

Abstract
Background:
Hepatocellular carcinoma (HCC) is the most common liver cancer and the mechanisms of hepatocarcinogenesis remain elusive.
Objective:
This study aims to mine hub genes associated with HCC using multiple databases.
Methods:
Data sets GSE45267, GSE60502, GSE74656 were downloaded from GEO database. Differentially expressed genes (DEGs) between HCC and control in each set were identified by limma software. The GO term and KEGG pathway enrichment of the DEGs aggregated in the datasets (aggregated DEGs) were analyzed using DAVID and KOBAS 3.0 databases. Protein-protein interaction (PPI) network of the aggregated DEGs was constructed using STRING database. GSEA software was used to verify the biological process. Association between hub genes and HCC prognosis was analyzed using patients’ information from TCGA database by survminer R package.
Results:
From GSE45267, GSE60502 and GSE74656, 7583, 2349, and 553 DEGs were identified respectively. A total of 221 aggregated DEGs, which were mainly enriched in 109 GO terms and 29 KEGG pathways, were identified. Cell cycle phase, mitotic cell cycle, cell division, nuclear division and mitosis were the most significant GO terms. Metabolic pathways, cell cycle, chemical carcinogenesis, retinol metabolism and fatty acid degradation were the main KEGG pathways. Nine hub genes (TOP2A, NDC80, CDK1, CCNB1, KIF11, BUB1, CCNB2, CCNA2 and TTK) were selected by PPI network and all of them were associated with prognosis of HCC patients.
Conclusion:
TOP2A, NDC80, CDK1, CCNB1, KIF11, BUB1, CCNB2, CCNA2 and TTK were hub genes in HCC, which may be potential biomarkers of HCC and targets of HCC therapy.
Keywords: Hepatocellular carcinoma, hub gene, bioinformatics, differentially expressed gene, database, mRNA
1. INTRODUCTION
Hepatocellular carcinoma (HCC), the most common type of primary liver cancer, is one of the most common malignant tumors of digestive system [1]. There were many causes of HCC, including viral infection [2], alcohol intake [3], obesity [4], and environmental pollution [5]. Though many efforts have been done, the molecular mechanisms of hepatocarcinogenesis remain elusive due to the complexity. With the advancement of precise cancer treatment, the research of HCC has turned into gene mutation and genome-wide studies. A large number of genes have been reported to play roles in the carcinogenesis of HCC, which may be useful for the development of effective prevention and treatment regimens for HCC. However, the identification of hub genes and potential underlying mechanisms of their associations with HCC from the information of various studies remain a challenge.
Bioinformatics, a new subject of genetic data collection, analysis and dissemination to the research community, combines medicine, statistics and mathematics and refers to database-like activities, involving persistent sets of data that are maintained in a consistent state over essentially indefinite periods of time. It has been rapidly applied in the field of oncogene. Resources of the public databases can be fully used to screen useful information from a large number of data by bioinformatics, improving the efficiency and accuracy of research [6]. The Cancer Genome Atlas (TCGA), Gene Expression Omnibus (GEO) and Oncomine are the commonly used databases for cancer research.
In this study, we used integrated bioinformatics to explore genes, especially the hub genes, related to the pathogenesis of HCC and the underlying mechanisms of the associations of the hub genes with HCC. The identification of hub genes and the potential mechanisms such as signal pathways associated with HCC may shed light on the understanding of HCC pathogenesis and thus provide new information for the precise management of HCC.
2. METHODS
2.1. Gene mRNA Expression Data
GEO (Gene Expression Omnibus) database [7] (https://www.ncbi.nlm.nih.gov/geo/) is currently the largest and most comprehensive public database containing gene mRNA expression data resources. We obtained the datasets in recent 5 years (from 2014 to 2018) from the GEO database by setting the keyword as “hepatocellular carcinoma”, organism as “Homo sapiens”, and study type as “expression profiling by array”. Clinical samples should include HCC and non-HCC liver tissues. Finally, the data sets GSE45267 [8], GSE60502 [9], and GSE74656 (Yin HY, unpublished data, 2015), which provided data of both HCC and non-HCC samples, were selected. There were 46 HCC tissue samples and 41 non-HCC tissue samples in GSE45267, and the gene detection platform was GPL570 [HG-U133_Plus_2] Affymetrix Human Genome U133 Plus 2.0 Array. A total of 18 HCC tissue samples and 18 non-HCC tissue samples were selected from GSE60502, and the platform was GPL96 [HG-U133A] Affymetrix Human Genome U133A Array. Five HCC tissue samples and 5 non-HCC tissue samples were chosen from GSE74656, and the platform was GPL16043 GeneChip® PrimeView™ Human Gene Expression Array (with External spike-in RNAs). HCC tissue samples were regarded as HCC group, and non-HCC tissue samples were considered as control group. We downloaded the raw data from website, and used R language to process the data. Subsequently, the differentially expressed genes (DEGs) in the HCC group compared with control group from the 3 datasets were identified using the “limma software” package [10]. Adjusted p value (adj_pval) < 0.01 and logFC (log fold change) > 0 were considered to be significant. The genes with logFC > 0 were thought to be up-regulated genes, and those with logFC < 0 were regarded as down-regulated genes. Finally, we obtained the DEGs of each dataset. The DEGs aggregated in the three datasets (aggregated DEGs) were identified by the “Robust Rank Aggregation (RRA)” package [11].
2.2. Functional Enrichment Analysis
The aggregated DEGs were further applied for functional and enrichment analysis. GO (Gene Ontology) terms were analyzed by the online tools DAVID 6.7 [12] (https://david-d.ncifcrf.gov/), and the terms with adj_pval < 0.01 were chosen. KEGG (Kyoto Encyclopedia of Genes and Genomes) pathways of the aggregated DEGs were analyzed on KOBAS 3.0 website [13] (http://kobas.cbi.pku.edu.cn/), and pathways with p < 0.01 (unadjusted p value) were selected.
2.3. Protein-protein Interaction (PPI) Network
The protein-protein interaction (PPI) network of the aggregated DEGs was constructed using STRING (version 10.5) [14] (https://string-db.org/) and visualized using Cytoscape 3.6.1 software. The nodes with degree ≥ 1 were reserved in the PPI network. Genes of their proteins with degree ≥ 50 were considered as hub genes. Proteins encoded by these hub genes may have high interaction with other proteins, and are at central positions in PPI network.
2.4. Gene Set Enrichment Analysis (GSEA)
GSEA V2.2.4.jar software [15] was used to perform GSEA analysis. The HCC and control samples from GSE45267 were chosen. The Hallmarks in MSigDB (Molecular Signatures Database) were selected as reference gene set. The number of permutations was 1,000. The gene sets with normalized enrichment score (NES) of > 1, normal p value (NOM p-val) < 0.05 and false discovery rate value (FDR) of < 0.25 were regarded as significant enrichment gene sets.
2.5. Overall Survival Analysis
We downloaded the data of HCC patients’ clinical information from TCGA (The Cancer Genome Atlas) database [16] (https://cancergenome.nih.gov/). These data included the diagnosis time, time of death, and gene mRNA expression levels of the patients. We processed these data using perl language for further analysis. The survival time and survival status of all HCC patients and the mRNA expression data of each hub gene were obtained using the “hash” package [17]. The association between each hub gene and prognosis of HCC patients was analyzed using “survminer” package [18] and p < 0.05 (unadjusted p value) was considered significant.
3. RESULTS
3.1. Screening of DEGs Aggregated in the Datasets
Datasets GSE45267, GSE60502, and GSE74656 which were submitted within 5 years (from 2014 to 2018) and have data of both HCC and non-HCC samples were chosen for study. The boxplots of the three datasets were shown in Supplementary Fig. (S1 (679.8KB, pdf) ), which confirmed that the expression values in the three datasets had small deviations in distribution. The analysis of the three datasets and comparison of HCC group with control group identified 7,583 (4,020 up-regulated and 3,563 down-regulated), 2,349 (1,247 up-regulated and 1,102 down-regulated) and 553 (297 up-regulated and 256 down-regulated) DEGs from GSE45267, GSE60502 and GSE74656, respectively. Distributions of the DEGs in the three data sets were shown in Fig. 1 (A-C), respectively. The top 20 up-regulated and down-regulated DEGs of GSE45267, GSE60502 and GSE74656 were shown in Supplementary Tables S1-S3 (679.8KB, pdf) , respectively. A total of 221 aggregated DEGs were obtained by further analysis of the DEGs. The top 20 up- and down-regulated aggregated DEGs were displayed in Table 1. In addition, we showed the data by a heatmap (Fig. 2). The trend of the mRNA expression of the aggregated DEGs screened by RRA method was consistent in all the three datasets, indicating that the aggregated DEGs we selected were representative.
Fig. (1).
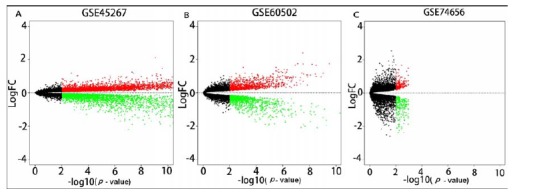
The volcano of GSE45267 DEGs (A), GSE60502 DEGs (B) and GSE74656 DEGs (C).
Table 1. The top 20 up-regulated and down-regulated DEGs aggregated in GSE45267, GSE60502 and GSE74656.
| Up-regulated Genes | Score | Down-regulated Genes | Score |
|---|---|---|---|
| NDC80 | 3.81E-08 | FCN3 | 1.16E-08 |
| SPINK1 | 4.89E-07 | HAMP | 1.47E-07 |
| RACGAP1 | 7.81E-06 | APOF | 2.20E-07 |
| ACSL4 | 7.81E-06 | CYP1A2 | 2.64E-07 |
| GINS1 | 1.09E-05 | SLC22A1 | 4.30E-07 |
| PLVAP | 2.10E-05 | CRHBP | 9.44E-07 |
| EZH2 | 4.16E-05 | GYS2 | 9.44E-07 |
| GPC3 | 5.91E-05 | HGFAC | 5.23E-06 |
| CEP55 | 6.25E-05 | AKR1D1 | 6.32E-06 |
| DLGAP5 | 7.43E-05 | CLEC4M | 6.32E-06 |
| CENPE | 8.95E-05 | MT1F | 1.05E-05 |
| CDC20 | 0.00011 | CFHR4 | 1.77E-05 |
| TOP2A | 0.000125 | LPA | 2.09E-05 |
| TTK | 0.000141 | DNASE1L3 | 2.65E-05 |
| KNTC1 | 0.000187 | MT1E | 2.75E-05 |
| ZWILCH | 0.000194 | GNMT | 3.54E-05 |
| NUSAP1 | 0.000258 | KCNN2 | 3.66E-05 |
| CTHRC1 | 0.000258 | CFP | 3.79E-05 |
| SHCBP1 | 0.000278 | CYP2B6 | 4.32E-05 |
| NCAPG | 0.000281 | CYP2E1 | 4.98E-05 |
Abbreviation: DEGs, Differentially Expressed Genes.
Fig. (2).
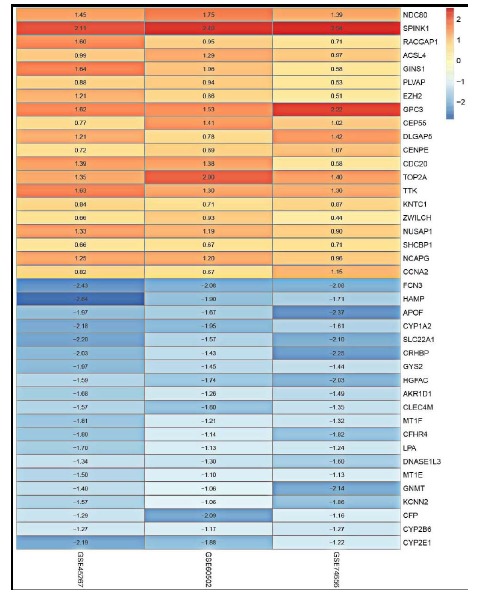
The heatmap of the top 20 up-regulated and down-regulated DEGs aggregated in GSE45267, GSE60502 and GSE74656. The values in each box represents logFC values, logFC > 0 were up-regulated genes, and logFC < 0 were down-regulated genes.
3.2. GO Analysis
We performed GO analysis using the aggregated DEGs. A total of 109 significant GO terms were obtained (adj_pval < 0.01). GO terms contained BP (Biological Process), MF (Molecular Function), and CC (Cellular Component) categories. The number of target genes enriched in the top 5 of the three GO categories were shown in Fig. (3A). Detailed information was shown in Table 2. We used circle diagram to demonstrate the gene distribution of the main enriched GO terms (Fig. 3B). Cell cycle phase (adj_pval = 5.57E-14), mitotic cell cycle (adj_pval = 1.45E-13), cell division (adj_pval = 2.62E-13), nuclear division (adj_pval = 2.76E-13), and mitosis (adj_pval = 2.76E-13) were the top 5 BP terms. Electron carrier activity (adj_pval = 1.35E-11), oxygen binding (adj_pval = 1.78E-10), heme binding (adj_pval = 7.11E-08), cadmium ion binding (adj_pval = 9.47E-08), and tetrapyrrole binding (adj_pval = 1.45E-07) were the top 5 MF terms. Spindle (adj_pval = 1.40E-11), microsome (adj_pval = 2.37E-08), vesicular fraction (adj_pval = 3.64E-08), condensed chromosome kinetochore (adj_pval = 7.84E-08), and condensed chromosome, centromeric region (adj_pval = 2.48E-07) were the top 5 CC terms. All these GO terms were enriched with many genes of the DEGs we selected. Cell cycle phase, mitotic cell cycle and cell division were the most significant enriched GO terms of the DEGs selected.
Fig. (3).
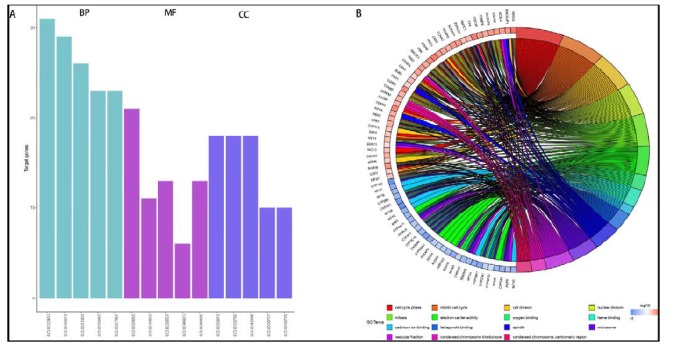
The number of genes enriched in the top five biological process (BP), molecular function (MF) and cellular component (CC) terms (A) and the circle diagram of the top five GO enriched biological process (BP), molecular function (MF) and cellular component (CC) terms (B).
Table 2. The main GO terms of the DEGs aggregated in GSE45267, GSE60502 and GSE74656.
| Category | ID | Term | Count | adj_pval | Genes |
|---|---|---|---|---|---|
| BP | GO:0022403 | cell cycle phase | 31 | 5.57E-14 | PRC1, NEK2, DBF4, KNTC1, TTK, CEP55, KIF2C, SAC3D1, NCAPG, BUB1, ZWILCH, CCNA2, ASPM, CDCA3, CDC7, CDK1, KIF11, DLGAP5, TPX2, NUSAP1, CENPE, NDC80, CDC20, PBK, TACC3, CDK4, CDKN3, CCNB1, CCNB2, CKS2, BUB1B |
| BP | GO:0000278 | mitotic cell cycle | 29 | 1.45E-13 | PRC1, NEK2, DBF4, KNTC1, TTK, CEP55, KIF2C, SAC3D1, NCAPG, BUB1, ZWILCH, CCNA2, ASPM, CDCA3, CDC7, CDK1, KIF11, DLGAP5, TPX2, NUSAP1, CENPE, NDC80, CDC20, PBK, CDK4, CDKN3, CCNB1, CCNB2, BUB1B |
| BP | GO:0051301 | cell division | 26 | 2.62E-13 | PRC1, NEK2, KNTC1, CEP55, SAC3D1, NCAPG, BUB1, ZWILCH, CCNA2, ASPM, CDCA3, CDC7, CDK1, KIF11, NUSAP1, CENPE, NDC80, CDC20, CDK4, RACGAP1, MCM5, CCNB1, CCNB2, CKS2, BUB1B, CDCA7L |
| BP | GO:0000280 | nuclear division | 23 | 2.76E-13 | CDK1, KIF11, NEK2, DLGAP5, TPX2, KNTC1, NUSAP1, NDC80, CENPE, CDC20, PBK, CEP55, CCNB1, KIF2C, CCNB2, SAC3D1, NCAPG, BUB1, BUB1B, ZWILCH, CCNA2, ASPM, CDCA3 |
| BP | GO:0007067 | mitosis | 23 | 2.76E-13 | CDK1, KIF11, NEK2, DLGAP5, TPX2, KNTC1, NUSAP1, NDC80, CENPE, CDC20, PBK, CEP55, CCNB1, KIF2C, CCNB2, SAC3D1, NCAPG, BUB1, BUB1B, ZWILCH, CCNA2, ASPM, CDCA3 |
| MF | GO:0009055 | electron carrier activity | 21 | 1.35E-11 | CYP3A4, STEAP3, GCDH, CYP2C19, CYP2B6, ADH6, CYP26A1, KMO, CYP4F12, CYP2E1, CYP1A2, CYP4A11, CYP39A1, HAAO, CYP2A6, AKR7A3, CYP2A7, CYP4F2, RDH16 |
| MF | GO:0019825 | oxygen binding | 11 | 1.78E-10 | CYP3A4, CYP4A11, CYP2C19, CYP2B6, HAAO, CYP26A1, CYP2A7, CYP4F12, CYP2E1, CYP1A2, CYP4F2 |
| MF | GO:0020037 | heme binding | 13 | 7.11E-08 | CYP3A4, CYP4A11, CYP39A1, CYP27A1, CYP2C19, CYP2B6, CYP26A1, CYP2A6, CYP2A7, CYP4F12, CYP2E1, CYP1A2, CYP4F2 |
| MF | GO:0046870 | cadmium ion binding | 6 | 9.47E-08 | MT1M, MT1E, MT1H, MT1G, MT1X, MT1F |
| MF | GO:0046906 | tetrapyrrole binding | 13 | 1.45E-07 | CYP3A4, CYP4A11, CYP39A1, CYP27A1, CYP2C19, CYP2B6, CYP26A1, CYP2A6, CYP2A7, CYP4F12, CYP2E1, CYP1A2, CYP4F2 |
| CC | GO:0005819 | spindle | 18 | 1.40E-11 | CDK1, KIF4A, KIF11, PRC1, NEK2, DLGAP5, KNTC1, TPX2, NUSAP1, TTK, CENPE, CDC20, CBX1, RACGAP1, SAC3D1, BUB1, BUB1B, ASPM |
| CC | GO:0005792 | microsome | 18 | 2.37E-08 | CYP3A4, AQP9, CYP2C19, CYP2B6, IGFALS, CYP26A1, CYP4F12, CYP2E1, CYP1A2, CYP4A11, CYP39A1, CYP2A6, SORT1, CYP2A7, SRD5A2, CYP4F2, ACSL4, RDH16 |
| CC | GO:0042598 | vesicular fraction | 18 | 3.64E-08 | CYP3A4, AQP9, CYP2C19, CYP2B6, IGFALS, CYP26A1, CYP4F12, CYP2E1, CYP1A2, CYP4A11, CYP39A1, CYP2A6, SORT1, CYP2A7, SRD5A2, CYP4F2, ACSL4, RDH16 |
| CC | GO:0000777 | condensed chromosome kinetochore | 10 | 7.84E-08 | KIF2C, CENPM, HJURP, KNTC1, BUB1, BUB1B, CENPE, NDC80, CENPK, ZWILCH |
| CC | GO:0000779 | condensed chromosome, centromeric region | 10 | 2.48E-07 | KIF2C, CENPM, HJURP, KNTC1, BUB1, BUB1B, CENPE, NDC80, CENPK, ZWILCH |
Abbreviations: DEGs, Differentially Expressed Genes; BP, Biological Process; MF, Molecular Function; CC, Cellular Component.
3.3. Enriched KEGG Pathways
We converted gene symbol of all the aggregated DEGs to ensemble ID by DAVID database, and then analyzed the KEGG pathways by KOBAS 3.0. Finally, we obtained 29 significant enriched KEGG pathways (p < 0.01, Table 3). Metabolic pathways (hsa01100, p = 8.71E-12), cell cycle (hsa04110, p = 1.48E-10), chemical carcinogenesis (hsa05204, p = 5.45E-09), retinol metabolism (hsa00830, p = 1.18E-08), and fatty acid degradation (hsa00071, p = 1.36E-08) were the most significant enrichment pathways. With visualization by Cytoscape software, we demonstrated the relationship between the enriched KEGG pathways and the aggregated DEGs (Fig. 4A). We found that the down-regulated DEGs enriched in the most KEGG pathways, and mainly focused on metabolic pathways (hsa01100), chemical carcinogenesis (hsa05204), metabolism of xenobiotics by cytochrome P450 (hsa00980), retinol metabolism (hsa00830), and drug metabolism - cytochrome P450 (hsa00982). The up-regulated DEGs mainly concentrated on cell cycle (hsa04110), oocyte meiosis (hsa04114), p53 signaling pathway (hsa04115), and progesterone-mediated oocyte maturation (hsa04914). CYP3A4, CYP1A2, ADH1C, ADH6, and CYP2E1 were the genes participated in the most KEGG pathways and they were all down-regulated genes in HCC.
Table 3. The KEGG pathways of the DEGs aggregated in GSE45267, GSE60502 and GSE74656.
| S. No. | Term | ID | Input Number | p | ||
|---|---|---|---|---|---|---|
| 1 | Metabolic pathways | hsa01100 | 38 | 8.71E-12 | ||
| 2 | Cell cycle | hsa04110 | 13 | 1.48E-10 | ||
| 3 | Chemical carcinogenesis | hsa05204 | 10 | 5.45E-09 | ||
| 4 | Retinol metabolism | hsa00830 | 9 | 1.18E-08 | ||
| 5 | Fatty acid degradation | hsa00071 | 8 | 1.36E-08 | ||
| 6 | Metabolism of xenobiotics by cytochrome P450 | hsa00980 | 9 | 2.65E-08 | ||
| 7 | Drug metabolism - cytochrome P450 | hsa00982 | 8 | 2.46E-07 | ||
| 8 | Tyrosine metabolism | hsa00350 | 6 | 1.13E-06 | ||
| 9 | p53 signaling pathway | hsa04115 | 7 | 3.51E-06 | ||
| 10 | Mineral absorption | hsa04978 | 6 | 9.06E-06 | ||
| 11 | Progesterone-mediated oocyte maturation | hsa04914 | 7 | 2.80E-05 | ||
| 12 | Caffeine metabolism | hsa00232 | 3 | 3.15E-05 | ||
| 13 | Glycolysis / Gluconeogenesis | hsa00010 | 6 | 3.43E-05 | ||
| 14 | Tryptophan metabolism | hsa00380 | 5 | 3.66E-05 | ||
| 15 | Oocyte meiosis | hsa04114 | 7 | 0.0001 | ||
| 16 | Linoleic acid metabolism | hsa00591 | 4 | 0.000182 | ||
| 17 | Steroid hormone biosynthesis | hsa00140 | 5 | 0.000216 | ||
| 18 | Arachidonic acid metabolism | hsa00590 | 5 | 0.000286 | ||
| 19 | Prion diseases | hsa05020 | 4 | 0.000313 | ||
| 20 | Bile secretion | hsa04976 | 5 | 0.000448 | ||
| 21 | PPAR signaling pathway | hsa03320 | 5 | 0.000505 | ||
| 22 | Primary bile acid biosynthesis | hsa00120 | 3 | 0.000594 | ||
| 23 | Complement and coagulation cascades | hsa04610 | 5 | 0.00071 | ||
| 24 | Drug metabolism - other enzymes | hsa00983 | 4 | 0.000815 | ||
| 25 | Melanoma | hsa05218 | 4 | 0.004018 | ||
| 26 | AMPK signaling pathway | hsa04152 | 5 | 0.004832 | ||
| 27 | FoxO signaling pathway | hsa04068 | 5 | 0.006584 | ||
| 28 | HTLV-I infection | hsa05166 | 7 | 0.007323 | ||
| 29 | Valine, leucine and isoleucine degradation | hsa00280 | 3 | 0.008979 | ||
Fig. (4).
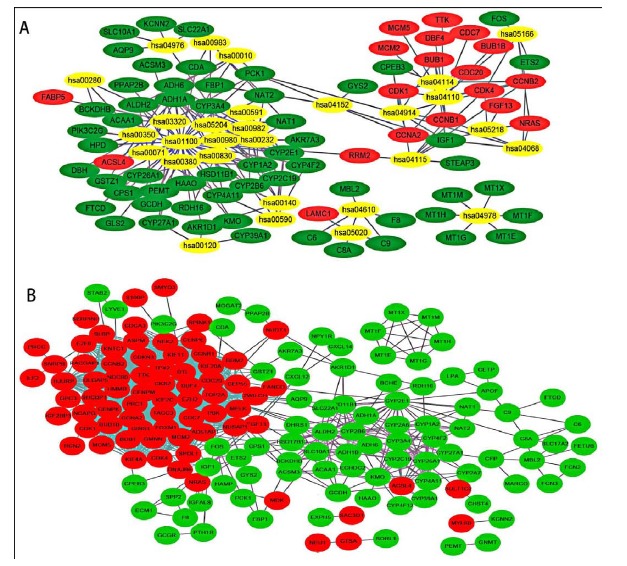
The relationship between the enriched KEGG pathways and the DEGs aggregated in GSE45267, GSE60502 and GSE74656 (A) and the protein-protein interaction (PPI) network of the these DEGs (B). Yellow (gray) represents KEGG pathways ID; Red (deep color) nodes indicate up-regulated DEGs; Green (light dark) nodes represent down-regulated DEGs; The lines between nodes represent the interactions between proteins.
3.4. Protein-protein Interaction Network Construction
STRING website was used to establish the PPI network of all the aggregated DEGs. A total of 161 proteins had been identified to interact with other proteins. Of the 161 proteins, 87 were up-regulated and 74 were down-regulated. We observed that there existed close connection between the up-regulated proteins (Fig. 4B). We considered the genes of the protein with degree ≥ 50 as hub genes. TOP2A, NDC80, CDK1, CCNB1, KIF11, BUB1, CCNB2, CCNA2, and TTK were selected as hub genes with degrees of 65, 54, 53, 52, 51, 50, 50, 50, and 50, respectively. They were all up-regulated genes.
3.5. GSEA Analysis
GSE45267 was chosen as a validation dataset to verify the biological process associated with HCC. By comparing HCC with control, we found that the mainly enriched gene sets associated with HCC were mitotic spindle (NOM p-val = 0.002, FDR = 0.214, Fig. 5A), G2M checkpoint (NOM p-val = 0.004, FDR = 0.125, Fig. 5B), E2F targets (NOM p-val = 0.004, FDR = 0.070, Fig. 5C), spermatogenesis (NOM p-val = 0.020, FDR = 0.097, Fig. 5D) and DNA repair (NOM p-val = 0.032, FDR = 0.151, Fig. 5E). The top ten core enriched genes in each enriched gene set were presented in Table 4.
Fig. (5).
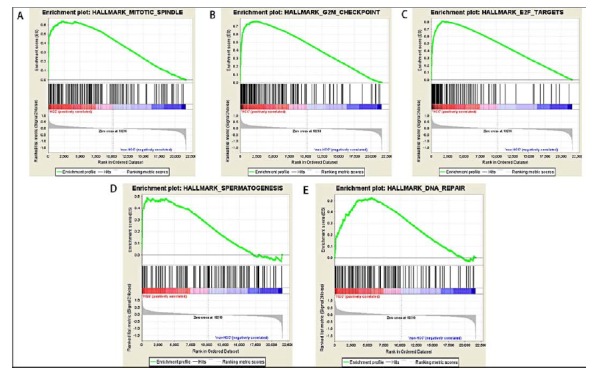
HCC enrichment gene sets by GSEA database. (A) Mitotic spindle; (B) G2M checkpoint; (C) E2F targets; (D) Spermatogenesis; (E) DNA repair.
Table 4. The HCC enrichment gene sets in GSE45267 by GSEA database.
| Enrichment Gene Set | NES |
NOM
p-val |
FDR | Top Ten Core Enriched Genes |
|---|---|---|---|---|
| MITOTIC_SPINDLE | 1.611 | 0.002 | 0.214 | TTK, NDC80, RACGAP1, KIF4A, PRC1, TOP2A, ANLN, DLGAP5, KIF11, NUSAP1 |
| G2M_CHECKPOINT | 1.553 | 0.004 | 0.125 | TTK, NDC80, CDKN3, RACGAP1, KIF4A, PRC1, TOP2A, PBK, CDC20, HMMR |
| E2F_TARGETS | 1.540 | 0.004 | 0.070 | GINS1, CDKN3, RACGAP1, KIF4A, TOP2A, BUB1B, CDC20, HMMR, EZH2, DLGAP5 |
| SPERMATOGENESIS | 1.550 | 0.020 | 0.097 | TTK, CDKN3, EZH2, NEK2, AURKA, CDK1, RFC4, CLGN, KIF2C, RPL39L |
| DNA_REPAIR | 1.578 | 0.032 | 0.151 | ZWINT, RFC4, PRIM1, SAC3D1, FEN1, POLA1, TYMS, RFC5, PCNA, POLR3C |
Abbreviations: NES, Enrichment Score; NOM p-val, Normal p value; FDR, False Discovery Rate Value.
3.6. Overall Survival Analysis
A total of 373 HCC patients’ clinical information data were downloaded from TCGA database. The high expression group (n = 187) and low expression group (n = 186) of each hub gene were obtained based on the median mRNA expression value of each hub gene in all HCC samples. The median mRNA expression values of the hub genes were 1290.13 (TOP2A), 286.75 (NDC80), 529.68 (CDK1), 739.87 (CCNB1), 285.39 (KIF11), 295.75 (BUB1), 341.56 (CCNB2), 370.13 (CCNA2) and 154.84 (TTK), respectively. Those with the values of mRNA expression greater than or equal to the median value were included in high expression group, and those with the values of mRNA expression less than the median value were included in low expression group. Subsequently, the overall survival analyses of HCC patients with different mRNA expression levels of the hub genes we selected were performed. The results were shown in Fig. (6). The p values of the survival curves of the nine hub genes were TOP2A, 0.0042 (Fig. 6A), NDC80, 0.0012 (Fig. 6B), CDK1, 0.0012 (Fig. 6C), CCNB1, 0.00024 (Fig. 6D), KIF11, 0.00044, (Fig. 6E), BUB1, 0.00015 (Fig. 6F), CCNB2, 0.049 (Fig. 6G), CCNA2, 0.0065 (Fig. 6H) and TTK, 0.0016 (Fig. 6I), respectively. Among these genes, BUB1, CCNB1 and KIF11 were most significant.
Fig. (6).
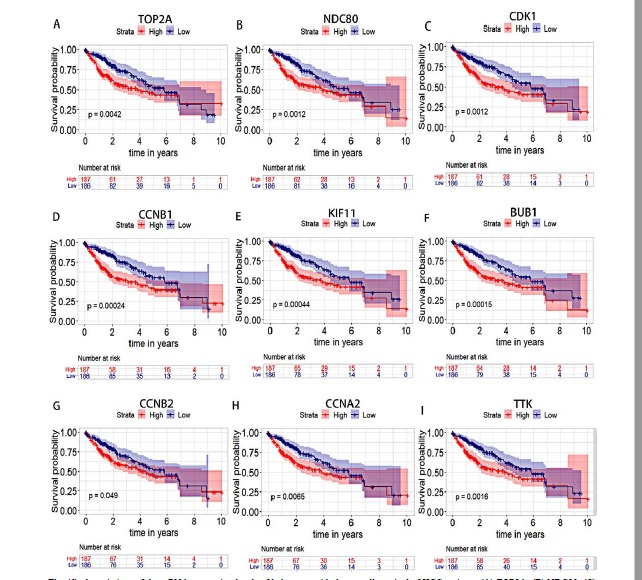
Associations of the mRNA expression levels of hub genes with the overall survival of HCC patients. (A) TOP2A; (B) NDC80; (C) CDK1; (D) CCNB1; (E) KIF11; (F) BUB1; (G) CCNB2; (H) CCNA2; (I) TTK. The area around each survival curve represents the confidence interval of the survival curve.
4. DISCUSSION
The alteration of gene mRNA expression and gene mutation play important roles in the occurrence and progression of HCC. Clarification the changes of genes in HCC and the underlying mechanisms of action will provide a basis for the biomarkers of HCC diagnosis and prognosis and the molecular targets of anti-tumor therapy of HCC. Gene chip detection provides a new method for screening specific genes of HCC, and for investigating the pathogenesis and treatment of HCC. This study screened genes with significant differences in mRNA expression levels between HCC and normal tissues by examining three chip datasets of GEO database, and clarified their specific biological mechanisms in HCC by analyzing multiple datasets of HCC.
In this study, we first got the DEGs of each dataset we selected, and then, to avoid the differences of each dataset in measurement platforms and laboratory conditions, we used the “RRA” package to get the DEGs aggregated in the datasets (aggregated DEGs). After obtaining the aggregated DEGs, we analyzed the biological processes which these genes participate in. The results showed that there were 109 GO terms significantly associated with these DEGs. The top five BP terms with the most significant statistical difference included cell cycle phase, mitotic cell cycle, cell division, nuclear division and mitosis. The top five MF terms with the most significant statistical difference consisted of electron carrier activity, oxygen binding, heme binding, cadmium ion binding and tetrapyrrole binding. The top five CC terms with the most significant statistical difference included spindle, microsome, vesicular fraction, condensed chromosome kinetochore and condensed chromosome, centromeric region. We observed that all these GO terms play important roles in maintaining the normal growth and metabolism of the organism. Through the KOBAS website, we got 29 KEGG pathways with statistically significant differences. The 10 pathways with the most obvious statistical differences included metabolic pathways, cell cycle, chemical carcinogenesis, retinol metabolism, fatty acid degradation, metabolism of xenobiotics by cytochrome P450, drug metabolism - cytochrome P450, tyrosine metabolism, p53 signaling pathway and mineral absorption. Through STRING database, we constructed the PPI network of the aggregated DEGs, and found that TOP2A, NDC80, CDK1, CCNB1, KIF11, BUB1, CCNB2, CCNA2 and TTK were the hub genes of the network. All the hub genes were up-regulated genes. To further verify that the aggregated DEGs were involved in important biological processes in HCC pathogenesis, we chose GSE45267 as a validation dataset to conduct GSEA analysis. The results showed that mitotic spindle, G2M checkpoint, E2F targets, spermatogenesis, and DNA repair were the most significantly enriched gene sets potentially associated with HCC. These findings, together with the results of GO and KEGG analysis, confirmed that the development of HCC implicated important biological processes. The core genes of each enrichment gene set in GSEA were obtained. Some important core genes, such as TTK, NDC80, TOP2A, KIF11, and CDK1, also belong to the hub genes selected by PPI network, further confirming the key roles of the hub genes we selected from the three datasets. Moreover, survival analyses showed that the expressions of all the hub genes (TOP2A, NDC80, CDK1, CCNB1, KIF11, BUB1, CCNB2, CCNA2, and TTK) were associated with the prognosis of HCC patients.
TOP2A (DNA Topoisomerase II Alpha) plays a role in DNA transcription. The mRNA expression of TOP2A was abnormal in many tumors, mainly breast cancer [19] and malignant peripheral nerve sheath tumors [20]. TOP2A mRNA expression was shown to be distinct between HCC tumors and adjacent non-tumoral liver, and microarray analysis of 172 cases of HCC tissues found that the increased mRNA expression of TOP2A was related to advanced histological grading, microvascular invasion, an early age onset of the malignancy and chemoresistance [21]. In this study, we demonstrated that TOP2A had the highest degree in the PPI network although TOP2A was not enriched in the main GO terms and KEGG pathways. NDC80 (Nuclear division cycle 80), also called Hec1, is a newly discovered gene that plays a role in tumorigenesis. Studies have confirmed its association with HCC progression, mainly by reducing apoptosis and cell cycle arrest at S-phase [22, 23]. Our analysis also showed that NDC80 was highly expressed in HCC, ranked second in all hub genes, and participated in many GO terms. CDK1 (Cyclin dependent kinase 1) was the third hub gene in our study, and was also enriched in multiple GO terms and KEGG pathways. Previous study has demonstrated that cell cycle was the main biological pathway of CDK1 participation [24]. CDK1 was found to play a particularly important role in KRAS mutant tumours [25], and be an important partner in the regulation of apoptin-induced apoptosis [26]. In the development of HCC, CDK1 was also found to be closely related to miR-378 [27] and miR-582-5p [28]. CCNB1 (Cyclin B1), CCNB2 (Cyclin B2), and CCNA2 (Cyclin A2) were also hub genes in the PPI network and their degrees were 52, 50, and 50, respectively. They are all members of the cyclin family. Cyclin expression and degradation play an important role in cell mitosis [29], mainly by regulating CDK kinase [30]. All the three genes, CCNB1 [31], CCNB2 [32] and CCNA2 [33], have been shown to play roles in HCC. The mechanism of action of cyclin family in HCC deserves further exploration. KIF11 (Kinesin family member 11) has also been shown to play a role in the pathogenesis of cancer [34], but few studies have been done on HCC. BUB1 [35] and TTK [36] were proven to be elevated in HCC tissues, and may promote the progression and metastasis of HCC, but relevant studies are still scarce. Overall survival analysis of the hub genes in our study confirmed that their expressions were all associated with the prognosis of HCC patients, and the most significant of them were the expressions of BUB1, CCNB1 and KIF11. All of these results provide clues and directions for the further study of these genes in HCC.
The present study chose GSE45267, GSE60502, and GSE74656 datasets for performing analyses. The numbers of samples between the three datasets were different. However, the boxplots of the three datasets indicated high quality of the data. In the screening of DEGs in each dataset, adj_p val < 0.01 and logFC > 0 were considered to be significant. The value of logFC was set at a low level so that genes with mRNA expression of small differences could also be included in our study. This may avoid the overlook of some genes with important biological role but with no significant difference in expression. “RRA” package was used to identify the genes which had different mRNA expression between HCC tissues and normal liver tissues in the three datasets. The “aggregated DEGs” were proposed, because the DEGs identified by “RRA” method were different from previous “overlapping DEGs” and “aggregated DEGs” can be a better indication of the meaning of the DEGs we screened. “RRA” is a method of using probability model to aggregate sorting list [11, 37], which can make a proper aggregation of gene sets from different microarray platforms, and the results we get finally represent the ranking after aggregating analysis but not the ranking in each dataset.
In this study, a total of 221 aggregated DEGs were obtained. Among the top 20 up- and down-regulated aggregated DEGs (Table 1), 5 hub genes (CDK1, CCNB1, KIF11, BUB1 and CCNB2) obtained from STRING did not list at the top of the aggregated DEGs ranking. The hub genes encode key proteins which have stronger interaction with other proteins in our queried gene list but may not necessarily exhibit significant difference in mRNA expression between tumor and normal tissues. In GO term and KEGG pathway analyses, TOP2A which was the top 1 hub gene with the highest interaction score in our study did not enrich in the most significant GO terms and KEGG pathways, and the other hub genes also enriched in a few KEGG pathways, but the GSEA analyses confirmed the involvement of TOP2A and other hub genes in important pathways. This inconsistency may be caused by the different principles between the GO/KEGG enrichment analysis and GSEA analysis. GO/KEGG enrichment analysis needs to set a threshold before analyzing, and only focuses on the genes with obvious expression differences. It is possible to omit some genes that have important biological functions but are not significantly different in expression. Regulatory networks between genes may be overlooked. However, GSEA analysis concerns about gene set, emphasizes identifying synergistic gene sets from all target genes, and can include the genes that were left out by GO/KEGG enrichment analysis. In the present study, p < 0.01 was set as the threshold of KEGG enrichment analysis, and this could lead to the overlook of some minimally significant biological pathways that the hub genes involved in. This might also be the reason why some hub genes we selected, such as TOP2A, did not enrich in the most significant KEGG pathways, but enriched in the results of GSEA analyses. From the findings of this study, we speculated that there could be important regulatory networks between TOP2A and other proteins which have potentially important roles in HCC. Actually, our GSEA analysis showed that TOP2A was among the mainly enriched gene sets associated with HCC including mitotic spindle, G2M checkpoint and E2F targets. Survival analysis showed that the expression levels of TOP2A were associated with the overall survival of HCC patients. Previous studies have also shown the overexpression of TOP2A in HCC [21, 38]. Therefore, TOP2A may play an important role in HCC by interaction with other proteins although further study will be needed to confirm this hypothesis.
In this study, we chose the most recent datasets for analysis. Boxplots of the datasets suggested high quality of the data. Data analysis was performed using the RRA method, which is considered to be highly appropriate for analyzing datasets from multiple databases [11, 37]. We also validated the findings using a validation dataset. All of these may reflex the reliability and comprehensiveness of the hub genes identified. Among the hub genes identified in the present study, TOP2A [39-45], CDK1 [39, 42, 43, 45-48], CCNB1 [41, 43, 45, 46], BUB1 [39-41, 46, 49], CCNB2 [40, 41, 43, 44], CCNA2 [39, 43], and TTK [45] have been reported to be hub genes that participate in HCC in previous studies. The present study confirmed the core roles of these genes in HCC. However, NDC80 and KIF11 have not been described as hub genes involved in HCC in previous studies. Therefore, the findings of the present study enriched the information of hub genes associated with HCC.
The present study was based on bioinformatics methods, some studies have confirmed the roles of certain hub genes mined in HCC but the specific mechanisms of the involvement of these hub genes in HCC remain mostly unexplored. Therefore, laboratory and clinical studies will be required to verify the reliability of our results and find out the genes most closely related to the pathogenesis of HCC in order to provide new and precise information for the prevention and control of HCC.
CONCLUSION
Our study found some DEGs and explored the main GO terms and KEGG pathways involved in HCC. We selected nine hub genes (TOP2A, NDC80, CDK1, CCNB1, KIF11, BUB1, CCNB2, CCNA2, and TTK) and confirmed that their mRNA expressions were all related to the prognosis of HCC. These findings may shed light on future investigations pertaining to the diagnosis and therapy of HCC.
ACKNOWLEDGEMENTS
Declared none.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Not applicable.
HUMAN AND ANIMAL RIGHTS
No Animals/Humans were used for studies that are the basis of this research.
CONSENT FOR PUBLICATION
Not applicable.
AVAILABILITY OF DATA AND MATERIALS
The authors confirm that the data supporting the results and findings of this study are available within the article.
FUNDING
None.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
SUPPLEMENTARY MATERIAL
Supplementary material is available on the publisher’s web site along with the published article.
REFERENCES
- 1.Forner A., Reig M., Bruix J. Hepatocellular carcinoma. Lancet. 2018;391(10127):1301–1314. doi: 10.1016/S0140-6736(18)30010-2. [DOI] [PubMed] [Google Scholar]
- 2.Xie M., Yang Z., Liu Y., Zheng M. The role of HBV-induced autophagy in HBV replication and HBV related-HCC. Life Sci. 2018;205:107–112. doi: 10.1016/j.lfs.2018.04.051. [DOI] [PubMed] [Google Scholar]
- 3.Kunnathuparambil S.G., Anoobjohn K., Zameer P.K.M., Sreesh S., Narayan P., Vinayakumar K.R. Is alcohol abstinence a risk factor for development of hepatocellular carcinoma (HCC) in alcohol related cirrhosis? J. Clin. Exp. Hepatol. 2013;3(1):S104–S104. doi: 10.1016/j.jceh.2013.03.176. [DOI] [Google Scholar]
- 4.Kühn T., Nonnenmacher T., Sookthai D., Schübel R., Quintana Pacheco D.A., von Stackelberg O., Graf M.E., Johnson T., Schlett C.L., Kirsten R., Ulrich C.M., Kaaks R., Kauczor H.U., Nattenmüller J. Anthropometric and blood parameters for the prediction of NAFLD among overweight and obese adults. BMC Gastroenterol. 2018;18(1):113. doi: 10.1186/s12876-018-0840-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Deng H., Eckel S.P., Liu L., Lurmann F.W., Cockburn M.G., Gilliland F.D. Particulate matter air pollution and liver cancer survival. Int. J. Cancer. 2017;141(4):744–749. doi: 10.1002/ijc.30779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gentleman R.C., Carey V.J., Bates D.M., Bolstad B., Dettling M., Dudoit S., Ellis B., Gautier L., Ge Y., Gentry J., Hornik K., Hothorn T., Huber W., Iacus S., Irizarry R., Leisch F., Li C., Maechler M., Rossini A.J., Sawitzki G., Smith C., Smyth G., Tierney L., Yang J.Y., Zhang J. Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol. 2004;5(10):R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barrett T., Troup D.B., Wilhite S.E., Ledoux P., Evangelista C., Kim I.F., Tomashevsky M., Marshall K.A., Phillippy K.H., Sherman P.M., Muertter R.N., Holko M., Ayanbule O., Yefanov A., Soboleva A. NCBI GEO: Archive for functional genomics data sets--10 years on. Nucleic Acids Res. 2011;39(Database issue):D1005–D1010. doi: 10.1093/nar/gkq1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen C.L., Tsai Y.S., Huang Y.H., Liang Y.J., Sun Y.Y., Su C.W., Chau G.Y., Yeh Y.C., Chang Y.S., Hu J.T., Wu J.C. Lymphoid enhancer factor 1 contributes to hepatocellular carcinoma progression through transcriptional regulation of epithelial-mesenchymal transition regulators and stemness genes. Hepatol Commun. 2018;2(11):1392–1407. doi: 10.1002/hep4.1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang Y.H., Cheng T.Y., Chen T.Y., Chang K.M., Chuang V.P., Kao K.J. Plasmalemmal Vesicle Associated Protein (PLVAP) as a therapeutic target for treatment of hepatocellular carcinoma. BMC Cancer. 2014;14:815. doi: 10.1186/1471-2407-14-815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kolde R., Laur S., Adler P., Vilo J. Robust rank aggregation for gene list integration and meta-analysis. Bioinformatics. 2012;28(4):573–580. doi: 10.1093/bioinformatics/btr709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang W., Sherman B.T., Lempicki R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 13.Ai C., Kong L. CGPS: A machine learning-based approach integrating multiple gene set analysis tools for better prioritization of biologically relevant pathways. J. Genet. Genomics. 2018;45(9):489–504. doi: 10.1016/j.jgg.2018.08.002. [DOI] [PubMed] [Google Scholar]
- 14.Szklarczyk D., Morris J.H., Cook H., Kuhn M., Wyder S., Simonovic M., Santos A., Doncheva N.T., Roth A., Bork P., Jensen L.J., von Mering C. The STRING database in 2017: Quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017;45(D1):D362–D368. doi: 10.1093/nar/gkw937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102(43):15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grossman R.L., Heath A.P., Ferretti V., Varmus H.E., Lowy D.R., Kibbe W.A., Staudt L.M. Toward a shared vision for cancer genomic data. N. Engl. J. Med. 2016;375(12):1109–1112. doi: 10.1056/NEJMp1607591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brown C. hash: Full Feature Implementation of Hash/Associated Arrays/Dictionaries. Available from: https://CRAN.R-project.org/package=hash [Accessed on: June 27, 2019]
- 18.Kassambara A., Kosinski M., Biecek P., Fabian S. Survminer: Drawing survival curves using ‘ggplot2’R package version 0.4.4. Available from: https://CRAN.R-project.org/package=survminer [Accessed on: June 27, 2019]
- 19.Slamon D.J., Press M.F. Alterations in the TOP2A and HER2 genes: Association with adjuvant anthracycline sensitivity in human breast cancers. J. Natl. Cancer Inst. 2009;101(9):615–618. doi: 10.1093/jnci/djp092. [DOI] [PubMed] [Google Scholar]
- 20.Amirnasr A., Verdijk R.M., van Kuijk P.F., Taal W., Sleijfer S., Wiemer E.A.C. Expression and inhibition of BRD4, EZH2 and TOP2A in neurofibromas and malignant peripheral nerve sheath tumors. PLoS One. 2017;12(8):e0183155. doi: 10.1371/journal.pone.0183155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wong N., Yeo W., Wong W.L., Wong N.L., Chan K.Y., Mo F.K., Koh J., Chan S.L., Chan A.T., Lai P.B., Ching A.K., Tong J.H., Ng H.K., Johnson P.J., To K.F. TOP2A overexpression in hepatocellular carcinoma correlates with early age onset, shorter patients survival and chemoresistance. Int. J. Cancer. 2009;124(3):644–652. doi: 10.1002/ijc.23968. [DOI] [PubMed] [Google Scholar]
- 22.Ju L.L., Chen L., Li J.H., Wang Y.F., Lu R.J., Bian Z.L., Shao J.G. Effect of NDC80 in human hepatocellular carcinoma. World J. Gastroenterol. 2017;23(20):3675–3683. doi: 10.3748/wjg.v23.i20.3675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu B., Yao Z., Hu K., Huang H., Xu S., Wang Q., Yang Y., Ren J. ShRNA-mediated silencing of the Ndc80 gene suppress cell proliferation and affected hepatitis B virus-related hepatocellular carcinoma. Clin. Res. Hepatol. Gastroenterol. 2016;40(3):297–303. doi: 10.1016/j.clinre.2015.08.002. [DOI] [PubMed] [Google Scholar]
- 24.Santamaría D., Barrière C., Cerqueira A., Hunt S., Tardy C., Newton K., Cáceres J.F., Dubus P., Malumbres M., Barbacid M. CDK1 is sufficient to drive the mammalian cell cycle. Nature. 2007;448(7155):811–815. doi: 10.1038/nature06046. [DOI] [PubMed] [Google Scholar]
- 25.Costa-Cabral S., Brough R., Konde A., Aarts M., Campbell J., Marinari E., Riffell J., Bardelli A., Torrance C., Lord C.J., Ashworth A. CDK1 is a synthetic lethal target for KRAS mutant tumours. PLoS One. 2016;11(2):e0149099. doi: 10.1371/journal.pone.0149099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhao J., Han S.X., Ma J.L., Ying X., Liu P., Li J., Wang L., Zhang Y., Ma J., Zhang L., Zhu Q. The role of CDK1 in apoptin-induced apoptosis in hepatocellular carcinoma cells. Oncol. Rep. 2013;30(1):253–259. doi: 10.3892/or.2013.2426. [DOI] [PubMed] [Google Scholar]
- 27.Zhou J., Han S., Qian W., Gu Y., Li X., Yang K. Metformin induces miR-378 to downregulate the CDK1, leading to suppression of cell proliferation in hepatocellular carcinoma. OncoTargets Ther. 2018;11:4451–4459. doi: 10.2147/OTT.S167614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang Y., Huang W., Ran Y., Xiong Y., Zhong Z., Fan X., Wang Z., Ye Q. miR-582-5p inhibits proliferation of hepatocellular carcinoma by targeting CDK1 and AKT3. Tumour Biol. 2015;36(11):8309–8316. doi: 10.1007/s13277-015-3582-0. [DOI] [PubMed] [Google Scholar]
- 29.Glotzer M., Murray A.W., Kirschner M.W. Cyclin is degraded by the ubiquitin pathway. Nature. 1991;349(6305):132–138. doi: 10.1038/349132a0. [DOI] [PubMed] [Google Scholar]
- 30.Sherr C.J., Roberts J.M. Living with or without cyclins and cyclin-dependent kinases. Genes Dev. 2004;18(22):2699–2711. doi: 10.1101/gad.1256504. [DOI] [PubMed] [Google Scholar]
- 31.Chai N., Xie H.H., Yin J.P., Sa K.D., Guo Y., Wang M., Liu J., Zhang X.F., Zhang X., Yin H., Nie Y.Z., Wu K.C., Yang A.G., Zhang R. FOXM1 promotes proliferation in human hepatocellular carcinoma cells by transcriptional activation of CCNB1. Biochem. Biophys. Res. Commun. 2018;500(4):924–929. doi: 10.1016/j.bbrc.2018.04.201. [DOI] [PubMed] [Google Scholar]
- 32.Gao C.L., Wang G.W., Yang G.Q., Yang H., Zhuang L. Karyopherin subunit-α 2 expression accelerates cell cycle progression by upregulating CCNB2 and CDK1 in hepatocellular carcinoma. Oncol. Lett. 2018;15(3):2815–2820. doi: 10.3892/ol.2017.7691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang F., Gong J., Wang G., Chen P., Yang L., Wang Z. Waltonitone inhibits proliferation of hepatoma cells and tumorigenesis via FXR-miR-22-CCNA2 signaling pathway. Oncotarget. 2016;7(46):75165–75175. doi: 10.18632/oncotarget.12614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Asbaghi Y., Thompson L.L., Lichtensztejn Z., McManus K.J. KIF11 silencing and inhibition induces chromosome instability that may contribute to cancer. Genes Chromosomes Cancer. 2017;56(9):668–680. doi: 10.1002/gcc.22471. [DOI] [PubMed] [Google Scholar]
- 35.Xu B., Xu T., Liu H., Min Q., Wang S., Song Q. MiR-490-5p suppresses cell proliferation and invasion by targeting BUB1 in hepatocellular carcinoma cells. Pharmacology. 2017;100(5-6):269–282. doi: 10.1159/000477667. [DOI] [PubMed] [Google Scholar]
- 36.Liu X., Liao W., Yuan Q., Ou Y., Huang J. TTK activates Akt and promotes proliferation and migration of hepatocellular carcinoma cells. Oncotarget. 2015;6(33):34309–34320. doi: 10.18632/oncotarget.5295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Võsa U., Kolde R., Vilo J., Metspalu A., Annilo T. Comprehensive meta-analysis of microRNA expression using a robust rank aggregation approach. Methods Mol. Biol. 2014;1182:361–373. doi: 10.1007/978-1-4939-1062-5_28. [DOI] [PubMed] [Google Scholar]
- 38.Watanuki A., Ohwada S., Fukusato T., Makita F., Yamada T., Kikuchi A., Morishita Y. Prognostic significance of DNA topoisomerase IIalpha expression in human hepatocellular carcinoma. Anticancer Res. 2002;22(2B):1113–1119. [PubMed] [Google Scholar]
- 39.Zhang L., Huang Y., Ling J., Zhuo W., Yu Z., Shao M., Luo Y., Zhu Y. Screening and function analysis of hub genes and pathways in hepatocellular carcinoma via bioinformatics approaches. Cancer Biomark. 2018;22(3):511–521. doi: 10.3233/CBM-171160. [DOI] [PubMed] [Google Scholar]
- 40.Xing T., Yan T., Zhou Q. Identification of key candidate genes and pathways in hepatocellular carcinoma by integrated bioinformatical analysis. Exp. Ther. Med. 2018;15(6):4932–4942. doi: 10.3892/etm.2018.6075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen Z., Chen J., Huang X., Wu Y., Huang K., Xu W., Xie L., Zhang X., Liu H. Identification of potential key genes for hepatitis B Virus-associated hepatocellular carcinoma by bioinformatics analysis. J. Comput. Biol. 2019;26(5):485–494. doi: 10.1089/cmb.2018.0244. [DOI] [PubMed] [Google Scholar]
- 42.Ni W., Zhang S., Jiang B., Ni R., Xiao M., Lu C., Liu J., Qu L., Ni H., Zhang W., Zhou P. Identification of cancer-related gene network in hepatocellular carcinoma by combined bioinformatic approach and experimental validation. Pathol. Res. Pract. 2019;215(6):152428. doi: 10.1016/j.prp.2019.04.020. [DOI] [PubMed] [Google Scholar]
- 43.Wu M., Liu Z., Li X., Zhang A., Lin D., Li N. Analysis of potential key genes in very early hepatocellular carcinoma. World J. Surg. Oncol. 2019;17(1):77. doi: 10.1186/s12957-019-1616-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li C., Zhou D., Jiang X., Liu M., Tang H., Mei Z. Identifying hepatocellular carcinoma-related hub genes by bioinformatics analysis and CYP2C8 is a potential prognostic biomarker. Gene. 2019;698:9–18. doi: 10.1016/j.gene.2019.02.062. [DOI] [PubMed] [Google Scholar]
- 45.Wu M., Liu Z., Zhang A., Li N. Identification of key genes and pathways in hepatocellular carcinoma: A preliminary bioinformatics analysis. Medicine (Baltimore) 2019;98(5):e14287. doi: 10.1097/MD.0000000000014287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jin B., Wang W., Du G., Huang G.Z., Han L.T., Tang Z.Y., Fan D.G., Li J., Zhang S.Z. Identifying hub genes and dysregulated pathways in hepatocellular carcinoma. Eur. Rev. Med. Pharmacol. Sci. 2015;19(4):592–601. [PubMed] [Google Scholar]
- 47.Zhu Q., Sun Y., Zhou Q., He Q., Qian H. Identification of key genes and pathways by bioinformatics analysis with TCGA RNA sequencing data in hepatocellular carcinoma. Mol. Clin. Oncol. 2018;9(6):597–606. doi: 10.3892/mco.2018.1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang Y., Wang S., Xiao J., Zhou H. Bioinformatics analysis to identify the key genes affecting the progression and prognosis of hepatocellular carcinoma. Biosci. Rep. 2019;39(2):BSR20181845. doi: 10.1042/BSR20181845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li L., Lei Q., Zhang S., Kong L., Qin B. Screening and identification of key biomarkers in hepatocellular carcinoma: Evidence from bioinformatic analysis. Oncol. Rep. 2017;38(5):2607–2618. doi: 10.3892/or.2017.5946. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material is available on the publisher’s web site along with the published article.
Data Availability Statement
The authors confirm that the data supporting the results and findings of this study are available within the article.