

Abstract
Purpose of review
The past decade in LGL leukemia research has seen increased pairing of clinical data with molecular markers, shedding new insights on LGL leukemia pathogenesis and heterogeneity. This review summarizes the current standard of care of LGL leukemia, updates from clinical trials, and our congruent improved understanding of LGL pathogenesis.
Recent findings
Various clinical reports have identified associations between stem, bone-marrow, and solid organ transplants and incidence of LGL leukemia. There is also a potential for underdiagnosis of LGL leukemia within the rheumatoid arthritis patient population, emphasizing our need for continued study. Preliminary results from the BNZ-1 clinical trial, which targets IL-15 along with IL-2 and IL-9 signaling pathways, show some evidence of clinical response.
Summary
With advances in our understanding of LGL pathogenesis from both the bench and the clinic, exciting avenues for investigations lie ahead for LGL leukemia.
Keywords: Large granular lymphocyte leukemia, STAT3, autoimmunity, organ transplant, BNZ-1
Introduction
Large granular lymphocyte (LGL) leukemia is a rare chronic lymphoproliferative disease of T-cell and natural killer (NK) cell lineage. [1] Since its initial description in 1985, the World Health Organization (WHO) has recognized T and NK-cell LGL leukemia under the subgroup of mature peripheral T-cell neoplasms in 1999, and further distinguished chronic NK-cell lymphocytosis from the more aggressive form of NK–cell LGL leukemia in 2008. [2, 3] The most recent WHO classification from 2016 highlights the discovery of signal transducer and activator of transcription 3 (STAT3) and STAT5b mutations. [2, 4–6] This review will discuss epidemiology, clinical presentation, diagnosis, management, pathogenesis, and future therapeutic options of LGL leukemia. Active research is underway to better understand pathogenesis of LGL leukemia and to develop effective therapeutic regimens for this rare disease.
Epidemiology and Disease Prevalence
Roughly 85% of reported LGL leukemia cases are the T-LGL subtype, with less than 10% of cases described as chronic NK-LGL leukemia. The aggressive NK-cell disease typically presents in young Asian populations with Epstein Barr virus infection suspected as a contributing etiologic agent.[1] In fact, overall disease prevalence differs across ethnicities as well. In North America and Europe, LGL leukemia accounts for 2–5% of chronic lymphoproliferative disorders, whereas in Asia, it accounts for a higher proportion at 5–6%.[7] Recent demographic studies of European and North American cohorts place the average incidence of LGL leukemia at 0.2–0.72 per million persons per year (Table 1).[8, 9]
Table 1.
Demographics and secondary diseases in LGL leukemia.
| Category | Further details and references |
|---|---|
| Age at diagnosis | |
| Typically in the 6th decade of life | Median 60 years, rare pediatric, <25% of adult patients <50 years old [1]; 66.5 years median age, 14% <50 years [9]; median age 67 [8] |
| Time to treatment | |
| Treatment at diagnosis is ~60% [12] | 45% required systemic therapy (of these, 60% within 1 month of dx, 37% between 1–6 months of dx) [9] |
| Survival rate | |
| ~10 years | Median overall survival 9–10 years [9]; overall survival 10 years ~70% [1] |
| Incidence | |
| Overall incidence: 2–5% North America and Europe, 5–6% Asia; roughly same incidence for males and females [1] | Overall average=0.2 cases per 1,000,000 (US population) Male-to-female ratio 1.05 with females diagnosed at a younger age (65 vs. 68 years) [9] 53% male, 47% female [8] Aggressive NK-LGL leukemia incidence is mainly in Asia and affects younger patients [1] |
| Ethnicity incidence | |
| Not significantly different among ethnic groups | White 0.2, Black 0.14, American Indian/Alaska Native 0.24, Asian/Pacific Islander 0.15 [9] |
| Subtypes of LGL leukemia | |
| T-cell (αβ or γδ phenotype) | 85% of cases, majority are TCRαβ+ phenotype [1] 85% of cases [9] 91% of cases [8] |
| NK-cell, chronic | <10% of cases [1] 6% of cases [8] |
| NK-cell, aggressive | <5% of cases [1] |
| Secondary diseases | |
| Most common autoimmune disease: Rheumatoid arthritis (RA) | 10–18% [1] 17–36% [14] 11–36% [12, 13] |
| Rarer occurring autoimmune diseases: systemic lupus erythematosus (SLE), Felty’s syndrome, Sjogren’s syndrome, and others. | Occasional (<1%) [1] Sjogren 2–27%, Felty 40%, SLE rare; thyroid disorders 2–3% [14] Case report [13] |
| Blood-related autoimmune diseases: pure red cell aplasia (PRCA), acquired autoimmune hemolytic anemia (AIHA), immune thrombocytopenia (ITP), and others | 5%; PRCA 5–27%, AIHA 5–9% [1] PRCA 5%, AIHA <2% [14] PRCA 5%, AIHA 3%, ITP rare [12, 13] |
| Blood disorders/bone marrow failure: aplastic anemia, myelodysplastic syndrome, B-cell malignancies | AA: Occasional association [12, 73] MDS: 3–10% [1]; <4% myelodysplasia [12, 13] B-cell malignancies: 5% [1]; 5–7% [13]; 27–43% [34]; 5–7% [12] |
| Other: Pulmonary artery hypertension | <1%[1]; Rare [13] |
Similar demographic information has been reported across cohorts with the median age at diagnosis in the sixth decade of life, with more than 75% of patients over the age of 50. The most recent population study of T-LGL leukemia showed that incidence did not differ across ethnicities or sexes, although females tend to be diagnosed 3 years earlier than males. LGL leukemia is a relatively indolent disease with 45% of patients requiring systemic treatment at time of diagnosis.[9] However, most patients will eventually require treatment at some point during the disease course, and the overall median survival is approximately 9 years. [8, 10] Overall survival is impacted by presence of comorbidities and age at diagnosis.[9] Clinical features and responses to treatment are similar between chronic NK- and T-LGL leukemias.[10] On the other hand, aggressive NK-LGL leukemia tends to be refractory to treatment with an overall survival as low as 58 days. [11]
Clinical Presentation
About one-third of patients are asymptomatic at the time of diagnosis. [1] These asymptomatic patients are often referred to hematology specialists by their primary care physician due to abnormal complete blood count (CBC) values, such as increased lymphocyte count. Another common incidental finding is low neutrophil counts, or neutropenia. Some patients with severe neutropenia may display no symptoms or infections for a very long period of time, whereas others may exhibit symptoms related to neutropenia such as recurrent oral aphthous ulcerations and fever due to bacterial infections. In serious cases, sepsis may occur. Splenomegaly is reported at 20–50% frequency, and recurrent infection is seen in 15–39% of patients [1]. Anemia is also a frequent symptom seen in patients, and 10–30% of anemic patients are transfusion dependent [1].
LGL leukemia has a long history of association with autoimmune diseases, with many patients presenting with an autoimmune or hematological disorder before the eventual diagnosis of LGL leukemia (Table 1). Rheumatoid arthritis (RA) is one of the most commonly associated autoimmune disorders with 10–36% of LGL leukemia patients suffering from concomitant RA [1, 12, 13]. In addition, due to more prominent RA symptoms in LGL leukemia and overlapping immunosuppressive treatments for both RA and LGL leukemia, it is possible that LGL leukemia is underdiagnosed in the RA patient population. In fact, in a study of 529 RA patients, roughly 3.6% exhibited clonal T-LGL expansions as determined by polymerase chain reaction (PCR). Given that RA affects millions of people worldwide, there is the distinct possibility that LGL leukemia affects a much larger population than previously thought, thus emphasizing the need for continued study.
Additionally, LGL leukemia patients may also have other less common diseases such as Sjӧgren syndrome, Behҫet disease, systemic lupus erythematosus, chronic inflammatory bowel disease, autoimmune thyroid disorders, and immune thrombocytopenia. [1, 13–15] Felty’s syndrome (FS) is yet another autoimmune disorder common to LGL leukemia patients, affecting up to 40% of patients, that exhibits a strong symptomatic overlap with LGL leukemia. [14, 15] Both diseases commonly present with RA, splenomegaly, and neutropenia as well as with a high prevalence of DR4 haplotype and a favorable response to methotrexate (MTX) therapy, thus suggesting a common pathogenesis. [15]
Hematological disorders are also often associated with LGL leukemia. Indeed, there is a higher prevalence of not only neutropenia, but also autoimmune hemolytic anemia (AIHA), myelodysplastic syndrome, B-cell malignancies, and aplastic anemia (AA) in LGL leukemia patients. [14] Of note, pure red cell aplasia (PRCA) has been documented in 5–68% of cases depending on the cohort. [13, 14, 16] This widely varied range is likely due to differential presentation depending on ethnicity, with PRCA much more prevalent amongst East Asian LGL leukemia patient populations. [17].
Diagnosis
A definitive diagnosis of LGL leukemia is established when an expanded clonal T- or NK-LGL population is found with appropriate clinical context. Increased number of LGL cells (> 2×109/L; normal: <0.3×109/L) or LGL count between (0.4 – 2×109/L) with paired clinical or hematological features such as RA or cytopenia are compatible with the diagnosis. Leukemic LGL cells can easily be identified on a blood smear, as they are larger than circulating peripheral lymphocytes with ample cytoplasm that contains azurophilic granules (Figure 1A). However, LGL cells are not cytologically distinguishable from activated cytotoxic lymphocytes nor are T- and NK-LGL cells distinguishable.
Figure 1:
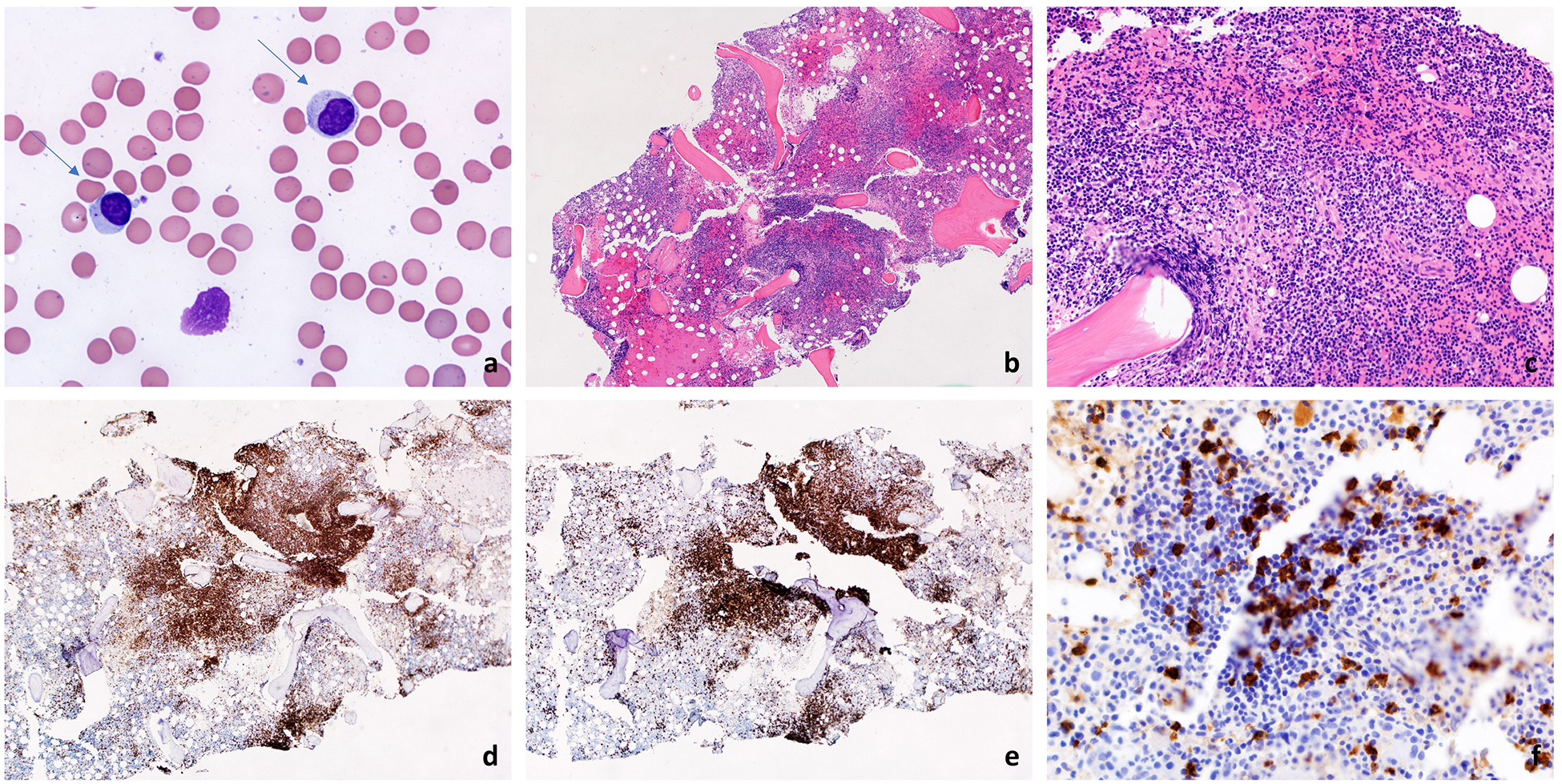
Large granular lymphocytes are medium sized cells containing abundant granular cytoplasm, highlighted with arrows (A, 1000x). A bone marrow biopsy shows hypercellularity (B, 100x) and an interstitial non-paratrabecular lymphocytic infiltrate (C, 200x). The infiltrate is positive for CD3 (D, 100x) and CD8 (E, 100x). Many of the lymphoid cells are also positive for CD57 (F, 400x).
Clonality of the LGL cells is essential to categorize this lymphoproliferation as a leukemia and distinguish it from autoimmune or infectious disease. Clonality of T-LGL cells can be established with (PCR) with probes for T-cell receptor (TCR)γ. TCRγ can be used for both αβ and γδ T-cell lineages, as this gene is rearranged at an early stage of T-cell development. Flow cytometric analysis assessing Vβ TCR cell surface protein repertoire can also be used to gauge clonality, with the caveat that it may be less sensitive than the PCR tests. [1] Identifing clonality of NK-LGL is a more difficult task as NK cells lack TCR recombination or expression. In this case, immunophenotyping assays measuring expression patterns of killer cell immunoglobulin-like receptors (KIRs) prove useful in diagnosis. [1]
Flow cytometry is utilized to assess expression of cell surface markers and is important to establish an expanded T- or NK cell population. T-LGL cells exhibit a mature post-thymic phenotype and, in most cases display CD3+, TCRαβ+, CD8+, CD16+, CD45RA+, and CD57+ and are CD4−, CD5dim, CD27−, CD28−, CD45RO−. NK-LGL cells display CD2+, CD3−, TCRαβ−, CD4−, CD8+, CD16+, CD94+, and CD56+.[1] Although clonal, LGL leukemic cells are heterogeneous between patients and exhibit different marker phenotypes. For example some patients with T-LGL cells expressing CD56+ may have more aggressive clinical behavior and have been associated withmutations of the Stat5b gene. [5, 18] Of interest, there is also a distinct LGL leukemia phenotype, CD3+CD4+CD8+CD56+, that may also have characteristic STAT5b mutatations, but have an indolent clinical course. [5]
Bone marrow aspirate or biopsy is not routinely performed as blood studies can readily establish the diagnosis. However, bone marrow can be evaluated when the diagnosis is difficult to establish and other diagnoses are on the differential, such as MDS. A typical LGL leukemia bone marrow features hypercellularity (Figure 1B–F) with CD3+, CD8+, and CD57+ infiltrate. As morphologic infiltration may be subtle, immunohistochemistry studies can be useful. The characteristic pattern is an interstitial/intra-sinusoidal distribution of cytotoxic T cells expressing cytotoxic markers such as perforin, granzyme B, and/or TIA-1. [19] The degree of LGL infiltration does not necessarily correlate with severity of clinical presentation [1]. Extensive details on diagnosis of LGL leukemia are well-illustrated in Lamy, Moignet, and Loughran 2017 [1].
Current treatment options of LGL leukemia
Prognosis
T- and NK-LGL leukemias are chronic diseases that require management as the disease evolves. Overall survival at 10 years is about 70%, and disease-related death is mainly due to severe infection that occurs in less than 10% of the LGL patient population. [1] Conversely, aggressive NK-LGL leukemia has a much lower survival rate because patients do not respond to currently available treatments. [1]
Indication for treatment
Treatment is indicated for patients with symptomatic anemia, reduced neutrophil counts with recurrent infections, or autoimmune conditions such as RA. However, once the diagnosis is established, patients should be routinely monitored for symptoms and disease progression regardless of treatment status. For a patient with an absolute neutrophil count (ANC) above 0.5 × 109/L and no associated symptoms, treatment is not indicated and patients are monitored instead. [13]
Therapeutic approach
Historically, immunosuppressive drugs such as methotrexate (MTX), cyclophosphamide, and cyclosporine remain the standard treatment options. Here we review the three canonical immunosuppressive agents and another non-canonical immunosuppressant in detail for clinical management of LGL leukemia (Table 2).
Table 2.
Current immunosuppressive therapies for LGL leukemia.
| Type of treatment | Typical treatment dose & duration | Additional information |
|---|---|---|
| Immunosuppressive therapy | ||
| Methotrexate | 10 mg/m2 per week; taken indefinitely | First-line; especially for neutropenic patients |
| Cyclophosphamide | 100 mg per day; taken up to 12 months | Typically used for patients with anemia, especially PRCA (typically non-responders to methotrexate). |
| Cyclosporine A | 3 mg/kg per day; taken indefinitely | Prescribed if patients fail the previous two treatments. |
| Prednisone | 1 mg/kg orally daily 30 days then tapered off 24 days | Typically given in combination with methotrexate [20] |
Canonical immunosuppressive treatments
MTX is the proposed first-line therapy, which was established after prospective studies on MTX with or without prednisone. [13, 20, 21] MTX is a treatment of choice for primary RA, and for this reason its use especially benefits LGL patients who also have RA as an associated disease. MTX is administered orally at 10 mg/m2 per week.[1] Although MTX is generally well-tolerated, patients are monitored for hepatic and lung function. Patients may continue taking this treatment as long as they are responsive. Treatment response is evaluated after four months according to the blood count criteria described above.
It is important to note that the largest prospective clinical trial, to date, in LGL leukemia showed that patients with STAT3 mutations are more likely to respond to MTX treatment and have favorable outcome.[20] This trial also showed that the overall response rate for MTX was 38%, which includes both partial and complete responders. This response rate is seemingly lower than cyclophosphamide (described below). For this reason a randomized prospective trial is ongoing to directly compare the efficacy of MTX and cyclophosphamide, with study completion date in November 2019 (NCT01976182). [22]
Cyclophosphamide is administered orally at 50–100 mg per day and appears to be a good treatment choice for LGL leukemia patients with PRCA or predominant anemia.[1] Cyclophosphamide should not be used for more than 12 months due to associated toxicities and increased risk for developing MDS and acute myeloid leukemia (AML).[23, 24] A retrospective study of a cohort of 45 patients reported a 71% overall response rate.[25] Because the response rate of cyclophosphamide is similar to if not higher than MTX, it can also be considered as a first-line treatment for LGL leukemia.
Cyclosporine is administered orally at 3 mg/kg per day [1] and is often reserved for patients who fail both MTX and cyclophosphamide treatment. This is because it must be taken indefinitely if patients are responsive, as cessation causes symptoms to rapidly recur. [13] The complete response rate for cyclosporine is <5%. [13] Patients should be carefully monitored for renal function and hypertension while on cyclosporine. [26]
Primary treatment options for LGL leukemia largely depend on initial clinical presentation of the patient. Previous treatment history, autoimmune manifestations and other co-morbidities, as well as the side-effect profile of the therapeutics should be considered when establishing treatment. If the primary treatment fails, a switch between MTX and cyclophosphamide is recommended, reserving cyclosporine for those who fail both treatments. [13]
Non-canonical immunosuppressive treatment
Prednisone as a primary first-line therapy is ineffective in treating neutropenia and reducing LGL counts, with just 2 out of 22 patients having either complete or partial response.[10] However, utilizing prednisone as an adjunctive therapy provides rapid stabilization of blood counts while MTX or other primary immunosuppressive treatments are being implemented, as primary immunosuppressives such as MTX and cyclophosphamide often are slow-acting.[13, 20]
Evaluation of treatment response
With the initiation of treatment, the patient should be followed to assess treatment efficacy. At minimum, evaluation of peripheral CBC should be conducted after four months. There are five distinctive levels of treatment response: 1) complete molecular response, 2) hematologic complete response, 3) hematologic partial response, 4) treatment failure or stable disease, and 5) progressive disease.
Complete molecular response is defined as no clonal T-cell detection by PCR and also meets criterion for hematologic complete response. Hematologic complete response is noted when CBC values normalize to ANC > 1.5 × 109/L, platelets > 150 × 109/L, and lymphocyte count < 4 × 109/L. The number of circulating LGL cells should also be in the normal range, which can be detected using flow cytometry. Hematologic partial response is defined when there is an improvement in the blood counts, but the patient still does not meet the threshold for complete remission (ANC > 0.5 × 109/L but < 1.5 × 109/L), or a reduction in number of transfusion requirements but still transfusion dependent. When the patient does not meet the above criteria within four months of treatment initiation, but there is no worsening of CBC values, stable disease or treatment failure is ascribed. Progressive disease is noted when CBC values worsen or findings of organomegaly such as hepatosplenomegaly are detected.
Current understanding of LGL pathogenesis and potential treatment targets
Although the etiology of LGL leukemia is not fully elucidated, many of the underlying mechanisms and co-occurring disorders have been characterized. Survival network analysis [27] of LGL leukemia suggests the existence of many dysregulated pathways including apoptosis, proliferation and general immune dysfunction, which were further validated by others.[28–30]
These molecular and systemic pathologies collectively characterize a complex etiology of LGL leukemia, which is likely to include neoplastic,[31] viral,[32] and/or auto-origin mechanisms.[14, 33] Several studies correlated LGL leukemia development and/or progression with various malignancies, however, no causative link has been established to date.[31, 34, 35] Patient sero-reactivity to conserved HIV-1 and HTLV epitopes motivated investigation to uncover whether a retroviral component, and in particular human retroviruses, could contribute to the pathogenesis. [36–38][32, 39, 40] Extensive analysis of long insert mate pair next-generation sequencing data failed to detect clonal integration of an exogenous retrovirus within 11 LGL leukemia samples. [40] Additionally, LGL leukemia was reported following bone marrow, stem cell and solid organ transplantation (SOT), likely related to exposure to a potential alloantigen or infectious agent.[41–43] Given the limited size of the patient population, a mechanistic link and more clear definition of the prevalence of secondary T-LGL leukemia in SOT recipients await further investigations.[44]
Leukemic cells are phenotypically similar to terminally differentiated effector memory T cells. This suggests that their expansion may be initiated in the same manner as typical antigen-activated cytotoxic killer cells.[33] The currently accepted model of LGL pathogenesis[1] hypothesizes that an unidentified antigen is an initial stimulus for oligoclonal proliferation of LGL cells. Failure to clear the inciting event results in chronic STAT1 and STAT3 activation[45] that fuels the emergence of a dominant clone. Subsequent clonal drift is observed in 37% of LGL leukemia patients[46] and could further support the persisting antigen model as LGL cells could be responding to different epitopes of the same origin over time.
Altogether, molecular and systemic abnormalities as well as the autoimmune and inflammatory nature of LGL leukemia-associated manifestations suggest that this malignant lymphoproliferation most likely results from multiple, non-mutually exclusive pathologies.
JAK/STAT and STAT3 dysregulation
Dysregulation of the Janus kinase/signal transducer and activator of transcription (JAK/STAT) pathway is a hallmark of LGL leukemia pathobiology[45] that promotes survival and sustains the abnormal persistence of LGL clones with effector functionality.[27]
STAT3 sequence alterations are present in about 40% of LGL patients. [6] These somatic activation mutations can accelerate clonal expansion and evolution of the leukemic cell population by enhanced activation from physiological stimulation.[47] It is believed that STAT3 activating mutations modulate the transcription of survival components such as anti-apoptotic Mcl-1[45] and these genetic lesions fuel LGL expansions.[20] Somatic activating STAT5b mutations are found at a 2% frequency in LGL leukemia and are thought to contribute in some rare patients to a more aggressive disease. [5]
Consequently, dysregulated JAK/STAT signaling and lymphocyte activation leads to the production of pro-inflammatory cytokines such as IFN-γ, IL-8, IL-10, IL-1β, IL-12p35, IL-18, IL-1Ra, RANTES, MIP-1α/β that can contribute to cytopenia and autoimmune disease development.[48, 20, 49, 14] On the other hand, cytokines such as IFN-γ, IL-6, and IL-15 are elevated in LGL leukemic sera, initiate JAK/STAT signaling, and may explain constitutive STAT3 activation in patients who do not habor somatic activating mutations.[50]
Pro-survival signaling in LGL leukemia
An in silico model of LGL leukemia pathway network signaling proposed IL-15 and platelet-derived growth factor (PDGF), together with antigenic stimulation, to be necessary master switches promoting leukemic survival. Their constitutive activation recapitulates known molecular abnormalities characteristic of LGL leukemia.[27]
These predictions were validated experimentally and it was found that PDGF mediates leukemic clone survival via an autocrine loop.[51] The previous knowledge of the role of pro-inflammatory IL-15 in controlling both the proliferation and cytotoxicity of T- and NK-LGL cells[52] was expanded with the observation of high levels of IL-15Rα transcripts detectable in purified clones.[53] In addition to mediating pro-survival signaling, IL-15 also contributes to cell death inhibition. Not only does IL-15 alter the expression of Mcl-1 but it promotes degradation of pro-apoptotic BID.[54] BNZ-1, a multi-cytokine inhibitor that antagonizes the binding of IL-15, IL-2, and IL-9 to the common gamma chain receptor, has been tested ex vivo on LGL leukemic cells and cell lines. This peptide antagonist blocked survival signaling, reduced viability and enhanced apoptosis [55]. Results of an active phase I/II clinical trial (NCT03239392) have shown preliminary evidence of clinical response in some LGL leukemia patients treated with BNZ-1[56].
Interestingly, an IL-15 transgenic mouse model showed that prolonged IL-15 exposure induces an aggressive leukemia with LGL-like features that exhibits Myc/NF-κB signaling as well as subsequent DNMT3b overexpression and DNA hypermethylation.[57] As concurrent mutations in DNMT3A, TET2, and STAT3 were recently identified in a T-LGL leukemia case study [58], these findings may implicate DNA methylation as a potential driver and therapeutic target in LGL leukemia.
The network model also identified sphingosine kinase 1 (SPHK1) and sphingolipid metabolism as key players in leukemic LGL survival.[27] The physiological balance between pro-apoptotic and pro-survival sphingolipids is maintained by the interconversion of pro-apoptotic ceramide to sphingosine and pro-survival sphingosine-1-phosphate (S1P). SPHK1 catalyzes the production of S1P, shifting the balance towards cell survival. This process can be reversed with use of SPHK1 inhibitors that were shown to downregulate the JAK/STAT pathway and induce apoptosis in NK-LGL leukemia.[59]
Molecular abnormalities characteristic of LGL clones are intertwined in a complex survival network driven by intrinsic and extrinsic stimuli. Multiple dysregulated pathways orchestrate both pro-survival and anti-apoptotic signaling, and their overlap makes the identification of potential therapeutic targets more challenging. However, mechanistic understanding of this dysregulation and interplay both at the molecular and cellular level is necessary. Just as malignant cell survival results from compounded and multifactorial cell signaling dysregulation, successful therapeutic approaches may result from the combined targeting of several pathways.
Future Directions for Therapy
Drug design and discovery heavily relies on the fundamental understanding of complex signaling networks and their perturbations within a given tumor type. Thus, the studies described above contribute to a knowledge base that is essential to future therapeutic development in LGL leukemia. However, meticulous validation of target, off-target, and toxicity effects in in vitro, in vivo and ex vivo models are just as crucial to the sustainable development of effective therapeutics.[60]
Unfortunately, 97% of drugs tested in early clinical stages fail trials[61] and are never approved by the U.S. Food and Drug Administration for their intended indications, usually as a result of dose-limiting toxicities and lack of anticipated efficacy. A recent study by the Sheltzer group identifies the pitfalls associated with cancer drug validation at early pre-clinical stages. Usually, an initial screen of an investigational agent’s protein target is predominantly based on RNA interference and small molecule inhibitors. Their notorious off-target effects and the lack of genetic knock-out validation leads to reporting of inaccurate mechanisms of action of investigated agents, and contributes to the high rate of clinical trial failures at early phases.[62]
Although several promising agents are being validated in various stages of pre- and clinical testing, MTX and cyclophosphamide remain as first-line therapies for LGL leukemia and there is currently no cure available for this lymphoproliferative disease.[13, 20, 21, 25] Some of the presumed MTX mechanisms of action involve inhibition of purine and pyrimidine synthesis, accumulation of polyamines as well as ROS generation, suppression of alarmin function, and more recently proposed, modulation of JAK/STAT and NF-κB pathways.[63, 64]
MTX, on the other hand, has been approved for use in RA for over 30 years and has been efficacious in treating various heme malignancies since the 1940s, albeit at much higher treatment doses.[65, 64] With the initial 5-year survival rate of 10% in the first half of nineteenth century to over 90% of complete or partial remission rate in pediatric acute leukemias nowadays, MTX is a perfect example of an “old but gold” drug.[66, 67] Its efficacy in childhood heme malignancy treatment, however, can be attributed to a number of clinical trials that tested dose intensification regimens and pairing with irradiation or with other drugs such as prednisone. The use of clinical and biological prognostic variables such as early treatment response or genetic subtype helped stratify pediatric patients by risk and assign the optimal therapy with an appropriate intensity.[66, 68]
Overall, as exemplified by childhood acute leukemias, the use of conventional agents with utilization optimized over the course of many clinical trials has led to dramatic improvement of patient survival.[69] However, due to the mostly indolent course of LGL leukemia and its rarity, large cohort trials with treatment randomization and sufficient power to detect significant differences between treatment groups are challenging, and multi-center collaborations are necessary. This is certainly not a unique challenge in LGL-leukemia, as many other rare and orphan diseases face similar issues. One analytical strategy to addres the low sample size issue of rare disease is to employ a case matched control study, using a database of historical controls. [70]
In a prospective setting, a multi-drug approach may improve LGL leukemia patient outcomes given that leukemic clone expansion and persistence relies on multiple, often redundant and overlapping pathways.[27, 30, 71, 29] Precisely for these reasons, a better understanding of dysregulated signaling involved in LGL leukemia pathogenesis should not be underappreciated as it may identify novel treatment targets. Increased understanding of synergistic therapeutic vulnerabilities may justify the combinatorial use of novel and FDA-approved agents in clinical trials to come.
Conclusion
Comprehensive dynamic and structural analysis of network modeling as well as gene set enrichment analyses and meticulous experimental validation are needed to deepen our knowledge of the intricate molecular interplay in this lymphoproliferative disease. Just as the identification of aberrant signaling pathways may be predictive of changes in global gene expression patterns and impact on inciting mutations, identification and characterization of novel somatic mutations may be reciprocally informative of signaling pathways contributing to the molecular pathology of the LGL leukemia.[6, 58, 71, 72]
Undoubtedly, close collaboration between clinical and basic science research on a multi-center scale is indispensable to improve available knowledge on LGL leukemia pathogenesis, current therapeutic approaches and management. In an era of a globally ageing population and large potential for the underdiagnosis of this malignant lymphoproliferation, there is an urgency to better the patient outcomes. To help us achieve this goal we ask physicians to encourage their patients to join the LGL Leukemia Registry at the University of Virginia.
Acknowledgments:
We thank Bryna Shemo and Rachel Stidham for LGL Leukemia Registry support. We wish to extend a special thanks to the LGL Leukemia Registry patients for their enthusiastic support and interest in our research.
Funding: LGL leukemia research in the Loughran lab is supported by the National Cancer Institute of the National Institutes of Health under award number R01CA178393 and P30CA044579 (to TPL) and the NIH Cancer Research Training in Molecular Biology Award T32CA009109 (to KBM). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Additional support was provided by the Bess Family Charitable Fund, the LGL Leukemia Foundation and a generous anonymous donor.
Footnotes
Disclosures: Thomas P. Loughran, Jr. is on the Scientific Advisory Board and has stock options for Keystone Nano and Bioniz Therapeutics. Thomas P. Loughran and David J. Feith have received honoraria from Kymera Therapeutics. There are no conflicts of interest with the work presented in this manuscript.
References
- 1.Lamy T, Moignet A, Loughran TP (2017) LGL leukemia: from pathogenesis to treatment. Blood 129:1082–1094 [DOI] [PubMed] [Google Scholar]
- 2.Swerdlow SH, Campo E, Pileri SA, et al. (2016) The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 127:2375–2390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Loughran TP, Kadin ME, Starkebaum G, Abkowitz JL, Clark EA, Disteche C, Lum LG, Slichter SJ (1985) Leukemia of large granular lymphocytes: association with clonal chromosomal abnormalities and autoimmune neutropenia, thrombocytopenia, and hemolytic anemia. Ann Intern Med 102:169–175 [DOI] [PubMed] [Google Scholar]
- 4.Jerez A, Clemente MJ, Makishima H, et al. (2012) STAT3 mutations unify the pathogenesis of chronic lymphoproliferative disorders of NK cells and T-cell large granular lymphocyte leukemia. Blood 120:3048–3057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rajala HLM, Eldfors S, Kuusanmäki H, et al. (2013) Discovery of somatic STAT5b mutations in large granular lymphocytic leukemia. Blood 121:4541–4550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koskela HLM, Eldfors S, Ellonen P, et al. (2012) Somatic STAT3 mutations in large granular lymphocytic leukemia. N Engl J Med 366:1905–1913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Loughran TP (1993) Clonal diseases of large granular lymphocytes. Blood 82:1–14 [PubMed] [Google Scholar]
- 8.Dinmohamed AG, Brink M, Visser O, Jongen-Lavrencic M (2016) Population-based analyses among 184 patients diagnosed with large granular lymphocyte leukemia in the Netherlands between 2001 and 2013. Leukemia 30:1449–1451 [DOI] [PubMed] [Google Scholar]
- 9.Shah MV, Hook CC, Call TG, Go RS (2016) A population-based study of large granular lymphocyte leukemia. Blood Cancer J 6:e455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bareau B, Rey J, Hamidou M, et al. (2010) Analysis of a French cohort of patients with large granular lymphocyte leukemia: a report on 229 cases. Haematologica 95:1534–1541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Suzuki R, Suzumiya J, Nakamura S, et al. (2004) Aggressive natural killer-cell leukemia revisited: large granular lymphocyte leukemia of cytotoxic NK cells. Leukemia 18:763–770 [DOI] [PubMed] [Google Scholar]
- 12.Moignet A, Lamy T (2018) Latest advances in the diagnosis and treatment of large granular lymphocytic leukemia. Am Soc Clin Oncol Educ Book 38:616–625 [DOI] [PubMed] [Google Scholar]
- 13.Lamy T, Loughran TP (2011) How I treat LGL leukemia. Blood 117:2764–2774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bockorny B, Dasanu CA (2012) Autoimmune manifestations in large granular lymphocyte leukemia. Clin Lymphoma Myeloma Leuk 12:400–405 [DOI] [PubMed] [Google Scholar]
- 15.Liu X, Loughran TP (2011) The spectrum of large granular lymphocyte leukemia and Felty’s syndrome. Curr Opin Hematol 18:254–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Qiu Z-Y, Qin R, Tian G-Y, Wang Y, Zhang Y-Q (2019) Pathophysiologic mechanisms and management of large granular lymphocytic leukemia associated pure red cell aplasia. Onco Targets Ther 12:8229–8240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kawakami T, Sekiguchi N, Kobayashi J, et al. (2018) Frequent STAT3 mutations in CD8+ T cells from patients with pure red cell aplasia. Blood Adv 2:2704–2712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gentile TC, Uner AH, Hutchison RE, Wright J, Ben-Ezra J, Russell EC, Loughran TP (1994) CD3+, CD56+ aggressive variant of large granular lymphocyte leukemia. Blood 84:2315–2321 [PubMed] [Google Scholar]
- 19.Morice WG, Jevremovic D, Hanson CA (2007) The expression of the novel cytotoxic protein granzyme M by large granular lymphocytic leukaemias of both T-cell and NK-cell lineage: an unexpected finding with implications regarding the pathobiology of these disorders. Br J Haematol 137:237–239 [DOI] [PubMed] [Google Scholar]
- 20.Loughran TP, Zickl L, Olson TL, et al. (2015) Immunosuppressive therapy of LGL leukemia: prospective multicenter phase II study by the Eastern Cooperative Oncology Group (E5998). Leukemia 29:886–894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Loughran TP, Kidd PG, Starkebaum G (1994) Treatment of large granular lymphocyte leukemia with oral low-dose methotrexate. Blood 84:2164–2170 [PubMed] [Google Scholar]
- 22.Lamy T, Pastoret C, Houot R, et al. (2019) Prospective, multicentric phase II randomized trial comparing the efficacy of methotrexate or cyclophosphamide in large granular lymphocytic leukemia: A french national study. report on the interim analysis. Blood 134:1545–1545 [Google Scholar]
- 23.Fraiser LH, Kanekal S, Kehrer JP (1991) Cyclophosphamide toxicity. Characterising and avoiding the problem. Drugs 42:781–795 [DOI] [PubMed] [Google Scholar]
- 24.Smith RE, Bryant J, DeCillis A, Anderson S, National Surgical Adjuvant Breast and Bowel Project Experience (2003) Acute myeloid leukemia and myelodysplastic syndrome after doxorubicin-cyclophosphamide adjuvant therapy for operable breast cancer: the National Surgical Adjuvant Breast and Bowel Project Experience. J Clin Oncol 21:1195–1204 [DOI] [PubMed] [Google Scholar]
- 25.Moignet A, Hasanali Z, Zambello R, et al. (2014) Cyclophosphamide as a first-line therapy in LGL leukemia. Leukemia 28:1134–1136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Graham RM (1994) Cyclosporine: mechanisms of action and toxicity. Cleve Clin J Med 61:308–313 [DOI] [PubMed] [Google Scholar]
- 27.Zhang R, Shah MV, Yang J, Nyland SB, Liu X, Yun JK, Albert R, Loughran TP (2008) Network model of survival signaling in large granular lymphocyte leukemia. Proc Natl Acad Sci USA 105:16308–16313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sun H, Wei S, Yang L (2019) Dysfunction of immune system in the development of large granular lymphocyte leukemia. Hematology Am Soc Hematol Educ Program 24:139–147 [DOI] [PubMed] [Google Scholar]
- 29.Kallemeijn MJ, de Ridder D, Schilperoord-Vermeulen J, van der Klift MY, Sandberg Y, van Dongen JJM, Langerak AW (2017) Dysregulated signaling, proliferation and apoptosis impact on the pathogenesis of TCRγδ+ T cell large granular lymphocyte leukemia. PLoS One 12:e0175670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leblanc F, Zhang D, Liu X, Loughran TP (2012) Large granular lymphocyte leukemia: from dysregulated pathways to therapeutic targets. Future Oncol 8:787–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Viny AD, Maciejewski JP (2015) High rate of both hematopoietic and solid tumors associated with large granular lymphocyte leukemia. Leuk Lymphoma 56:503–504 [DOI] [PubMed] [Google Scholar]
- 32.Nyland SB, Feith DJ, Poss M, Olson TL, Krissinger DJ, Poiesz BJ, Ruscetti FW, Loughran TP (2019) Retroviral sero-reactivity in LGL leukaemia patients and family members. Br J Haematol. doi: 10.1111/bjh.16223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wlodarski MW, Nearman Z, Jankowska A, Babel N, Powers J, Leahy P, Volk H-D, Maciejewski JP (2008) Phenotypic differences between healthy effector CTL and leukemic LGL cells support the notion of antigen-triggered clonal transformation in T-LGL leukemia. J Leukoc Biol 83:589–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Viny AD, Lichtin A, Pohlman B, Loughran T, Maciejewski J (2008) Chronic B-cell dyscrasias are an important clinical feature of T-LGL leukemia. Leuk Lymphoma 49:932–938 [DOI] [PubMed] [Google Scholar]
- 35.Skarbnik APZ, Portell CA, Maciejewski JP, et al. (2013) Association Of Large Granular Lymphocytic Leukemia (LGL) With B-Cell Lymphoproliferative Disorders. Blood 122:1387–1387 [Google Scholar]
- 36.Starkebaum G, Loughran TP, Kalyanaraman VS, Kadin ME, Kidd PG, Singer JW, Ruscetti FW (1987) Serum reactivity to human T-cell leukaemia/lymphoma virus type I proteins in patients with large granular lymphocytic leukaemia. Lancet 1:596–599 [DOI] [PubMed] [Google Scholar]
- 37.Loughran T, Coyle T, Sherman M, Starkebaum G, Ehrlich G, Ruscetti F, Poiesz B Detection of Human T-cel l Leukemia/Lymphoma Virus, Type 11, in a Patient With Large Granular Lymphocyte Leukemia. [PubMed]
- 38.Pulik M, Lionnet F, Genet P, Petitdidier C, Jary L, Fourcade C (1997) CD3+ CD8+ CD56- clonal large granular lymphocyte leukaemia and HIV infection. Br J Haematol 98:444–445 [DOI] [PubMed] [Google Scholar]
- 39.Perzova R, Graziano E, Sanghi S, et al. (2013) Increased seroreactivity to HERV-K10 peptides in patients with HTLV myelopathy. Virol J 10:360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li W, Yang L, Harris RS, Lin L, Olson TL, Hamele CE, Feith DJ, Loughran TP, Poss M (2019) Retrovirus insertion site analysis of LGL leukemia patient genomes. BMC Med Genomics 12:88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chang H, Kamel-Reid S, Hussain N, Lipton J, Messner HA (2005) T-cell large granular lymphocytic leukemia of donor origin occurring after allogeneic bone marrow transplantation for B-cell lymphoproliferative disorders. Am J Clin Pathol 123:196–199 [PubMed] [Google Scholar]
- 42.Gill H, Ip AHW, Leung R, So JCC, Pang AWK, Tse E, Leung AYH, Lie AKW, Kwong YL (2012) Indolent T-cell large granular lymphocyte leukaemia after haematopoietic SCT: a clinicopathologic and molecular analysis. Bone Marrow Transplant 47:952–956 [DOI] [PubMed] [Google Scholar]
- 43.Gentile TC, Hadlock KG, Uner AH, Delal B, Squiers E, Crowley S, Woodman RC, Foung SK, Poiesz BJ, Loughran TP (1998) Large granular lymphocyte leukaemia occurring after renal transplantation. Br J Haematol 101:507–512 [DOI] [PubMed] [Google Scholar]
- 44.Alfano G, Fontana F, Colaci E, Mori G, Cerami C, Messerotti A, Potenza L, Luppi M, Cappelli G (2019) T-cell large granular lymphocyte leukemia in solid organ transplant recipients: case series and review of the literature. Int J Hematol 110:313–321 [DOI] [PubMed] [Google Scholar]
- 45.Epling-Burnette PK, Liu JH, Catlett-Falcone R, et al. (2001) Inhibition of STAT3 signaling leads to apoptosis of leukemic large granular lymphocytes and decreased Mcl-1 expression. J Clin Invest 107:351–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Clemente MJ, Wlodarski MW, Makishima H, et al. (2011) Clonal drift demonstrates unexpected dynamics of the T-cell repertoire in T-large granular lymphocyte leukemia. Blood 118:4384–4393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kerr CM, Clemente MJ, Chomczynski PW, et al. (2019) Subclonal STAT3 mutations solidify clonal dominance. Blood Adv 3:917–921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shvidel L, Duksin C, Tzimanis A, Shtalrid M, Klepfish A, Sigler E, Haran M, Eilat E, Berrebi A (2002) Cytokine release by activated T-cells in large granular lymphocytic leukemia associated with autoimmune disorders. Hematol J 3:32–37 [DOI] [PubMed] [Google Scholar]
- 49.Kothapalli R, Nyland SB, Kusmartseva I, Bailey RD, McKeown TM, Loughran TP (2005) Constitutive production of proinflammatory cytokines RANTES, MIP-1beta and IL-18 characterizes LGL leukemia. Int J Oncol 26:529–535 [PubMed] [Google Scholar]
- 50.Teramo A, Gattazzo C, Passeri F, et al. (2013) Intrinsic and extrinsic mechanisms contribute to maintain the JAK/STAT pathway aberrantly activated in T-type large granular lymphocyte leukemia. Blood 121:3843–54, S1 [DOI] [PubMed] [Google Scholar]
- 51.Yang J, Liu X, Nyland SB, Zhang R, Ryland LK, Broeg K, Baab KT, Jarbadan NR, Irby R, Loughran TP (2010) Platelet-derived growth factor mediates survival of leukemic large granular lymphocytes via an autocrine regulatory pathway. Blood 115:51–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zambello R, Facco M, Trentin L, et al. (1997) Interleukin-15 triggers the proliferation and cytotoxicity of granular lymphocytes in patients with lymphoproliferative disease of granular lymphocytes. Blood 89:201–211 [PubMed] [Google Scholar]
- 53.Chen J, Petrus M, Bamford R, Shih JH, Morris JC, Janik JE, Waldmann TA (2012) Increased serum soluble IL-15Rα levels in T-cell large granular lymphocyte leukemia. Blood 119:137–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hodge DL, Yang J, Buschman MD, et al. (2009) Interleukin-15 enhances proteasomal degradation of bid in normal lymphocytes: implications for large granular lymphocyte leukemias. Cancer Res 69:3986–3994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang TT, Yang J, Zhang Y, et al. (2019) IL-2 and IL-15 blockade by BNZ-1, an inhibitor of selective γ-chain cytokines, decreases leukemic T-cell viability. Leukemia 33:1243–1255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Brammer JE, Sokol L, Tagaya Y, et al. (2019) Blockade of IL-15 Utilizing Bnz-1, a Selective γ-Chain Inhibiting Peptide, Is Safe and Has Clinical Activity in Patients with T-Cell Large Granular Lymphocytic Leukemia (T-LGLL): Results of a Phase I/II Multi-Center Clinical Trial. Blood 134:2835–2835 [Google Scholar]
- 57.Mishra A, Liu S, Sams GH, et al. (2012) Aberrant overexpression of IL-15 initiates large granular lymphocyte leukemia through chromosomal instability and DNA hypermethylation. Cancer Cell 22:645–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Raess PW, Cascio MJ, Fan G, Press R, Druker BJ, Brewer D, Spurgeon SE (2017) Concurrent STAT3, DNMT3A, and TET2 mutations in T-LGL leukemia with molecularly distinct clonal hematopoiesis of indeterminate potential. Am J Hematol 92:E6–E8 [DOI] [PubMed] [Google Scholar]
- 59.LeBlanc FR, Liu X, Hengst J, Fox T, Calvert V, Petricoin EF, Yun J, Feith DJ, Loughran TP (2015) Sphingosine kinase inhibitors decrease viability and induce cell death in natural killer-large granular lymphocyte leukemia. Cancer Biol Ther 16:1830–1840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kaelin WG (2017) Common pitfalls in preclinical cancer target validation. Nat Rev Cancer 17:425–440 [DOI] [PubMed] [Google Scholar]
- 61.Wong CH, Siah KW, Lo AW (2019) Estimation of clinical trial success rates and related parameters. Biostatistics 20:273–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lin A, Giuliano CJ, Palladino A, et al. (2019) Off-target toxicity is a common mechanism of action of cancer drugs undergoing clinical trials. Sci Transl Med. doi: 10.1126/scitranslmed.aaw8412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Thomas S, Fisher KH, Snowden JA, Danson SJ, Brown S, Zeidler MP (2015) Methotrexate is a JAK/STAT pathway inhibitor. PLoS One 10:e0130078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bedoui Y, Guillot X, Sélambarom J, Guiraud P, Giry C, Jaffar-Bandjee MC, Ralandison S, Gasque P (2019) Methotrexate an Old Drug with New Tricks. Int J Mol Sci. doi: 10.3390/ijms20205023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Farber S, Diamond LK (1948) Temporary remissions in acute leukemia in children produced by folic acid antagonist, 4-aminopteroyl-glutamic acid. N Engl J Med 238:787–793 [DOI] [PubMed] [Google Scholar]
- 66.Hunger SP, Lu X, Devidas M, Camitta BM, Gaynon PS, Winick NJ, Reaman GH, Carroll WL (2012) Improved survival for children and adolescents with acute lymphoblastic leukemia between 1990 and 2005: a report from the children’s oncology group. J Clin Oncol 30:1663–1669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gamis AS, Alonzo TA, Perentesis JP, Meshinchi S, COG Acute Myeloid Leukemia Committee (2013) Children’s Oncology Group’s 2013 blueprint for research: acute myeloid leukemia. Pediatr Blood Cancer 60:964–971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hunger SP, Loh ML, Whitlock JA, Winick NJ, Carroll WL, Devidas M, Raetz EA, COG Acute Lymphoblastic Leukemia Committee (2013) Children’s Oncology Group’s 2013 blueprint for research: acute lymphoblastic leukemia. Pediatr Blood Cancer 60:957–963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Vitanza NA (2016) 50 Years Ago in TheJournal ofPediatrics: Induction of Remission in Acute Leukemia of Childhood by Combination of Prednisone and Either 6-Mercaptopurine or Methotrexate. J Pediatr 173:100. [DOI] [PubMed] [Google Scholar]
- 70.O’Connor OA, Marchi E, Volinn W, Shi J, Mehrling T, Kim WS (2018) Strategy for assessing new drug value in orphan diseases: an international case match control analysis of the PROPEL study. JNCI Cancer Spectr 2:pky038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Moosic KB, Paila U, Olson KC, Dziewulska K, Wang TT, Xing JC, Ratan A, Feith DJ, Loughran TP, Olson TL (2019) Genomics of LGL leukemia and select other rare leukemia/lymphomas. Best Pract Res Clin Haematol 32:196–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Andersson EI, Rajala HLM, Eldfors S, et al. (2013) Novel somatic mutations in large granular lymphocytic leukemia affecting the STAT-pathway and T-cell activation. Blood Cancer J 3:e168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Go RS, Lust JA, Phyliky RL (2003) Aplastic anemia and pure red cell aplasia associated with large granular lymphocyte leukemia. Semin Hematol 40:196–200 [DOI] [PubMed] [Google Scholar]