

Abstract
The prevalence of high blood pressure (also known as hypertension) has steadily increased over the last few decades. Known as a silent killer, hypertension increases the risk for cardiovascular disease and can lead to stroke, heart attack, kidney failure and associated sequela. While numerous hypertensive therapies are currently available, it is estimated that only half of medicated patients exhibit blood pressure control. This signifies the need for a better understanding of the underlying cause of disease and for more effective therapies. While blood pressure homeostasis is very complex and involves the integrated control of multiple body systems, smooth muscle contractility and arterial resistance are important contributors. Strong evidence from pre-clinical animal models and genome-wide association studies indicate that smooth muscle contraction and BP homeostasis are governed by the small GTPase RhoA and its downstream target, Rho kinase. In this review, we summarize the signaling pathways and regulators that impart tight spatial-temporal control of RhoA activity in smooth muscle cells and discuss current therapeutic strategies to target these RhoA pathway components. We also discuss known allelic variations in the RhoA pathway and consider how these polymorphisms may affect genetic risk for hypertension and its clinical manifestations.
Keywords: blood pressure, smooth muscle, RhoA, Rho Kinase (ROCK), Guanine nucleotide exchange factor (GEF), GTPase activating protein (GAP)
l. Introduction
Hypertension (HTN) is a major cardiovascular risk factor that significantly increases the incidence of stroke, myocardial infarction, heart failure, retinopathy, and kidney disease (Lim, et al., 2012). Although HTN is one of the most modifiable cardiovascular risk factors, the number of individuals with HTN is increasing world-wide. Further amplifying the importance of HTN, the American Heart Association has recently revised its definition of Stage 1 HTN to include individuals with systolic blood pressure (BP) between 130 and 139 mmHg or diastolic BP between 80 and 89 mmHg. This change was prompted by studies demonstrating beneficial effects of lowering BP below the 120/80 mmHg threshold (Guo, et al., 2013; Lewington, et al., 2003; Whelton, et al., 2017) and effectively increased the number of Americans categorized as hypertensive from 32% to 46% (Whelton, et al., 2017). It is also becoming clear that many people suffer from masked HTN (normal readings in the clinic, but hypertensive outside the clinic) and non-dipping HTN (steady BP through the day but no decrease in BP at night) (Booth, et al., 2016; Peacock, et al., 2014; Viera & Shimbo, 2014), suggesting that more intensive BP monitoring would identify additional at-risk individuals (Hinderliter, et al., 2018).
A number of relatively inexpensive first-line therapies are available to treat HTN including diuretics, angiotensin-converting enzyme (ACE) inhibitors, angiotensin II (A-II) receptor blockers, and calcium channel blockers. However, these drugs are usually prescribed empirically and are often ineffective. Indeed, over 50% of adults who are being treated for HTN still do not have their BP under control (S. Yoon, et al., 2015). Although treatment can be improved by multidrug regimens that target different BP control mechanisms, thirteen percent of treated patients have drug-resistant HTN and remain hypertensive even after taking three 3 or more medications, or require 4 medications for adequate BP control (Achelrod, et al., 2015; Persell, 2011). Taking multiple BP medications also increases the risk of unwanted side effects and drug-drug interactions. While incomplete health history and poor patient compliance contribute to the difficulties in treating HTN, our lack of understanding of the etiology of HTN is also a major factor.
Many of the difficulties of treating HTN stem from the fact that BP is an extremely complex trait regulated by many organ systems. Although the major determinants of BP are cardiac output and systemic vascular resistance, BP homeostasis requires proper regulation of heart and vasculature function by the autonomic nervous system, kidneys, and endocrine organs. The fact that these systems are tightly integrated by many feedback loops further complicates our understanding of the development of HTN and its treatment. Nearly all heritable genetic mutations that cause HTN affect kidney function and/or salt balance, but these variants only explain about 10% of HTN cases. More recent genome wide association studies (GWAS) have identified many genetic loci that correlate with relatively small differences in BP between populations. However, because most of these variations are within or near genes with no known connection to BP regulation, our understanding of how they affect the development of HTN is limited. A number of the genes identified by GWAS are highly expressed in endothelial and smooth muscle cells (SMCs), highlighting the importance of the vasculature as a major regulator of BP and as a target for potential therapies (Ehret, et al., 2016; Ehret, et al., 2011; Padmanabhan, et al., 2015; Wain, et al., 2011).
Recent advancements suggest that RhoA signaling in the vasculature is a particularly attractive target for therapeutic intervention in the treatment of HTN. Extensive studies have shown RhoA signaling enhances Ca2+-dependent, myosin- based force production in vascular SMCs and recent studies from our lab and others have implicated several components of the RhoA signaling pathway in the development of HTN in mouse models (Guilluy, et al., 2010; Loirand & Pacaud, 2010; A. Wirth, et al., 2008). Moreover, human genetic studies have identified BP- associated variants in several additional Rho-related genes further implicating this pathway in human HTN (Loirand, 2015). The goals of the current review are to summarize the data supporting the role of RhoA signaling in the development of HTN and to provide an overview of current and potential therapeutic targets within this pathway that could lead to better and perhaps novel HTN therapies.
2. Impact of the RhoA pathway on BP homeostasis
The 22 members of the Rho family of small GTPases can be divided into three major subfamilies, Rac, RhoA, and Cdc42. The RhoA family GTPases (RhoA, RhoB, and RhoC) are widely expressed and share 85% amino acid homology including a C-terminal cysteine residue that is the target of geranylgeranylation, a posttranslational modification that anchors RhoA family proteins to the plasma membrane. As discussed below, RhoA is by far the most studied member of this subfamily and has been shown to regulate a variety of cellular processes including (but not limited to) the modulation of actin and microtubule dynamics, cell force, cell shape and polarity, endocytosis, exocytosis, cell adhesion and migration, proliferation, and differentiation (Narumiya & Thumkeo, 2018). Because of its well-recognized role in mediating BP homeostasis, the remainder of this review will be focused on RhoA.
Like all GTPases, RhoA is regulated by guanosine triphosphate (GTP) binding and cycles between the active GTP-bound form and the inactive guanosine diphosphate (GDP)-bound form and this cycle is under the direct control of three groups of regulatory proteins. Guanine dissociation inhibitors (GDIs) sequester RhoA into an inactive cytoplasmic fraction, guanine nucleotide exchange factors (GEFs) activate RhoA by facilitating exchange of GDP for GTP, and GTPase activating proteins (GAPs) promote RhoA’s intrinsic GTPase activity to hydrolyze GTP to GDP and efficiently turn off (or limit) RhoA-dependent signaling. When GTP-bound, RhoA interacts with a variety of effector molecules that mediate its varied functions including the Rho-associated coiled-coil domain containing protein kinases (ROCK I and II), the diaphanous-related formins (mDial and mDia2), protein kinase N, citron kinase, rhophilin, and the rhotekins I and II among other enzymes (Narumiya & Thumkeo, 2018).
2.1. Rho A-dependent SMC contractility and peripheral vascular resistance.
Vascular resistance is a major determinant of BP and is controlled, in large part, by SMC contraction within small peripheral arterioles (Cowley, 2006; Davis & Hill, 1999; Davis, et al., 2001; Hall, 2003; Lifton, et al., 2001). Importantly, studies in genetically engineered mice revealed that germline deletion of the Rho- specific GEF, LARG, significantly attenuated salt-induced HTN, while SMC- specific knockout of the related GEF, pll5RhoGEF, inhibited the development of HTN in response to A-II. In addition, we recently showed that depletion of the SMC-selective, Rho-specific GAP, GRAF3 (ArhGAP42) in mice leads to basal HTN, increased pressor responses to A-II, endothelin-1 (ET1), and phenylephrine (PE), and elevated deoxycorticosterone-acetate (DOCA)-salt induced HTN. Collectively, these studies strongly support a critical role for RhoA in governing BP by modulating SMC tone.
Mechanistically, excitation-contraction coupling in SMC is mediated by Ca2+-dependent activation of myosin light chain kinase (MLCK), and SMC tension is directly proportional to myosin light chain (MLC) phosphorylation at S19 (Figure 1). By mechanisms that are still somewhat unclear, MLC phosphorylation enables myosin-actin cross-bridge cycling to enhance force generation (Budzyn, et al., 2006; Etienne-Manneville & Hall, 2002). Besides promoting an inositol triphosphate-mediated increase in intracellular calcium, many circulating vasoconstrictors that activate G-protein coupled receptors including A-II, ET1, PE, and sphingosine-1 -phosphate (SIP) also stimulate RhoA activity which further enhances Ca2+-dependent SMC contractility (Guilluy, et al, 2010; Loirand & Pacaud, 2010; A. Wirth, et al., 2008) (Figure 1). RhoA activation also regulates constriction of the pre-glomerular afferent arterioles that control kidney perfusion. In this tubuloglomerular feedback system, increased kidney perfusion (Carlstrom, et al., 2015) results in increased NaCl delivery from the loop of Henle to the macula densa, a cluster of epithelial cells that are adjacent to the abluminal SMCs of the afferent arterioles. Increased NaCl uptake by macula densa cells results in the secretion of ATP and adenosine that stimulate RhoA activity in afferent arteriole SMC via the P2Y4/P2Y6 and A2 G-protein coupled receptors, respectively (Homma, et al., 2014; Inscho, 2009; Nakamura, et al., 2003; Roos, et al., 2006; Y. Shi, et al., 2006; Yano, et al., 1995). Since the kidneys typically receive about 25% of total cardiac output, increased contractility of renal arterioles has a significant impact on systemic BP. Taken together these studies indicate that aberrant activation or loss of tight temporal control of the RhoA pathway in resistance vessels can lead high basal BP and exaggerated responses to vasoconstrictors.
Figure 1. RhoA pathway in smooth muscle cells (SMC).
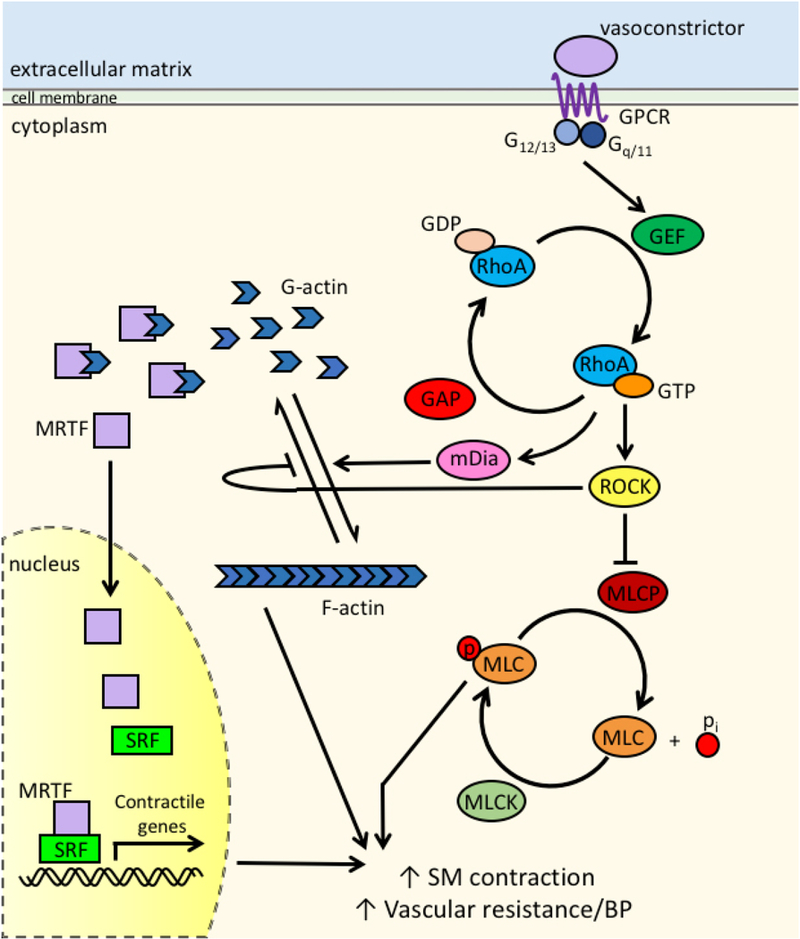
In addition to Ca2+− dependent activation of MLCK (not shown here), vasoconstrictors such as All, PE, and ET-1 stimulate G-protein signaling in SMC via GPCRs. G-proteins influence the activity of RhoA by activating GEFs, which facilitate the dissociation of GDP from RhoA, allowing GTP to bind. This activation is balanced by GAPs, which increase the efficiency of GTP hydrolysis. When RhoA is in its active, GTP-bound form, it’s able to interact with any of it’s downstream effectors, such as ROCK and mDia. ROCK inhibits MLCP, allowing for prolonged phosphorylation of MLC and enhanced SMC contraction, increased vascular resistance, and increased blood pressure. Simultaneously, mDia and ROCK promote the polymerization of F-actin from G-actin, releasing G-actin bound MRTF. Free MRTF translocates to the nucleus where it induces SRF-dependent transcription of contractile genes. These concurrency of these latter events further enhance SMC contraction and vascular resistance.
RhoA regulates several effector molecules that impact SMC contractility (Figure 1). Direct phosphorylation of MLC by ROCKs I and II promotes actin- myosin crossbridge cycling as does ROCK dependent inhibition of MLC phosphatase (MLCP). RhoA activity is also critical for de novo formation of actin filaments and formation of focal adhesions that are required for myosin-dependent force development and transmission, respectively. The Rho effectors mDia 1 and 2 are the most potent regulators of actin filament formation as these proteins function to directly catalyze actin polymerization in cooperation with the actin binding protein, profilin. ROCKs also inhibit actin de-polymerization by phosphorylating and activating LIM-kinase 1 and 2 (on Thr 508 or 505 respectively), which in turn, phosphorylate and inhibit the actin filament severing protein, cofilin (Ohashi, et al., 2000; Sumi, et al., 2001; Vardouli, et al., 2005; Yang, et al., 1998). Finally, ROCK-dependent phosphorylation of ezrin-radixin-moesin (ERM) proteins promotes their tethering to integral plasma membrane proteins effectively stabilizing actin filaments and increasing force transmission (Matsui, et al., 1998).
In addition to promoting acute changes in SMC contractility, recent studies indicate that RhoA signaling also controls the transcription of numerous contractile genes by modulating serum response factor (SRF) activity. SRF binds to CArG (CC(A/T)6GG) cis elements located within the promoters of nearly all SMC contractile genes (including SM myosin heavy chain, SM22, calponin, and SM a- actin). SRF activity is modulated by transcription cofactors of the myocardin family (Chang, et al, 2003; C. Y Chen & Schwartz, 1996; Dalton & Treisman, 1992; Hill & Treisman, 1995; Mack, et al., 2000) and two such co-factors, myocardin transcription factor A and B (MRTF-A and MRTF-B) mediate strong trans-activation of SMC contractile genes (Hinson, et al., 2007; D. Z. Wang & Olson, 2004). We have previously demonstrated that RhoA promotes SMC contractile gene expression through actin polymerization-dependent regulation of MRTF-A and MRTF-B nuclear localization (Hinson, et al., 2007; Lockman, et al., 2004; Miralles, et al., 2003; Sotiropoulos, et al., 1999; Staus, et al., 2007). Cytoplasmic monomeric G-actin is abundant when RhoA activity is low (for example in SMC under low tension (Albinsson, et al., 2004)), and under these conditions, G-actin binds to MRTF and masks an N-terminal nuclear localization sequence, resulting in cytoplasmic sequestration of these SRF co-factors. Upon RhoA activation, G-actin is recruited into growing F-actin filaments and MRTF-G- actin association decreases. As a consequence, MRTF nuclear localization sequence is un-masked, and MRTF accumulates in the nucleus and promotes SRF- dependent gene expression (Mack, 2011). Thus, signaling through RhoA in small arteriolar SMC enhances Ca2+ sensitivity, promotes actin remodeling and induces expression of contractile proteins each of which increase SMC tone and peripheral vascular resistance.
2.2. Non-vascular RhoA responses associated with BP homeostasis
Although Rho signaling components are relatively strongly expressed in vascular SMCs, nearly all, with the exception of the RhoGAP GRAF3 (see section 6 below), are expressed in many other tissues. Thus, when evaluating Rho signaling molecules as targets of anti-HTN therapy, it is important to consider the potential impact of modulating Rho-signaling in other organ systems. Interestingly, with respect to BP regulation, studies using pre-clinical models indicate that attenuating RhoA signaling in the vasculature, kidney, myocardium, and CNS could all lead to the desired outcome of lowering BP. For example, blocking RhoA activity in endothelial cells can indirectly inhibit SMC contractility by increasing the secretion of the potent vasodilator, nitric oxide (Laufs & Liao, 1998; Ming, et al., 2004; Wolfrum, et al., 2004; Zhou & Liao, 2009). Some evidence suggests that blocking RhoA activity in tubular epithelial cells can alter sodium channel activity, limit Na+ reabsorption, and aid in maintaining blood volume homeostasis (Hayashi, et al., 2004; Karpushev, et al., 2010; Loirand & Pacaud, 2014; Nishiki, et al., 2003; Pochynyuk, et al., 2006; Staruschenko, et al, 2004; Szaszi, et al., 2000). Moreover, investigators have shown that inhibiting RhoA activity in the nucleus tractucs solitarius within the central nervous system reduced sympathetic nerve activity, heart rate, and BP in normotensive rats and these effects are even more pronounced in spontaneously hypertensive rats (Ito, et al., 2005; Ito, et al., 2003). Likewise, while infusion of A-II into the neural cistern of rats promoted a significant rise in BP, co-infusion of A-II and the ROCK inhibitor, Y27632 did not (Sagara, et al., 2007). On the other hand, some studies in cells and invertebrate model systems indicate that inhibition of Rho/Rho kinase signaling in motor neurons antagonized the secretion of parasympathetic relaxation factors (including acetylcholine) and promoted the secretion of sympathetic contractile agonists (including dopamine) (Hiley, et al., 2006; Yamaguchi, et al., 2000). Since perivascular nerves play a major role in the control of resistance arteriole tone, such outcomes may limit the therapeutic efficacy of RhoA/ROCK inhibitors as future anti-hypertensive therapies. Moreover, inhibition of RhoA in the heart leads to conduction defects (Wei, et al., 2004; Yatani, et al., 2005). Thus, as described in further detail below, targeting vascular SMC specific regulators of the RhoA pathway may provide a better avenue for pharmacological BP control.
3. Targeting the RhoA pathway for Therapeutic BP Control
In agreement with the pre-clinical animal studies highlighted above, several lines of evidence strongly implicate RhoA signaling in the development of human HTN. First, increased Rho-kinase activity has been observed in some hypertensive patient populations and as reviewed in section 3.1 Rho kinase inhibitors have been successful in reducing systemic HTN in these cases, although current formulations exhibit relatively short-term effects (Feng, et al, 2016; Loirand, 2015; Zhou, et al., 2011). Second, an autosomal dominant mutation in the E3 ligase, Cullin-3 (which targets RhoA), has been shown to cause high BP in patients with Gordon’s Syndrome (pseudohypoaldosteronism type HE). Importantly, the identical mutation in pre-clinical mouse models and led to decreased Cullin-3 activity, reduced ubiquitin-mediated RhoA degradation in vascular SMCs, and increased BP (Boyden, et al., 2012; Ibeawuchi, et al., 2015). Third, many GWAS conducted over the past decade have identified common BP-associated genetic variations in coding and non-coding regions within or near genes linked to the RhoA signaling cascade. For example, in one study that used HTN as a dichotomous trait, two of the eight BP-associated loci were located in RhoA-related genes. One was within RhoBTB 1, which functions with the aforementioned Cullin-3 complex to maintain low RhoA levels (Boyden, et al., 2012; Pelham, et al., 2012), while the other was within rhotekin-2 (RTKN2), a RhoA effector with as yet unknown function. Two separate GWAS identified a BP-associated locus within PLEKHA7 (Plekstrin Homology domain containing family A member 7) (Levy, et al., 2009; Lin, et al., 2011) which interacts with the junctional proteins cingulin and paracingulin to regulate several Rho family GTPases, including RhoA in the heart and kidneys (Citi, et al., 2012). Importantly, PLEKHA7 was subsequently shown to be required for the development of salt-induced HTN in mice (Endres, et al., 2014). Moreover, a few variants in ROCK II have been associated with the regulation of BP. Of particular interest was the identification of a common nonsynonymous ROCK II variant 43 IN (versus 43 IT) that was associated with an increase in BP in twins (Liao, et al., 2015; L. Liu, et al., 2013; Loirand & Pacaud, 2014; Rankinen, et al., 2008; Seasholtz, et al., 2006). This result was supported by Liao et al., who showed that the 43 IN variant had increased kinase activity and was associated with enhanced arterial stiffening, a vascular property strongly associated with HTN (Liao, et al., 2015; L. Liu, et al., 2013; Loirand & Pacaud, 2014; Rankinen, et al., 2008; Seasholtz, et al., 2006). This group identified a second variation in the ROCK II 3’UTR (rs9789060) that was also associated with increased stiffening and went on to show it affected ROCK II expression by interfering with miR-1183- dependent degradation of ROCK II mRNA levels. It is important to note that a third study failed to find an association between rs9789060 and BP (L. Liu, et al., 2013). In another study on 586 normotensive and 607 hypertensive Caucasians, Rankinen et al. identified a minor allele locus within the ROCK II gene that lowered the risk of HTN by 85%. (Rankinen, et al, 2008). Finally, as discussed in further detail below, three separate GWAS for BP and cardiovascular disease endpoints identified a novel BP associated locus within the Rho-specific GAP, GRAF3/ArhGAP42 (Ehret, et al., 2011; Kato, et al., 2015; Li, et al., 2016; Wain, et al., 2011). Recent causality studies from our group demonstrated that GRAF3 is selectively expressed in SMC and is required for BP homeostasis in mice (Bai, et al., 2013; Bai, et al., 2017a; Bai, et al., 2017b).
Collectively, these studies reveal that the RhoA signaling axis may provide tractable targets for the treatment of human HTN and related cardiovascular sequela. Indeed, some commonly used anti-hypertensives (i.e. ACE inhibitors, A- II blockers, and statins) likely function by interfering with RhoA signaling, supporting the clinical utility of inhibiting this pathway. (Brandes, 2005; Carbone, et al., 2015; Guilluy, et al., 2010; Kanaki, et al., 2013). Nonetheless, despite the importance of the RhoA pathway in the pathogenesis of HTN and several other debilitating diseases including amyotrophic lateral sclerosis, mental retardation, hepatocellular, lung, and colorectal carcinomas (Boettner & Van Aelst, 2002; Jansen, et al., 2018; Sahai & Marshall, 2002), surprisingly few treatments are available to directly target this pathway. In fact, of the nearly 300,000 ongoing clinical trials, only a handful involve compounds that target RhoA signaling components (clinicaltrials.gov). This could be due to the fact that many members of the RhoA pathway (with exception of Rho kinase) have traditionally been regarded as “undruggable” However, as described below, significant advancements in high resolution crystal structures, structure-function analyses and drug development technology are beginning to overcome these challenges and provide hope for the development of new therapies to target this critical pathway.
3.1. Targeting Rho Kinase
The serine/threonine kinases, ROCK I and ROCK II, are the best studied RhoA effectors and have been implicated in a variety of diseases including HTN (Loirand, 2015). Since the development of kinase inhibitors has proven to be a very successful therapeutic approach, it is not surprising that ROCK has been an attractive target in the search for RhoA signaling inhibitors and that ROCK inhibitors are the furthest along in regard to clinical testing. Because ROCK I and II share 60% identity overall, 90% identity within the kinase domain identity, and 100% identity within the ATP binding pocket (Nakagawa, et al., 1996) they share many substrates and promote many of the same downstream cell functions (Feng, et al., 2016). Although ROCK I and ROCK II expression can vary somewhat between tissues, both of these kinases are widely expressed (Nakagawa, et al., 1996; Y. Wang, et al., 2009). Differences in subcellular localization have been noted with ROCK I localizing more readily to microtubule-organizing centers (Chevrier, et al., 2002) and catenin/E-cadherin containing complexes at the plasma membrane (Smith, et al, 2012) and ROCK II to vimentin (Sin, et al., 1998) and actin fibers (X. Q. Chen, et al., 2002) (Schofield & Bernard, 2013; Y Wang, et al., 2009). Isoform-specific inhibitors are being developed that could have differential effects depending upon the disease treated or end-point measured (Boerma, et al., 2008; Hyun Lee, et al., 2014; Loirand, 2015; Zanin-Zhorov, et al., 2014; Zanin- Zhorov, et al., 2017)(clinicaltrials.gov).
While over 30 common downstream targets of ROCK have been identified (Adducin, Diaphanous (mDia), LIM kinase (LIMK), NHE1, MARCKS, NF-L, CRMP2,FAK, c-Jun N-terminal kinase (JNK), MLC, MLCK, MLCP, ezrin/radixin/moesin (ERM), rhophilin, rhotekin, citron kinase, and Tau) (Feng, et al., 2016; Loirand, 2015; Schofield & Bernard, 2013; Siehler, 2009), the most pertinent in regard to SM contraction and BP regulation is the myosin binding subunit (MYPT-1) of the MLCP complex. ROCK-dependent phosphorylation of MYPT-1 at T696, T853, and S854 inhibits MLCP activity resulting in increased MLC phosphorylation and hence increased contraction (Y Wang, et al., 2009; Angela Wirth, 2010). ROCK has also been shown to directly phosphorylate MLC at S19.
As reviewed more thoroughly elsewhere (Feng, et al., 2016; Loirand, 2015) over 170 ROCK inhibitors are in various stages of development (Table 1). Given the involvement of actin-based processes in many pathologies (adhesion, migration, cell division, force generation, etc.), ROCK inhibitors are being studied as treatments for a wide variety of disease states including central nervous system disorders (subarachnoid hemorrhage and vasospasm, cerebral stroke, spinal cord injury), neurodegenerative diseases (Alzheimer’s disease, Parkinson’s, Huntington disease, and amyotrophic lateral sclerosis [ALS]), cardiovascular disease (systemic HTN, pulmonary arterial HTN (PAH), spastic and stable angina, atherosclerosis, Raynaud syndrome), asthma, glaucoma, autoimmune diseases, cancer, erectile dysfunction, and kidney disease. Although the results of these studies and clinical trials provide important information on ROCK and its role in disease progression, we will focus our discussion on the major ROCK inhibitors that have been most commonly used to treat vascular diseases.
Table 1. ROCK inhibitors currently in clinical trials.
ROCK inhibitor compounds and presumptive applications are outlined here, along with the phase of the study, the status of the study, and the registered clinical trials identifier number. NA, not applicable
| Drug | Disease | Phase | Status |
clinicaltrials.gov ID |
|---|---|---|---|---|
| Fasudil | Cerebral vasospasm, brain ischemia, stable angina, pulmonary HTN | NA | Approved in Japan and China | NA |
| Raynaud/Scleroderma | 3 | completed | NCT00498615 | |
| Carotid Stenosis | 2 | terminated | NCT00670202 | |
| Atherosclerosis, Hypercholesterolemia | 2 | completed | NCT00120718 | |
| Amyotrophic Lateral Sclerosis | 2 | uknown | NCT01935518 | |
| Cardiovascular Disease | 2 | recruiting | NCT03404843 | |
| Ripasudil | Glaucoma and ocular HTN | NA | Approved in Japan | NA |
| Fuchs’ Endothelial Dystrophy | 4 | recruiting | NCT03249337 | |
| AR-12286 | Chronic Angle-closure Glaucoma | 2 | unknown | NCT02152774 |
| Advanced Glaucoma | 2 | unknown | NCT02173223 | |
| Glaucoma | 2 | unknown | NCT02174991 | |
| Exfoliation Syndrome, Ocular HTN, Open Angle Glaucoma | 2 | completed | NCT01936389 | |
| AR-13324 | Open-angle glaucoma, Ocular HTN | NA | FDA approved | NA |
| SAR-407899 | Erectile dysfunction | 2 | completed | NCT00914277 |
| Microvascular Coronary Artery Disease | 2 | recruiting | NCT03236311 | |
| Chronic Kidney Disease | 1 | completed | NCT01485900 | |
| KD-025 | Psoriasis Vulgaris | 3 | completed | NCT02317627 |
| Psoriasis | 2 | recruiting | NCT02852967 | |
| Graft vs. Host Disease | 2 | recruiting | NCT02841995 | |
| Idiopathic Pulmonary Fibrosis | 2 | recruiting | NCT02688647 | |
| Fibrotic Disease | 1 | recruiting | NCT03530995 |
Fasudil (also known as HA-1077) was the first ROCK inhibitor described and was also the first to be tested clinically. This isoquinoline-based drug is classified as a class I ROCK inhibitor because it reversibly competes with ATP binding to the ROCK kinase domain. Fasudil inhibits both isoforms of ROCK with an IC50 of ΙμΜ while hydroxyfasudil, the major active metabolite, is slightly more potent with an IC50 of ~0.7μΜ (Nagumo, et al., 2000). Like most other ROCK inhibitors of this class fasudil also inhibits other members of the broad AGC kinase family including PKA and PKC, albeit with less potency (IC50 of 5μΜ and 37μΜ, respectively) (Bain, et al., 2007; Nagumo, et al., 2000). Fasudil was shown to reduce BP by attenuating the Rho-mediated inhibition of MLCP in SMC (Nagumo, et al., 2000), by increasing endothelial nitric oxide synthase expression (Takemoto, et al, 2002), and by reducing circulating ACE and A-II levels (Ocaranza, et al, 2011). Fasudil also inhibited pulmonary artery SMC proliferation, a process important for vessel stiffening, by a mechanism that likely involved downstream inhibition of c-Jun N-terminal kinase (JNK) and ERK- dependent activation of c-jun and c-fos expression (X. Y Chen, et al., 2009).
Fasudil hydrochloride hydrate under the trade name Eril® was approved in Japan in 1995 to treat vasospasm-induced cerebral ischemia that can occur following surgery for subarachnoid hemorrhage. In 2002, Masumoto et.al. showed that intracoronary fasudil was effective as an acute treatment of vasospastic angina (Masumoto, et al., 2002), and additional clinical trials showed that long-term treatment with oral fasudil reduced stable effort-induced angina and improved exercise tolerance with no major adverse effects (Shimokawa, et al., 2002; Vicari, et al., 2005). In a rat model, moderate doses of fasudil decreased PAH while higher doses also decreased mean systemic arterial pressure (Jiang, et al., 2007). By 2011, intravenous and inhaled fasudil were approved to treat PAH (Y Fukumoto, et al., 2005; Nagaoka, et al., 2005), and a new extended release formulation of fasudil, AT-877ER, was shown to reduce PAH in patients after three months of use (Y Fukumoto, et al., 2013). Fasudil decreased forearm vascular resistance more dramatically in hypertensive patients than in normotensive controls (Masumoto, et al., 2001). Fasudil has been shown to have beneficial effects on kidney function in diabetic rats (Komers, et al, 2011), suggesting that it might be useful for treating diabetic patients who are frequently hypertensive and have kidney disease. Ongoing phase III clinical trials are also assessing whether fasudil is an effective treatment for Reynaud’s syndrome and carotid stenosis (clinicaltrials.gov). Fasudil has been approved in China, but not in the United States or Europe.
Several fasudil derivatives that are more potent and specific inhibitors of ROCK have been developed and are being used to treat glaucoma and ocular HTN. Ripasudil (Glanatec®), approved in Japan in 2014, affects the trabecular meshwork in the eye and reduces intraocular pressure by facilitating the outflow of aqueous humor through Schlemm’s canal The most common side effect of ripasudil treatment is conjunctival hyperemia, which subsides over time or with discontinued use (Garnock-Jones, 2014). Netarsudil (Rhopressa ®) has similar effects and indications, and in 2017, became the first ROCK inhibitor approved by the United States (Lu, et al., 2017; R. Ren, et al., 2016). Netarsudil also decreased the amount of aqueous humour produced, a feature attributed to additional effects of netarsudil as a norepinephrine transport inhibitor (Lu, et al., 2017; R. Ren, et al., 2016).
The classic ROCK inhibitor, Y-27632, is a pyridine-based class I inhibitor that has been tested in a number of animal and human disease models including HTN. In a landmark study, Uehata et.al. demonstrated that Y-27632 decreased BP in spontaneously hypertensive rats (SHR), DOCA-salt treated rats, and rats made hypertensive by clipping of the renal artery (Uehata, et al., 1997). Although mesenteric and cerebral arteries in SHR or DOCA/salt treated rats were more responsive to Y-27632 than those in normotensive rats (Asano & Nomura, 2003; Weber & Webb, 2001), Y-27632 likely affected HTN by multiple mechanisms. For example, Y-27632 also reversed the decrease in renal sodium excretion observed in the SHR model (Nishiki, et al., 2003), most likely by affecting the activity and location of Na+/H+ exchanger, NHE3 (Hayashi, et al., 2004; Szaszi, et al., 2000). In addition, local infusion of Y-27632 into the nucleus tractus solitarius of the brainstem caused a reduction in BP, heart rate, and renal sympathetic nerve activity, and the magnitude of these effects was greater in the SHR model (Ito, et al., 2005; Ito, et al., 2003). Subsequent studies by the same group demonstrated that fasudil had similar effects (Ito, et al., 2005; Ito, et al., 2003). Although Y- 27632’s poor potency and kinase selectivity limit its use clinically, Y-27632 derivatives with better pharmacologic properties are being developed and tested.
SAR407899 is a promising relatively new isoquinoline-based class I ROCK inhibitor that is significantly more potent than older generation drugs (IC50 between 122 and 280nM) (Lohn, et al., 2009). In vitro studies demonstrated that SAR407899 inhibited myosin phosphatase phosphorylation, stress fiber formation, cell proliferation, and monocyte chemotaxis (Lohn, et al., 2009). SAR407899 dose-dependently lowered BP in SHR, stroke-prone SHR, L-NG-Nitroarginine methyl ester (L-NAME), and DOCA-salt rat models, and its effects in some models was superior to ACE inhibitors and calcium channel blockers (Lohn, et al., 2009). SAR407899 inhibited pressor responses to PE, A-II, and vasopressin in rats more strongly than Fasudil or Y-27632, and it inhibited ETl-induced vasoconstriction of renal arteries isolated from diabetic rats more strongly than Y- 27632 (Grisk, et al., 2012). In spite of its efficient antihypertensive effects and the fact that long term treatment of rats with SAR407899 was well-tolerated, development of SAR407899 as an anti-hypertensive has been discontinued (Loirand, 2015). SAR407899 is still being tested in clinical trials as a treatment for kidney disease and microvascular coronary artery disease and it may prove useful for treatment of erectile dysfunction in diabetic and hypertensive patients where the eNO system is impaired (Guagnini, et al., 2012).
Although ROCK inhibition has been shown to reduce BP and vascular resistance in many models, there are concerns about the suitability of ROCK inhibitors as viable long-term treatments for systemic HTN. Although most ROCK inhibitors seem to be fairly well-tolerated, the ubiquitous nature of ROCK I and ROCK II expression coupled with the relative lack of specificity of most ROCK inhibitors (especially the class I drugs) makes potentially unknown side- effects a significant drawback. While local delivery strategies can sometimes mitigate this concern (i.e. to the eye or specific vascular beds) (Bodor & Buchwald, 2000; Feng, et al., 2016), systemic hypotension can be a serious problem for patients being treated in this manner. Another potential problem is that the systemic BP lowering effects of ROCK inhibitors frequently decrease after 7–10 days of chronic treatment (Feng, et al., 2016). Moreover, ROCK inhibitors did not affect systolic BP in some long-term studies of salt-sensitive hypertensive Dahl rats (Loirand & Pacaud, 2014; Nishiki, et al., 2003). The precise causes of this tolerance or inactivity are unknown, but likely involve compensation by the many feedback pathways that regulate BP. Finally many ROCK inhibitors have short half-lives, which is not ideal for the treatment of a chronic diseases like HTN (Surma, et al., 2011).
3.2. Targeting RhoA directly
Numerous failed attempts to identify small molecule inhibitors of H-Ras have led to the concept that small GTPases per se do not make good drug targets due to their globular structure and lack of surface moieties required for high affinity binding of small molecules (Jansen, et al., 2018; Kristelly, et al., 2004; Shang, et al., 2012). However, such direct targeting may be the most effective means to reduce signal output given that many disease-associated mutations of small GTPases enable GEF-independent activation (Olson, 2018; Porter, et al., 2016). Moreover, in the case of HTN, direct targeting could provide an added benefit over ROCK inhibitors, as such an approach would block additional downstream pathways implicated in SMC contractility (i.e. mDial and mDia2, MRTF etc).
3.2.1. GTP-binding inhibitors
Although direct targeting of the GTP binding site of RhoA (or related small GTPases) is challenging due to its high affinity for GTP (pico to nano-molar range) and the high concentration of GTP in cells (~0.5 mM), some studies support the validity of this approach. Indeed, using an in silico virtual docking approach followed by surface-plasmon resonance validation of synthesized chemicals, Deng et. al. identified lead compounds that inhibited Rho-GTP binding in a dose- dependent fashion with IC50 values ranging from 1.24–2.05 μΜ (Deng, et al., 2011). After further structural modifications to increase water solubility, one compound ((A)-3-(3-(ethyl(quinolin-2-yl)amino)phenyl)acrylic acid) was shown to both attenuate PE-induced contraction in thoracic aorta rings ex vivo and to reduce cerebral vasospasm in a subarachnoid hemorrhage model in rats (Ma S and Li J 2014). While the in vivo efficacy was similar to that of fasudil, this second generation inhibitor still exhibited relatively low potency (IC50 71 μΜ in the contractile assay). Future studies will be necessary to determine whether these or related compounds exhibit specificity for RhoA versus other Rho family GTPases and other GTP-binding proteins.
In support of the possibility of identifying GTP binding inhibitors that can specifically target particular Rho-related family members, simultaneous multiplex screening for small molecules in the 200,000 Molecular Libraries Screening Center Network library has already identified a CDC42 specific inhibitor as well as a broad Ras family inhibitor. These inhibitors prevented GTP binding in a dose- dependent fashion (as noncompetitive allosteric inhibitors) and are active in cell based assays (Hong, et al., 2013; Surviladze, et al., 2010; Surviladze, et al., 2012). The subsequent application of this technology to screen the Prestwick Chemical Library of off patent and FDA approved drugs for inhibitors of eight Ras-related GTPases (but not RhoA, B, or C) led to the identification of R-enantiomers of naproxen and ketorolac (approved NSAIDs) as GTP binding inhibitors of Racl and Cdc42 (Oprea, et al., 2015). Although not biochemically confirmed, in silico docking analyses predicted that these drugs bind to an allosteric site near the GTP binding site that alters Mg2+ ion coordination and results in stabilizing the GTPase in its GDP-bound form. To our knowledge, this approach has not been used successfully to identify RhoA specific inhibitors, but these results provide strong proof-of-concept for this approach.
3.2.2. Selective non-covalent RhoA modifiers
The potential for identifying allosteric modifiers to inhibit RhoA has been demonstrated by the fact that a number of bacterial toxins are highly potent (though non-selective) inhibitors of Rho family members. For example, Histophilus somni and Vibrio parahemolyticus produce toxins that inhibit Rho proteins by promoting the covalent attachment of an AMP molecule to tyrosine 34, while toxin B produced by Clostridium difficile induces glucosylation of nearby threonine 37. These residues lie within the regulatory switch-I domain of Rho family members and addition of these bulky modifications in this domain inactivate the Rho GTPases by multiple mechanisms that include inhibition of GTPase cycling (by blocking GEF and GAP association), inhibition of cytosol- membrane cycling (by blocking Rho GDI interactions) and inhibition of Rho effector coupling (Lemichez, 2017; Pommier & Cherfils, 2005). Similarly, Clostridium botulinum exoenzyme C3 transferase toxin inhibits RhoA, RhoB, and RhoC in vitro and in vivo by promoting ADP-ribosylation of asparagine 41 which blocks GEF binding (Jansen, et al., 2018; Vogelsgesang, et al., 2007). Such pathogenic agents have been useful as pharmacological tools to completely block Rho-dependent signaling pathways in cells and in pre-clinical animal models. However, their potential as therapeutic agents is limited because of their difficult delivery, their non-specific actions, and their sometimes covalent and irreversible effects (Jansen, et al., 2018; Marchioni & Zheng, 2009). Nonetheless, the future exploitation of derivatives or mimetics of such enzymes could lead to the development of new inhibitors with tremendous clinical utility.
Finally, Rho family inactivation can be achieved by blocking plasma membrane targeting. For example, Rho GTPases are isoprenylated on carboxy- terminal Cysteine residues (within a so-called CAAX box) and this modification is important for membrane targeting and activation as evidenced by the fact that proteolytic cleavage of this site by Yersinia spp.-derived toxins effectively block Rho, Rac and CdC42 activation (Lemichez, 2017). Likewise, as RhoA is geranylgeranylated at this site, geranylgeranyl-transferase inhibitors and HMG- CoA-reductase inhibitors (which block both cholesterol and isoprenyl biogenesis) block RhoA membrane association and activation. In fact the clinical utility of this approach is highlighted by the fact HMG-CoA reductase inhibitors such as simvastatin and atorvastatin used to treat high cholesterol, also have antihypertensive properties (Kanaki, et al., 2013) and their BP lowering effects have been attributed to their ability to block RhoA signaling (Brandes, 2005; Kanaki, et al., 2013; Strazzullo, et al., 2007). However, the BP lowering effects of statins are modest (2 mmHg decrease systolic BP) and these isoprenoid pathway inhibitors display poor selectivity for individual Rho GTPases. Thus, further advancements in drug development are needed to realize the full potential of this approach.
4. RhoA GEFs and BP Control
Another potential strategy for inhibiting RhoA signaling is inhibition of the GEFs that control RhoA activity. Over 24 Rho-specific GEFs have been identified, and to date, at least 5 of these have been implicated in regulating SMC differentiation and/or contractility. This list includes three members of the RGS- GEF subfamily (P115-RhoGEF, PDZ-RhoGEF, LARG), p63RhoGEF and lymphoid blast crisis (Lbc) (Cario-Toumaniantz, et al., 2012; Hilgers, et al., 2007; Jin, et al., 2006; A. Wirth, et al., 2008; Ying, et al., 2009). Each of these RhoGEFs contains a Dbl homology (DH) domain (also known as the RhoGEF domain) followed by a pleckstrin homology (PH) domain (Table 2). The DH domain serves as both the catalytic site and the major binding interface for RhoA, while the PH domain facilitates membrane binding and cooperates with DH domains to fully activate RhoA. Other common domains include a regulator of G-protein signaling (RGS) domain that binds heterotrimeric G proteins, or the density 95, disk large, zona occludes-1 (PDZ) domain that binds to specific transmembrane receptors (including the Lysophosphatidic Acid (LPA)receptor among others (Yamada, et al., 2005). Importantly, these 5 GEFs are all highly expressed in conductance and resistance arteries of rats, mice, and humans (Cario-Toumaniantz, et al., 2012; Hilgers, et al., 2007; Jin, et al., 2006; A. Wirth, et al., 2008; Ying, et al., 2009)(Genotype-Tissue Expression [GTEx] Portal, accessed on 07/12/2018).
Table 2. Domain Structure of RhoGEF proteins associated with blood pressure.
The RhoGEF domain (also known as the Dbl homology [DH] domain), is the catalytically active domain of RhoGEFs and major binding site for RhoA GTPase. Immediately downstream of the RhoGEF domain is a pleckstrin homology (PH) domain, which works cooperatively with the RhoGEF domain to fully activate the RhoA GTPase. Other functional domains in these RhoGEFs are the Regulators of G protein signaling homology (RH) domain and the postsynaptic density 95, disk large, zona occludens-1 (PDZ) domain. Potential blood pressure therapeutics are being developed to target the interaction interface between RhoGEF domain and RhoA. Length is shown in amino acids (AA).
| GEF Protein | Length (AA) | Domain | |||
|---|---|---|---|---|---|
| PDZ | RH | Rho-GEF (DH) | PH | ||
| pll5-RhoGEF | 927 | ✓ | ✓ | ✓ | |
| PDZ-RhoGEF | 1562 | ✓ | ✓ | ✓ | ✓ |
| LARG | 1544 | ✓ | ✓ | ✓ | ✓ |
| p63-RhoGEF | 580 | ✓ | ✓ | ||
The regulator of G-protein signaling (RGS) family of Rho GEFs (LARG, pll5RhoGEF, and PDZRhoGEF) (Suzuki, et al., 2003) has received a lot of attention in the BP field because these proteins are activated by Gα12 and Gα13. coupled receptors which transduce signals mediated by major contractile agonists that include A-II, PE, ET1, and thromboxane A2 (Fukuhara, et al., 2001). pll5RhoGEF (pi 15) is the critical GEF that mediates A-II-dependent RhoA activity in SMC and small arterioles, and importantly, Guilluy et. al. showed that SM-specific deletion of pi 15 rendered mice resistant to A-II-dependent HTN (Guilluy et al., 2010b). However, A-II-dependent activation of RhoA in SMC has also been linked to activation of LARG (Ying, et al., 2006), PDZ-RhoGEF (Hilgers, et al., 2007; Ying, et al., 2009), and p63RhoGAP (Wuertz, et al., 2010) and inhibition of pl90RhoGAP (see below; (Bregeon, et al., 2009)), suggesting significant overlap between these pathways. For example, Ying et. al. showed that Ca2+/PYK2-dependent activation of PDZ-RhoGEF was necessary for maximal A-II induced RhoA activation in SMC (Ying, et al., 2009). Alternatively, p63-RhoGEF, which is highly expressed in arterial SM, was shown to be important for the early phase of A-II-dependent vessel contractility (Wuertz, et al., 2010) and for maximal pressor response to other vasoconstrictors such as PE and ET1 that act through Gα q/11 (Momotani, et al., 2011). Interestingly, p115 mutant mice exhibited normal pressor responses to ET1 and PE, but did not respond fully to A-II and had a partial reduction in DOCA/salt- induced HTN. On the other hand, LARG knockout mice were fully resistant to salt-induced HTN (A. Wirth, et al, 2008), which is consistent with subsequent findings that LARG regulates RhoA activity and SMC contractility in response to mechanical forces—which are known to be applied to small vessels in this volume overload model (Guilluy, et al, 2011). While not yet confirmed in pre-clinical animal models, the non-RGS Rho GEF termed lymphoid blast crisis (Lbc) is necessary for serotonin-dependent activation of RhoA and contractility in vascular SMC (Bear, et al, 2010). Thus, specific vasoconstrictors can lead to activation of distinct but overlapping sets of RhoGEFs to enable the fine-tuning of vessel tone and BP homeostasis.
With respect to pharmacological treatments, these findings indicate that targeting a critical SM GEF might provide greater therapeutic benefit than targeting RhoA or Rho kinase, because it could lead to a more modest (and cell type restricted) reduction of RhoA activity and result in fewer side effects. Moreover, the use of validated inhibitors could provide ‘personalized’ approaches that would better target the underlying pathophysiology (i.e. to limit HTN due to volume overload or elevated sympathetic activity). On the other hand, known functional redundancies also suggest that it may be necessary to target multiple GEFs to achieve therapeutic efficacy.
4.1. Targeting RhoGEFs
GEF protein-dependent nucleotide exchange involves a multi-step reaction. Upon binding to the GDP-bound form of the GTPase, GEFs facilitate release of GDP resulting in the formation of a nucleotide-free GTPase-GEF transition complex. The reaction then terminates with the binding of GTP (which is in a 10:1 molar excess in the cell) and dissociation of the GEF from the now active GTPase. The implication of this multi-step process for drug discovery is that these transition-binding intermediates tend to have the characteristics of druggable hotspots (i.e. exposed hydrophobic surfaces, unpaired polar groups, deeply curved surfaces, etc.) while the unbound, inactive proteins do not (Beglov, et al., 2018). Thus, it may be possible to identify small molecules that specifically target the GEF-GTPase interface (i.e. inhibit GEF binding or stabilize the GEF-GTPase transition state) or directly interfere with GEF activity.
4.1.1. Targeting the RhoA-GEF interface
Facilitated by high resolution crystal structures and sophisticated in silico screening, some recent drug discovery approaches support the possibility of targeting the RhoA-GEF interface. For example, Shang et. al. virtually screened 4 million compounds for their ability to pack into the Rho-GEF binding surface groove of RhoA. One of the drugs identified (termed Rhosin) contains two aromatic rings tethered by a linker which wraps around RhoA AA58 (a critical Tryptophan that is essential for GEF binding) and prevents RhoA from interacting with LARG, DBL, LBC, p115RhoGEF and PDZ RhoGEF without interfering with RhoGAP, RhoGDI, or RhoA effector binding (Shang, et al., 2012). Rhosin reversibly inhibited serum-induced RhoA, RhoB and RhoC activity, but did not inhibit other Rho GTPases (Racl or Ccd42) (Shang, et al, 2012). Rhosin also significantly and dose-dependently inhibited several RhoA-dependent functions in cells including MLC and PAK phosphorylation and stress fiber and focal adhesion formation (Shang, et al., 2012). Rhosin suppressed invasion and mammary sphere formation in breast cancer cells (Shang, et al., 2012), and also mitigated the acquisition of drug resistance in cancer stem cells (C. Yoon, et al., 2016). The ability of Rhosin to impact either BP homeostasis (or cancer growth) has not yet been tested in animal models. Since Rhosin exhibits a relatively low binding affinity, (Kd ~0.4 μΜ for RhoA binding in vitro) the identification of derivatives with better pharmacologic properties will be needed before Rhosin compounds can be tested clinically (Shang, et al., 2012). Moreover, since Rhosin binds to RhoA and prevents all GEFs from binding to this site, this drug is likely to have more off- target effects than one that could interfere with specific GEF complexes.
The first proof-of-concept for targeting a specific GEF-GTPase interface was a series of elegant studies that unveiled the mechanism by which the fungal toxin, Brefeldin A (BFA), inhibited Arfl- dependent trafficking of proteins from the endoplasmic reticulum to the Golgi. Biochemical studies revealed that BFA selectively blocked the ability of the ArflGEF, Sec7, from catalyzing GDP release. Importantly co-crystalization studies revealed that BFA binds to a hydrophobic pocket that does not exist in ArflGDP but is created upon Sec7 binding. Since this energetically unfavorable hydrophobic pocket drives the conformational changes necessary for nucleotide dissociation, BFA binding effectively locks the complex in a GDP bound conformation (Cherfils & Melancon, 2005; Mossessova, et al., 2003; Zeghouf, et al., 2005). In spite of high sequence homology among the ArflGEFs, only the Sec7 complex is targeted by BFA, suggesting that ‘interfaciaF small molecule inhibitors could be identified that specifically target RhoA-GEF interactions.
These studies have inspired a new line of investigation that seeks to exploit novel pockets at small GTPase-GEF interfaces. The most productive approach so far used NMR-based fragment screening combined with high resolution structure/functional analyses to identify small molecule inhibitors of kRas and its GEF, Sosl. These screens identified compounds that either block Sosl binding to kRAS or that bind to a site adjacent to the functionally important switch I/II regions of KRas and (like BFA) block SOS 1-dependent nucleotide exchange. A similar approach was used to identify a ligand that binds to the cavity adjacent to the switch II region of RhoA and inhibits the interaction between RhoAGDP and the LARG DH domain (Gao, et al, 2014). Small molecules have also been identified that inhibit TrioGEFs interaction with Racl and RhoG and Cdc42’s interaction with the GEF, intersectin-1 (Smithers & Overduin, 2016). Clearly much work needs to be done to realize the potential for such ligands. The inhibitors identified exhibit very low potency (in the high micromolar range) and their ability to block additional GEFs has not yet been evaluated. In fact, in silico evaluation of aforementioned SOS1 inhibitor suggests that the hydrogen bonds formed by this ligand in the Ras-SOSl complex would likely be conserved in the complex of Ras and another cognate GEF, RasGRFl (Gao, et al., 2014). While challenging, further medicinal chemistry and molecular dynamics approaches should facilitate our ability to target RhoA’s interaction with specific GEFs.
4.2.2. Targeting GEF activity
Another viable approach is to directly target the activity of specific RhoGEFs before they interact with the small GTPases. Using a high-throughput screen, Shang et.al. identified a compound, Y16, that binds LARG at the junction between the DH and PH domains with a Kd of ~80nM (Shang, et al., 2013). Y16 prevented LARG from interacting with RhoA in vitro and reversibly attenuated serum-induced RhoA activity in NIH 3T3 cells and mammary sphere formation MCF7 cells with little to no toxicity (Shang, et al., 2013). A separate fluorescent ligand-based screen of ten thousand compounds yielded five additional selective inhibitors of LARG-dependent RhoA GTPase activity, though their mechanisms of action have not yet been determined (Evelyn, et al., 2009). Again, to date, many of these lead compounds exhibit low potency, but theoretically, combination therapy strategies which pair Y16 with some of these compounds and/or their derivatives may lead to a highly efficacious approach to limit LARG-mediated signaling.
Finally, since many GEFs are regulated by protein-protein interactions and post-translational modifications, it may be possible to identify drugs that block these mechanisms. In an excellent example of this approach, cAMP is a potent activator of the Rapl GEFs, Exchange Proteins directly Activated by cAMP (EPAC1 and 2) and high throughput screens to identify small molecules that displace cAMP binding led to the discovery of several inhibitors or partial agonists for these GEFs (Brown, et al., 2014; Z. Liu, et al., 2017; Parnell, et al., 2017). Such studies support the concept that identification of small molecules that prevent physiological activation of specific GEFs could prove therapeutically useful. Along these lines, several studies have shown that RGSRhoGEFs are regulated by phosphorylation. Guilluy et al. demonstrated that A-II-dependent activation of pll5GEF was mediated by phosphorylation of the PH domain (Tyr738) by Janus tyrosine kinase (Guilluy, et al., 2010). Importantly, phosphorylation mimetic and deficient variants of Tyr738 elevated and reduced pll5’s GEF activity, respectively. PDZRhoGEF and LARG are also activated by tyrosine phosphorylation. Focal adhesion kinase (FAK) and its related family member, proline-rich tyrosine kinase 2 (PYK2) phosphorylate and activate PDZRhoGEF (Chikumi, et al, 2002; Ying, et al, 2009), while FAK, Tec and the serine- threonine kinase, p90 ribosomal kinase-2 each phosphorylate and activate LARG (Chikumi, et al., 2002; G. X. Shi, et al., 2018; Suzuki, et al., 2003; Ying, et al., 2009). Drugs that specifically inhibit these phosphorylation events could prove to be useful RhoA signaling inhibitors.
5. GTPase activating proteins (GAPs)
Although GEFs have classically been considered to be the major regulators of RhoGTPase activity, increasing evidence suggest the GAPs are also critically important. As their name implies, GAPs enhance the intrinsic GTPase activity of the Rho GTPases by several orders of magnitude thus decreasing the length of time that GTPases are in the active form (Bos, et al., 2007; Puetz, et al., 2009; Rittinger, et al., 1997). The 66 RhoGAPs in the human genome comprise a broad and diverse family that can be further subdivided based upon the presence of a variety of functional domains (Tcherkezian & Lamarche-Vane, 2007) (Table 3). By mediating interactions with different membrane and protein components these domains are critical for the selective function of the GAP proteins and the dynamic inhibition of small GTPase signaling. Several Rho-selective GAPs including pl90ARhoGAP, ArhGAPl, Myr5, GRAF1, and GRAF3 have been shown to inhibit RhoA activity in cultured vascular SMC and could be potential targets for therapeutic interventions that affect vascular function and BP (Bai, et al, 2013; Mori, et al, 2009). However, GRAF3 is the only RhoGAP that has been directly linked to the regulation of SM contractility and BP homeostasis (Bai, et al, 2013; Ehret, et al, 2011; Kato, et al, 2015; Li, et al, 2016; Wain, et al., 2011).
Table 3. Domain Structure of RhoGAP proteins associated with blood pressure.
The GAP domain is the catalytically active domain of RhoGAPs. GRAF3 also contains a Bin/amphiphysin/RVS (BAR) domain, a pleckstrin homology (PH) domain, and a SRC homology 3 (SH3) domain, which sense and induce membrane curvature, aid in lipid binding, and promote protein-protein interactions, respectively. In addition to the catalytic GAP domain, pl90-RhoGAP also contains a GTP-binding domain (GBD), four diphenylalanine motifs (FF), and a polybasic region (PBR). These regions aid in GTP binding, assist in protein- protein binding, and allow for membrane association, respectively. Potential blood pressure therapeutics involving these RhoGAPs focus on activating the GAP domain. Length is shown in amino acids (AA)
| GAP Protein | Length (AA) | Domain | |||||||
|---|---|---|---|---|---|---|---|---|---|
| BAR | PH | GBD | FF | PBR | GBD | Rho GAP | SH3 | ||
| GRAF 3 | 874 | ✓ | ✓ | ✓ | ✓ | ||||
| PI 90-RhoGAP | 1499 | ✓ | ✓ | ✓ | ✓ | ✓ | |||
| ✓ | |||||||||
| ✓ | |||||||||
| ✓ | |||||||||
5.1. GRAF3 and Hypertension
We originally identified the founding member of the GRAF (GTPase Regulator Associated with FAK) family by screening an embryonic Agt 11 expression library for proteins that interacted with the carboxyl-terminal domain of FAK (Hildebrand, et al., 1996; Taylor, et al., 1998; Taylor, et al., 1999). This family which is now known to comprise 3 members, GRAF1 (ArhGAP26), GRAF2 (ArhGAPIO) and GRAF3 (ArhGAP42) contain an N-terminal BAR (Bin/amphiphysin/Rvs) domain, a phosphatidylserine (PS)-binding PH domain, a central Rho-GAP domain, a serine/proline rich domain, and a C-terminal FAEC- binding SH3 domain (Hildebrand, et al., 1996). GRAF1 is expressed predominantly in the brain and striated muscle (cardiac and skeletal), and our studies in GRAF 1-depleted Xenopus and mice revealed that GRAF 1-dependent inhibition of RhoA activity promoted mammalian muscle growth by facilitating myoblast fusion and injury repair (Doherty, et al., 2011; Lenhart, et al., 2014; Lenhart, et al., 2015; Taylor, et al., 1998). GRAF2 is more ubiquitously expressed (X. R. Ren, et al., 2001) and could partially compensate for the loss of GRAF1 during myotube formation, supporting at least some functional redundancy within this family (Lenhart, et al, 2014). Evolutionarily, GRAF3 is the youngest family member and is the most recently annotated. Importantly, we found that GRAF3 was highly and selectively expressed in SMC with particularly high expression in resistance vessels (Bai, et al., 2013) suggesting that its expression levels might be a major determinant of RhoA activity, SM contraction, and vessel tone.
Over the last several years, studies from our laboratory confirmed that GRAF3 is a SMC-selective Rho GAP protein that imparts tight control of BP homeostasis by modulating vascular resistance. Specifically, we showed that mice with gene-trap-mediated reductions in GRAF3 levels exhibited significant basal HTN and elevated pressor responses to A-II, ET1, PE, and DOC A salt (Bai, et al., 2013). Notably, the hypertensive phenotype in this model was fully reversed by treatment with ROCK inhibitors or by Cre-mediated re-expression of GRAF3 in SMC, strongly supporting the contention that GRAF3 control of SMC RhoA activity is necessary for homeostatic control of basal and pressor-induced BP. Mechanistically, our data supported a model wherein GRAF3 controls BP homeostasis by limiting RhoA-dependent MLC phosphorylation and blunting acute Ca2+’mediated SMC contractility in resistance vessels. Moreover, depletion of endogenous GRAF3 from vascular SMC enhanced MRTF-A nuclear accumulation, and stimulated expression of contractile proteins including SM a- actin, SM-myosin heavy chain, calponin and SM-22, indicating that changing the levels of GRAF3 would likely have a long-lasting impact on vessel tone (Bai, et al., 2017a). Interestingly, GRAF3 mRNA was significantly upregulated in SMC cultures subjected to cyclic stretch and in isolated portal vein segments subjected to static stretch and we showed that these effects were mediated via the RhoA/MRTF/SRF pathway (Bai, et al., 2017a). Since similar physical forces are known to be increased in the vessel wall under hypertensive conditions (Albinsson, et al., 2004), we postulated that GRAF3 might serve as a transcriptionally mediated negative feedback loop of the RhoA signaling axis. In support of this possibility, arterial GRAF3 mRNA levels were significantly increased in mice made hypertensive by L-NAME or DOCA-salt regimens (Bai, et al., 2013). In fact, taking advantage of an elegant mouse model developed by the Smithies lab in which plasma volumes of mice range from ~50% below normal to ~50% above normal (Kakoki, et al., 2013), we showed that GRAF3 expression increased in parallel to and was strongly correlated with plasma volume (r2=0.94) (Bai, et al., 2017a). Taken together, these findings indicate that GRAF3 serves as a mechanical strain-sensitive rheostat that acts to prevent excessive feed-forward activation of the RhoA signaling axis in order to control SMC tone and BP.
Of clinical importance, three separate GWAS studies identified a blood-pressure associated allele within the GRAF3 locus. Notably, our recently published follow-up studies revealed that GRAF3 mRNA levels were approximately 3-fold higher in arteries from individuals homozygous for the protective allele and our human genetic data from well-characterized untreated patients confirmed that the protective allele was associated with a 5mm Hg decrease in BP (Bai, et al, 2017b; Ehret, et al, 2011; Kato, et al, 2015; Wain, et al, 2011). We went on to identify a novel mechanism for the BP-associated locus which mapped to the first intron of the GRAF3 gene (.ARHGAP42) (Bai, et al., 2017b). Our studies revealed that rs604723 is the causative SNP at this locus and that the minor T allele variation increases ARHGAP42 expression by promoting SRF binding to a SMC-selective intronic regulatory element. Our demonstration that the minor ARHGAP42 allele is more highly expressed in HuAoSMCs, when coupled with similar data from human artery samples, strongly supports our hypothesis that this variation reduces BP by inhibiting RhoA-dependent constriction of resistance vessels. Our data add to a growing body of evidence that common noncoding variants alter cardiovascular risk by altering transcription factor binding and gene expression and support previous studies implicating RhoA signaling in the regulation of BP homeostasis in mice. Moreover, when coupled with the remarkable SMC-selective expression pattern of GRAF3, these studies provide strong evidence for GRAF3 as a novel therapeutic target for HTN.
5.2. Druggability ofRhoGAPs
To date, targeting GAPs for therapeutic advances has been largely overlooked. This is due, in part, because GAPs have little to no effect on promoting GTP hydrolysis of oncogenic Ras and Rho mutants, and therefore would not be good targets for cancer treatment (Holderfield, 2018). GAPs have also been considered less attractive targets for anti-hypertensive therapies because the drug would need to enhance GAP activity and it is traditionally thought to be more difficult to develop small molecule activators than small molecule inhibitors. Nonetheless, due to the multi-domain nature and the varied physiological regulation of RhoGAP proteins, there are several possibilities for allosteric modulation of GAP activity. Indeed, the activities of several RhoGAPs including the GRAFs, OPHN1, β- chimerin, DLC1, and p50 Rho GAP are regulated by intramolecular auto- inhibition. For GRAF1 and similarly structured Oligophrenin and ASAP1, the BAR and PH domains physically associate with the GAP domain to sterically inhibit its function (Eberth, et al., 2009; Jian, et al., 2009). We and others have shown that this mechanism also controls the activity of GRAF3 (unpublished; (Luo, et al., 2017)) (Figure 2). Interestingly, FAK/Src-dependent phosphorylation of GRAF3 at Tyr376 modulated its GAP activity (Luo, et al., 2017) and we have shown that a phosphomimetic E376 variant exhibited elevated activity (unpublished observations). By defining the structural interactions that control GRAF3 activity, these results could lead to the development of GRAF3 activators that target the BAR-PH/GAP interface that should be useful for reducing arterial tone.
Figure 2. Domain Structure and therapeutic strategy for targeting the SMC- specific RhoGAP GRAF3.
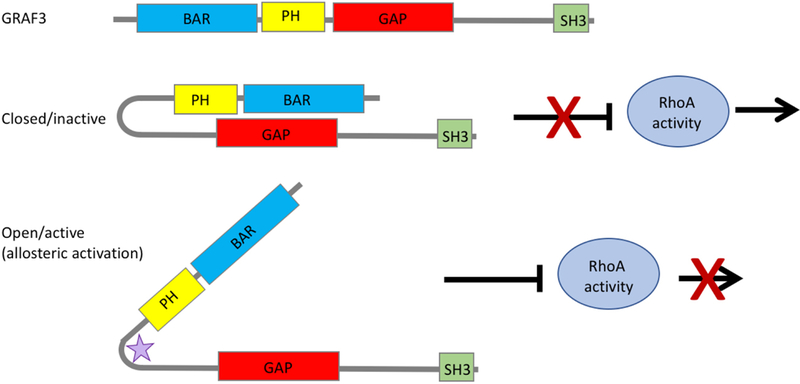
Like other BAR-PH-GAP containing proteins, the BAR and PH domains of GRAF3 act as a functional unit to autoinhibit the GAP domain, preventing GRAF3’s interaction with RhoA. One potential therapeutic strategy is to use allosteric activation (either post-translational modifications or small molecules, represented here by the purple star) to lock GRAF3 in its open conformation, enhancing hydrolysis of GTP to GDP, thereby decreasing the activity of RhoA and promoting SMC relaxation and decreased blood pressure.
6. Conclusions
In conclusion, the search for new approaches to control high BP remains dramatically important for reducing global health burden, and targeting RhoA- mediated SM contractility is a promising avenue (Figure 3). Indeed, strong evidence from pre-clinical animal models indicate that modulating the activity of SM Rho-GEFs, Rho-GAPs, or ROCK has a major impact on systemic BP homeostasis. While new advances in drug development have led to potent and specific ROCK inhibitors that can be safely used in patients, whether any of these compounds exhibit the necessary selectivity and pharmacological profiles required for BP management in patients requires further study. Although efforts to target GEF and GAPs or the RhoA binding interface for these enzymes are lagging behind, recent advances in drug discovery indicate that it will likely soon be possible to engineer clinically-relevant small molecule regulators of these enzymes that could be effective anti-hypertensive therapies. To ensure success in this regard, we will need to continue to gain a better understanding of the mechanisms that regulate these enzymes. Moreover, based on the fact that BP is a highly variable trait among individuals, a better understanding of the genetic mechanisms regulating this disease is critical for a more personalized treatment plan for patients. Given that genetic variations in both upstream activators and downstream mediators of RhoA have been linked to BP regulation, screening for such variants could potentially be used to tailor more effective individualized treatment regimens.
Figure 3. Therapeutic potential of the RhoA pathway to treat HTN.
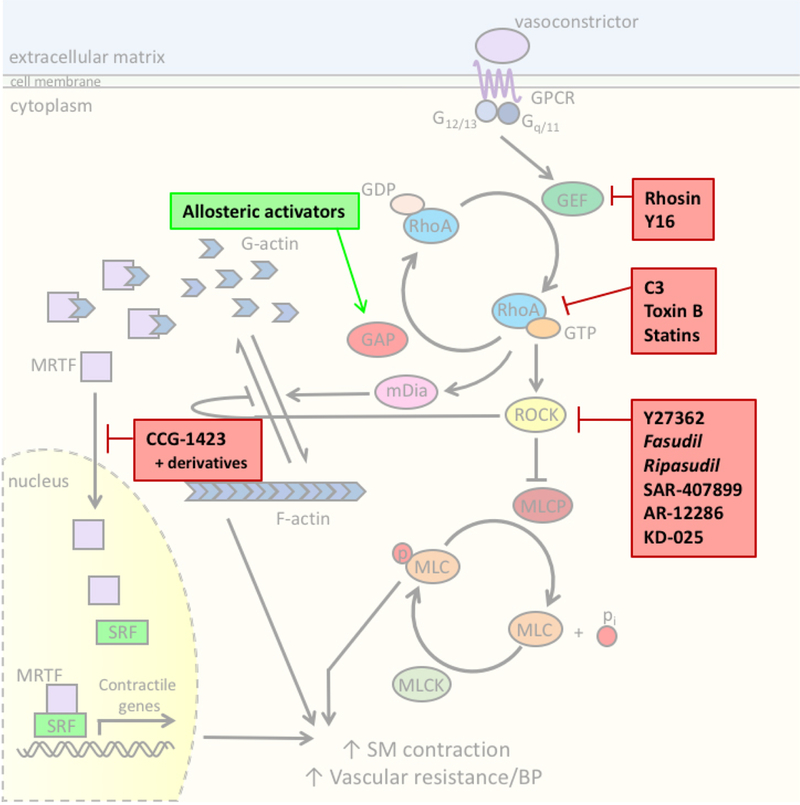
There are many components of the SMC RhoA contractile pathway that could be targeted to reduce systemic blood pressure. While not an exhaustive list, shown here in bold boxes are compounds discussed in this article, including those that are proof-of- concept, in clinical development, or currently used in the clinic (italicized).
Abbreviations
- ACE
angiotensin converting enzyme
- A-II
angiotensin II
- BP
blood pressure
- DOCA
deoxycorticosterone-acetate
- ET1
endothelin-1
- GAP
GTPase activating protein
- GDP
guanosine diphosphate
- GEF
guanine nucleotide exchange factor
- GTP
guanosine triphosphate
- GWAS
genome-wide association studies
- HTN
hypertension
- L-NAME L-NG
Nitroarginine methyl ester
- MLC
myosin light chain
- MLCK
myosin light chain kinase
- MLCP
myosin light chain phosphatase
- MRTF-A
myocardin related transcription factor-A
- MRTF-B
myocardin related transcription factor-B
- PAH
pulmonary arterial hypertension
- PE
phenylephrine
- ROCK
Rho Kinase or Rho-associated coiled-coil domain containing protein kinases
- SHR
spontaneously hypertensive rat
- SM
smooth muscle
- SMC
smooth muscle cell
- SRF
serum response factor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Statement
The authors declare that there are no conflicts of interest.
References
- Achelrod D, Wenzel U, & Frey S (2015). Systematic Review and Meta- Analysis of the Prevalence of Resistant Hypertension in Treated Hypertensive Populations. American Journal of Hypertension, 28, 355–361. [DOI] [PubMed] [Google Scholar]
- Albinsson S, Nordstrom I, & Hellstrand P (2004). Stretch of the vascular wall induces smooth muscle differentiation by promoting actin polymerization. J Biol Chem, 279, 34849–34855. [DOI] [PubMed] [Google Scholar]
- Asano M, & Nomura Y (2003). Comparison of inhibitory effects of Y-27632, a Rho kinase inhibitor, in strips of small and large mesenteric arteries from spontaneously hypertensive and normotensive Wistar-Kyoto rats. Hypertens Res, 26,97–106. [DOI] [PubMed] [Google Scholar]
- Bai X, Lenhart KC, Bird KE, Suen AA, Rojas M, Kakoki M, et al. (2013). The smooth muscle-selective RhoGAP GRAF3 is a critical regulator of vascular tone and hypertension. Nature communications, 4, 2910–2910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai X, Mangum K, Kakoki M, Smithies O, Mack CP, & Taylor JM (2017a). GRAF3 serves as a blood volume-sensitive rheostat to control smooth muscle contractility and blood pressure. Small GTPases, 00–00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai X, Mangum KD, Dee RA, Stouffer GA, Lee CR, Oni-Orisan A, et al. (2017b). Blood pressure-associated polymorphism controls ARHGAP42 expression via serum response factor DNA binding. The Journal of Clin ical Investigation, 127, 670–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bain J, Plater L, Elliott M, Shpiro N, Hastie CJ, Mclauchlan EL, et al. (2007). The selectivity of protein kinase inhibitors: a further update. Biochemical Journal, 408, 297–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bear MD, Li M, Liu Y, Giel-Moloney MA, Fanburg BL, & Toksoz D (2010). The Lbc Rho guanine nucleotide exchange factor alpha-catulin axis functions in serotonin-induced vascular smooth muscle cell mitogenesis and RhoA/ROCK activation. J Biol Chem, 285, 32919–32926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beglov D, Hall DR, Wakefield AE, Luo L, Allen KN, Kozakov D, et al. (2018). Exploring the structural origins of cryptic sites on proteins. Proc Natl Acad Sci US A, 115, E3416–E3425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodor N, & Buchwald P (2000). Soft drug design: general principles and recent applications. Med Res Rev, 20, 58–101. [DOI] [PubMed] [Google Scholar]
- Boerma M, Fu Q, Wang J, Loose DS, Bartolozzi A, Ellis JL, et al. (2008). Comparative gene expression profiling in three primary human cell lines after treatment with a novel inhibitor of Rho kinase or atorvastatin. Blood Coagul Fibrinolysis, 19, 709–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boettner B, & Van Aelst L (2002). The role of Rho GTPases in disease development. Gene, 286, 155–174. [DOI] [PubMed] [Google Scholar]
- Booth JN 3rd, Muntner P, Diaz KM, Viera AJ, Bello NA, Schwartz JE, et al. (2016). Evaluation of Criteria to Detect Masked Hypertension. J Clin Hypertens (Greenwich), 18, 1086–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bos JL, Rehmann H, & Wittinghofer A (2007). GEFs and GAPs: Critical Elements in the Control of Small G Proteins. Cell, 129, 865–877. [DOI] [PubMed] [Google Scholar]
- Boyden LM, Choi M, Choate KA, Nelson-Williams CJ, Farhi A, Toka HR, et al. (2012). Mutations in kelch-like 3 and cullin 3 cause hypertension and electrolyte abnormalities. Nature, 482, 98–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandes RP (2005). Statin-mediated inhibition of Rho: only to get more NO? Circ Res, 96, 927–929. [DOI] [PubMed] [Google Scholar]
- Bregeon J, Loirand G, Pacaud P, & Rolli-Derkinderen M (2009). Angiotensin II induces RhoA activation through SHP2-dependent dephosphorylation of the RhoGAP p190A in vascular smooth muscle cells. Am J Physiol Cell Physiol, 297, C1062–1070. [DOI] [PubMed] [Google Scholar]
- Brown LM, Rogers KE, Aroonsakool N, McCammon JA, & Insel PA (2014). Allosteric inhibition of Epac: computational modeling and experimental validation to identify allosteric sites and inhibitors. J Biol Chem, 289, 29148–29157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budzyn K, Marley PD, & Sobey CG (2006). Targeting Rho and Rho-kinase in the treatment of cardiovascular disease. Trends Pharmacol Sci, 27, 97–104. [DOI] [PubMed] [Google Scholar]
- Carbone ML, Bregeon J, Devos N, Chadeuf G, Blanchard A, Azizi M, et al. (2015). Angiotensin II activates the RhoA exchange factor Arhgef1 in humans. Hypertension, 65, 1273–1278. [DOI] [PubMed] [Google Scholar]
- Cario-Toumaniantz C, Ferland-McCollough D, Chadeuf G, Toumaniantz G, Rodriguez M, Galizzi JP, et al. (2012). RhoA guanine exchange factor expression profile in arteries: evidence for a Rho kinase-dependent negative feedback in angiotensin Il-dependent hypertension. Am JPhysiol Cell Physiol, 302, C1394–1404. [DOI] [PubMed] [Google Scholar]
- Carlstrom M, Wilcox CS, & Arendshorst WJ (2015). Renal autoregulation in health and disease. Physiol Rev, 95, 405–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang DF, Belaguli NS, Iyer D, Roberts WB, Wu SP, Dong XR, et al. (2003). Cysteine-rich LIM-only proteins CRP1 and CRP2 are potent smooth muscle differentiation cofactors. Dev Cell, 4, 107–118. [DOI] [PubMed] [Google Scholar]
- Chen CY, & Schwartz RJ (1996). Recruitment of the tinman homolog Nkx- 2.5 by serum response factor activates cardiac alpha-actin gene transcription. Mol Cell Biol, 16, 6372–6384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen XQ, Tan I, Ng CH, Hall C, Lim L, & Leung T (2002). Characterization of RhoA-binding kinase ROKalpha implication of the pleckstrin homology domain in ROKalpha function using region-specific antibodies. J Biol Chem, 277, 12680–12688. [DOI] [PubMed] [Google Scholar]
- Chen XY, Dun JN, Miao QF, & Zhang YJ (2009). Fasudil hydrochloride hydrate, a Rho-kinase inhibitor, suppresses 5-hydroxytryptamine-induced pulmonary artery smooth muscle cell proliferation via JNK and ERK1/2 pathway. Pharmacology, 83, 67–79. [DOI] [PubMed] [Google Scholar]
- Cherfils J, & Melancon P (2005). On the action of Brefeldin A on Sec 7- [DOI] [PubMed] [Google Scholar]
- stimulated membrane-recruitment and GDP/GTP exchange of Arf proteins. Biochem Soc Trans, 33, 635–638. [DOI] [PubMed] [Google Scholar]
- Chevrier V, Piel M, Collomb N, Saoudi Y, Frank R, Paintrand M, et al. (2002). The Rho-associated protein kinase p160ROCK is required for centrosome positioning. J Cell Biol, 157, 807–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chikumi H, Fukuhara S, & Gutkind JS (2002). Regulation of G protein-linked guanine nucleotide exchange factors for Rho, PDZ-RhoGEF, and LARG by tyrosine phosphorylation: evidence of a role for focal adhesion kinase. J Biol Chem, 277, 12463–12473. [DOI] [PubMed] [Google Scholar]
- Citi S, Pulimeno P, & Paschoud S (2012). Cingulin, paracingulin, and PLEKHA7: signaling and cytoskeletal adaptors at the apical junctional complex. Annals of the New York Academy of Sciences, 1257, 125–132. [DOI] [PubMed] [Google Scholar]
- Cowley AW Jr. (2006). The genetic dissection of essential hypertension. Nat Rev Genet, 7, 829–840. [DOI] [PubMed] [Google Scholar]
- Dalton S, & Treisman R (1992). Characterization of SAP-1, a protein recruited by serum response factor to the c-fos serum response element. Cell, 68, 597–612. [DOI] [PubMed] [Google Scholar]
- Davis MJ, & Hill MA (1999). Signaling mechanisms underlying the vascular myogenic response. Physiol Rev, 79, 387–423. [DOI] [PubMed] [Google Scholar]
- Davis MJ, Wu X, Nurkiewicz TR, Kawasaki J, Davis GE, Hill MA, et al. (2001). Integrins and mechanotransduction of the vascular myogenic response. Am J Physiol Heart Circ Physiol, 280, H1427–1433. [DOI] [PubMed] [Google Scholar]
- Deng J, Feng E, Ma S, Zhang Y, Liu X, Li H, et al. (2011). Design and synthesis of small molecule RhoA inhibitors: a new promising therapy for cardiovascular diseases? J Med Chem, 54, 4508–4522. [DOI] [PubMed] [Google Scholar]
- Doherty JT, Lenhart KC, Cameron MV, Mack CP, Conlon FL, & Taylor JM (2011). Skeletal muscle differentiation and fusion are regulated by the BAR-containing Rho-GTPase-activating protein (Rho-GAP), GRAF1. J Biol Chem, 286, 25903–25921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberth A, Lundmark R, Gremer L, Dvorsky R, Koessmeier KT, McMahon HT, et al. (2009). A BAR domain-mediated autoinhibitory mechanism for RhoGAPs of the GRAF family. Biochem J, 417, 371–377. [DOI] [PubMed] [Google Scholar]
- Ehret GB, Ferreira T, Chasman DI, Jackson AU, Schmidt EM, Johnson T, et al. (2016). The genetics of blood pressure regulation and its target organs from association studies in 342,415 individuals. Nat Genet, 48, 1171–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehret GB, Munroe PB, Rice KM, Bochud M, Johnson AD, Chasman DI, et al. (2011). Genetic variants in novel pathways influence blood pressure and cardiovascular disease risk. Nature, 478, 103–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endres BT, Priestley JR, Palygin O, Flister MJ, Hoffman MJ, Weinberg BD, et al. (2014). Mutation of Plekha7 attenuates salt-sensitive hypertension in the rat. Proceedings of the National Academy of Sciences of the United States of America, 111, 12817–12822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etienne-Manneville S, & Hall A (2002). Rho GTPases in cell biology. Nature, 420, 629–635. [DOI] [PubMed] [Google Scholar]
- Evelyn CR, Ferng T, Rojas RJ, Larsen MJ, Sondek J, & Neubig RR (2009). High-throughput screening for small-molecule inhibitors of LARG- stimulated RhoA nucleotide binding via a novel fluorescence polarization assay. J Biomol Screen, 14, 161–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, LoGrasso PV, Defert O, & Li R (2016). Rho Kinase (ROCK) Inhibitors and Their Therapeutic Potential. Journal of Medicinal Chemistry, 59, 2269–2300. [DOI] [PubMed] [Google Scholar]
- Fukuhara S, Chikumi H, & Gutkind JS (2001). RGS-containing RhoGEFs: the missing link between transforming G proteins and Rho? Oncogene, 20, 1661–1668. [DOI] [PubMed] [Google Scholar]
- Fukumoto Y, Matoba T, Ito A, Tanaka H, Kishi T, Hayashidani S, et al. (2005). Acute vasodilator effects of a Rho-kinase inhibitor, fasudil, in patients with severe pulmonary hypertension. Heart, 91, 391–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukumoto Y, Yamada N, Matsubara H, Mizoguchi M, Uchino K, Yao A, et al. (2013). Double-blind, placebo-controlled clinical trial with a rho-kinase inhibitor in pulmonary arterial hypertension. Circ J, 77, 2619–2625. [DOI] [PubMed] [Google Scholar]
- Gao J, Ma R, Wang W, Wang N, Sasaki R, Snyderman D, et al. (2014). Automated NMR fragment based screening identified a novel interface blocker to the LARG/RhoA complex. PLoS One, 9, e88098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garnock-Jones KP (2014). Ripasudil: first global approval. Drugs, 74, 2211–2215. [DOI] [PubMed] [Google Scholar]
- Grisk O, Schluter T, Reimer N, Zimmermann U, Katsari E, Plettenburg O, et al. (2012). The Rho kinase inhibitor SAR407899 potently inhibits endothelin-1-induced constriction of renal resistance arteries. J Hypertens, 30, 980–989. [DOI] [PubMed] [Google Scholar]
- Guagnini F, Ferazzini M, Grasso M, Blanco S, & Croci T (2012). Erectile properties of the Rho-kinase inhibitor SAR407899 in diabetic animals and human isolated corpora cavernosa. J Transl Med, 10, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilluy C, Bregeon J, Toumaniantz G, Rolli-Derkinderen M, Retailleau K, Loufrani L, et al. (2010). The Rho exchange factor Arhgef1 mediates the effects of angiotensin II on vascular tone and blood pressure. Nat Med, 16, 183–190. [DOI] [PubMed] [Google Scholar]
- Guilluy C, Swaminathan V, Garcia-Mata R, Timothy O’Brien E, Superfine R, & Burridge K (2011). The Rho GEFs LARG and GEF-H1 regulate the mechanical response to force on integrins. Nat Cell Biol, 13, 724–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Zhang X, Guo L, Li Z, Zheng L, Yu S, et al. (2013). Association between pre-hypertension and cardiovascular outcomes: a systematic review and meta-analysis of prospective studies. Curr Hypertens Rep, 15, 703–716. [DOI] [PubMed] [Google Scholar]
- Hall JE (2003). The kidney, hypertension, and obesity. Hypertension, 41, 625–633. [DOI] [PubMed] [Google Scholar]
- Hayashi H, Szaszi K, Coady-Osberg N, Furuya W, Bretscher AP, Orlowski J, et al. (2004). Inhibition and redistribution of NHE3, the apical Na+/H+ exchanger, by Clostridium difficile toxin B. J Gen Physiol, 123, 491–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrand JD, Taylor JM, & Parsons JT (1996). An SH3 domain- containing GTPase-activating protein for Rho and Cdc42 associates with focal adhesion kinase. Mol Cell Biol, 16, 3169–3178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiley E, McMullan R, & Nurrish SJ (2006). The Galpha12-RGS RhoGEF- RhoA signalling pathway regulates neurotransmitter release in C. elegans. Embo J, 25, 5884–5895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilgers RH, Todd J Jr., & Webb RC (2007). Increased PDZ- RhoGEF/RhoA/Rho kinase signaling in small mesenteric arteries of angiotensin II-induced hypertensive rats. JHypertens, 25, 1687–1697. [DOI] [PubMed] [Google Scholar]
- Hill CS, & Treisman R (1995). Differential activation of c-fos promoter elements by serum, lysophosphatidic acid, G proteins and polypeptide growth factors. Embo J, 14, 5037–5047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinderliter AL, Voora RA, & Viera AJ (2018). Implementing ABPM into Clinical Practice. Curr Hypertens Rep, 20, 5. [DOI] [PubMed] [Google Scholar]
- Hinson JS, Medlin MD, Lockman K, Taylor JM, & Mack CP (2007). Smooth muscle cell-specific transcription is regulated by nuclear localization of the myocardin-related transcription factors. Am J Physiol Heart Circ Physiol, 292, H1170–1180. [DOI] [PubMed] [Google Scholar]
- Holderfield M (2018). Efforts to Develop KRAS Inhibitors. Cold Spring Harb Perspect Med, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homma K, Hayashi K, Wakino S, Tokuyama H, Kanda T, Tatematsu S, et al. (2014). Rho-kinase contributes to pressure-induced constriction of renal microvessels. Keio J Med, 63, 1–12. [DOI] [PubMed] [Google Scholar]
- Hong L, Kenney SR, Phillips GK, Simpson D, Schroeder CE, Noth J, et al. (2013). Characterization of a Cdc42 protein inhibitor and its use as a molecular probe. J Biol Chem, 288, 8531–8543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyun Lee J, Zheng Y, von Bomstadt D, Wei Y, Balcioglu A, Daneshmand A, et al. (2014). Selective ROCK2 inhibition in focal cerebral ischemia. Annals of Clinical and Translational Neurology, 1, 2–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibeawuchi SR, Agbor LN, Quelle FW, & Sigmund CD (2015). Hypertension-causing Mutations in Cullin3 Protein Impair RhoA Protein Ubiquitination and Augment the Association with Substrate Adaptors. The Journal of biological chemistry, 290, 19208–19217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inscho EW (2009). ATP, P2 receptors and the renal microcirculation. Purinergic Signal, 5, 447–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Hirooka Y, Kimura Y, Shimokawa H, & Takeshita A (2005). Effects of hydroxyfasudil administered to the nucleus tractus solitarii on blood pressure and heart rate in spontaneously hypertensive rats. Clin Exp Hypertens, 27, 269–277. [PubMed] [Google Scholar]
- Ito K, Hirooka Y, Sakai K, Kishi T, Kaibuchi K, Shimokawa H, et al. (2003). Rho/Rho-kinase pathway in brain stem contributes to blood pressure regulation via sympathetic nervous system: possible involvement in neural mechanisms of hypertension. Circ Res, 92, 1337–1343. [DOI] [PubMed] [Google Scholar]
- Jansen S, Gosens R, Wieland T, & Schmidt M (2018). Paving the Rho in cancer metastasis: Rho GTPases and beyond. Pharmacol Ther, 183, 1–21. [DOI] [PubMed] [Google Scholar]
- Jian X, Brown P, Schuck P, Gruschus JM, Balbo A, Hinshaw JE, et al. (2009). Autoinhibition of Arf GTPase-activating protein activity by the BAR domain in ASAP1. J Biol Chem, 284, 1652–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang BH, Tawara S, Abe K, Takaki A, Fukumoto Y, & Shimokawa H (2007). Acute vasodilator effect of fasudil, a Rho-kinase inhibitor, in monocrotaline-induced pulmonary hypertension in rats. J Cardiovasc Pharmacol, 49, 85–89. [DOI] [PubMed] [Google Scholar]
- Jin L, Ying Z, Hilgers RH, Yin J, Zhao X, Imig JD, et al. (2006). Increased RhoA/Rho-kinase signaling mediates spontaneous tone in aorta from angiotensin II-induced hypertensive rats. J Pharmacol Exp Ther, 318, 288–295. [DOI] [PubMed] [Google Scholar]
- Kakoki M, Pochynyuk OM, Hathaway CM, Tomita H, Hagaman JR, Kim HS, et al. (2013). Primary aldosteronism and impaired natriuresis in mice underexpressing TGFbetal. Proc Natl Acad Sci U S A, 110, 5600–5605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanaki AI, Sarafidis PA, Georgianos PI, Kanavos K, Tziolas IM, Zebekakis PE, et al. (2013). Effects of low-dose atorvastatin on arterial stiffness and central aortic pressure augmentation in patients with hypertension and hypercholesterolemia. Am JHypertens, 26, 608–616. [DOI] [PubMed] [Google Scholar]
- Karpushev AV, Ilatovskaya DV, Pavlov TS, Negulyaev YA, & Staruschenko A (2010). Intact Cytoskeleton Is Required for Small G Protein Dependent Activation of the Epithelial Na(+) Channel. PLoS One, 5, e8827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato N, Loh M, Takeuchi F, Verweij N, Wang X, Zhang W, et al. (2015). Trans-ancestry genome-wide association study identifies 12 genetic loci influencing blood pressure and implicates a role for DNA methylation. Nat Genet, 47, 1282–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komers R, Oyama TT, Beard DR, & Anderson S (2011). Effects of systemic inhibition of Rho kinase on blood pressure and renal haemodynamics in diabetic rats. British Journal of Pharmacology, 162, 163–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristelly R, Gao G, & Tesmer JJ (2004). Structural determinants of RhoA binding and nucleotide exchange in leukemia-associated Rho guanine- nucleotide exchange factor. J Biol Chem, 279, 47352–47362. [DOI] [PubMed] [Google Scholar]
- Laufs U, & Liao JK (1998). Post-transcriptional regulation of endothelial nitric oxide synthase mRNA stability by Rho GTPase. J Biol Chem, 273, 24266–24271. [DOI] [PubMed] [Google Scholar]
- Lemichez E (2017). New Aspects on Bacterial Effectors Targeting Rho GTPases. Curr Top Microbiol Immunol, 399, 155–174. [DOI] [PubMed] [Google Scholar]
- Lenhart KC, Becherer AL, Li J, Xiao X, McNally EM, Mack CP, et al. (2014). GRAF1 promotes ferlin-dependent myoblast fusion. Dev Biol, 393, 298–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenhart KC, O’Neill TJ, Cheng Z, Dee R, Demonbreun AR, Li J, et al. (2015). GRAF1 deficiency blunts sarcolemmal injury repair and exacerbates cardiac and skeletal muscle pathology in dystrophin-deficient mice. Skeletal Muscle, 5, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy D, Ehret GB, Rice K, Verwoert GC, Launer LJ, Dehghan A, et al. (2009). Genome-wide association study of blood pressure and hypertension. Nature genetics, 41, 677–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewington S, Clarke R, Qizilbash N, Petro R,R,C & Collaboration PS (2003). Age-specific relevance of usual blood pressure to vascular mortality: a meta-analysis of individual data for one million adults in 61 prospective studies. The Lancet, 360, 1903–1913. [DOI] [PubMed] [Google Scholar]
- Li C, He J, Chen J, Zhao J, Gu D, Hixson JE, et al. (2016). Genome-Wide Gene-Sodium Interaction Analyses on Blood Pressure: The Genetic Epidemiology Network of Salt-Sensitivity Study. Hypertension, 68, 348–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao YC, Liu PY, Lin HF, Lin WY, Liao JK, & Juo SH (2015). Two functional polymorphisms of ROCK2 enhance arterial stiffening through inhibiting its activity and expression. JMol Cell Cardiol, 79, ISO- 186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lifton RP, Gharavi AG, & Geller DS (2001). Molecular mechanisms of human hypertension. Cell, 104, 545–556. [DOI] [PubMed] [Google Scholar]
- Lim SS, Vos T, Flaxman AD, Danaei G, Shibuya K, Adair-Rohani H, et al. (2012). A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990–2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet, 380, 2224–2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Lai X, Chen B, Xu Y, Huang B, Chen Z, et al. (2011). Genetic variations in CYP17A1, CACNB2 and PLEKHA7 are associated with blood pressure and/or hypertension in She ethnic minority of China. Atherosclerosis, 219, 709–714. [DOI] [PubMed] [Google Scholar]
- Liu L, Cao Y, Cui G, Li Z, Sun J, Zhang L, et al. (2013). Association analysis of polymorphisms in ROCK2 with cardiovascular disease in a Chinese population. PLoS One, 8, e53905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Zhu Y, Chen H, Wang P, Mei FC, Ye N, et al. (2017). Structure- activity relationships of 2-substituted phenyl-N-phenyl-2- oxoacetohydrazonoyl cyanides as novel antagonists of exchange proteins directly activated by cAMP (EPACs). BioorgMed Chem Lett, 27, 5163–5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockman K, Hinson JS, Medlin MD, Morris D, Taylor JM, & Mack CP (2004). Sphingosine 1-phosphate stimulates smooth muscle cell differentiation and proliferation by activating separate serum response factor co-factors. J Biol Chem, 279, 42422–42430. [DOI] [PubMed] [Google Scholar]
- Lohn M, Plettenburg O, Ivashchenko Y, Kannt A, Hofmeister A, Kadereit D, et al. (2009). Pharmacological characterization of SAR407899, a novel rho-kinase inhibitor. Hypertension, 54, 676–683. [DOI] [PubMed] [Google Scholar]
- Loirand G (2015). Rho Kinases in Health and Disease: From Basic Science to Translational Research. Pharmacological Reviews, 67, 1074–1095. [DOI] [PubMed] [Google Scholar]
- Loirand G, & Pacaud P (2010). The role of Rho protein signaling in hypertension. Nat Rev Cardiol, 7, 637–647. [DOI] [PubMed] [Google Scholar]
- Loirand G, & Pacaud P (2014). Involvement of Rho GTPases and their regulators in the pathogenesis of hypertension. Small GTPases, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu LJ, Tsai JC, & Liu J (2017). Novel Pharmacologic Candidates for Treatment of Primary Open-Angle Glaucoma. The Yale Journal of Biology and Medicine, 90, 111–118. [PMC free article] [PubMed] [Google Scholar]
- Luo W, Janostiak R, Tolde O, Ryzhova LM, Koudelkova L, Dibus M, et al. (2017). ARHGAP42 is activated by Src-mediated tyrosine phosphorylation to promote cell motility. JCell Sci, 130, 2382–2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack CP (2011). Signaling mechanisms that regulate smooth muscle cell differentiation. Arterioscler Thromb Vasc Biol, 31, 1495–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack CP, Thompson MM, Lawrenz-Smith S, & Owens GK (2000). Smooth muscle alpha-actin CArG elements coordinate formation of a smooth muscle cell-selective, serum response factor-containing activation complex. Circ Res, 86, 221–232. [DOI] [PubMed] [Google Scholar]
- Marchioni F, & Zheng Y (2009). Targeting rho GTPases by peptidic structures. Curr Pharm Des, 15, 2481–2487. [DOI] [PubMed] [Google Scholar]
- Masumoto A, Hirooka Y, Shimokawa H, Hironaga K, Setoguchi S, & Takeshita A (2001). Possible involvement of Rho-kinase in the pathogenesis of hypertension in humans. Hypertension, 38, 1307–1310. [DOI] [PubMed] [Google Scholar]
- Masumoto A, Mohri M, Shimokawa H, Urakami L, Usui M, & Takeshita A (2002). Suppression of coronary artery spasm by the Rho-kinase inhibitor fasudil in patients with vasospastic angina. Circulation, 105, 1545–1547. [DOI] [PubMed] [Google Scholar]
- Matsui T, Maeda M, Doi Y, Yonemura S, Amano M, Kaibuchi K, et al. (1998). Rho-kinase phosphorylates COOH-terminal threonines of ezrin/radixin/moesin (ERM) proteins and regulates their head-to-tail association. J Cell Biol, 140, 647–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ming XF, Barandier C, Viswambharan H, Kwak BR, Mach F, Mazzolai L, et al. (2004). Thrombin stimulates human endothelial arginase enzymatic activity via RhoA/ROCK pathway: implications for atherosclerotic endothelial dysfunction. Circulation, 110, 3708–3714. [DOI] [PubMed] [Google Scholar]
- Miralles F, Posern G, Zaromytidou AI, & Treisman R (2003). Actin dynamics control SRF activity by regulation of its coactivator MAL. Cell, 113, 329–342. [DOI] [PubMed] [Google Scholar]
- Momotani K, Artamonov MV, Utepbergenov D, Derewenda U, Derewenda ZS, & Somlyo AV (2011). p63RhoGEF couples Galpha(q/11)-mediated signaling to Ca2+ sensitization of vascular smooth muscle contractility. Circ Res, 109, 993–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori K, Amano M, Takefuji M, Kato K, Morita Y, Nishioka T, et al. (2009). Rho-kinase contributes to sustained RhoA activation through phosphorylation of p190A RhoGAP. J Biol Chem, 284, 5067–5076. [DOI] [PubMed] [Google Scholar]
- Mossessova E, Corpina RA, & Goldberg J (2003). Crystal structure of ARF1*Sec7 complexed with Brefeldin A and its implications for the guanine nucleotide exchange mechanism. Mol Cell, 12, 1403–1411. [DOI] [PubMed] [Google Scholar]
- Nagaoka T, Fagan KA, Gebb SA, Morris KG, Suzuki T, Shimokawa FL, et al. (2005). Inhaled Rho Kinase Inhibitors Are Potent and Selective Vasodilators in Rat Pulmonary Hypertension. American Journal of Respiratory and Critical Care Medicine, 171, 494–499. [DOI] [PubMed] [Google Scholar]
- Nagumo H, Sasaki Y, Ono Y, Okamoto H, Seto M, & Takuwa Y (2000). Rho kinase inhibitor HA-1077 prevents Rho-mediated myosin phosphatase inhibition in smooth muscle cells. American Journal of Physiology-Cell Physiology, 278, C57–C65. [DOI] [PubMed] [Google Scholar]
- Nakagawa O, Fujisawa K, Ishizaki T, Saito Y, Nakao K, & Narumiya S (1996). ROCK-I and ROCK-II, two isoforms of Rho-associated coiled-coil forming protein serine/threonine kinase in mice. FEBS Letters, 392, 189–193. [DOI] [PubMed] [Google Scholar]
- Nakamura A, Hayashi K, Ozawa Y, Fujiwara K, Okubo K, Kanda T, et al. (2003). Vessel- and vasoconstrictor-dependent role of rho/rho-kinase in renal microvascular tone. J Vase Res, 40, 244–251. [DOI] [PubMed] [Google Scholar]
- Narumiya S, & Thumkeo D (2018). Rho signaling research: history, current status and future directions. FEBS Lett, 592, 1763–1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiki K, Tsuruoka S, Kawaguchi A, Sugimoto K, Schwartz GJ, Suzuki M, et al. (2003). Inhibition of Rho-kinase reduces renal Na-H exchanger activity and causes natriuresis in rat. J Pharmacol Exp Ther, 304, 723–728. [DOI] [PubMed] [Google Scholar]
- Ocaranza MP, Rivera P, Novoa U, Pinto M, Gonzalez L, Chiong M, et al. (2011). Rho kinase inhibition activates the homologous angiotensin converting enzyme-angiotensin-(1–9) axis in experimental hypertension. J Hypertens, 29, 706–715. [DOI] [PubMed] [Google Scholar]
- Ohashi K, Nagata K, Maekawa M, Ishizaki T, Narumiya S, & Mizuno K (2000). Rho-associated kinase ROCK activates LIM-kinase 1 by phosphorylation at threonine 508 within the activation loop. J Biol Chem, 275, 3577–3582. [DOI] [PubMed] [Google Scholar]
- Olson MF (2018). Rho GTPases, their post-translational modifications, disease- associated mutations and pharmacological inhibitors. Small GTPases, 9, 203–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oprea TI, Sklar LA, Agola JO, Guo Y, Silberberg M, Roxby J, et al. (2015). Novel Activities of Select NSAID R-Enantiomers against Rac1 and Cdc42 GTPases. PLoS One, 10, e0142182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padmanabhan S, Caulfield M, & Dominiczak AF (2015). Genetic and molecular aspects of hypertension. Circ Res, 116, 937–959. [DOI] [PubMed] [Google Scholar]
- Parnell E, McElroy SP, Wiejak J, Baillie GL, Porter A, Adams DR, et al. (2017). Identification of a Novel, Small Molecule Partial Agonist for the Cyclic AMP Sensor, EPAC1. Sci Rep, 7, 294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peacock J, Diaz KM, Viera AJ, Schwartz JE, & Shimbo D (2014). Unmasking masked hypertension: prevalence, clinical implications, diagnosis, correlates and future directions. Journal Of Human Hypertension, 28, 521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelham CJ, Ketsawatsomkron P, Groh S, Grobe JL, de Lange WJ, Ibeawuchi SR, et al. (2012). Cullin-3 regulates vascular smooth muscle function and arterial blood pressure via PPARgamma and RhoA/Rho-kinase. Cell metabolism, 16, 462–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persell SD (2011). Prevalence of Resistant Hypertension in the United States, 2003–2008. Hypertension, 57, 1076–1080. [DOI] [PubMed] [Google Scholar]
- Pochynyuk O, Medina J, Gamper N, Genth H, Stockand JD, & Staruschenko A (2006). Rapid translocation and insertion of the epithelial Na+ channel in response to RhoA signaling. J Biol Chem, 281, 26520–26527. [DOI] [PubMed] [Google Scholar]
- Pommier Y, & Cherfils J (2005). Interfacial inhibition of macromolecular interactions: nature’s paradigm for drug discovery. Trends Pharmacol Sci, 26, 138–145. [DOI] [PubMed] [Google Scholar]
- Porter AP, Papaioannou A, & Malliri A (2016). Deregulation of Rho GTPases in cancer. Small GTPases, 7, 123–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puetz S, Lubomirov LT, & Pfitzer G (2009). Regulation of Smooth Muscle Contraction by Small GTPases (Vol. 24). [DOI] [PubMed] [Google Scholar]
- Rankinen T, Church T, Rice T, Markward N, Blair SN, & Bouchard C (2008). A major haplotype block at the rho-associated kinase 2 locus is associated with a lower risk of hypertension in a recessive manner: the HYPGENE study. Hypertens Res, 31, 1651–1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren R, Li G, Le TD, Kopczynski C, Stamer WD, & Gong H (2016). Netarsudil Increases Outflow Facility in Human Eyes Through Multiple Mechanisms. Investigative Ophthalmology & Visual Science, 57, 6197–6209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren XR, Du QS, Huang YZ, Ao SZ, Mei L, & Xiong WC (2001). Regulation of CDC42 GTPase by proline-rich tyrosine kinase 2 interacting with PSGAP, a novel pleckstrin homology and Src homology 3 domain containing rhoGAP protein. J Cell Biol, 152, 971–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rittinger K, Walker PA, Eccleston JF, Smerdon SJ, & Gamblin SJ (1997). Structure at 1.65 A of RhoA and its GTPase-activating protein in complex with a transition-state analogue. Nature, 389, 758–762. [DOI] [PubMed] [Google Scholar]
- Roos MH, van Rodijnen WF, van Lambalgen AA, ter Wee PM, & Tangelder GJ (2006). Renal microvascular constriction to membrane depolarization and other stimuli: pivotal role for rho-kinase. Pflugers Arch, 452, 471–477. [DOI] [PubMed] [Google Scholar]
- Sagara Y, Hirooka Y, Nozoe M, Ito K, Kimura Y, & Sunagawa K (2007). Pressor response induced by central angiotensin II is mediated by activation of Rho/Rho-kinase pathway via ATI receptors. JHypertens, 25, 399–406. [DOI] [PubMed] [Google Scholar]
- Sahai E, & Marshall CJ (2002). RHO-GTPases and cancer. Nature Reviews Cancer, 2, 133. [DOI] [PubMed] [Google Scholar]
- Schofield AV, & Bernard O (2013). Rho-associated coiled-coil kinase (ROCK) signaling and disease. Crit Rev Biochem Mol Biol, 48, 301–316. [DOI] [PubMed] [Google Scholar]
- Seasholtz TM, Wessel J, Rao F, Rana BK, Khandrika S, Kennedy BP, et al. (2006). Rho kinase polymorphism influences blood pressure and systemic vascular resistance in human twins: role of heredity. Hypertension, 47, 937–947. [DOI] [PubMed] [Google Scholar]
- Shang X, Marchioni F, Evelyn CR, Sipes N, Zhou X, Seibel W, et al. (2013). Small-molecule inhibitors targeting G-protein-coupled Rho guanine nucleotide exchange factors. Proc Natl Acad Sci USA, 110, 3155–3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang X, Marchioni F, Sipes N, Evelyn CR, Jerabek-Willemsen M, Duhr S, et al. (2012). Rational design of small molecule inhibitors targeting RhoA subfamily Rho GTPases. Chem Biol, 19, 699–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi GX, Yang WS, Jin L, Matter ML, & Ramos JW (2018). RSK2 drives cell motility by serine phosphorylation of LARG and activation of Rho GTPases. Proc Natl Acad Sci USA, 115, E190–E199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Wang X, Chon KH, & Cupples WA (2006). Tubuloglomerular feedback-dependent modulation of renal myogenic autoregulation by nitric oxide. Am JPhysiol Regul Integr Comp Physiol, 290, R982–991. [DOI] [PubMed] [Google Scholar]
- Shimokawa H, Hiramori K, Iinuma H, Hosoda S, Kishida H, Osada H, et al. (2002). Anti-anginal effect of fasudil, a Rho-kinase inhibitor, in patients with stable effort angina: a multicenter study. J Cardiovasc Pharmacol, 40, 751–761. [DOI] [PubMed] [Google Scholar]
- Siehler S (2009). Regulation of RhoGEF proteins by G12/13-coupled receptors. Br J Pharmacol, 158, 41–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sin WC, Chen XQ, Leung T, & Lim L (1998). RhoA-binding kinase alpha translocation is facilitated by the collapse of the vimentin intermediate filament network. Mol Cell Biol, 18, 6325–6339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AL, Dohn MR, Brown MV, & Reynolds AB (2012). Association of Rho-associated protein kinase 1 with E-cadherin complexes is mediated by p120-catenin. Mol Biol Cell, 23, 99–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smithers CC, & Overduin M (2016). Structural Mechanisms and Drug Discovery Prospects of Rho GTPases. Cells, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotiropoulos A, Gineitis D, Copeland J, & Treisman R (1999). Signal- regulated activation of serum response factor is mediated by changes in actin dynamics. Cell, 98, 159–169. [DOI] [PubMed] [Google Scholar]
- Staruschenko A, Nichols A, Medina JL, Camacho P, Zheleznova NN, & Stockand JD (2004). Rho small GTPases activate the epithelial Na(+) channel. J Biol Chem, 279, 49989–49994. [DOI] [PubMed] [Google Scholar]
- Staus DP, Blaker AL, Taylor JM, & Mack CP (2007). Diaphanous 1 and 2 regulate smooth muscle cell differentiation by activating the myocardin- related transcription factors. Arterioscler Thromb Vasc Biol, 27, 478–486. [DOI] [PubMed] [Google Scholar]
- Strazzullo P, Kerry SM, Barbato A, Versiero M, D’Elia L, & Cappuccio FP (2007). Do statins reduce blood pressure?: a meta-analysis of randomized, controlled trials. Hypertension, 49, 792–798. [DOI] [PubMed] [Google Scholar]
- Sumi T, Matsumoto K, & Nakamura T (2001). Specific activation of LIM kinase 2 via phosphorylation of threonine 505 by ROCK, a Rho-dependent protein kinase. J Biol Chem, 276, 670–676. [DOI] [PubMed] [Google Scholar]
- Surma M, Wei L, & Shi J (2011). Rho kinase as a therapeutic target in cardiovascular disease. Future Cardiol, 7, 657–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surviladze Z, Waller A, Wu Y, Romero E, Edwards BS, Wandinger-Ness A, et al. (2010). Identification of a small GTPase inhibitor using a high- throughput flow cytometry bead-based multiplex assay. J Biomol Screen, 15, 10–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surviladze Z, Young SM, & Sklar LA (2012). High-throughput flow cytometry bead-based multiplex assay for identification of Rho GTPase inhibitors. Methods Mol Biol, 827, 253–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki N, Nakamura S, Mano H, & Kozasa T (2003). Gal2 activates Rho GTPase through tyrosine-phosphorylated leukemia-associated RhoGEF. Proceedings of the National Academy of Sciences of the United States of America, 100, 733–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szaszi K, Kurashima K, Kapus A, Paulsen A, Kaibuchi K, Grinstein S, et al. (2000). RhoA and rho kinase regulate the epithelial Na+/H+ exchanger NHE3. Role of myosin light chain phosphorylation. J Biol Chem, 275, 28599–28606. [DOI] [PubMed] [Google Scholar]
- Takemoto M, Sun J, Hiroki J, Shimokawa H, & Liao JK (2002). Rho- Kinase Mediates Hypoxia-Induced Downregulation of Endothelial Nitric Oxide Synthase. Circulation, 106, 57–62. [DOI] [PubMed] [Google Scholar]
- Taylor JM, Hildebrand JD, Mack CP, Cox ΜE, & Parsons JT (1998). Characterization of graf, the GTPase-activating protein for rho associated with focal adhesion kinase. Phosphorylation and possible regulation by mitogen-activated protein kinase. J Biol Chem, 273, 8063–8070. [DOI] [PubMed] [Google Scholar]
- Taylor JM, Macklem MM, & Parsons JT (1999). Cytoskeletal changes induced by GRAF, the GTPase regulator associated with focal adhesion kinase, are mediated by Rho. J Cell Sci, 112 (Pt 2), 231–242. [DOI] [PubMed] [Google Scholar]
- Tcherkezian J, & Lamarche-Vane N (2007). Current knowledge of the large RhoGAP family of proteins. Biol Cell, 99, 67–86. [DOI] [PubMed] [Google Scholar]
- Uehata M, Ishizaki T, Satoh H, Ono T, Kawahara T, Morishita T, et al. (1997). Calcium sensitization of smooth muscle mediated by a Rho- associated protein kinase in hypertension. Nature, 389, 990–994. [DOI] [PubMed] [Google Scholar]
- Vardouli L, Moustakas A, & Stournaras C (2005). LIM-kinase 2 and cofilin phosphorylation mediate actin cytoskeleton reorganization induced by transforming growth factor-beta. J Biol Chem, 280, 11448–11457. [DOI] [PubMed] [Google Scholar]
- Vicari RM, Chaitman B, Keefe D, Smith WB, Chrysant SG, Tonkon MJ, et al. (2005). Efficacy and Safety of Fasudil in Patients With Stable Angina: A Double-Blind, Placebo-Controlled, Phase 2 Trial. Journal of the American College of Cardiology, 46, 1803–1811. [DOI] [PubMed] [Google Scholar]
- Viera AJ, & Shimbo D (2014). Ambulatory blood pressure phenotypes and the risk for hypertension. Curr Hypertens Rep, 16, 481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelsgesang M, Pautsch A, & Aktories K (2007). C3 exoenzymes, novel insights into structure and action of Rho-ADP-ribosylating toxins. Naunyn Schmiedebergs Arch Pharmacol, 374, 347–360. [DOI] [PubMed] [Google Scholar]
- Wain LV, Verwoert GC, O’Reilly PF, Shi G, Johnson T, Johnson AD, et al. (2011). Genome-wide association study identifies six new loci influencing pulse pressure and mean arterial pressure. Nat Genet, 43, 1005–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang DZ, & Olson EN (2004). Control of smooth muscle development by the myocardin family of transcriptional coactivators. Curr Opin Genet Dev, 14, 558–566. [DOI] [PubMed] [Google Scholar]
- Wang Y, Zheng XR, Riddick N, Bryden M, Baur W, Zhang X, et al. (2009). ROCK isoform regulation of myosin phosphatase and contractility in vascular smooth muscle cells. Circ Res, 104, 531–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber DS, & Webb RC (2001). Enhanced relaxation to the rho-kinase inhibitor Y-27632 in mesenteric arteries from mineralocorticoid hypertensive rats. Pharmacology, 63, 129–133. [DOI] [PubMed] [Google Scholar]
- Wei L, Taffet GE, Khoury DS, Bo J, Li Y, Yatani A, et al. (2004). Disruption of Rho signaling results in progressive atrioventricular conduction defects while ventricular function remains preserved. Faseb J, 18, 857–859. [DOI] [PubMed] [Google Scholar]
- Whelton PK, Carey RM, Aronow WS, Casey DE, Collins KJ, Dennison Himmelfarb C, et al. (2017). 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the Prevention, Detection, Evaluation, and Management of High Blood Pressure in Adults. A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. [Google Scholar]
- Wirth A (2010). Rho kinase and hypertension. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease, 1802, 1276–1284. [DOI] [PubMed] [Google Scholar]
- Wirth A, Benyo Z, Lukasova M, Leutgeb B, Wettschureck N, Gorbey S, et al. (2008). G12-G13-LARG-mediated signaling in vascular smooth muscle is required for salt-induced hypertension. Nat Med, 14, 64–68. [DOI] [PubMed] [Google Scholar]
- Wolfrum S, Dendorfer A, Rikitake Y, Stalker TJ, Gong Y, Scalia R, et al. (2004). Inhibition of Rho-kinase leads to rapid activation of phosphatidylinositol 3-kinase/protein kinase Akt and cardiovascular protection. Arterioscler Thromb Vasc Biol, 24, 1842–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuertz CM, Lorincz A, Vettel C, Thomas MA, Wieland T, & Lutz S (2010). p63RhoGEF--a key mediator of angiotensin II-dependent signaling and processes in vascular smooth muscle cells. Faseb J, 24, 4865–4876. [DOI] [PubMed] [Google Scholar]
- Yamada T, Ohoka Y, Kogo M, & Inagaki S (2005). Physical and functional interactions of the lysophosphatidic acid receptors with PDZ domain- containing Rho guanine nucleotide exchange factors (RhoGEFs). J Biol Chem, 280, 19358–19363. [DOI] [PubMed] [Google Scholar]
- Yamaguchi Y, Katoh H, Yasui H, Aoki J, Nakamura K, & Negishi M (2000). Galpha(12) and galpha(13) inhibit Ca(2+)-dependent exocytosis through Rho/Rho-associated kinase-dependent pathway. J Neurochem, 75, 708–717. [DOI] [PubMed] [Google Scholar]
- Yang N, Higuchi O, Ohashi K, Nagata K, Wada A, Kangawa K, et al. (1998). Cofilin phosphorylation by LIM-kinase 1 and its role in Rac- mediated actin reorganization. Nature, 393, 809–812. [DOI] [PubMed] [Google Scholar]
- Yano H, Hayashi K, Momiyama T, Saga H, Haruna M, & Sobue K (1995). Transcriptional regulation of the chicken caldesmon gene. Activation of gizzard-type caldesmon promoter requires a CArG box-like motif. J Biol Chem, 270, 23661–23666. [DOI] [PubMed] [Google Scholar]
- Yatani A, Irie K, Otani T, Abdellatif M, & Wei L (2005). RhoA GTPase regulates L-type Ca2+ currents in cardiac myocytes. Am J Physiol Heart Circ Physiol, 288, H650–659. [DOI] [PubMed] [Google Scholar]
- Ying Z, Giachini FR, Tostes RC, & Webb RC (2009). PYK2/PDZ- RhoGEF links Ca2+ signaling to RhoA. Arterioscler Thromb Vasc Biol, 29, 1657–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying Z, Jin L, Palmer T, & Webb RC (2006). Angiotensin II up-regulates the leukemia-associated Rho guanine nucleotide exchange factor (RhoGEF), a regulator of G protein signaling domain-containing RhoGEF, in vascular smooth muscle cells. Mol Pharmacol, 69, 932–940. [DOI] [PubMed] [Google Scholar]
- Yoon C, Cho SJ, Aksoy BA, Park DJ, Schultz N, Ryeom SW, et al. (2016). Chemotherapy Resistance in Diffuse-Type Gastric Adenocarcinoma Is Mediated by RhoA Activation in Cancer Stem-Like Cells. Clin Cancer Res, 22, 971–983. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Yoon S, Fryar C, & Carroll M (2015). Hypertension prevalence and control among adults: United States, 2011–2014 In NCHS data brief (Vol. ). Hyattsville, MD: National Center for Health Statistics. [PubMed] [Google Scholar]
- Zanin-Zhorov A, Flynn R, Luznik L, Panoskaltsis-Mortari A, Jing D, Goodman K, et al. (2014). A Selective and Potent Rock 2 Inhibitor (KD025) Decreases Human STAT3-Dependent IL-21 and IL-17 Production and Experimental Chronic Graft-Versus-Host Disease (cGVHD). Blood, 124, 540–540. [Google Scholar]
- Zanin-Zhorov A, Weiss JM, Trzeciak A, Chen W, Zhang J, Nyuydzefe MS, et al. (2017). Cutting Edge: Selective Oral ROCK2 Inhibitor Reduces Clinical Scores in Patients with Psoriasis Vulgaris and Normalizes Skin Pathology via Concurrent Regulation of IL-17 and IL-10. The Journal of Immunology, 198, 3809–3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeghouf M, Guibert B, Zeeh JC, & Cherfils J (2005). Arf, Sec7 and Brefeldin A: a model towards the therapeutic inhibition of guanine nucleotide-exchange factors. Biochem Soc Trans, 33, 1265–1268. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Gensch C, & Liao JK (2011). Rho-associated coiled-coil-forming kinases (ROCKs): potential targets for the treatment of atherosclerosis and vascular disease. Trends Pharmacol Sci, 32, 167–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, & Liao JK (2009). Rho kinase: an important mediator of atherosclerosis and vascular disease. Curr Pharm Des, 15, 3108–3115. [DOI] [PMC free article] [PubMed] [Google Scholar]