

Graphical abstract
Keywords: Anti-carcinogenesis activity, Genome sequencing, HR-ESI-MS, Inoscavin A, Phellinus gilvus, Phenylpropanoids biosynthesis
Abstract
Phellinus gilvus (Schwein.) Pat, a species of ‘Sanghuang’, has been well-documented for various medicinal uses, but the genome information and active constituents are largely unknown. Here, we sequenced the whole-genome of P. gilvus, identified phenylpropanoids as its key anti-cancer components, and deduced their biosynthesis pathways. A 41.11-Mb genome sequence was assembled and the heatmap created with high-throughput chromosome conformation capture techniques data suggested all bins could be clearly divided into 11 pseudochromosomes. Cellular experiments showed that P. gilvus fruiting body was more effective to inhibit hepatocellular carcinoma cells than mycelia. High resolution electrospray ionization mass spectroscopy (HR-ESI-MS) analysis revealed P. gilvus fruiting body was rich in phenylpropanoids, and several unique phenylpropanoids in Phellinus spp. exhibited potent anti-carcinogenesis activity. Based on genomic, HR-ESI-MS information and differentially expressed genes in transcriptome analysis, we deduced the biosynthesis pathway of four major phenylpropanoids in P. gilvus. Transcriptome analysis revealed the deduced genes expressions were synergistically changed with the production of phenylpropanoids. The optimal candidate genes of phenylpropanoids’ synthesis pathway were screened by molecular docking analysis. Overall, our results provided a high-quality genomic data of P. gilvus and inferred biosynthesis pathways of four phenylpropanoids with potent anti-carcinogenesis activities. These will be a valuable resource for further genetic improvement and effective use of the P. gilvus.
Introduction
Phellinus gilvus (Schwein.) Pat, a species of ‘Sanghuang’, belongs to the family Hymenochaetaceae in Basidomycota. Sanghuang (Phellinus spp.) have been used as a Chinese traditional medicine for over 2000 years to treat various diseases such as stomachache, inflammation and tumors. Recent investigations demonstrated that Phellinus spp. have multifunctional bioactivities, including anti-carcinogenesis, anti-inflammatory, anti-oxidative, anti-fungal and immunomodulatory activities, as well as anti-diabetic, hepatoprotective and neuroprotective effects [1]. Phytochemical studies showed that Phellinus spp. are rich in phenylpropanoids and triterpenoids [2]. But the main active components of P. gilvus are still largely unknown.
Sanghuang includes several species of Phellinus spp., which have similar phenotype, but the bioactivities of different origins were distinct. Due to the similar phenotype and limited genomic information, the accurate identification and classification of Sanghuang is difficult [3]. The rapid development of next generation sequencing techniques makes it possible to further explore the fungi at the molecular level. Up to now, many genomes of fungi have been published in the Ensembl fungus database (http://fungi.ensembl.org/index.html). However, the genome information P. gilvus had not been reported. Here we sequenced the whole-genome of P. gilvus strain S12, which was a wild strain gathered from mulberry. A 41.11-Mb genome sequence was obtained, a total of 9982 gene models were annotated, and high-throughput chromosome conformation capture (Hi-C) data suggested that the majority of contigs could map onto 11 pseudochromosomes. High resolution electrospray ionization mass spectroscopy (HE-ESI-MS) and methyl thiazolyl tetrazolium (MTT) assays identified phenylpropanoids as the key components for the anti-carcinogenesis effect of P. gilvus, and their biosynthesis pathways were deduced based on genomic, transcriptomic data and molecular docking analysis.
Materials and methods
Fungal isolation and DNA preparation
P. gilvus strain S12 was isolated from the fruiting body grown in a mulberry tree in Zhejiang province of China (Fig. 1A), which was identified as P. gilvus by Institute of Microbiology, Chinese Academy of Sciences (Beijing, China) based on culture characteristics, microscopic characteristics and rRNA gene sequence analysis. Briefly, it was based on the growth speed, color and morphology of vegetative mycelia, and the gene sequence of rRNA. The rRNA sequence of S12 have been submitted to NCBI (Accession No. MT275660). P. gilvus strain S12 has been deposited in our laboratory (Accession No. 2010S12). A patch of the fruiting body was inoculated into potato dextrose agar (PDA) medium at 28 °C (Fig. 1B). The grown mycelia were inoculated into potato dextrose broth (PDB) medium. Mycelium pellets 10 mm in diameter (Fig. 1C) were removed from media for extraction of genomic DNA. Genomic-tip kit (QIAGEN, Germany) was used for DNA extraction. Briefly, 1 mL of buffer B1 were add to the 20 mg mycelium pellets frozen in liquid nitrogen, and mix well with vortex. 2 μL RNaseA, 40 μL lysozyme and 45 μL Protesae K were added and reverse blending, 0.35 mL buffer B2 was added after incubation 60 min at 37 °C. And centrifugation at 12,000 rpm for 5 min after incubation 60 min at 50 °C. The supernatant were added to tips for DNA purification according to the handbook. The purified DNA were used for library construction.
Fig. 1.

The separation and cultivation of P. gilvus. (A) Fruiting body grown on the mulberry tree; (B) Mycelia to colony; (C) Proliferating mycelium pellets in PDA liquid medium; (D) Artificial media used to culture of P. gilvus.
Genome size and heterozygosity estimation
The 350-bp library was constructed according to Illumina’s standard protocol and 2100 Bioanalyzer (Agilent, USA) was used for quantifying. The 350-bp library was subjected to paired-ended 150-bp sequencing by Illumina NovaSeq 6000 (CA, USA). The genome size of P. gilvus was estimated by the k-mer method [4]. Quality-filtered reads were subjected to 17-mer frequency distribution analysis using the Jellyfish program. The genome size was estimated using the following formula: Genome size = k-mer number/average k-mer depth. The average k-mer depth is the k-mer depth corresponding to the main peak. The sequence where the k-mer depth appears at half of the corresponding depth of the main peak is a heterozygous sequence, and the sequences where the k-mer depth is more than twice the corresponding depths of the main peak are repetitive sequences.
Genome sequencing and assembly
P. gilvus strain S12 genome was sequenced by Single Molecule Real-Time (SMRT) method [5]. DNA libraries with 20-kb inserts were constructed. The 20-kb library was constructed according to PacBio’s standard methods and 2100 Bioanalyzer was used for quantifying. The sequencing data (filtered reads: 13.99-Gb, sequencing depth: 340.28×) was assembled by CANU (version 1.5) with default parameters [6]. Quiver software is then used to polish the assembled genome [5]. Finally, Illumina reads above were used for error correction and gap filling with Pilon [7]. The integrity of the fungal genome assembly were evaluated by Benchmarking Universal Single-Copy Orthologs (BUSCO, version 2.0) [8].
Hi-C assembly
To anchor scaffolds onto the chromosome, we conducted Hi-C auxiliary assembly. Sample preparation was according to Belton et al and Liu et al [9], [10]. Fresh mycelium pellets 10 mm in diameter (Fig. 1C) were removed from media for nuclei isolation. The purified nuclei were digested by Hind III and then were incubated with biotin-14-dCTP. The ligated DNA was cut into 300–700-bp fragments, and then was purified by biotin-streptavidin-mediated pull down as Hi-C library. Finally, we obtained sequencing data (5.68-Gb, 138.17×) via the NovaSeq 6000 platform (Illumina, CA, USA). The valid interaction pairs were acquired using Bowtie2 in HiC-Pro version 2.9.0 [11]. BWA (version 0.7.15) [12] was used to map the Hi-C short reads obtained from the Illumina HiSeq platform against the draft genome. The comparison mode was “aln” and the other parameters were set to the defaults. Division, sorting and orientation of the genome sequences were evaluated by LACHESIS, with parameters CLUSTER_MIN_RE_SITES = 41, CLUSTER_MAX_ LINK_DENSITY = 1, CLUSTER_NONINFORMATIVE_RATIO = 5, ORDER_MIN _N_RES_IN_TRUN = 10 and ORDER_MIN_N_RES_IN_SHREDS = 10, Keep the defaults for all the other parameters [13], [14]. We used the Hi-C interacted signals to correct assembling errors. The genome assembled to the chromosome was cut into 20-Kb of one bin, the number of Hi-C mapped read pairs between any two bin was the signal intensity of the interaction. The heatmap was drawn according to the signal intensity of any two bins’ interaction. The intensity signal was measured by the number of mapped read pairs between the two bins.
Genetic component analysis
Tandem repeat sequences were identified using LTR_FINDER version 1.05 [15], MITE-Hunter [16], RepeatScout version 1.0.5 [17] and PILER-DF version 2.4 [18]. PASTE Classifier [19] was used to classify the database, which was then combined with the database of Repbase as the final repeat sequence database [20]. RepeatMasker version 4.0.6 software was used to predict the repeat sequence of the fungus based on the established repeat sequence database [21]. The software tRNAscan-SE was used to predict the tRNA in the genome [22], and the software Infernal version 1.1 was used to predict the rRNA in the genome [23] and other ncRNAs based on the Rfam database [24]. The predicted protein sequences were compared with the protein sequences in the swiss-prot database [25], and the homologous gene sequences (possible genes) were searched in the genome by using the software GenBlastA [26], and then the immature termination codon and code shift mutations in the gene sequences were searched by using the software GeneWise to obtain the pseudogenes [27].
Genome annotations
The prediction of gene structure was mainly based on ab initio prediction and homologous protein prediction. Genscan, Augustus (version 2.4), GlimmerHMM (version 3.0.4), GeneID (version 1.4) and SNAP (version 2006-07-28) were used for ab initio prediction [28], [29], [30]. GeMoMa (version 1.3.1) was used for prediction based on homologous proteins [31]. Finally, EVidenceModeler (version 1.1.1) was used [32]. The predicted gene sequences were annotated functionally by COG, KEGG, swiss-prot, TrEMBL and Nr databases [25], [33], [34], [35]. Based on Nr database, GO databases were annotated by Blast2GO [36]. Hmmer software (version 3.2.1) was used for function annotation based on Pfam database [37], [38]. The predicted gene protein sequences were annotated by functional databases antiSMASH (version 5.0, https://antismash.secondarymetabolites.org/) with default parameter [39].
Homologous gene analysis and phylogenetic tree construction
OrthoVenn2 (https://orthovenn2.bioinfotoolkits.net/) was used to compare and annotate the orthologous gene clusters among the four fungi in Hymenochaetaceae [40]. The parameter settings of E-value = 1e−5, Inflation value = 1.5. OrthoMCL (version 2.0.9) was used to cluster the protein sequences [41]. All-vs-all blastp reciprocal best hit was used for all proteins, and E-value = 1e−5. According to the results of gene family clustering, single copy orthologous genes were extracted, and multiple sequence alignment of single copy homologous proteins were conducted by MUSCLE software. Finally, phylogenetic tree is constructed by PhyML, with the JTT model and 100 bootstrap replicates [42]. We then used MCMCTREE to estimate the divergence times [43], and the parameter were as follows: clock = 2, model = 3, BDparas = 110, kappa gamma = 62, alpha gamma = 11, rgene gamma = 11 and sigma2 gamma = 14.5. The sequence information of these fungi were acquired from Ensembl fungus database, the accession numbers were GCA_002287475 (Phellinus noxius), GCA_001020605 (Schizopora paradoxa), GCA_000271605 (Fomitiporia mediterranea), GCA_002760635 (Ganoderma sinense), GCA_000344655 (Fomitopsis pinicola), GCA_000143185 (Schizophyllum commune), GCA_002003045 (Lentinula edodes) and GCA_001632375 (Exidia_glandulosa).
Transcriptome sequencing
The mycelia (Fig. 1C) and fruiting body (Fig. 1D) were prepared for the transcriptome sequencing. There were three biological replicates for each sample. TRIzol reagent (Invitrogen, USA) were used for isolation of Total RNA. Agilent 2100 Bioanalyzer, a NanoDrop (NanoDrop, USA), and a Qubit 2.0 (Invitrogen, USA) were used to assess the integrity, purity and concentration of total RNA. The libraries’ construction and RNA-Seq were conducted by the Biomarker Biotechnology Corporation (Beijing, China). The HISTAT2 was used to map clean reads (>100-bp) to the genome with the default parameters [44]. The mapped reads were assembled to transcripts via StringTie [45]. The transcript or gene expression levels were measured by FPKM (Fragments Per Kilobase of transcript per Million fragments mapped) [46]. The Fold Change (FC) ≥ 2 and False Discovery Rate (FDR) less than 0.01 were identified as differentially expressed genes (DEGs).
Preparation of extracts of P. gilvus mycelia and fruiting body
The dried fruiting body and mycelia powder was extracted with boiling water for 2 h. The aqueous extracts were concentrated by rotary evaporator and then were mixed with three volumes of ethanol. The supernatant was collected after centrifugation. The supernatant was lyophilized to obtain extracts powders.
High performance liquid chromatography (HPLC) and HE-ESI-MS analysis
The extracts (1 g) of power were desalted through a C18 column (H2O, methanol), and then dissolved in 75% methanol. HPLC was performed on an Agilent 1260 HPLC system. Chromatographic separation was performed on an Exsil Plus EPS C18 (5 μM, 4.6 × 250 mm). The sample size was 10 µL. The mobile phase A was waterwith 0.3% formic acid and mobile phase B was acetonitrile with 0.3% formic acid. A mobile phase gradient was used with the percentage of B in A varying as follows: initial concentration (0 min), 5% B; 15 min, 10% B; 20 min, 20% B; 35 min, 30% B; 60 min, 50% B. The flow rate was 1 mL/min, and the column temperature was maintained at 30 °C. The eluted fractions of each peaks were collected and analyzed by HE-ESI-MS (an Agilent G6224A TOF spectrometer). The TOF mass spectrometer with an ESI interface (Agilent, USA) were used for HE-ESI-MS experiments. The positive ESI conditions were as follows: gas temperature = 350 °C, drying gas = 9 L/min, nebulizer = 40 psig, capillary = 3500.
MTT assays
The inhibitory effects of extracts on the human hepatocellular carcinoma (HepG2) cells were measured by the analysis of live cells number determined with a MTT-based colorimetric assay according to Li et al [47]. Briefly, the cells (1 × 105 cells/well) were added to a 96-well plate, with different final concentration of extracts or dimethyl sulfoxide (DMSO, negative control). After cultivation for 48 h, the percentage of living cells was determined by reading absorbance at 570 nm with a Benchmark microplate reader (Bio-Rad, USA). The inhibition rate (%) = 1 − (mean absorbency in test wells)/(mean absorbency in control wells) × 100%.
Molecular docking
The possible interactions between each isolated compound and the corresponding enzyme binding sites were analyzed by Molecular Operating Environment (MOE) software package (Chemical Computing Group, Canada). A molecular modeling study was performed using the docking program named Induced-Fit, a refinement method in software MOE. To eliminate any bond length and bond angle biases, the ligands were subjected to an “energy minimize” prior to docking. The binding affinities (S-values) in MOE were used to evaluate the interactions between proteins and ligands. The scores (binding affinities) were obtained based on the virtual calculation of various interactions of the ligands with the targeted receptors [48].
Results
Genome size and heterozygosity estimation
P. gilvus strain S12 was used for genome sequencing. 2.69-Gb high-quality data (~80×) were obtained. Based on the k-mer genome survey analysis, P. gilvus was estimated to have a genome size of 33.36-Mb (Table 1). k-mer analysis with a length of 17 (17-mers) indicated the genome had a heterozygosity of 0.52% and a repetitive sequence content of ~16.63% (Fig. 2A), which indicated that P. gilvus strain S12 was a binuclear fungus.
Table 1.
Statistics for the P. gilvus Genome.
| Features | Number | Length | Percentage (%) |
|---|---|---|---|
| Estimated genome size | 33.36-Mb | ||
| Assembled contig sequences | 73 | 41.11-Mb | |
| Contig N50 | 2.37-Mb | ||
| Contig N90 | 132.69-Kb | ||
| Contig Max | 4.15-Mb | ||
| Total read pairs (Hi-C) | 19,013,106 | ||
| Mapped reads (Hi-C) | 30,520,075 | 80.26 | |
| Unique mapped read pairs(Hi-C) | 9,838,035 | 51.74 | |
| Assembled scaffold sequences | 79 | 41.11-Mb | |
| Scaffold N50 | 2.42-Mb | ||
| Scaffold N90 | 119.48-Kb | ||
| Scaffold Max | 6.61-Mb | ||
| Anchored scaffolds | 67 | 35.09-Mb | 85.36% |
| Gap length | 2.30-Kb | ||
| GC content (%) | 47.89 | ||
| Chromosome | 11 |
Fig. 2.

General features of the P. gilvus genome sequencing. (A) The k-mer distribution. (B) Subreads length distribution. The abscissa represents the length of the subreads (bp); the left ordinate represents the number of subreads, which corresponds to the green bar graph; the right ordinate represents the total number of bases (Mb) of subreads larger than the corresponding length; and the red dashed line represents the length of the N50 of the subreads.
Genome sequencing, assembly and annotation
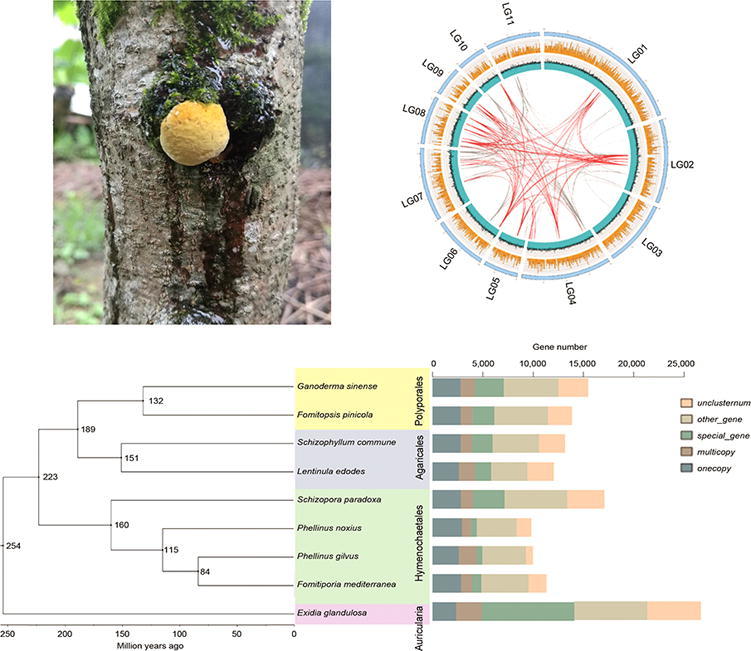
For accurate assembly information of the reference genome, the P. gilvus strain S12 genome was sequenced using a de novo assembly strategy combining PacBio single-molecule long reads with Hi-C auxiliary assembly. A 41.11-Mb genome sequence was obtained by assembling 982,223 PacBio reads (~340×), which is larger than the estimated genome size of 33.36-Mb obtained from the k-mer analysis (Fig. 2B). The draft genome assembly consisted of 73 contigs with a contig N50 of ~2.37-Mb (Table 1). 273 complete conservative core genes (94.14%) were found in fungal BUSCO. In order to improve the draft genome from genome-assembly level to chromosome level, we used LACHESIS to assemble contigs onto 11 pseudochromosomes based on 5.68-Gb, (~138×) of Hi-C data (Fig. 3A). The alignment efficiency between mapped reads and the assembled genome is 80.26%, among which the ratio of unique mapped read pairs is 51.74%. A heatmap created with Hi-C data suggested that all bins could be clearly divided into the 11 pseudochromosomes, which was consistent to the LACHESIS assembly. Within each group, the interaction intensity at diagonal position is higher than that at non-diagonal position were observed, which indicated that the interaction intensity between adjacent sequences was high (Supporting Information Fig. S1). In turn, parts of the contigs were interrupted because of lacking interaction signals in heatmap. As a result, the genome assembly by Hi-C consisted of 79 scaffolds with a scaffold N50 of ~2.42-Mb, the gap was 2.3-Kb. A total of 35.09-Mb (85.36%) sequence was anchored to the 11 pseudochromosomes. We finally generated a 41.11-Mb of P. gilvus genome, with a contig N50 of ~2.37-Mb and scaffold N50 of ~2.42-Mb (Table 1).
Fig. 3.

Genomic characterization and comparative genomics of P. gilvus. (A) The outer circle is the chromosome marked with the number, the scale value unit is Mb; The second circle (yellow) is gene density, which counts the number of genes per 10-kb of genome length. The third circle (blue) is GC content, and the GC percentage per 1-kb length of genome is calculated, the minimum scale value is 20%, and the maximum scale value is 80%. The innermost line is the corresponding relation of repeated sequences. The gray line is the repeated sequence of 5–10-kb, and the red line is the repeated sequence of more than 10-kb. (B) The Venn diagram indicates the numbers of genes shared among the four Hymenochaetales; (C) Phylogenetic tree of P. gilvus with 8 other fungi species genome covering 4 fungi orders (Hymenochaetales, Polyporales, Agaricales, and Auricularia) in Agaricus. The histogram is the statistics of gene types.
A total of 9982 gene models were predicted (Supporting Information Table S1), with an average sequence length of 2367.12-bp. On average, each predicted gene contains 7.31 exons and 6.31 introns (Supporting Information Table S2). 9521 genes could be annotated by functional databases (GO, KEGG, COG, Pfam, Swiss-prot, TrEMBL and Nr) (Supporting Information Table S3). According to GO database, annotated genes were mainly distributed in 4 functional entries, “Catalytic activity”, “Binding”, “Metabolic process” and “Cellular process” (Supporting Information Fig. S2A). By NCBI COG mapping, 6056 proteins were assigned to COG categories (Supporting Information Fig. S2B). “General functional prediction only” had the highest number of genes (1028), which were not unambiguously assigned to a particular group. This was followed by “posttranslational modification, protein turnover, chaperones” (542) and “signal transduction mechanisms” (488) as the most gene-rich classes in the COG groupings. 288 genes involved in “Secondary metabolites biosynthesis, transport and catabolism” were identified. These findings are consistent with the results of P. gilvus rich in secondary metabolites.
In total, 8.57-Mb (20.85%) of repetitive sequences were identified, the majority of the repeats are Class 1 (18.21% of the genome; Supporting Information Table S4). 8230 (82.45%) genes were anchored to the 11 pseudochromosomes. In addition, 149 noncoding RNAs and 525 pseudogenes were predicted.
Comparative genomic and phylogenomic analyses
The OrthoVenn2 was used to identify orthologous genes among P. gilvus and 3 other fungi species in Hymenochaetales, and a total of 5099 orthologous genes and 268 unique genes were identified (Fig. 3B). Among the 268 unique genes, 214 genes could be functionally annotated by GO database (Supporting Information Fig. S3). According to the annotation, these genes mainly distributed in biological process (25), metabolic process (18) and cellular metabolic process (13). The molecular function mainly involved in oxidoreductase activity (11), binding (6) and transferase activity (6). The most genes (7) of GO enrichment is involved in oxidoreductase activity, acting on paired donors, with incorporation or reduction of molecular oxygen, which were involved in secondary metabolites biosynthesis. All these data suggested P. gilvus might have better medicinal activity.
To investigate the evolutionary position of P. gilvus in Agaricus, we compared and identified orthologous genes among P. gilvus and 8 other fungi species covering 4 fungi orders (Hymenochaetales, Polyporales, Agaricales and Auricularia) in Agaricus by OrthoMCL. A total of 1538 single-copy orthologous genes were identified. We inferred a phylogeny and divergence estimate using 13,842 orthologous genes. The phylogenetic analysis showed that P. gilvus was more closely related to Fomitiporia mediterranea, and the estimated divergence time of P. gilvus and F. mediterranea was 84 millions of years ago (Mya). P. gilvus diverged from Polyporales and Agaricales was about 223 Mya (Fig. 3C).
Phenylpropanoids may be the key anti-carcinogenesis components in P. gilvus
In order to further investigate the medicinal values of P. gilvus, we carried out HepG2 cells inhibitory assay among mycelia and fruiting body. The inhibitory effects of extracts were tested on HepG2 cells under dosages of 400 and 800 μg/mL. As shown in Fig. 4A, P. gilvus fruiting body exhibited a more potent inhibition on HepG2 cells growth than mycelia. At the doses of 400 and 800 μg/mL, the inhibition rate of P. gilvus fruiting body extracts were 34.14% and 77.38% on HepG2 cells, compared to 29.14% and 41.37% of mycelia, respectively.
Fig. 4.

Analysis of medicinal values of P. gilvus. (A) The inhibitory efficiency of extracts on the proliferation of HepG2 hepatocarcinoma cells. (B) HPLC analyses of different medical fungi extracts. The indicated compounds are as follows: (I) P. gilvus mycelia, (1) (10E, 12Z, 15Z)-9-hydroxy-10, 12, 15-octadecatrienoic, (2) 9-deoxy-11β-hydroxyprostratin, (3) unknown compound and (4) unknown compound; (II) P. gilvus fruiting body, (1) β-Adenosine, (2) 5,6-Dihydrouracil, (3) 3,4-Dihydroxybenzalacetone, (4) Hydroxycinnamic acid; (5) Phellibaumin D, (6) Interfungin B, (7) Phelligridimer A, (8) Gliotoxin and (9) Inoscavin A. (C) The inhibitory efficiency of five phenylpropanoids from P. gilvus on the proliferation of HepG2 hepatocarcinoma cells. (D) Morphology of HepG2 cells after treatment with different phenylpropanoids from P. gilvus. The dosage of P3, P5, P6 and P7 was 80 μg/mL, P9 was 20 μg/mL. Date are presented as the mean ± SE (n = 5).
To clarify the cause for the differential anti-carcinogenesis efficacies, the compounds were analyzed by HPLC and HE-ESI-MS. As shown in Fig. 4B, the main bioactive compounds of P. gilvus fruiting body were phenylpropanoids (3, 4-dihydroxybenzalacetone, hydroxycinnamic acid, phellibaumin D, interfungin B, phelligridimer A and inoscavin A), and P. gilvus mycelia possessed scarce secondary metabolites (Supporting Information Tables S5 and S6, Data S1).
To confirm the anti-carcinogenesis activity of phenylpropanoids in P. gilvus fruiting body, five phenylpropanoids compounds (3, 4-dihydroxybenzalacetone, phellibaumin D, interfungin B, phelligridimer A and inoscavin A) were isolated for MTT assays, some of which were unique in Phellinus spp. [1]. As is shown in Fig. 4C and D, all five compounds exhibited adequate and dose-dependent anti-carcinogenesis activities in vitro. At the dose of 80 μg/ml, the inhibition rate of the five compounds were 47.33%, 55.06%, 44.50%, 24.68% and 82.27%, respectively. These data suggested that phenylpropanoids might be the key anti-carcinogenesis component in P. gilvus fruiting body.
Synthesis pathway of phenylpropanoids in P. gilvus
Some studies have found that secondary metabolic-related synthesis genes exist in clusters [49], so we annotated the genome by AntiSMASH database, which allows the rapid identification secondary metabolite biosynthesis gene clusters in fungal genomes [39], as the Table S7 shown, the gene clusters of P. gilvus were classified into terpene, NPRS-like and T1PKS, but there were no phenylpropanoids biosynthetic gene clusters were annotated.
In order to further explore the biosynthetic pathways of the active phenylpropanoids, RNA-Seq analysis of P. gilvus samples collected from mycelia (Fig. 1C) and fruiting bodies were performed (Fig. 1D). A total of 53.17-Gb clean data were obtained after mRNA sequencing for 6 samples with at least 8.35-Gb clean data for each sample. Percentage of mapped clean reads compared to all clean reads varied from 89.29% to 96.46% (Supporting Information Table S8). A total of 3435 DEGs (1741 enriched in fruiting body and 1694 enriched in mycelia) were identified. By NCBI COG mapping, of the 1741 fruiting body-enriched genes, “General functional prediction only” (125), “Carbohydrate transport and metabolism” (106) and “Secondary metabolites biosynthesis, transport and catabolism” (85) are the most gene-rich classes (Supporting Information Fig. S4).
HPLC and HE-ESI-MS results showed that P. gilvus fruiting body contained all the five phenylpropanoids while the mycelia contained none of them (Fig. 4B, Supporting Information Tables S5 and S6, Data S1). PAL, cinnamate-4-hydroxylase (C4H), p-coumarate 3-hydroxylase (C3H) and 4CL are four key enzyme genes, which are responsible for caffeoyl-CoA from phenylalanine. Caffeoyl-CoA are important precursor for various phenylpropanoids biosynthesis [50]. Therefore we examined the expression levels of genes that are involved in phenylpropanoids biosynthesis. Among the phenylpropanoids-biosynthetic genes, 4 were enriched in the fruiting body (PAL, 3/3; 4CL, 1/3). There no rigorous cinnamate-4-hydroxylase (C4H), p-coumarate 3-hydroxylase (C3H) genes could be annotated by KEGG. Because both C4H and C3H belong to cytochrome P450 monooxygenase superfamily [51], we identified several cytochrome P450 monooxygenase genes (P450s, 11/33) that may be involved in phenylpropanoids biosynthesis, which were enriched in the fruiting body (Supporting Information Table S10). These results suggested that the above genes might be involved in the synthesis of phenylpropanoids.
According to Lee and Yun, Hispidin, hispolon and 3, 4-dihydroxybenzalacetone were derived from caffeoyl-CoA, which were important intermediate products for biosynthesis of phenylpropanoids. Caffeoyl-CoA was biosynthesized via cinnamic acid, p-coumaric acid pathway [50]. Hispidin might be biosynthesized via enlongation and self-cyclization of caffeoyl-CoA, with cooperation of polyketide synthase (PKS) and palmitoyl protein thioesterase (PPT) genes. And hispolon might be biosynthesized via enlongation, decarboxylation and dehydrogenation of caffeoyl-CoA, with cooperation of PKS, PPT and decarboxylase genes. Under the action of dehydrogenase, hispolon turns to its quinone structure. Then, additional reaction happened between these two compounds, later intramolecular addition reaction happened to get a more stable compound inoscavin A [50], [52]. The putative genes involved in inoscavin A biosynthesis, that are PAL and 4CL, could be annotated in P. gilvus genome (Supporting Information Table S9), and four intermediates were found in our study and other researches [1], [50]. Cytochrome P450 monooxygenase genes, PKS, PPT, decarboxylase and dehydrogenase genes, which might be involved in biosynthesis of inoscavin A, were also enriched in the fruiting body (Supporting Information Table S9). Based on the data above, we deduced the biosynthetic pathways of inoscavin A (Scheme 1). In the same way, we inferred that 3, 4-dihydroxybenzalacetone might be synthesized from caffeoyl-CoA by elongation and decarboxylation (Supporting Information Scheme S1), phellibaumin D was synthesized by the coupling of hispidin and oxidation product of caffeic acid (Supporting Information Scheme S2), interfungin B was synthesized by the coupling of hispidin and oxidation product of 3, 4-dihydroxybenzalacetone (Supporting Information Scheme S3).
Scheme 1.

Key steps in inoscavin A biosynthesis. Catalyzed by the enzymes phenylalanine ammonia-lyase (PAL), cytochrome P450 monooxygenase (P450), 4-coumarate-CoA ligase (4CL), polyketide synthase (PKS), palmitoyl protein thioesterase (PPT) and some presumed enzymes. Several representative intermediates are shown.
In order to narrow the scope of candidate genes in the synthesis pathway, the possible interactions between each isolated compound and the corresponding enzyme binding sites were analyzed by molecular docking. Since the major sequence similarities of the P. gilvus-derived enzymes to the proteins with known structures are less than 20%, we could not use homology modeling methods for docking analysis. So we had to use the most similar proteins from protein data bank (PDB) database to conduct molecular docking analysis, thus providing clues of the potential binding of the compounds to their catalytic enzymes. The S-values were obtained based on the virtual calculation of the interaction of ligands with the targeted proteins. Higher absolute value of S score indicates stronger binding between compound and protein. The |S score| > 5 indicates adequate binding affinity while |S score| > 6 suggests a strong interaction [48]. As the Table 2 shown, the optimal candidate enzymes, such as P450, PKS, PPT, decarboxylase and dehydrogenase, were filtrated with S-values −5.29, −5.01, −8.47, −10.91, −6.75 and −6.61 respectively (Table 2, Fig. 5 and Supporting Information Data S2).
Table 2.
The optimal candidate genes obtained by molecular docking analysis in Inoscavin A biosynthesis.
| Gene ID | Gene name | PDB ID | Compound ID | S score |
|---|---|---|---|---|
| EVM0000919 | PAL | 1W27 | 1 | −4.63 |
| EVM0001849 | P450 | 2Q9F | 2 | −5.29 |
| EVM0002405 | P450 | 2Q9F | 2 | −5.29 |
| EVM0003813 | P450 | 2Q9F | 2 | −5.29 |
| EVM0004734 | P450 | 2Q9F | 2 | −5.29 |
| EVM0006460 | P450 | 2Q9F | 2 | −5.29 |
| EVM0007121 | P450 | 2Q9F | 2 | −5.29 |
| EVM0009092 | P450 | 2Q9F | 2 | −5.29 |
| EVM0009176 | P450 | 2Q9F | 2 | −5.29 |
| EVM0001849 | P450 | 2Q9F | 3 | −5.01 |
| EVM0002405 | P450 | 2Q9F | 3 | −5.01 |
| EVM0003813 | P450 | 2Q9F | 3 | −5.01 |
| EVM0004734 | P450 | 2Q9F | 3 | −5.01 |
| EVM0006460 | P450 | 2Q9F | 3 | −5.01 |
| EVM0007121 | P450 | 2Q9F | 3 | −5.01 |
| EVM0009092 | P450 | 2Q9F | 3 | −5.01 |
| EVM0009176 | P450 | 2Q9F | 3 | −5.01 |
| EVM0006638 | 4CL | 2D1S | 4 | −6.2 |
| EVM0007106 | PKS | 2QO3 | 5 | −8.47 |
| EVM0004239 | PPT | 1C8U | 6/7 | −10.91 |
| EVM0001166 | Decarboxylase | 2P1F | 9 | −6.75 |
| EVM0002689 | Dehydrogenase | 4QI3 | 10 | −6.61 |
| EVM0002854 | Dehydrogenase | 4QI3 | 10 | −6.61 |
Fig. 5.

Molecular docking analysis of Inoscavin A biosynthesis related genes. (A) Molecular docking analysis of polyketide synthase (PKS, EVM0007106) and its corresponding substrate. (B) Molecular docking analysis of palmitoyl protein thioesterase (PPT, EVM0004239) and its corresponding substrate.
Discussion
Sanghuang are traditional medicinal fungi, mainly including P. igniarius, P. baumii, P. linteus and so on [3]. Here we found a wild species parasitic on mulberry, the fungus was identified as P. gilvus by Institute of Microbiology, Chinese Academy of Sciences (Beijing, China) based on culture characteristics, microscopic characteristics and rRNA gene sequence analysis. We estimated the genome size by k-mer analysis and found that the estimated genome size was 33.36-Mb and it was a binuclear fungus (Fig. 2A). Because of the high heterozygosity, the short reads produced by Illumina platform might be assembled incorrectly. In detail, binuclear fungus may contain two sets of highly homologous genomes, the short reads produced by Illumina platform might be assembled the two homologous genomes into one genomes by mistake, resulting a smaller and incorrect genome size was estimated. The SMRT technology conducted by PacBio platform can deliver more highly accurate long reads [5]. Therefore we conducted genome sequencing combining PacBio long-reads sequencing with Hi-C auxiliary assembly. The sequencing data generated by PacBio platform showed the draft genome size was 41.11-Mb, consisting of 73 contigs with a contig N50 of ~2.37-Mb (Fig. 2B, Table 1). Genome assembly de novo from PacBio’s reads also might exist a few incorrect assembly because of the high heterozygosity. Hi-C data is a rich source of long-range information for assigning, ordering and orienting draft-genome sequences to chromosomes, and it can also interrupt incorrect assembly according to the interaction intensity [14]. It can perfectly cooperate with PacBio to complete high-quality genome assembly. As a result, 73 contigs were re-assembled into 79 scaffolds. The scaffold N50 was ~2.42-Mb and the gap length was 2.30-Kb. Hi-C auxiliary assembly suggested that P. gilvus had 11 pseudochromosomes. A total of 35.09-Mb (85.36%) sequence was anchored to the 11 pseudochromosomes (Table 1). 273 complete conservative core genes (94.14%) were found in fungal BUSCO. All these proved that PacBio platform with Hi-C auxiliary assembly helped us to obtain a more high-quality genome data. It is also the first report of P. gilvus genome.
Comparative genomics analysis in Hymenochaetales showed P. gilvus had 268 unique genes (Fig. 3B). The richest genes were related to oxidoreductase activity, which might involve in secondary metabolites biosynthesis (Supporting Information Fig. S3). This might be the reason why C4H and C3H genes, which had oxidoreductase activity, could not be annotated in P. gilvus genome. These founding implied that P. gilvus had unique medicinal values.
Phenylpropanoids are the most representative and predominant type of bioactive compounds in Phellinus spp. [1]. Chao et al [53] found that 3, 4-dihydroxybenzalacetone could suppress human non-small cell lung carcinoma cells metastasis. Hispidin exerted antitumor effects against pancreatic cancer cells, gastric cancer cells, HepG2 and human erythroleukemia cells [54], [55], [56]. Hispolon could induce apoptosis in melanoma cells and inhibit the growth of human breast cancer cells [57], [58]. Phellifuropyranone A, meshimakobnol A and meshimakobnol B showed anti-proliferative activity against mouse melanoma cells and human lung cancer cells in vitro [59]. Zhang et al [60] found that 3, 4-dihydroxybenzalacetone and inoscavin A from P. baumii showed proliferation inhibition activity on tumor cells. However, there is no report on anti-carcinogenesis activity of the other phenylpropanoids compounds in Phellinus spp. Here we isolated five major phenylpropanoids (3, 4-dihydroxybenzalacetone, phellibaumin D, interfungin B, phelligridimer A and inoscavin A) in P. gilvus and tested their anti-carcinogenesis activity. Four compounds exhibited higher anti-carcinogenesis activities in vitro. Some of them were newly high activity anti-carcinogenesis compounds in P. gilvus.
It has not been reported for the synthetic pathways of phenylpropanoids in P. gilvus so far. Nambudiri et al [61] proposed that caffeic acid was the precursors of hispidin, which is an important intermediate in the synthesis of other phenylpropanoids. Lee and Yun [50] reviewed that biosynthesis of hispidin was the incorporation of two acetate units and L-phenylalanine in Polyporus schweinizii. Tracer studies showed incorporation of D, L-phenylalanine, D, L-tyrosine, cinnamate, p-coumarate and caffeate into the styryl unit, and efficient incorporation of acetate and malonate into the pyrone ring [62]. According to the results of the integrated metabolome and proteome study in P. gilvus, the increase of polyphenol content was caused by upregulation of the phenylpropanoid biosynthesis, such as PAL, C4H and 4CL [56]. PAL, tyrosine ammonia-lyase (TAL), C4H, and C3H are key genes for caffeic acid biosynthesis. PAL, could be identified in genome of P. gilvus, however we could not identify TAL according to the gene annotation of P. gilvus, suggested that the phenylpropanoids were phenylalanine-derived in P. gilvus (Supporting Information Table S9). Hispidin and hispolon derived from caffeyl-CoA and were the result of elongation by malonyl-CoA [50], [63]. The PKS and PPT are key enzymes for the elongation (Supporting Information Table S9). Inoscavin A was considered to be synthesized by the coupling of hispidin and quinone structure of hispolon [50], [64].
Phenylpropanoids biosynthetic gene clusters could not be identified according to antiSMASH analysis, excepting several NPRS and T1PKS clusters, which might involve in elongation of caffeyl-CoA (Supporting Information Table S7). So we conducted RNA-seq for further exploring the biosynthetic pathways of the bioactive phenylpropanoids. DEGs analysis showed that plenty of genes including PAL, 4CL, P450, PKS, PPT, decarboxylase and dehydrogenase genes probably involved in the bioactive phenylpropanoids biosynthesis were enriched (Supporting Information Table S9), which were synergetically changed with the production of these phenylpropanoids. Based on the above information, we speculated on the biosynthesis pathway of inoscavin A in P. gilvus (Scheme 1). In the same way, we also deduced the biosynthetic pathways of the other three high bioactive phenylpropanoids (3, 4-dihydroxybenzalacetone, phellibaumin D and interfungin B; Supporting Information Schemes S1–S3).
In order to screening for optimal candidate genes in the phenylpropanoids’ synthesis pathway, we conducted molecular docking analysis. The S-values of PKS, PPT, decarboxylase and dehydrogenase enzymes, were −8.47, −10.91, −6.75 and −6.61 respectively (Table 2, Fig. 5 and Supporting Information Data S2), which suggested a strong interaction between each isolated compound and the corresponding enzyme. These data proved that the synthetic pathways of these phenylpropanoids were reliable, and these will provide important resources for the exploration of the synthetic pathway of phenylpropanoids in P. gilvus.
Conclusions
This paper reports a high quality whole-genome sequence of a new species of Sanghuang, P. gilvus strain S12, for the first time. We also confirmed that phenylpropanoids were the key anti-carcinogenesis components in P. gilvus. And four high bioactive phenylpropanoids’ biosynthetic pathways were deduced. These data generated in this work will be a valuable resource for further genetic improvement and effective use of the P. gilvus. And it also provided theoretical bases for the development of new anti-carcinogenesis drugs and synthetic biology.
Compliance with ethics requirements
This article does not contain any studies with human or animal subjects.
Data availability statement
The NCBI SRA Bioproject (PRJNA608208) included whole-genome assembly sequence, raw data of genome-seq, Hi-C and RNA-seq. All sequencing data involved in this work can be acquired at NCBI.
Author contributions
Huo J, Wu C and Li Y contributed to the conception of the study. Zhong S, Du X, Cao Y, Wang W, Sun Y., Tian Y., Zhu J and Chen J carried out the experiments. Huo J, Wu C and Li Y contributed significantly to data analysis and manuscript preparation. Xuan L helped perform the analysis with constructive discussions.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was supported financially by Science and Technology Department of Zhejiang Province (2018C02003).
Footnotes
Peer review under responsibility of Cairo University.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jare.2020.04.011.
Appendix A. Supplementary material
The following are the Supplementary data to this article:
References
- 1.Chen W., Tan H., Liu Q., Zheng X., Zhang H., Liu Y. A review: The bioactivities and pharmacological applications of Phellinus linteus. Molecules. 2019;24(10):1888. doi: 10.3390/molecules24101888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Suabjakyong P., Saiki R., Van Griensven L.J., Higashi K., Nishimura K., Igarashi K. Polyphenol extract from Phellinus igniarius protects against acrolein toxicity in vitro and provides protection in a mouse stroke model. PLoS ONE. 2015;10(3) doi: 10.1371/journal.pone.0122733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wu S., Dai Y., Hattori T., Yu T., Wang D., Parmasto E. Species clarification for the medicinally valuable 'sanghuang' mushroom. Bot Stu. 2012;53(1):135–149. http://ejournal.sinica.edu.tw/bbas/content/2012/1/Bot531-13.pdf [Google Scholar]
- 4.Marcais G., Kingsford C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics. 2011;27(6):764–770. doi: 10.1093/bioinformatics/btr011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chin C., Alexander D.H., Marks P., Klammer A.A., Drake J., Heiner C. Nonhybrid, finished microbial genome assemblies from long-read smrt sequencing data. Nat Methods. 2013;10(6):563–569. doi: 10.1038/nmeth.2474. [DOI] [PubMed] [Google Scholar]
- 6.Koren S., Walenz B., Berlin K., Miller J., Bergman N., Phillippy A. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017;27:722–736. doi: 10.1101/gr.215087.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Walker B.J., Abeel T., Schea T., Priest M., Abouelliel A., Sakthikumar S. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE. 2014;9 doi: 10.1371/journal.pone.0112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Simão F.A., Waterhouse R.M., Ioannidis P., Kriventseva E.V., Zdobnov E.M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31(19):3210–3212. doi: 10.1093/bioinformatics/btv351. [DOI] [PubMed] [Google Scholar]
- 9.Belton J.M., Mccord R.P., Gibcus J.H., Naumova N., Zhan Y., Dekker J. Hi–C: a comprehensive technique to capture the conformation of genomes. Methods. 2012;58:268–276. doi: 10.1016/j.ymeth.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu Y., Tang Q., Cheng P., Zhu M., Zhang H., Liu J. Whole-genome sequencing and analysis of the Chinese herbal plant Gelsemium elegans. Acta Pharm Sinica B. 2019 doi: 10.1016/j.apsb.2019.08.004(Pre-proof). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Servant N., Varoquaux N., Lajoie B.R., Viara E., Chen C., Vert J. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 2015;16:259. doi: 10.1186/s13059-015-0831-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler Transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Imakaev M., Fudenberg G., McCord R.P., Naumova N., Goloborodko A., Lajoie B.R. Iterative correction of Hi-C data reveals hallmarks of chromosome organization. Nat Methods. 2012;9:999–1003. doi: 10.1038/nmeth.2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burton J.N., Adey A., Patwardhan R.P., Qiu R., Kitzman J.O., Shendure J. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat Biotechnol. 2013;31:1119–1125. doi: 10.1038/nbt.2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xu Z., Wang H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 2007;35:W265–W268. doi: 10.1093/nar/gkm286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Han Y., Wessler S.R. MITE-Hunter: a program for discovering miniature inverted-repeat transposable elements from genomic sequences. Nucleic Acids Res. 2010;38:1–8. doi: 10.1093/nar/gkq862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Price A.L., Jones N.C., Pevzner P.A. De novo identification of repeat families in large genomes. Bioinformatics. 2005;21:i351–i358. doi: 10.1093/bioinformatics/bti1018. [DOI] [PubMed] [Google Scholar]
- 18.Edgar R.C., Myers E.W. PILER: identification and classification of genomic repeats. Bioinformatics. 2005;21:i152–i158. doi: 10.1093/bioinformatics/bti1003. [DOI] [PubMed] [Google Scholar]
- 19.Wicker T., Sabot F., Hua-Van A., Bennetzen J.L., Capy P., Chalhoub B. A unified classification system for eukaryotic transposable elements. Nat Rev Genet. 2007;8(12):973–982. doi: 10.1038/nrg2165. [DOI] [PubMed] [Google Scholar]
- 20.Jurka J., Kapitonov V.V., Pavlicek A., Klonowski P., Kohany O., Walichiewicz J. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet Genome Res. 2005;110:462–467. doi: 10.1159/000084979. [DOI] [PubMed] [Google Scholar]
- 21.Chen N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr Protoc Bioinformatics. 2009;4:10. doi: 10.1002/0471250953.bi0410s25. [DOI] [PubMed] [Google Scholar]
- 22.Lowe T.M., Eddy S.R. tRNAscan-SE: a program for improved detection of transfer rna genes in genomic sequence. Nucleic Acids Res. 1997;5:955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nawrocki E.P., Eddy S.R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics. 2013;29:2933–2935. doi: 10.1093/bioinformatics/btt509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gardner P.P., Daub J., Tate J.G., Nawrocki E.P., Kolbe D.L., Lindgreen S. Rfam: updates to the RNA families database. Nucleic Acids Res. 2009;37:D136–D140. doi: 10.1093/nar/gkn766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boeckmann B., Bairoch A., Apweiler R., Blatter M., Estreicher A., Gasteiger E. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 2003;31:365–370. doi: 10.1093/nar/gkg095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.She R., Chu J.S., Wang K., Pei J., Chen N. genBlastA: Enabling BLAST to identify homologous gene sequences. Genome Res. 2009;19:143–149. doi: 10.1101/gr.082081.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Birney E., Clamp M., Durbin R. GeneWise and Genomewise. Genome Res. 2004;14:988–995. doi: 10.1101/gr.1865504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stanke M., Waack S. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics. 2003;19:i215–i225. doi: 10.1093/bioinformatics/btg1080. [DOI] [PubMed] [Google Scholar]
- 29.Majoros W.H., Pertea M., Salzberg S.L. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics. 2004;20:2878–2879. doi: 10.1093/bioinformatics/bth315. [DOI] [PubMed] [Google Scholar]
- 30.Alioto T., Blanco E., Parra G., Guigo R. Using geneid to identify genes. Curr Protoc Bioinformatics. 2018;64 doi: 10.1002/cpbi.56. [DOI] [PubMed] [Google Scholar]
- 31.Keilwagen J., Wenk M., Erickson J.L., Schattat M.H., Grau J., Hartung F. Using intron position conservation for homology-based gene prediction. Nucleic Acids Res. 2016;44:e88–e89. doi: 10.1093/nar/gkw092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Haas B.J., Salzbery S.L., Zhu W., Pertea M., Allen J.E., Orvis J. Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 2008;9:R7. doi: 10.1186/gb-2008-9-1-r7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kanehisa M., Goto S., Kawashima S., Okuno Y., Hattori M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004;32:D277–D280. doi: 10.1093/nar/gkh063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Deng Y., Li J., Wu S., Zhu Y., Chen Y., He F. Integrated nr database in protein annotation system and its localization. Comput Eng. 2006;32(5):71–74. doi: 10.1109/INFOCOM.2006.241. [DOI] [Google Scholar]
- 35.Tatusov R.L., Galperin M.Y., Natale D.A., Koonin E.V. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000;28(1):33–36. doi: 10.1093/nar/28.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Altschul S.F., Madden T.L., Schäffer A.A., Zhang J., Zhang Z., Miller W. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25(17):3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Eddy S.R. Profile hidden Markov models. Bioinformatics. 1998;14(9):755–763. doi: 10.1093/bioinformatics/14.9.755. [DOI] [PubMed] [Google Scholar]
- 38.Finn R.D., Coggill P., Eberhardt R.Y., Eddy S.R., Mistry J., Mitchell A.L. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 2016;44(D1):D279–D285. doi: 10.1093/nar/gkv1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Blin K., Shaw S., Steinke K., Villebro R., Ziemert N., Lee S.Y. antiSMASH 5.0: updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res. 2019;47(W1):W81–W87. doi: 10.1093/nar/gkz310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xu L., Dong Z., Fang L., Luo Y., Wei Z., Guo H. OrthoVenn2: a web server for whole-genome comparison and annotation of orthologous clusters across multiple species. Nucleic Acids Res. 2019;47(W1):W52–W58. doi: 10.1093/nar/gkv487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li L., Stoeckert C.J., Jr, Roos D.S. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13:2178–2189. doi: 10.1101/gr.1224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Edgar R.C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- 44.Kim D., Langmead B., Salzberg S.L. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12(4):357–360. doi: 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pertea M., Pertea G.M., Antonescu C.M., Chang T., Mendell J.T., Salzberg S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol. 2015;33(3):290–295. doi: 10.1038/nbt.3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Florea Liliana, Song Li, Salzberg Steven L. Thousands of exon skipping events differentiate among splicing patterns in sixteen human tissues. F1000Res. 2013;2:188. doi: 10.12688/f1000research10.12688/f1000research.2-188.v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li Y., Ji D., Zhong S., Zhu J., Chen S., Hu G. Anti-tumor effects of proteoglycan from Phellinus linteus by immunomodulating and inhibiting Reg IV/EGFR/Akt signaling pathway in colorectal carcinoma. Int J Biol Macromol. 2011;48:511–517. doi: 10.1016/j.ijbiomac.2011.01.014. [DOI] [PubMed] [Google Scholar]
- 48.Wang S., Tian Y., Zhang J.Y., Xu H.B., Zhou P., Wang M. Targets fishing and identification of calenduloside E as Hsp90AB1: design, synthesis, and evaluation of clickable activity-based probe. Front Pharmacol. 2018;9:532. doi: 10.3389/fphar.2018.00532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chavali A.K., Rhee S.Y. Bioinformatics tools for the identification of gene clusters that biosynthesize specialized metabolites. Brief Bioinform. 2018;19(5):1022–1034. doi: 10.1093/bib/bbx020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee I.K., Yun B.S. Styrylpyrone-class compounds from medicinal fungi Phellinus and Inonotus spp., and their medicinal importance. J Antibiot. 2011;64:349–359. doi: 10.1038/ja.2011.2. [DOI] [PubMed] [Google Scholar]
- 51.Kahn R.A., Durst F. Function and evolution of plant cytochrome P450. Recent Adv Phytochem. 2000;34:151–190. doi: 10.1016/S0079-9920(00)80007-6. [DOI] [Google Scholar]
- 52.Lee I.K., Yun B.S. Highly oxygenated and unsaturated metabolites providing adiversity of hispidin class antioxidant in the medicinal mushrooms Inonotus and Phellinus. Bioorg Med Chem. 2007;15:3309–3314. doi: 10.1016/j.bmc.2007.03.039. [DOI] [PubMed] [Google Scholar]
- 53.Chao W., Deng J.S., Li P.Y., Liang Y.C., Huang G.J. 3,4-Dihydroxybenzalactone suppresses human non-small cell lung carcinoma cells metastasis via suppression of epithelial to mesenchymal transition, ROS-mediated PI3K/AKT/MAPK/MMP and NFκB signaling pathways. Molecules. 2017;22:537. doi: 10.3390/molecules22040537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chandimali N., Huynh D.L., Jin W.Y., Kwon T. Combination effects of hispidin and gemcitabine via inhibition of stemness in pancreatic cancer stem cells. Anticancer Res. 2018;38:3967–3975. doi: 10.21873/anticanres.12683. [DOI] [PubMed] [Google Scholar]
- 55.Lv L., Zhou Z., Zhou Z., Zhang L., Yan R., Zhao Z. Hispidin induces autophagic and necrotic death in sgc-7901 gastric cancer cells through lysosomal membrane permeabilization by inhibiting tubulin polymerization. Oncotarget. 2017;8(16):26992–27006. doi: 10.18632/oncotarget.15935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang H., Chen R., Zhang J., Bu Q., Wang W., Liu Y. The integration of metabolome and proteome reveals bioactive polyphenols and hispidin in ARTP mutagenized Phellinus baumii. Sci Rep. 2019;9(1):1–12. doi: 10.1038/s41598-019-52711-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen Y., Lee S., Lin C., Liu C. Hispolon decreases melanin production and induces apoptosis in melanoma cells through the downregulation of tyrosinase and microphthalmia-associated transcription factor (MITF) expressions and the activation of caspase-3, -8 and -9. Int J Mol Sci. 2014;15(1):1201–1215. doi: 10.3390/ijms15011201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jang E.H., Jang S.Y., Cho I., Hong D., Jung B., Park M. Hispolon inhibits the growth of estrogen receptor positive human breast cancer cells through modulation of estrogen receptor alpha. Biochem Biophys Res Commun. 2015;463(4):917–922. doi: 10.1016/j.bbrc.2015.06.035. [DOI] [PubMed] [Google Scholar]
- 59.Kojima K., Ohno T., Inoue M., Mizukami H., Nagatsu A. Phellifuropyranone A: a new furopyranone compound isolated from fruit bodies of wild Phellinus linteus. Chem Pham Bull. 2008;56(2):173–175. doi: 10.1248/cpb.56.173. [DOI] [PubMed] [Google Scholar]
- 60.Zhang H., Shao Q., Wang W., Zhang J., Zhang Z., Liu Y. Characterization of compounds with tumor–cell proliferation inhibition activity from mushroom (Phellinus baumii) mycelia produced by solid-state fermentation. Molecules. 2017;22:698. doi: 10.3390/molecules22050698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nambudiri M.D., Vance C.P., Towers G.H.N. Effect of light on enzymes of phenylpropanoid metabolism and hispidin biosynthesis in Polyporus hispidus. Biochem J. 1973;134:891–897. doi: 10.1007/BF00487345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Perrin P.W., Towers G.H.N. Hispidin biosynthesis in cultures of Polyporus hispidus. Phytochemistry. 1973;12:589–592. doi: 10.1016/S0031-9422(00)84448-9. [DOI] [Google Scholar]
- 63.Wangun H.V., Härtl A., Kiet T.T., Hertweck C. Inotilone and related phenylpropanoid polyketides from Inonotus sp. and their identification as potent COX and XO inhibitors. Org Biomol Chem. 2006;4:2545–2548. doi: 10.1039/b604505g. [DOI] [PubMed] [Google Scholar]
- 64.Gill M. Pigments of fungi (Macromycetes) Nat Prod Rep. 2003;20:615–639. doi: 10.1039/B202267M. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The NCBI SRA Bioproject (PRJNA608208) included whole-genome assembly sequence, raw data of genome-seq, Hi-C and RNA-seq. All sequencing data involved in this work can be acquired at NCBI.