

Abstract
This review is focusing on the understanding of various factors and components governing and controlling the occurrence of ventricular arrhythmias including (i) the role of various ion channel-related changes in the action potential (AP), (ii) electrocardiograms (ECGs), (iii) some important arrhythmogenic mediators of reperfusion, and pharmacological approaches to their attenuation. The transmembrane potential in myocardial cells is depending on the cellular concentrations of several ions including sodium, calcium, and potassium on both sides of the cell membrane and active or inactive stages of ion channels. The movements of Na+, K+, and Ca2+ via cell membranes produce various currents that provoke AP, determining the cardiac cycle and heart function. A specific channel has its own type of gate, and it is opening and closing under specific transmembrane voltage, ionic, or metabolic conditions. APs of sinoatrial (SA) node, atrioventricular (AV) node, and Purkinje cells determine the pacemaker activity (depolarization phase 4) of the heart, leading to the surface manifestation, registration, and evaluation of ECG waves in both animal models and humans. AP and ECG changes are key factors in arrhythmogenesis, and the analysis of these changes serve for the clarification of the mechanisms of antiarrhythmic drugs. The classification of antiarrhythmic drugs may be based on their electrophysiological properties emphasizing the connection between basic electrophysiological activities and antiarrhythmic properties. The review also summarizes some important mechanisms of ventricular arrhythmias in the ischemic/reperfused myocardium and permits an assessment of antiarrhythmic potential of drugs used for pharmacotherapy under experimental and clinical conditions.
Keywords: genetics, ischemia—reperfusion, electrocardiogram (ECG), arrhythmia < cardiovascular, therapy -, action potential (AP)
The Heart and Arrhythmias
The heart is the key center of the blood circulation in fish, reptiles, birds, and mammals, an organ contracts autonomously and rhythmically, and functioning in conjunction with an extensive network of blood vessels to supply all cells and organs with oxygen and nutrients for serving their physiological function. Do we really need another review on cardiac arrhythmias in a special issue, someone may ask as you browse through several publications in cardiovascular journals. The answer is yes, and the mission of the current review is to present a short and comprehensive analysis of what is currently known about ischemia- and reperfusion-induced arrhythmias and their mechanisms, focusing on changes in shapes and waves in action potential (AP) and electrocardiogram (ECG), respectively, in animal models and human beings. The human heart produces left ventricular contraction about 70 times in a minute under physiological conditions. The duration and shape of APs are controlled and determined by the function and activity of various ion channels and genes in an individual cardiac cell. In various myocardial cell types, the duration of AP is varied from species to species and the beat-to-beat variability for the AP duration is an important arrhythmogenic predictor, determining the intensity of cell-to-cell coupling (Magyar et al., 2016; Nanasi et al., 2017).
Various techniques for AP recordings and ion current measurements started about 70 years ago and became more sophisticated decade by decade, including the use of monophasic, two-electrode voltage clamp, and whole cell configuration of patch clamp. The recording of resting membrane potential and AP duration have initially been reported by Woodbury et al. (Woodbury et al., 1950), Burgen and Terroux (Burgen and Terroux, 1953), Coraboeuf and Weidmann (Coraboeuf and Weidmann, 1954), and Vaughan Williams (Vaughan Williams, 1959) in cardiac muscle fiber under in vitro conditions. Particularly, ion movements via voltage-regulated ion channels and pore-regulated proteins across cardiac cell membranes primarily determine the morphology and the duration of the AP in myocardial cells (Sankaranarayanan et al., 2017; Smith and Eisner, 2019). The resting transmembrane potential in an intact cardiac cell is about 85–90 mV negative to the exterior, and the move of Na+ into the resting cell leads to the change of the membrane potential and the propagation of the cellular AP. Thus, this movement of Na+ determines Ca2+ and K+ release and exchange mechanisms via outer and inner cell membranes and organelles.
Ventricular arrhythmias are the leading cause of sudden cardiac death during and/or following an ischemic episode, acute heart failure, and recovery from myocardial infarction. Life threatening arrhythmias are very common and unpredictable in the failing myocardium; thus, the primary objective of their prevention is to eliminate various environmental risk factors, following the application of appropriate pharmacotherapies or nonpharmacological interventions. Heart failure and sudden ventricular arrhythmia caused deaths are likely to occur before an intervention can be implemented (Ramirez et al., 2014; Ramirez et al., 2017). The tool used for the detection and diagnosis of cardiac arrhythmias is the ECG, thus, the aim of antiarrhythmic therapy could be considered for acute termination or long-term treatment to prevent recurrence of various types of arrhythmias. However, antiarrhythmic drugs have multiple effects on the generation of AP and the waves of ECG, and their mechanisms are very complex (Alvarez et al., 2019; Paar et al., 2019). In addition, an antiarrhythmic agent could modulate other targets and tissues in the different parts of the myocardium in comparison with its primary site of action. In other words, an arrhythmia type and its origin could result from multiple underlying mechanisms at the same time, which may result in either from increased or decreased various types of K+, Na+, and Ca2+ currents via cell membranes and cell organelles. Thus, pharmacological therapies of cardiac arrhythmias should be focused on the most relevant underlying mechanism of arrhythmias, where it's known, and interventions may be antiarrhythmic by suppressing the initiating arrhythmogenic mechanism. Atrial fibrillation (AF) is caused by various mechanisms and one of the most common cardiac rhythm disturbances (Chua et al., 2019; Sutanto et al., 2019) associated with increased risk of heart failure, stroke (Kostopoulou et al., 2019), and death. More than dozens of genetic loci have been associated with AF, but the current review does not attempt to focus on these genetic mechanisms (Kalsto et al., 2019) and clinical management (Sutanto et al., 2019) of AF in detail.
The Action Potential
Ion currents through cell membranes critically determine the shape of the AP and the waves of the ECG, which serve for the recognition and diagnosis of various types of cardiac arrhythmias. Namely, Na+ currents, L- and T-type Ca2+ currents, transient outward currents, inward rectifier and pacemaker currents, delayed rectifier K+ currents, Na+–Ca2+-exchanger (NCX), and Na+–K+-ATPase pump are the basic components of the AP development. In each AP, Na+ entry and K+ efflux and exchanges are the initial processes for determining of other ion concentrations and functions inside the cardiac cell. The ATP-depending exchange mechanism, called Na+–K+-pump, serves to determine and maintain intracellular ion homeostasis, which is electrogenic, and generating a net K+ current. Under physiological conditions, intracellular Ca2+ is maintained at very low level in cardiac cells, and the entry of Ca2+ during AP via L-type Ca2+ channels is the primary signal to the sarcoplasmic reticulum (SR) to release Ca2+ ions from its store, leading to the initiation of Ca2+-dependent contraction termed excitation–contraction coupling. Ca2+ efflux from SR into the cytoplasm happens via ryanodine receptor release channels (RyRc), and the subsequent elimination of intracellular Ca2+ from cytoplasm occurs by SERCA, which pumps Ca2+ ions back to the SR. In the meantime, the electrogenic NCX located on the cardiac cell membrane surface exchanges three Na+ from the exterior for each Ca2+ ion extruded. During the past three decades, intensive investigation focused on the ion currents in AP phases, which were connected to specific channel-controlled genes, encoding the major pore-regulated proteins.
The AP, Ion Channels, and Genes
The identification of specific ion channels and their encoded proteins has contributed to a better understanding of the physiological and pathological APs and their changes, which affect the shape of ECG waves, leading to the mechanisms and diagnosis of various types of arrhythmias and discovery of potential new antiarrhythmic drug targets.
SCN5A
The primary gene (SCN5A) encodes the pore-forming alpha subunit of the primary Na+ channel (Nav1.5) in the human heart (Lieve et al., 2017; Verkerk et al., 2018), and this so-called “gain-of-function mutation” determines the late Na+ current (INa), which leads to (i) the development of one type of long-QT syndrome (LQTS), (ii) the prolongation of the duration of the AP, and (iii) the genesis of premature ventricular contractions in myocardial cells (Keating and Sanguinetti, 2001; Splawski et al., 2002; Wang et al., 2004; Amin et al., 2018). In addition, it was found that genetic mutations of SCN5A variants, including Nav1.5, determine INa and the development of both the LQTS and Brugada syndromes (Tse et al., 2017; Li et al., 2018). Therefore, interventions, which inhibit the mutated gene's (SCN5A) function and/or the abnormal late Na+ current (Figure 1) may prevent the development of LQTS. In contrast, drugs enhance the late Na+ current may cause both atrial and ventricular arrhythmias; called proarrhythmic agents.
Figure 1.
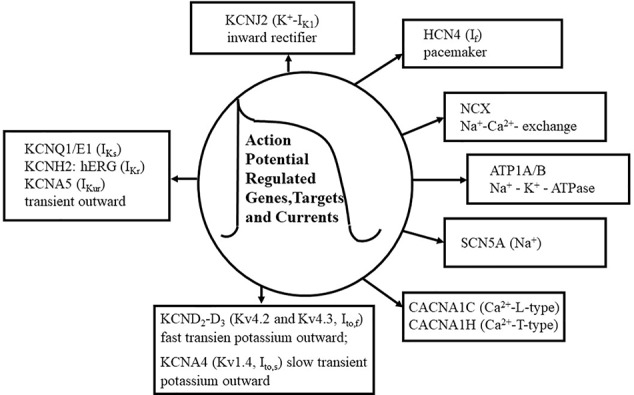
Action potential (AP) genes and ion channels. The figure shows the summary of basic ion currents, genes and their targets in the AP. Various types of Ca2+ currents are shown, including L-type and T-type Ca2+ currents (CACNA1C, CACNA1H). KCND2-D3 genes (Kv4.2, Kv4.3), as voltage-gated fast transient outward potassium (Ito,f) and slow transient K+ outward (Ito,s) channels are also shown. Additionally, Na+ and K+ channels and their pore-regulated proteins and voltage regulations are also depicted. The Na+ current (SCN5A) is about 50 times lager as any other current during the depolarization phase (phase 1) of the AP, although its portion persists in the plateau phase. Several types of Ca2 + currents e.g., CACNA1C (L-type) and CACNA1H (T-type), are activated during the phases 0, 1, and 2 of the AP. Fast transient outward potassium currents (KCND2-D3, encoding Kv4.2-Kv4.3) are functioning in the phases of 0, 1, and 2 of the AP. The activation of KCDN2 is rapidly terminated in the “notch” phase, phase 1, of the AP. Rectifier potassium channels include IKs (KCNQ1, KCNE1), IKr (KCNH2, hERG), and IKur (KCNA5), which are also activated in the phases of 0, 1, and 2 of the AP. Inward rectifier current (KCNJ2), pacemaker current (HCN4, If), and Na+–K+-ATPase (ATP1A/B) are activated in the phase 4 of the AP. Na+–Ca2+ exchange mechanism (NCX) is functioning in the phases of 1, 2, and 3 of the AP.
KCND2-D3
Transient outward potassium currents (KCND2-D3, Kv4.2, and Kv4.3) determine the development of the phases (0, 1, and 2) of the AP, however, the activation of KCND2 (Kv4.2) is rapidly terminated in the “notch” phase of the AP. It has been reported that KCND2 and KCND3 (Frank-Hansen et al., 2005), encoding of the voltage-gated potassium channel alpha subunits, Kv4.2 and Kv4.3, conducting the fast transient outward K+ current (Ito,f) in the prolongation of AP duration, which may be associated with the development of LQTS in the myocardium. These authors (Frank-Hansen et al., 2005) concluded that mutations of KCND2 and KCND3 are not a frequent cause of LQTS, however, some variants of these two genes may play a significant role in the development of LQTS phenotype. In another study (Perrin et al., 2014), it was shown that KCND2 gene, as a susceptibility gene, contributes to the sudden cardiac death in an anterior J-wave ECG pattern in patients. The results of the aforementioned publication (Perrin et al., 2014) were the first to implicate KCND2 gene (Kv4.2) as a potential cause of an atypical J-wave pattern related to the sudden cardiac death. Considerable attention has been paid to the understanding of the roles of two functionally distinct K+ channels; Kv4.2 and Kv4.3 subunits (Niwa and Nerbonne, 2010), which encode the fast transient outward potassium channels (Ito,f), while Kv1.4 (KCNA4) encodes the function of the slow potassium channels (Ito,s). Thus, it could be concluded that several signaling cascade mechanisms and regulatory proteins have been implicated in the control and regulation of Ito,f and Ito,s proteins/channels, regarding the mechanisms of arrhythmogenesis under physiological and various pathological conditions. Although cardiac Ito,f and Ito,s channels are differently expressed in various mammalian species and contribute to the heterogeneity of the AP and its waveforms, they have a potential importance in the genesis of cardiac arrhythmias. In addition, it has recently been demonstrated by Zhang et al. (2019a), that jingzhaotoxin-V (JZTX-V), a selective inhibitor of A-type potassium channel, plays a crucial role in the regulation of Ito potassium channels, supporting its use as a new potential and novel antiarrhythmic agent.
KCNQ1/E1 and KCNA5
K+ currents so-called delayed rectifier currents, including slow (IKs) and ultrarapid (IKur) channels (Zareba et al., 2003), have been dissociated on the basis how quickly they are activated during the AP. However, cardiac KCNQ1/E1 encodes only slow delayed rectifier channels and does not IKr (rapid delayed rectifier K+ current). These K+ rectifier channel activations are increased with the time, whereas Ca2+ currents are inactivated, leading to the phase-3 (repolarization phase) of AP (Figure 1). It was described (Christophersen et al., 2013) that KCNA5 (IKur) has a high frequency of rare variants related to the development of AF, emphasizing that KCNA5 (Figure 1) is probably one of the most predominant genes to produce AF (Colman et al., 2017). KCNA5 also encodes hKv1.5, and this genetic alteration increases the electrical activity of cells, therefore, the controlling of hKv1.5 expression and function may result in a proper physiological electrical activity, suggesting that hKv1.5 may be a potential target for the treatment of AF (Xie et al., 2019). Thus, the selective inhibition of hKv1.5 function could be related to the prolongation of atrial AP without an increase in the duration of ventricular AP in human beings.
KCNH2
KCNH2, initially described and termed hERG, regulates IKr (rapid delayed rectifier K+ channel) and the blocking of IKr still remains a major issue in the development of antiarrhythmic drugs (Chen et al., 2000; Mitcheson et al., 2000; Mehta et al., 2018) in connection with the pharmacotherapy of LQTS at genetic levels (Mehta et al., 2018; Yin et al., 2018; Cortez et al., 2019; Kerr et al., 2019). The voltage activated potassium channel (Kv11.1) is encoded by the humane-ether-a-go-go related gene (hERG), which predominantly contributes to the electrical activity of the myocardium (Vandenberg et al., 2012; Vasseur et al., 2019). This channel mediates IKr current (Figure 1) during the repolarization phase of the AP (Orvos et al., 2019) and its malfunction plays a critical role in the development of LQTS (Perissinotti et al., 2018; Ng et al., 2019). In an additional elegant study, Wu et al. (2019) described that KCNQ1 mutation interferes with intracellular hERG transport processes, leading to the development of the phenotype LQTS.
KCNJ2
It was published in an elegant review (Kubo et al., 2005) that inward rectifier K+ channel (Kir) subfamilies, based on their amino acid sequence alignments, play important roles in various human diseases, including arrhythmogenesis (Figure 1). Therefore, pharmacological aspects of Kir channel mediated functions could be expected as therapeutic tools for the treatment of various cardiac arrhythmias. It was suggested that (Yue et al., 2011; Qi et al., 2015) myofibroblasts also play a critical role in arrhythmogenesis in chronic heart failure by increasing extracellular matrix protein production and having a direct electric interaction with myocytes. However, fibroblasts are not excitable cells electrically, but they are able to express various ion channels including IK1, and having resting membrane potential about −40 mV (Kamkin et al., 2003). The study by Qi et al. (2015), demonstrated that KCNJ2 (inward rectifier potassium channel; IK1) subunit is present in fibroblasts and cardiomyocytes as well. Thus, drug therapies, which alter the function of KCNJ2 ion channels in connection with Ca2+ entry may be an important target for the development of other new antiarrhythmic agents, as inward rectifier potassium channel blockers, in failing myocardium (Szuts et al., 2013).
HCN4, “Funny” Current
The pacemaker current (If or IKf) is called “funny” current or pacemaker channel, which is an electric current through the HCN4 and subtype genes regulated proteins in the myocardium (Figure 1). The “funny” current is a component of the electrical conduction system producing the natural pacemaker activity of the heart. The If was first described in Purkinje fibers and atrial cells, and has been extensively studied for several decades (Difrancesco and Ojeda, 1980; Mesirca et al., 2013; Mengesha et al., 2017; Monfredi et al., 2018), however, its function is not completely understood in arrhythmogenesis. HCN4 gene-related arrhythmias, including atrioventricular block and AF, have still an unexplored role in arrhythmogenesis, although HCN4 gene (cyclic nucleotide gated 4 channel) is generally accepted as a genetic marker in myocardial pacemaker tissues (Difrancesco, 2015). Thus, myocardial structural abnormalities could be closely associated with HCN4 mutations, leading to the development of cardiac arrhythmias via phosphatidyl-inositol 3,4,5-trisphosphate (PIP3) signal transduction mechanism; regulating the function of funny current, controlling the pacemaker activity and heart rate in the myocardium (Difrancesco, 2019). The term of “funny” refers to a mixed Na+ and K+ permeability through cell membranes and having a very slow physiological kinetic. In addition, an increase in the activation of If augments Na+ influx in the cell, and paradoxically, in a “reverse mode,” elevates intracellular Ca2+ accumulation leading to elevated systolic calcium transients. Consequently, the inhibition of If reduces intracellular Ca2+ rise, thus, alters several processes of ventricular remodeling and structural cardiac function, including apoptosis, cardiomyopathy, and arrhythmias (Yampolsky et al., 2019), which may have an important target in clinical managements of arrhythmogenesis.
NCX
The pharmacological inhibition of NCX protein expression could be a further target and may be an additional advantage for the therapy of heart failure and cardiac arrhythmias under both experimental and clinical conditions (Sipido et al., 2002; Jost et al., 2013; Schwartz et al., 2013; Bogeholz et al., 2016; Devalla et al., 2016; Kohajda et al., 2016; Javidanpour et al., 2018). During the period of diastole in the myocardium, cytosolic free Ca2+ is removed by both the reuptake of SERCA (Ca2+-ATPase) and transmembrane extrusion by NCX (Voigt et al., 2012). The basic role of NCX has been recognized in two major types of triggered arrhythmias including delayed afterdepolarization (DAD) and early afterdepolarization (EAD). DAD is developed by Ca2+ release from the SR under cellular Ca2+ overload. The increased Ca2+ overload in the cytosol is removed by NCX, causing additional Na+ influx and cell membrane depolarization, which leads to DAD. Thus, interventions, which are able to interfere with the NCX could be an effective antiarrhythmic pharmacotherapy in patients, showing DAD signs on AP and/or on ECG recordings. EAD interrupts AP in the phase of repolarization, and not only various Ca2+ channel-related intracellular Ca2+ overload but even other ion channels and transporters, particularly Na+ and K+ channels and exchangers, could also contribute to the development of EAD. Under clinical conditions, EAD triggering is a very common event in human, if the heart rate is reduced and extracellular K+ concentration is low. Basically, the duration of AP is longer in Purkinje and endocardial cells than in epicardial tissues, therefore, EAD is more frequently induced in Purkinje and endocardial cells in comparison with epicardial cells. Cardiac arrhythmias showing the sign of EAD could be treated by antiarrhythmic drugs, which shorten the AP duration, e.g., antiarrhythmic agents belong to class IB (Vaughan Williams, 1992).
ATP1alpha/Beta (Na+–K+-ATP-ase, Na+–K+-Pump)
It is of interest to note that calcium channels include the structurally homologous family of voltage-gated ion channels, such as CACNA1C-encoded L-type calcium channel (Cav1.2, LTCC), which transports calcium ions into myocytes, regulating the excitation–contraction coupling process, and leading to the development of LQTS phenotype (Estes et al., 2019; Ye et al., 2019). The results of the aforementioned recent publications reveal that a new population of Ca2+ channels represents a new “pathological substrates” for LQTS-related arrhythmias. During each AP the cell gains intracellular Na+ and loses intracellular K+ content. At the end of each AP, for returning to the physiological membrane potential of −90 mV in the myocyte, the Na+–K+-ATP-ase serves the energy to extrude three Na+ ions and enter two K+ ions. This process is electrogenic, generating a net outward Na+ current during the phases 3 and 4 of the AP. Several decades ago, it was measured and described, using the whole-cell patch-clamp technique that the aforementioned exchange mechanism is generated by the Na+–K+-pump, leading to the outward membrane current (Ip) in a single myocyte (Gadsby et al., 1985). Several groups published that peak Ip may be the clearing of excess of Na+, which is accumulated close to the inner cardiac cell membrane leaflet, if the Na+–K+-pump is inhibited (Carmeliet, 1992; Aronsen et al., 2013; Garcia et al., 2016). Thus, a restricted Na+ diffusion at a subsarcolemmal area in the cardiomyocyte may be a reasonable therapeutic implication (Han et al., 2010; Lu and Hilgemann, 2017) in the treatment of cardiac arrhythmias.
The ECG and Inherited Syndromes
The duration of APs from different regions of the myocardium, including the sinus node, AV node, and His-Purkinje system, determines the coordinated ventricular contraction, and the electrical activation of the heart, which is normally represented by ECG recordings. In the ECG, P wave, QRS complex, and T wave show the physiological or pathological electrical activity of the myocardium. P wave represents the depolarization phase of the atria, QRS complex indicates the depolarization of the left and right ventricles, while T wave shows the repolarization phase of both ventricles in the ECG (Figure 2).
Figure 2.
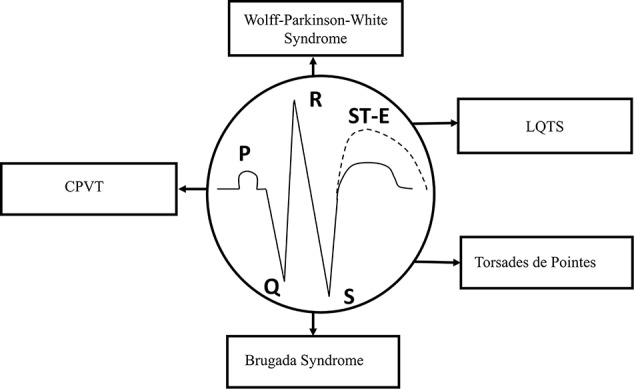
Schematic representation of the ECG. Types of syndromes originated from various gene mutations. Gene mutation-related arrhythmias include long-QT and Brugada syndromes, catecholaminergic polymorphic ventricular tachycardia (CPVT), “torsade de pointes” arrhythmias, and Wolff–Parkinson–White (WPW) syndrome. ST-E, ST-segment elevation.
At least, four major inherited syndromes (Figure 2) are currently known, which may lead to sudden cardiac death without any structural myocardial diseases, including LQTS, catecholaminergic polymorphic ventricular tachycardia (CPVT), Brugada syndrome, and “torsades de pointes” arrhythmias (Savastano et al., 2014; Delise et al., 2018; Michowitz et al., 2019). However, Wolff–Parkinson–White (WPW) syndrome, which may also be an inherited cardiac disease and leads to reentry ventricular arrhythmias, has some significant structural changes in the myocardial conduction system. Although, these syndromes and sudden consequences were first published several decades ago, the investigation of their genetic origins started about only in the last decade of the twentieth century.
Sodium channels and cardiac structural abnormalities lead to changes in ECG signs and pathological cardiac consequences, which may be associated with SCN5A gene mutation in Brugada syndrome (Coronel et al., 2005). It was suggested that a reduced expression in the number of Nav1.5 sodium channels may result in Brugada syndrome in myocardial cells and heterozygous patients (Mohler et al., 2004). Thus, interventions, which could increase the expression of Nav1.5 channels in the myocardium may prevent the development of lethal arrhythmias in “Brugada patients,” however, this mechanism should be further investigated.
The increased length of ventricular AP prolongs the QT interval on the ECG, which could be associated with the LQTS and the development of ventricular tachycardia. Four of the aforementioned syndromes (LQT, CPVT, Brugada syndrome, torsades de pointes) appear in patients who did not previously show any cardiac symptoms (Singh et al., 2019) or gross structural changes except syncope before the sudden cardiac death. The ventricular tachycardia with prolonged QT interval is termed “torsades de points” arrhythmia (Bossu et al., 2018). LQT and Brugada syndromes are the most common inherited arrhythmias, while CPVT occurs much rarely.
Dessertenne published (Dessertenne, 1966) initially several original articles about polymorphic ventricular tachycardia in the 1960s, and termed “torsades de pointes” arrhythmia based on ECG signs. Then, it has been the subject of debate for decades, whether “torsades de pointes” is a syndrome or an arrhythmia (Curtis, 1991), and it was concluded that this type of arrhythmia can be a type of arrhythmias and also a syndrome. The final outcomes of the “torsades de pointes” arrhythmia may be syncope, self-terminating, or ventricular fibrillation (VF) leading to sudden cardiac death. The QT interval's prolongation could be genetic origin or acquired (Thomas and Behr, 2016). It was also under extensive investigation (Naksuk et al., 2019) and published that LQTS is a type of arrhythmias, in which “torsades de pointes” arrhythmia causes ventricular tachycardia, VF, and sudden death by the mutation of various genes (KCNQ1/E1 and KCNH2/hERG) underlying potassium repolarization currents, including IKr and IKs (Anson et al., 2004; Nerbonne and Kass, 2005; Daubert et al., 2007; Vink et al., 2018; Zhou et al., 2019). In conclusion, clinical management of “torsades de pointes” arrhythmia associated with prolonged QT interval can be pharmacologically treated with intravenous magnesium and potassium infusions for the correction of electrolyte imbalance, and administration of isoproterenol to increase heart rate (Narang and Ozcan, 2019). The application of antiarrhythmic drugs, which prolong the repolarization phase of AP in myocardial cells should be avoided.
The CPVT, which is relatively a rare disease in comparison with the appearance of Brugada and LQTS, applies to an autosomal inherited familial disorder characterized by exercise-induced adrenergic polymorphic ventricular tachycardia, leading to VF and sudden cardiac death (Marks et al., 2002). These “arrhythmogenic” genes are located to the region of genetic locus of chromosome 1q42–q43 as described by Marks et al. (2002), Swan et al. (1999), and Rampazzo et al. (1995). Genetic analyses and findings led to the conclusion that chromosomal area of 1q42–q43 genes is probably closely connected to the function of ryanodine receptor (RyR)–calcium-release channels in cardiac tissues (Priori et al., 2001; Marks et al., 2002). Based on these aforementioned publications, it was suggested that potential inhibitors of RyR, especially RyR2, could be an important antiarrhythmic target for the development of new therapeutic agents (Bongianino et al., 2017; Batiste et al., 2019).
WPW syndrome is defined as the prototype of reentry mechanism, having an accessory connection between the left atrium and left ventricle, and this connection results in a pathological characteristic in QRS complexes. WPW syndrome, based on ECG recordings, was described by these authors in details (Wolff et al., 1930) about ninety years ago. Gollob et al. (2001) first published the gene in familial WPW syndrome, which could be responsible for this type of arrhythmia and sudden cardiac deaths. The authors (Gollob et al., 2001) described a mutation in the encoding gene of gamma subunit of AMP-activated protein kinase in families, which is associated with WPW syndrome, since gamma subunit of AMP-activated protein kinase has a substantial impact in the phosphorylation of several metabolic pathways, including the control of energy substrates in myocardial tissues. Currently, relatively little information is available about the mechanism of AV conduction system and the role of gamma subunit of AMP-activated protein kinase on the control and regulation of various ion channels in familial WPW syndrome (Miyamoto, 2018), although the genetic origin of WPW syndrome and gamma subunit of AMP-activated protein kinase in connection with the reentry mechanism was supported by some recent publications (Bowles et al., 2015; Ben Jehuda et al., 2018). Thus, these findings suggest that gamma subunit of AMP-activated protein kinase and glycogen regulation could be a potent therapeutic target in the treatment of familial WPW syndrome.
Clinical Management of Prolonged QT interval
Several variations are clinically available for the management of QT prolongation-related arrhythmias (Antoniou et al., 2017). The acceleration of the electrical conduction system in both under experimental and clinical conditions is generally accepted for the management of arrhythmias with prolonged QT interval. Thus, interventions that modify the function of various ion channels, especially Na+, K+, Ca2+, and Mg2+ and their exchange mechanisms, could be useful tools for pharmacotherapies of arrhythmias with prolonged QT interval. The QRS wave until the end of the T wave represents the ventricular depolarization and repolarization phases on the ECG. Abnormalities of electrolytes should be corrected with K+ infusion monitored continuously, following by intravenous Mg2+ sulfate infusion to terminate the prolonged QT interval. In some case, if it is indicated, lidocaine and phenytoin as Na+ channel blockers successfully can be also used for the management of LQTS. The beta1/beta2 nonselective adrenergic receptor agonist agent, isoproterenol, shortens QT interval, which could be an effective therapy in humans unresponsive to magnesium sulfate infusion. However, other antiarrhythmic agents initially classified by Vaughan Williams (1975), which are used in clinical therapy against hypoxia- or ischemia-induced arrhythmias e.g., beta adrenergic receptor blockers and calcium channel antagonists, should be avoided for the therapeutic management of arrhythmias with prolonged QT interval. A promising future approach, under experimental conditions to manage the CPVT-initiated QT prolongation, was published by Batiste et al. (2019) showing that an unnatural verticilide enantiomer inhibits the function of RyR2-mediated calcium leak, which inhibits QT prolongation, and prevents life threatening arrhythmias.
The left cardiac sympathetic denervation was also proposed as a useful therapy for LQTS including CPVT (De Ferrari et al., 2015; Desimone et al., 2015; Sgro et al., 2019) in addition to the application of anti‐arrhythmic agents and implantable cardioverter defibrillators. Thus, the combination of these interventions can significantly improve the final outcome of the clinical management of LQTS including CPVT in patients, and the prevention of sudden cardiac death.
Reperfusion-Induced Injury and Arrhythmias: Major Components
The response of the ischemic myocardium to reperfusion depends on the condition of the tissue at the time it is reperfused. The condition of the myocardium depends on the severity of the ischemia and the duration of the ischemic event. Substantially altered physiological processes in ultrastructural and metabolic changes during ischemia are required for the understanding of the myocardial function to reperfusion. The major ultrastructural and metabolic changes observed in the ischemic myocardium destined to die develop because of the lack of nutrition and oxygen supply and the depressed coronary flow. By definition, the myocardium is considered to be in VF, the most severe life-threatening arrhythmias, if an irregular undulating baseline is observed on the ECG (Walker et al., 1988). The term of “reperfusion-induced” injury caused by VF is originated from the definition itself of the “reperfusion”. Thousands of basic and clinical studies revealed that electrophysiological changes of the ischemic/reperfused myocardium is a dysfunction of cellular homeostasis, which includes the depletion and/or accumulation of various biochemical substances, altering the function of various ion channels and cell-to-cell coupling. Reperfusion-induced arrhythmias include the ectopic beat, tachycardia and fibrillation. The incidence and severity of reperfusion-induced arrhythmias is more frequently occurred under experimental conditions in comparison with human beings (Jennings and Reimer, 1983; Manning and Hearse, 1984; Nakata et al., 1990; Kloner, 1993; Yellon and Hausenloy, 2007; Van Der Weg et al., 2019). Under clinical conditions, reperfusion-induced arrhythmias could mainly be observed during the process of thrombolysis following myocardial ischemia and infarction, after an insult of angina and percutaneous coronary intervention (Corr and Witkowski, 1983; Tzivoni et al., 1983; Tolg et al., 2006; Yu et al., 2017; Van Der Weg et al., 2019). Thrombolytic agents, including urokinase, ataplase, tissue plasminogen activator, are used to dissolve coronary thrombus during early hours of cardiac infarction. The basic arrhythmogenic or proarrhythmogenic role of different mediators, biochemical components, and chemical substances, which accumulate or decompose by virtue of several pathological processes during ischemia/reperfusion in the cellular milieu of the myocardium could be evaluated by various criteria. Therefore, it is important to prove that the antiarrhythmic effect of a drug is primarily originated from its receptor binding activities, and not secondary, e.g., by modifying the coronary blood flow rate. The mission of this chapter is to present a brief summary of what is currently known about the origin of reperfusion-induced arrhythmias, their most important mechanisms, and final outcomes preventing the processes of necrosis, apoptosis and autophagy caused cell deaths under experimental and clinical conditions. The potential importance of major arrhythmogenic components and mediators in reperfusion is shown in Figure 3.
Figure 3.
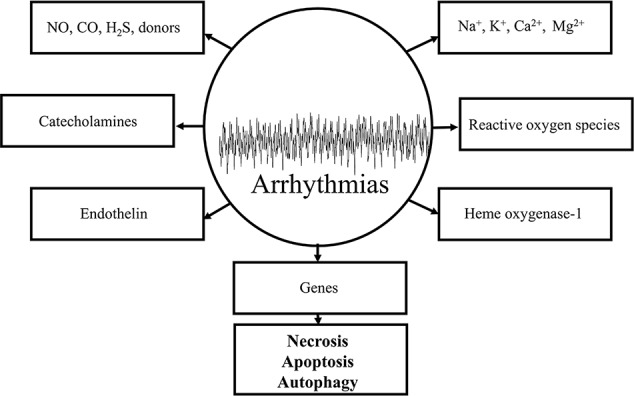
Schematic representation of some important arrhythmogenic components and mediators in the genesis of reperfusion-induced arrhythmias. All of the depicted mechanisms are very complex and each of them significantly contributes to the maldistribution of Na+, K+, Ca2+ exchange mechanisms by virtue of causing damages in cell membranes and receptors leading to necrotic-, apoptotic-, and autophagic-induced cell deaths.
The potential mechanisms underlying the genesis of reperfusion-induced arrhythmias have been extensively studied by thousands of investigators. The several pathological processes include the elevation of cAMP (Podzuweit et al., 1978; Oikawa et al., 2013), stimulation of adrenergic receptors (Sheridan et al., 1980; Amirahmadi et al., 2008; Robertson et al., 2014), abnormal lipid metabolism involving lysophosphatides' production (Akita et al., 1986; Axelsen et al., 2015), maldistribution of ionic components through cell membranes, particularly that of Na+, Ca2+, K+, and Mg2+, and the genesis of reactive oxygen species (ROS). While a huge variety of pathways may have separately been responsible for the reperfusion-induced arrhythmogenesis, it is reasonable to speculate that all of the aforementioned substances could be directly or indirectly connected to each other, and together they play a crucial final outcome, leading to irreversible arrhythmias, heart failure, and cardiac death.
K+, Na+, Ca2+, and Mg2+
If cardiac tissue is successfully reperfused in the early phase of reversible injury, its reaction is much different from that of the ischemia-induced injury. Myocytes explosively swell, thus, total tissue water content increases about by 20% following the first two min of reperfusion period (Whalen et al., 1974), and tissue electrolyte changes associate with the swelling are striking. As a consequence, there is a significant increase in cellular Na+, Ca2+, and Cl−, while cellular K+ and Mg2+ are substantially decreased (Whalen et al., 1974).
K+
Ischemia causes functional disturbance in K+ homeostasis in the myocardium and plays a critical role in the genesis of ischemia/reperfusion-induced ventricular arrhythmias (Harris et al., 1958; Tosaki et al., 1988; Curtis and Hearse, 1989). Various mechanisms of drugs by which cellular K+ loss causes electrophysiological disturbances, including the function and expression of ATP-dependent K+ channels, may involve the blocking and/or opening of these K+ channels (Noma, 1983; Tosaki et al., 1995; Das and Sarkar, 2005; Kaya et al., 2019) at various degrees, which may aggravate or diminish the severity of ventricular arrhythmias in the ischemic/reperfused myocardium. Thus, the modification of the expression of KATP channels by KATP channel opener or blocker agents, respectively, can increase or decrease the incidence of ischemia/reperfusion-induced arrhythmias. The results of the papers cited above clearly show that interventions, which modify K+ transport mechanisms via cell membranes, can even increase or reduce the incidence of ischemia/reperfusion-induced arrhythmias.
Na+
The sodium ion plays a basic process determining the physiological or pathological shape of APs and ECGs in the myocardium. Several mechanisms are currently known, which lead to intracellular Na+ accumulation under both physiological and pathological conditions, and the degree of the sodium channel blockade critically determines the membrane potential of the cell and the heart rate. Under pathological conditions, such as myocardial ischemia, the effects of Na+/H+, Na+/K+, and Na+/Ca2+ exchange mechanisms increase the intracellular Ca2+ entry, and as a consequence, the ischemic load with additional Ca2+ results in a further activation of Na+/Ca2+ mechanism during reperfusion, leading to the development of reperfusion-induced arrhythmias (Tani and Neely, 1989; Belardinelli et al., 2006). At the early minutes of reperfusion, the substantial accumulation of intracellular Na+ during ischemia contributes to a significant increase of cellular edema formation, heart failure, and ventricular arrhythmias. All of these complex mechanisms, without any pharmacological or nonpharmacological interventions, produce an excessive intracellular Ca2+ overload leading to necrotic, apoptotic, and autophagic cell deaths. Antiarrhythmic agents, which can modify these pathologic processes, including Na+-induced Ca2+ overload (Wang et al., 2007), Na+/H+ exchange mechanism, and Na+-induced K+ loss could prevent the genesis of reperfusion-induced ventricular arrhythmias and sudden cardiac death. In this respect, the Na+/H+ exchange mechanism is extensively studied to prevent reperfusion-induced arrhythmias in a variety of experimental models (Karmazyn, 1996; Ravens and Himmel, 1999; Wirth et al., 2001; Woodcock et al., 2001; Ono et al., 2004; Yamada et al., 2005; Szepesi et al., 2015; Hegyi et al., 2018). It can be concluded that Na+/H+ exchange mechanism, which is not species specific, is a basic target for pharmacological prevention in the attenuation of ischemia/reperfusion-induced cardiac damage, and may emerge as an effective basic therapeutic strategy in human beings. Finally, it is of interest to note that a direct manipulation in extracellular Na+ concentration in isolated buffer perfused hearts, a significant decrease in the incidence of reperfusion-induced VF and ventricular tachycardia was detected (Tosaki et al., 1989).
Ca2+
Under physiological conditions, intracellular Ca2+ content is maintained at very low levels, less than 100 nmol. Upon reperfusion, the massive osmotic load in ischemic myocytes, which contain very low level of ATP to fuel the ion channel proteins and pumps results in explosive cell swelling with membrane disruption and myocyte death. Reperfusion-induced arrhythmias are associated with sudden ECG changes, in which there is a Ca2+ overload at sufficient level to produce an elevated oscillatory release of Ca2+, reflecting in a significant ATP depletion (Coetzee et al., 1987; Coetzee et al., 1988). Thus, the preservation of cell membrane integrity includes maintenance of Ca2+ homeostasis, various types of ion exchange receptor-mediated mechanisms, osmotic control, and the preservation of the rich-energy phosphates. Therefore, interventions, e.g., Ca2+ channel antagonists (Tosaki et al., 1987a; Otani et al., 2013; Becerra et al., 2016), beta-adrenergic receptor blockers (Tosaki et al., 1987b; Winchester and Pepine, 2014; Gatzke et al., 2018), and several other newly discovered molecular structures and interventions (Wilder et al., 2016; Bukhari et al., 2018; Gatzke et al., 2018), which directly or indirectly contribute to the preservation of the aforementioned processes can diminish the incidence of reperfusion-induced arrhythmias, heart failure, and sudden cardiac death. In addition, during the past three decades several new mechanisms have been proved and implicated based on molecular biological studies, as endogenous mediators, into the pathways of ischemia/reperfusion-induced injury and arrhythmogenesis, including gaseous molecules; nitric monoxide, carbon dioxide, and hydrogen sulfide.
Mg2+
Magnesium is a “natural” Ca2+ channel antagonists (Iseri and French, 1984), a potent cofactor for the function of several enzymes, and plays a crucial role in protein synthesis. Mg2+ is also necessary for the insertion of various proteins into cell membranes, and stabilizes the structure of ribosomes (Flatman, 1984). A variety of disorders including diabetes, hypertension, renal tubular disease, hyperthyroidism, steatorrhea, hepatic and cardiac failure may deplete Mg2+ stores causing severe ventricular arrhythmias (Iseri and French, 1984; Lazzerini et al., 2018). It was demonstrated about three decades ago by Tzivoni et al. (1988) that “torsade de pointes” ventricular arrhythmias, which were previously resistant to conventional antiarrhythmic therapies, were prevented and controlled by the infusion of magnesium sulfate. In addition, it was also published by other investigators (Tosaki et al., 1993b; Ying et al., 2007; Amoni et al., 2017) that application of Mg2+ is a useful tool for the prevention in the development of reperfusion-induced arrhythmias. Although, the precise mechanism of Mg2+-induced antiarrhythmic effect is unclear, it has recently been supported that inhibition of the upregulation of P-selectin expression (Ying et al., 2007) and G-protein-coupled receptors (Ye et al., 2018) may be involved in Mg2+-induced cardiac protection. The antiarrhythmic effect of Mg2+ may be related to the mediation of a number of cellular processes, including particularly the reduction of cellular K+ loss, displacement of intracellular Ca2+, and maintaining the physiological Mg2+ level in myocardial cells. In addition, the possibility that Mg2+ could reduce cellular Na+ accumulation during ischemia and hence limit intracellular Ca2+ overload, could be of considerable importance to its antiarrhythmic effect (Tosaki et al., 1993b).
Reactive Oxygen Species
Studies focusing on the role of ROS in arrhythmogenesis were first published by Manning and Hearse (Manning and Hearse, 1984) and Woodward and Zakaria (Woodward and Zakaria, 1985). The authors described that ROS, which particularly derive from oxygen, cause ventricular tachycardia and VF during the first minute of the reperfusion period. The arrhythmogenic ROS may arise from several sources in the ischemic/reperfused myocardium including the degradation of catecholamines, the metabolism of arachidonic acid, mitochondrial electron transport processes, and the conversion of xanthine to hypoxanthine by xanthine oxidase. Direct evidence for the burst of ROS generation during the first few minutes of reperfusion has been initially proved by several investigators (Garlick et al., 1987; Zweier and Kuppusamy, 1988; Zweier et al., 1988; Blasig et al., 1990; Tosaki and Braquet, 1990; Samouilov et al., 2019), using electron paramagnetic resonance spectroscopy and its modification. The association between the role of sudden burst of ROS and arrhythmogenesis came also first from electrophysiological studies, proving that ROS generating systems can be potentially arrhythmogenic (Kusama et al., 1989).
The presented results strongly suggest that ROS generation plays a basic role in arrhythmogenesis in reperfusion, and the possibility also exists that ROS formation could interact with other potential pathological mechanisms to facilitate arrhythmogenesis during reperfusion. Therefore, several groups of natural and synthetic pharmacological agents could significantly diminish and/or suppress the incidence of reperfusion-induced arrhythmias. Thus, beta adrenergic receptor blockers (Manning and Hearse, 1984; Lujan et al., 2007; Kloner et al., 2011), calcium channel antagonists (Guc et al., 1993; Podesser et al., 1995; Kato et al., 2004; Bukhari et al., 2018), spin traps (Hearse and Tosaki, 1987; Tosaki et al., 1993a; Zuo et al., 2009; Pisarenko et al., 2019), and natural plants or their extracts (Tosaki et al., 1996; Shen et al., 1998; Broskova et al., 2013; Sedighi et al., 2018; Zhang et al., 2019b) alone, or in their combinations can significantly reduce the severity of ischemia/reperfusion-induced arrhythmias. Several natural products originated from plants, fruits, and/or vegetables showing antiarrhythmic activity by reducing the incidence of reperfusion-induced arrhythmias and preserving myocardial function, include mainly polyphenol and flavonoid molecular structures (Hung et al., 2000; Pataki et al., 2002; Bak et al., 2006; Broskova et al., 2013; Ma et al., 2014; Woodman et al., 2018; Kaya et al., 2019). Natural antioxidant activities and features of flavonoids/polyphenols are important examples of myocardial protection and preservation in ischemic/reperfused hearts, and being not merely a matter of various action mechanisms but showing that differences in molecular formulations may exist among them, even when they are of similar structures.
Heme Oxygenase-1 and Carbon Monoxide System
Heme oxygenase (HO) catalyzes the first step of heme degradation to carbon monoxide (CO) and biliverdin, and subsequently releases heme iron (Lee et al., 1997; Ryter et al., 1998). CO is an endogenous gaseous molecule and having an important role in hemodynamic regulation in vascular smooth muscle cells (Ishizaka and Griendling, 1997), thus, HO is able to modulate vessel tone, and regulate the changes in blood pressure via the increase of cellular cGMP contents by the activation of soluble guanylate cyclase (Johnson et al., 1995).
Two major isozymes of HO exist and produced by two distinct genes (Choi and Alam, 1996); HO-1 is inducible and can be found in various mammalian cells, and HO-2 is constitutively mainly expressed in neurons of the central nervous system. HO-1 expression or repression can be occurred in various disorders (Dulak and Jozkowicz, 2014; Loboda et al., 2016; Haines and Tosaki, 2018) and tissues subjected to oxidative and/or hemodynamic stress (Ottani et al., 2015; Cheng and Rong, 2017). Studies demonstrated that HO-1 expression can afford cellular protection via different mechanisms against oxidant stress under both in vitro (Lee et al., 1996) and in vivo (Nath et al., 1992; Waldman et al., 2019; Yu et al., 2019) conditions. It has been suggested that HO-1-mediated CO production, as a signal transduction molecule, prevents the development of reperfusion-induced VF (Csonka et al., 1999b; Pataki et al., 2001; Bak et al., 2003; Bak et al., 2010). Liang et al. (2014) published that CO prolongs the duration of AP by the inhibition of inward rectifying K+ channels. These authors also demonstrated that CO inhibits the function of Kir2.2 and Kir2.3 inward rectifier channels in myocardial cells, and directly affects Kir2.3 function via the phosphatidylinositol(4,5)-bisphosphate system. The aforementioned findings show that the inhibition of Kir2.2 and Kir2.3 leads to a substantial prolongation of AP, which may be responsible for the development of reperfusion-induced VF. In addition, Ottani A et al. (Ottani et al., 2015) demonstrated the beneficial effect of NDP-α-MSH, which associated with the overexpression of HO-1 and Bcl-XL, reducing the incidence of arrhythmias and infarct size in the ischemic/reperfused myocardium. It was also shown that very low concentration of CO added directly to the perfusion buffer protects the ischemic/reperfused myocardium via cGMP signaling in isolated hearts (Bak et al., 2005). It is reasonable to believe that manipulation of various pathways with pharmacological interventions in the HO-1/CO system plays an important and preventive role in the genesis of the development of reperfusion-induced arrhythmias and myocardial cell death.
NO, CO, H2S (Gaseous Molecules)
NO
Nitric oxide (NO) is an endogenous free-radical-type cell signaling molecule with several therapeutic applications. Under physiological conditions, NO is originated from the terminal nitrogen of L-arginine and produced by NO synthase, which has three different isozymes, including endothelial, neural, and inducible encoded by three different genes. NO activates guanylyl cyclase enzyme leading to the increase of cellular cGMP levels by the dephosphorylation of myosin light chains and reducing cytosolic Ca2+ concentration, thereby the relaxation of smooth muscles in a board range of organs in mammalians. Thus, NO as a soluble gas molecule is having several pharmacological potentials and therapeutic applications in various diseases including the central nervous and cardiovascular systems, treatment of pulmonary and arterial hypertension, vasospastic angina, and myocardial infarction. NO is able also to inhibit the aggregation of thrombocytes. The signaling and physiological importance of NO was recognized by the awarding of Nobel Prize in Medicine and Physiology to R. Furchgott, L. Ignaro, and F. Murad in 1998.
The role of NO was also intensively studied in ischemia/reperfusion-induced injury in the myocardium during the past two and half decades (Pabla and Curtis, 1995; Liu et al., 1997; Csonka et al., 1999a; Masini et al., 1999; Varga et al., 1999; Fauconnier et al., 2011; Bienvenu et al., 2017; Van Der Weg et al., 2019). The substantial role and investigation of precise mechanisms of NO, both under physiological or pathological conditions, are not a question of debate in cell signaling and myocardial function. A number of studies show that interventions contributed to NO production via different mechanisms significantly reduced the reperfusion-induced injury including the incidence of arrhythmias (Shen et al., 1998; Kawahara et al., 2003; Imaizumi et al., 2008; Egom et al., 2011; Kisvari et al., 2015; Enayati et al., 2018; Jones et al., 2018). Various protective NO-mediated signal mechanisms, including the mitoK-(ATP) (Kawahara et al., 2003), the nuclear factor erythroid 2-related factor (NrF2), the extracellular-signaling-regulated-kinase (ERK) (Imaizumi et al., 2008), and Pak1/Akt1 signaling cascade (Egom et al., 2011) have also been reported.
In addition, organic nitrates used for the therapy of myocardial ischemia as NO donors, activate the soluble guanylyl cyclase increasing cGMP and subsequently reducing the intracellular Ca2+ level, and prevent the genesis of ischemia and reperfusion-induced arrhythmias. This latest pharmacologically controlled NO-mediated mechanism by organic nitrates is currently probably one of the most important interventions in the clinical management of myocardial ischemia/reperfusion-induced damages. However, it is of also interest to note that other synthetic molecules, e.g., aspirin and molsidomine, which are currently used in clinical medication, and their modified molecular structures (Bertuglia et al., 2004; Szoke et al., 2019) could release NO, leading to cardiac protection in the ischemic/reperfused myocardium.
CO
CO is another vasoactive gaseous molecule in close connection with heme oxygenase-1 system discussed in one of the previous chapters, and having substantial toxicological and physiological importance in living subjects (Peers and Steele, 2012). Several tissues including cardiac myocytes express both HO-1 and HO-2 proteins, and HO-1 expression can be particularly increased by several stress factors (Ewing et al., 1994; Wu et al., 2011; Haines and Tosaki, 2018) including myocardial ischemia/reperfusion (Maulik et al., 1996; Lakkisto et al., 2002) as demonstrated at mRNA and protein levels. Interestingly, homozygote (HO-1−/−) knockout mouse hearts subjected to ischemia/reperfusion period produced significantly higher cardiac damage and the incidence of VF in comparison with that of heterozygote (HO-1+/−) and wild-type mice (HO-1+/+) (Bak et al., 2010; Juhasz et al., 2011). Substantial scientific evidence suggests that several of the beneficial and harmful effects of CO arise from its ability, at dose-dependent manner, to modify signal transduction pathways and activities of ion channel proteins. It is apparent from these publications reviewed here that HO-1 and HO-1-mediated endogenous cellular CO levels have cardioprotective effects against myocardial ischemia/reperfusion-induced arrhythmias, and the pharmacological manipulation of HO-1 expression under severely controlled conditions may have clinical application as an antiarrhythmic agent.
H2S
Hydrogen sulfide was initially considered as a toxic molecule due to its property to inhibit cytochrome-oxidase c in mitochondrial respiration processes. H2S, like CO and NO, is endogenously generated gaseous molecule having substantial metabolic and physiological effects in various tissues including neuronal and myocardial cells. In mammalian tissues, three different enzymes encoded by three distinct genes produce H2S: cystathionine-gamma-lyase, cystathionine-beta-synthase, and 3-mercaptopiruvate sulfur transferase. In most cells, cystathionine-gamma-lyase and cystathionine-beta-synthase utilize homocysteine and L-cysteine as substrates to liberate pyruvate, ammonium, and H2S (Wang, 2002). A number of studies have reported the cardioprotective effect of H2S in myocardial ischemia/reperfusion-induced injury via different actions, which include ATP-sensitive K+-channel opening (Johansen et al., 2006), cGMP generation (Salloum et al., 2012), Akt-mTORC2 (Zhou et al., 2014), and microRNAs (Ren et al., 2019) mediated mechanisms, lead to the significant reduction of apoptotic and necrotic cellular deaths. Consequently, later on, it was also reported that pharmacological interventions serving as H2S donors produced cardiac protection against ischemic/reperfused cellular deaths (Sun et al., 2017; Sun et al., 2018). Currently, relatively little is known about H2S-mediated cardiac protection and remodeling against reperfusion-induced arrhythmias, however, growing evidence indicates that H2S donors could play a role in the reduction of the incidence of reperfusion-induced arrhythmias (Sun et al., 2015; Testai et al., 2016). Thus, H2S releasing molecules may particularly be also considered as potential pharmacological option in the prevention of the development of reperfusion-induced arrhythmias, and may have the ability to protect the myocardium against necrosis- and apoptosis-induced cell death.
Catecholamines
The role of catecholamines in arrhythmogenesis is not a question of debate, however, sometimes, depending on the experimental conditions, it can be controversial, and these endogenous mediators may have different effects in the genesis of ischemia-induced arrhythmias in comparison with reperfusion-induced rhythm disturbances (Opie and Coetzee, 1988; Kurz et al., 1991; Vanoli et al., 1994). The results of several published papers support the fact that stimulation of adrenergic receptors, especially alpha-1 and beta-1, is arrhythmogenic under ischemic/reperfused conditions (Sheridan et al., 1980; Bolli et al., 1984; Thandroyen et al., 1987; Tosaki et al., 1987b; Yasutake and Avkiran, 1995), and the alpha-1 adrenergic receptor stimulation could be due to the elevated transsarcolemmal Ca2+ influx and increased release of this ion from the SR (Thandroyen et al., 1987). Furthermore, the pharmacological blockade of alpha-1 adrenergic stimulation could mediate Na+/H+ exchange mechanism during reperfusion, leading to the reduction of the incidence of reperfusion-induced VF (Yasutake and Avkiran, 1995). Beta adrenergic receptor blockers could also be effective against arrhythmias by virtue of their ability to directly inhibit catecholamine-induced arrhythmogenesis on beta receptors (Ibanez et al., 2012; Winchester and Pepine, 2014). If this is so, the stimulation of beta adrenergic receptors and the concomitant activation of adenyl cyclase enzyme would appear be directly involved in the development of reperfusion-induced dysrhythmias. Therefore, these observations would suggest that stimulation of beta adrenergic receptors could be involved in the genesis of reperfusion-induced arrhythmias and the beta receptor blockade may be pharmacologically expected to be potentially protective.
Endothelin
Endothelin (ET) is one of numerous endogenous substances generating and accumulating in the ischemic/reperfused heart, or washed out during reperfusion periods, has been implicated as a potential arrhythmogenic factor. Thus, ET could function as an endogenous peptide-structure mediator in arrhythmogenesis, and has a potentially strong vascular action capable of exacerbating myocardial ischemia and heart failure. ET (“big endothelin,” endothelin-1) was first isolated from aortic endothelial cells (Yanagisawa et al., 1988), and is a potent vasoconstrictor agent, which specifically binds to ET-1 receptors, modulating the function of sodium, calcium and potassium ion channels, leading to pathologic changes on the ECG (Curtis et al., 1993; Brunner and Kukovetz, 1996; Wang et al., 2002; Vago et al., 2004; Adlbrecht et al., 2014; Falk et al., 2018). Hundreds of antiarrhythmic mechanisms, synthetic and nonsynthetic pharmacological agents were proposed, with more or less success, to protect directly or indirectly the myocardium against ET-1-related cardiac dysfunction (Isaka et al., 2007; Na et al., 2007; Du et al., 2008; Chan et al., 2016; Lee et al., 2017). The aforementioned approaches of ET-elicited vascular activities lead to multiple theories without precise and very specific discrimination between mechanisms in terms of their importance in reperfusion-induced arrhythmogenesis. However, it is reasonable to suppose that the cardiac protection is probably originated from (i) the inhibition of ET synthesis (Ceylan et al., 2018), (ii) the direct pharmacological blockade of ET receptors (Wainwright et al., 2005; Kaoukis et al., 2013), (iii) downregulation of various microRNAs (Wang et al., 2019), and (iv) their multiple combinations (i–iii).
Genetic Origin of Reperfusion-Induced Arrhythmias
At the time of writing, there is no substantial scientific evidence available, opposite to ischemia-induced arrhythmias, about the direct genetic origin of reperfusion-induced arrhythmias, since their origin itself is the “reperfusion event,” and such an approach has not been yet intensively investigated based on its genetic-mediated mechanisms (Szendrei et al., 2002; Kovacs et al., 2013; Ravingerova et al., 2013; Bienvenu et al., 2017; Paar et al., 2019). Therefore, it is reasonable to suppose that some gene encoded proteins and channels are already present upon reperfusion in their inactive forms in the myocardium, which may be immediately or quickly activated upon a reperfusion event, leading to the rapid maldistribution of ionic homeostasis, producing other arrhythmogenic components, and the genesis of reperfusion-induced arrhythmias. In majority of experimental studies, drugs and interventions were added prior to the induction of myocardial ischemia, thereby slowing the rate of ischemia-induced damages so that, less degree of cellular injury is present at the onset of reperfusion in the myocardium in drug-treated groups. Therefore, about a drug-afforded protection or a rapid activation of an inactive gene at the moment of reperfusion can only be obtained from observations, in which drug-induced ion channel activities or gene expression-related changes are immediately studied (or even later) at the onset of reperfusion. Therefore, it is important to consider all the possible inactive/active stages of various genes and function of ion channels at the onset of reperfusion, by which a drug induced cardiac protection might be successfully achieved.
Conclusion
Myocardial ischemia/reperfusion-induced alterations resulting in heart failure, conduction abnormalities and arrhythmias, in view of structural and electrical remodeling, which are important processes in the development various myocardial diseases. Multiple mechanisms responsible for the development of reperfusion-induced VF, which lead to the sudden cardiac death without pharmacological or nonpharmacological intervention in animal models and post-myocardial infarcted patients.
Potential mechanisms depicted in Figure 3 show that reperfusion of the ischemic myocardium intensifies the incidence of reperfusion-induced arrhythmias, which are followed by necrosis-, apoptosis-, and autophagy-induced cell deaths (Osipov et al., 2009; Zhang et al., 2012; Chang et al., 2013; Czegledi et al., 2019; Gyongyosi et al., 2019; Yasuda et al., 2019). From a different point of view, autophagic processes may be even beneficial, depending on their intensity in reversible injured myocardial cells. However, necrosis-induced cell death could dominate and mask autophagic signal transduction processes and autophagy-induced cell death in myocardial tissues (Czegledi et al., 2019; Gyongyosi et al., 2019). Cellular biology and genetics have a real value and significant impact to try better to define the generation of cardiovascular diseases, including life threatening cardiac arrhythmias. Nowadays, the research direction and available modern techniques in cell biology are intensively productive, but should do much to clarify gaps between the basic molecular science and clinical relevance in arrhythmogenesis.
Limitation
Cardiac muscle contraction and relaxation are very complex and primarily determined by several intracellular processes that control the force activation and inactivation, which include Na+, K+, and Ca2+ exchange mechanisms, including the ATP-dependent rate and Ca2+ sequestration capacity of the SR. The immediate extrapolation of AP and ECG changes obtained under experimental conditions to an actual clinical situation must be viewed with some caution because of the presence or absence of the blood and its elements (e.g., leukocytes, platelets), signal transductions, and possible interspecies differences in cardiac metabolism. In addition, the duration and shape of APs are different in His-Purkinje cells, AV node, and ventricular tissues, and the refractoriness is basically determined by the voltage-dependent recovery of Na+ channels from inactivation returning to activation in close connection with the function of calcium channels. Thus, the duration of effective refractory period is varied in different cells of the heart and the longest interval at which a premature stimuli fails to generate a propagated response, frequently is used to estimate drug effects in physiologically functioning cardiac tissues. Under physiological and pathological experimental conditions, to investigate the effect of an antiarrhythmic agent, the key aim is related to study various changes in the duration of the effective refractory period, which could modify the activation/inactivation stage of Na+ channels in close connection with different types of Ca2+ channels and genes e.g., Na+-induced Ca2+ release from the SR. Despite the limitation of several antiarrhythmic drugs can possess protective effects against LQTS and reperfusion-induced arrhythmias even some of them may be of particular concern. Additional experimental and clinical prospective studies powered to determine differences in ventricular APs in connection with ion channels and ECGs to confirm these observations are now warranted.
Author Contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Funding
This work was supported by grants of NKFIH-K-124719 and the European Union and the State of Hungary, co-financed by the European Social Fund in the framework of GINOP-2.3.2-15-2016-00043.
Conflict of Interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Adlbrecht C., Wurm R., Pezawas T., Andreas M., Redwan B., Distelmaier K., et al. (2014). Effects of endothelin A receptor blockade in patients with ST-elevation acute coronary syndrome–a rhythmologic substudy. Life Sci. 118, 430–434. 10.1016/j.lfs.2014.02.015 [DOI] [PubMed] [Google Scholar]
- Akita H., Creer M. H., Yamada K. A., Sobel B. E., Corr P. B. (1986). Electrophysiologic effects of intracellular lysophosphoglycerides and their accumulation in cardiac lymph with myocardial ischemia in dogs. J. Clin. Invest. 78, 271–280. 10.1172/JCI112561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez C. K., Cronin E., Baker W. L., Kluger J. (2019). Heart failure as a substrate and trigger for ventricular tachycardia. J. Interv Card Electrophysiol 56, 229–247. 10.1007/s10840-019-00623-x [DOI] [PubMed] [Google Scholar]
- Amin A. S., Reckman Y. J., Arbelo E., Spanjaart A. M., Postema P. G., Tadros R., et al. (2018). SCN5A mutation type and topology are associated with the risk of ventricular arrhythmia by sodium channel blockers. Int. J. Cardiol. 266, 128–132. 10.1016/j.ijcard.2017.09.010 [DOI] [PubMed] [Google Scholar]
- Amirahmadi F., Turnbull L., Du X. J., Graham R. M., Woodcock E. A. (2008). Heightened alpha1A-adrenergic receptor activity suppresses ischaemia/reperfusion-induced Ins(1,4,5)P3 generation in the mouse heart: a comparison with ischaemic preconditioning. Clin. Sci. (Lond) 114, 157–164. 10.1042/CS20070110 [DOI] [PubMed] [Google Scholar]
- Amoni M., Kelly-Laubscher R., Petersen M., Gwanyanya A. (2017). Cardioprotective and Anti-arrhythmic Effects of Magnesium Pretreatment Against Ischaemia/Reperfusion Injury in Isoprenaline-Induced Hypertrophic Rat Heart. Cardiovasc. Toxicol. 17, 49–57. 10.1007/s12012-015-9355-6 [DOI] [PubMed] [Google Scholar]
- Anson B. D., Ackerman M. J., Tester D. J., Will M. L., Delisle B. P., Anderson C. L., et al. (2004). Molecular and functional characterization of common polymorphisms in HERG (KCNH2) potassium channels. Am. J. Physiol. Heart Circ. Physiol. 286, H2434–H2441. 10.1152/ajpheart.00891.2003 [DOI] [PubMed] [Google Scholar]
- Antoniou C. K., Dilaveris P., Manolakou P., Galanakos S., Magkas N., Gatzoulis K., et al. (2017). QT Prolongation and Malignant Arrhythmia: How Serious a Problem? Eur. Cardiol. 12, 112–120. 10.15420/ecr.2017:16:1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aronsen J. M., Swift F., Sejersted O. M. (2013). Cardiac sodium transport and excitation-contraction coupling. J. Mol. Cell Cardiol. 61, 11–19. 10.1016/j.yjmcc.2013.06.003 [DOI] [PubMed] [Google Scholar]
- Axelsen L. N., Calloe K., Braunstein T. H., Riemann M., Hofgaard J. P., Liang B., et al. (2015). Diet-induced pre-diabetes slows cardiac conductance and promotes arrhythmogenesis. Cardiovasc. Diabetol. 14, 87. 10.1186/s12933-015-0246-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bak I., Szendrei L., Turoczi T., Papp G., Joo F., Das D. K., et al. (2003). Heme oxygenase-1-related carbon monoxide production and ventricular fibrillation in isolated ischemic/reperfused mouse myocardium. FASEB J. 17, 2133–2135. 10.1096/fj.03-0032fje [DOI] [PubMed] [Google Scholar]
- Bak I., Varadi J., Nagy N., Vecsernyes M., Tosaki A. (2005). The role of exogenous carbon monoxide in the recovery of post-ischemic cardiac function in buffer perfused isolated rat hearts. Cell Mol. Biol. (Noisy-le-grand) 51, 453–459. [PubMed] [Google Scholar]
- Bak I., Lekli I., Juhasz B., Nagy N., Varga E., Varadi J., et al. (2006). Cardioprotective mechanisms of Prunus cerasus (sour cherry) seed extract against ischemia-reperfusion-induced damage in isolated rat hearts. Am. J. Physiol. Heart Circ. Physiol. 291, H1329–H1336. 10.1152/ajpheart.01243.2005 [DOI] [PubMed] [Google Scholar]
- Bak I., Czompa A., Juhasz B., Lekli I., Tosaki A. (2010). Reduction of reperfusion-induced ventricular fibrillation and infarct size via heme oxygenase-1 overexpression in isolated mouse hearts. J. Cell Mol. Med. 14, 2268–2272. 10.1111/j.1582-4934.2010.01142.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batiste S. M., Blackwell D. J., Kim K., Kryshtal D. O., Gomez-Hurtado N., Rebbeck R. T., et al. (2019). Unnatural verticilide enantiomer inhibits type 2 ryanodine receptor-mediated calcium leak and is antiarrhythmic. Proc. Natl. Acad. Sci. U. S. A 116, 4810–4815. 10.1073/pnas.1816685116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becerra R., Roman B., Di Carlo M. N., Mariangelo J. I., Salas M., Sanchez G., et al. (2016). Reversible redox modifications of ryanodine receptor ameliorate ventricular arrhythmias in the ischemic-reperfused heart. Am. J. Physiol. Heart Circ. Physiol. 311, H713–H724. 10.1152/ajpheart.00142.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belardinelli L., Shryock J. C., Fraser H. (2006). Inhibition of the late sodium current as a potential cardioprotective principle: effects of the late sodium current inhibitor ranolazine. Heart 92 Suppl 4, iv6–iv14. 10.1136/hrt.2005.078790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben Jehuda R., Eisen B., Shemer Y., Mekies L. N., Szantai A., Reiter I., et al. (2018). CRISPR correction of the PRKAG2 gene mutation in the patient's induced pluripotent stem cell-derived cardiomyocytes eliminates electrophysiological and structural abnormalities. Heart Rhythm. 15, 267–276. 10.1016/j.hrthm.2017.09.024 [DOI] [PubMed] [Google Scholar]
- Bertuglia S., Giusti A., Del Soldato P. (2004). Antioxidant activity of nitro derivative of aspirin against ischemia-reperfusion in hamster cheek pouch microcirculation. Am. J. Physiol. Gastrointest. Liver Physiol. 286, G437–G443. 10.1152/ajpgi.00339.2003 [DOI] [PubMed] [Google Scholar]
- Bienvenu L. A., Morgan J., Reichelt M. E., Delbridge L. M. D., Young M. J. (2017). Chronic in vivo nitric oxide deficiency impairs cardiac functional recovery after ischemia in female (but not male) mice. J. Mol. Cell Cardiol. 112, 8–15. 10.1016/j.yjmcc.2017.08.012 [DOI] [PubMed] [Google Scholar]
- Blasig I. E., Ebert B., Hennig C., Pali T., Tosaki A. (1990). Inverse relationship between ESR spin trapping of oxyradicals and degree of functional recovery during myocardial reperfusion in isolated working rat heart. Cardiovasc. Res. 24, 263–270. 10.1093/cvr/24.4.263 [DOI] [PubMed] [Google Scholar]
- Bogeholz N., Pauls P., Kaese S., Schulte J. S., Lemoine M. D., Dechering D. G., et al. (2016). Triggered activity in atrial myocytes is influenced by Na(+)/Ca(2+) exchanger activity in genetically altered mice. J. Mol. Cell Cardiol. 101, 106–115. 10.1016/j.yjmcc.2016.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolli R., Fisher D. J., Taylor A. A., Young J. B., Miller R. R. (1984). Effect of alpha-adrenergic blockade on arrhythmias induced by acute myocardial ischemia and reperfusion in the dog. J. Mol. Cell Cardiol. 16, 1101–1117. 10.1016/S0022-2828(84)80037-1 [DOI] [PubMed] [Google Scholar]
- Bongianino R., Denegri M., Mazzanti A., Lodola F., Vollero A., Boncompagni S., et al. (2017). Allele-Specific Silencing of Mutant mRNA Rescues Ultrastructural and Arrhythmic Phenotype in Mice Carriers of the R4496C Mutation in the Ryanodine Receptor Gene (RYR2). Circ. Res. 121, 525–536. 10.1161/CIRCRESAHA.117.310882 [DOI] [PubMed] [Google Scholar]
- Bossu A., Houtman M. J. C., Meijborg V. M. F., Varkevisser R., Beekman H. D. M., Dunnink A., et al. (2018). Selective late sodium current inhibitor GS-458967 suppresses Torsades de Pointes by mostly affecting perpetuation but not initiation of the arrhythmia. Br. J. Pharmacol. 175, 2470–2482. 10.1111/bph.14217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowles N. E., Jou C. J., Arrington C. B., Kennedy B. J., Earl A., Matsunami N., et al. (2015). Exome analysis of a family with Wolff-Parkinson-White syndrome identifies a novel disease locus. Am. J. Med. Genet. A 167A, 2975–2984. 10.1002/ajmg.a.37297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broskova Z., Drabikova K., Sotnikova R., Fialova S., Knezl V. (2013). Effect of plant polyphenols on ischemia-reperfusion injury of the isolated rat heart and vessels. Phytother. Res. 27, 1018–1022. 10.1002/ptr.4825 [DOI] [PubMed] [Google Scholar]
- Brunner F., Kukovetz W. R. (1996). Postischemic antiarrhythmic effects of angiotensin-converting enzyme inhibitors. Role of suppression of endogenous endothelin secretion. Circulation 94, 1752–1761. 10.1161/01.cir.94.7.1752 [DOI] [PubMed] [Google Scholar]
- Bukhari I. A., Almotrefi A. A., Mohamed O. Y., Al-Masri A. A., Sheikh S. A. (2018). Protective effect of fenofibrate against ischemia-/reperfusion-induced cardiac arrhythmias in isolated rat hearts. Fundam. Clin. Pharmacol. 32, 141–146. 10.1111/fcp.12342 [DOI] [PubMed] [Google Scholar]
- Burgen A. S., Terroux K. G. (1953). The membrane resting and action potentials of the cat auricle. J. Physiol. 119, 139–152. 10.1113/jphysiol.1953.sp004834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeliet E. (1992). A fuzzy subsarcolemmal space for intracellular Na+ in cardiac cells? Cardiovasc. Res. 26, 433–442. 10.1093/cvr/26.5.433 [DOI] [PubMed] [Google Scholar]
- Ceylan A. F., Wang S., Kandadi M. R., Chen J., Hua Y., Pei Z., et al. (2018). Cardiomyocyte-specific knockout of endothelin receptor a attenuates obesity cardiomyopathy. Biochim. Biophys. Acta Mol. Basis. Dis. 1864, 3339–3352. 10.1016/j.bbadis.2018.07.020 [DOI] [PubMed] [Google Scholar]
- Chan E. A., Buckley B., Farraj A. K., Thompson L. C. (2016). The heart as an extravascular target of endothelin-1 in particulate matter-induced cardiac dysfunction. Pharmacol. Ther. 165, 63–78. 10.1016/j.pharmthera.2016.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang R., Li Y., Yang X., Yue Y., Dou L., Wang Y., et al. (2013). Protective role of deoxyschizandrin and schisantherin A against myocardial ischemia-reperfusion injury in rats. PloS One 8, e61590. 10.1371/journal.pone.0061590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J., Mitcheson J. S., Lin M., Sanguinetti M. C. (2000). Functional roles of charged residues in the putative voltage sensor of the HCN2 pacemaker channel. J. Biol. Chem. 275, 36465–36471. 10.1074/jbc.M007034200 [DOI] [PubMed] [Google Scholar]
- Cheng Y., Rong J. (2017). Therapeutic Potential of Heme Oxygenase-1/carbon Monoxide System Against Ischemia-Reperfusion Injury. Curr. Pharm. Des. 23, 3884–3898. 10.2174/1381612823666170413122439 [DOI] [PubMed] [Google Scholar]
- Choi A. M., Alam J. (1996). Heme oxygenase-1: function, regulation, and implication of a novel stress-inducible protein in oxidant-induced lung injury. Am. J. Respir. Cell Mol. Biol. 15, 9–19. 10.1165/ajrcmb.15.1.8679227 [DOI] [PubMed] [Google Scholar]
- Christophersen I. E., Olesen M. S., Liang B., Andersen M. N., Larsen A. P., Nielsen J. B., et al. (2013). Genetic variation in KCNA5: impact on the atrial-specific potassium current IKur in patients with lone atrial fibrillation. Eur. Heart J. 34, 1517–1525. 10.1093/eurheartj/ehs442 [DOI] [PubMed] [Google Scholar]
- Chua W., Easter C. L., Guasch E., Sitch A., Casadei B., Crijns H., et al. (2019). Development and external validation of predictive models for prevalent and recurrent atrial fibrillation: a protocol for the analysis of the CATCH ME combined dataset. BMC Cardiovasc. Disord. 19, 120. 10.1186/s12872-019-1105-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coetzee W. A., Opie L. H., Saman S. (1987). Proposed role of energy supply in the genesis of delayed afterdepolarizations–implications for ischemic or reperfusion arrhythmias. J. Mol. Cell Cardiol. 19 Suppl 5, 13–21. 10.1016/S0022-2828(87)80606-5 [DOI] [PubMed] [Google Scholar]
- Coetzee W., Biermans G., Callewaert G., Vereecke J., Opie L. H., Carmeliet E. (1988). The effect of inhibition of mitochondrial energy metabolism on the transient inward current of isolated guinea-pig ventricular myocytes. J. Mol. Cell Cardiol. 20, 181–185. 10.1016/S0022-2828(88)80051-8 [DOI] [PubMed] [Google Scholar]
- Colman M. A., Ni H., Liang B., Schmitt N., Zhang H. (2017). In silico assessment of genetic variation in KCNA5 reveals multiple mechanisms of human atrial arrhythmogenesis. PloS Comput. Biol. 13, e1005587. 10.1371/journal.pcbi.1005587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coraboeuf E., Weidmann S. (1954). Temperature effects on the electrical activity of Purkinje fibres. Helv. Physiol. Pharmacol. Acta 12, 32–41. [PubMed] [Google Scholar]
- Coronel R., Casini S., Koopmann T. T., Wilms-Schopman F. J., Verkerk A. O., De Groot J. R., et al. (2005). Right ventricular fibrosis and conduction delay in a patient with clinical signs of Brugada syndrome: a combined electrophysiological, genetic, histopathologic, and computational study. Circulation 112, 2769–2777. 10.1161/CIRCULATIONAHA.105.532614 [DOI] [PubMed] [Google Scholar]
- Corr P. B., Witkowski F. X. (1983). Potential electrophysiologic mechanisms responsible for dysrhythmias associated with reperfusion of ischemic myocardium. Circulation 68, I16–I24. -. 6305533. [PubMed] [Google Scholar]
- Cortez D., Zareba W., Mcnitt S., Polonsky B., Rosero S. Z., Platonov P. G. (2019). Quantitative T-wave morphology assessment from surface ECG is linked with cardiac events risk in genotype-positive KCNH2 mutation carriers with normal QTc values. J. Cardiovasc. Electrophysiol 30, 2907–2913. 10.1111/jce.14210 [DOI] [PubMed] [Google Scholar]
- Csonka C., Szilvassy Z., Fulop F., Pali T., Blasig I. E., Tosaki A., et al. (1999. a). Classic preconditioning decreases the harmful accumulation of nitric oxide during ischemia and reperfusion in rat hearts. Circulation 100, 2260–2266. 10.1161/01.cir.100.22.2260 [DOI] [PubMed] [Google Scholar]
- Csonka C., Varga E., Kovacs P., Ferdinandy P., Blasig I. E., Szilvassy Z., et al. (1999. b). Heme oxygenase and cardiac function in ischemic/reperfused rat hearts. Free Radic. Biol. Med. 27, 119–126. 10.1016/S0891-5849(99)00077-5. 10443928. [DOI] [PubMed] [Google Scholar]
- Curtis M. J., Hearse D. J. (1989). Ischaemia-induced and reperfusion-induced arrhythmias differ in their sensitivity to potassium: implications for mechanisms of initiation and maintenance of ventricular fibrillation. J. Mol. Cell Cardiol. 21, 21–40. 10.1016/0022-2828(89)91490-9 [DOI] [PubMed] [Google Scholar]
- Curtis M. J., Pugsley M. K., Walker M. J. (1993). Endogenous chemical mediators of ventricular arrhythmias in ischaemic heart disease. Cardiovasc. Res. 27, 703–719. 10.1093/cvr/27.5.703 [DOI] [PubMed] [Google Scholar]
- Curtis M. J. (1991). Torsades de pointes: arrhythmia, syndrome, or chimera? A perspective in the light of the Lambeth Conventions. Cardiovasc. Drugs Ther. 5, 191–200. 10.1007/BF03029820 [DOI] [PubMed] [Google Scholar]
- Czegledi A., Tosaki A., Gyongyosi A., Zilinyi R., Tosaki A., Lekli I. (2019). Electrically-Induced Ventricular Fibrillation Alters Cardiovascular Function and Expression of Apoptotic and Autophagic Proteins in Rat Hearts. Int. J. Mol. Sci. 20, 1628. 10.3390/ijms20071628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das B., Sarkar C. (2005). Is the sarcolemmal or mitochondrial K(ATP) channel activation important in the antiarrhythmic and cardioprotective effects during acute ischemia/reperfusion in the intact anesthetized rabbit model? Life Sci. 77, 1226–1248. 10.1016/j.lfs.2004.12.042 [DOI] [PubMed] [Google Scholar]
- Daubert J. P., Zareba W., Rosero S. Z., Budzikowski A., Robinson J. L., Moss A. J. (2007). Role of implantable cardioverter defibrillator therapy in patients with long QT syndrome. Am. Heart J. 153, 53–58. 10.1016/j.ahj.2007.01.025 [DOI] [PubMed] [Google Scholar]
- De Ferrari G. M., Dusi V., Spazzolini C., Bos J. M., Abrams D. J., Berul C. I., et al. (2015). Clinical Management of Catecholaminergic Polymorphic Ventricular Tachycardia: The Role of Left Cardiac Sympathetic Denervation. Circulation 131, 2185–2193. 10.1161/CIRCULATIONAHA.115.015731 [DOI] [PubMed] [Google Scholar]
- Delise P., Allocca G., Sitta N., Migliore F., Dagradi F., Spazzolini C., et al. (2018). Cardiac arrest and Brugada syndrome: Is drug-induced type 1 ECG pattern always a marker of low risk? Int. J. Cardiol. 254, 142–145. 10.1016/j.ijcard.2017.10.118 [DOI] [PubMed] [Google Scholar]
- Desimone C. V., Bos J. M., Bos K. M., Liang J. J., Patel N. A., Hodge D. O., et al. (2015). Effects on repolarization using dynamic QT interval monitoring in long-QT patients following left cardiac sympathetic denervation. J. Cardiovasc. Electrophysiol 26, 434–439. 10.1111/jce.12609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dessertenne F. (1966). [Ventricular tachycardia with 2 variable opposing foci]. Arch. Mal. Coeur Vaiss 59, 263–272. [PubMed] [Google Scholar]
- Devalla H. D., Gelinas R., Aburawi E. H., Beqqali A., Goyette P., Freund C., et al. (2016). TECRL, a new life-threatening inherited arrhythmia gene associated with overlapping clinical features of both LQTS and CPVT. EMBO Mol. Med. 8, 1390–1408. 10.15252/emmm.201505719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Difrancesco D., Ojeda C. (1980). Properties of the current if in the sino-atrial node of the rabbit compared with those of the current iK, in Purkinje fibres. J. Physiol. 308, 353–367. 10.1113/jphysiol.1980.sp013475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Difrancesco D. (2015). HCN4, Sinus Bradycardia and Atrial Fibrillation. Arrhythm. Electrophysiol Rev. 4, 9–13. 10.15420/aer.2015.4.1.9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Difrancesco D. (2019). Comparing pathways for long-term heart rate modulation by the funny current. J. Gen. Physiol. 151, 1066–1069. 10.1085/jgp.201912409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du R. H., Yi H. W., Dai D. Z., Tang W. H., Dai Y. (2008). Inflammatory factors that contribute to upregulation of ERG and cardiac arrhythmias are suppressed by CPU86017, a class III antiarrhythmic agent. J. Pharm. Pharmacol. 60, 1089–1095. 10.1211/jpp.60.8.0015 [DOI] [PubMed] [Google Scholar]
- Dulak J., Jozkowicz A. (2014). Novel faces of heme oxygenase-1: mechanisms and therapeutic potentials. Antioxid. Redox Signal 20, 1673–1676. 10.1089/ars.2013.5761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egom E. E., Mohamed T. M., Mamas M. A., Shi Y., Liu W., Chirico D., et al. (2011). Activation of Pak1/Akt/eNOS signaling following sphingosine-1-phosphate release as part of a mechanism protecting cardiomyocytes against ischemic cell injury. Am. J. Physiol. Heart Circ. Physiol. 301, H1487–H1495. 10.1152/ajpheart.01003.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enayati A., Yassa N., Mazaheri Z., Rajaei M., Pourabouk M., Ghorghanlu S., et al. (2018). Cardioprotective and anti-apoptotic effects of Potentilla reptans L. root via Nrf2 pathway in an isolated rat heart ischemia/reperfusion model. Life Sci. 215, 216–226. 10.1016/j.lfs.2018.11.021 [DOI] [PubMed] [Google Scholar]
- Estes S. I., Ye D., Zhou W., Dotzler S. M., Tester D. J., Bos J. M., et al. (2019). Characterization of the CACNA1C-R518C Missense Mutation in the Pathobiology of Long-QT Syndrome Using Human Induced Pluripotent Stem Cell Cardiomyocytes Shows Action Potential Prolongation and L-Type Calcium Channel Perturbation. Circ. Genom. Precis Med. 12, e002534. 10.1161/CIRCGEN.119.002534 [DOI] [PubMed] [Google Scholar]
- Ewing J. F., Raju V. S., Maines M. D. (1994). Induction of heart heme oxygenase-1 (HSP32) by hyperthermia: possible role in stress-mediated elevation of cyclic 3':5'-guanosine monophosphate. J. Pharmacol. Exp. Ther. 271, 408–414. 10.1006/abbi.1997.0169 [DOI] [PubMed] [Google Scholar]
- Falk M., Huhn R., Behmenburg F., Ritz-Timme S., Mayer F. (2018). Biomechanical stress in myocardial infarctions: can endothelin-1 and growth differentiation factor 15 serve as immunohistochemical markers? Int. J. Legal. Med. 132, 509–518. 10.1007/s00414-017-1726-z [DOI] [PubMed] [Google Scholar]
- Fauconnier J., Meli A. C., Thireau J., Roberge S., Shan J., Sassi Y., et al. (2011). Ryanodine receptor leak mediated by caspase-8 activation leads to left ventricular injury after myocardial ischemia-reperfusion. Proc. Natl. Acad. Sci. U. S. A 108, 13258–13263. 10.1073/pnas.1100286108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flatman P. W. (1984). Magnesium transport across cell membranes. J. Membr. Biol. 80, 1–14. 10.1007/BF01868686 [DOI] [PubMed] [Google Scholar]
- Frank-Hansen R., Larsen L. A., Andersen P., Jespersgaard C., Christiansen M. (2005). Mutations in the genes KCND2 and KCND3 encoding the ion channels Kv4.2 and Kv4.3, conducting the cardiac fast transient outward current (ITO,f), are not a frequent cause of long QT syndrome. Clin. Chim. Acta 351, 95–100. 10.1016/j.cccn.2004.08.017 [DOI] [PubMed] [Google Scholar]
- Gadsby D. C., Kimura J., Noma A. (1985). Voltage dependence of Na/K pump current in isolated heart cells. Nature 315, 63–65. 10.1038/315063a0 [DOI] [PubMed] [Google Scholar]
- Garcia A., Liu C. C., Cornelius F., Clarke R. J., Rasmussen H. H. (2016). Glutathionylation-Dependence of Na(+)-K(+)-Pump Currents Can Mimic Reduced Subsarcolemmal Na(+) Diffusion. Biophys. J. 110, 1099–1109. 10.1016/j.bpj.2016.01.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garlick P. B., Davies M. J., Hearse D. J., Slater T. F. (1987). Direct detection of free radicals in the reperfused rat heart using electron spin resonance spectroscopy. Circ. Res. 61, 757–760. 10.1161/01.res.61.5.757 [DOI] [PubMed] [Google Scholar]
- Gatzke N., Guc N., Hillmeister P., Dulsner A., Le Noble F., Buschmann E. E., et al. (2018). Cardiovascular drugs attenuated myocardial resistance against ischaemia-induced and reperfusion-induced injury in a rat model of repetitive occlusion. Open Heart 5, e000889. 10.1136/openhrt-2018-000889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gollob M. H., Green M. S., Tang A. S., Gollob T., Karibe A., Ali Hassan A. S., et al. (2001). Identification of a gene responsible for familial Wolff-Parkinson-White syndrome. N. Engl. J. Med. 344, 1823–1831. 10.1056/NEJM200106143442403 [DOI] [PubMed] [Google Scholar]
- Guc M. O., Boachie-Ansah G., Kane K. A., Wadsworth R. M. (1993). Comparison of antiarrhythmic and electrophysiologic effects of diltiazem and its analogue siratiazem. J. Cardiovasc. Pharmacol. 22, 681–686. 10.1097/00005344-199311000-00002 [DOI] [PubMed] [Google Scholar]
- Gyongyosi A., Zilinyi R., Czegledi A., Tosaki A., Tosaki A., Lekli I. (2019). The role of autophagy and death pathways in dose-dependent isoproterenol-induced cardiotoxicity. Curr. Pharm. Des. 25 (19), 2192–2198. 10.2174/1381612825666190619145025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haines D. D., Tosaki A. (2018). Role of Heme Oxygenases in Cardiovascular Syndromes and Co-morbidities. Curr. Pharm. Des. 24, 2322–2325. 10.2174/1381612824666180727110353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han F., Bossuyt J., Martin J. L., Despa S., Bers D. M. (2010). Role of phospholemman phosphorylation sites in mediating kinase-dependent regulation of the Na+-K+-ATPase. Am. J. Physiol. Cell Physiol. 299, C1363–C1369. 10.1152/ajpcell.00027.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris A. S., Toth L. A., Tan E. H. (1958). Arrhythmic and antiarrhythmic effects of sodium, potassium, and calcium salts and of glucose injected into coronary arteries of infarcted and normal hearts. Circ. Res. 6, 570–579. 10.1161/01.res.6.5.570 [DOI] [PubMed] [Google Scholar]
- Hearse D. J., Tosaki A. (1987). Free radicals and reperfusion-induced arrhythmias: protection by spin trap agent PBN in the rat heart. Circ. Res. 60, 375–383. 10.1161/01.res.60.3.375 [DOI] [PubMed] [Google Scholar]
- Hegyi B., Bossuyt J., Griffiths L. G., Shimkunas R., Coulibaly Z., Jian Z., et al. (2018). Complex electrophysiological remodeling in postinfarction ischemic heart failure. Proc. Natl. Acad. Sci. U. S. A 115, E3036–E3044. 10.1073/pnas.1718211115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung L. M., Chen J. K., Huang S. S., Lee R. S., Su M. J. (2000). Cardioprotective effect of resveratrol, a natural antioxidant derived from grapes. Cardiovasc. Res. 47, 549–555. 10.1016/s0008-6363(00)00102-4 [DOI] [PubMed] [Google Scholar]
- Ibanez B., Fuster V., Macaya C., Sanchez-Brunete V., Pizarro G., Lopez-Romero P., et al. (2012). Study design for the “effect of METOprolol in CARDioproteCtioN during an acute myocardial InfarCtion” (METOCARD-CNIC): a randomized, controlled parallel-group, observer-blinded clinical trial of early pre-reperfusion metoprolol administration in ST-segment elevation myocardial infarction. Am. Heart J. 164, 473–480 e475. 10.1016/j.ahj.2012.07.020 [DOI] [PubMed] [Google Scholar]
- Imaizumi S., Miura S., Nakamura K., Kiya Y., Uehara Y., Zhang B., et al. (2008). Antiarrhythmogenic effect of reconstituted high-density lipoprotein against ischemia/reperfusion in rats. J. Am. Coll. Cardiol. 51, 1604–1612. 10.1016/j.jacc.2007.12.040 [DOI] [PubMed] [Google Scholar]
- Isaka M., Kudo A., Imamura M., Kawakami H., Yasuda K. (2007). Endothelin receptors, localized in sympathetic nerve terminals of the heart, modulate norepinephrine release and reperfusion arrhythmias. Basic Res. Cardiol. 102, 154–162. 10.1007/s00395-006-0623-2 [DOI] [PubMed] [Google Scholar]
- Iseri L. T., French J. H. (1984). Magnesium: nature's physiologic calcium blocker. Am. Heart J. 108, 188–193. 10.1016/0002-8703(84)90572-6 [DOI] [PubMed] [Google Scholar]
- Ishizaka N., Griendling K. K. (1997). Heme oxygenase-1 is regulated by angiotensin II in rat vascular smooth muscle cells. Hypertension 29, 790–795. 10.1161/01.HYP.29.3.790 [DOI] [PubMed] [Google Scholar]
- Javidanpour S., Dianat M., Badavi M., Mard S. A. (2018). The inhibitory effect of rosmarinic acid on overexpression of NCX1 and stretch- induced arrhythmias after acute myocardial infarction in rats. Biomed. Pharmacother. 102, 884–893. 10.1016/j.biopha.2018.03.103 [DOI] [PubMed] [Google Scholar]
- Jennings R. B., Reimer K. A. (1983). Factors involved in salvaging ischemic myocardium: effect of reperfusion of arterial blood. Circulation 68, I25–I36. [PubMed] [Google Scholar]
- Johansen D., Ytrehus K., Baxter G. F. (2006). Exogenous hydrogen sulfide (H2S) protects against regional myocardial ischemia-reperfusion injury–Evidence for a role of K ATP channels. Basic Res. Cardiol. 101, 53–60. 10.1007/s00395-005-0569-9 [DOI] [PubMed] [Google Scholar]
- Johnson R. A., Lavesa M., Askari B., Abraham N. G., Nasjletti A. (1995). A heme oxygenase product, presumably carbon monoxide, mediates a vasodepressor function in rats. Hypertension 25, 166–169. 10.1161/01.HYP.25.2.166 [DOI] [PubMed] [Google Scholar]
- Jones D. A., Rathod K. S., Williamson A., Harrington D., Andiapen M., Van Eijl S., et al. (2018). The effect of intracoronary sodium nitrite on the burden of ventricular arrhythmias following primary percutaneous coronary intervention for acute myocardial infarction. Int. J. Cardiol. 266, 1–6. 10.1016/j.ijcard.2018.01.029 [DOI] [PubMed] [Google Scholar]
- Jost N., Nagy N., Corici C., Kohajda Z., Horvath A., Acsai K., et al. (2013). ORM-10103, a novel specific inhibitor of the Na+/Ca2+ exchanger, decreases early and delayed afterdepolarizations in the canine heart. Br. J. Pharmacol. 170, 768–778. 10.1111/bph.12228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juhasz B., Varga B., Czompa A., Bak I., Lekli I., Gesztelyi R., et al. (2011). Postischemic cardiac recovery in heme oxygenase-1 transgenic ischemic/reperfused mouse myocardium. J. Cell Mol. Med. 15, 1973–1982. 10.1111/j.1582-4934.2010.01153.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalsto S. M., Siland J. E., Rienstra M., Christophersen I. E. (2019). Atrial Fibrillation Genetics Update: Toward Clinical Implementation. Front. Cardiovasc. Med. 6, 127. 10.3389/fcvm.2019.00127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamkin A., Kiseleva I., Isenberg G. (2003). Activation and inactivation of a non-selective cation conductance by local mechanical deformation of acutely isolated cardiac fibroblasts. Cardiovasc. Res. 57, 793–803. 10.1016/s0008-6363(02)00775-7 [DOI] [PubMed] [Google Scholar]
- Kaoukis A., Deftereos S., Raisakis K., Giannopoulos G., Bouras G., Panagopoulou V., et al. (2013). The role of endothelin system in cardiovascular disease and the potential therapeutic perspectives of its inhibition. Curr. Top. Med. Chem. 13, 95–114. 10.2174/1568026611313020003 [DOI] [PubMed] [Google Scholar]
- Karmazyn M. (1996). The sodium-hydrogen exchange system in the heart: its role in ischemic and reperfusion injury and therapeutic implications. Can. J. Cardiol. 12, 1074–1082. [PubMed] [Google Scholar]
- Kato M., Dote K., Sasaki S., Takemoto H., Habara S., Hasegawa D. (2004). Intracoronary verapamil rapidly terminates reperfusion tachyarrhythmias in acute myocardial infarction. Chest 126, 702–708. 10.1378/chest.126.3.702 [DOI] [PubMed] [Google Scholar]
- Kawahara K., Takase M., Yamauchi Y. (2003). Increased vulnerability to ischemia/reperfusion-induced ventricular tachyarrhythmias by pre-ischemic inhibition of nitric oxide synthase in isolated rat hearts. Cardiovasc. Pathol. 12, 49–56. 10.1016/S1054-8807(02)00155-2 [DOI] [PubMed] [Google Scholar]
- Kaya S. T., Bozdogan O., Ozarslan T. O., Taskin E., Eksioglu D., Erim F., et al. (2019). The protection of resveratrol and its combination with glibenclamide, but not berberine on the diabetic hearts against reperfusion-induced arrhythmias: the role of myocardial KATP channel. Arch. Physiol. Biochem. 125, 114–121. 10.1080/13813455.2018.1440409 [DOI] [PubMed] [Google Scholar]
- Keating M. T., Sanguinetti M. C. (2001). Molecular and cellular mechanisms of cardiac arrhythmias. Cell 104, 569–580. 10.1016/s0092-8674(01)00243-4 [DOI] [PubMed] [Google Scholar]
- Kerr S. M., Klaric L., Halachev M., Hayward C., Boutin T. S., Meynert A. M., et al. (2019). An actionable KCNH2 Long QT Syndrome variant detected by sequence and haplotype analysis in a population research cohort. Sci. Rep. 9, 10964. 10.1038/s41598-019-47436-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisvari G., Kovacs M., Seprenyi G., Vegh A. (2015). The activation of PI 3-kinase/Akt pathway is involved in the acute effects of simvastatin against ischaemia and reperfusion-induced arrhythmias in anaesthetised dogs. Eur. J. Pharmacol. 769, 185–194. 10.1016/j.ejphar.2015.11.016 [DOI] [PubMed] [Google Scholar]
- Kloner R. A., Dow J. S., Bhandari A. (2011). First direct comparison of the late sodium current blocker ranolazine to established antiarrhythmic agents in an ischemia/reperfusion model. J. Cardiovasc. Pharmacol. Ther. 16, 192–196. 10.1177/1074248410386485 [DOI] [PubMed] [Google Scholar]
- Kloner R. A. (1993). Does reperfusion injury exist in humans? J. Am. Coll. Cardiol. 21, 537–545. 10.1016/0735-1097(93)90700-b [DOI] [PubMed] [Google Scholar]
- Kohajda Z., Farkas-Morvay N., Jost N., Nagy N., Geramipour A., Horvath A., et al. (2016). The Effect of a Novel Highly Selective Inhibitor of the Sodium/Calcium Exchanger (NCX) on Cardiac Arrhythmias in In Vitro and In Vivo Experiments. PloS One 11, e0166041. 10.1371/journal.pone.0166041. PMC5104402 Pharma, have been involved in the development of ORM-10962. Other authors have nothing to declare. The commercial adherence of the Orion Pharma does not alter our adherence to PLOS ONE policies on sharing data and materials. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostopoulou A., Zeljko H. M., Bogossian H., Ciudin R., Costa F., Heijman J., et al. (2019). Atrial fibrillation-related stroke in women: Evidence and inequalities in epidemiology, mechanisms, clinical presentation, and management. Clin. Cardiol. 43 (1), 14–23. 10.1002/clc.23284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs M., Gonczi M., Kovacs E., Vegh A. (2013). Time course analysis of cardiac pacing-induced gene expression changes in the canine heart. Mol. Cell Biochem. 372, 257–266. 10.1007/s11010-012-1467-8 [DOI] [PubMed] [Google Scholar]
- Kubo Y., Adelman J. P., Clapham D. E., Jan L. Y., Karschin A., Kurachi Y., et al. (2005). International Union of Pharmacology. LIV. Nomenclature and molecular relationships of inwardly rectifying potassium channels. Pharmacol. Rev. 57, 509–526. 10.1124/pr.57.4.11 [DOI] [PubMed] [Google Scholar]
- Kurz T., Yamada K. A., Datorre S. D., Corr P. B. (1991). Alpha 1-adrenergic system and arrhythmias in ischaemic heart disease. Eur. Heart J. 12 Suppl F, 88–98. 10.1093/eurheartj/12.suppl_f.88 [DOI] [PubMed] [Google Scholar]
- Kusama Y., Bernier M., Hearse D. J. (1989). Singlet oxygen-induced arrhythmias. Dose- and light-response studies for photoactivation of rose bengal in the rat heart. Circulation 80, 1432–1448. 10.1161/01.cir.80.5.1432 [DOI] [PubMed] [Google Scholar]
- Lakkisto P., Palojoki E., Backlund T., Saraste A., Tikkanen I., Voipio-Pulkki L. M., et al. (2002). Expression of heme oxygenase-1 in response to myocardial infarction in rats. J. Mol. Cell Cardiol. 34, 1357–1365. 10.1006/jmcc.2002.2094 [DOI] [PubMed] [Google Scholar]
- Lazzerini P. E., Bertolozzi I., Finizola F., Acampa M., Natale M., Vanni F., et al. (2018). Proton Pump Inhibitors and Serum Magnesium Levels in Patients With Torsades de Pointes. Front. Pharmacol. 9, 363. 10.3389/fphar.2018.00363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee P. J., Alam J., Wiegand G. W., Choi A. M. (1996). Overexpression of heme oxygenase-1 in human pulmonary epithelial cells results in cell growth arrest and increased resistance to hyperoxia. Proc. Natl. Acad. Sci. U. S. A 93, 10393–10398. 10.1073/pnas.93.19.10393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee P. J., Jiang B. H., Chin B. Y., Iyer N. V., Alam J., Semenza G. L., et al. (1997). Hypoxia-inducible factor-1 mediates transcriptional activation of the heme oxygenase-1 gene in response to hypoxia. J. Biol. Chem. 272, 5375–5381. 10.1074/jbc.272.9.5375 [DOI] [PubMed] [Google Scholar]
- Lee T. M., Chang N. C., Lin S. Z. (2017). Inhibition of infarction-induced sympathetic innervation with endothelin receptor antagonism via a PI3K/GSK-3beta-dependent pathway. Lab. Invest. 97, 243–255. 10.1038/labinvest.2016.138 [DOI] [PubMed] [Google Scholar]
- Li W., Yin L., Shen C., Hu K., Ge J., Sun A. (2018). SCN5A Variants: Association With Cardiac Disorders. Front. Physiol. 9, 1372. 10.3389/fphys.2018.01372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang S., Wang Q., Zhang W., Zhang H., Tan S., Ahmed A., et al. (2014). Carbon monoxide inhibits inward rectifier potassium channels in cardiomyocytes. Nat. Commun. 5, 4676. 10.1038/ncomms5676 [DOI] [PubMed] [Google Scholar]
- Lieve K. V., Verkerk A. O., Podliesna S., Van Der Werf C., Tanck M. W., Hofman N., et al. (2017). Gain-of-function mutation in SCN5A causes ventricular arrhythmias and early onset atrial fibrillation. Int. J. Cardiol. 236, 187–193. 10.1016/j.ijcard.2017.01.113 [DOI] [PubMed] [Google Scholar]
- Liu P., Hock C. E., Nagele R., Wong P. Y. (1997). Formation of nitric oxide, superoxide, and peroxynitrite in myocardial ischemia-reperfusion injury in rats. Am. J. Physiol. 272, H2327–H2336. 10.1152/ajpheart.1997.272.5.H2327 [DOI] [PubMed] [Google Scholar]
- Loboda A., Damulewicz M., Pyza E., Jozkowicz A., Dulak J. (2016). Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: an evolutionarily conserved mechanism. Cell Mol. Life Sci. 73, 3221–3247. 10.1007/s00018-016-2223-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu F. M., Hilgemann D. W. (2017). Na/K pump inactivation, subsarcolemmal Na measurements, and cytoplasmic ion turnover kinetics contradict restricted Na spaces in murine cardiac myocytes. J. Gen. Physiol. 149, 727–749. 10.1085/jgp.201711780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lujan H. L., Kramer V. J., Dicarlo S. E. (2007). Sex influences the susceptibility to reperfusion-induced sustained ventricular tachycardia and beta-adrenergic receptor blockade in conscious rats. Am. J. Physiol. Heart Circ. Physiol. 293, H2799–H2808. 10.1152/ajpheart.00596.2007 [DOI] [PubMed] [Google Scholar]
- Ma Y., Wang Y., Gao Y., Fu Y., Li J. (2014). Total flavonoids from Ganshanbian (Herba Hyperici Attenuati) effect the expression of CaL-alpha1C and K(ATP)-Kir6.1 mRNA of the myocardial cell membrane in myocardial ischemia-reperfusion arrhythmia rats. J. Tradit. Chin. Med. 34, 357–361. 10.1016/S0254-6272(14)60102-3 [DOI] [PubMed] [Google Scholar]
- Magyar J., Kistamas K., Vaczi K., Hegyi B., Horvath B., Banyasz T., et al. (2016). Concept of relative variability of cardiac action potential duration and its test under various experimental conditions. Gen. Physiol. Biophys. 35, 55–62. 10.4149/gpb_2015019 [DOI] [PubMed] [Google Scholar]
- Manning A. S., Hearse D. J. (1984). Reperfusion-induced arrhythmias: mechanisms and prevention. J. Mol. Cell Cardiol. 16, 497–518. 10.1016/S0022-2828(84)80638-0 [DOI] [PubMed] [Google Scholar]
- Marks A. R., Priori S., Memmi M., Kontula K., Laitinen P. J. (2002). Involvement of the cardiac ryanodine receptor/calcium release channel in catecholaminergic polymorphic ventricular tachycardia. J. Cell Physiol. 190, 1–6. 10.1002/jcp.10031 [DOI] [PubMed] [Google Scholar]
- Masini E., Salvemini D., Ndisang J. F., Gai P., Berni L., Moncini M., et al. (1999). Cardioprotective activity of endogenous and exogenous nitric oxide on ischaemia reperfusion injury in isolated guinea pig hearts. Inflamm. Res. 48, 561–568. 10.1007/s000110050504 [DOI] [PubMed] [Google Scholar]
- Maulik N., Sharma H. S., Das D. K. (1996). Induction of the haem oxygenase gene expression during the reperfusion of ischemic rat myocardium. J. Mol. Cell Cardiol. 28, 1261–1270. 10.1006/jmcc.1996.0116 [DOI] [PubMed] [Google Scholar]
- Mehta A., Ramachandra C. J. A., Singh P., Chitre A., Lua C. H., Mura M., et al. (2018). Identification of a targeted and testable antiarrhythmic therapy for long-QT syndrome type 2 using a patient-specific cellular model. Eur. Heart J. 39, 1446–1455. 10.1093/eurheartj/ehx394 [DOI] [PubMed] [Google Scholar]
- Mengesha H. G., Tafesse T. B., Bule M. H. (2017). If Channel as an Emerging Therapeutic Target for Cardiovascular Diseases: A Review of Current Evidence and Controversies. Front. Pharmacol. 8, 874. 10.3389/fphar.2017.00874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesirca P., Marger L., Toyoda F., Rizzetto R., Audoubert M., Dubel S., et al. (2013). The G-protein-gated K+ channel, IKACh, is required for regulation of pacemaker activity and recovery of resting heart rate after sympathetic stimulation. J. Gen. Physiol. 142, 113–126. 10.1085/jgp.201310996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michowitz Y., Milman A., Andorin A., Sarquella-Brugada G., Gonzalez Corcia M. C., Gourraud J. B., et al. (2019). Characterization and Management of Arrhythmic Events in Young Patients With Brugada Syndrome. J. Am. Coll. Cardiol. 73, 1756–1765. 10.1016/j.jacc.2019.01.048 [DOI] [PubMed] [Google Scholar]
- Mitcheson J. S., Chen J., Lin M., Culberson C., Sanguinetti M. C. (2000). A structural basis for drug-induced long QT syndrome. Proc. Natl. Acad. Sci. U. S. A. 97, 12329–12333. 10.1073/pnas.210244497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto L. (2018). Molecular Pathogenesis of Familial Wolff-Parkinson-White Syndrome. J. Med. Invest. 65, 1–8. 10.2152/jmi.65.1 [DOI] [PubMed] [Google Scholar]
- Mohler P. J., Rivolta I., Napolitano C., Lemaillet G., Lambert S., Priori S. G., et al. (2004). Nav1.5 E1053K mutation causing Brugada syndrome blocks binding to ankyrin-G and expression of Nav1.5 on the surface of cardiomyocytes. Proc. Natl. Acad. Sci. U. S. A 101, 17533–17538. 10.1073/pnas.0403711101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monfredi O., Tsutsui K., Ziman B., Stern M. D., Lakatta E. G., Maltsev V. A. (2018). Electrophysiological heterogeneity of pacemaker cells in the rabbit intercaval region, including the SA node: insights from recording multiple ion currents in each cell. Am. J. Physiol. Heart Circ. Physiol. 314, H403–H414. 10.1152/ajpheart.00253.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Na T., Huang Z. J., Dai D. Z., Zhang Y., Dai Y. (2007). Abrupt changes in FKBP12.6 and SERCA2a expression contribute to sudden occurrence of ventricular fibrillation on reperfusion and are prevented by CPU86017. Acta Pharmacol. Sin. 28, 773–782. 10.1111/j.1745-7254.2007.00580.x [DOI] [PubMed] [Google Scholar]
- Nakata T., Hearse D. J., Curtis M. J. (1990). Are reperfusion-induced arrhythmias caused by disinhibition of an arrhythmogenic component of ischemia? J. Mol. Cell Cardiol. 22, 843–858. 10.1016/0022-2828(90)90116-J [DOI] [PubMed] [Google Scholar]
- Naksuk N., Sugrue A. M., Padmanabhan D., Kella D., Desimone C. V., Kapa S., et al. (2019). Potentially modifiable factors of dofetilide-associated risk of torsades de pointes among hospitalized patients with atrial fibrillation. J. Interv. Card. Electrophysiol. 54, 189–196. 10.1007/s10840-018-0476-2 [DOI] [PubMed] [Google Scholar]
- Nanasi P. P., Magyar J., Varro A., Ordog B. (2017). Beat-to-beat variability of cardiac action potential duration: underlying mechanism and clinical implications. Can. J. Physiol. Pharmacol. 95, 1230–1235. 10.1139/cjpp-2016-0597 [DOI] [PubMed] [Google Scholar]
- Narang A., Ozcan C. (2019). Severe Torsades de Pointes with acquired QT prolongation. Eur. Heart J. Acute Cardiovasc. Care 8, 775–776. 10.1177/2048872616649473 [DOI] [PubMed] [Google Scholar]
- Nath K. A., Balla G., Vercellotti G. M., Balla J., Jacob H. S., Levitt M. D., et al. (1992). Induction of heme oxygenase is a rapid, protective response in rhabdomyolysis in the rat. J. Clin. Invest. 90, 267–270. 10.1172/JCI115847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nerbonne J. M., Kass R. S. (2005). Molecular physiology of cardiac repolarization. Physiol. Rev. 85, 1205–1253. 10.1152/physrev.00002.2005 [DOI] [PubMed] [Google Scholar]
- Ng C. A., Perry M. D., Liang W., Smith N. J., Foo B., Shrier A., et al. (2019). High-throughput phenotyping of heteromeric human ether-a-go-go-related gene potassium channel variants can discriminate pathogenic from rare benign variants. Heart Rhythm. 17 (3), 492–500. 10.1016/j.hrthm.2019.09.020 [DOI] [PubMed] [Google Scholar]
- Niwa N., Nerbonne J. M. (2010). Molecular determinants of cardiac transient outward potassium current (I(to)) expression and regulation. J. Mol. Cell Cardiol. 48, 12–25. 10.1016/j.yjmcc.2009.07.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noma A. (1983). ATP-regulated K+ channels in cardiac muscle. Nature 305, 147–148. 10.1038/305147a0 [DOI] [PubMed] [Google Scholar]
- Oikawa M., Wu M., Lim S., Knight W. E., Miller C. L., Cai Y., et al. (2013). Cyclic nucleotide phosphodiesterase 3A1 protects the heart against ischemia-reperfusion injury. J. Mol. Cell Cardiol. 64, 11–19. 10.1016/j.yjmcc.2013.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono M., Nakamura M., Koga Y. (2004). KB-R9032, newly developed Na(+)/H(+) exchange inhibitor, attenuates reperfusion-induced arrhythmias in isolated perfused rat heart. J. Anesth. 18, 196–202. 10.1007/s00540-004-0232-x [DOI] [PubMed] [Google Scholar]
- Opie L. H., Coetzee W. A. (1988). Role of calcium ions in reperfusion arrhythmias: relevance to pharmacologic intervention. Cardiovasc. Drugs Ther. 2, 623–636. 10.1007/BF00054202 [DOI] [PubMed] [Google Scholar]
- Orvos P., Kohajda Z., Szlovák J., Gazdag P., Árpádffy-Lovas T., Tóth D., et al. (2019). Evaluation of Possible Proarrhythmic Potency: Comparison of the Effect of Dofetilide, Cisapride, Sotalol, Terfenadine, and Verapamil on hERG and Native IKr Currents and on Cardiac Action Potential. Toxicol. Sci. 168 (2), 365–380. 10.1093/toxsci/kfy299 [DOI] [PubMed] [Google Scholar]
- Osipov R. M., Bianchi C., Feng J., Clements R. T., Liu Y., Robich M. P., et al. (2009). Effect of hypercholesterolemia on myocardial necrosis and apoptosis in the setting of ischemia-reperfusion. Circulation 120, S22–S30. 10.1161/CIRCULATIONAHA.108.842724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otani N., Matsuda R., Oda K., Nishino S., Inoue T., Kaneko N. (2013). Protective effect of K201 on isoproterenol-induced and ischemic-reperfusion-induced ventricular arrhythmias in the rat: comparison with diltiazem. J. Cardiovasc. Pharmacol. Ther. 18, 184–190. 10.1177/1074248412465489 [DOI] [PubMed] [Google Scholar]
- Ottani A., Giuliani D., Neri L., Calevro A., Canalini F., Vandini E., et al. (2015). NDP-alpha-MSH attenuates heart and liver responses to myocardial reperfusion via the vagus nerve and JAK/ERK/STAT signaling. Eur. J. Pharmacol. 769, 22–32. 10.1016/j.ejphar.2015.10.022 [DOI] [PubMed] [Google Scholar]
- Paar V., Jirak P., Larbig R., Zagidullin N. S., Brandt M. C., Lichtenauer M., et al. (2019). Pathophysiology of Calcium Mediated Ventricular Arrhythmias and Novel Therapeutic Options with Focus on Gene Therapy. Int. J. Mol. Sci. 20, 5304. 10.3390/ijms20215304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pabla R., Curtis M. J. (1995). Effects of NO modulation on cardiac arrhythmias in the rat isolated heart. Circ. Res. 77, 984–992. 10.1161/01.res.77.5.984 [DOI] [PubMed] [Google Scholar]
- Pataki T., Bak I., Csonka C., Kovacs P., Varga E., Blasig I. E., et al. (2001). Regulation of ventricular fibrillation by heme oxygenase in ischemic/reperfused hearts. Antioxid. Redox Signal 3, 125–134. 10.1089/152308601750100623 [DOI] [PubMed] [Google Scholar]
- Pataki T., Bak I., Kovacs P., Bagchi D., Das D. K., Tosaki A. (2002). Grape seed proanthocyanidins improved cardiac recovery during reperfusion after ischemia in isolated rat hearts. Am. J. Clin. Nutr. 75, 894–899. 10.1093/ajcn/75.5.894 [DOI] [PubMed] [Google Scholar]
- Peers C., Steele D. S. (2012). Carbon monoxide: a vital signalling molecule and potent toxin in the myocardium. J. Mol. Cell Cardiol. 52, 359–365. 10.1016/j.yjmcc.2011.05.013 [DOI] [PubMed] [Google Scholar]
- Perissinotti L. L., De Biase P. M., Guo J., Yang P. C., Lee M. C., Clancy C. E., et al. (2018). Determinants of Isoform-Specific Gating Kinetics of hERG1 Channel: Combined Experimental and Simulation Study. Front. Physiol. 9, 207. 10.3389/fphys.2018.00207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrin M. J., Adler A., Green S., Al-Zoughool F., Doroshenko P., Orr N., et al. (2014). Evaluation of genes encoding for the transient outward current (Ito) identifies the KCND2 gene as a cause of J-wave syndrome associated with sudden cardiac death. Circ. Cardiovasc. Genet. 7, 782–789. 10.1161/CIRCGENETICS.114.000623 [DOI] [PubMed] [Google Scholar]
- Pisarenko O., Studneva I., Timoshin A., Veselova O. (2019). Protective efficacy of dinitrosyl iron complexes with reduced glutathione in cardioplegia and reperfusion. Pflugers. Arch. 471, 583–593. 10.1007/s00424-018-02251-2 [DOI] [PubMed] [Google Scholar]
- Podesser B. K., Schwarzacher S., Zwoelfer W., Binder T. M., Wolner E., Seitelberger R. (1995). Comparison of perioperative myocardial protection with nifedipine versus nifedipine and metoprolol in patients undergoing elective coronary artery bypass grafting. J. Thorac. Cardiovasc. Surg. 110, 1461–1469. 10.1016/S0022-5223(95)70069-2 [DOI] [PubMed] [Google Scholar]
- Podzuweit T., Dalby A. J., Cherry G. W., Opie L. H. (1978). Cyclic AMP levels in ischaemic and non-ischaemic myocardium following coronary artery ligation: relation to ventricular fibrillation. J. Mol. Cell Cardiol. 10, 81–94. 10.1016/0022-2828(78)90008-1 [DOI] [PubMed] [Google Scholar]
- Priori S. G., Napolitano C., Tiso N., Memmi M., Vignati G., Bloise R., et al. (2001). Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation 103, 196–200. 10.1161/01.cir.103.2.196 [DOI] [PubMed] [Google Scholar]
- Qi X. Y., Huang H., Ordog B., Luo X., Naud P., Sun Y., et al. (2015). Fibroblast inward-rectifier potassium current upregulation in profibrillatory atrial remodeling. Circ. Res. 116, 836–845. 10.1161/CIRCRESAHA.116.305326 [DOI] [PubMed] [Google Scholar]
- Ramirez J., Laguna P., Bayes De Luna A., Malik M., Pueyo E. (2014). QT/RR and T-peak-to-end/RR curvatures and slopes in chronic heart failure: relation to sudden cardiac death. J. Electrocardiol. 47, 842–848. 10.1016/j.jelectrocard.2014.08.013 [DOI] [PubMed] [Google Scholar]
- Ramirez J., Orini M., Minchole A., Monasterio V., Cygankiewicz I., Bayes De Luna A., et al. (2017). Sudden cardiac death and pump failure death prediction in chronic heart failure by combining ECG and clinical markers in an integrated risk model. PloS One 12, e0186152. 10.1371/journal.pone.0186152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rampazzo A., Nava A., Erne P., Eberhard M., Vian E., Slomp P., et al. (1995). A new locus for arrhythmogenic right ventricular cardiomyopathy (ARVD2) maps to chromosome 1q42-q43. Hum. Mol. Genet. 4, 2151–2154. 10.1093/hmg/4.11.2151 [DOI] [PubMed] [Google Scholar]
- Ravens U., Himmel H. M. (1999). Drugs preventing Na+ and Ca2+ overload. Pharmacol. Res. 39, 167–174. 10.1006/phrs.1998.0416 [DOI] [PubMed] [Google Scholar]
- Ravingerova T., Carnicka S., Ledvenyiova V., Barlaka E., Galatou E., Chytilova A., et al. (2013). Upregulation of genes involved in cardiac metabolism enhances myocardial resistance to ischemia/reperfusion in the rat heart. Physiol. Res. 62 Suppl 1, S151–S163. [DOI] [PubMed] [Google Scholar]
- Ren L., Wang Q., Chen Y., Ma Y., Wang D. (2019). Involvement of MicroRNA-133a in the Protective Effect of Hydrogen Sulfide against Ischemia/Reperfusion-Induced Endoplasmic Reticulum Stress and Cardiomyocyte Apoptosis. Pharmacology 103, 1–9. 10.1159/000492969 [DOI] [PubMed] [Google Scholar]
- Robertson S., Thomson A. L., Carter R., Stott H. R., Shaw C. A., Hadoke P. W., et al. (2014). Pulmonary diesel particulate increases susceptibility to myocardial ischemia/reperfusion injury via activation of sensory TRPV1 and beta1 adrenoreceptors. Part. Fibre Toxicol. 11, 12. 10.1186/1743-8977-11-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryter S., Kvam E., Richman L., Hartmann F., Tyrrell R. M. (1998). A chromatographic assay for heme oxygenase activity in cultured human cells: application to artificial heme oxygenase overexpression. Free Radic. Biol. Med. 24, 959–971. 10.1016/S0891-5849(97)00380-8 [DOI] [PubMed] [Google Scholar]
- Salloum F. N., Das A., Samidurai A., Hoke N. N., Chau V. Q., Ockaili R. A., et al. (2012). Cinaciguat, a novel activator of soluble guanylate cyclase, protects against ischemia/reperfusion injury: role of hydrogen sulfide. Am. J. Physiol. Heart Circ. Physiol. 302, H1347–H1354. 10.1152/ajpheart.00544.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samouilov A., Ahmad R., Boslett J., Liu X., Petryakov S., Zweier J. L. (2019). Development of a fast-scan EPR imaging system for highly accelerated free radical imaging. Magn. Reson. Med. 82, 842–853. 10.1002/mrm.27759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankaranarayanan R., Kistamas K., Greensmith D. J., Venetucci L. A., Eisner D. A. (2017). Systolic [Ca(2+) ]i regulates diastolic levels in rat ventricular myocytes. J. Physiol. 595, 5545–5555. 10.1113/JP274366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savastano S., Rordorf R., Vicentini A., Petracci B., Taravelli E., Castelletti S., et al. (2014). A comprehensive electrocardiographic, molecular, and echocardiographic study of Brugada syndrome: validation of the 2013 diagnostic criteria. Heart Rhythm. 11, 1176–1183. 10.1016/j.hrthm.2014.04.010 [DOI] [PubMed] [Google Scholar]
- Schwartz P. J., Ackerman M. J., George A. L., Jr., Wilde A. A. M. (2013). Impact of genetics on the clinical management of channelopathies. J. Am. Coll. Cardiol. 62, 169–180. 10.1016/j.jacc.2013.04.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedighi M., Nazari A., Faghihi M., Rafieian-Kopaei M., Karimi A., Moghimian M., et al. (2018). Protective effects of cinnamon bark extract against ischemia-reperfusion injury and arrhythmias in rat. Phytother. Res. 32, 1983–1991. 10.1002/ptr.6127 [DOI] [PubMed] [Google Scholar]
- Sgro A., Drake T. M., Lopez-Ayala P., Phan K. (2019). Left cardiac sympathetic denervation in the management of long QT syndrome and catecholaminergic polymorphic ventricular tachycardia: A meta-regression. Congenit. Heart Dis. 14 (6), 1102–1112. 10.1111/chd.12855 [DOI] [PubMed]
- Shen J., Wang J., Zhao B., Hou J., Gao T., Xin W. (1998). Effects of EGb 761 on nitric oxide and oxygen free radicals, myocardial damage and arrhythmia in ischemia-reperfusion injury in vivo. Biochim. Biophys. Acta 1406, 228–236. 10.1016/s0925-4439(98)00007-6 [DOI] [PubMed] [Google Scholar]
- Sheridan D. J., Penkoske P. A., Sobel B. E., Corr P. B. (1980). Alpha adrenergic contributions to dysrhythmia during myocardial ischemia and reperfusion in cats. J. Clin. Invest. 65, 161–171. 10.1172/JCI109647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh M., Morin D. P., Link M. S. (2019). Sudden cardiac death in Long QT syndrome (LQTS), Brugada syndrome, and catecholaminergic polymorphic ventricular tachycardia (CPVT). Prog. Cardiovasc. Dis. 62, 227–234. 10.1016/j.pcad.2019.05.006 [DOI] [PubMed] [Google Scholar]
- Sipido K. R., Volders P. G., Vos M. A., Verdonck F. (2002). Altered Na/Ca exchange activity in cardiac hypertrophy and heart failure: a new target for therapy? Cardiovasc. Res. 53, 782–805. 10.1016/s0008-6363(01)00470-9 [DOI] [PubMed] [Google Scholar]
- Smith G. L., Eisner D. A. (2019). Calcium Buffering in the Heart in Health and Disease. Circulation 139, 2358–2371. 10.1161/CIRCULATIONAHA.118.039329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Splawski I., Timothy K. W., Tateyama M., Clancy C. E., Malhotra A., Beggs A. H., et al. (2002). Variant of SCN5A sodium channel implicated in risk of cardiac arrhythmia. Science 297, 1333–1336. 10.1126/science.1073569 [DOI] [PubMed] [Google Scholar]
- Sun Y. G., Wang X. Y., Chen X., Shen C. X., Li Y. G. (2015). Hydrogen sulfide improves cardiomyocytes electrical remodeling post ischemia/reperfusion injury in rats. Int. J. Clin. Exp. Pathol. 8, 474–481. [PMC free article] [PubMed] [Google Scholar]
- Sun X., Wang W., Dai J., Jin S., Huang J., Guo C., et al. (2017). A Long-Term and Slow-Releasing Hydrogen Sulfide Donor Protects against Myocardial Ischemia/Reperfusion Injury. Sci. Rep. 7, 3541. 10.1038/s41598-017-03941-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X., Wang W., Dai J., Huang J., Shi M., Chu X., et al. (2018). Donor heart preservation with a novel long-term and slow-releasing hydrogen sulfide system. Nitric. Oxide 81, 1–10. 10.1016/j.niox.2018.09.001 [DOI] [PubMed] [Google Scholar]
- Sutanto H., Laudy L., Clerx M., Dobrev D., Crijns H., Heijman J. (2019). Maastricht antiarrhythmic drug evaluator (MANTA): A computational tool for better understanding of antiarrhythmic drugs. Pharmacol. Res. 148, 104444. 10.1016/j.phrs.2019.104444 [DOI] [PubMed] [Google Scholar]
- Swan H., Piippo K., Viitasalo M., Heikkila P., Paavonen T., Kainulainen K., et al. (1999). Arrhythmic disorder mapped to chromosome 1q42-q43 causes malignant polymorphic ventricular tachycardia in structurally normal hearts. J. Am. Coll. Cardiol. 34, 2035–2042. 10.1016/s0735-1097(99)00461-1 [DOI] [PubMed] [Google Scholar]
- Szendrei L., Turoczi T., Kovacs P., Vecsernyes M., Das D. K., Tosaki A. (2002). Mitochondrial gene expression and ventricular fibrillation in ischemic/reperfused nondiabetic and diabetic myocardium. Biochem. Pharmacol. 63, 543–552. 10.1016/s0006-2952(01)00913-3 [DOI] [PubMed] [Google Scholar]
- Szepesi J., Acsai K., Sebok Z., Prorok J., Pollesello P., Levijoki J., et al. (2015). Comparison of the efficiency of Na+/Ca2+ exchanger or Na+/H+ exchanger inhibition and their combination in reducing coronary reperfusion-induced arrhythmias. J. Physiol. Pharmacol. 66, 215–226. [PubMed] [Google Scholar]
- Szoke K., Czompa A., Lekli I., Szabados-Furjesi P., Herczeg M., Csavas M., et al. (2019). A new, vasoactive hybrid aspirin containing nitrogen monoxide-releasing molsidomine moiety. Eur. J. Pharm. Sci. 131, 159–166. 10.1016/j.ejps.2019.02.020 [DOI] [PubMed] [Google Scholar]
- Szuts V., Menesi D., Varga-Orvos Z., Zvara A., Houshmand N., Bitay M., et al. (2013). Altered expression of genes for Kir ion channels in dilated cardiomyopathy. Can. J. Physiol. Pharmacol. 91, 648–656. 10.1139/cjpp-2012-0413 [DOI] [PubMed] [Google Scholar]
- Tani M., Neely J. R. (1989). Role of intracellular Na+ in Ca2+ overload and depressed recovery of ventricular function of reperfused ischemic rat hearts. Possible involvement of H+-Na+ and Na+-Ca2+ exchange. Circ. Res. 65, 1045–1056. 10.1161/01.res.65.4.1045 [DOI] [PubMed] [Google Scholar]
- Testai L., Marino A., Piano I., Brancaleone V., Tomita K., Di Cesare Mannelli L., et al. (2016). The novel H2S-donor 4-carboxyphenyl isothiocyanate promotes cardioprotective effects against ischemia/reperfusion injury through activation of mitoKATP channels and reduction of oxidative stress. Pharmacol. Res. 113, 290–299. 10.1016/j.phrs.2016.09.006 [DOI] [PubMed] [Google Scholar]
- Thandroyen F. T., Flint N. S., Worthington M. G., Opie L. H. (1987). Arrhythmogenic action of alpha 1-adrenoceptor stimulation in normoxic rat ventricular myocardium: influence of nisoldipine, reduced extracellular Ca2+ and ryanodine. J. Mol. Cell Cardiol. 19, 841–851. 10.1016/S0022-2828(87)80613-2 [DOI] [PubMed] [Google Scholar]
- Thomas S. H., Behr E. R. (2016). Pharmacological treatment of acquired QT prolongation and torsades de pointes. Br. J. Clin. Pharmacol. 81, 420–427. 10.1111/bcp.12726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolg R., Witt M., Schwarz B., Kurz T., Kurowski V., Hartmann F., et al. (2006). Comparison of carvedilol and metoprolol in patients with acute myocardial infarction undergoing primary coronary intervention–the PASSAT Study. Clin. Res. Cardiol. 95, 31–41. 10.1007/s00392-006-0317-7 [DOI] [PubMed] [Google Scholar]
- Tosaki A., Braquet P. (1990). DMPO and reperfusion injury: arrhythmia, heart function, electron spin resonance, and nuclear magnetic resonance studies in isolated working guinea pig hearts. Am. Heart J. 120, 819–830. 10.1016/0002-8703(90)90197-6 [DOI] [PubMed] [Google Scholar]
- Tosaki A., Szekeres L., Hearse D. J. (1987. a). Diltiazem and the reduction of reperfusion-induced arrhythmias in the rat: protection is secondary to modification of ischemic injury and heart rate. J. Mol. Cell Cardiol. 19, 441–451. 10.1016/S0022-2828(87)80396-6 [DOI] [PubMed] [Google Scholar]
- Tosaki A., Szekeres L., Hearse D. J. (1987. b). Metoprolol reduces reperfusion-induced fibrillation in the isolated rat heart: protection is secondary to bradycardia. J. Cardiovasc. Pharmacol. 10, 489–497. 10.1097/00005344-198711000-00001 [DOI] [PubMed] [Google Scholar]
- Tosaki A., Balint S., Szekeres L. (1988). Protective effect of lidocaine against ischemia and reperfusion-induced arrhythmias and shifts of myocardial sodium, potassium, and calcium content. J. Cardiovasc. Pharmacol. 12, 621–628. 10.1097/00005344-198812000-00001 [DOI] [PubMed] [Google Scholar]
- Tosaki A., Koltai M., Braquet P. (1989). Effects of low extracellular sodium concentration on reperfusion induced arrhythmias: changes in the myocardial sodium, potassium and calcium contents in isolated guinea pig hearts. Cardiovasc. Res. 23, 993–1000. 10.1093/cvr/23.12.993 [DOI] [PubMed] [Google Scholar]
- Tosaki A., Bagchi D., Pali T., Cordis G. A., Das D. K. (1993. a). Comparisons of ESR and HPLC methods for the detection of OH. radicals in ischemic/reperfused hearts. A relationship between the genesis of free radicals and reperfusion arrhythmias. Biochem. Pharmacol. 45, 961–969. 10.1016/0006-2952(93)90182-v [DOI] [PubMed] [Google Scholar]
- Tosaki A., Szerdahelyi P., Engelman R. M., Das D. K. (1993. b). Effects of extracellular magnesium manipulation on reperfusion-induced arrhythmias and myocardial ion shifts in isolated ischemic reperfused rat hearts. J. Pharmacol. Exp. Ther. 267, 1045–1053. [PubMed] [Google Scholar]
- Tosaki A., Engelman D. T., Engelman R. M., Das D. K. (1995). Diabetes and ATP-sensitive potassium channel openers and blockers in isolated ischemic/reperfused hearts. J. Pharmacol. Exp. Ther. 275, 1115–1123. [PubMed] [Google Scholar]
- Tosaki A., Pali T., Droy-Lefaix M. T. (1996). Effects of Ginkgo biloba extract and preconditioning on the diabetic rat myocardium. Diabetologia 39, 1255–1262. 10.1007/s001250050567 [DOI] [PubMed] [Google Scholar]
- Tse G., Chan Y. W., Keung W., Yan B. P. (2017). Electrophysiological mechanisms of long and short QT syndromes. Int. J. Cardiol. Heart Vasc. 14, 8–13. 10.1016/j.ijcha.2016.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzivoni D., Keren A., Granot H., Gottlieb S., Benhorin J., Stern S. (1983). Ventricular fibrillation caused by myocardial reperfusion in Prinzmetal's angina. Am. Heart J. 105, 323–325. 10.1016/0002-8703(83)90534-3 [DOI] [PubMed] [Google Scholar]
- Tzivoni D., Banai S., Schuger C., Benhorin J., Keren A., Gottlieb S., et al. (1988). Treatment of torsade de pointes with magnesium sulfate. Circulation 77, 392–397. 10.1161/01.cir.77.2.392 [DOI] [PubMed] [Google Scholar]
- Vago H., Soos P., Zima E., Geller L., Keltai K., Roka A., et al. (2004). Changes of endothelin-1 and big endothelin-1 levels and action potential duration during myocardial ischemia-reperfusion in dogs with and without ventricular fibrillation. J. Cardiovasc. Pharmacol. 44 Suppl 1, S376–S379. 10.1097/01.fjc.0000166299.10443.0c [DOI] [PubMed] [Google Scholar]
- Van Der Weg K., Prinzen F. W., Gorgels A. P. (2019). Editor's Choice- Reperfusion cardiac arrhythmias and their relation to reperfusion-induced cell death. Eur. Heart J. Acute Cardiovasc. Care 8, 142–152. 10.1177/2048872618812148 [DOI] [PubMed] [Google Scholar]
- Vandenberg J. I., Perry M. D., Perrin M. J., Mann S. A., Ke Y., Hill A. P. (2012). hERG K(+) channels: structure, function, and clinical significance. Physiol. Rev. 92, 1393–1478. 10.1152/physrev.00036.2011 [DOI] [PubMed] [Google Scholar]
- Vanoli E., Hull S. S., Jr., Foreman R. D., Ferrari A., Schwartz P. J. (1994). Alpha 1-adrenergic blockade and sudden cardiac death. J. Cardiovasc. Electrophysiol. 5, 76–89. 10.1111/j.1540-8167.1994.tb01116.x [DOI] [PubMed] [Google Scholar]
- Varga E., Bodi A., Ferdinandy P., Droy-Lefaix M. T., Blasig I. E., Tosaki A. (1999). The protective effect of EGb 761 in isolated ischemic/reperfused rat hearts: a link between cardiac function and nitric oxide production. J. Cardiovasc. Pharmacol. 34, 711–717. 10.1097/00005344-199911000-00013 [DOI] [PubMed] [Google Scholar]
- Vasseur L., Cens T., Wagner R., Saint N., Kugler V., Chavanieu A., et al. (2019). Importance of the Choice of a Recombinant System to Produce Large Amounts of Functional Membrane Protein hERG. Int. J. Mol. Sci. 20, 3181. 10.3390/ijms20133181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan Williams E. M. (1959). Relation of extracellular to intracellular potential records from single cardiac muscle fibres. Nature 183, 1341–1342. 10.1038/1831341a0 [DOI] [PubMed] [Google Scholar]
- Vaughan Williams E. M. (1975). Classification of antidysrhythmic drugs. Pharmacol. Ther. B 1, 115–138. 10.1016/0306-039x(75)90019-7 [DOI] [PubMed] [Google Scholar]
- Vaughan Williams E. M. (1992). Classifying antiarrhythmic actions: by facts or speculation. J. Clin. Pharmacol. 32, 964–977. 10.1002/j.1552-4604.1992.tb03797.x [DOI] [PubMed] [Google Scholar]
- Verkerk A. O., Amin A. S., Remme C. A. (2018). Disease Modifiers of Inherited SCN5A Channelopathy. Front. Cardiovasc. Med. 5, 137. 10.3389/fcvm.2018.00137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vink A. S., Neumann B., Lieve K. V. V., Sinner M. F., Hofman N., El Kadi S., et al. (2018). Determination and Interpretation of the QT Interval. Circulation 138, 2345–2358. 10.1161/CIRCULATIONAHA.118.033943 [DOI] [PubMed] [Google Scholar]
- Voigt N., Li N., Wang Q., Wang W., Trafford A. W., Abu-Taha I., et al. (2012). Enhanced sarcoplasmic reticulum Ca2+ leak and increased Na+-Ca2+ exchanger function underlie delayed afterdepolarizations in patients with chronic atrial fibrillation. Circulation 125, 2059–2070. 10.1161/CIRCULATIONAHA.111.067306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wainwright C. L., Mccabe C., Kane K. A. (2005). Endothelin and the ischaemic heart. Curr. Vasc. Pharmacol. 3, 333–341. 10.2174/157016105774329417 [DOI] [PubMed] [Google Scholar]
- Waldman M., Nudelman V., Shainberg A., Zemel R., Kornwoski R., Aravot D., et al. (2019). The Role of Heme Oxygenase 1 in the Protective Effect of Caloric Restriction against Diabetic Cardiomyopathy. Int. J. Mol. Sci. 20, 2427. 10.3390/ijms20102427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker M. J., Curtis M. J., Hearse D. J., Campbell R. W., Janse M. J., Yellon D. M., et al. (1988). The Lambeth Conventions: guidelines for the study of arrhythmias in ischaemia infarction, and reperfusion. Cardiovasc. Res. 22, 447–455. 10.1093/cvr/22.7.447 [DOI] [PubMed] [Google Scholar]
- Wang Q. D., Pernow J., Sjoquist P. O., Ryden L. (2002). Pharmacological possibilities for protection against myocardial reperfusion injury. Cardiovasc. Res. 55, 25–37. 10.1016/s0008-6363(02)00261-4 [DOI] [PubMed] [Google Scholar]
- Wang Q., Chen S., Chen Q., Wan X., Shen J., Hoeltge G. A., et al. (2004). The common SCN5A mutation R1193Q causes LQTS-type electrophysiological alterations of the cardiac sodium channel. J. Med. Genet. 41, e66. 10.1136/jmg.2003.013300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S., Radhakrishnan J., Ayoub I. M., Kolarova J. D., Taglieri D. M., Gazmuri R. J. (2007). Limiting sarcolemmal Na+ entry during resuscitation from ventricular fibrillation prevents excess mitochondrial Ca2+ accumulation and attenuates myocardial injury. J. Appl. Physiol. (1985) 103, 55–65. 10.1152/japplphysiol.01167.2006 [DOI] [PubMed] [Google Scholar]
- Wang S., Guo X., Long C. L., Li C., Zhang Y. F., Wang J., et al. (2019). SUR2B/Kir6.1 channel openers correct endothelial dysfunction in chronic heart failure via the miR-1-3p/ET-1 pathway. Biomed. Pharmacother. 110, 431–439. 10.1016/j.biopha.2018.11.135 [DOI] [PubMed] [Google Scholar]
- Wang R. (2002). Two's company, three's a crowd: can H2S be the third endogenous gaseous transmitter? FASEB J. 16, 1792–1798. 10.1096/fj.02-0211hyp [DOI] [PubMed] [Google Scholar]
- Whalen D. A., Jr., Hamilton D. G., Ganote C. E., Jennings R. B. (1974). Effect of a transient period of ischemia on myocardial cells. I. Effects on cell volume regulation. Am. J. Pathol. 74, 381–397. -. PMC1910794. [PMC free article] [PubMed] [Google Scholar]
- Wilder C. D., Masoud R., Yazar D., O'brien B. A., Eykyn T. R., Curtis M. J. (2016). Contractile function assessment by intraventricular balloon alters the ability of regional ischaemia to evoke ventricular fibrillation. Br. J. Pharmacol. 173, 39–52. 10.1111/bph.13332. PMC4813384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winchester D. E., Pepine C. J. (2014). Usefulness of Beta blockade in contemporary management of patients with stable coronary heart disease. Am. J. Cardiol. 114, 1607–1612. 10.1016/j.amjcard.2014.08.026 [DOI] [PubMed] [Google Scholar]
- Wirth K. J., Maier T., Busch A. E. (2001). NHE1-inhibitor cariporide prevents the transient reperfusion-induced shortening of the monophasic action potential after coronary ischemia in pigs. Basic Res. Cardiol. 96, 192–197. 10.1007/s003950170070 [DOI] [PubMed] [Google Scholar]
- Wolff L., Parkinson J., White D. P. (1930). Bundle-branch block with short P-R interval in healthy young people prone to paroxysmal tachycardia. Am. Heart J. 5, 685–704. 10.1016/S0002-8703(30)90086-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodbury L. A., Woodbury J. W., Hecht H. H. (1950). Membrane resting and action potentials of single cardiac muscle fibers. Circulation 1, 264–266. 10.1161/01.cir.1.2.264 [DOI] [PubMed] [Google Scholar]
- Woodcock E. A., Arthur J. F., Harrison S. N., Gao X. M., Du X. J. (2001). Reperfusion-induced Ins(1,4,5)P(3) generation and arrhythmogenesis require activation of the Na(+)/Ca(2+) exchanger. J. Mol. Cell Cardiol. 33, 1861–1869. 10.1006/jmcc.2001.1450 [DOI] [PubMed] [Google Scholar]
- Woodman O. L., Chin K. Y., Thomas C. J., Ng D. C. H., May C. N. (2018). Flavonols and Flavones - Protecting Against Myocardial Ischemia/Reperfusion Injury by Targeting Protein Kinases. Curr. Med. Chem. 25, 4402–4415. 10.2174/0929867325666180326161730 [DOI] [PubMed] [Google Scholar]
- Woodward B., Zakaria M. N. (1985). Effect of some free radical scavengers on reperfusion induced arrhythmias in the isolated rat heart. J. Mol. Cell Cardiol. 17, 485–493. 10.1016/S0022-2828(85)80053-5 [DOI] [PubMed] [Google Scholar]
- Wu M. L., Ho Y. C., Yet S. F. (2011). A central role of heme oxygenase-1 in cardiovascular protection. Antioxid. Redox Signal 15, 1835–1846. 10.1089/ars.2010.3726 [DOI] [PubMed] [Google Scholar]
- Wu J., Sakaguchi T., Takenaka K., Toyoda F., Tsuji K., Matsuura H., et al. (2019). A trafficking-deficient KCNQ1 mutation, T587M, causes a severe phenotype of long QT syndrome by interfering with intracellular hERG transport. J. Cardiol. 73, 343–350. 10.1016/j.jjcc.2018.10.011 [DOI] [PubMed] [Google Scholar]
- Xie Y., Ding W. G., Liu Y., Yu M., Sun X., Matsuura H. (2019). Long-term 4-AP treatment facilitates functional expression of human Kv1.5 channel. Eur. J. Pharmacol. 844, 195–203. 10.1016/j.ejphar.2018.12.022 [DOI] [PubMed] [Google Scholar]
- Yamada C., Xue Y., Chino D., Hashimoto K. (2005). Effects of KB-R9032, a new Na+/H+ exchange inhibitor, on canine coronary occlusion/reperfusion-induced ventricular arrhythmias. J. Pharmacol. Sci. 98, 404–410. 10.1254/jphs.FP0050278 [DOI] [PubMed] [Google Scholar]
- Yampolsky P., Koenen M., Mosqueira M., Geschwill P., Nauck S., Witzenberger M., et al. (2019). Augmentation of myocardial If dysregulates calcium homeostasis and causes adverse cardiac remodeling. Nat. Commun. 10, 3295. 10.1038/s41467-019-11261-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagisawa M., Kurihara H., Kimura S., Tomobe Y., Kobayashi M., Mitsui Y., et al. (1988). A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature 332, 411–415. 10.1038/332411a0 [DOI] [PubMed] [Google Scholar]
- Yasuda J., Okada M., Yamawaki H. (2019). Protective effect of T3 peptide, an active fragment of tumstatin, against ischemia/reperfusion injury in rat heart. J. Pharmacol. Sci. 139, 193–200. 10.1016/j.jphs.2019.01.010 [DOI] [PubMed] [Google Scholar]
- Yasutake M., Avkiran M. (1995). Exacerbation of reperfusion arrhythmias by alpha 1 adrenergic stimulation: a potential role for receptor mediated activation of sarcolemmal sodium-hydrogen exchange. Cardiovasc. Res. 29, 222–230. [PubMed] [Google Scholar]
- Ye L., Neale C., Sljoka A., Lyda B., Pichugin D., Tsuchimura N., et al. (2018). Mechanistic insights into allosteric regulation of the A2A adenosine G protein-coupled receptor by physiological cations. Nat. Commun. 9, 1372. 10.1038/s41467-018-03314-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye D., Tester D. J., Zhou W., Papagiannis J., Ackerman M. J. (2019). A pore-localizing CACNA1C-E1115K missense mutation, identified in a patient with idiopathic QT prolongation, bradycardia, and autism spectrum disorder, converts the L-type calcium channel into a hybrid nonselective monovalent cation channel. Heart Rhythm. 16, 270–278. 10.1016/j.hrthm.2018.08.030 [DOI] [PubMed] [Google Scholar]
- Yellon D. M., Hausenloy D. J. (2007). Myocardial reperfusion injury. N. Engl. J. Med. 357, 1121–1135. 10.1056/NEJMra071667 [DOI] [PubMed] [Google Scholar]
- Yin C., Zhang P., Yang J., Zhang L. (2018). Unique ECG presentations and clinical management of a symptomatic LQT2 female carrying a novel de novo KCNH2 mutation. J. Electrocardiol. 51, 111–116. 10.1016/j.jelectrocard.2017.08.022 [DOI] [PubMed] [Google Scholar]
- Ying S. Q., Fang L., Xiang M. X., Xu G., Shan J., Wang J. A. (2007). Protective effects of magnesium against ischaemia-reperfusion injury through inhibition of P-selectin in rats. Clin. Exp. Pharmacol. Physiol. 34, 1234–1239. 10.1111/j.1440-1681.2007.04697.x [DOI] [PubMed] [Google Scholar]
- Yu L., Huang B., Po S. S., Tan T., Wang M., Zhou L., et al. (2017). Low-Level Tragus Stimulation for the Treatment of Ischemia and Reperfusion Injury in Patients With ST-Segment Elevation Myocardial Infarction: A Proof-of-Concept Study. JACC Cardiovasc. Interv. 10, 1511–1520. 10.1016/j.jcin.2017.04.036 [DOI] [PubMed] [Google Scholar]
- Yu L. M., Dong X., Xue X. D., Zhang J., Li Z., Wu H. J., et al. (2019). Protection of the myocardium against ischemia/reperfusion injury by punicalagin through an SIRT1-NRF-2-HO-1-dependent mechanism. Chem. Biol. Interact. 306, 152–162. 10.1016/j.cbi.2019.05.003 [DOI] [PubMed] [Google Scholar]
- Yue L., Xie J., Nattel S. (2011). Molecular determinants of cardiac fibroblast electrical function and therapeutic implications for atrial fibrillation. Cardiovasc. Res. 89, 744–753. 10.1093/cvr/cvq329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zareba W., Moss A. J., Sheu G., Kaufman E. S., Priori S., Vincent G. M., et al. (2003). Location of mutation in the KCNQ1 and phenotypic presentation of long QT syndrome. J. Cardiovasc. Electrophysiol. 14, 1149–1153. 10.1046/j.1540-8167.2003.03177.x. 14678125. [DOI] [PubMed] [Google Scholar]
- Zhang T., Li Z., Dang H., Chen R., Liaw C., Tran T. A., et al. (2012). Inhibition of Mas G-protein signaling improves coronary blood flow, reduces myocardial infarct size, and provides long-term cardioprotection. Am. J. Physiol. Heart Circ. Physiol. 302, H299–H311. 10.1152/ajpheart.00723.2011 [DOI] [PubMed] [Google Scholar]
- Zhang Y., Luo J., He J., Rong M., Zeng X. (2019. a). JZTX-V Targets the Voltage Sensor in Kv4.2 to Inhibit Ito Potassium Channels in Cardiomyocytes. Front. Pharmacol. 10, 357. 10.3389/fphar.2019.00357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Wu J., Guo S., Lin W., Zhang B., Chen X., et al. (2019. b). The clinical efficacy and safety of the Chinese herbal medicine Astragalus (Huangqi) preparation for the treatment of acute myocardial infarction: A systematic review of randomized controlled trials. Med. (Baltimore) 98, e15256. 10.1097/MD.0000000000015256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y., Wang D., Gao X., Lew K., Richards A. M., Wang P. (2014). mTORC2 phosphorylation of Akt1: a possible mechanism for hydrogen sulfide-induced cardioprotection. PloS One 9, e99665. 10.1371/journal.pone.0099665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X., Bueno-Orovio A., Schilling R. J., Kirkby C., Denning C., Rajamohan D., et al. (2019). Investigating the Complex Arrhythmic Phenotype Caused by the Gain-of-Function Mutation KCNQ1-G229D. Front. Physiol. 10, 259. 10.3389/fphys.2019.00259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo L., Chen Y. R., Reyes L. A., Lee H. L., Chen C. L., Villamena F. A., et al. (2009). The radical trap 5,5-dimethyl-1-pyrroline N-oxide exerts dose-dependent protection against myocardial ischemia-reperfusion injury through preservation of mitochondrial electron transport. J. Pharmacol. Exp. Ther. 329, 515–523. 10.1124/jpet.108.143479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zweier J. L., Kuppusamy P. (1988). Electron paramagnetic resonance measurements of free radicals in the intact beating heart: a technique for detection and characterization of free radicals in whole biological tissues. Proc. Natl. Acad. Sci. U. S. A 85, 5703–5707. 10.1073/pnas.85.15.5703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zweier J. L., Kuppusamy P., Lutty G. A. (1988). Measurement of endothelial cell free radical generation: evidence for a central mechanism of free radical injury in postischemic tissues. Proc. Natl. Acad. Sci. U. S. A 85, 4046–4050. 10.1073/pnas.85.11.4046 [DOI] [PMC free article] [PubMed] [Google Scholar]