 65(clinicaltrials.gov), e.g., BAN2401 (Biogen/Eisai), gantenerumab (Hoffman-La Roche), crenezumab (Genentech/Hoffman-La Roche), and KHK6640 (Kyowa Hakko Kirin), anti-Aβ monoclonal antibodies that bind with higher affinity to more aggregated insoluble conformations of Aβ such as protofibrils and/or fibrils compared to soluble Aβ monomers and/or oligomers; aducanumab (Biogen), an anti-Aβ monoclonal antibody that binds with higher affinity to soluble oligomeric as well as insoluble fibrillar Aβ aggregates compared to monomeric Aβ; LY3002813 (Eli Lilly), an anti-Aβ monoclonal antibody that recognizes the pyroglutamate Aβ monomer Aβp3–42; and intravenous immunoglobulin (IVIg; Octapharm), an immunoglobulin serum fraction obtained from healthy donors, used to supplement/replace the immunoglobulin fraction in AD patients.
Anti-tau antibodies:
Similar to anti-amyloid therapeutic interventions, anti-tau antibodies targeting hyperphosphorylated toxic tau conformations are being investigated as potential passive immunotherapies for AD 163,164. Since antibodies are obviously most efficacious when targeted against extracellular antigens, immunotherapy approaches aimed at the typically intracellular tau aggregates 165 initially appeared to face many challenges. However, emerging evidence has suggested that secreted extracellular tau species may initiate the spread of pathology and act as seeds for further tau aggregation 166–168, providing a clear rationale for tau immunotherapy approaches.
Anti-tau passive immunotherapy trials that are still under clinical investigation 65 include: RO7105705 (Hoffman-La Roche), an anti-tau antibody that specifically recognizes a phosphorylated serine residue (Tau/pS409) present in intracellular pre-NFTs as well as extracellular neuropil threads and mature NFTs; LY3303560 (Eli Lilly), an anti-tau antibody that specifically binds to the N-terminus of tau aggregates over monomers; ABBV-8E12 (AbbVie), an anti-tau antibody that has high affinity for all forms of extracellular aggregated tau; and BIIB092 (Bristol-Myers Squibb and Biogen), an anti-tau antibody that has high affinity for tau residues 15–24 and specificity for extracellular secreted forms of tau, as well as tau aggregates. Gene therapy strategies to target intracellular tau are also being investigated; these include the use of anti-tau intracellular antibodies or ‘intrabodies’ 169. Intrabodies are antibody fragments (e.g., single chain variable fragments) that can recognize specific antigens such as tau and are expressed intracellularly using viral gene therapy approaches to transduce desired cell populations 169–171.
Anti-BACE1 antibodies:
Another strategy to reduce the production of Aβ is to inhibit BACE1, one of the enzymes that cleaves APP to produce Aβ. Anti-BACE1 antibodies are being investigated to inhibit APP cleavage by either sterically blocking the BACE1 active site or by blocking the allosteric site that regulates enzyme activity. Preclinical testing has shown that this strategy holds promise 172,173 and clinical investigation will likely follow.
Anti-ApoE antibodies:
Apolipoprotein E (ApoE) is the primary carrier of lipids and cholesterol within the brain 174 and the ε4 isoform of ApoE has been identified as one of the strongest genetic risk factors for late-onset AD 175,176. Recent studies have highlighted the potential of anti-ApoE antibodies as passive immunotherapy candidates in AD 177,178 and clinical investigation is likely to follow.
Anti-inflammatory antibodies:
Several preclinical studies have shown that systemic inflammatory stimuli in the periphery can trigger an adverse central immune response, which subsequently leads to neurotoxicity 179,180. Indeed, AD patients with elevated levels of the pro-inflammatory cytokine TNF-α have a typically faster cognitive decline 181. Etanercept (Amgen and Pfizer) is a fusion protein consisting of human IgG1 Fc portion linked to a dimeric ligand-binding region of tumor necrosis factor alpha cell surface receptor (p75 TNF-α) 182 that was under clinical investigation as a potential passive immunotherapy for AD. However, the inability to meet clinical endpoints has currently halted further investigation of this strategy.
Current strategies and challenges in delivering passive immunotherapies for Alzheimer’s disease.
Pathological changes in AD initiate as localized protein aggregation but spread globally throughout the course of disease progression 186,187. Hence, whole brain delivery of antibodies will eventually become crucial to obtain widespread CNS effects and acceptable clinical efficacy. Currently, most AD passive immunotherapy clinical trials utilize the systemic route of administration. Unfortunately, it is likely that systemically administered exogenous antibodies do not cross the BBB or BCSFBs to an appreciable extent and often remain restricted to the endothelial compartment where they cannot engage target antigens 156. High doses of systemic exogenous antibodies, often administered in order to attempt overcoming poor delivery to the CNS, have been linked to adverse events such as vasogenic edema and microhemorrhages (ARIA-E or ARIA-H respectively) 188. Strategies to enhance delivery of systemically administered exogenous antibodies to the CNS such as transient disruption of the BBB with focused ultrasound (e.g., BAM-10 189), or shuttling antibodies across CNS barriers using bispecific antibodies (e.g., anti-BACE-1/TfR 190–192) are also being tested.
Strategies exploring the administration of passive immunotherapies for AD directly into the central compartment have also received increasing interest. For example, preclinical studies have shown that ICV administration of anti-amyloid antibodies results in widespread brain delivery and reduces parenchymal plaque burden 193–196. ICV administration of passive immunotherapies for AD also outperforms systemic delivery approaches, both in efficacy and safety (reduced incidence of ARIAs) 197. Perispinal administration has received renewed attention as a potential means to deliver drugs to the intracranial venous system, which is potentially in communication with the CSF 198. Perispinal injection involves injecting the drug between the spinous processes of the lower dorsal vertebrae, outside the spinal canal, and posterior to the ligamentum flavum with the expectation that the drug is rapidly absorbed by local vertebral venous vasculature and eventually drains into the external vertebral venous plexus (EVVP) 198. Vertebral veins are valveless and are in communication with intracranial veins allowing drug in the EVVP to access the intracranial venous system, and eventually the CSF 198 potentially via communication between the intracranial venous system, arachnoid villi 199, dural lymphatics 200,201, and other extracellular pathways 202,203. Perispinal administration of etanercept has showed rapid anti-inflammatory response in some studies 204–207; however the outcomes of this route are somewhat controversial 208. Intranasal delivery is also emerging as a promising non-invasive central delivery approach to target passive immunotherapies to the CNS; indeed, delivery of antibodies 209–211, as well as antibody fragments 212, have been reported to reduce pathology in rodent models of AD. However, the detailed CNS distribution, mechanisms responsible for transport from the nasal epithelia to the CNS, and strategies to optimize CNS delivery of intranasally applied antibodies have only recently been explored 213. Further work is clearly needed to better define alternative delivery approaches for targeting antibodies to the CNS.
PARKINSON’S DISEASE
Parkinson’s disease (PD) affects nearly 10 million individuals worldwide and nearly 1 million individuals in the United States alone and, like other neurodegenerative disorders, PD poses a significant financial burden due to large healthcare costs and lost earning potential associated with those afflicted and their caregivers (e.g., it may be estimated that PD accounts for over $20 billion in direct and indirect costs in the U.S. today with PD prevalence / costs expected to rise dramatically by 2040) 214,215. Bradykinesia, postural instability, rigidity, and tremor are the major clinical symptoms observed in Parkinsonian disorders 216. Accumulation of an abnormal form of the presynaptic neuronal protein alpha synuclein within neuronal perikarya as Lewy bodies is a hallmark of idiopathic PD 216. As with AD, there are currently no disease-modifying therapeutics for the treatment of PD; strategies that can target different alpha synuclein aggregation profiles and other pathological targets observed with PD progression will be crucial for success. Passive immunotherapies are well suited to this challenge and are therefore currently being investigated for the treatment of PD 217.
Passive immunotherapies for Parkinson’s disease.
In this section, we will limit the discussion to antibodies as potential therapies for PD (Figure. 9) in the context of three broad categories based on their antigen/target: (i) anti-alpha synuclein antibodies, (ii) fusion proteins, and (iii) anti-LAG3 antibodies.
Figure 9. Passive immunotherapy strategies to treat Parkinson’s disease.

Disease pathology in Parkinson’s disease typically entails the accumulation and aggregation of abnormal alpha synuclein protein, subsequently leading to neuronal cell death and cognitive decline. Anti-alpha synuclein antibodies may be used to block the intracellular aggregation of abnormal alpha synuclein which typically leads to the formation of intracellular Lewy bodies (thus the most likely strategy would be to use intrabodies) or prevent the cell-to-cell transmission of extracellular abnormal alpha synuclein (using conventional antibodies). Extracellular anti-alpha synuclein antibodies may prevent abnormal alpha synuclein monomers and oligomers from aggregating further and may recruit microglia to phagocytose abnormal protein via Fc mediated interactions. Lymphocyte activation gene 3 (LAG3) protein was recently implicated in the internalization of pathologic alpha synuclein during cell-to-cell transmission so an anti-LAG3 antibody strategy may therefore be promising to prevent the spread of alpha synuclein pathology. Abbreviations: ECS – extracellular space. Adapted from: 217,231.
Anti-alpha synuclein antibodies:
A study by Masliah and coworkers showing a reduction in alpha-synuclein pathology in the CNS was the first preclinical study to investigate passive immunotherapy targeting alpha-synuclein for PD treatment 218. Several other preclinical studies followed to investigate the efficacy of anti-alpha synuclein antibodies in PD therapy 219. These antibodies demonstrated varied specificity for epitopes and conformations of alpha synuclein and included the C-terminus 220,221, N-terminus 222, or central region of alpha synuclein 222, as well as alpha synuclein protofibrils 223. Clinical investigation of passive immunotherapies for PD has been fairly limited thus far. A monoclonal anti-alpha synuclein antibody PRX002 (Prothena Corp.) has been shown to be safe in humans but its efficacy remains to be demonstrated in a clinical setting 224.
Fusion proteins:
Glial-derived neurotrophic factor (GDNF) has been shown to promote neuronal cell survival and has long been thought to be a promising potential therapy for PD. However, GDNF cannot appreciably cross the barriers of the CNS following systemic administration; several different strategies have been tried in the hope of successful central GDNF therapy over the years (e.g., intraventricular or intraparenchymal GDNF infusions) but these have so far met with challenges 225. This initially spurred efforts to engineer an immunoglobulin fusion protein that might utilize a putative BBB transcytosis system (e.g., the transferrin receptor or the human insulin receptor) to shuttle GDNF from the blood circulation into the brain parenchyma 226; however, despite initially positive pre-clinical findings, systemic delivery of a GDNF-human insulin receptor antibody fusion protein ultimately did not show behavioral or anatomical efficacy in a macaque PD model and, further, produced metaplastic and neoplastic pancreatic lesions in rhesus monkeys that caution against use of such a systemically applied growth factor-insulin receptor antibody conjugate for future clinical trials 227.
Anti-LAG3 antibodies:
Although Lewy bodies and other alpha synuclein aggregates typically occur intracellularly, a secreted form of abnormal alpha synuclein has also been reported to contribute to the spread of pathology to other brain regions in a prion-like manner 228–230. This transfer of abnormal alpha synuclein between neurons was recently reported to involve the lymphocyte-activation gene 3 (LAG3) transmembrane protein 231. LAG3 is a transmembrane protein that structurally resembles the T cell co-receptor CD4, which binds MHC class II molecules and is expressed by neurons in the cortex and cerebellum, as well as cells/cellular processes in developing white matter and the choroid plexus 232. Although the physiological function of LAG3 remains largely unknown, it has been demonstrated that LAG3 binds to abnormal alpha synuclein preformed fibrils (PFFs) but not monomers and facilitates the entry of pathologic alpha synuclein PFFs into neurons via clathrin-mediated endocytosis 231; based on these findings an anti-LAG3 passive immunotherapy approach to inhibit the spread of pathologic alpha synuclein within the CNS may be promising and warrants future investigation.
Current strategies and challenges in delivering passive immunotherapies for Parkinson’s disease.
Among the innovative approaches that have been considered to enable anti-alpha synuclein antibodies to access and engage intracellular aggregates, is the use of intracellular antibodies (i.e., intrabodies) 233,234. Expression of antigen specific intrabodies within the CNS requires that brain cells be transfected with anti-alpha synuclein scFv cDNA containing plasmids or viruses 171. However, delivering plasmids and viral vectors to brain tissue remains challenging due to their highly limited capacity to cross CNS barriers following systemic delivery 235 and their limited spread away from the site of administration following central delivery approaches such as direct intraparenchymal or intrathecal administration 235–237. Delivering viral vectors to the CNS also may pose safety concerns with certain vector types 238. Another unique challenge for intrabodies to engage their target is the instability of antibody disulfide bonds in the reducing environment of the cell cytoplasm 217. Endogenous immunoglobulin disulfide bonds are formed under highly controlled redox potential conditions within the endoplasmic reticulum; these conditions favor the formation and stability of disulfide bonds 239. Endogenous immunoglobulins remain protected from the reducing environment of the cell cytoplasm by vesicles until they are secreted 240 into a physiological fluid (e.g., blood or CSF), which has a redox potential that can sustain disulfide bond stability 241; intrabodies may fail to fully access this complex intracellular protein trafficking pathway.
In general, the challenges faced for delivering passive therapies to the CNS for the treatment of PD faces some of the same challenges as those for other neurodegenerative disorders. Systemically administered exogenous therapeutic antibodies may not appreciably cross CNS barriers to engage their pathologic target 156, while most central routes of delivery are limited by their invasiveness and, at least so far, a suspected inability to provide global drug delivery 236,237.
HUNTINGTON’S DISEASE
Huntington’s disease (HD) is a hereditary neurodegenerative disease marked by progressive cognitive, behavioral, and motor decline. HD prevalence worldwide is around 3 per 100,000 people, ranging from a high in Europe, North America, and Australia (~6 per 100,000) to a low in Asia (<1 per 100,000) 242. Late-stage HD brains reveal severe atrophy of the cortex and striatum 243–245. Pathology in HD is caused by an expanded trinucleotide repeat pattern CAG (>36–40 repeats) encoding an abnormally long string of the amino acid glutamine (polyQ tract) in exon 1 of the huntingtin gene (HTT) thereby producing a misfolded mutant huntingtin protein (mHTT) 246. While the precise function of the Huntingtin protein remains unknown, it has been hypothesized that Huntingtin is a membrane-associated protein that is involved in vesicular trafficking 247. Approved treatment options for HD are currently quite limited and only address the symptoms of the disease (i.e., symptomatic, not disease-modifying therapies). Among the currently approved therapies are small molecule therapeutics that suppress involuntary movement and anti-psychotic drugs. Potential new HD therapies under preclinical and clinical investigation include macromolecules with a variety of targets implicated in HD pathology.
Passive immunotherapies for Huntington’s disease.
Although there are currently no approved antibody-based therapeutics for HD, there are a number of different antibodies in the preclinical pipeline that target mHTT as well as other proteins involved in neuronal cell survival and neuroinflammation. In this section, we will discuss antibodies as potential therapies for HD (Figure. 10) in the context of three broad categories based on their antigen/target: (i) anti-mHTT antibodies, (ii) anti-inflammatory antibodies, and (iii) BDNF mimetics.
Figure 10. Passive immunotherapy strategies to treat Huntington’s disease.

Huntington’s disease (HD) pathology is characterized by the intracellular accumulation and aggregation of the mutant huntingtin protein (mHTT), which results in subsequent cell death, and the spread of pathology due to cell-to-cell transmission of extracellular mHTT. Other hallmarks of HD pathology include pro-inflammatory signals (e.g., SEM4D/plexinB1 signaling pathway), and down-regulation of cell survival/neurotrophic signals (e.g., BDNF/TrkB signaling pathway). HD progression may potentially be blocked by passive immunotherapy strategies that target one or more aspects of this pathology. For example, anti-HTT antibodies (e.g., intrabodies) may be used to target intracellular mHTT. It is important to note that anti-HTT intrabodies typically bind to both normal HTT and mHTT. The ratio of mHTT to normal HTT is indicative of HD pathology and mHTT mRNA transcripts were found to exceed normal HTT in the cortex and striatum of nearly 75% patients in an HD clinical study 257. The increase in mHTT compared to normal HTT may be attributed to increased transcription of the mHTT allele, or decreased clearance of mHTT, or both 257. Therefore, engineering antibodies that recognize and bind with higher affinity to mHTT than normal HTT may be important since an equimolar inhibition of mHTT and normal HTT may increase the mHTT to normal HTT ratio 257. Additionally, normal HTT is thought to play a role in promoting cell survival and depleting it may further exacerbate disease pathology and clinical outcome 258.
Anti-mHTT antibodies:
Antibody-based therapeutics targeting both the intracellular and extracellular forms of mHTT are potential strategies for HD intervention that are still in the preclinical stage of investigation 248. Targeting intracellular and/or membrane bound mHTT has the potential to slow down and/or prevent cell death 249, while targeting extracellular secreted mHTT has the potential to slow down and/or prevent cell-to-cell transmission and spread of pathology 250. Indeed, an anti-mHTT monoclonal antibody developed by AFFiRis that binds to extracellular mHTT has been shown to reduce levels of the abnormal Huntingtin protein in plasma and organs in the YAC128 mouse model of Huntington’s disease after intraperitoneal administration 251.
Anti-inflammatory antibodies:
Immune dysfunction has emerged as an early hallmark in HD pathology; indeed, proinflammatory signals have been shown to exacerbate HD progression in humans 248,252. One such proinflammatory signal is the semaphorin 4D (SEMA4D) protein, which is expressed by infiltrating immune cells while its receptor is expressed by neurons, endothelial cells, and oligodendrocytes 253. Expressions of both SEMA4D and its CNS receptor plexin-B1 have been shown to be upregulated in HD, suggesting a possible correlation between the SEMA4D proinflammatory signal and HD pathology 254. Importantly, preclinical studies have shown that anti-SEMA4D antibodies dampen neuroinflammation and can rescue the disease phenotype in a transgenic mouse model of HD 255. Vaccinex is currently investigating the efficacy of anti-SEMA4D monoclonal antibody (VX15/2503) for the treatment of HD in clinical trials and received a fast-track designation from the FDA in 2016 for the development of this therapy.
BDNF mimetics:
Given that HD causes cortical and striatal atrophy, another therapeutic target for HD is an important signaling pathway for neuronal survival activated by brain-derived neurotrophic factor (BDNF). Pfizer has identified two mouse monoclonal antibodies, known as 38B8 and 29D7, that act as BDNF mimetics and activate the Tropomyosin receptor kinase B (TrkB) signaling pathway leading to cell survival. These two antibodies have been shown to have some neuroprotective effects in rat primary striatal neurons in vitro 256, although in vivo efficacy has to our knowledge not yet been established.
Conventional anti-HTT antibodies may be used to target extracellular mHTT and prevent its cell-to-cell transmission. Additionally, antibodies that mimic BDNF may be used to activate the TrkB signaling pathway to promote neuronal survival, while anti-SEM4D antibodies may be used to interrupt the SEM4D/plexinB1 pro-inflammatory signaling pathway. Adapted from: 248,250,251. Abbreviations: BDNF – brain derived neurotrophic factor; SEM4D - semaphorin 4D; TrkB – Tropomyosin receptor kinase B.
Current strategies and challenges in delivering passive immunotherapies for Huntington’s disease.
Intrabodies are typically used to target intracellular mHTT pathology; however, this strategy faces the same challenges posed by gene therapy delivery and safety as in other CNS disease contexts. Typically intrabodies are smaller antibody fragments such as scFvs, in order to simplify protein expression 248. Intrabodies for HD passive immunotherapy are currently under preclinical investigation. While intrabodies targeting the abnormally expanded polyQ tract unfortunately caused rapid cell death and worsened mHTT aggregation in preclinical studies 259, intrabodies targeting other mHTT domains have demonstrated a reduction in aggregates 248,260.
As with most other CNS disorders, passive immunotherapies for HD have typically been administered via the systemic route and face the challenge of inadequate access to the brain parenchyma (i.e., the site of target engagement) due to the presence of the CNS barriers 156. Central routes of delivery, while invasive, may be promising for delivery directly to the most vulnerable brain regions such as the striatum and cerebral cortex. Non-invasive routes of central delivery such as intranasal administration also hold some promise. For example, intranasal application of a small molecule BDNF mimetic was found reduce motor dysfunction and pathology by acting via the TrkB signaling pathway in a mouse model of HD 261; these studies may be extended to investigate the efficacy of HD passive immunotherapies in the near future.
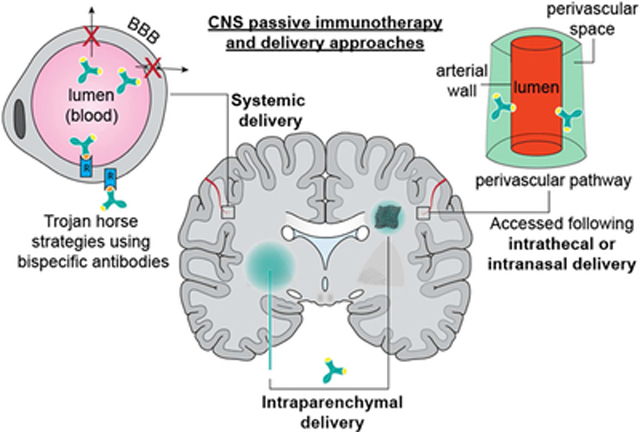
SUMMARY OF SYSTEMIC ADMINISTRATION STRATEGIES TO DELIVER PASSIVE IMMUNOTHERAPIES FOR CNS DISORDERS
Systemic administration (i.e., delivery of drugs via the blood circulation) of passive immunotherapies to investigate potential treatments for CNS disorders has historically been the primary focus of both industry and academic studies for several reasons. First, most biologics, such as antibody-based therapeutics are susceptible to protease degradation and permeate poorly across physiological barriers (e.g., the gastrointestinal mucosa) due to their large size and charge 262,263. Second, the pharmacokinetics of the most typical parenteral routes of administration (i.e., intravenous, intramuscular, or subcutaneous) are relatively simpler and better understood than that for other routes of administration such as oral or intranasal where an often complex initial absorption step must be accounted for. Third, the brain is a highly vascularized organ with capillary density as high as several thousand mm/mm3 (total capillary length per tissue volume) 264. The typical distance between capillaries and neurons within the brain ranges between –25 μm 264. However, a major hurdle to systemic drug delivery to the CNS is the existence of the BBB and BCSFBs 265–267. While it has been reported that a small fraction of endogenous IgG circulating in the blood may access the CNS 268,269 via sites where the BBB is absent (e.g., the circumventricular organs) 202,203 the capacity of these pathways to allow entry of exogenous systemically administered antibodies into the CNS at therapeutically relevant levels is limited 156. Indeed only 0.009% of IVIg has been detected in the brain and a large portion of this fraction has been observed to be sequestered within the endothelial compartment of cerebral microvessels, i.e., it is unable to access the brain parenchyma to engage with target antigens 156. The difficulty in being able to distinguish between the systemically administered exogenous antibody fraction sequestered within the cerebral endothelial cells versus the antibody fraction that truly gains access to the brain parenchyma has resulted in an overall poor quantitative estimation of antibody CNS levels following systemic delivery 156,270–272. Fourth, it is often assumed that the BBB is compromised under pathological conditions and that its ability to restrict systemically administered drugs from entering the brain is altered under such conditions. However, the degree of BBB disruption varies greatly depending on the stage of disease progression and may be heterogeneous in different brain regions 273; indeed, careful study of BBB permeability to systemically applied human IgG in several common mouse models of AD (mutant PS2-APP, tau and APOE lines) and amyotrophic lateral sclerosis (mutant superoxide dismutase 1(SOD1) line) revealed no change in IgG levels in cortex, cerebellum, or spinal cord 274.
The poor outcomes of clinical trials investigating systemically administered passive immunotherapies for CNS disorders and a better understanding of the limited levels of exogenous IgG capable of passively reaching the CNS has spurred new efforts to enhance transport across or around the CNS barriers. For example, one such strategy has involved the transient disruption of the BBB via methods such as MRI-guided focused ultrasound with microbubbles 275,276 or systemic infusion of hyperosmolar solutions (e.g., hyperosmolar mannitol) 277–279. However, it has long been appreciated that disruption of the BBB poses a risk since it non-specifically allows entry of not just drugs but other serum macromolecules into the CNS 280. A more specific approach being investigated to enhance the CNS delivery of antibodies across the BBB is the application of methods (sometimes referred to as ‘Trojan Horse’ strategies) that utilize endogenous receptor-mediated vesicular transport systems (primarily clathrin-coated vesicles 281) to shuttle their ligands (nutrients, metabolites, proteins etc.) from the luminal to the abluminal surface of the brain endothelium, i.e., from the blood to the brain 282. Endogenous receptors involved in putative receptor-mediated transport (RMT) at the BBB include the transferrin receptor, insulin receptor, and low-density lipoprotein-receptor related protein, among others 282,283. Although RMT in brain endothelium is relatively downregulated compared to endothelium in other parts of the body, it may be crucial for macromolecule transport across the BBB 281. Such BBB-crossing strategies typically involve a drug consisting of an antibody, antibody-fusion protein, or antibody-decorated nanoparticle with two main components, the first of which targets one of the aforementioned RMT pathways in brain endothelial cells while the second consists of a therapeutic payload (e.g., lysosomal enzyme) or disease-modifying Fab portion directed against CNS pathology (e.g., amyloid beta or alpha-synuclein) 281,282,284,285. However, ‘molecular hitchhiking’ of therapeutic molecules across the BBB by harnessing endogenous transport mechanisms 285 may sometimes come at a price, e.g., the expression of transcytosis receptor targets (e.g., transferrin receptor) in other regions of the body poses the risk of off-target side effects, depending on the nature of the antibody 106,107,286. Additionally, it has been suggested that antibodies directed against BBB RMT systems should ideally demonstrate low affinity binding in order to allow the antibody to be successfully released following transit across endothelial cells of the BBB; such low affinity interactions will often require larger systemic doses to be administered in order to achieve therapeutically relevant levels within the brain parenchyma 107,190. Lastly, exogenous antibodies targeted to an endogenous RMT system at the BBB may compete with the endogenous ligand of the receptor in some cases, a situation that may cause complications over time depending on the nature of the receptor system being targeted 100. Identification of new RMT pathways at the BBB is under investigation 191 in order find delivery mechanisms with a larger transport capacity and fewer off-target side effects. An important caveat to such approaches is that any antibody or antibody conjugate that first crosses the brain endothelium must then navigate endothelial-, pericyte-, and astrocyte-associated basement membranes in crossing the perivascular compartment (which may consist of fused basement membranes or a potential pericapillary space), before finally moving beyond astrocytic endfeet to reach the brain extracellular spaces and then diffuse to target neurons 122,287,288. These steps may pose a particular barrier to larger macromolecule therapeutics like antibodies, which may not always easily escape the perivascular spaces to enter the parenchyma 122,213; however, further studies are needed to better understand the distribution of endothelial cell-crossing therapeutics between the PVS and brain ECS.
SUMMARY OF CENTRAL ADMINISTRATION STRATEGIES TO DELIVER PASSIVE IMMUNOTHERAPIES FOR CNS DISORDERS
Central administration strategies that bypass the BBB and BCSFBs entirely are emerging as a necessary tool to facilitate the delivery of large biologics like therapeutic antibodies to the CNS. One approach to bypass the BBB is direct injection or infusion of substances into the brain parenchyma; this may be particularly suitable when narrow, focal delivery to specific brain regions is desired. Transport within the brain extracellular spaces is particularly limited to short range distribution by diffusion 236,237, a size-dependent process that may be slow and inefficient over longer distances for large macromolecules like antibodies 289. This transport limitation may be desirable to ‘target’ drugs to a small area of the brain, e.g., a brain tumor (although invading cancer cells migrating away from tumors to other brain sites may still be beyond reach). Treatment of whole brain disorders that may require chronic drug administration paradigms (e.g., Alzheimer’s disease, neuropathic lysosomal storage disorders, Parkinson’s disease, and Huntington’s disease, to name a few) with direct parenchymal injections will likely be neither practical nor feasible due to the number of injection sites required for therapeutic delivery.
One well-described drug delivery method for direct brain injection is CED 114. It was originally thought that this technique could accomplish delivery to a larger brain volume than simple injections due to a resulting pressure gradient imposed between the catheter tip and the tissue interstitium that might force an infusate to flow through the extracellular space. However, it is now better appreciated that parenchymally-infused substances are much more likely to distribute via faster bulk flow along low-resistance pathways in the brain, e.g., cerebral perivascular spaces and white matter tracts 122,290–296, than along narrow gray matter extracellular spaces that exhibit high hydraulic resistance to flow of any kind 236; indeed, these low-resistance pathways associated with the perivascular spaces and white matter are now more commonly credited for the large area of tracer distribution following CED 295,297. Important considerations for CED include optimal injection volumes and rates that ideally achieve the desired target volume of distribution. In keeping with these considerations, the FDA approved the ‘iPlan Flow’ software to help target therapies more accurately to specific brain regions and without significantly losing the drug to the CSF via white matter tracts or the pial/ependymal surfaces 298. Improved cannula designs 299,300 that may help prevent backflow are also under active investigation. Recent work includes the investigation of multifunctional microfabricated devices (e.g., a miniaturized neural drug delivery system, or MiNDS) with smaller catheters that can more precisely deliver smaller volumes at lower flow rates (e.g., an order of magnitude lower than typical CED) and with rapid on/off dosing; such devices have recently been tested for feasibility and functionality in both rodents and non-human primates 301. Though the above strategies are invasive, and the transport of large molecules may still be limited with some of them, intraparenchymal delivery methods nonetheless continue to hold promise for certain types of targeted therapies. To date there are no approved macromolecule therapeutics delivered directly into the brain parenchyma; a recent FDA approval (the Cleveland Multiport Catheter CED device) 302 may open the way for further studies (this particular device has so far been utilized in clinical trials administering topotecan, a small molecule chemotherapeutic, intratumorally; 65).
Another central delivery approach to bypass the BBB and BCSFBs involves administration directly into the CSF within and surrounding the brain and spinal cord. Possible routes of administration include lumbar intrathecal (into the lower/caudal spinal CSF space), cisternal intrathecal (into the cisterna magna, near the brainstem, the reason this route is rarely used clinically), or ICV into the lateral, third, or fourth ventricles in the brain’s interior) 303. A recent study reviewed the safety and usage of ICV devices in patients and suggested that it is a reasonable long-term drug delivery strategy 304. CSF-administration shows some promise for larger macromolecules, though clinically almost all are still in trials 124. It is important to note that many clinical trials listing ‘intrathecal’ as the route of delivery actually administer the therapeutic agent into the CSF by intrathecal injection/lumbar puncture or by administration into the ventricles using a device such as the Ommaya reservoir or the Rickham device (subcutaneous implanted reservoirs with a catheter placed into a ventricle or surgically created resection cavity that can be accessed through the skin); indeed, some clinical trials require an implanted ventricular access device for eligibility. Intraventricular delivery is not technically equivalent to intrathecal, (with obvious differences in location and application) so caution should be exercised when assuming the actual route of delivery based on use of the term ‘intrathecal’; the implications of an entirely different site of delivery may be important for drug distribution between the cranial and spinal CSF compartments and critical for understanding the pharmacokinetics. There are currently three biologics approved in the United States by the FDA for CSF-administration: (i) ziconotide, a 2.6 kDa peptide delivered via lumbar intrathecal infusion approved in 2004 for chronic pain 305,306, (ii) nusinersen, a 7.5 kDa antisense oligonucleotide delivered via lumbar intrathecal injection approved in 2016 for spinal muscular atrophy 307–310, and (iii) cerliponase alfa, a 66 kDa enzyme delivered via intracerebroventricular infusion approved in 2017 for late infantile neuronal ceroid lipofuscinosis type 2 (CLN2) 311. The latter disorder is unarguably a whole-brain disease, for which delivery of the enzyme to every cell is desired; however, the actual distribution of this enzyme throughout the brain is has not been fully described. The lack of published studies describing detailed distribution profiles for intracerebroventricularly and intrathecally applied macromolecules is something that needs to be addressed in the future; indeed, there is an urgent need for preclinical studies focused on the mechanisms governing macromolecule transport to and distribution within the brain so that the potential translatability of different methods can be better understood across different therapeutic classes (e.g., peptide versus oligonucleotide versus larger proteins).
CSF administration to target drugs to the brain relies on communication between the CSF and the brain interstitial fluid (ISF), the governing physiology of which is an area of great interest but also a topic with significant questions and some recent controversies 122,312 Modern studies have confirmed that diffusion appears to hinder the transport of substances between the CSF and bordering brain extracellular spaces, for a variety of macromolecules, including antibodies 122; this finding is somewhat in line with older work where CSF-administered macromolecule delivery into the brain was previously thought to be minimal 313,314. However, the low-resistance pathways (the cerebral perivascular spaces and white matter tracts, discussed above) have increasingly been appreciated to play perhaps a key role in rapid exchange between the CSF and brain ISF 122,291–295. Transport along the perivascular space (defined here as the fluid-filled vascular connective tissue space of the vessel adventitia and also possibly the extracellular space associated with the smooth muscle basement membrane of the tunica media) has been suggested to occur in part due to vessel pulsatility driving convective flow along the vessel wall 290,295,315 or alternatively, dispersion 316. Most importantly, substantial entry into the perivascular compartment from the CSF may theoretically provide access to the whole brain (by reaching down to the level of the capillaries) so it is critically important to better understand what factors (physiological and physicochemical) govern this access and how to tune it for better delivery 122.
The intranasal route of administration has also received recent attention as a potentially non-invasive method to deliver biologics to the CNS 126,127,317–321. Several groups have demonstrated that intranasal administration of specific full length IgG antibodies 209–211, and smaller antibody fragments 212, may result in CNS delivery sufficient to show efficacy in rodent models of AD. Recent published work from our laboratory shows that intranasally administered IgG can rapidly access the CNS at therapeutically relevant levels via transport along extracellular perineural and perivascular pathways associated the olfactory and trigeminal nerves with further widespread distribution within the brain via the perivascular spaces of cerebral blood vessels 213. Evidence supporting access to olfactory and trigeminal pathways in the nasal lamina propria and subsequent transport to and distribution within the CNS following intranasal delivery has now been demonstrated for untargeted antibodies 213, and targeted antibodies 209, as well as other protein 127,321 and dextran tracers 319. This accumulating evidence suggests (i) the transport pathways from the nasal mucosa to the CNS are unique to the nasal route of administration and (ii) that these unique anatomical pathways themselves do not appear to vary considerably for different intranasally administered macromolecules. However, specific binding (e.g., binding to antigens or Fc receptors) or non-specific binding (e.g., binding to extracellular matrix components) interactions will likely affect the efficiency of transport of different antibodies along these nose-to-brain pathways. Intranasal delivery to the CNS may be influenced by several factors: formulation 322,323, molecular size 319,324, use of nasal epithelial permeability enhancers 319,325, and body position 326,327 among others. Size-dependent aspects of intranasal antibody delivery to the CNS can now be addressed due to significant advances in protein engineering and the rise of antibody fragment-based therapies 10. Molecular size may potentially influence intranasal delivery to the CNS at several stages of the transport process. Intranasally administered molecules must first cross the nasal epithelium via transcellular or paracellular pathways 126,318, prior to accessing perineural and perivascular pathways to the CNS. However, tight junction proteins expressed at the nasal epithelium (e.g., occludin, claudins-1, −3, and −5, and zonula occludens-1 and −2 expressed in the apical olfactory epithelium 328) impose a size-dependent barrier to paracellular transport 126,318,319. Smaller molecules are thus able to cross the nasal epithelium to reach the underlying lamina propria and access pathways to the CNS more efficiently than larger molecules; such a size-dependence has been demonstrated using fluorophore-labeled 3 and 10 kDa dextrans 319. Preliminary studies from our laboratory also suggest a similar trend for intranasally administered antibodies, with better brain delivery for smaller sdAbs (hydrodynamic diameter (dH) ~ 4.5 nm) and Fab fragments (dH ~ 6.5 nm) than for full length IgG (~ 10 nm) (213 & unpublished observations). It has been suggested that IgG transport across the nasal epithelium following intranasal administration may also be attributed to FcRn-dependent transcytosis 213,329,330. Once in the nasal lamina propria, intranasally administered molecules would need to escape clearance into the sink of the systemic circulation or nasal lymphatics in order to access perineural and perivascular pathways to the CNS 317. Larger hydrophilic molecules are more likely to escape clearance into the systemic circulation and lymphatics than smaller molecules 317. Entry of intranasally administered macromolecules into the CSF has also been shown to be size-dependent 324. As with nasal epithelial transport, IgG access to the CSF following intranasal delivery may partly be attributed to FcRn-dependent transport processes at lining cells of nerves and leptomeningeal vessels 122,213,287, although this will require further study. Our work has shown that intranasally administered IgG accesses the brain more efficiently than the CSF compartment, suggesting that some degree of CNS entry/distribution can occur without access to the CSF first 213. Taken together, the size-dependent entry of intranasally administered antibodies to the CNS appears most likely attributed to size-dependent transport across the nasal epithelium, although other mechanisms influenced by molecular size may also play important roles.
Our work investigating the distribution of antibodies within the CNS following intraparenchymal 289, intrathecal 122, and intranasal administration 213 (Figure. 11) emphasizes that drug access to perivascular spaces and subsequent distribution along these spaces into the brain may likely govern whole brain delivery and thus therapeutic efficacy 122,236,287. Several physiological factors may influence the capacity of antibodies to access the perivascular compartments of cerebral blood vessels and subsequently diffuse from the perivascular space and into the brain parenchyma. First, size-dependent entry of protein tracers into the perivascular spaces of cerebral blood vessels has been shown to play a large role in brain distribution from the CSF, with smaller sdAbs having greater access to the perivascular spaces compared to full length IgG 122. Size-dependent access to the perivascular spaces likely serves additional physiological roles; e.g., insulin-like growth factor 1 (IGF1, 7.6 kDa) binding to insulin-like growth factor binding protein 2 (IGFBP-2; 32 kDa) in the CSF may prevent substantial brain access of IGF-1 under some conditions, and release of IGF-1 from this same binding protein may allow free IGF-1 to enter the brain more easily 331, possibly via non-saturable perivascular pathways 332. Second, binding of immunoglobulin G to Fc receptors (e.g., the Fcγ receptor 2b) in the brain at key interfaces (e.g., the pial surfaces, the glia limitans, and around cerebral blood vessels) may impede the ability of IgG to enter the brain from the CSF and hinder its exit from the perivascular compartment by diffusion. Surprisingly, very little information is available on Fc receptor distribution in the CNS so such a ‘binding’ barrier cannot yet be fully appreciated. Third, the astrocyte basement membrane may also pose a barrier to substances attempting to enter the extracellular space from the perivascular spaces 288. Indeed, previous studies have demonstrated that full-length IgG exhibits limited diffusion out of the perivascular spaces and into the surrounding neuropil 122,288. Finally, the impact of administration parameters, e.g., infusion rates, delivery volumes 269, and body positioning 326,333, etc. may ultimately prove quite significant when carefully studied across the different routes.
Figure 11. Immunoglobulin G (IgG) access to the perivascular space (PVS) surrounding cerebral blood vessels following intrathecal and intranasal delivery.

(A, B) Examples of blood vessels at the rat the cortical surface and in the striatum respectively showing intrathecally administered rat IgG accessing the PVS. (C, D) Examples of blood vessels in the rat olfactory bulb and at the cortical surface respectively showing intranasally administered rat IgG accessing the PVS. Abbreviations: IT – intrathecal; IN – intranasal; RECA-1 – rat endothelial cell antigen-1 (endothelial cell marker); GFAP – glial fibrillary acidic protein (astrocyte marker); DAPI – 4’, 6-diamidino-2-phenylindole (cell nucleus marker).
Pathological changes that occur with the progression of CNS disorders may also have an important, disease-specific impact on the ability of centrally administered drugs to access the perivascular compartment of cerebral blood vessels and diffuse within the brain target regions. For example, brain tumors often have an astrocytic border that likely serves to hinder diffusion between surrounding brain and the tumor. Increased laminin content in the tumor extracellular matrix may also further impede antibody diffusion within the tumor microenvironment 334. Perivascular access and transport may also be physically blocked by cancer cells that have been shown to reside and disperse along cerebral perivascular compartments 335–338, the subpial spaces 335, nerves 335, and white matter tracts 335; remarkably, and perhaps not coincidentally, such pathways are likely the same ones important for drug transport to and from deeper brain regions following central delivery 292–294. The disease pathology in Alzheimer’s disease and related dementias has been shown to influence the architecture of cerebral perivascular compartments due to either deposition of hyperphosphorylated tau or Aβ40 within the perivascular compartment 339, sometimes accompanied by enlargement of the perivascular space 340. Similar abnormally enlarged cerebral perivascular spaces have also been demonstrated in patients with traumatic brain injuries 341, acute ischemic stroke 342, and certain lysosomal storage disorders 343. Whether such a physiologically enlarged perivascular space affects perivascular drug transport remains an open question.
A final important consideration is systemic exposure, which is often relatively low with intra-CSF administration compared to systemic or intranasal routes of administration 122,126,318. However, CSF-administered substances do eventually drain from the central compartment via arachnoid granulations into the blood of the dural sinuses 344 as well as via lymphatic pathways that ultimately drain into systemic circulation as well 200,201.
CONCLUSIONS AND FUTURE PERSPECTIVES
The development of passive immunotherapies for CNS disorders has lagged behind that of other non-CNS indications 345. The reasons for this development lag for CNS indications are varied but they are thought to include insufficient mechanistic understanding of the brain, a paucity of reliable biomarkers to monitor disease progression and drug efficacy, and perhaps most significantly, the tremendous challenge of effective drug delivery to target brain regions 12. However, this review emphasizes that we have nonetheless made significant strides toward better understanding the challenges in overcoming the various physiological and pathological barriers that may impede therapeutic antibody delivery to the CNS. Creative strategies that harness the capacity of pre-existing anatomical pathways and mechanisms to deliver antibodies to their target site, coupled with rapid advances in antibody engineering that facilitate customization of antibody target selectivity, effector functions, and distribution properties, provide a promising outlook for the future of passive immunotherapies to treat CNS disorders.
In this review, we primarily focused on passive immunotherapies for the treatment of proteinopathies (e.g., PD, AD, and HD) and brain cancers. However, passive immunotherapies are also being investigated and applied for treating other CNS disorders such as multiple sclerosis, stroke, and traumatic brain injury among others. Multiple sclerosis (MS), an autoimmune neuroinflammatory disorder of unknown etiology causes demyelination of axons within the CNS. There are currently no treatments that can entirely stop disease progression or reverse existing disabilities in MS patients 346–348. Acute inflammatory episodes in MS are typically treated with intravenously administered corticosteroids that cause immunosuppression; however, the symptomatic relief and response to corticosteroids diminishes with repeated use 346.
Several passive immunotherapies that largely target immune responses in the periphery have been approved for treatment in MS 346,348: (i) natalizumab (Tysabri; Biogen/Élan; FDA approval – 2004) targets the cell adhesion molecule α4-integrin and blocks lymphocyte migration into the CNS 348–351; (ii) alemtuzumab (Lemtrada; Genzyme/Sanofi; FDA approval – 2014) depletes lymphocytes via antibody-mediated cell cytotoxicity (ADCC) and by activating the complement system 348,352–354; (iii) daclizumab (Zinbryta; Biogen/Abbvie; FDA approval – 2016) targets the IL2 receptor 348,355 but was recently withdrawn (April, 2018) due to complex and significant adverse events; and (iv) ocrelizumab (Ocrevus; Genentech/Roche; FDA approval – 2017) targets CD20+ B cells and is the first and only therapy currently that has been approved for the treatment of both primary and progressive forms of MS 348,356–58. However, therapies that directly target the inflammation in the central compartment in MS could potentially be more effective 347. The BBB appears to be intact during the first 6 weeks of lesion formation in MS when activation of microglia and macrophages begins with very few peripheral lymphocytes infiltrating the brain at this stage 347,359. Thus, strategies targeting passive immunotherapies to the CNS at the earliest stages of MS to prevent steps that lead to lesion formation could potentially alter the course of the disease but face the challenge of delivering antibodies across the BBB. Systemic immunosuppressive therapies that deplete lymphocyte populations also pose a risk of CNS infections 360 and investigating unique passive immunotherapy targets that prevent pathological infiltration of lymphocytes into the CNS without hampering CNS immune surveillance is crucial.
Ischemic strokes have the highest incidence among all cases of stroke and are caused due to obstruction of blood flow within cerebral vessels that may be attributed to one or more factors, e.g., fatty deposits along vessel walls that narrow the vessel lumen (atherosclerosis), elevated blood pressure, diabetes, and genetic predisposition. 361,362 The obstruction in the cerebral blood vessel may be caused either due to the formation of a blood clot (thrombus) within the brain itself (referred to as cerebral thrombosis) or a blood clot that migrates from the periphery into the brain (referred to as a cerebral embolism) 363. Arterial occlusion in stroke triggers a sequence of events leading to cell death and subsequent cognitive damage due to ischemia and neuroinflammatory responses in the brain 361. Elimination of the cerebral blood clot either by dissolving it using a thrombolytic agent such as tissue plasminogen activator (tPA) or by surgical removal (embolectomy) are currently the most effective treatments available for ischemic stroke 362. However, due to the narrow therapeutic time window (e.g., less than 4.5 hours post-stroke for tPA treatment) and invasiveness (e.g., embolectomy), currently available treatment options only benefit a small number of stroke patients 362. Passive immunotherapies are an attractive means to modulate neuroinflammation and neuroplasticity in stroke patients both as a means to widen the therapeutic window of current revascularization strategies and to minimize brain damage 362. For example, antibodies against myelin-associated proteins that inhibit neurite growth (such as Nogo-A and its receptor Nogo-66) are being explored as a way to increase neuroplasticity following stroke 364–366. tPA therapy has side-effects such as cerebral edema and hemorrhage that may partly be attributed to its interaction with the N-methyl-D-aspartate (NMDA) receptor, resulting in Ca2+ ion influx and the triggering of cell death signaling cascades 367. Antibodies that block the interaction between tPA and NMDA receptors are being investigated as a neuroprotective strategy in stroke 362. Since the BBB often appears compromised following ischemic stroke 368, it may pose less of a barrier for therapeutic antibody delivery to brain target sites. However, it may very well be crucial to administer the antibody-based therapy at specific points in the time course of post-stroke pathological events for central delivery and efficacy. For instance, systemic administration of anti-Nogo-A antibodies immediately post-stroke during the hyperacute phase (when tissue damage pathways are more active than tissue repair) may result in deleterious rather than beneficial effects 364.
Traumatic brain injuries are typically grouped based on severity of the impact/injury as mild, moderate, or severe. Mild traumatic brain injury (TBI), commonly referred to as a concussion, is often caused by a blunt non-penetrating trauma and its repeated occurrence (often seen in athletes playing high impact contact sports) has been linked to neurodegenerative conditions such as chronic traumatic encephalopathy (CTE) 369,370. Although primary mechanical injury in TBI may cause hematoma, edema, hemorrhage, and axonal injury, it is becoming apparent that secondary injury due to sterile neuroinflammation, excitotoxicity, and oxidative stress may ultimately result in more progressive long-term detrimental effects 371,372. Passive immunotherapies that can inhibit neurotoxic innate and adaptive immune responses without disrupting physiological CNS immune surveillance are therefore being investigated as potential treatment strategies for TBI 372. These strategies include intravenous immunoglobulin (IVIg) to ‘normalize’ the immune environment and monoclonal antibodies that can inhibit lymphocyte trafficking to the CNS (e.g., anti-CD20 and anti-CXCL10 antibodies) 372. Finally, it bears noting that BBB disruption observed in TBI correlates with adverse neurological effects, so restoration of normal BBB function is also being investigated as a therapeutic strategy 373.
An important physiological distinction between proteinopathies such as AD, PD, and HD versus conditions such as MS, stroke, and TBI appears related to differences in the intactness of the CNS barriers. For example, work with experimental models and clinical studies have demonstrated that the BBB may be profoundly compromised over the course of disease progression in conditions like MS but remain relatively intact during the course of pathological processes associated with AD 274. Future endeavors to translate passive immunotherapies for the treatment of CNS disorders will therefore have to take into consideration the spatial and temporal heterogeneity in the extent the BBB and the BCSFBs restrict antibody transport to brain target sites in different diseases and stages of pathology.
Table 1:
Strategies for delivering passive immunotherapies to the CNS (adapted from 263).
| Delivery strategy for CNS penetration/distribution | Advantages | Disadvantages |
|---|---|---|
| Parenteral systemic administration (IV infusion, SC/ IM injection) | ||
| Passive systemic delivery: passive transport across the BBB and BCSFBs and subsequent diffusion within the brain parenchyma | Well characterized and commonly used route of administration; global brain delivery; conventional antibody production | Less than 0.1% of systemically administered exogenous antibody penetrates the brain parenchyma 156; high doses required; possible side-effects (microhemorrhages, edema); pleiotropic effects/no targeted region specific delivery; neutralizing anti-drug antibodies may affect pharmacokinetics and diminish effects with chronic dosing |
| Receptor-mediated transport (RMT): bispecific antibodies and fusion proteins targeting transcytosis receptors expressed at the BBB allow the antibody/fusion protein to be shuttled across the BBB 282,285 | Well characterized and commonly used route of administration; global brain delivery; enhancement of delivery compared to passive transport across CNS barriers | Antibody engineering and selection of an appropriate RMT pathway is required; expression of transcytosis receptors in the periphery poses a risk of off-target side effects 106,286; antibodies may need to have a low affinity to the transcytosis receptor to escape the endothelial compartment and enter the brain parenchyma consequently requiring high doses 190; pleiotropic effects/no targeted region specific delivery; neutralizing anti-drug antibodies may affect pharmacokinetics and diminish effects with chronic dosing |
| BBB disruption strategies (MRI-guided focused ultrasound with microbubbles; hyperosmolar mannitol) | Enhancement of delivery compared to passive transport across CNS barriers; global brain delivery (hyperosmolar mannitol infusion) or region specific delivery (MRIguided focused ultrasound with microbubbles) | Requires expertise and/or additional equipment to apply and monitor BBB disruption strategies; BBB disruption poses a risk since it allows non-specific entry of plasma proteins/macromolecules into the CNS which may subsequently cause neurotoxicity 280 |
| Central administration (bypassing BBB and BCSFBs) | ||
| Intraparenchymal administration (infusion via convection enhanced delivery; implantation of polymer release system) | Relatively targeted delivery at site of administration (e.g., potentially advantageous while targeting brain tumors); limited peripheral side effects | Limited diffusion away from the site of administration prevents global delivery; Surgically invasive; restricted volume of administration necessitates reloading for chronic dosing; risk of infection |
| Intracerebroventricular (ICV) administration | Duration and rate of administration into the CSF can be controlled using a device (e.g., a Rickham reservoir or Ommaya reservoir) that allows isovolumetric and chronic dosing into the ventricles via a port implanted under the scalp in the subgaleal space 304; limited peripheral exposure; antibody-based therapeutic access to cerebral perivascular spaces may allow more global and rapid distribution within the brain relative to diffusion alone 122 | Surgically invasive; therapeutic needs to cross the ependyma, perivascular lining cells, and/or pia and glia limitans to access the brain parenchyma; reservoir reloading runs a risk of infection; possibility of device failure; diffusion out of the cerebral perivascular compartment and into the brain parenchyma may be restricted and dependent on several factors such as antibody size and interactions with receptors and extracellular matrix components, among others |
| Intrathecal administration | Duration and rate of administration into the CSF can be controlled using an infusion pump (e.g., Synchromed, Medtronic); antibody-based therapeutic access to cerebral perivascular spaces may allow more global and rapid distribution within the brain relative to diffusion alone 122 | Surgically invasive; therapeutic needs to cross the perivascular lining cells and/or pia and glia limitans to access the brain parenchyma; diffusion out of the cerebral perivascular compartment and into the brain parenchyma may be restricted and dependent on several factors such as antibody size and interactions with receptors and extracellular matrix components, among others; device failure and risk of infection |
| Intranasal administration | Non-invasive; some targeting to the CNS via direct nasal mucosae-to-brain pathways 213; rapid distribution within the brain via access to cerebral perivascular compartment; lower costs due to ease of self-administration | Limited transport across the nasal epithelial barriers, rapid clearance from the nasal mucosae, inter- and intra-patient variability in CNS delivery efficiency; enzymatic degradation in the nasal passages; therapeutic needs to cross the perivascular lining cells and/or pia and glia limitans to access the brain parenchyma; diffusion out of the cerebral perivascular compartment and into the brain parenchyma may be restricted and dependent on several factors such as antibody size and interactions with receptors and extracellular matrix components, among others; novel route of administration with preliminary characterization; pleiotropic effects/no targeted region specific delivery; potential immunogenicity |
ACKNOWLEDGEMENTS:
This work was generously supported by the Michael J. Fox Foundation for Parkinson’s Research, the Clinical and Translational Science Award program administered through the NIH National Center for Advancing Translational Sciences (NIH UL1TR000427 and KL2TR000428), the Wisconsin Alumni Research Foundation Accelerator Program, the University of Wisconsin-Madison School of Pharmacy, the Graduate School at the University of Wisconsin-Madison, fellowships through the National Science Foundation Graduate Research Fellowship under Grant No. DGE-1256259 (MEP) and (BWR), NIH fellowship (NRSA T32 EBO11434–MEP), Parkinson’s Foundation-American Parkinson’s Disease Association Summer Student Fellowship (PF-APDA-SFW-1730 – SB), and the Hilldale Undergraduate Research Fellowship from the University of Wisconsin-Madison (SB). Laser scanning confocal microscopy was performed with training and guidance from Dr. Michael Taylor (Nikon A1R) and Dr. Arash Bashirullah (Olympus FV1000) at the University of Wisconsin-Madison School of Pharmacy.
CONFLICTS OF INTEREST:
RGT acknowledges periodically receiving honoraria/speaking fees from organizations within academia, foundations and the biotechnology and pharmaceutical industry (Abbott, Biogen Idec, BioMarin, Denali Therapeutics, Genentech, Merck, Pfizer Neuroscience, and Shire). RGT also acknowledges occasional service as a consultant to industry on CNS drug delivery (Denali Therapeutics, Genentech, and Shire). NNK acknowledges July 2018 employment with Merck following completion of her PhD. RGT and MEP are co-inventors on patents and/or patent applications related to CNS drug delivery.
ABBREVIATIONS:
- CNS
central nervous system
- Ig
immunoglobulin
- IgG
immunoglobulin G
- Fc
crystallizable fragment
- Fab
antigen binding fragment
- FcRn
neonatal Fc receptor
- sdAb
single domain antibody
- BBB
blood-brain barrier
- BCSFBs
blood-cerebrospinal fluid barriers
- GBM
glioblastoma multiforme
- VEGF
vascular endothelial growth factor
- MMP-9
matrix metalloproteinase-9
- FDA
Food and Drug Administration
- PFS
progression free survival
- BELOB
bevacizumab and lomustine for recurrent GBM
- BTB
blood-tumor barrier
- TCR
T cell receptor
- MHC
major histocompatibility complex
- APC
antigen presenting cell
- Treg
regulatory T cell
- CTLA-4
cytotoxic T-lymphocyte-associated protein 4
- PD-L1
programmed cell death ligand-1
- PD-1
programmed cell death-1
- CD
classification determinant
- CSF
cerebrospinal fluid
- PCNSL
primary CNS lymphoma
- HD-MTX
high dose methotrexate
- WBRT
whole brain radiotherapy
- ADC
antibody drug conjugate
- EGFR
epithelial growth factor receptor
- MMAF
cytotoxin monomethyl auristatin F
- IL
interleukin
- HER2
human epidermal growth factor receptor 2
- CED
convection-enhanced delivery
- ICV
intracerebroventricular
- PE38
Pseudomonas exotoxin
- AD
Alzheimer’s disease
- Aβ
amyloid β-peptide
- APP
amyloid precursor protein
- sAPP
secreted amyloid precursor protein
- CTF
carboxy-terminal fragment
- AICD
amino-terminal APP intracellular domain
- BACE1
beta-site APP-cleaving enzyme 1
- CAA
cerebral amyloid angiopathy
- PHF
paired helical filament
- NFT
neurofibrillary tangle
- APOE
apolipoprotein E
- DIAN
Dominantly Inherited Alzheimer Network
- ARIA
amyloid-related imaging abnormalities
- TNF-α
tumor necrosis factor alpha
- EVVP
external vertebral venous plexus
- PD
Parkinson’s disease
- GDNF
glial-derived neurotrophic factor
- LAG3
lymphocyte-activation gene 3
- PFF
preformed fibril
- ECS
extracellular space
- HD
Huntington’s disease
- HTT
huntingtin gene
- mHTT
mutant huntingtin protein
- SEMA4D
semaphorin 4D protein
- TrkB
Tropomyosin receptor kinase B
- BDNF
brain-derived neurotrophic factor
- SOD
superoxide dismutase
- RMT
receptor-mediated transport
- MiNDS
miniaturized neural drug delivery system
- CLN2
ceroid lipofuscinosis type 2
- ISF
interstitial fluid
- IGF1
insulin-like growth factor 1
- IGFBP-2
insulin-like growth factor binding protein 2
- PVS
perivascular space
- IT
intrathecal
- IN
intranasal
- RECA-1
rat endothelial cell antigen-1
- GFAP
glial fibrillary acidic protein
- tPA
tissue plasminogen activator
- DAPI
4’, 6-diamidino-2-phenylindole
- NMDA
N-methyl-D-aspartate
REFERENCES:
- 1.Kindt TJ, Goldsby RA & Osborne BA Kuby Immunology. 6th Ed edn, (W. H. Freeman and Co., 2007). [Google Scholar]
- 2.Köhler G. & Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 256, 495–497 (1975). [DOI] [PubMed] [Google Scholar]
- 3.Urquhart L. Market watch: Top drugs and companies by sales in 2017. Nat Rev Drug Discov 17, 232, doi: 10.1038/nrd.2018.42 (2018). [DOI] [PubMed] [Google Scholar]
- 4.Chiu ML & Gilliland GL Engineering antibody therapeutics. Curr Opin Struct Biol 38, 163–173, doi: 10.1016/j.sbi.2016.07.012 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Kuo TT & Aveson VG Neonatal Fc receptor and IgG-based therapeutics. MAbs 3, 422–430, doi: 10.4161/mabs.3.5.16983 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nimmerjahn F. & Ravetch JV Fc-receptors as regulators of immunity. Adv Immunol 96, 179–204, doi: 10.1016/S0065-2776(07)96005-8 (2007). [DOI] [PubMed] [Google Scholar]
- 7.Nimmerjahn F. & Ravetch JV Analyzing antibody-Fc-receptor interactions. Methods Mol Biol 415, 151–162, doi: 10.1007/978-1-59745-570-1_9 (2008). [DOI] [PubMed] [Google Scholar]
- 8.Nimmerjahn F. & Ravetch JV Fcgamma receptors as regulators of immune responses. Nat Rev Immunol 8, 34–47, doi: 10.1038/nri2206 (2008). [DOI] [PubMed] [Google Scholar]
- 9.Nimmerjahn F. & Ravetch JV FcγRs in health and disease. Curr Top Microbiol Immunol 350, 105–125, doi: 10.1007/82_2010_86 (2011). [DOI] [PubMed] [Google Scholar]
- 10.Holliger P. & Hudson PJ Engineered antibody fragments and the rise of single domains. Nat Biotechnol 23, 1126–1136, doi: 10.1038/nbt1142 (2005). [DOI] [PubMed] [Google Scholar]
- 11.Wu AM & Senter PD Arming antibodies: prospects and challenges for immunoconjugates. Nat Biotechnol 23, 1137–1146, doi: 10.1038/nbt1141 (2005). [DOI] [PubMed] [Google Scholar]
- 12.Hammarlund-Udenaes M, de Lange E. & Thorne R. Drug Delivery to the Brain. 1 edn, (AAPSPress. Springer., 2014). [Google Scholar]
- 13.Reese TS & Karnovsky MJ Fine structural localization of a blood-brain barrier to exogenous peroxidase. J Cell Biol 34, 207–217 (1967). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Becker NH, Novikoff AB & Zimmerman HM Fine structure observations of the uptake of intravenously injected peroxidase by the rat choroid plexus. J Histochem Cytochem 15, 160–165 (1967). [DOI] [PubMed] [Google Scholar]
- 15.Nabeshima S, Reese TS, Landis DM & Brightman MW Junctions in the meninges and marginal glia. J Comp Neurol 164, 127–169, doi: 10.1002/cne.901640202 (1975). [DOI] [PubMed] [Google Scholar]
- 16.Mokri B. The Monro-Kellie hypothesis: applications in CSF volume depletion. Neurology 56, 1746–1748 (2001). [DOI] [PubMed] [Google Scholar]
- 17.Chamberlain MC, Baik CS, Gadi VK, Bhatia S. & Chow LQ Systemic therapy of brain metastases: non-small cell lung cancer, breast cancer, and melanoma. Neuro Oncol 19, i1–i24, doi: 10.1093/neuonc/now197 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hall WA, Djalilian HR, Nussbaum ES & Cho KH Long-term survival with metastatic cancer to the brain. Med Oncol 17, 279–286 (2000). [DOI] [PubMed] [Google Scholar]
- 19.Maher EA, Mietz J, Arteaga CL, DePinho RA & Mohla S. Brain metastasis: opportunities in basic and translational research. Cancer Res 69, 6015–6020, doi: 10.1158/0008-5472.CAN-08-4347 (2009). [DOI] [PubMed] [Google Scholar]
- 20.Black P. & Loeffler J. Cancer of the Nervous System. 2nd edn, (Lippincott Williams & Wilkins, 2004). [Google Scholar]
- 21.Russel DC & Rubinstein LJ Pathology of tumors of the nervous system. 5th edn, (1989). [Google Scholar]
- 22.Johnson DR & O’Neill BP Glioblastoma survival in the United States before and during the temozolomide era. J Neurooncol 107, 359–364, doi: 10.1007/s11060-011-0749-4 (2012). [DOI] [PubMed] [Google Scholar]
- 23.Stupp R. et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 352, 987–996, doi: 10.1056/NEJMoa043330 (2005). [DOI] [PubMed] [Google Scholar]
- 24.Stupp R. et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol 10, 459–466, doi: 10.1016/S1470-2045(09)70025-7 (2009). [DOI] [PubMed] [Google Scholar]
- 25.Miyauchi JT & Tsirka SE Advances in immunotherapeutic research for glioma therapy. J Neurol, doi: 10.1007/s00415-017-8695-5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sampson JH, Maus MV & June CH Immunotherapy for Brain Tumors. J Clin Oncol 35, 2450–2456, doi: 10.1200/JCO.2017.72.8089 (2017). [DOI] [PubMed] [Google Scholar]
- 27.Patel AP et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 344, 1396–1401, doi: 10.1126/science.1254257 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Claes A, Idema AJ & Wesseling P. Diffuse glioma growth: a guerilla war. Acta Neuropathol 114, 443–458, doi: 10.1007/s00401-007-0293-7 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Green AL & Kieran MW Pediatric brainstem gliomas: new understanding leads to potential new treatments for two very different tumors. Curr Oncol Rep 17, 436, doi: 10.1007/s11912-014-0436-7 (2015). [DOI] [PubMed] [Google Scholar]
- 30.Ferrara N, Hillan KJ, Gerber HP & Novotny W. Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat Rev Drug Discov 3, 391–400, doi: 10.1038/nrd1381 (2004). [DOI] [PubMed] [Google Scholar]
- 31.Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med 285, 1182–1186, doi: 10.1056/NEJM197111182852108 (1971). [DOI] [PubMed] [Google Scholar]
- 32.Folkman J, Merler E, Abernathy C. & Williams G. Isolation of a tumor factor responsible for angiogenesis. J Exp Med 133, 275–288 (1971). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Leung DW, Cachianes G, Kuang WJ, Goeddel DV & Ferrara N. Vascular endothelial growth factor is a secreted angiogenic mitogen. Science 246, 1306–1309 (1989). [DOI] [PubMed] [Google Scholar]
- 34.Senger DR et al. Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science 219, 983–985 (1983). [DOI] [PubMed] [Google Scholar]
- 35.Senger DR, Perruzzi CA, Feder J. & Dvorak HF A highly conserved vascular permeability factor secreted by a variety of human and rodent tumor cell lines. Cancer Res 46, 5629–5632 (1986). [PubMed] [Google Scholar]
- 36.Plate KH, Breier G, Weich HA & Risau W. Vascular endothelial growth factor is a potential tumour angiogenesis factor in human gliomas in vivo. Nature 359, 845–848, doi: 10.1038/359845a0 (1992). [DOI] [PubMed] [Google Scholar]
- 37.Brem S, Cotran R. & Folkman J. Tumor angiogenesis: a quantitative method for histologic grading. J Natl Cancer Inst 48, 347–356 (1972). [PubMed] [Google Scholar]
- 38.Brem SS et al. Inhibition of angiogenesis and tumor growth in the brain. Suppression of endothelial cell turnover by penicillamine and the depletion of copper, an angiogenic cofactor. Am J Pathol 137, 1121–1142 (1990). [PMC free article] [PubMed] [Google Scholar]
- 39.Field KM, Jordan JT, Wen PY, Rosenthal MA & Reardon DA Bevacizumab and glioblastoma: scientific review, newly reported updates, and ongoing controversies. Cancer 121, 997–1007, doi: 10.1002/cncr.28935 (2015). [DOI] [PubMed] [Google Scholar]
- 40.Vredenburgh JJ et al. Phase II trial of bevacizumab and irinotecan in recurrent malignant glioma. Clin Cancer Res 13, 1253–1259, doi: 10.1158/1078-0432.CCR-06-2309 (2007). [DOI] [PubMed] [Google Scholar]
- 41.Vredenburgh JJ et al. Bevacizumab plus irinotecan in recurrent glioblastoma multiforme. J Clin Oncol 25, 4722–4729, doi: 10.1200/JCO.2007.12.2440 (2007). [DOI] [PubMed] [Google Scholar]
- 42.Friedman HS et al. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J Clin Oncol 27, 4733–4740, doi: 10.1200/JCO.2008.19.8721 (2009). [DOI] [PubMed] [Google Scholar]
- 43.Kreisl TN et al. Phase II trial of single-agent bevacizumab followed by bevacizumab plus irinotecan at tumor progression in recurrent glioblastoma. J Clin Oncol 27, 740–745, doi: 10.1200/JCO.2008.16.3055 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cohen MH, Shen YL, Keegan P. & Pazdur R. FDA drug approval summary: bevacizumab (Avastin) as treatment of recurrent glioblastoma multiforme. Oncologist 14, 1131–1138, doi: 10.1634/theoncologist.2009-0121 (2009). [DOI] [PubMed] [Google Scholar]
- 45.Taal W. et al. Single-agent bevacizumab or lomustine versus a combination of bevacizumab plus lomustine in patients with recurrent glioblastoma (BELOB trial): a randomised controlled phase 2 trial. Lancet Oncol 15, 943–953, doi: 10.1016/S1470-2045(14)70314-6 (2014). [DOI] [PubMed] [Google Scholar]
- 46.Gilbert MR et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N Engl J Med 370, 699–708, doi: 10.1056/NEJMoa1308573 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Treps L, Perret R, Edmond S, Ricard D. & Gavard J. Glioblastoma stem-like cells secrete the pro-angiogenic VEGF-A factor in extracellular vesicles. J Extracell Vesicles 6, 1359479, doi: 10.1080/20013078.2017.1359479 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stefanini MO, Wu FT, Mac Gabhann F. & Popel AS The presence of VEGF receptors on the luminal surface of endothelial cells affects VEGF distribution and VEGF signaling. PLoS Comput Biol 5, e1000622, doi: 10.1371/journal.pcbi.1000622 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Armstrong TS, Wen PY, Gilbert MR & Schiff D. Management of treatment-associated toxicites of anti-angiogenic therapy in patients with brain tumors. Neuro Oncol 14, 1203–1214, doi: 10.1093/neuonc/nor223 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yu Z. et al. Efficacy and safety of bevacizumab for the treatment of glioblastoma. Exp Ther Med 11, 371–380, doi: 10.3892/etm.2015.2947 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hudson N. et al. Differential apicobasal VEGF signaling at vascular blood-neural barriers. Dev Cell 30, 541–552, doi: 10.1016/j.devcel.2014.06.027 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Driessens G, Kline J. & Gajewski TF Costimulatory and coinhibitory receptors in anti-tumor immunity. Immunol Rev 229, 126–144, doi: 10.1111/j.1600-065X.2009.00771.x (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Buchbinder EI & Desai A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am J Clin Oncol 39, 98–106, doi: 10.1097/COC.0000000000000239 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Huang J. et al. Immune Checkpoint in Glioblastoma: Promising and Challenging. Front Pharmacol 8, 242, doi: 10.3389/fphar.2017.00242 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Huang B. et al. Advances in Immunotherapy for Glioblastoma Multiforme. J Immunol Res 2017, 3597613, doi: 10.1155/2017/3597613 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Brunet JF et al. A new member of the immunoglobulin superfamily--CTLA-4. Nature 328, 267–270, doi: 10.1038/328267a0 (1987). [DOI] [PubMed] [Google Scholar]
- 57.Azuma M. et al. B70 antigen is a second ligand for CTLA-4 and CD28. Nature 366, 76–79, doi: 10.1038/366076a0 (1993). [DOI] [PubMed] [Google Scholar]
- 58.Carter T, Shaw H, Cohn-Brown D, Chester K. & Mulholland P. Ipilimumab and Bevacizumab in Glioblastoma. Clin Oncol (R Coll Radiol) 28, 622–626, doi: 10.1016/j.clon.2016.04.042 (2016). [DOI] [PubMed] [Google Scholar]
- 59.Ishida Y, Agata Y, Shibahara K. & Honjo T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J 11, 3887–3895 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dong H, Zhu G, Tamada K. & Chen L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat Med 5, 1365–1369, doi: 10.1038/70932 (1999). [DOI] [PubMed] [Google Scholar]
- 61.Zang X. & Allison JP The B7 family and cancer therapy: costimulation and coinhibition. Clin Cancer Res 13, 5271–5279, doi: 10.1158/1078-0432.CCR-07-1030 (2007). [DOI] [PubMed] [Google Scholar]
- 62.Xue S, Hu M, Iyer V. & Yu J. Blocking the PD-1/PD-L1 pathway in glioma: a potential new treatment strategy. J Hematol Oncol 10, 81, doi: 10.1186/s13045-017-0455-6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Long GV et al. Combination nivolumab and ipilimumab or nivolumab alone in melanoma brain metastases: a multicentre randomised phase 2 study. Lancet Oncol 19, 672–681, doi: 10.1016/S1470-2045(18)30139-6 (2018). [DOI] [PubMed] [Google Scholar]
- 64.Goldberg SB et al. Pembrolizumab for patients with melanoma or non-small-cell lung cancer and untreated brain metastases: early analysis of a non-randomised, open-label, phase 2 trial. Lancet Oncol 17, 976–983, doi: 10.1016/S1470-2045(16)30053-5 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.clinicaltrials.gov. (2018).
- 66.Sallusto F. et al. T-cell trafficking in the central nervous system. Immunol Rev 248, 216–227, doi: 10.1111/j.1600-065X.2012.01140.x (2012). [DOI] [PubMed] [Google Scholar]
- 67.Engelhardt B. et al. Vascular, glial, and lymphatic immune gateways of the central nervous system. Acta Neuropathol 132, 317–338, doi: 10.1007/s00401-016-1606-5 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Seabrook TJ, Johnston M. & Hay JB Cerebral spinal fluid lymphocytes are part of the normal recirculating lymphocyte pool. J Neuroimmunol 91, 100–107 (1998). [DOI] [PubMed] [Google Scholar]
- 69.Mrugala MM, Rubenstein JL, Ponzoni M. & Batchelor TT Insights into the biology of primary central nervous system lymphoma. Curr Oncol Rep 11, 73–80 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pfannl R. & Harris F. Pathology of primary central nervous system lymphoma and related conditions. 29–41 (Butterworth-Heinemann, 2004). [Google Scholar]
- 71.Neuwelt EA et al. Primary CNS lymphoma treated with osmotic blood-brain barrier disruption: prolonged survival and preservation of cognitive function. J Clin Oncol 9, 1580–1590, doi: 10.1200/JCO.1991.9.9.1580 (1991). [DOI] [PubMed] [Google Scholar]
- 72.Coté TR, Manns A, Hardy CR, Yellin FJ & Hartge P. Epidemiology of brain lymphoma among people with or without acquired immunodeficiency syndrome. AIDS/Cancer Study Group. J Natl Cancer Inst 88, 675–679 (1996). [DOI] [PubMed] [Google Scholar]
- 73.Ferry J. & Harris N. in Pathology and Rejection Diagnosis in Solid Organ Transplantation (eds Solez K, Racusen L, & Billingham M) 277–301 (Marcel Dekker, 1994). [Google Scholar]
- 74.Aho R, Ekfors T, Haltia M. & Kalimo H. Pathogenesis of primary central nervous system lymphoma: invasion of malignant lymphoid cells into and within the brain parenchyme. Acta Neuropathol 86, 71–76 (1993). [DOI] [PubMed] [Google Scholar]
- 75.Phillips EH, Fox CP & Cwynarski K. Primary CNS lymphoma. Curr Hematol Malig Rep 9, 243–253, doi: 10.1007/s11899-014-0217-2 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Löscher W. & Potschka H. Drug resistance in brain diseases and the role of drug efflux transporters. Nat Rev Neurosci 6, 591–602, doi: 10.1038/nrn1728 (2005). [DOI] [PubMed] [Google Scholar]
- 77.Li L, Agarwal S. & Elmquist WF Brain efflux index to investigate the influence of active efflux on brain distribution of pemetrexed and methotrexate. Drug Metab Dispos 41, 659–667, doi: 10.1124/dmd.112.049254 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Citterio G, Reni M, Gatta G. & Ferreri AJM Primary central nervous system lymphoma. Crit Rev Oncol Hematol 113, 97–110, doi: 10.1016/j.critrevonc.2017.03.019 (2017). [DOI] [PubMed] [Google Scholar]
- 79.Batchelor TT et al. Rituximab monotherapy for patients with recurrent primary CNS lymphoma. Neurology 76, 929–930, doi: 10.1212/WNL.0b013e31820f2d94 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Angelov L. et al. Blood-brain barrier disruption and intra-arterial methotrexate-based therapy for newly diagnosed primary CNS lymphoma: a multi-institutional experience. J Clin Oncol 27, 3503–3509, doi: 10.1200/JCO.2008.19.3789 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gan HK, van den Bent M, Lassman AB, Reardon DA & Scott AM Antibody-drug conjugates in glioblastoma therapy: the right drugs to the right cells. Nat Rev Clin Oncol 14, 695–707, doi: 10.1038/nrclinonc.2017.95 (2017). [DOI] [PubMed] [Google Scholar]
- 82.Casacó A. et al. Phase I single-dose study of intracavitary-administered Nimotuzumab labeled with 188 Re in adult recurrent high-grade glioma. Cancer Biol Ther 7, 333–339 (2008). [DOI] [PubMed] [Google Scholar]
- 83.Riva P. et al. 131I radioconjugated antibodies for the locoregional radioimmunotherapy of high-grade malignant glioma--phase I and II study. Acta Oncol 38, 351–359 (1999). [DOI] [PubMed] [Google Scholar]
- 84.Sivasankaran B. et al. Tenascin-C is a novel RBPJkappa-induced target gene for Notch signaling in gliomas. Cancer Res 69, 458–465, doi: 10.1158/0008-5472.CAN-08-2610 (2009). [DOI] [PubMed] [Google Scholar]
- 85.Zalutsky MR et al. Clinical experience with alpha-particle emitting 211At: treatment of recurrent brain tumor patients with 211At-labeled chimeric antitenascin monoclonal antibody 81C6. J Nucl Med 49, 30–38, doi: 10.2967/jnumed.107.046938 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Phillips AC et al. ABT-414, an Antibody-Drug Conjugate Targeting a Tumor-Selective EGFR Epitope. Mol Cancer Ther 15, 661–669, doi: 10.1158/1535-7163.MCT-15-0901 (2016). [DOI] [PubMed] [Google Scholar]
- 87.Lopus M. et al. Maytansine and cellular metabolites of antibody-maytansinoid conjugates strongly suppress microtubule dynamics by binding to microtubules. Mol Cancer Ther 9, 2689–2699, doi: 10.1158/1535-7163.MCT-10-0644 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Parrish KE, Sarkaria JN & Elmquist WF Improving drug delivery to primary and metastatic brain tumors: strategies to overcome the blood-brain barrier. Clin Pharmacol Ther 97, 336–346, doi: 10.1002/cpt.71 (2015). [DOI] [PubMed] [Google Scholar]
- 89.Gavrilovic IT & Posner JB Brain metastases: epidemiology and pathophysiology. J Neurooncol 75, 5–14, doi: 10.1007/s11060-004-8093-6 (2005). [DOI] [PubMed] [Google Scholar]
- 90.Valiente M. et al. Serpins promote cancer cell survival and vascular co-option in brain metastasis. Cell 156, 1002–1016, doi: 10.1016/j.cell.2014.01.040 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hudis CA Trastuzumab--mechanism of action and use in clinical practice. N Engl J Med 357, 39–51, doi: 10.1056/NEJMra043186 (2007). [DOI] [PubMed] [Google Scholar]
- 92.Paik S. et al. Pathologic findings from the National Surgical Adjuvant Breast and Bowel Project: prognostic significance of erbB-2 protein overexpression in primary breast cancer. J Clin Oncol 8, 103–112, doi: 10.1200/JCO.1990.8.1.103 (1990). [DOI] [PubMed] [Google Scholar]
- 93.Kirsch DG & Hochberg FH Targeting HER2 in brain metastases from breast cancer. Clin Cancer Res 9, 5435–5436 (2003). [PubMed] [Google Scholar]
- 94.Park WY et al. Intrathecal Trastuzumab Treatment in Patients with Breast Cancer and Leptomeningeal Carcinomatosis. Cancer Res Treat 48, 843–847, doi: 10.4143/crt.2014.234 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lu NT et al. Intrathecal trastuzumab: immunotherapy improves the prognosis of leptomeningeal metastases in HER-2+ breast cancer patient. J Immunother Cancer 3, 41, doi: 10.1186/s40425-015-0084-y (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Watanabe H. et al. The effect of nivolumab treatment for central nervous system metastases in non-small cell lung cancer. Journal of clinical oncology 35 (2017). [Google Scholar]
- 97.Margolin K. Ipilimumab in a Phase II trial of melanoma patients with brain metastases. Oncoimmunology 1, 1197–1199, doi: 10.4161/onci.20687 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Margolin K. et al. Ipilimumab in patients with melanoma and brain metastases: an open-label, phase 2 trial. Lancet Oncol 13, 459–465, doi: 10.1016/S1470-2045(12)70090-6 (2012). [DOI] [PubMed] [Google Scholar]
- 99.Wang W, Wang EQ & Balthasar JP Monoclonal antibody pharmacokinetics and pharmacodynamics. Clin Pharmacol Ther 84, 548–558, doi: 10.1038/clpt.2008.170 (2008). [DOI] [PubMed] [Google Scholar]
- 100.Groothuis DR The blood-brain and blood-tumor barriers: a review of strategies for increasing drug delivery. Neuro Oncol 2, 45–59 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Schlageter KE, Molnar P, Lapin GD & Groothuis DR Microvessel organization and structure in experimental brain tumors: microvessel populations with distinctive structural and functional properties. Microvasc Res 58, 312–328, doi: 10.1006/mvre.1999.2188 (1999). [DOI] [PubMed] [Google Scholar]
- 102.Lockman PR et al. Heterogeneous blood-tumor barrier permeability determines drug efficacy in experimental brain metastases of breast cancer. Clin Cancer Res 16, 5664–5678, doi: 10.1158/1078-0432.CCR-10-1564 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sarin H. Physiologic upper limits of pore size of different blood capillary types and another perspective on the dual pore theory of microvascular permeability. J Angiogenes Res 2, 14, doi: 10.1186/2040-2384-2-14 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Michel CC Transport of macromolecules through microvascular walls. Cardiovasc Res 32, 644–653 (1996). [PubMed] [Google Scholar]
- 105.Razpotnik R, Novak N, Čurin Šerbec V. & Rajcevic U. Targeting Malignant Brain Tumors with Antibodies. Front Immunol 8, 1181, doi: 10.3389/fimmu.2017.01181 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Gatter KC, Brown G, Trowbridge IS, Woolston RE & Mason DY Transferrin receptors in human tissues: their distribution and possible clinical relevance. J Clin Pathol 36, 539–545 (1983). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Yu YJ & Watts RJ Developing therapeutic antibodies for neurodegenerative disease. Neurotherapeutics 10, 459–472, doi: 10.1007/s13311-013-0187-4 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.van Tellingen O. et al. Overcoming the blood-brain tumor barrier for effective glioblastoma treatment. Drug Resist Updat 19, 1–12, doi: 10.1016/j.drup.2015.02.002 (2015). [DOI] [PubMed] [Google Scholar]
- 109.Zünkeler B. et al. Quantification and pharmacokinetics of blood-brain barrier disruption in humans. J Neurosurg 85, 1056–1065, doi: 10.3171/jns.1996.85.6.1056 (1996). [DOI] [PubMed] [Google Scholar]
- 110.Jones PL, Gallagher BM & Sands H. Autoradiographic analysis of monoclonal antibody distribution in human colon and breast tumor xenografts. Cancer Immunol Immunother 22, 139–143 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Munson JM & Shieh AC Interstitial fluid flow in cancer: implications for disease progression and treatment. Cancer Manag Res 6, 317–328, doi: 10.2147/CMAR.S65444 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Baxter LT & Jain RK Transport of fluid and macromolecules in tumors. I. Role of interstitial pressure and convection. Microvasc Res 37, 77–104 (1989). [DOI] [PubMed] [Google Scholar]
- 113.Jain RK & Baxter LT Mechanisms of heterogeneous distribution of monoclonal antibodies and other macromolecules in tumors: significance of elevated interstitial pressure. Cancer Res 48, 7022–7032 (1988). [PubMed] [Google Scholar]
- 114.Bobo RH et al. Convection-enhanced delivery of macromolecules in the brain. Proc Natl Acad Sci U S A 91, 2076–2080 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Patel SJ et al. Safety and feasibility of convection-enhanced delivery of Cotara for the treatment of malignant glioma: initial experience in 51 patients. Neurosurgery 56, 1243–1252; discussion 1252–1243 (2005). [DOI] [PubMed] [Google Scholar]
- 116.Sampson JH et al. Comparison of intratumoral bolus injection and convection-enhanced delivery of radiolabeled antitenascin monoclonal antibodies. Neurosurg Focus 20, E14, doi: 10.3171/foc.2006.20.4.9 (2006). [DOI] [PubMed] [Google Scholar]
- 117.Ivasyk I, Morgenstern PF, Wembacher-Schroeder E. & Souweidane MM Influence of an intratumoral cyst on drug distribution by convection-enhanced delivery: case report. J Neurosurg Pediatr 20, 256–260, doi: 10.3171/2017.5.PEDS1774 (2017). [DOI] [PubMed] [Google Scholar]
- 118.Larson SM, Carrasquillo JA, Cheung NK & Press OW Radioimmunotherapy of human tumours. Nat Rev Cancer 15, 347–360, doi: 10.1038/nrc3925 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Bao X, Pastan I, Bigner DD & Chandramohan V. EGFR/EGFRvIII-targeted immunotoxin therapy for the treatment of glioblastomas via convection-enhanced delivery. Receptors Clin Investig 3, doi: 10.14800/rci.1430 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Bigner DD et al. Phase I studies of treatment of malignant gliomas and neoplastic meningitis with 131I-radiolabeled monoclonal antibodies anti-tenascin 81C6 and anti-chondroitin proteoglycan sulfate Me1–14 F (ab’)2--a preliminary report. J Neurooncol 24, 109–122 (1995). [DOI] [PubMed] [Google Scholar]
- 121.Brown MT et al. Intrathecal 131I-labeled antitenascin monoclonal antibody 81C6 treatment of patients with leptomeningeal neoplasms or primary brain tumor resection cavities with subarachnoid communication: phase I trial results. Clin Cancer Res 2, 963–972 (1996). [PubMed] [Google Scholar]
- 122.Pizzo ME et al. Intrathecal antibody distribution in the rat brain: surface diffusion, perivascular transport, and osmotic enhancement of delivery. J Physiol, doi: 10.1113/JP275105 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Perissinotti AJ & Reeves DJ Role of intrathecal rituximab and trastuzumab in the management of leptomeningeal carcinomatosis. Ann Pharmacother 44, 1633–1640, doi: 10.1345/aph.1P197 (2010). [DOI] [PubMed] [Google Scholar]
- 124.Calias P, Banks WA, Begley D, Scarpa M. & Dickson P. Intrathecal delivery of protein therapeutics to the brain: a critical reassessment. Pharmacol Ther 144, 114–122, doi: 10.1016/j.pharmthera.2014.05.009 (2014). [DOI] [PubMed] [Google Scholar]
- 125.Pastan I, Hassan R, Fitzgerald DJ & Kreitman RJ Immunotoxin therapy of cancer. Nat Rev Cancer 6, 559–565, doi: 10.1038/nrc1891 (2006). [DOI] [PubMed] [Google Scholar]
- 126.Lochhead JJ & Thorne RG Intranasal delivery of biologics to the central nervous system. Adv Drug Deliv Rev 64, 614–628, doi: 10.1016/j.addr.2011.11.002 (2012). [DOI] [PubMed] [Google Scholar]
- 127.Thorne RG, Pronk GJ, Padmanabhan V. & Frey WH Delivery of insulin-like growth factor-I to the rat brain and spinal cord along olfactory and trigeminal pathways following intranasal administration. Neuroscience 127, 481–496, doi: 10.1016/j.neuroscience.2004.05.029 (2004). [DOI] [PubMed] [Google Scholar]
- 128.Goodwin CR et al. Local delivery methods of therapeutic agents in the treatment of diffuse intrinsic brainstem gliomas. Clin Neurol Neurosurg 142, 120–127, doi: 10.1016/j.clineuro.2016.01.007 (2016). [DOI] [PubMed] [Google Scholar]
- 129.Prince, M., Comas-Herrera, A., Knapp, M., Guerchet, M. & Karagiannidou, M. (2016). [Google Scholar]
- 130.(Alzheimer’s association, 2018).
- 131.McKhann G. et al. Clinical diagnosis of Alzheimer’s disease: report of the NINCDSADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 34, 939–944 (1984). [DOI] [PubMed] [Google Scholar]
- 132.Serrano-Pozo A, Frosch MP, Masliah E. & Hyman BT Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med 1, a006189, doi: 10.1101/cshperspect.a006189 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.McKhann GM et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7, 263–269, doi: 10.1016/j.jalz.2011.03.005 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Jack CR et al. Introduction to the recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7, 257–262, doi: 10.1016/j.jalz.2011.03.004 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Selkoe DJ & Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med 8, 595–608, doi: 10.15252/emmm.201606210 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Chow VW, Mattson MP, Wong PC & Gleichmann M. An overview of APP processing enzymes and products. Neuromolecular Med 12, 1–12, doi: 10.1007/s12017-009-8104-z (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Haass C, Kaether C, Thinakaran G. & Sisodia S. Trafficking and proteolytic processing of APP. Cold Spring Harb Perspect Med 2, a006270, doi: 10.1101/cshperspect.a006270 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Finder VH & Glockshuber R. Amyloid-beta aggregation. Neurodegener Dis 4, 13–27, doi: 10.1159/000100355 (2007). [DOI] [PubMed] [Google Scholar]
- 139.Tarasoff-Conway JM et al. Clearance systems in the brain-implications for Alzheimer disease. Nat Rev Neurol 11, 457–470, doi: 10.1038/nrneurol.2015.119 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Hook G, Yu J, Toneff T, Kindy M. & Hook V. Brain pyroglutamate amyloid-β is produced by cathepsin B and is reduced by the cysteine protease inhibitor E64d, representing a potential Alzheimer’s disease therapeutic. J Alzheimers Dis 41, 129–149, doi: 10.3233/JAD-131370 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Musiek ES & Holtzman DM Three dimensions of the amyloid hypothesis: time, space and ‘wingmen’. Nat Neurosci 18, 800–806, doi: 10.1038/nn.4018 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Benilova I, Karran E. & De Strooper B. The toxic Aβ oligomer and Alzheimer’s disease: an emperor in need of clothes. Nat Neurosci 15, 349–357, doi: 10.1038/nn.3028 (2012). [DOI] [PubMed] [Google Scholar]
- 143.Ahmed M. et al. Structural conversion of neurotoxic amyloid-beta(1–42) oligomers to fibrils. Nat Struct Mol Biol 17, 561–567, doi: 10.1038/nsmb.1799 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Roher AE et al. Oligomerizaiton and fibril asssembly of the amyloid-beta protein. Biochim Biophys Acta 1502, 31–43 (2000). [DOI] [PubMed] [Google Scholar]
- 145.Sengupta U, Nilson AN & Kayed R. The Role of Amyloid-β Oligomers in Toxicity, Propagation, and Immunotherapy. EBioMedicine 6, 42–49, doi: 10.1016/j.ebiom.2016.03.035 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Iqbal K, Liu F. & Gong CX Tau and neurodegenerative disease: the story so far. Nat Rev Neurol 12, 15–27, doi: 10.1038/nrneurol.2015.225 (2016). [DOI] [PubMed] [Google Scholar]
- 147.Lannfelt L. et al. Perspectives on future Alzheimer therapies: amyloid-β protofibrils - a new target for immunotherapy with BAN2401 in Alzheimer’s disease. Alzheimers Res Ther 6, 16, doi: 10.1186/alzrt246 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.St-Amour I, Cicchetti F. & Calon F. Immunotherapies in Alzheimer’s disease: Too much, too little, too late or off-target? Acta Neuropathol 131, 481–504, doi: 10.1007/s00401-015-1518-9 (2016). [DOI] [PubMed] [Google Scholar]
- 149.Cummings J, Lee G, Mortsdorf T, Ritter A. & Zhong K. Alzheimer’s disease drug development pipeline: 2017. Alzheimers Dement (N Y) 3, 367–384, doi: 10.1016/j.trci.2017.05.002 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Bard F. et al. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med 6, 916–919, doi: 10.1038/78682 (2000). [DOI] [PubMed] [Google Scholar]
- 151.DeMattos RB et al. Peripheral anti-A beta antibody alters CNS and plasma A beta clearance and decreases brain A beta burden in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A 98, 8850–8855, doi: 10.1073/pnas.151261398 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Panza F, Solfrizzi V, Imbimbo BP & Logroscino G. Amyloid-directed monoclonal antibodies for the treatment of Alzheimer’s disease: the point of no return? Expert Opin Biol Ther 14, 1465–1476, doi: 10.1517/14712598.2014.935332 (2014). [DOI] [PubMed] [Google Scholar]
- 153.Salloway S. et al. Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N Engl J Med 370, 322–333, doi: 10.1056/NEJMoa1304839 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Doody RS et al. Phase 3 trials of solanezumab for mild-to-moderate Alzheimer’s disease. N Engl J Med 370, 311–321, doi: 10.1056/NEJMoa1312889 (2014). [DOI] [PubMed] [Google Scholar]
- 155.Doody RS, Farlow M, Aisen PS & Committee A. s. D. C. S. D. A. a. P. Phase 3 trials of solanezumab and bapineuzumab for Alzheimer’s disease. N Engl J Med 370, 1460, doi: 10.1056/NEJMc1402193 (2014). [DOI] [PubMed] [Google Scholar]
- 156.St-Amour I. et al. Brain bioavailability of human intravenous immunoglobulin and its transport through the murine blood-brain barrier. J Cereb Blood Flow Metab 33, 1983–1992, doi: 10.1038/jcbfm.2013.160 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Siemers ER et al. Phase 3 solanezumab trials: Secondary outcomes in mild Alzheimer’s disease patients. Alzheimers Dement 12, 110–120, doi: 10.1016/j.jalz.2015.06.1893 (2016). [DOI] [PubMed] [Google Scholar]
- 158.Honig LS et al. Trial of Solanezumab for Mild Dementia Due to Alzheimer’s Disease. N Engl J Med 378, 321–330, doi: 10.1056/NEJMoa1705971 (2018). [DOI] [PubMed] [Google Scholar]
- 159.Panza F. et al. The potential of solanezumab and gantenerumab to prevent Alzheimer’s disease in people with inherited mutations that cause its early onset. Expert Opin Biol Ther 18, 25–35, doi: 10.1080/14712598.2018.1389885 (2018). [DOI] [PubMed] [Google Scholar]
- 160.Sperling RA et al. The A4 study: stopping AD before symptoms begin? Sci Transl Med 6, 228fs213, doi: 10.1126/scitranslmed.3007941 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Andreasen N. et al. First administration of the Fc-attenuated anti-β amyloid antibody GSK933776 to patients with mild Alzheimer’s disease: a randomized, placebo-controlled study. PLoS One 10, e0098153, doi: 10.1371/journal.pone.0098153 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Delnomdedieu M. et al. First-In-Human safety and long-term exposure data for AAB-003 (PF-05236812) and biomarkers after intravenous infusions of escalating doses in patients with mild to moderate Alzheimer’s disease. Alzheimers Res Ther 8, 12, doi: 10.1186/s13195-016-0177-y (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Pedersen JT & Sigurdsson EM Tau immunotherapy for Alzheimer’s disease. Trends Mol Med 21, 394–402, doi: 10.1016/j.molmed.2015.03.003 (2015). [DOI] [PubMed] [Google Scholar]
- 164.Asuni AA, Boutajangout A, Quartermain D. & Sigurdsson EM Immunotherapy targeting pathological tau conformers in a tangle mouse model reduces brain pathology with associated functional improvements. J Neurosci 27, 9115–9129, doi: 10.1523/JNEUROSCI.2361-07.2007 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Iqbal K. et al. Tau pathology in Alzheimer disease and other tauopathies. Biochim Biophys Acta 1739, 198–210, doi: 10.1016/j.bbadis.2004.09.008 (2005). [DOI] [PubMed] [Google Scholar]
- 166.Yamada K. Extracellular Tau and Its Potential Role in the Propagation of Tau Pathology. Front Neurosci 11, 667, doi: 10.3389/fnins.2017.00667 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Yamada K. In Vivo Microdialysis of Brain Interstitial Fluid for the Determination of Extracellular Tau Levels. Methods Mol Biol 1523, 285–296, doi: 10.1007/978-1-4939-6598-4_17 (2017). [DOI] [PubMed] [Google Scholar]
- 168.de Calignon A. et al. Propagation of tau pathology in a model of early Alzheimer’s disease. Neuron 73, 685–697, doi: 10.1016/j.neuron.2011.11.033 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Ising C. et al. AAV-mediated expression of anti-tau scFvs decreases tau accumulation in a mouse model of tauopathy. J Exp Med 214, 1227–1238, doi: 10.1084/jem.20162125 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Marasco WA Intrabodies: turning the humoral immune system outside in for intracellular immunization. Gene Ther 4, 11–15, doi: 10.1038/sj.gt.3300346 (1997). [DOI] [PubMed] [Google Scholar]
- 171.Chen SY, Bagley J. & Marasco WA Intracellular antibodies as a new class of therapeutic molecules for gene therapy. Hum Gene Ther 5, 595–601, doi: 10.1089/hum.1994.5.5-595 (1994). [DOI] [PubMed] [Google Scholar]
- 172.Atwal JK et al. A therapeutic antibody targeting BACE1 inhibits amyloid-β production in vivo. Sci Transl Med 3, 84ra43, doi: 10.1126/scitranslmed.3002254 (2011). [DOI] [PubMed] [Google Scholar]
- 173.Arbel M, Yacoby I. & Solomon B. Inhibition of amyloid precursor protein processing by beta-secretase through site-directed antibodies. Proc Natl Acad Sci U S A 102, 7718–7723, doi: 10.1073/pnas.0502427102 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Puglielli L, Tanzi RE & Kovacs DM Alzheimer’s disease: the cholesterol connection. Nat Neurosci 6, 345–351, doi: 10.1038/nn0403-345 (2003). [DOI] [PubMed] [Google Scholar]
- 175.Corder EH et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261, 921–923 (1993). [DOI] [PubMed] [Google Scholar]
- 176.Strittmatter WJ et al. Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci U S A 90, 1977–1981 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Luz I, Liraz O. & Michaelson DM An Anti-apoE4 Specific Monoclonal Antibody Counteracts the Pathological Effects of apoE4 In Vivo. Curr Alzheimer Res 13, 918–929 (2016). [DOI] [PubMed] [Google Scholar]
- 178.Liao F. et al. Targeting of nonlipidated, aggregated apoE with antibodies inhibits amyloid accumulation. J Clin Invest, doi: 10.1172/JCI96429 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Perry VH & Holmes C. Microglial priming in neurodegenerative disease. Nat Rev Neurol 10, 217–224, doi: 10.1038/nrneurol.2014.38 (2014). [DOI] [PubMed] [Google Scholar]
- 180.Cunningham C, Wilcockson DC, Campion S, Lunnon K. & Perry VH Central and systemic endotoxin challenges exacerbate the local inflammatory response and increase neuronal death during chronic neurodegeneration. J Neurosci 25, 9275–9284, doi: 10.1523/JNEUROSCI.2614-05.2005 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Perry VH, Cunningham C. & Holmes C. Systemic infections and inflammation affect chronic neurodegeneration. Nat Rev Immunol 7, 161–167, doi: 10.1038/nri2015 (2007). [DOI] [PubMed] [Google Scholar]
- 182.Decourt B, Lahiri DK & Sabbagh MN Targeting Tumor Necrosis Factor Alpha for Alzheimer’s Disease. Curr Alzheimer Res 14, 412–425, doi: 10.2174/1567205013666160930110551 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Citron M. Alzheimer’s disease: strategies for disease modification. Nat Rev Drug Discov 9, 387–398, doi: 10.1038/nrd2896 (2010). [DOI] [PubMed] [Google Scholar]
- 184.Weiner HL & Frenkel D. Immunology and immunotherapy of Alzheimer’s disease. Nat Rev Immunol 6, 404–416, doi: 10.1038/nri1843 (2006). [DOI] [PubMed] [Google Scholar]
- 185.Taguchi H. et al. Catalytic antibodies to amyloid beta peptide in defense against Alzheimer disease. Autoimmun Rev 7, 391–397, doi: 10.1016/j.autrev.2008.03.004 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Braak H, Thal DR, Ghebremedhin E. & Del Tredici K. Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J Neuropathol Exp Neurol 70, 960–969, doi: 10.1097/NEN.0b013e318232a379 (2011). [DOI] [PubMed] [Google Scholar]
- 187.Thal DR, Rüb U, Orantes M. & Braak H. Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology 58, 1791–1800 (2002). [DOI] [PubMed] [Google Scholar]
- 188.Sperling RA et al. Amyloid-related imaging abnormalities in amyloid-modifying therapeutic trials: recommendations from the Alzheimer’s Association Research Roundtable Workgroup. Alzheimers Dement 7, 367–385, doi: 10.1016/j.jalz.2011.05.2351 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Jordão JF et al. Antibodies targeted to the brain with image-guided focused ultrasound reduces amyloid-beta plaque load in the TgCRND8 mouse model of Alzheimer’s disease. PLoS One 5, e10549, doi: 10.1371/journal.pone.0010549 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Yu YJ et al. Boosting brain uptake of a therapeutic antibody by reducing its affinity for a transcytosis target. Sci Transl Med 3, 84ra44, doi: 10.1126/scitranslmed.3002230 (2011). [DOI] [PubMed] [Google Scholar]
- 191.Zuchero YJ et al. Discovery of Novel Blood-Brain Barrier Targets to Enhance Brain Uptake of Therapeutic Antibodies. Neuron 89, 70–82, doi: 10.1016/j.neuron.2015.11.024 (2016). [DOI] [PubMed] [Google Scholar]
- 192.Kanodia JS et al. Prospective Design of Anti-Transferrin Receptor Bispecific Antibodies for Optimal Delivery into the Human Brain. CPT Pharmacometrics Syst Pharmacol 5, 283–291, doi: 10.1002/psp4.12081 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Chauhan NB, Siegel GJ & Lichtor T. Distribution of intraventricularly administered antiamyloid-beta peptide (Abeta) antibody in the mouse brain. J Neurosci Res 66, 231–235, doi: 10.1002/jnr.1215 (2001). [DOI] [PubMed] [Google Scholar]
- 194.Chauhan NB, Siegel GJ & Lichtor T. Effect of age on the duration and extent of amyloid plaque reduction and microglial activation after injection of anti-Abeta antibody into the third ventricle of TgCRND8 mice. J Neurosci Res 78, 732–741, doi: 10.1002/jnr.20298 (2004). [DOI] [PubMed] [Google Scholar]
- 195.Chauhan NB & Siegel GJ Intracerebroventricular passive immunization in transgenic mouse models of Alzheimer’s disease. Expert Rev Vaccines 3, 717–725, doi: 10.1586/14760584.3.6.717 (2004). [DOI] [PubMed] [Google Scholar]
- 196.Chauhan NB & Siegel GJ Efficacy of anti-Abeta antibody isotypes used for intracerebroventricular immunization in TgCRND8. Neurosci Lett 375, 143–147, doi: 10.1016/j.neulet.2004.10.090 (2005). [DOI] [PubMed] [Google Scholar]
- 197.Thakker DR et al. Intracerebroventricular amyloid-beta antibodies reduce cerebral amyloid angiopathy and associated micro-hemorrhages in aged Tg2576 mice. Proc Natl Acad Sci U S A 106, 4501–4506, doi: 10.1073/pnas.0813404106 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Tobinick EL Perispinal Delivery of CNS Drugs. CNS Drugs 30, 469–480, doi: 10.1007/s40263-016-0339-2 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Sakka L, Coll G. & Chazal J. Anatomy and physiology of cerebrospinal fluid. Eur Ann Otorhinolaryngol Head Neck Dis 128, 309–316, doi: 10.1016/j.anorl.2011.03.002 (2011). [DOI] [PubMed] [Google Scholar]
- 200.Louveau A. et al. Structural and functional features of central nervous system lymphatic vessels. Nature 523, 337–341, doi: 10.1038/nature14432 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Aspelund A. et al. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J Exp Med 212, 991–999, doi: 10.1084/jem.20142290 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Broadwell RD & Sofroniew MV Serum proteins bypass the blood-brain fluid barriers for extracellular entry to the central nervous system. Exp Neurol 120, 245–263, doi: 10.1006/exnr.1993.1059 (1993). [DOI] [PubMed] [Google Scholar]
- 203.Balin BJ, Broadwell RD, Salcman M. & el-Kalliny M. Avenues for entry of peripherally administered protein to the central nervous system in mouse, rat, and squirrel monkey. J Comp Neurol 251, 260–280, doi: 10.1002/cne.902510209 (1986). [DOI] [PubMed] [Google Scholar]
- 204.Tobinick E. Perispinal etanercept produces rapid improvement in primary progressive aphasia: identification of a novel, rapidly reversible TNF-mediated pathophysiologic mechanism. Medscape J Med 10, 135 (2008). [PMC free article] [PubMed] [Google Scholar]
- 205.Tobinick EL & Gross H. Rapid cognitive improvement in Alzheimer’s disease following perispinal etanercept administration. J Neuroinflammation 5, 2, doi: 10.1186/1742-2094-5-2 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Tobinick EL & Gross H. Rapid improvement in verbal fluency and aphasia following perispinal etanercept in Alzheimer’s disease. BMC Neurol 8, 27, doi: 10.1186/1471-237-78-27 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Tobinick E. Perispinal etanercept for neuroinflammatory disorders. Drug Discov Today 14, 168–177, doi: 10.1016/j.drudis.2008.10.005 (2009). [DOI] [PubMed] [Google Scholar]
- 208.Roerink ME et al. Central delivery of iodine-125-labeled cetuximab, etanercept and anakinra after perispinal injection in rats: possible implications for treating Alzheimer’s disease. Alzheimers Res Ther 7, 70, doi: 10.1186/s13195-015-0149-7 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Chauhan MB & Chauhan NB Brain Uptake of Neurotherapeutics after Intranasal versus Intraperitoneal Delivery in Mice. J Neurol Neurosurg 2 (2015). [PMC free article] [PubMed] [Google Scholar]
- 210.Kolobov VV, Davydova TV, Zakharova IA, Gorbatov V. I. u. & Fomina VG [Repressional effects of the glutamate antibodies on expression of Dffb gene in the brain of rats with experimental Alzheimer’s disease]. Mol Biol (Mosk) 46, 757–765 (2012). [PubMed] [Google Scholar]
- 211.Kolobov VV, Zakharova IA, Fomina VG, Gorbatov VY & Davydova TV Effect of antibodies to glutamate on caspase-3 activity in brain structures of rats with experimental Alzheimer’s disease. Bull Exp Biol Med 154, 425–427 (2013). [DOI] [PubMed] [Google Scholar]
- 212.Cattepoel S, Hanenberg M, Kulic L. & Nitsch RM Chronic intranasal treatment with an anti-Aβ(30–42) scFv antibody ameliorates amyloid pathology in a transgenic mouse model of Alzheimer’s disease. PLoS One 6, e18296, doi: 10.1371/journal.pone.0018296 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Kumar NN et al. Delivery of immunoglobulin G antibodies to the rat nervous system following intranasal administration: Distribution, dose-response, and mechanisms of delivery. J Control Release 286, 467–484, doi: 10.1016/j.jconrel.2018.08.006 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Kowal SL, Dall TM, Chakrabarti R, Storm MV & Jain A. The current and projected economic burden of Parkinson’s disease in the United States. Mov Disord 28, 311–318, doi: 10.1002/mds.25292 (2013). [DOI] [PubMed] [Google Scholar]
- 215.Elkouzi, A. <http://www.parkinson.org/understanding-parkinsons/causes-andstatistics?gclid=Cj0KCQjw_ODWBRCTARIsAE2_EvWJiyWiK83o98QlssnoQnYrGt5Im8vlVdVj_N1RrJVNL3WboPcxK0UaAq5ZEALw_wcB> Accessed: (2018).
- 216.Dickson DW Parkinson’s disease and parkinsonism: neuropathology. Cold Spring Harb Perspect Med 2, doi: 10.1101/cshperspect.a009258 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Valera E. & Masliah E. Immunotherapy for neurodegenerative diseases: focus on α-synucleinopathies. Pharmacol Ther 138, 311–322, doi: 10.1016/j.pharmthera.2013.01.013 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Masliah E. et al. Passive immunization reduces behavioral and neuropathological deficits in an alpha-synuclein transgenic model of Lewy body disease. PLoS One 6, e19338, doi: 10.1371/journal.pone.0019338 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 219.George S. & Brundin P. Immunotherapy in Parkinson’s Disease: Micromanaging Alpha-Synuclein Aggregation. J Parkinsons Dis 5, 413–424, doi: 10.3233/JPD-150630 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Bae EJ et al. Antibody-aided clearance of extracellular α-synuclein prevents cell-to-cell aggregate transmission. J Neurosci 32, 13454–13469, doi: 10.1523/JNEUROSCI.1292-12.2012 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Games D. et al. Reducing C-terminal-truncated alpha-synuclein by immunotherapy attenuates neurodegeneration and propagation in Parkinson’s disease-like models. J Neurosci 34, 9441–9454, doi: 10.1523/JNEUROSCI.5314-13.2014 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Shahaduzzaman M. et al. Anti-human α-synuclein N-terminal peptide antibody protects against dopaminergic cell death and ameliorates behavioral deficits in an AAV-α-synuclein rat model of Parkinson’s disease. PLoS One 10, e0116841, doi: 10.1371/journal.pone.0116841 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Fagerqvist T. et al. Monoclonal antibodies selective for α-synuclein oligomers/protofibrils recognize brain pathology in Lewy body disorders and α-synuclein transgenic mice with the disease-causing A30P mutation. J Neurochem 126, 131–144, doi: 10.1111/jnc.12175 (2013). [DOI] [PubMed] [Google Scholar]
- 224.Schenk DB et al. First-in-human assessment of PRX002, an anti-α-synuclein monoclonal antibody, in healthy volunteers. Mov Disord 32, 211–218, doi: 10.1002/mds.26878 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Sherer TB, Fiske BK, Svendsen CN, Lang AE & Langston JW Crossroads in GDNF therapy for Parkinson’s disease. Mov Disord 21, 136–141, doi: 10.1002/mds.20861 (2006). [DOI] [PubMed] [Google Scholar]
- 226.Zhou QH, Boado RJ, Lu JZ, Hui EK & Pardridge WM Monoclonal antibody-glial-derived neurotrophic factor fusion protein penetrates the blood-brain barrier in the mouse. Drug Metab Dispos 38, 566–572, doi: 10.1124/dmd.109.031534 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Emborg ME et al. Intracerebral transplantation of differentiated human embryonic stem cells to hemiparkinsonian monkeys. Cell Transplant 22, 831–838, doi: 10.3727/096368912X647144 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Tyson T, Steiner JA & Brundin P Sorting out release, uptake and processing of alpha-synuclein during prion-like spread of pathology. J Neurochem 139 Suppl 1, 275–289, doi: 10.1111/jnc.13449 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Tyson T. et al. Novel animal model defines genetic contributions for neuron-to-neuron transfer of α-synuclein. Sci Rep 7, 7506, doi: 10.1038/s41598-017-07383-6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Dehay B, Vila M, Bezard E, Brundin P. & Kordower JH Alpha-synuclein propagation: New insights from animal models. Mov Disord 31, 161–168, doi: 10.1002/mds.26370 (2016). [DOI] [PubMed] [Google Scholar]
- 231.Mao X. et al. Pathological α-synuclein transmission initiated by binding lymphocyte-activation gene 3. Science 353, doi: 10.1126/science.aah3374 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Workman CJ, Rice DS, Dugger KJ, Kurschner C. & Vignali DA Phenotypic analysis of the murine CD4-related glycoprotein, CD223 (LAG-3). Eur J Immunol 32, 2255–2263, doi: (2002). [DOI] [PubMed] [Google Scholar]
- 233.Zhou C, Emadi S, Sierks MR & Messer A. A human single-chain Fv intrabody blocks aberrant cellular effects of overexpressed alpha-synuclein. Mol Ther 10, 1023–1031, doi: 10.1016/j.ymthe.2004.08.019 (2004). [DOI] [PubMed] [Google Scholar]
- 234.Lynch SM, Zhou C. & Messer A. An scFv intrabody against the nonamyloid component of alpha-synuclein reduces intracellular aggregation and toxicity. J Mol Biol 377, 136–147, doi: 10.1016/j.jmb.2007.11.096 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Gray SJ, Woodard KT & Samulski RJ Viral vectors and delivery strategies for CNS gene therapy. Ther Deliv 1, 517–534 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Wolak DJ & Thorne RG Diffusion of Macromolecules in the Brain: Implications for Drug Delivery. Mol Pharm, doi: 10.1021/mp300495e (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Syková E. & Nicholson C. Diffusion in brain extracellular space. Physiol Rev 88, 1277–1340, doi: 10.1152/physrev.00027.2007 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Raper SE et al. A pilot study of in vivo liver-directed gene transfer with an adenoviral vector in partial ornithine transcarbamylase deficiency. Hum Gene Ther 13, 163–175, doi: 10.1089/10430340152712719 (2002). [DOI] [PubMed] [Google Scholar]
- 239.Hwang C, Sinskey AJ & Lodish HF Oxidized redox state of glutathione in the endoplasmic reticulum. Science 257, 1496–1502 (1992). [DOI] [PubMed] [Google Scholar]
- 240.Baumal R. & Scharff MD Synthesis, assembly and secretion of mouse immunoglobulin. Transplant Rev 14, 163–183 (1973). [DOI] [PubMed] [Google Scholar]
- 241.Ouellette D. et al. Studies in serum support rapid formation of disulfide bond between unpaired cysteine residues in the VH domain of an immunoglobulin G1 molecule. Anal Biochem 397, 37–47, doi: 10.1016/j.ab.2009.09.027 (2010). [DOI] [PubMed] [Google Scholar]
- 242.Pringsheim T. et al. The incidence and prevalence of Huntington’s disease: a systematic review and meta-analysis. Mov Disord 27, 1083–1091, doi: 10.1002/mds.25075 (2012). [DOI] [PubMed] [Google Scholar]
- 243.Vonsattel JP et al. Neuropathological classification of Huntington’s disease. J Neuropathol Exp Neurol 44, 559–577 (1985). [DOI] [PubMed] [Google Scholar]
- 244.Waldvogel HJ, Kim EH, Tippett LJ, Vonsattel JP & Faull RL The Neuropathology of Huntington’s Disease. Curr Top Behav Neurosci 22, 33–80, doi: 10.1007/7854_2014_354 (2015). [DOI] [PubMed] [Google Scholar]
- 245.de la Monte SM, Vonsattel JP & Richardson EP Morphometric demonstration of atrophic changes in the cerebral cortex, white matter, and neostriatum in Huntington’s disease. J Neuropathol Exp Neurol 47, 516–525 (1988). [DOI] [PubMed] [Google Scholar]
- 246.Bates GP et al. Huntington disease. Nat Rev Dis Primers 1, 15005, doi: 10.1038/nrdp.2015.5 (2015). [DOI] [PubMed] [Google Scholar]
- 247.Truant R, Atwal R. & Burtnik A. Hypothesis: Huntingtin may function in membrane association and vesicular trafficking. Biochem Cell Biol 84, 912–917, doi: 10.1139/o06-181 (2006). [DOI] [PubMed] [Google Scholar]
- 248.Denis HL, Lauruol F. & Cicchetti F. Are immunotherapies for Huntington’s disease a realistic option? Mol Psychiatry, doi: 10.1038/s41380-018-0021-9 (2018). [DOI] [PubMed] [Google Scholar]
- 249.Lecerf JM et al. Human single-chain Fv intrabodies counteract in situ huntingtin aggregation in cellular models of Huntington’s disease. Proc Natl Acad Sci U S A 98, 4764–4769, doi: 10.1073/pnas.071058398 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Masnata M. & Cicchetti F. The Evidence for the Spread and Seeding Capacities of the Mutant Huntingtin Protein in. Front Neurosci 11, 647, doi: 10.3389/fnins.2017.00647 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Smrzka O. et al. (CHDI Foundation, Annual Huntington’s Disease Therapeutics conference, 2017). [Google Scholar]
- 252.Politis M. et al. Microglial activation in regions related to cognitive function predicts disease onset in Huntington’s disease: a multimodal imaging study. Hum Brain Mapp 32, 258–270, doi: 10.1002/hbm.21008 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Okuno T. et al. Roles of Sema4D-plexin-B1 interactions in the central nervous system for pathogenesis of experimental autoimmune encephalomyelitis. J Immunol 184, 1499–1506, doi: 10.4049/jimmunol.0903302 (2010). [DOI] [PubMed] [Google Scholar]
- 254.Hodges A. et al. Regional and cellular gene expression changes in human Huntington’s disease brain. Hum Mol Genet 15, 965–977, doi: 10.1093/hmg/ddl013 (2006). [DOI] [PubMed] [Google Scholar]
- 255.Southwell AL et al. Anti-semaphorin 4D immunotherapy ameliorates neuropathology and some cognitive impairment in the YAC128 mouse model of Huntington disease. Neurobiol Dis 76, 46–56, doi: 10.1016/j.nbd.2015.01.002 (2015). [DOI] [PubMed] [Google Scholar]
- 256.Todd D. et al. A monoclonal antibody TrkB receptor agonist as a potential therapeutic for Huntington’s disease. PLoS One 9, e87923, doi: 10.1371/journal.pone.0087923 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Liu W. et al. Increased Steady-State Mutant Huntingtin mRNA in Huntington’s Disease Brain. J Huntingtons Dis 2, 491–500, doi: 10.3233/JHD-130079 (2013). [DOI] [PubMed] [Google Scholar]
- 258.Cattaneo E, Zuccato C. & Tartari M. Normal huntingtin function: an alternative approach to Huntington’s disease. Nat Rev Neurosci 6, 919–930, doi: 10.1038/nrn1806 (2005). [DOI] [PubMed] [Google Scholar]
- 259.Khoshnan A, Ko J. & Patterson PH Effects of intracellular expression of anti-huntingtin antibodies of various specificities on mutant huntingtin aggregation and toxicity. Proc Natl Acad Sci U S A 99, 1002–1007, doi: 10.1073/pnas.022631799 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Snyder-Keller A, McLear JA, Hathorn T. & Messer A. Early or late-stage anti-N-terminal Huntingtin intrabody gene therapy reduces pathological features in B6.HDR6/1 mice. J Neuropathol Exp Neurol 69, 1078–1085, doi: 10.1097/NEN.0b013e3181f530ec (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Simmons DA et al. A small molecule TrkB ligand reduces motor impairment and neuropathology in R6/2 and BACHD mouse models of Huntington’s disease. J Neurosci 33, 18712–18727, doi: 10.1523/JNEUROSCI.1310-13.2013 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Jitendra Sharma, P. K., Bansal, S. & Banik, A. Noninvasive routes of proteins and peptides drug delivery. Indian J Pharm Sci 73, 367–375, doi: 10.4103/0250-474X.95608 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Thorne RG & Frey WH Delivery of neurotrophic factors to the central nervous system: pharmacokinetic considerations. Clin Pharmacokinet 40, 907–946, doi: 10.2165/00003088-200140120-00003 (2001). [DOI] [PubMed] [Google Scholar]
- 264.Thorne R. in Drug delivery to the brain - Physiological concepts, Methodologies and Approaches Advances in the Pharmaceutical Sciences (eds Hammarlund-Udenaes, de Lange, & Thorne) Ch. Appendix, 685–707 (Springer, 2014). [Google Scholar]
- 265.Abbott NJ, Patabendige AA, Dolman DE, Yusof SR & Begley DJ Structure and function of the blood-brain barrier. Neurobiol Dis 37, 13–25, doi: 10.1016/j.nbd.2009.07.030 (2010). [DOI] [PubMed] [Google Scholar]
- 266.Abbott NJ Blood-brain barrier structure and function and the challenges for CNS drug delivery. J Inherit Metab Dis 36, 437–449, doi: 10.1007/s10545-013-9608-0 (2013). [DOI] [PubMed] [Google Scholar]
- 267.Abbott J. in Drug Delivery to the Brain. Advances in the Pharmaceutical Sciences Series. (eds Hammarlund-Udenaes Margareta, de Lange Elizabeth, & Thorne Robert) Ch. 1, 4 (AAPSPress. Springer; , 2014). [Google Scholar]
- 268.Poduslo JF, Curran GL & Berg CT Macromolecular permeability across the blood-nerve and blood-brain barriers. Proc Natl Acad Sci U S A 91, 5705–5709 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Davson H. & Segal M. Physiology of the CSF and Blood-brain barriers, 506–507 (CRC Press, 1995). [Google Scholar]
- 270.Banks WA et al. Passage of amyloid beta protein antibody across the blood-brain barrier in a mouse model of Alzheimer’s disease. Peptides 23, 2223–2226 (2002). [DOI] [PubMed] [Google Scholar]
- 271.Petereit HF & Rubbert-Roth A. Rituximab levels in cerebrospinal fluid of patients with neurological autoimmune disorders. Mult Scler 15, 189–192, doi: 10.1177/1352458508098268 (2009). [DOI] [PubMed] [Google Scholar]
- 272.Levites Y. et al. Insights into the mechanisms of action of anti-Abeta antibodies in Alzheimer’s disease mouse models. FASEB J 20, 2576–2578, doi: 10.1096/fj.06-6463fje (2006). [DOI] [PubMed] [Google Scholar]
- 273.de Lange E. in Drug delivery to the brain - Physiological concepts, Methodologies and Approaches (eds Hammarlund-Udenaes M, Thorne R, & de Lange E) (Springer, 2014). [Google Scholar]
- 274.Bien-Ly N. et al. Lack of Widespread BBB Disruption in Alzheimer’s Disease Models: Focus on Therapeutic Antibodies. Neuron 88, 289–297, doi: 10.1016/j.neuron.2015.09.036 (2015). [DOI] [PubMed] [Google Scholar]
- 275.Fan CH et al. Contrast-enhanced ultrasound imaging for the detection of focused ultrasound-induced blood-brain barrier opening. Theranostics 4, 1014–1025, doi: 10.7150/thno.9575 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Chen CC et al. Targeted drug delivery with focused ultrasound-induced blood-brain barrier opening using acoustically-activated nanodroplets. J Control Release 172, 795–804, doi: 10.1016/j.jconrel.2013.09.025 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Pappius HM, Savaki HE, Fieschi C, Rapoport SI & Sokoloff L. Osmotic opening of the blood-brain barrier and local cerebral glucose utilization. Ann Neurol 5, 211–219, doi: 10.1002/ana.410050302 (1979). [DOI] [PubMed] [Google Scholar]
- 278.Rapoport SI, Bachman DS & Thompson HK Chronic effects of osmotic opening of the blood-brain barrier in the monkey. Science 176, 1243–1245 (1972). [DOI] [PubMed] [Google Scholar]
- 279.Rapoport SI, Hori M. & Klatzo I. Testing of a hypothesis for osmotic opening of the blood-brain barrier. Am J Physiol 223, 323–331 (1972). [DOI] [PubMed] [Google Scholar]
- 280.Kovacs ZI et al. Disrupting the blood-brain barrier by focused ultrasound induces sterile inflammation. Proc Natl Acad Sci U S A 114, E75–E84, doi: 10.1073/pnas.1614777114 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Preston JE, Joan Abbott N. & Begley DJ Transcytosis of macromolecules at the blood-brain barrier. Adv Pharmacol 71, 147–163, doi: 10.1016/bs.apha.2014.06.001 (2014). [DOI] [PubMed] [Google Scholar]
- 282.Lajoie JM & Shusta EV Targeting receptor-mediated transport for delivery of biologics across the blood-brain barrier. Annu Rev Pharmacol Toxicol 55, 613–631, doi: 10.1146/annurev-pharmtox-010814-124852 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Fishman JB, Rubin JB, Handrahan JV, Connor JR & Fine RE Receptor-mediated transcytosis of transferrin across the blood-brain barrier. J Neurosci Res 18, 299–304, doi: 10.1002/jnr.490180206 (1987). [DOI] [PubMed] [Google Scholar]
- 284.Watts RJ & Dennis MS Bispecific antibodies for delivery into the brain. Curr Opin Chem Biol 17, 393–399, doi: 10.1016/j.cbpa.2013.03.023 (2013). [DOI] [PubMed] [Google Scholar]
- 285.Bell RD & Ehlers MD Breaching the blood-brain barrier for drug delivery. Neuron 81, 1–3, doi: 10.1016/j.neuron.2013.12.023 (2014). [DOI] [PubMed] [Google Scholar]
- 286.Couch JA et al. Addressing safety liabilities of TfR bispecific antibodies that cross the blood-brain barrier. Sci Transl Med 5, 183ra157, 181–112, doi: 10.1126/scitranslmed.3005338 (2013). [DOI] [PubMed] [Google Scholar]
- 287.Abbott NJ, Pizzo ME, Preston JE, Janigro D. & Thorne RG The role of brain barriers in fluid movement in the CNS: is there a ‘glymphatic’ system? Acta Neuropathol 135, 387–407, doi: 10.1007/s00401-018-1812-4 (2018). [DOI] [PubMed] [Google Scholar]
- 288.Hannocks MJ et al. Molecular characterization of perivascular drainage pathways in the murine brain. J Cereb Blood Flow Metab 38, 669–686, doi: 10.1177/0271678X17749689 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Wolak DJ, Pizzo ME & Thorne RG Probing the extracellular diffusion of antibodies in brain using in vivo integrative optical imaging and ex vivo fluorescence imaging. J Control Release 197, 78–86, doi: 10.1016/j.jconrel.2014.10.034 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Rennels ML, Gregory TF, Blaumanis OR, Fujimoto K. & Grady PA Evidence for a ‘paravascular’ fluid circulation in the mammalian central nervous system, provided by the rapid distribution of tracer protein throughout the brain from the subarachnoid space. Brain Res 326, 47–63 (1985). [DOI] [PubMed] [Google Scholar]
- 291.Iliff JJ et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid β. Sci Transl Med 4, 147ra111, doi: 10.1126/scitranslmed.3003748 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Cserr HF, Cooper DN & Milhorat TH Flow of cerebral interstitial fluid as indicated by the removal of extracellular markers from rat caudate nucleus. Exp Eye Res 25 Suppl, 461–473 (1977). [DOI] [PubMed] [Google Scholar]
- 293.Szentistványi I, Patlak CS, Ellis RA & Cserr HF Drainage of interstitial fluid from different regions of rat brain. Am J Physiol 246, F835–844, doi: 10.1152/ajprenal.1984.246.6.F835 (1984). [DOI] [PubMed] [Google Scholar]
- 294.Ichimura T, Fraser PA & Cserr HF Distribution of extracellular tracers in perivascular spaces of the rat brain. Brain Res 545, 103–113 (1991). [DOI] [PubMed] [Google Scholar]
- 295.Hadaczek P. et al. The “perivascular pump” driven by arterial pulsation is a powerful mechanism for the distribution of therapeutic molecules within the brain. Mol Ther 14, 69–78, doi: 10.1016/j.ymthe.2006.02.018 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Carare RO et al. Solutes, but not cells, drain from the brain parenchyma along basement membranes of capillaries and arteries: significance for cerebral amyloid angiopathy and neuroimmunology. Neuropathol Appl Neurobiol 34, 131–144, doi: 10.1111/j.1365-2990.2007.00926.x (2008). [DOI] [PubMed] [Google Scholar]
- 297.Salegio EA et al. Distribution of nanoparticles throughout the cerebral cortex of rodents and non-human primates: Implications for gene and drug therapy. Front Neuroanat 8, 9, doi: 10.3389/fnana.2014.00009 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Buonerba C. et al. A comprehensive outlook on intracerebral therapy of malignant gliomas. Crit Rev Oncol Hematol 80, 54–68, doi: 10.1016/j.critrevonc.2010.09.001 (2011). [DOI] [PubMed] [Google Scholar]
- 299.Yin D. et al. Cannula placement for effective convection-enhanced delivery in the nonhuman primate thalamus and brainstem: implications for clinical delivery of therapeutics. J Neurosurg 113, 240–248, doi: 10.3171/2010.2.JNS091744 (2010). [DOI] [PubMed] [Google Scholar]
- 300.Yin D, Forsayeth J. & Bankiewicz KS Optimized cannula design and placement for convection-enhanced delivery in rat striatum. J Neurosci Methods 187, 46–51, doi: 10.1016/j.jneumeth.2009.12.008 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Dagdeviren C. et al. Miniaturized neural system for chronic, local intracerebral drug delivery. Sci Transl Med 10, doi: 10.1126/scitranslmed.aan2742 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Vogelbaum M. Novel convection enhanced drug delivery device, <https://consultqd.clevelandclinic.org/novel-convection-enhanced-delivery-device-wins-fda-clearance/> (2017).
- 303.Kaufman H. Cerebrospinal fluid collections. (Thieme, 1998). [Google Scholar]
- 304.Cohen-Pfeffer JL et al. Intracerebroventricular Delivery as a Safe, Long-Term Route of Drug Administration. Pediatr Neurol 67, 23–35, doi: 10.1016/j.pediatrneurol.2016.10.022 (2017). [DOI] [PubMed] [Google Scholar]
- 305.Wallace MS et al. Intrathecal ziconotide in the treatment of chronic nonmalignant pain: a randomized, double-blind, placebo-controlled clinical trial. Neuromodulation 9, 75–86, doi: 10.1111/j.1525-1403.2006.00055.x (2006). [DOI] [PubMed] [Google Scholar]
- 306.Wallace MS Ziconotide: a new nonopioid intrathecal analgesic for the treatment of chronic pain. Expert Rev Neurother 6, 1423–1428, doi: 10.1586/14737175.6.10.1423 (2006). [DOI] [PubMed] [Google Scholar]
- 307.Scoto M, Finkel RS, Mercuri E. & Muntoni F. Therapeutic approaches for spinal muscular atrophy (SMA). Gene Ther 24, 514–519, doi: 10.1038/gt.2017.45 (2017). [DOI] [PubMed] [Google Scholar]
- 308.Chiriboga CA et al. Results from a phase 1 study of nusinersen (ISIS-SMN(Rx)) in children with spinal muscular atrophy. Neurology 86, 890–897, doi: 10.1212/WNL.0000000000002445 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Finkel RS et al. Treatment of infantile-onset spinal muscular atrophy with nusinersen: a phase 2, open-label, dose-escalation study. Lancet 388, 3017–3026, doi: 10.1016/S0140-6736(16)31408-8 (2016). [DOI] [PubMed] [Google Scholar]
- 310.Haché M. et al. Intrathecal Injections in Children With Spinal Muscular Atrophy: Nusinersen Clinical Trial Experience. J Child Neurol 31, 899–906, doi: 10.1177/0883073815627882 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Kaufman MB Pharmaceutical Approval Update. P T 42, 562–580 (2017). [PMC free article] [PubMed] [Google Scholar]
- 312.Hladky SB & Barrand MA Mechanisms of fluid movement into, through and out of the brain: evaluation of the evidence. Fluids Barriers CNS 11, 26, doi: 10.1186/2045-8118-11-26 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Pardridge WM Drug delivery to the brain. J Cereb Blood Flow Metab 17, 713–731, doi: 10.1097/00004647-199707000-00001 (1997). [DOI] [PubMed] [Google Scholar]
- 314.Billiau A. et al. Tissue distribution of human interferons after exogenous administration in rabbits, monkeys, and mice. Arch Virol 68, 19–25 (1981). [DOI] [PubMed] [Google Scholar]
- 315.Iliff JJ et al. Cerebral arterial pulsation drives paravascular CSF-interstitial fluid exchange in the murine brain. J Neurosci 33, 18190–18199, doi: 10.1523/JNEUROSCI.1592-13.2013 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Asgari M, de Zélicourt D. & Kurtcuoglu V. Glymphatic solute transport does not require bulk flow. Sci Rep 6, 38635, doi: 10.1038/srep38635 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Kumar NN et al. Relative vascular permeability and vascularity across different regions of the rat nasal mucosa: implications for nasal physiology and drug delivery. Sci Rep 6, 31732, doi: 10.1038/srep31732 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Lochhead J. & Thorne R. in Drug delivery to the brain - Physiological concepts, Methodologies and Approaches Advances in the Pharmaceutical Sciences (eds Hammarlund-Udenaes, deLange, & Thorne) Ch. 14, 401–431 (Springer, 2014). [Google Scholar]
- 319.Lochhead JJ, Wolak DJ, Pizzo ME & Thorne RG Rapid transport within cerebral perivascular spaces underlies widespread tracer distribution in the brain after intranasal administration. J Cereb Blood Flow Metab 35, 371–381, doi: 10.1038/jcbfm.2014.215 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Thorne RG, Emory CR, Ala TA & Frey WH Quantitative analysis of the olfactory pathway for drug delivery to the brain. Brain Res 692, 278–282 (1995). [DOI] [PubMed] [Google Scholar]
- 321.Thorne RG, Hanson LR, Ross TM, Tung D. & Frey WH Delivery of interferon-beta to the monkey nervous system following intranasal administration. Neuroscience 152, 785–797, doi: 10.1016/j.neuroscience.2008.01.013 (2008). [DOI] [PubMed] [Google Scholar]
- 322.Illum L. Nasal drug delivery - recent developments and future prospects. J Control Release 161, 254–263, doi: 10.1016/j.jconrel.2012.01.024 (2012). [DOI] [PubMed] [Google Scholar]
- 323.Costantino HR, Illum L, Brandt G, Johnson PH & Quay SC Intranasal delivery: physicochemical and therapeutic aspects. Int J Pharm 337, 1–24, doi: 10.1016/j.ijpharm.2007.03.025 (2007). [DOI] [PubMed] [Google Scholar]
- 324.Sakane T. et al. Direct drug transport from the rat nasal cavity to the cerebrospinal fluid: the relation to the molecular weight of drugs. J Pharm Pharmacol 47, 379–381 (1995). [DOI] [PubMed] [Google Scholar]
- 325.Davis SS & Illum L. Absorption enhancers for nasal drug delivery. Clin Pharmacokinet 42, 1107–1128, doi: 10.2165/00003088-200342130-00003 (2003). [DOI] [PubMed] [Google Scholar]
- 326.Merkus P, Ebbens FA, Muller B. & Fokkens WJ Influence of anatomy and head position on intranasal drug deposition. Eur Arch Otorhinolaryngol 263, 827–832, doi: 10.1007/s00405-006-0071-5 (2006). [DOI] [PubMed] [Google Scholar]
- 327.Djupesland PG Nasal drug delivery devices: characteristics and performance in a clinical perspective-a review. Drug Deliv Transl Res 3, 42–62, doi: 10.1007/s13346-012-0108-9 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Wolburg H. et al. Epithelial and endothelial barriers in the olfactory region of the nasal cavity of the rat. Histochem Cell Biol 130, 127–140, doi: 10.1007/s00418-008-0410-2 (2008). [DOI] [PubMed] [Google Scholar]
- 329.Ye L, Zeng R, Bai Y, Roopenian DC & Zhu X. Efficient mucosal vaccination mediated by the neonatal Fc receptor. Nat Biotechnol 29, 158–163, doi: 10.1038/nbt.1742 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Heidl S, Ellinger I, Niederberger V, Waltl EE & Fuchs R. Localization of the human neonatal Fc receptor (FcRn) in human nasal epithelium. Protoplasma 253, 1557–1564, doi: 10.1007/s00709-015-0918-y (2016). [DOI] [PubMed] [Google Scholar]
- 331.Loddick SA et al. Displacement of insulin-like growth factors from their binding proteins as a potential treatment for stroke. Proc Natl Acad Sci U S A 95, 1894–1898 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Guan J, Beilharz EJ, Skinner SJ, Williams CE & Gluckman PD Intracerebral transportation and cellular localisation of insulin-like growth factor-1 following central administration to rats with hypoxic-ischemic brain injury. Brain Res 853, 163–173 (2000). [DOI] [PubMed] [Google Scholar]
- 333.Lee H. et al. The Effect of Body Posture on Brain Glymphatic Transport. J Neurosci 35, 11034–11044, doi: 10.1523/JNEUROSCI.1625-15.2015 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Vargová L. et al. Diffusion parameters of the extracellular space in human gliomas. Glia 42, 77–88, doi: 10.1002/glia.10204 (2003). [DOI] [PubMed] [Google Scholar]
- 335.Scherer H. Structural development in gliomas. The American Journal of Cancer 34, 333–351 (1938). [Google Scholar]
- 336.Calabrese C. et al. A perivascular niche for brain tumor stem cells. Cancer Cell 11, 69–82, doi: 10.1016/j.ccr.2006.11.020 (2007). [DOI] [PubMed] [Google Scholar]
- 337.Gilbertson RJ & Rich JN Making a tumour’s bed: glioblastoma stem cells and the vascular niche. Nat Rev Cancer 7, 733–736, doi: 10.1038/nrc2246 (2007). [DOI] [PubMed] [Google Scholar]
- 338.Thorsen F. & Tysnes BB Brain tumor cell invasion, anatomical and biological considerations. Anticancer Res 17, 4121–4126 (1997). [PubMed] [Google Scholar]
- 339.Merlini M, Wanner D. & Nitsch RM Tau pathology-dependent remodelling of cerebral arteries precedes Alzheimer’s disease-related microvascular cerebral amyloid angiopathy. Acta Neuropathol 131, 737–752, doi: 10.1007/s00401-016-1560-2 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340.Chen W, Song X, Zhang Y. & Initiative A. s. D. N. Assessment of the Virchow-Robin Spaces in Alzheimer disease, mild cognitive impairment, and normal aging, using high-field MR imaging. AJNR Am J Neuroradiol 32, 1490–1495, doi: 10.3174/ajnr.A2541 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341.Inglese M. et al. Dilated perivascular spaces: hallmarks of mild traumatic brain injury. AJNR Am J Neuroradiol 26, 719–724 (2005). [PMC free article] [PubMed] [Google Scholar]
- 342.Doubal FN, MacLullich AM, Ferguson KJ, Dennis MS & Wardlaw JM Enlarged perivascular spaces on MRI are a feature of cerebral small vessel disease. Stroke 41, 450–454, doi: 10.1161/STROKEAHA.109.564914 (2010). [DOI] [PubMed] [Google Scholar]
- 343.Zafeiriou DI & Batzios SP Brain and spinal MR imaging findings in mucopolysaccharidoses: a review. AJNR Am J Neuroradiol 34, 5–13, doi: 10.3174/ajnr.A2832 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Pollay M. The function and structure of the cerebrospinal fluid outflow system. Cerebrospinal Fluid Res 7, 9, doi: 10.1186/1743-8454-7-9 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Pangalos MN, Schechter LE & Hurko O. Drug development for CNS disorders: strategies for balancing risk and reducing attrition. Nat Rev Drug Discov 6, 521–532, doi: 10.1038/nrd2094 (2007). [DOI] [PubMed] [Google Scholar]
- 346.Wootla B. et al. Recent Advances in Monoclonal Antibody Therapies for Multiple Sclerosis. Expert Opin Biol Ther 16, 827–839, doi: 10.1517/14712598.2016.1158809 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Hemmer B, Nessler S, Zhou D, Kieseier B. & Hartung HP Immunopathogenesis and immunotherapy of multiple sclerosis. Nat Clin Pract Neurol 2, 201–211, doi: 10.1038/ncpneuro0154 (2006). [DOI] [PubMed] [Google Scholar]
- 348.Baecher-Allan C, Kaskow BJ & Weiner HL Multiple Sclerosis: Mechanisms and Immunotherapy. Neuron 97, 742–768, doi: 10.1016/j.neuron.2018.01.021 (2018). [DOI] [PubMed] [Google Scholar]
- 349.Engelhardt B. & Kappos L. Natalizumab: targeting alpha4-integrins in multiple sclerosis. Neurodegener Dis 5, 16–22, doi: 10.1159/000109933 (2008). [DOI] [PubMed] [Google Scholar]
- 350.Polman CH et al. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med 354, 899–910, doi: 10.1056/NEJMoa044397 (2006). [DOI] [PubMed] [Google Scholar]
- 351.Sellebjerg F. et al. Exploring potential mechanisms of action of natalizumab in secondary progressive multiple sclerosis. Ther Adv Neurol Disord 9, 31–43, doi: 10.1177/1756285615615257 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.Coles AJ et al. Alemtuzumab vs. interferon beta-1a in early multiple sclerosis. N Engl J Med 359, 1786–1801, doi: 10.1056/NEJMoa0802670 (2008). [DOI] [PubMed] [Google Scholar]
- 353.Jones JL & Coles AJ Mode of action and clinical studies with alemtuzumab. Exp Neurol 262 Pt A, 37–43, doi: 10.1016/j.expneurol.2014.04.018 (2014). [DOI] [PubMed] [Google Scholar]
- 354.Havrdova E, Horakova D. & Kovarova I. Alemtuzumab in the treatment of multiple sclerosis: key clinical trial results and considerations for use. Ther Adv Neurol Disord 8, 31–45, doi: 10.1177/1756285614563522 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Gross CC et al. Impaired NK-mediated regulation of T-cell activity in multiple sclerosis is reconstituted by IL-2 receptor modulation. Proc Natl Acad Sci U S A 113, E2973–2982, doi: 10.1073/pnas.1524924113 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356.Hauser SL, Belachew S. & Kappos L. Ocrelizumab in Primary Progressive and Relapsing Multiple Sclerosis. N Engl J Med 376, 1694, doi: 10.1056/NEJMc1702076 (2017). [DOI] [PubMed] [Google Scholar]
- 357.Hauser SL et al. Ocrelizumab versus Interferon Beta-1a in Relapsing Multiple Sclerosis. N Engl J Med 376, 221–234, doi: 10.1056/NEJMoa1601277 (2017). [DOI] [PubMed] [Google Scholar]
- 358.Montalban X. et al. Ocrelizumab versus Placebo in Primary Progressive Multiple Sclerosis. N Engl J Med 376, 209–220, doi: 10.1056/NEJMoa1606468 (2017). [DOI] [PubMed] [Google Scholar]
- 359.Engelhardt B. The blood-central nervous system barriers actively control immune cell entry into the central nervous system. Curr Pharm Des 14, 1555–1565 (2008). [DOI] [PubMed] [Google Scholar]
- 360.Hemmer B, Frohman E, Hartung HP & Stüve O. Central nervous system infections - a potential complication of systemic immunotherapy. Curr Opin Neurol 19, 271–276, doi: 10.1097/01.wco.0000227037.70329.b0 (2006). [DOI] [PubMed] [Google Scholar]
- 361.Lo EH, Dalkara T. & Moskowitz MA Mechanisms, challenges and opportunities in stroke. Nat Rev Neurosci 4, 399–415, doi: 10.1038/nrn1106 (2003). [DOI] [PubMed] [Google Scholar]
- 362.Yu CY, Ng G. & Liao P. Therapeutic antibodies in stroke. Transl Stroke Res 4, 477–483, doi: 10.1007/s12975-013-0281-2 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363.Tomura N. et al. Differentiation between cerebral embolism and thrombosis on sequential CT scans. J Comput Assist Tomogr 14, 26–31 (1990). [DOI] [PubMed] [Google Scholar]
- 364.Kilic E. et al. Role of Nogo-A in neuronal survival in the reperfused ischemic brain. J Cereb Blood Flow Metab 30, 969–984, doi: 10.1038/jcbfm.2009.268 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365.Gillani RL et al. Cognitive recovery in the aged rat after stroke and anti-Nogo-A immunotherapy. Behav Brain Res 208, 415–424, doi: 10.1016/j.bbr.2009.12.015 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366.Ma J. et al. An experimental test of stroke recovery by implanting a hyaluronic acid hydrogel carrying a Nogo receptor antibody in a rat model. Biomed Mater 2, 233–240, doi: 10.1088/1748-6041/2/4/005 (2007). [DOI] [PubMed] [Google Scholar]
- 367.Nicole O. et al. The proteolytic activity of tissue-plasminogen activator enhances NMDA receptor-mediated signaling. Nat Med 7, 59–64, doi: 10.1038/83358 (2001). [DOI] [PubMed] [Google Scholar]
- 368.Haley MJ & Lawrence CB The blood-brain barrier after stroke: Structural studies and the role of transcytotic vesicles. J Cereb Blood Flow Metab 37, 456–470, doi: 10.1177/0271678X16629976 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369.McKee AC, Stein TD, Kiernan PT & Alvarez VE The neuropathology of chronic traumatic encephalopathy. Brain Pathol 25, 350–364, doi: 10.1111/bpa.12248 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370.Mckee AC & Daneshvar DH The neuropathology of traumatic brain injury. Handb Clin Neurol 127, 45–66, doi: 10.1016/B978-0-444-52892-6.00004-0 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371.Blennow K. et al. Traumatic brain injuries. Nat Rev Dis Primers 2, 16084, doi: 10.1038/nrdp.2016.84 (2016). [DOI] [PubMed] [Google Scholar]
- 372.Putatunda R, Bethea JR & Hu WH Potential immunotherapies for traumatic brain and spinal cord injury. Chin J Traumatol 21, 125–136, doi: 10.1016/j.cjtee.2018.02.002 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373.Chodobski A, Zink BJ & Szmydynger-Chodobska J. Blood-brain barrier pathophysiology in traumatic brain injury. Transl Stroke Res 2, 492–516, doi: 10.1007/s12975-011-0125-x (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]