

Abstract
Cardiac troponin I (cTnI), the inhibitory-unit, and cardiac troponin T (cTnT), the tropomyosin-binding unit together with the Ca-binding unit (cTnC) of the hetero-trimeric troponin complex signal activation of the sarcomeres of the adult cardiac myocyte. The unique structure and heart myocyte restricted expression of cTnI and cTnT led to their worldwide use as biomarkers for acute myocardial infarction (AMI) beginning more than 30 years ago. Over these years, high sensitivity antibodies (hs-cTnI and hs-cTnT) have been developed. Together with careful determination of history, physical examination, and EKG, determination of serum levels using hs-cTnI and hs-cTnT permits risk stratification of patients presenting in the Emergency Department (ED) with chest pain. With the ability to determine serum levels of these troponins with high sensitivity came the question of whether such measurements may be of diagnostic and prognostic value in conditions beyond AMI. Moreover, the finding of elevated serum troponins in physiological states such as exercise and pathological states where cardiac myocytes may be affected requires understanding of how troponins may be released into the blood and whether such release may be benign. We consider these questions by relating membrane stability to the complex biology of troponin with emphasis on its sensitivity to the chemo-mechanical and micro-environment of the cardiac myocyte. We also consider the role determinations of serum troponins play in the precise phenotyping in personalized and precision medicine approaches to promote cardiac health.
Keywords: Troponin I, Troponin T, Mechano-signaling, Programmed cell death, Post-translational modifications, Precision medicine
Graphical abstract
Highlights
-
•
Serum levels of cardiac TnI and cardiac TnT permit stratification of patients with chest pain.
-
•
Release of troponins into blood involves not only frank necrosis but also programmed necroptosis.
-
•
Genome wide analysis of serum troponin levels in the general population may be prognostic about cardiovascular health.
-
•
Significant levels of serum troponins with exhaustive exercise may not be benign.
-
•
Troponin in serum can lead to important data related to personalized and precision medicine.
1. Introduction
Early recognition of unique domains and restricted expression of the cardiac troponin (cTn) protein complex in the heart led to development of sensitive antibodies for use as serum biomarkers in disorders such as acute myocardial infarction (AMI) and coronary artery disease (CAD) [[1], [2], [3], [4], [5]]. Cummins and colleagues testing cardiac troponin I (cTnI) [2] and Katus et al. [3] testing cardiac troponin T (cTnT) first proposed these proteins as serum biomarkers for myocyte injury. Although there is the general reference to “troponin” (we refer to the complex here as cTn) as a biomarker, it exists as a highly regulated protein complex situated on the sarcomere thin filament and consisting of cTnC, a Ca2+ sensor, cTnI, an inhibitor, and cTnT, a tropomyosin (Tm) binder [[6], [7], [8]]. Capture and detection of serum cTn have improved greatly with the development and use of high sensitivity antibodies (hs-cTnI and hs-cTnT). Although useful in AMI, there remains unsolved issues related to the use of these antibodies in measurements of cTnI and cTnT in stratification of patients with other etiologies and in exercise. A current applicable example is recommendations and value of use hs-TnI antibodies for stratification of patients in patients infected with SARS-CoV2 [9]. Recent studies report a prevalence of myocardial injury as a cause in hospital deaths in these patients [10]. To provide further understanding of possible mechanisms and interpretations of elevations of cTn beyond AMI, we discuss cTn functioning in the cardiac myocyte responding not only to activating Ca2+ ions and chemical stresses, but also to “inside-out signaling” via stresses and mechano-transduction arising from within the myocyte at the Z-disk, M-Band and cytoskeletal network [6,[11], [12], [13]]. Sarcomeres and cTn also sense “outside-in” signals arising from the extracellular matrix and integrin signaling to the cytoskeleton [[14], [15], [16]]. These inside-out stresses may arise from mutations of sarcomeric proteins linked to common cardiomyopathies [17], whereas outside-in stresses may arise from elevations in end diastolic volume as in hypertension and heart failure [18]. Before considering the complex mechanical environment of the sarcomeres as an important determinant of the release of cTn to the serum, we describe current views of sarcomere structure and function followed by the general view of mechanisms of release of cTn in AMI together with a typical clinical experience using hs-TnT to stratify patients with suspected AMI. We then discuss important aspects of the state of cTn released into serum followed by consideration of other mechanisms of release. It is generally held that cTn is a marker mainly for AMI. Yet, we and others think this is a narrow perspective and that understanding of the biology of cTn and performing more research on mechanism of release are important.
Whereas cTnC is common to the heart and slow skeletal (ss) muscle, cTnI and cTnT have unique and divergent stretches of amino acids forming epitopes that have been important in the development of antibodies for specific capture and detection in serum [6]. Beyond this major difference, there are significant variations in domains critical to the function of cTnI and cTnT in the heart. Fig. 1 illustrates and its legend details these significant thin filament functions employing earlier evidence [6,19] and recent electron cryomicroscopy of cardiac thin filaments reported by Yamada et al. [8]. Of additional significance is that together with extracellular matrix proteins, passive tension during diastole is due to the stretch of spring-like elements in cardiac titin, a giant protein extending from the M-band to the Z-disk with extensive interactions with other cytoskeletal elements. An important aspect of diastolic state is the radial position of cross-bridges modifying the local concentration of myosin heads poised for interaction with actins. Moving myosin heads closer to the thin filament occurs with stretch of titin [20], and phosphorylation of myosin light chain 2 (MLC2) [21] and myosin binding protein C (MyBP-C) [22]. It is also important that despite a single regulatory Ca-binding site on cTnC, activation of thin filaments is highly cooperative. The cooperative response is due to interactions among contiguous actins and Tms promoted by feed-back effects of strongly bound cross-bridges [23] and interactions of MyBP-C with the thin filament [24]. These cooperative interactions are critical in maintaining systolic elastance during cardiac ejection [23] and, as will be discussed, important in mechano-signaling from myofilament proteins to the cytoskeletal network responsible in part for membrane integrity.
Fig. 1.

Thin filament molecular signaling in ventricular cardiac myocytes. Top panel. Immune-histochemical staining of a ventricular myocyte carried out as described in Ke et al. [7] of an adult rat cardiomyocyte probed with an anti-p21-activated kinase (Pak1) antibody (labeling shown in green) plus rhodamine-conjugated phalloidin (actin labeling in red with yellow indicating localization of Pak1 at the Z-disk. Middle panel. Cartoon illustrating sarcomere thin filaments, central thick filaments with myosin heads as side arms, and titin molecules running through the center of the thick filaments. Thin filaments and titin attach at the vertical Z-disk sarcomere boundaries. Bottom panel. Functional unit of cardiac sarcomere in diastole and systole based on previous studies reviewed in [6,18,22] and on recent electron cryomicroscopy reported by Yamada et al. [8]. Note the elongated state of cTn stretching across a string of 7 actins. In diastole, with no Ca-bound to a regulatory site on cTnC, Tm is held in a locked position impeding the reaction of cross-bridges with actin. Holding Tm in this position requires an interaction of cTnI inhibitory peptide (Ip) with actin as well cTnI C-terminal regions with Tm. In this state cTnT also interacts with Tm rendering it relatively immobile. With Ca-binding to cTnC there is opening a hydrophobic, sticky patch, promoting an interaction of a switch peptide (Sw) of cTnI with cTnC and releasing the thin filament from the inhibited state and actively pushing Tm. Associated with this release are significant movements of the cTnI mobile domain away from the thin filament and loss of cTnT's hold on Tm. Tm is now in an unlocked state permitting the actin-cross-bridge reaction powered by MgATP. See text for further discussion of thin filament regulatory processes and of titin, myosin light chains (MLC) and myosin binding protein C (MyBP-C) in the diastolic/systolic transition (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
2. cTn release in AMI, and a clinical experience with conventional and hs-cTnT assay in risk stratification for patients with chest pain
2.1. Frank necrosis and release of cTn in AMI
The release of cTn from the myocytes of AMI patients has been commonly attributed to a necrotic injury involving membrane instabilities in the myocyte phospho-lipid bilayer. These instabilities induce membrane blebs or so-called wounds in AMI in which membranes bubble out, burst and release cellular contents. The presence of a rapidly releasable cytoplasmic pools of cTnI and cTnT [25,26] accessible to the serum support this mechanism of transient release in AMI. Studies in the laboratory of Robert Jennings [27] brought attention to translocation of cellular contents via the loss of membrane integrity and formation of blebs in ischemic injury to the heart. They proposed that stress on the membrane is related to cell swelling associated with the hypotonic cytoplasmic environment in hypoxic and anaerobic states during ischemia. The Jennings lab demonstrated using electron-microscopy that the blebs are membrane-enclosed, bubble-like structures extending from the myocyte sarcolemma and contain cellular constituents. A study by Remppis and Katus [28] provided evidence supporting the concept of bleb-related loss of cTnT. They showed that in transient ischemia, the appearance of cTnT in the blood has a significantly shorter half-life than long duration ischemia with possible cellular necrosis. These data fit with the idea that the transient ischemia promotes bleb formation as proposed by Jennings and collaborators. Studies employing synthetic polymers (Poloxamer 188) to block Tn release during endo-vascular occlusion of the LAD in a pig model, provide strong evidence for a prominent role of membrane integrity in ischemic stress [29,30]. Compared to controls treated with polyethylene glycol, intra-coronary infusion of Poloxamer resulted in a significant reduction in TnI in the serum following reperfusion. Although not investigated in the context of AMI, it appears likely that the loss of membrane integrity and formation of blebs is an early event that may be similarly blocked by use membrane stabilizers.
2.2. Clinical experience with conventional and high-sensitivity troponin T assay
Before considering the complexities in the use of cTn as a biomarker, we discuss the successes with their widespread use in emergency medicine. To illustrate the use of serum cTn levels in a typical clinical setting, we present data derived from the experience of Blessing Health System, a regional referral center for western Illinois, northeastern Missouri and southeastern Iowa. The hospital, which has been in operation for 144 years, is a 307-bed acute care facility served by over 300 physicians and other providers. Each year the Blessing Emergency Department is visited by approximately 40,000 patients which includes a population representative of the 6 million patients in the US presenting to emergency departments with chest pain. As with the larger population, only a minority of patients with chest pain presenting to the Blessing Health System have an acute MI.
We focus here on the use of high sensitivity TnT (hs-cTnT) which was first approved for use in the United States by the Federal Drug Administration in January 2017 (Roche Elecsys Troponin T Gen 5 Stat Assay). The use of hs-cTnT is the latest advance in the evolution of biomarkers used in diagnosis of AMI. Early markers included creatine kinase (CK) and lactate dehydrogenase (LDH), both intracellular molecules, followed by identification of more specific CK isoforms that improved specificity. Following early studies by Katus et al. [3] which introduced the use of cTnT as a biomarker, the use of cardiac specific cTn assays in clinical risk stratification came into focus by the mid-1990s. The ability to detect circulating levels of cTnT in greater than or equal to 50% of a healthy population together with performance at high precision (coefficient of variation ≤ 10% at the 99th percentile URL) characterize the hs-cTnT assay as highly sensitive [16]. The Gen 5 hs-cTnT assay has a level of detection of 4 ng/L and a 99th percentile upper limit of normal in healthy individuals of 19 ng/L. In contrast older, conventional cTnT assays have both a higher limit of detection and lower precision. The diagnostic accuracy of hs-cTnT has been extensively analyzed by Reichlin et al. [31] and Neuman et al. [32].
It has been a long-standing challenge in the practice of emergency medicine to tease out the subset of patients presenting with chest pain who have a true AMI from those patients with other sources of chest pain. Patients with AMI should be rapidly identified and receive immediate treatment including hospitalization while patients with benign causes of chest pain should also be rapidly identified but can often be safely discharged home. An essential element in the diagnostic criteria for this decision, apart from a careful history, physical exam and EKG is the detection of a rise and fall in biomarkers of myocardial cell death, generally ascribed to necrosis. The Fourth Universal Definition of Myocardial Infarction differentiates myocardial infarction from injury [33]. Acute myocardial injury is defined by a rise and/or fall of cTn with at least one value above the 99th percentile upper reference limit. Acute myocardial infarction is established when acute myocardial injury is accompanied by evidence of acute myocardial ischemia including at least one of the following: symptoms of myocardial ischemia, new ischemic EKG changes including pathologic Q waves, imaging evidence of new myocardial injury consistent with ischemia and/or identification of a coronary thrombus.
Myocardial infarctions are classified into several different types, depending on the mechanism or events leading to the infarct [33]. A Type 1 myocardial infarction occurs when an atheromatous plaque ruptures acutely causing coronary thrombosis. In contrast, a Type 2 MI results from a mismatch of oxygen demand and supply not related to acute thrombosis. Type 2 MI may occur, for example, in sustained tachyarrhythmia or shock. Note that diagnosis of both Type 1 and Type 2 MI requires the rise and/or fall of cTnT as described above. As the sensitivity of the cTn assay increases, we can anticipate more patients falling into the diagnostic categories of either Type 1 or especially Type 2 MI where, in the past, with less sensitive biomarkers, these infarcts were less easily detected. Stable patterns of cTn elevation are also more readily detected with higher sensitivity assays and reflect chronic myocardial injury due, for example, to structural heart disease or chronic kidney disease.
An additional tool to help risk stratify patients presenting with chest pain is the History, Electrocardiogram, Age, Risk Factors, and Troponin (HEART) score (Mahler et al. [17]) which has been adopted by Blessing Health System as part of an accelerated diagnostic protocol. The HEART score is quickly computed at the bedside and estimates the 30-day risk for major adverse cardiac events (MACE), namely, AMI, percutaneous coronary intervention, coronary bypass grafting, or death. Specifically, the HEART score assigns 0–2 points for each of the following: history (H), EKG interpretation (E), age (A), presence of cardiac risk factors (R) and cTn levels (T). A score of 0–3 indicates low risk for MACE (0.9–1.7%), scoring 4–6 is moderate risk (12–16.6%) and a score of 7–10 indicates high risk for MACE (50–65%).
The following clinical vignette illustrates a typical approach to risk stratification using hs-cTnT and the HEART score as part of an accelerated diagnostic approach: A 43-year-old woman presents to the emergency department with chest pain radiating to the neck and arm. The pain began one hour prior to arrival, is sharp in nature and is worse with deep breaths and movement of her neck and arm. Her past medical history includes hypertension, but she is otherwise healthy. Her EKG is normal. Initial measurement of hs-cTnT is 7 ng/L which is within normal range. Her HEART score is calculated by adding 1 point for history, 1 point for hypertension as a cardiac risk factor, but zero points for age, normal EKG and a normal hs-cTnT. Her total score of 2 places her at low risk for MACE. This low risk is confirmed by measurement of a second, normal level of hs-TnT of 9 ng/L at 3 h. She has ruled out for AMI and, barring any other significant non-cardiac causes of her symptoms, she may be discharged home safely with appropriate outpatient follow up. This approach has been backed by extensive clinical trials [[18], [19], [20], [21], [22]]. Establishing rule out of AMI has also been documented with use of a single hs-cTnT measurement below detection limits of 5 ng/L together with a non-ischemic EKG [23].
The time course of the rise and fall of serum cTn is an important consideration in the diagnosis of AMI and depends on both blood flow and how soon after symptom onset a sample is obtained. This timing is also assay-dependent. In the case of an acute MI, cTn levels will generally rise to abnormally high levels in 3–12 h after symptom onset and then decline at a much slower rate declining over 5–14 days. It is crucial to time sampling such that the chance of detecting significant, dynamic changes in cTn levels is optimized. For patients presenting to an ED, cTn is sampled at time of presentation and an additional sample is obtained several hours later. Current guidelines recommend at least a 3 h interval between samples while emerging data suggest that shorter intervals may be used for hs-cTnT [34]. Very late sampling may require additional, serial measurements to differentiate cnhronic myocardial injury from the slow rate of decline in cTn levels with AMI. Conversely, a single, normal hs-cTnT level sampled greater than 6 h after development of symptoms may be enough to rule out AMI.
After implementation of hs-cTnT and the HEART score, physicians at Blessing Hospital were able to show that a larger percentage of patients who were low risk for MACE could be safely discharged from the ED. Fig. 2 shows the percentage of patients presenting to the ED identified as low risk for MACE who were subsequently discharged using a conventional cTnT assay (blue line) and after implementation of the Roche Gen 5 hs-cTnT assay (yellow line). The data were obtained from retrospective chart analysis and represent a total of 1315 patients (conventional cTnT) and 2902 patients (hs-cTnT) studied 6 months before and 14 months after implementation of hs-cTnT, respectively. The data collection and analysis are in compliance with the Ethical Committee Guidelines of the Blessing Hospital. The two populations were similar in age, gender mix and ethnicity (Fig. 2). The dashed line represents an average monthly value over the time period indicated. Our data indicate that, on average, a larger percentage of low risk patients (54% vs 42%) were managed on an outpatient basis after implementation of the hs-cTnT assay. For low-risk patients discharged from the ED, both the 30-day readmission rate and 30-day all-cause mortality rates were not increased, and, in fact, decreased slightly after implementation of the hs-cTnT assay. The 30-day readmission rate was 0.31% with the hs-cTnT vs 0.46% using the conventional cTnT assay. In addition, the 30-day mortality rate was 0.17% for patients with hs-cTnT and 0.23% using the conventional cTnT assay. These findings support the notion that the increased rate of ED discharge for low-risk patients is both safe and appropriate. For low risk patients admitted to the hospital, implementation of the hs-cTnT assay revealed a slightly increased rate of stress testing (40% vs 34% with conventional cTnT) but a decreased rate of cardiac catheterization (7.4% vs 10.1% with the conventional cTnT). These findings suggest that additional risk stratification of low risk patients admitted to the hospital does not translate into an increased rate of cardiac catheterization. In summary, we conclude that adoption of hs-cTnT coupled with the HEART score has improved our ability to rapidly and effectively differentiate patients with AMI from those who have a more benign condition and, for those patients at low risk for MACE, when appropriate, safely discharge them from the ED avoiding unnecessary testing and costly hospitalization. Other, recent studies have similarly concluded that the use of hs-CnT and a validated risk score improves the ability to safely discharge low-risk patients with chest pain from the ED [35].
Fig. 2.
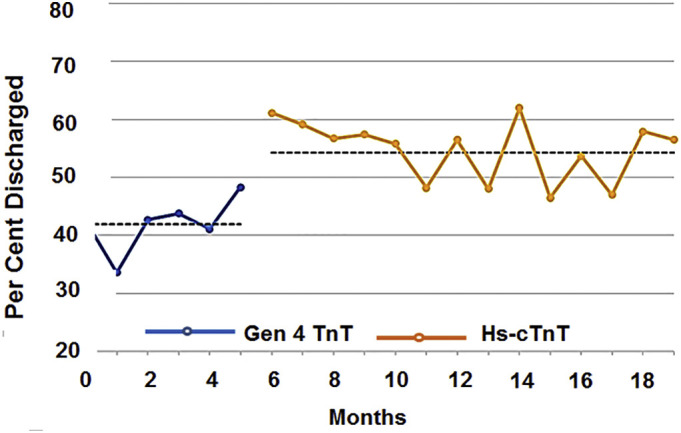
Percent of low-risk patients discharged from the emergency department before and after implemenation of hs-cTnT. Measurement with cTnT (hs-cTnT) were in 1315 (2902) patients, who were age 53 ± 18 years (52 ± 18 years) mean ± SD, 42% (45%) male, 58% (55%) female, 92% (91%) caucasison, 7% (8%) African American, 2% (2%) Hispanic or other. See text for discussion.
3. Domains, epitopes, and post-translational modification in capture and detection of serum cTnI and cTnT
3.1. Domains useful as epitopes for capture and detection of serum cTn
The use of hs-cTnI and hs-TnI is considered the gold standard for determination if patients with chest pain have an AMI. Yet more thorough interpretation of elevations of serum cTn in AMI patients and non-AMI individuals requires discussion of the domains useful in capture and detection as well the post-translation protein modifications Analysis of the primary structures of cTnI and cTnT revealed domain differences between these proteins in the heart and their counterparts in skeletal muscle. The slow skeletal isoform of TnI (ssTnI), which is important in cardiac protection, is expressed in the fetal and neonatal heart and replaced by cTnI [36,37] early in development. A major difference between cardiac and skeletal isoforms is the presence of an N-terminal extension in cTnI consisting of ~30 amino acids with sites of phosphorylation by protein kinase A [5]. Fig. 3 left panels summarize functional domains of cTnI and cTnT with emphasis on unique features employed as epitopes for analysis in serum. The domains are those defined most recently by Yamada et al. [8]. The epitope examples are taken from Collinson et al. [38], who reported an extensive summary of cTnI and cTnT epitopes with links to tables published by the International Society for Clinical Chemistry. In view of their potential influence on immune-capture (C) and detection (D), we also include in the right panels of Fig. 3 a summary of post-translational modifications by phosphorylation and proteolysis of cTnI and cTnT. Epitopes for C and D stretch across the N-terminal unique domain of cTnI Other regions show conservative variations (numbering using accession NP-000354), for example the Roche peptide of the cTnI-190-196C-epitope is DWRKNID corresponding to DWRKNVE in ssTnI-160-166. In Roche antibodies (numbering using accession AAK92231), the peptide of cTnT-125-131C-epitope is DRIEKRR corresponding to ERIEKRR in fsTnT; the D epitope cTnT-136-147 is EQQRIRNEREKE corresponding to fsTnT, EQQRIRAEKERE. As illustrated in Fig. 3 in the case of cTnI, epitopes are within regions in which phosphorylation may affect the epitope, whereas with cTnT antibodies the epitopes are in regions unmodified by protein phosphorylation. Below, we discuss the potential value of determination of phosphorylation of cTnI and cTnT in relation to personalized medicine.
Fig. 3.

Domains and epitopes of cTnI and cTnT employed for capture (C) and detection (D). Left panels: Numbers on the structure of the thin filament correspond to cTnI and cTnT domains and indicate regions recognized by antibodies. Right panels: Sites of phosphorylation and proteolytic cleavage of cTnI and cTnT. Amino acid residue numbers use cTnI accession CAA62301 and cTnT AAK92231. See Fig. 1 and text for further information.
Use of antibodies against adult cTnI epitopes in fetal and newborn patients may be complicated by the expression of ssTnI, which lacks the N-terminal extension and sites of phosphorylation. A substantial expression of ssTnI has been reported in cardiac tissue of 20-day old infants with congenital heart disease [37]. Even so, hs-cTnI has been useful in determination of persistent high levels of serum cTn in predicting postoperative mortality and cardiac injury in infants undergoing cardiac surgery [39]. Never-the-less it would seem prudent to employ hs-cTnT with epitopes present across isoforms as has been done in preterm infants with hemodynamically significant patent ductus [40].
3.2. Proteolysis, interactions among cTn proteins and detection in serum
Proteolysis [[41], [42], [43], [44]] and protein-protein interactions among cTn components [45,46] are important considerations in the development of antibodies for immune-capture and detection in serum. A study by Katrukha et al. [47] considered degradation in providing a rational approach for choice of cTnI epitopes. These investigators employed a large set of antibodies to determine which regions of cTnI may be best to detect both full length cTnI, its fragments, and its complexes with cTnC. They detected full length cTnI, but there was degradation mainly at the N- and C-terminal regions both of which are highly mobile and without protection of protein-protein interactions. cTnI-30-110 demonstrated the most stability. The ratio of proteolytic products remained relatively constant with time, suggesting the degradation occurred in the myocyte rather than blood. After extensive analysis, they concluded that antibodies recognizing epitopes at cTnI-23-40 and/or cTnI-140-196 provide the best chance to avoid diagnostic underestimation due to proteolysis, autoantibodies, or interactions with cTnC. These results are reflected in epitopes employed for capture and detection of serum cTnI listed in Fig. 3. In the case of cTnT, proteolytic degradation has also been considered in epitope selection. It is known that N-terminal cleavage of cTnT by μ-calpain [42,48,49] and caspase [44] occurs with ischemic injury. Streng et al. [49] applying robust liquid chromatography mass spectrometry (MS) to analyze serum from AMI patients identified full length cTnT as well as fragments of cTnT consisting mainly of cTnT-S69-W287 and cTnT-S69-Q189, which was the most abundant. As indicated in Fig. 3, epitopes in current use for capture and detection are in these regions. Published [45,50,51] and future studies that include the application of robust immuno-enrichment together with MS offer more detailed understanding of the state of cTnI and cTnT in serum.
Determination of the presence of complexes of cTn units in serum assays is important in the diagnostic applications. It has been suggested that differences in these complexes may differ in Type 1 and 2 AMI. Van Wijk et al. [46] proposed that the presence of the full cTn complex may indicate severe chronic CAD, whereas a high level of free cTnI or cTnT may be more indicative of AMI. The basis for this proposal is that there is evidence that 5–10% of cTn in viable cells exists as the full complex [25,26] and may be released by bleb formation in conditions associated with CAD without frank necrosis [52]. We will extend discussion of this and other
mechanisms for cTn release below. Several studies have determined that serum cTnI and cTnT exist in complexes with cTnC. Early investigations [53] of the state of immunoreactive serum cTn in a cohort of 10 AMI patients determined that cTnI exists as a complex with cTnC, whereas cTnT was largely free. More recently van Wijk et al. [46] confirmed these studies and reported that other etiologies (demand ischemia and non-type I MI) also demonstrated complexed forms of cTnI and cTnT in serum. The complexes were mainly cTnI-cTnC but there was also the complete cTn complex. Phillips-Electronics Netherlands have employed antibodies to peptide cTnI-41-49 as C-epitope and cTnI-20-100 as D-epitope together with a monoclonal antibody for cTnC [38].
4. The sarcomere cytoskeletal network, mechano-transduction, and release of cTn into serum
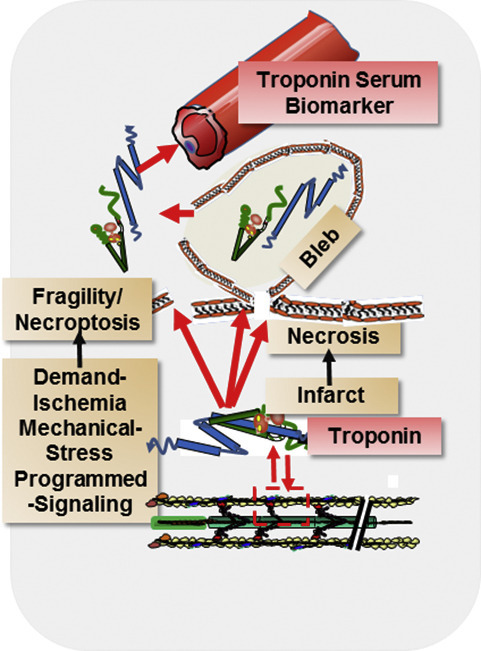
Although it is apparent that the major mechanism of serum appearance of cTn with AMI is the formation of blebs with cell rupture, there is substantial evidence that potential mechanisms of release are varied and complex. Evidence for a role of mechanical forces to and from the sarcomeres of cardiac myocytes provides insights into the complexity of the mechanisms of bleb formation, membrane integrity, and release of cTn. Fig. 4 illustrates these complexities. and their relation to membrane integrity. Illustrated is the microenvironment in which cardiac thin filaments function as part of an extensive mechano-transduction network connecting the functional unit of the sarcomere to stress sensors contiguous with the extracellular matrix (ECM), integrins, the Z-disk proteins, M-band proteins, costamere proteins, and microtubules [11,12,15]. Fig. 4 also shows a blood vessel and a part of the lymphatic vessel network demonstrating the path cTnI and cTnT must take to get from the thin filament or cytoplasmic locations to the serum. As noted in Fig. 4, there is also evidence for transit of cTn components across the nuclear envelope, where they act as epigenetic regulators of gene transcription [48,54], as discussed below. Early work indicated that a mechanism of cardiac injury involves changes in cytoskeleton proteins that promote adverse mechano-transduction and a loss in membrane stability. It follows that prognostic and diagnostic interpretation of changes in serum levels of cTnI and cTnT requires a discussion of mechano-transduction in cardiac myocytes. Mechano-transduction refers to the contribution of physical forces and mechanical state to the induction of biochemical and biophysical processes important in homeostasis and disorders of the myocardium. As discussed below, there is ample direct and indirect evidence connecting cTn release to membrane injury, via maladaptive outside/in and inside/out mechano-transduction in cardiac myocytes.
Fig. 4.

Membrane integrity and the micro-environment of cardiac sarcomeres. Sarcomeres are depicted in association with mechano-sensitive elements in a network of stress sensors including microtubules, Z-disk (with docked signaling molecules), M-band (with a terminal titin kinase), and costamere proteins(vinculin (Vin), paxillin (Pax),focal adhesion kinase (FAK), contiguous with extra-cellular matrix (ECM) -integrin signaling networks (via tensin and tailin). The dystroglycan/sarcoglycan complex is linked to these networks as shown and with dysferlin/caveolin at the t-tubule are major controllers of sarcolemma fragility. Included in the sarcomere micro-environment are mitochondria, and membrane elements (sarcomlemma, t-tubules and sarcoplasmic reticulum) controlling excitation and Ca-fluxes to and from the myofilaments. The illustration depicts the transit of cTn from thin filaments and sarcoplasm to blood and lymphatic capillaries related to AMI-induced membrane bleb formation and rupture and to membrane fragility associated with mechanical stressors. Note that mechnano-sensitive transcription factors at the Z-disk shuttle to and from the nucleus. There is also entry of cTnI and cTnT into the nuclear compartment, where they appear to function in epigenetic regulation that may be maladaptive. See text for discussion.
.
4.1. High levels of serum cTnI and cTnT without frank necrosis
A turning point in the understanding of cTn as a biomarker occurred with the report by Hickman et al. [52] evidence for an association of cTn release with formation of surface blebs that occur without frank cellular necrosis. The data were interpreted to indicate that the blebs could form and then be shed without cellular rupture, and that a possible mechanism involved ischemia related modifications in the mechanics of the cytoskeletal network, calpain, and integrin activation. Early work had suggested this mode of membrane fragility may be associated with the sarcomere/cytoskeletal/ECM network illustrated in Fig. 4. Sage and Jennings [55,56] made the important observation that mechanisms of injury and bleb formation involved the loss of the integrity of Z-band and cytoskeletal attachments, which they proposed contributed to membrane stabilization. Mechanical forces and blebs had been emphasized by the studies of Steenbergen [57], who reported a decrease in membrane stability in cells stressed by swelling. Later work reported by Armstrong et al. [58] showed evidence that spectrin and dystrophin associated proteins were translocated out of cardiac myocytes in concert with bleb formation. They proposed that spectrin and dystrophin associated proteins are important to the homeostasis of membrane integrity, and that their loss resulted in membrane fragility. In our studies [59], providing the first evidence of breakdown products of cTnI from isolated perfused hearts stressed by ischemia/reperfusion, we found a loss of α-actinin, a key element of the Z-disk and connections among cytoskeletal elements in the sarcomere compartment.
4.2. Outside/in stresses and serum cTnI and cTnT
Measurements using hs-cTnI and hs-TnT may provide diagnostic and prognostic insights into common disorders such as hypertension and heart failure with preserved ejection fraction (HFpEF) that promote outside/in stresses. Outside/in stresses likely to be related to cTn release involve a complex, potentially maladaptive, interaction among the increased contraction strain of regions remote from MI, elements of the sub-sarcolemma and transmembrane cytoskeleton, micro-tubules, proteases, and cytokines. A potential result of these interactions is loss of membrane stability, bleb formation and cTn release. Feng et al. [18] demonstrated that elevated end diastolic stress with increased pre-load promotes degradation and release of cTnI into serum and proposed a role for calpain. Work in our laboratory [60] reported an increase Ca-sensitivity of sarcomeres in regions remote from the infarcted zone of a mouse MI model. Evidence indicated an increased myofilament Ca-sensitivity resulting from oxidative stress and altered protein phosphorylation of sarcomeric proteins. Such an increase in myofilament Ca-sensitivity would be expected to increase the stress on membrane blebs and lead to cell death and cTn release from injured tissue as well as tissues stressed by oxygen demand in ischemia.
In their study of the effect of increased preload on cTn release, Feng et al. [18] implicated a role for calpains. The role of calpains is, however, complex with positive and negative effects on membrane stability. Data reported by Taneike et al. [61] explicitly connected calpain to preload-induced membrane injury in cardiac myocytes. Their data showed that pressure overload stress on mouse hearts deficient in calpain 4 induced uptake of membrane impermeant dye into the myocytes. Controls did not demonstrate this effect unless severely stressed. Moreover, induction of membrane injury by laser irradiation resealed in the controls but not in calpain deficient myocytes.
Evidence for an interaction between calpain and integrins, a prominent participant in outside/in mechano-transduction in cardiac myocyte injury, has been presented by Suryakumar et al. [62]. Their study compared the effect of moderate pressure overload by coarctation of the aorta in controls and beta-3-integrin KO mice. KO mice demonstrated an up-regulation of mu-calpain expression and induction of programmed cell death. They focused on apoptosis, but it is likely that necroptosis, which we consider below, was also involved as described in the work from the Kitsis laboratory [63,64]. As illustrated in Fig. 4, integrins are homo-dimeric proteins crossing the cardiac cell membrane interacting specifically with ECM sites at one end and with sites in the intracellular cytoskeleton particularly tailin [16,65]. Cytoplasmic regions of integrins have also been shown to interact with myocyte T-tubules and to modify Ca-release mechanisms [66]. Integrins are positioned to act as transducers of mechanical stresses and as stabilizers of the plasma membrane. Hessel et al. [67] directly tested a role for integrin signaling in release of cTnI by modifying integrin signaling in isolated neonatal rat cardiomyocytes (NRCM) by treatment with a peptide (GRGDS) that promotes integrin function as a stress sensor. Treatment with the peptide induced release of intact cTnI into the medium with no evidence of necrotic damage. They attributed this release to the presence of a releasable cytoplasmic pool leaving the cell by transient increases in membrane permeability or wounding. Follow up studies [68] reported that treatment of the NRCM with GRGDS also promoted uptake of membrane impermeant dyes. These studies with unloaded NRCM in culture are difficult to interpret with respect to the adult cardiomyocyte stressed by AMI or I/R injury. NRCM have poorly developed Ca-flux regulation and express the fetal TnI isoform, ss TnI, which has significant effects on function [36]. Nevertheless, there are data indicating a role for integrins in I/R injury, in which expression of the α7β1D isoform, the most abundant in the cardiac myocyte, is upregulated, whereas expression of the β1isoform is downregulated. Compared to controls, over expression of the α7β1D isoform in the heart protects the heart from I/R injury reducing infarct size [66]. This protection was attributed to the ability of the α7β1D isoform to activate Ca-release mechanism impaired by I/R. Although not studied explicitly, a potential for induction of necroptosis in these models is indicated by data showing a role for TNFα in integrin expression and actions [69].
Increasing evidence (see reviews by Caporizzo et al. [12]; Cooper [70]) emphasizes the significant role of microtubules in homeostatic and pathologic mechanisms as determinants of cardiac function includes their role in membrane stability and potentially in the release of cTn into blood. Dimers of α/β tubulin polymerize to form 25 nm microtubules localized in various distinct regions of the cardiac myocyte including at the plasma membrane. Interactions of the microtubules occur at the costamere with sites of interaction at the dystrophin/dystroglycan complex [71]. There is evidence of both increases and decreases in microtubule density associated with cardiac disorders. In the case of cardiac hypertrophy, there is an increase in microtubule density imposing a resistance to cell shortening [72]. Drum et al. [73] reported that oxidative stress such as that occurring with AMI induced shrinkage of microtubules and decreased the surface expression of K channels in cardiac myocytes. The expression of these channels at the surface membrane is related to membrane stability and protection in I/R injury. It is also highly relevant that, as with other elements in the cytoskeleton involved in membrane stability, proteolysis of microtubules occurs with the activation of calpain that occurs with AMI and with IR injury.
4.3. Inside/out stresses and serum cTnI and cTnT
As stressed in Fig. 4, inside/out stresses arising at the level of the cardiac sarcomere and cytoskeleton provoke multiple short and long-term regulatory processes involving mechano-transduction via extensive interactions with a network of strain sensitive elements. A common alteration with inside/out signaling is adverse mechano-transduction triggered by mutations in the sarcomere and cytoskeletal network that result in common hypertrophic cardiomyopathies (HCM) [74]. In HCM, short term responses to the mutations include increased sarcomere Ca-response, altered Ca-fluxes that cause diastolic dysfunction [75] [76], and a mismatch between energy demand and supply [77]. Longer term mechanisms include an effect of the biophysical signal to strain the Z-disk and promote release of transcription factors that shuttle to the nucleus inducing hypertrophic growth and remodeling [11]. Strain at the M-band induces altered protein quality control [13]. There is also evidence of early onset fibrosis and micro-vascular changes with induction of hypoxic stresses [78] [79].
Taken together this remodeling in HCM indicates that measurements of serum cTns may be a useful marker in the diagnosis and prognosis of HCM. Evidence indicates the presence of elevated cTnT in the serum of HCM mutation carriers exhibiting sub-clinical disorders and no indication of hypertension, angina or MI or abnormal left ventricular fractional shortening or end-diastolic diameter [80]. Cramer et al. [81] reported detection of an elevated hs-cTnT in 74% of HCM patients in a cohort of 62. Of these, 26% had significant increases to the 99th percentile reference limit of 14 ng/L. The strongest correlation to this increase was left ventricular mass normalized to body surface area and maximum wall thickness rather than long term cardiovascular disease risk. It has also been pointed out that elevations in serum cTnI exist in a high proportion of patients with stabilized HCM. To address the relation of the elevated cTnI to the cardiovascular profile and prognosis of these patients, Agarwal et al. [82] performed a retrospective study of 167 stable HCM patients using hs-cTnI with greater 4 ng/L as a threshold for a clinically significant finding. Of the 167 patients meeting the criteria of the study, 34% had positive levels of cTnI. The positive cTnI levels correlated closely with a depression in global longitudinal strain (GLS) as determined with echocardiography. Importantly, Reant et al. [83] concluded from a retrospective study of 472 patients with HCM that abnormal GLS independently predicted poor outcomes. Ho et al. [17] employed echocardiographic strain imaging to determine the effect of HCM linked sarcomere mutations on systolic function. Their studies found that pre-clinical patients, who demonstrated diastolic dysfunction did not demonstrate systolic dysfunction as gauged by strain imaging. On the other hand, measurements on patients with demonstrable pathology showed both diastolic and systolic dysfunction. These findings were also seen in a mouse model of HCM with a αMHC (403) mutation. With age and increasing severity of the disorder, there was a worsening of global strain rate [84]. Potential mechanisms for these elevations in serum cTnI or cTnT in HCM include ischemia associated with micro-vascular changes, energy demand/supply mismatch, increased wall thickness, and dysfunction of the failing cardiac myocyte with defects in membrane stability. We emphasize that these mechanisms involve changes in the stress on the myocytes, inducing strain in the sarcomere/cytoskeletal network leading to programmed necrosis, instabilities in the sarcolemma, and shedding of blebs and possibly exosomes, as discussed above. Moreover, these findings suggest that determinations of cTnI and cTnT for prognosis of HCM should be further investigated and possibly carried out routinely with progression of the myopathy. Changes in global strain and strain rate in hearts of HCM patients carrying mutations in myosin and cardiac myosin-binding protein C, but with no hypertrophy are different from the hypertrophy positive patients described so far. Studies with hypertrophy negative patients demonstrated an enhanced strain rate and twist. A conclusion from these studies was that a mechanical abnormality is likely to trigger early onset fibrosis and hypertrophy. We discuss these finding in the section below on precision medicine.
5. Interpretation of serum cTn levels beyond AMI and in the broad population
The development of highly sensitive assays engendered frustrations, complexities and challenges in understanding the meaning of high levels of serum cTns in apparently healthy individuals and the value of their use in a variety of conditions presumably affecting the viability of cardiac myocytes.
5.1. Serum cTnI and cTnI levels and inotropic interventions
There is evidence of release of cTn into serum associated with interventions including inotropic agents. These effects are particularly evident in patients with CAD suggesting a vulnerability to demand ischemia. Interventions include rapid atrial pacing [85], stress echo measurements, and inotropic interventions [86] all of which may elevate serum cTn levels. In the case of rapid atrial pacing, ischemia without frank infarction was documented in CAD patients [85]. However, in a test of this idea in healthy horses treated with atropine/dobutamine infusion there was a significant elevation in serum cTnI levels [87]. These effects may be related to the energy wasting effects of adrenergic stimulation that mismatch oxygen demand and supply [88].
Results of a trial (COSMIC-HF) treating stable heart failure patients with the inotropic agent (Omecamtiv Mecarbil), which prolongs systolic elastance by specifically altering myosin kinetics, presents a conundrum in relating measures of cardiac performance to serum cTnI levels [89]. Treatment with the myosin activator guided by phamacokinetic analysis in 136 patients improved systolic and diastolic function and lowered heart rate compared to the 133 placebo patients. In 25% of the cohort there were significant elevations in serum cTnI at baseline. There was a significant increase of serum cTnI over baseline values in the Omecamtiv group compared to placebo, but lower NT-proBNP with no difference in adverse events. Patients had no MI, no severe chronic kidney disease or unstable angina and a limited number had atrial fibrillation. We can only speculate that the increase in mechanical stress associated with the prolonged systolic time may have induced a strain promoting membrane fragility in compromised cardiac myocytes. In contrast, using a hs-cTnT antibody, Packer et al. and Gao et al. [90,91] reported an improvement in function and a decrease in serum cTnT in chronic heart failure patients treated with angiotensin/neprilysiin inhibition. The initial trial testing Omecamtiv is in the process of being significantly expanded (GACTIC-HF) with an end point of improving clinical outcomes [92].
5.2. Serum cTn in the broad population
Findings affecting clinical interpretation of serum Tns in the ED include reports of serum elevations in the circulation in the general population with no apparent necrotic damage to the cardiomyocytes [93,94]. In a study [93] of 3546 individuals comparing serum levels of cTnT employing hs-cTnT with an older generation antibody concluded that the more sensitive assay detected measurable cTnT in ~25% of the population whereas the standard assay detected 0.7%. In a follow up period of 6.0–6.8 years, the study reported that the hs-cTnT provided prognostic information indicating an association of higher levels of cTnT with left ventricular systolic dysfunction and hypertrophy, chronic kidney disease and all-cause mortality.
The ARIC (Atherosclerosis Risk in Communities) study provides additional evidence that determination of cTn in serum with hs-antibodies has value in CVD risk prediction in the general population [95]. ARIC included hs-cTn measurements in 8121 individuals (54–74 years old) with an extensive analysis of association between elevations in cTnI and incident CHD, MI, stroke, HF hospitalization, and global CVD. In an ~15 year follow up of the cohort, the investigators concluded that there is a strong correlation of (adjective) risk in the general population with elevations of hs-cTnI and even stronger risk with elevations in both hs-cTnI and hs-cTnT. There were also sex and race-related differences with white at greater risk than black individuals and men at greater risk than women.
5.3. Genome wide association (GWAS) studies
A genome wide association study by Walsh et al. [94] on a large general population provided insights into the relation between elevated cTn levels, cardiovascular disease, and outcome measures. They employed both hs-TnI and hs-TnT antibodies to measure levels in 19,501 individuals and analyzed the data by follow-up determinations of outcomes as well as a genome-wide association study. In the follow-up period of 7.8 years, as expected, Welsch et al. reported that elevations in both cTnI and cTnT levels were significantly associated with cardiovascular disease including death (n = 266) and heart failure (n = 216). There was also a close association with both cTnI and cTnT to non-cardiovascular death in 374 of the participants. High levels of cTnI rather than cTnT were associated with MI and coronary artery disease after adjustng for risk factors. Elevated levels of cTnT, however, were more closely associated with non-cardiovascular disease deaths. GWAS identified 53 single nucleotide polymorphisms (SNPs) that were in different loci in the case of cTnI and cTnT. In the associations with elevated cTnI, the SNPs were loci in genes controlling expression of proteins related to the cytoskeleton, including pre-kallirein related to bradykinin (important in stability of the actin cytoskeleton [96]), vinculin (critical in anchoring the actin cytoskeleton to membranes [14]) and anoctamin 5 (related to dysferlinopathy similar to limb girdle disease and with cardiomyopathy [97]). Associations with cTnT levels were SORBS2 associated with expression of sorbin (involved in integrin adhesions sites and membranes [98]) or ArgBP2 (strongly associated with the Z-Disk [99]), PTPRD expressing tyrosine phosphatase (related to integrin activation [100]), and TRABD2A expressing Tiki1, a WNT signaling protein. Thus, the majority of the SNPs were differentially related to cTnI and cTnT levels and located in genes expressing proteins related to cardiac cellular cytoskeletal network. This result fit with earlier work [101] demonstrating a weak correlation between cardiovascular risk factors when comparing hs-cTnI and hs-cTnT in the same large population. It is intriguing that serum levels of cTnI and cTnT associated with different SNPs indicate that, in the case of the GWAS, the cTnI biomarker may be more cardio-specific whereas cTnT may be more indicative of alterations of mechanical stresses associated with disorders such as hypertension, and chronic kidney disease. With declining costs, increasing accessibility, and improved bioinformatics, genome wide association studies (GWAS) employing biomarkers such as cTnI and cTnT are likely to continue, expand, and be better interpreted.
5.4. cTnI and cTnT serum levels in exercise
GWAS information may help interpret high serum cTn levels dectected with hs-cTnI and hs-cTnT in apparently healthy individuals who perform intense, strenuous or prolonged exercise. These elevations have been treated as benign, but more recently this view has been modified. Aengevaeren et al. [102] measured blood concentrations of cTnI in 54–69 year old patients, before and 10 min after walking 18.6–34.2 miles (30–55 km). The cohort included 759 participants with a majority with subclinical profiles (86%) and others (14%) with a history of cardiovascular disorders. Nine per cent had a post-exercise cTnI blood level of >40 ng/L (the upper reference limit at the 99th percentile). In follow up studies over a period of 23–77 months, 27% of those with elevated cTnI (vs. 7% with cTnI <40) reached an endpoint: 29 died and 33 had MACE with a hazard ratio of 2.48 (95%CI). The conclusion from this work is that elevated cTnI levels after exercise especially in older adults may be of significance in providing an early indication of the possibility of early loss of life and cardiovascular morbidity. Even so, questions remain as to whether this effect may be reduced by changes in lifestyle, for example, and whether the reduction would reduce these negative end points [103]. Use of hs-cTnI for determination of the effect of simulated fire suppression in physically fit firefighters offers interesting insights into exercise induced increases in serum cTn in otherwise healthy individuals [104]. There was a significant elevaion in cTnI levels even 24 h after the exercise. With CAD related death occuring in ~40–50% of on duty firefighters, these data are highly relevant [105] .
5.5. Chronic kidney disease (CKD)
An important challenge is the interpretation of tests using hs-TnI antibodies showing high levels of cTnI in patients with chronic kidney disease (reviewed in [106,107]). This is not a surprising finding given that CKD patients commonly have coronary artery calcium deposits with elevated mortality [108]. Moreover, patients on dialysis are likely to die of cardiovascular disorders at a rate 10–20 times the broad population [109]. Efforts are underway to evaluate the usefulness of hs-cTn assays in stratification of patients with renal impairment. For example, Miller-Hodges et al. [110] consecutively evaluated serum cTnI levels in 4726 patients, 19% of whom had renal impairment (estimated GFR <60 mL/min/1.73 M2). One year after testing, patients with cTnI levels >99th percentile (diagnostic threshold 16 (women) and 34 (men) ng/mL) had a greater risk of MI or cardiac related death. A question is whether rises in cTn levels occur due to an increase in the release from cardiomyocytes or due to impaired renal elimination. To test this issue, van der Linden et al. [111] took advantage of the diurnal variation in cTnT levels reported by Klinkenberg et al. [112]. Their premise was that a diminished diurnal variation termed fading would indicate an effect of impaired renal elimination on cTnT levels. However, in sampling patients with CKD over a 24 h period, van der Linden et al. found no statistical change in the diurnal rhythms compared to a reference population. These data support the concept that cardiac cellular responses to CKD, for example CAD and altered mechanical loading, are dominant in CKD. Studies aimed at understanding the detailed mechanisms of connection in a cardio-renal syndrome and myocyte death pathways are needed.
6. Programmed cell death and release of cTnI and cTnT
Seminal experiments (reviewed in [64,113]) provided evidence that necrotic cell death is not a passive process but involves a signaling cascade subject to inhibition by small molecules. This process is termed programmed cell death or necroptosis. Although not extensively investigated, there is evidence for a relation between necroptosis and release of cTn into serum. The cascade is triggered by cytokines such as TNFα, known to increase with AMI [114] and coronary artery disease [115]. A ligand-receptor interaction, for example with tumor necrosis factor receptor (TNFR-α), triggers receptor interacting protein kinases (RIPK1/RIPK3) ultimately stimulating downstream signals that inhibit membrane stability and increase necroptosis. In one pathway phosphorylation of members of the cascade promote cellular necrosis. In a second pathway, the signaling cascade promotes cell death via Ca-dependent opening of the mitochondrial permeability transition pore (mPTP). Evidence has been presented that the opening of the mPTP in mitochondrial necroptosis involves ischemia induced adverse alterations in cellular Ca2+, inducing phosphorylation and activation of Ca-calmodulin dependent protein kinase (CaMKII). CaMKII activation can also occur by oxidation via H2O2 or NADPH oxidase (Nox 2) [116,117]. In either case death results from a loss of ATP and membrane instability. As discussed below there is an elevation of myocardial CaMKII activity in mouse models of hypertrophic cardiomyopathy (HCM) that may be related to cell death and cTn release [76].
6.1. Inhibition of necroptosis
Key members of the complex cascade of signaling pathways believed to regulate necroptosis have been summarized [64] and a specific cardioprotective inhibitor, necrostatin (Nec-1), developed [118]. Guo et al. [119] focused their studies on TRAF2 (TNFR associated factor-2) in the necroptotic pathway and reported KO of TRAF2 induced cardiac dysfunction with myocyte death. Also investigated were cTnI levels in the serum of the mice stressed by AMI in controls and mice deficient in TRAF2. In the TRAF2 KO mice, serum TnI was approximately double the values in the hearts expressing TRAF2. We think this is strong evidence that the route of release of Tn into the serum may be through programmed necrosis. Release of cTnI has also been reported to be due to necroptosis in cardiomyocyte death in acute myocarditis. Zhou et al. [120] reported that with myocarditis, there was an increase in cTnI release associated with increased RIPK1/RIPK3 expression, which promotes necroptosis. Down regulation of RIPK1/RIPK3 with the inhibitor, Nec-1, was able to block cTnI release and other markers of cell damage. Below we discuss the potential value of use of hs-cTn in myocarditis associated with SARS-CoV2 infection.
6.2. Necroptosis, apoptosis, and serum cTnI and cTnT
Studies by Weil et al. [121] and an analysis of these studies by Amgalan et al. [63] provide an example of potential connections between apoptosis and necroptosis in myocyte death. Weil et al. investigated a well-controlled porcine model of ischemia/reperfusion injury and reported that a 10 min period of occlusion of the left anterior descending coronary artery induced a significant increase in serum cTn, detected with a hs-cTnI antibody. cTnI levels rose from 13 ng/L to >38 ng/L (the 99th percentile reference limit) at 10 min reperfusion and to 1021 ng/L after 24 h of reperfusion. Cardiac function measured by several approaches fell significantly with the ischemia, recovered to 30–50% depending on the parameter measured, and gradually recovered over the 24-h period of reperfusion. The authors attributed these findings to a transient induction of focal cell death, which they proposed was attributable to apoptosis. In an editorial Amgalan et al. [63] made an argument against this hypothesis. They stressed that the apoptotic loss of cTnI at the levels determined in the blood is unlikely due to the small number myocytes (estimated to be 1 in 6000) affected. Amgalan et al. [63] also argued that it is more likely that changes noted in this model are due to programmed necrosis, which they propose has signals in common with apoptosis. The common signal is caspase activation, a consequence of apoptotic signaling which also affects the mitochondrial inner membrane thereby facilitating opening of the mPTP and necroptosis. Work reported by Feng et al. [116] at about this same time demonstrated a role for activation of Ca-calmodulin kinase (CaMK II) and elevated cellular Ca2+ in opening the mPTP leading to necroptosis. CaMK II activation was demonstrated to occur either by phosphorylation via the TRAF, RIP pathway or by oxidative activation via NADP-oxidase2 (NOX2) activation. More recent work reported by Wu et al. [117] validated and extended a role for oxidatively modified CaMKII in cell death by its effect on sequestration of KATP channels, which provide a protective current during I/R. With current understanding of the signaling cascades for activation of CaMKII and for programmed necrosis, these ideas could be tested and possibly translated to therapies.
7. cTnI and cTnT as biomarkers in personalized and precision medicine
Increasing application of techniques in personalized and precision medicine has implications regarding the use of serum cTn as biomarkers for cardiac disorders. As summarized here, the techniques and analyses in precision medicine are applicable to prevention of an AMI, but the patient presenting with chest pain due to a possible AMI needs immediate attention. Thus, in the case of AMI together with other diagnoses, determination of serum cTnI and cTnT offers a valuable approach.
The premise of personalized and precision medicine is the application of techniques meant to provide a personalized course of action based on an individual's genome, epigenome, transcriptome, proteome, metabolome, and microbiome. Evidence described above indicates that serum cTn may be a valuable addition to the numerous measurements that already comprise precision medicine. Modern analytic techniques leading to network analysis are available to reduce and make sense of these large data sets with the hope of advancing understanding of factors promoting health and factors promoting diseases. For example, see Oldham et al. [122]. Using data that more precisely characterize a single person or family challenges the current paradigm, where it is assumed that individuals in a population have similar characteristics and that well-powered statistical tests can be applied to know whether data developed from a drug, a diagnostic test, or a biomarker can drive a clinical decision. Moreover, current approaches to determining disease prognosis are based on similar assumptions. On the other hand, as discussed in detail by Leopold and Loscalzo [123], with precision medicine, deep phenotyping with layers of information derived from clinical assessment and from DNA, RNA, proteins, and metabolic elements, offers a path to individualized therapeutic approaches and a better determination of prognosis. Data derived from precision medicine give information on various biological systems that are quantified as an endophenotype. As the name implies, endophenotype refers to characteristics generally found in an individual or family, but not observable on the surface as is the case with signs and symptoms defining a phenotype. Endophenotype and phenotype are connected and important aspects of the promise of precision medicine.
7.1. Familial cardiomyopathies, precision medicine, and serum levels of cTn
Familial cardiomyopathies provide an example of the application of data describing an endophenotype and its connection to a phenotype in which we think measurements of serum levels of cTnI and cTnT may play an important role. It is well known that a common cause of HCM is a single point mutation in a variety of sarcomere/cytoskeletal proteins; however, the course of the disorder manifests in multiple, variable clinical and pathological characteristics [74,76,79]. HCM is thus well suited to the application of precision medicine to better understand the evolution of the disease and to inform rational, personalized approaches that include addressing disease causing mechanisms apart from the biophysical defect that triggers HCM. Maron et al. [74] describe a detailed prospective for application to HCM patients with extensive determination of associations of clinical physical characteristics and omic measurements and formulation of a network medicine analysis guiding physicians and researchers in a path leading to better understanding of the diagnosis, prognosis, complexities of HCM progression, and ideas for therapies.
A study of sub-groups of HCM patients in the NHLRI registry provides an excellent example of the quest for patient specific risk prediction with measurements that included hs-cTn [124]. In this study 2755 patients had biomarker measurements (hs-cTnT and N-terminal pro-B type brain natriuretic peptide) together with demographic, genetic, imaging, and fibrotic analysis. In the case of hs-cTnT, normal levels were ≤ 22 ng/L (men) and 12 ng/L (women). There were elevated hs-cTnT levels in 15% of men and 24% of the women in the cohort. Individuals with hypertension and LVEF <55% showed the most severe levels of hs-cTnT. Although there were valid determinations of elevations of hs-TnT in sub-groups of the cohort, the authors emphasized that prediction of out-come events requires long-term follow up. They also emphasized the value of the measurement in developing a multi-variable model related to patient specific risk predictions. Along these lines, Kubo et al. studied a smaller group (183 HCM patients) to investigate the value of determinations of hs-cTnT in risk prediction for cardiovascular events [125]. Ninety-nine patients (32%) had elevated hs-cTnT levels (≥ 14 ng/L). In a follow-up period of 4.1 ± 2.0 years 7% of the patients with normal hs-cTnT levels were diagnosed with adverse CV events, whereas 32% (32 patients) of the patients with elevated hs-cTnT levels demonstrated CV events including cardiac death, heart failure admission with progression to NY Heart Association Functional Class III and IV, embolic arrhythmic events. Kubo et al. concluded that levels of hs-cTnT is a predictor of CV events in HCM with a correlation of high levels with increased risk [125]. The authors drew a similar conclusion in a previous study measuring hs-cTnT levels in a group of 167 HCM patients [126].
As is the case with idiopathic DCM, it is apparent that in the case of familial DCM linked to mutations in the sarcomere/cytoskeletal network that measurements of serum cTn may be informative [127]. DCM linked mutations in the dystrophin/glycoprotein complex (DGC) as well as dysferlin support this idea. In the case of the DGC, Armstrong et al. reported that a δ-sarcoglycan mutation linked to DCM had a dominant negative effect inducing membrane instability documented with increased dye permeability with mechanical strain of the myocytes. Serum cTn was not measured [128]. However, Han et al. reported direct evidence that a DCM linked to dysferlin deficiency, led to increased levels of serum cTnT, especially with exercise stress [129,130]. Importantly a a dysferlin mutation in a human patient demonstrated a DCM phenotype [131]. Dysferlin deficient mice demonstrating a DCM phenotype also exhibit a depression in relaxation kinetics and due to blunted response to β-adrenergic stimulation [132].
One of the considerations regarding the determination of serum cTn levels in DCM is related to studies reporting the localization of cTn in the nuclear compartment of cardiac myocytes [133]. Although nuclear Tns would not be expected to contribute to the early rise in serum Tn levels, there are data [54,134] showing epigenetic effects of cTnT, which are relevant to the interpretation of GWAS and precision medicine. Wu et al. investigated human tissue and cardiac myocytes derived from patient specific inducible pluripotent stem cells (HiPSC-CM) expressing a mutant cTnT, (TNTT2 R173W) linked to DCM, and demonstrated a nuclear localization of mutant cTnT, but not wild-type cTnT in the controls [54]. Further mechanistic investigations showed that the mutant cTnT modified genes regulating expression of phosphodiesterase 2A and 3A in β-adrenergic signaling and exacerbating the DCM phenotype. Ultimately this epigenetic effect could be a factor in other cardiac dysfunction and contribute to elevated serum levels of Tns reported in DCM. A finding related to these studies on genetic DCM is the finding reported by Baba et al., who compared measurements of hs-cTnI and hs-cTnT in a group of patients with idiopathic DCM [127]. In these patients a high measure of hs-cTnT was evident in the patients with high risk for adverse events, and the higher the level the greater the risk. Moreover, this risk stratification was weak in the case of hs-cTnI measurements.
7.2. Post-translational modifications of cTnI and cTnT in precision medicine and clinical decisions
A valuable extension of the use of cTnI and cTnT as biomarkers in HCM as well as other cardiovascular disorders is the determination of the state of cTn. Relevant measurements of serum cTnI and cTnT as biomarkers in personalized medicine are couched in determination not only of their abundance, degradation, and association with neighboring proteins in serum, but also in determination of post-translational modifications. Knowing how cTnI and cTnT are post-translationally modified may provide a clearer picture of the underlying cardiac disorder and add another dimension to omic measurements.
Effects of phosphorylation of sites on cTnI and cTnT have been extensively reviewed in physiological and pathological states and represent a constellation of modifiers of levels and dynamics of cardiac function [6,19,[135], [136], [137], [138]]. Identification of post-translational modifications other than phosphorylation offers more possibilities for personalized medicine. For example, recent studies [139] have demonstrated glycation of 13 cTnI residues and 3 cTnT residues. These modifications may be diagnostic or prognostic in diabetic cardiomyopathy. The challenge is the reality of determining post-translational modifications to cTn in the ED and other clinical settings. Modern, cutting edge mass spectrometry offers some confidence regarding the practical reality of making these measurements within the relatively near future. Van Eyk and colleagues have led the development of mass spectrometry workflows employing state of the art instrumentation that permit routine determination of various forms of cTnI in patients [51,140]. The promise of these approaches awaits validation in a large population of patients. A continuing challenge is the instrumentation availability and cost as well as acceptance by physician networks. Recent advances indicate promising use of this technology in Alzheimer's detection. Schindler et al. employed determination of biomarkers using immunoprecipitation and liquid-chromatography mass spectrometry to assay for plasma β-amyloid [141]. Moreover, progress in the nanomaterials and nanotechnology offers the promise of a new generation of biosensors, some of which have applied to detection of cTnI [142]. This technology has created intriguing possibilities for a new generation of biological sensors based on nanoscale solid-state devices, which hold the promise of redefining the boundary of biological detection limits in sensitivity, speed, and particularly, portability.
An application of knowledge of post-translational modifications in serum cTnI and cTnT may be of significance in the clinical decision to treat stable, ischemic, coronary artery disease with percutaneous coronary intervention (stenting) surgery along with medical therapy or to employ standard “conservative” medical therapy alone, especially statins. Results of a large study (ISCHEMIA) revealed that there was no reduction in risk over 3+ years of follow-up for CV events or death in patients treated with invasive therapy compared to patients treated with conservative therapy [143]. A related issue is the significant number of statin intolerant (SI) patients. Hai et al. [144] concluded from a study of 952 SI patients with stable CAD that those with relatively high serum cTnI were more likely to have MACE and should be treated with rigorous non-statin lipid lowering therapy including novel therapies such as PCSK9 inhibition [145].
Currently evaluation of Covid-19 patients show that myocardial injury indicated by elevated hs-cTnI is associated with higher in-hospital mortality [10]. Controversies associated with the use hs-Tn as a biomarker in patients infected with SARS-CoV2 emphasize the need for further understanding of mechanisms of cTn appearance in serum. A guideline disseminated by The American College of Cardiology advised not to employ hs-Tn unless AMI is indicated in COVID-19 patients. However, in citing this directive, Chapman et al. made the counter argument of the value of hs-Tn in COVID-19 patients, especially the elderly, and those with compromised cardiovascular function and/or immune response [9]. In their words, this knowledge would “inform the use of inotropes, vasopressors, and diuretics.” Moreover, they stressed that hs-Tn measurements aid in the identification of the endophenotype of myocarditis and its treatment. In this and the examples of clinical decisions described above, refined understanding of the abundance and forms of cTnI and cTnT would appear to be of benefit.
8. Conclusion and perspectives
Technical advances have improved the detection of cTn biomarkers and improved stratification of patients with AMI and coronary artery disease associated with metabolic strains on myocyte integrity. However, interpreting elevated cTn serum levels measured with high sensitivity antibodies in the general population with no overt CVD and in cardiovascular disorders such as HCM, DCM, CKD, and myocarditis has led to the need to integrate understanding of the complex biology of cTn with its appearance in serum. In their statement regarding cTn levels, Chapman and colleagues emphasized a need “to better understand the utility of this essential biomarker and to educate clinicians on its interpretation and implications for prognosis and clinical decision making [9].” Evidence indicates that perturbations in the mechanical micro-environment of cardiac myocytes, strains from within the myocytes (eg. HCM), or strains outside the myocytes (elevated EDV and EDP) affect membrane fragility and may lead to release of cTn without cell rupture. Increasing awareness of signaling in programmed cell death also needs to be considered and investigated in interpretation elevated cTn in the serum. Determination and outcomes measures with cTn levels are an important asset in the extensive multivariable analyses in personalized and precision medicine [9,123]. Technological advances in detection of various forms of cTnI in serum indicate more to come in this important field of investigation.
Funding
This work was funded by the United States of America National Instutes of Health, National Heart, Lung and Blood Institute Grant PO1 HL 06246 (RJS).
Disclosure
R. John Solaro is a member to the Scientific Advisory Board of Cytokinetics, Inc. A Consultant to Pfizer, Inc. and a member of the Heart Failure Advisory Board of Amgen.
References
- 1.Wilkinson J.M., Grand R.J. Comparison of amino acid sequence of troponin I from different striated muscles. Nature. 1978;271:31–35. doi: 10.1038/271031a0. [DOI] [PubMed] [Google Scholar]
- 2.Cummins B., Auckland M.L., Cummins P. Cardiac-specific troponin-I radioimmunoassay in the diagnosis of acute myocardial infarction. Am. Heart J. 1987;113:1333–1344. doi: 10.1016/0002-8703(87)90645-4. [DOI] [PubMed] [Google Scholar]
- 3.Katus H.A., Remppis A., Looser S., Hallermeier K., Scheffold T., Kubler W. Enzyme linked immuno assay of cardiac troponin T for the detection of acute myocardial infarction in patients. J. Mol. Cell. Cardiol. 1989;21:1349–1353. doi: 10.1016/0022-2828(89)90680-9. [DOI] [PubMed] [Google Scholar]
- 4.Bodor G.S., Porter S., Landt Y., Ladenson J.H. Development of monoclonal antibodies for an assay of cardiac troponin-I and preliminary results in suspected cases of myocardial infarction. Clin. Chem. 1992;38:2203–2214. [PubMed] [Google Scholar]
- 5.Solaro R.J., Moir A.J., Perry S.V. Phosphorylation of troponin I and the inotropic effect of adrenaline in the perfused rabbit heart. Nature. 1976;262:615–617. doi: 10.1038/262615a0. [DOI] [PubMed] [Google Scholar]
- 6.Solaro R.J., Henze M., Kobayashi T. Integration of troponin I phosphorylation with cardiac regulatory networks. Circ. Res. 2013;112:355–366. doi: 10.1161/CIRCRESAHA.112.268672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ke Y., Wang L., Pyle W.G., de Tombe P.P., Solaro R.J. Intracellular localization and functional effects of P21-activated kinase-1 (Pak1) in cardiac myocytes. Circ. Res. 2004;94:194–200. doi: 10.1161/01.RES.0000111522.02730.56. [DOI] [PubMed] [Google Scholar]
- 8.Yamada Y., Namba K., Fujii T. Cardiac muscle thin filament structures reveal calcium regulatory mechanism. Nat. Commun. 2020;11:153. doi: 10.1038/s41467-019-14008-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chapman A.R., Bularga A., Mills N.L. High-sensitivity cardiac troponin can be an ally in the fight against COVID-19. Circulation. 2020 doi: 10.1161/CIRCULATIONAHA.120.047008. Online ahead of print. [DOI] [PubMed] [Google Scholar]
- 10.Shi S., Qin M., Shen B., Cai Y., Liu T., Yang F. China; JAMA Cardiol: 2020. Association of Cardiac Injury with Mortality in Hospitalized Patients with COVID-19 in Wuhan. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pyle W.G., Solaro R.J. At the crossroads of myocardial signaling: the role of Z-discs in intracellular signaling and cardiac function. Circ. Res. 2004;94:296–305. doi: 10.1161/01.RES.0000116143.74830.A9. [DOI] [PubMed] [Google Scholar]
- 12.Caporizzo M.A., Chen C.Y., Prosser B.L. Cardiac microtubules in health and heart disease. Exp Biol Med (Maywood) 2019;244:255–1272. doi: 10.1177/1535370219868960. 1535370219868960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gautel M., Djinovic-Carugo K. The sarcomeric cytoskeleton: from molecules to motion. J. Exp. Biol. 2016;219:135–145. doi: 10.1242/jeb.124941. [DOI] [PubMed] [Google Scholar]
- 14.Zemljic-Harpf A., Manso A.M., Ross R.S. Vinculin and Talin: focus on the myocardium. J. Investig. Med. 2009;57:849–855. doi: 10.231/JIM.0b013e3181c5e074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Israeli-Rosenberg S., Manso A.M., Okada H., Ross R.S. Integrins and integrin-associated proteins in the cardiac myocyte. Circ. Res. 2014;114:572–586. doi: 10.1161/CIRCRESAHA.114.301275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen C.A., Manso A.M., Ross R.S. Talin and Kindlin as Integrin-Activating Proteins: Focus on the Heart. Pediatr. Cardiol. 2019;40:1401–1409. doi: 10.1007/s00246-019-02167-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ho C.Y., Carlsen C., Thune J.J., Havndrup O., Bundgaard H., Farrohi F. Echocardiographic strain imaging to assess early and late consequences of sarcomere mutations in hypertrophic cardiomyopathy. Circ. Cardiovasc. Genet. 2009;2:314–321. doi: 10.1161/CIRCGENETICS.109.862128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feng J., Schaus B.J., Fallavollita J.A., Lee T.C., Canty J.M., Jr. Preload induces troponin I degradation independently of myocardial ischemia. Circulation. 2001;103:2035–2037. doi: 10.1161/01.cir.103.16.2035. [DOI] [PubMed] [Google Scholar]
- 19.Kobayashi T., Solaro R.J. Calcium, thin filaments, and the integrative biology of cardiac contractility. Annu. Rev. Physiol. 2005;67:39–67. doi: 10.1146/annurev.physiol.67.040403.114025. [DOI] [PubMed] [Google Scholar]
- 20.Hidalgo C., Granzier H. Tuning the molecular giant titin through phosphorylation: role in health and disease. Trends Cardiovas. Med. 2013;23:165–171. doi: 10.1016/j.tcm.2012.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Scruggs S.B., Solaro R.J. The significance of regulatory light chain phosphorylation in cardiac physiology. Arch. Biochem. Biophys. 2011;510:129–134. doi: 10.1016/j.abb.2011.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kensler R.W., Craig R., Moss R.L. Phosphorylation of cardiac myosin binding protein C releases myosin heads from the surface of cardiac thick filaments. Proc. Natl. Acad. Sci. U. S. A. 2017;114:E1355–e64. doi: 10.1073/pnas.1614020114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hinken A.C., Solaro R.J. A dominant role of cardiac molecular motors in the intrinsic regulation of ventricular ejection and relaxation. Physiology (Bethesda) 2007;22:73–80. doi: 10.1152/physiol.00043.2006. [DOI] [PubMed] [Google Scholar]
- 24.Risi C., Belknap B., Forgacs-Lonart E., Harris S.P., Schroder G.F., White H.D. N-Terminal Domains of Cardiac Myosin Binding Protein C Cooperatively Activate the Thin Filament. Structure. 2018;26 doi: 10.1016/j.str.2018.08.007. 1604-11.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martin A.F. Turnover of cardiac troponin subunits. Kinetic evidence for a precursor pool of troponin-I. J. Biol. Chem. 1981;256:964–968. [PubMed] [Google Scholar]
- 26.Bleier J., Vorderwinkler K.P., Falkensammer J., Mair P., Dapunt O., Puschendorf B. Different intracellular compartmentations of cardiac troponins and myosin heavy chains: a causal connection to their different early release after myocardial damage. Clin. Chem. 1998;44:1912–1918. [PubMed] [Google Scholar]
- 27.Jennings R.B., Reimer K.A. The cell biology of acute myocardial ischemia. Annu. Rev. Med. 1991;42:225–246. doi: 10.1146/annurev.me.42.020191.001301. [DOI] [PubMed] [Google Scholar]
- 28.Remppis A., Scheffold T., Greten J., Haass M., Greten T., Kubler W. Intracellular compartmentation of troponin T: release kinetics after global ischemia and calcium paradox in the isolated perfused rat heart. J. Mol. Cell. Cardiol. 1995;27:793–803. doi: 10.1016/0022-2828(95)90086-1. [DOI] [PubMed] [Google Scholar]
- 29.Bartos J.A., Matsuura T.R., Tsangaris A., Olson M., McKnite S.H., Rees J.N. Intracoronary Poloxamer 188 prevents reperfusion injury in a porcine model of ST-segment elevation myocardial infarction. JACC Basic Transl Sci. 2016;1:224–234. doi: 10.1016/j.jacbts.2016.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Houang E.M., Bartos J., Hackel B.J., Lodge T.P., Yannopoulos D., Bates F.S. Cardiac muscle membrane stabilization in myocardial reperfusion injury. JACC Basic Transl Sci. 2019;4:275–287. doi: 10.1016/j.jacbts.2019.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reichlin T., Hochholzer W., Bassetti S., Steuer S., Stelzig C., Hartwiger S. Early diagnosis of myocardial infarction with sensitive cardiac troponin assays. N. Engl. J. Med. 2009;361:858–867. doi: 10.1056/NEJMoa0900428. [DOI] [PubMed] [Google Scholar]
- 32.Neumann J.T., Twerenbold R., Ojeda F., Sorensen N.A., Chapman A.R., Shah A.S.V. Application of high-sensitivity troponin in suspected myocardial infarction. N. Engl. J. Med. 2019;380:2529–2540. doi: 10.1056/NEJMoa1803377. [DOI] [PubMed] [Google Scholar]
- 33.Thygesen K., Alpert J.S., Jaffe A.S., Chaitman B.R., Bax J.J., Morrow D.A. Fourth universal definition of myocardial infarction (2018) Circulation. 2018;138:e618–e751. doi: 10.1161/CIR.0000000000000617. [DOI] [PubMed] [Google Scholar]
- 34.Tomaszewski C.A., Nestler D., Shah K.H., Sudhir A., Brown M.D. Clinical policy: critical issues in the evaluation and Management of Emergency Department Patients with Suspected non-ST-elevation acute coronary syndromes. Ann. Emerg. Med. 2018;72:e65–e106. doi: 10.1016/j.annemergmed.2018.07.045. [DOI] [PubMed] [Google Scholar]
- 35.Potomac W., Diercks D.B. Using high sensitivity troponins to rule out acute coronary syndrome and lower admission rates. Cardiol. Rev. 2019;27:314–321. doi: 10.1097/CRD.0000000000000275. [DOI] [PubMed] [Google Scholar]
- 36.Urboniene D., Dias F.A., Pena J.R., Walker L.A., Solaro R.J., Wolska B.M. Expression of slow skeletal troponin I in adult mouse heart helps to maintain the left ventricular systolic function during respiratory hypercapnia. Circ. Res. 2005;97:70–77. doi: 10.1161/01.RES.0000173849.68636.1e. [DOI] [PubMed] [Google Scholar]
- 37.Hunkeler N.M., Kullman J., Murphy A.M. Troponin I isoform expression in human heart. Circ. Res. 1991;69:1409–1414. doi: 10.1161/01.res.69.5.1409. [DOI] [PubMed] [Google Scholar]
- 38.Collinson P.O., Saenger A.K., Apple F.S. High sensitivity, contemporary and point-of-care cardiac troponin assays: educational aids developed by the IFCC committee on clinical application of cardiac bio-markers. Clin. Chem. Lab. Med. 2019;57:623–632. doi: 10.1515/cclm-2018-1211. [DOI] [PubMed] [Google Scholar]
- 39.Su J.A., Kumar S.R., Mahmoud H., Bowdish M.E., Toubat O., Wood J.C. Postoperative serum troponin trends in infants undergoing cardiac surgery. Semin. Thorac. Cardiovasc. Surg. 2019;31:244–251. doi: 10.1053/j.semtcvs.2018.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Asrani P., Aly A.M., Jiwani A.K., Niebuhr B.R., Christenson R.H., Jain S.K. High-sensitivity troponin T in preterm infants with a hemodynamically significant patent ductus arteriosus. J Perinatol: Official Journal of the California Perinatal Association. 2018;38:1483–1489. doi: 10.1038/s41372-018-0192-x. [DOI] [PubMed] [Google Scholar]
- 41.Katrukha I.A., Kogan A.E., Vylegzhanina A.V., Kharitonov A.V., Tamm N.N., Filatov V.L. Full-size cardiac troponin I and its Proteolytic fragments in blood of patients with acute myocardial infarction: antibody selection for assay development. Clin. Chem. 2018;64:1104–1112. doi: 10.1373/clinchem.2017.286211. [DOI] [PubMed] [Google Scholar]
- 42.Wei H., Jin J.P. NH2-terminal truncations of cardiac troponin I and cardiac troponin T produce distinct effects on contractility and calcium homeostasis in adult cardiomyocytes. Am J Physiol Cell Physiol. 2015;308:C397–C404. doi: 10.1152/ajpcell.00358.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Streng A.S., de Boer D., van Doorn W.P., Kocken J.M., Bekers O., Wodzig W.K. Cardiac troponin T degradation in serum is catalysed by human thrombin. Biochem. Biophys. Res. Commun. 2016;481:165–168. doi: 10.1016/j.bbrc.2016.10.149. [DOI] [PubMed] [Google Scholar]
- 44.Communal C., Sumandea M., de Tombe P., Narula J., Solaro R.J., Hajjar R.J. Functional consequences of caspase activation in cardiac myocytes. Proc. Natl. Acad. Sci. U. S. A. 2002;99:6252–6256. doi: 10.1073/pnas.092022999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Labugger R., Organ L., Collier C., Atar D., Van Eyk J.E. Extensive troponin I and T modification detected in serum from patients with acute myocardial infarction. Circulation. 2000;102:1221–1226. doi: 10.1161/01.cir.102.11.1221. [DOI] [PubMed] [Google Scholar]
- 46.van Wijk X.M.R., Claassen S., Enea N.S., Li P., Yang S., Brouwer M.A. Cardiac troponin I is present in plasma of type 1 myocardial infarction patients and patients with troponin I elevations due to other etiologies as complex with little free I. Clin. Biochem. 2019;73:35–43. doi: 10.1016/j.clinbiochem.2019.06.012. [DOI] [PubMed] [Google Scholar]
- 47.Katrukha A.G., Bereznikova A.V., Filatov V.L., Esakova T.V., Kolosova O.V., Pettersson K. Degradation of cardiac troponin I: implication for reliable immunodetection. Clin. Chem. 1998;44:2433–2440. [PubMed] [Google Scholar]
- 48.Zhang T., Pereyra A.S., Wang Z.M., Birbrair A., Reisz J.A., Files D.C. Calpain inhibition rescues troponin T3 fragmentation, increases Cav1.1, and enhances skeletal muscle force in aging sedentary mice. Aging Cell. 2016;15:488–498. doi: 10.1111/acel.12453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Streng A.S., de Boer D., van Doorn W.P., Bouwman F.G., Mariman E.C., Bekers O. Identification and characterization of cardiac troponin T fragments in serum of patients suffering from acute myocardial infarction. Clin. Chem. 2017;63:563–572. doi: 10.1373/clinchem.2016.261511. [DOI] [PubMed] [Google Scholar]
- 50.Schneck N.A., Phinney K.W., Lee S.B., Lowenthal M.S. Quantification of cardiac troponin I in human plasma by immunoaffinity enrichment and targeted mass spectrometry. Anal. Bioanal. Chem. 2018;410:2805–2813. doi: 10.1007/s00216-018-0960-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Soetkamp D., Raedschelders K., Mastali M., Sobhani K., Bairey Merz C.N., Van Eyk J. The continuing evolution of cardiac troponin I biomarker analysis: from protein to proteoform. Expert Rev Proteomics. 2017;14:973–986. doi: 10.1080/14789450.2017.1387054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hickman P.E., Potter J.M., Aroney C., Koerbin G., Southcott E., Wu A.H. Cardiac troponin may be released by ischemia alone, without necrosis. Clin. Chim. Acta. 2010;411:318–323. doi: 10.1016/j.cca.2009.12.009. [DOI] [PubMed] [Google Scholar]
- 53.Bates K.J., Hall E.M., Fahie-Wilson M.N., Kindler H., Bailey C., Lythall D. Circulating immunoreactive cardiac troponin forms determined by gel filtration chromatography after acute myocardial infarction. Clin. Chem. 2010;56:952–958. doi: 10.1373/clinchem.2009.133546. [DOI] [PubMed] [Google Scholar]
- 54.Wu H., Lee J., Vincent L.G., Wang Q., Gu M., Lan F. Epigenetic regulation of Phosphodiesterases 2A and 3A underlies compromised beta-adrenergic Signaling in an iPSC model of dilated cardiomyopathy. Cell Stem Cell. 2015;17:89–100. doi: 10.1016/j.stem.2015.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sage M.D., Jennings R.B. Cytoskeletal injury and subsarcolemmal bleb formation in dog heart during in vitro total ischemia. Am. J. Pathol. 1988;133:327–337. [PMC free article] [PubMed] [Google Scholar]
- 56.Sage M.D., Jennings R.B. Myocyte swelling and plasmalemmal integrity during early experimental myocardial ischemia in vivo. Scanning Microsc. 1988;2:477–484. [PubMed] [Google Scholar]
- 57.Steenbergen C., Hill M.L., Jennings R.B. Volume regulation and plasma membrane injury in aerobic, anaerobic, and ischemic myocardium in vitro. Effects of osmotic cell swelling on plasma membrane integrity. Circ. Res. 1985;57:864–875. doi: 10.1161/01.res.57.6.864. [DOI] [PubMed] [Google Scholar]
- 58.Armstrong S.C., Latham C.A., Shivell C.L., Ganote C.E. Ischemic loss of sarcolemmal dystrophin and spectrin: correlation with myocardial injury. J. Mol. Cell. Cardiol. 2001;33:1165–1179. doi: 10.1006/jmcc.2001.1380. [DOI] [PubMed] [Google Scholar]
- 59.Van Eyk J.E., Powers F., Law W., Larue C., Hodges R.S., Solaro R.J. Breakdown and release of myofilament proteins during ischemia and ischemia/reperfusion in rat hearts: identification of degradation products and effects on the pCa-force relation. Circ. Res. 1998;82:261–271. doi: 10.1161/01.res.82.2.261. [DOI] [PubMed] [Google Scholar]
- 60.Avner B.S., Shioura K.M., Scruggs S.B., Grachoff M., Geenen D.L., Helseth D.L., Jr. Myocardial infarction in mice alters sarcomeric function via post-translational protein modification. Mol. Cell. Biochem. 2012;363:203–215. doi: 10.1007/s11010-011-1172-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Taneike M., Mizote I., Morita T., Watanabe T., Hikoso S., Yamaguchi O. Calpain protects the heart from hemodynamic stress. J. Biol. Chem. 2011;286:32170–32177. doi: 10.1074/jbc.M111.248088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Suryakumar G., Kasiganesan H., Balasubramanian S., Kuppuswamy D. Lack of beta3 integrin signaling contributes to calpain-mediated myocardial cell loss in pressure-overloaded myocardium. J. Cardiovasc. Pharmacol. 2010;55:567–573. doi: 10.1097/FJC.0b013e3181d9f5d4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Amgalan D., Pekson R., Kitsis R.N. Troponin release following brief myocardial ischemia: apoptosis versus necrosis. JACC Basic Transl Sci. 2017;2:118–121. doi: 10.1016/j.jacbts.2017.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Del Re D.P., Amgalan D., Linkermann A., Liu Q., Kitsis R.N. Fundamental mechanisms of regulated cell death and implications for heart disease. Physiol. Rev. 2019;99:1765–1817. doi: 10.1152/physrev.00022.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Manso A.M., Okada H., Sakamoto F.M., Moreno E., Monkley S.J., Li R. Loss of mouse cardiomyocyte Talin-1 and Talin-2 leads to beta-1 integrin reduction, costameric instability, and dilated cardiomyopathy. Proc. Natl. Acad. Sci. U. S. A. 2017;114:E6250–e9. doi: 10.1073/pnas.1701416114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Okada H., Lai N.C., Kawaraguchi Y., Liao P., Copps J., Sugano Y. Integrins protect cardiomyocytes from ischemia/reperfusion injury. J. Clin. Invest. 2013;123:4294–4308. doi: 10.1172/JCI64216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hessel M.H., Atsma D.E., van der Valk E.J., Bax W.H., Schalij M.J., van der Laarse A. Release of cardiac troponin I from viable cardiomyocytes is mediated by integrin stimulation. Pflugers Arch. 2008;455:979–986. doi: 10.1007/s00424-007-0354-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Swildens J., de Vries A.A., Li Z., Umar S., Atsma D.E., Schalij M.J. Integrin stimulation favors uptake of macromolecules by cardiomyocytes in vitro. Cell. Physiol. Biochem. 2010;26:999–1010. doi: 10.1159/000324013. [DOI] [PubMed] [Google Scholar]
- 69.Sun Z., Guo S.S., Fassler R. Integrin-mediated mechanotransduction. J. Cell Biol. 2016;215:445–456. doi: 10.1083/jcb.201609037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cooper Gt. Proliferating cardiac microtubules. Am. J. Physiol. Heart Circ. Physiol. 2009;297:H510–H511. doi: 10.1152/ajpheart.00517.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Belanto J.J., Mader T.L., Eckhoff M.D., Strandjord D.M., Banks G.B., Gardner M.K. Microtubule binding distinguishes dystrophin from utrophin. Proc. Natl. Acad. Sci. U. S. A. 2014;111:5723–5728. doi: 10.1073/pnas.1323842111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sato H., Nagai T., Kuppuswamy D., Narishige T., Koide M., Menick D.R. Microtubule stabilization in pressure overload cardiac hypertrophy. J. Cell Biol. 1997;139:963–973. doi: 10.1083/jcb.139.4.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Drum B.M., Yuan C., Li L., Liu Q., Wordeman L., Santana L.F. Oxidative stress decreases microtubule growth and stability in ventricular myocytes. J. Mol. Cell. Cardiol. 2016;93:32–43. doi: 10.1016/j.yjmcc.2016.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Maron B.J., Maron M.S., Maron B.A., Loscalzo J. Moving beyond the sarcomere to explain heterogeneity in hypertrophic cardiomyopathy: JACC review topic of the week. J. Am. Coll. Cardiol. 2019;73:1978–1986. doi: 10.1016/j.jacc.2019.01.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Muthuchamy M., Pieples K., Rethinasamy P., Hoit B., Grupp I.L., Boivin G.P. Mouse model of a familial hypertrophic cardiomyopathy mutation in alpha-tropomyosin manifests cardiac dysfunction. Circ. Res. 1999;85:47–56. doi: 10.1161/01.res.85.1.47. [DOI] [PubMed] [Google Scholar]
- 76.Deranek A.E., Klass M.M., Tardiff J.C. Moving beyond simple answers to complex disorders in sarcomeric cardiomyopathies: the role of integrated systems. Pflugers Arch. 2019;471:661–671. doi: 10.1007/s00424-019-02269-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sequeira V., Bertero E., Maack C. Energetic drain driving hypertrophic cardiomyopathy. FEBS Lett. 2019;593:1616–1626. doi: 10.1002/1873-3468.13496. [DOI] [PubMed] [Google Scholar]
- 78.Ryba D.M., Li J., Cowan C.L., Russell B., Wolska B.M., Solaro R.J. Long-term biased beta-Arrestin Signaling improves cardiac structure and function in dilated cardiomyopathy. Circulation. 2017;135:1056–1070. doi: 10.1161/CIRCULATIONAHA.116.024482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Marszalek R.J., Solaro R.J., Wolska B.M. Coronary arterial vasculature in the pathophysiology of hypertrophic cardiomyopathy. Pflugers Arch. 2019;471:769–780. doi: 10.1007/s00424-018-2224-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sato Y., Taniguchi R., Nagai K., Makiyama T., Okada H., Yamada T. Measurements of cardiac troponin T in patients with hypertrophic cardiomyopathy. Heart. 2003;89:659–660. doi: 10.1136/heart.89.6.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cramer G., Bakker J., Gommans F., Brouwer M., Kurvers M., Fouraux M. Relation of highly sensitive cardiac troponin T in hypertrophic cardiomyopathy to left ventricular mass and cardiovascular risk. Am. J. Cardiol. 2014;113:1240–1245. doi: 10.1016/j.amjcard.2013.12.033. [DOI] [PubMed] [Google Scholar]
- 82.Agarwal A., Yousefzai R., Shetabi K., Samad F., Aggarwal S., Cho C. Relationship of cardiac troponin to systolic global longitudinal strain in hypertrophic cardiomyopathy. Echocardiography. 2017;34:1470–1477. doi: 10.1111/echo.13645. [DOI] [PubMed] [Google Scholar]
- 83.Reant P., Mirabel M., Lloyd G., Peyrou J., Lopez Ayala J.M., Dickie S. Global longitudinal strain is associated with heart failure outcomes in hypertrophic cardiomyopathy. Heart. 2016;102:741–747. doi: 10.1136/heartjnl-2015-308576. [DOI] [PubMed] [Google Scholar]
- 84.Luo H.C., Pozios I., Vakrou S., Sorensen L., Abraham R.M., Abraham T. Age-related changes in familial hypertrophic cardiomyopathy phenotype in transgenic mice and humans. J Huazhong Univ Sci Technolog Med Sci. 2014;34:634–639. doi: 10.1007/s11596-014-1329-6. [DOI] [PubMed] [Google Scholar]
- 85.Turer A.T., Addo T.A., Martin J.L., Sabatine M.S., Lewis G.D., Gerszten R.E. Myocardial ischemia induced by rapid atrial pacing causes troponin T release detectable by a highly sensitive assay: insights from a coronary sinus sampling study. J. Am. Coll. Cardiol. 2011;57:2398–2405. doi: 10.1016/j.jacc.2010.11.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Siriwardena M., Campbell V., Richards A.M., Pemberton C.J. Cardiac biomarker responses to dobutamine stress echocardiography in healthy volunteers and patients with coronary artery disease. Clin. Chem. 2012;58:1492–1494. doi: 10.1373/clinchem.2012.187682. [DOI] [PubMed] [Google Scholar]
- 87.Decloedt A., De Clercq D., Ven S., Vera L., van Loon G. Right ventricular function during pharmacological and exercise stress testing in horses. Vet. J. 2017;227:8–14. doi: 10.1016/j.tvjl.2017.08.001. [DOI] [PubMed] [Google Scholar]
- 88.Triposkiadis F., Parissis J.T., Starling R.C., Skoularigis J., Louridas G. Current drugs and medical treatment algorithms in the management of acute decompensated heart failure. Expert Opin. Investig. Drugs. 2009;18:695–707. doi: 10.1517/13543780902922660. [DOI] [PubMed] [Google Scholar]
- 89.Teerlink J.R., Felker G.M., McMurray J.J., Solomon S.D., Adams K.F., Jr., Cleland J.G. Chronic Oral study of myosin activation to increase contractility in heart failure (COSMIC-HF): a phase 2, pharmacokinetic, randomised, placebo-controlled trial. Lancet. 2016;388:2895–2903. doi: 10.1016/S0140-6736(16)32049-9. [DOI] [PubMed] [Google Scholar]
- 90.Packer M., McMurray J.J., Desai A.S., Gong J., Lefkowitz M.P., Rizkala A.R. Angiotensin receptor neprilysin inhibition compared with enalapril on the risk of clinical progression in surviving patients with heart failure. Circulation. 2015;131:54–61. doi: 10.1161/CIRCULATIONAHA.114.013748. [DOI] [PubMed] [Google Scholar]
- 91.Gao Y., Xing C., Hao W., Zhao H., Wang L., Luan B. The impact of Sacrubitril/valsartan on clinical treatment and hs-cTnT and NT-ProBNP serum levels and the left ventricular function in patients with chronic heart failure. Int. Heart J. 2020;61:1–6. doi: 10.1536/ihj.19-231. [DOI] [PubMed] [Google Scholar]
- 92.Teerlink J.R., Diaz R., Felker G.M., McMurray J.J.V., Metra M., Solomon S.D. Omecamtiv Mecarbil in chronic heart failure with reduced ejection fraction: rationale and design of GALACTIC-HF. JACC Heart Fail. 2020;8:329–340. doi: 10.1016/j.jchf.2019.12.001. [DOI] [PubMed] [Google Scholar]
- 93.de Lemos J.A., Drazner M.H., Omland T., Ayers C.R., Khera A., Rohatgi A. Association of troponin T detected with a highly sensitive assay and cardiac structure and mortality risk in the general population. Jama. 2010;304:2503–2512. doi: 10.1001/jama.2010.1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Welsh P., Preiss D., Hayward C., Shah A.S.V., McAllister D., Briggs A. Cardiac troponin T and troponin I in the general population. Circulation. 2019;139:2754–2764. doi: 10.1161/CIRCULATIONAHA.118.038529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Jia X., Sun W., Hoogeveen R.C., Nambi V., Matsushita K., Folsom A.R. High-sensitivity troponin I and incident coronary events, stroke, heart failure hospitalization, and mortality in the ARIC study. Circulation. 2019;139:2642–2653. doi: 10.1161/CIRCULATIONAHA.118.038772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Liu L.B., Xue Y.X., Liu Y.H., Wang Y.B. Bradykinin increases blood-tumor barrier permeability by down-regulating the expression levels of ZO-1, occludin, and claudin-5 and rearranging actin cytoskeleton. J. Neurosci. Res. 2008;86:1153–1168. doi: 10.1002/jnr.21558. [DOI] [PubMed] [Google Scholar]
- 97.Penttila S., Vihola A., Palmio J., Udd B., Adam M.P., Ardinger H.H., Pagon R.A., Wallace S.E., LJH Bean, Stephens K. University of Washington, Seattle; Seattle (WA): 1993. ANO5 Muscle Disease. [PubMed] [Google Scholar]
- 98.Loveless T., Qadota H., Benian G.M., Hardin J. Caenorhabditis elegans SORB-1 localizes to integrin adhesion sites and is required for organization of sarcomeres and mitochondria in myocytes. Mol. Biol. Cell. 2017;28:3621–3633. doi: 10.1091/mbc.E16-06-0455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wang B., Golemis E.A., Kruh G.D. ArgBP2, a multiple Src homology 3 domain-containing, Arg/Abl-interacting protein, is phosphorylated in v-Abl-transformed cells and localized in stress fibers and cardiocyte Z-disks. J. Biol. Chem. 1997;272:17542–17550. doi: 10.1074/jbc.272.28.17542. [DOI] [PubMed] [Google Scholar]
- 100.Radel C., Rizzo V. Integrin mechanotransduction stimulates caveolin-1 phosphorylation and recruitment of Csk to mediate actin reorganization. Am. J. Physiol. Heart Circ. Physiol. 2005;288:H936–H945. doi: 10.1152/ajpheart.00519.2004. [DOI] [PubMed] [Google Scholar]
- 101.Welsh P., Preiss D., Shah A.S.V., McAllister D., Briggs A., Boachie C. Comparison between high-sensitivity cardiac troponin T and cardiac troponin I in a large general population cohort. Clin. Chem. 2018;64:1607–1616. doi: 10.1373/clinchem.2018.292086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Aengevaeren V.L., Hopman M.T.E., Thompson P.D., Bakker E.A., George K.P., Thijssen D.H.J. Exercise-induced cardiac troponin I increase and incident mortality and cardiovascular events. Circulation. 2019;140:804–814. doi: 10.1161/CIRCULATIONAHA.119.041627. [DOI] [PubMed] [Google Scholar]
- 103.Omland T., Aakre K.M. Cardiac troponin increase after endurance exercise. Circulation. 2019;140:815–818. doi: 10.1161/CIRCULATIONAHA.119.042131. [DOI] [PubMed] [Google Scholar]
- 104.Hunter A.L., Shah A.S., Langrish J.P., Raftis J.B., Lucking A.J., Brittan M. Fire simulation and cardiovascular health in firefighters. Circulation. 2017;135:1284–1295. doi: 10.1161/CIRCULATIONAHA.116.025711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kales S.N., Soteriades E.S., Christoudias S.G., Christiani D.C. Firefighters and on-duty deaths from coronary heart disease: a case control study. Environ. Health. 2003;2:14. doi: 10.1186/1476-069X-2-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Shirani J., Meera S., Dilsizian V. The Cardiorenal Axis: myocardial perfusion, metabolism, and innervation. Curr. Cardiol. Rep. 2019;21:60. doi: 10.1007/s11886-019-1147-3. [DOI] [PubMed] [Google Scholar]
- 107.Szczykowska J., Hryszko T., Naumnik B. Cardiac troponins in chronic kidney disease patients with special emphasis on their importance in acute coronary syndrome. Adv. Med. Sci. 2019;64:131–136. doi: 10.1016/j.advms.2018.08.016. [DOI] [PubMed] [Google Scholar]
- 108.Chen J., Budoff M.J., Reilly M.P., Yang W., Rosas S.E., Rahman M. Coronary artery calcification and risk of cardiovascular disease and death among patients with chronic kidney disease. JAMA Cardiol. 2017;2:635–643. doi: 10.1001/jamacardio.2017.0363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sarnak M.J., Levey A.S., Schoolwerth A.C., Coresh J., Culleton B., Hamm L.L. Kidney disease as a risk factor for development of cardiovascular disease: a statement from the American Heart Association Councils on Kidney in Cardiovascular Disease, High Blood Pressure Research, Clinical Cardiology, and Epidemiology and Prevention. Hypertension (Dallas, Tex : 1979) 2003;42:1050–1065. doi: 10.1161/01.HYP.0000102971.85504.7c. [DOI] [PubMed] [Google Scholar]
- 110.Miller-Hodges E., Anand A., Shah A.S.V., Chapman A.R., Gallacher P., Lee K.K. High-sensitivity cardiac troponin and the risk stratification of patients with renal impairment presenting with suspected acute coronary syndrome. Circulation. 2018;137:425–435. doi: 10.1161/CIRCULATIONAHA.117.030320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.van der Linden N., Cornelis T., Kimenai D.M., Klinkenberg L.J.J., Hilderink J.M., Luck S. Origin of cardiac troponin T elevations in chronic kidney disease. Circulation. 2017;136:1073–1075. doi: 10.1161/CIRCULATIONAHA.117.029986. [DOI] [PubMed] [Google Scholar]
- 112.Klinkenberg L.J., Wildi K., van der Linden N., Kouw I.W., Niens M., Twerenbold R. Diurnal rhythm of cardiac troponin: consequences for the diagnosis of acute myocardial infarction. Clin. Chem. 2016;62:1602–1611. doi: 10.1373/clinchem.2016.257485. [DOI] [PubMed] [Google Scholar]
- 113.Mishra P.K., Adameova A., Hill J.A., Baines C.P., Kang P.M., Downey J.M. Guidelines for evaluating myocardial cell death. Am. J. Physiol. Heart Circ. Physiol. 2019;317:H891–h922. doi: 10.1152/ajpheart.00259.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Mahmoud A.H., Taha N.M., Zakhary M., Tadros M.S. PTEN gene & TNF-alpha in acute myocardial infarction. Int J Cardiol Heart Vasc. 2019;23:100366. doi: 10.1016/j.ijcha.2019.100366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kaptoge S., Seshasai S.R., Gao P., Freitag D.F., Butterworth A.S., Borglykke A. Inflammatory cytokines and risk of coronary heart disease: new prospective study and updated meta-analysis. Eur. Heart J. 2014;35:578–589. doi: 10.1093/eurheartj/eht367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Feng N., Anderson M.E. CaMKII is a nodal signal for multiple programmed cell death pathways in heart. J. Mol. Cell. Cardiol. 2017;103:102–109. doi: 10.1016/j.yjmcc.2016.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wu Y., Wang Q., Feng N., Granger J.M., Anderson M.E. Myocardial death and dysfunction after ischemia-reperfusion injury require CaMKIIdelta oxidation. Sci. Rep. 2019;9:9291. doi: 10.1038/s41598-019-45743-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Smith C.C., Davidson S.M., Lim S.Y., Simpkin J.C., Hothersall J.S., Yellon D.M. Necrostatin: a potentially novel cardioprotective agent? Cardiovasc. Drugs Ther. 2007;21:227–233. doi: 10.1007/s10557-007-6035-1. [DOI] [PubMed] [Google Scholar]
- 119.Guo X., Yin H., Li L., Chen Y., Li J., Doan J. Cardioprotective role of tumor necrosis factor receptor-associated factor 2 by suppressing apoptosis and Necroptosis. Circulation. 2017;136:729–742. doi: 10.1161/CIRCULATIONAHA.116.026240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zhou F., Jiang X., Teng L., Yang J., Ding J., He C. Necroptosis may be a novel mechanism for cardiomyocyte death in acute myocarditis. Mol. Cell. Biochem. 2018;442:11–18. doi: 10.1007/s11010-017-3188-5. [DOI] [PubMed] [Google Scholar]
- 121.Weil B.R., Young R.F., Shen X., Suzuki G., Qu J., Malhotra S. Brief myocardial ischemia produces cardiac troponin I release and focal Myocyte apoptosis in the absence of pathological infarction in swine. JACC Basic Transl Sci. 2017;2:105–114. doi: 10.1016/j.jacbts.2017.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Oldham W.M., Oliveira R.K.F., Wang R.S., Opotowsky A.R., Rubins D.M., Hainer J. Network analysis to risk stratify patients with exercise intolerance. Circ. Res. 2018;122:864–876. doi: 10.1161/CIRCRESAHA.117.312482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Leopold J.A., Loscalzo J. Emerging role of precision medicine in cardiovascular disease. Circ. Res. 2018;122:1302–1315. doi: 10.1161/CIRCRESAHA.117.310782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Neubauer S., Kolm P., Ho C.Y., Kwong R.Y., Desai M.Y., Dolman S.F. Distinct subgroups in hypertrophic cardiomyopathy in the NHLBI HCM registry. J. Am. Coll. Cardiol. 2019;74:2333–2345. doi: 10.1016/j.jacc.2019.08.1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kubo T., Kitaoka H., Yamanaka S., Hirota T., Baba Y., Hayashi K. Significance of high-sensitivity cardiac troponin T in hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2013;62:1252–1259. doi: 10.1016/j.jacc.2013.03.055. [DOI] [PubMed] [Google Scholar]
- 126.Kubo T., Kitaoka H., Okawa M., Yamanaka S., Hirota T., Baba Y. Combined measurements of cardiac troponin I and brain natriuretic peptide are useful for predicting adverse outcomes in hypertrophic cardiomyopathy. Circ. J. 2011;75:919–926. doi: 10.1253/circj.cj-10-0782. [DOI] [PubMed] [Google Scholar]
- 127.Baba Y., Kubo T., Yamanaka S., Hirota T., Tanioka K., Yamasaki N. Clinical significance of high-sensitivity cardiac troponin T in patients with dilated cardiomyopathy. Int. Heart J. 2015;56:309–313. doi: 10.1536/ihj.14-335. [DOI] [PubMed] [Google Scholar]
- 128.Campbell M.D., Witcher M., Gopal A., Michele D.E. Dilated cardiomyopathy mutations in delta-sarcoglycan exert a dominant-negative effect on cardiac myocyte mechanical stability. Am. J. Physiol. Heart Circ. Physiol. 2016;310:H1140–H1150. doi: 10.1152/ajpheart.00521.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Han R., Bansal D., Miyake K., Muniz V.P., Weiss R.M., McNeil P.L. Dysferlin-mediated membrane repair protects the heart from stress-induced left ventricular injury. J. Clin. Invest. 2007;117:1805–1813. doi: 10.1172/JCI30848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Han R., Campbell K.P. Dysferlin and muscle membrane repair. Curr. Opin. Cell Biol. 2007;19:409–416. doi: 10.1016/j.ceb.2007.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kuru S., Yasuma F., Wakayama T., Kimura S., Konagaya M., Aoki M. A patient with limb girdle muscular dystrophy type 2B (LGMD2B) manifesting cardiomyopathy. Clin. Neurol. 2004;44:375–378. [PubMed] [Google Scholar]
- 132.Wei B., Wei H., Jin J.P. Dysferlin deficiency blunts beta-adrenergic-dependent lusitropic function of mouse heart. J. Physiol. 2015;593:5127–5144. doi: 10.1113/JP271225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Chase P.B., Szczypinski M.P., Soto E.P. Nuclear tropomyosin and troponin in striated muscle: new roles in a new locale? J. Muscle Res. Cell Motil. 2013;34:275–284. doi: 10.1007/s10974-013-9356-7. [DOI] [PubMed] [Google Scholar]
- 134.Nunez Lopez Y.O., Messi M.L., Pratley R.E., Zhang T., Delbono O. Troponin T3 associates with DNA consensus sequence that overlaps with p53 binding motifs. Exp. Gerontol. 2018;108:35–40. doi: 10.1016/j.exger.2018.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Streng A.S., de Boer D., van der Velden J., van Dieijen-Visser M.P., Wodzig W.K. Posttranslational modifications of cardiac troponin T: an overview. J. Mol. Cell. Cardiol. 2013;63:47–56. doi: 10.1016/j.yjmcc.2013.07.004. [DOI] [PubMed] [Google Scholar]
- 136.Ramirez-Correa G.A., Martinez-Ferrando M.I., Zhang P., Murphy A.M. Targeted proteomics of myofilament phosphorylation and other protein posttranslational modifications. Proteomics Clin. Appl. 2014;8:543–553. doi: 10.1002/prca.201400034. [DOI] [PubMed] [Google Scholar]
- 137.Sumandea M.P., Pyle W.G., Kobayashi T., de Tombe P.P., Solaro R.J. Identification of a functionally critical protein kinase C phosphorylation residue of cardiac troponin T. J. Biol. Chem. 2003;278:35135–35144. doi: 10.1074/jbc.M306325200. [DOI] [PubMed] [Google Scholar]
- 138.van der Velden J., Stienen G.J.M. Cardiac disorders and pathophysiology of Sarcomeric proteins. Physiol. Rev. 2019;99:381–426. doi: 10.1152/physrev.00040.2017. [DOI] [PubMed] [Google Scholar]
- 139.Janssens J.V., Ma B., Brimble M.A., Van Eyk J.E., Delbridge L.M.D., Mellor K.M. Cardiac troponins may be irreversibly modified by glycation: novel potential mechanisms of cardiac performance modulation. Sci. Rep. 2018;8:16084. doi: 10.1038/s41598-018-33886-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Zhang P., Kirk J.A., Ji W., dos Remedios C.G., Kass D.A., Van Eyk J.E. Multiple reaction monitoring to identify site-specific troponin I phosphorylated residues in the failing human heart. Circulation. 2012;126:1828–1837. doi: 10.1161/CIRCULATIONAHA.112.096388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Schindler S.E., Bollinger J.G., Ovod V., Mawuenyega K.G., Li Y., Gordon B.A. High-precision plasma β-amyloid 42/40 predicts current and future brain amyloidosis. Neurology. 2019;93:e1647–ee1659. doi: 10.1212/WNL.0000000000008081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Cheng Y., Chen K.S., Meyer N.L., Yuan J., Hirst L.S., Chase P.B. Functionalized SnO(2) nanobelt field-effect transistor sensors for label-free detection of cardiac troponin. Biosens. Bioelectron. 2011;26:4538–4544. doi: 10.1016/j.bios.2011.05.019. [DOI] [PubMed] [Google Scholar]
- 143.Maron D.J., Hochman J.S., Reynolds H.R., Bangalore S., O’Brien S.M., Boden W.E. Initial invasive or conservative strategy for stable coronary disease. N. Engl. J. Med. 2020;382:1395–1407. doi: 10.1056/NEJMoa1915922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Hai J.J., Wong Y.K., Wong C.K., Un K.C., Chan P.H., Siu C.W. Prognostic implications of statin intolerance in stable coronary artery disease patients with different levels of high-sensitive troponin. BMC Cardiovasc. Disord. 2019;19:168. doi: 10.1186/s12872-019-1152-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Lee J.S., Mukhopadhyay P., Matyas C., Trojnar E., Paloczi J., Yang Y.R. PCSK9 inhibition as a novel therapeutic target for alcoholic liver disease. Sci. Rep. 2019;9:17167. doi: 10.1038/s41598-019-53603-6. [DOI] [PMC free article] [PubMed] [Google Scholar]