

Abstract

Coproheme decarboxylases (ChdCs) catalyze the final step in heme b biosynthesis of monoderm and some diderm bacteria. In this reaction, coproheme is converted to heme b via monovinyl monopropionate deuteroheme (MMD) in two consecutive decarboxylation steps. In Firmicutes decarboxylation of propionates 2 and 4 of coproheme depend on hydrogen peroxide and the presence of a catalytic tyrosine. Here we demonstrate that ChdCs from Actinobacteria are unique in using a histidine (H118 in ChdC from Corynebacterium diphtheriae, CdChdC) as a distal base in addition to the redox-active tyrosine (Y135). We present the X-ray crystal structures of coproheme-CdChdC and MMD-CdChdC, which clearly show (i) differences in the active site architecture between Firmicutes and Actinobacteria and (ii) rotation of the redox-active reaction intermediate (MMD) after formation of the vinyl group at position 2. Distal H118 is shown to catalyze the heterolytic cleavage of hydrogen peroxide (kapp = (4.90 ± 1.25) × 104 M–1 s–1). The resulting Compound I is rapidly converted to a catalytically active Compound I* (oxoiron(IV) Y135•) that initiates the radical decarboxylation reactions. As a consequence of the more efficient Compound I formation, actinobacterial ChdCs exhibit a higher catalytic efficiency in comparison to representatives from Firmicutes. On the basis of the kinetic data of wild-type CdChdC and the variants H118A, Y135A, and H118A/Y135A together with high-resolution crystal structures and molecular dynamics simulations, we present a molecular mechanism for the hydrogen peroxide dependent conversion of coproheme via MMD to heme b and discuss differences between ChdCs from Actinobacteria and Firmicutes.
Keywords: coproheme decarboxylase, prokaryotic heme biosynthesis, enzyme kinetics, X-ray crystallography, Compound I formation
1. Introduction
Coproheme decarboxylases (ChdCs) catalyze the ultimate step of the “coproporphyrin-dependent” heme biosynthesis pathway, which is mainly utilized in monoderm bacteria.1−3 The product heme b is generated by the stepwise decarboxylation of propionate groups at positions 2 and 4 of the pyrrole rings A and B of iron coproporphyrin III (coproheme), via the three-propionate intermediate monovinyl monopropionate deuteroheme (MMD).4,5 Coproheme and MMD are redox-active substrates.
Mechanistic studies on this reaction have mainly focused on representatives from the phylum Firmicutes, which forms a phylogenetically distinct clade (clade 1, Figure S1).1,3 The oxidative decarboxylation of coproheme is hydrogen peroxide mediated6 and requires 2 equiv of the oxidant for full conversion of one coproheme to heme b.7 A tyrosine residue was identified as the catalytically relevant radical site essential for both decarboxylation reactions in ChdC from the Firmicutes Staphylococcus aureus(8) and Listeria monocytogenes.9 This tyrosyl radical is formed from a Compound I (oxoiron-(IV) Por•+) intermediate and manifests as Compound I*. Compound I formation was verified recently in an inactive variant of LmChdC, with chlorite as alternative oxidant.9
Actinobacterial representatives (clade 2, Figure S1) were highly important in the process of identifying the coproporphyrin-dependent heme biosynthesis pathway, as ChdCs from Mycobacterium tuberculosis, Streptomyces coelicolor, and Propionibacterium acnes were shown to be essential to form heme b.4,10 However, detailed mechanistic studies on actinobacterial ChdCs are scarce. We previously demonstrated that the actinobacterial ChdC from Corynebacterium diphtheriae is more efficient in coproheme decarboxylation in comparison to firmicute ChdCs.3 So far the structural basis for this drastic difference in catalytic power remains unclear. From sequence alignments and homology modeling the most striking structural difference appeared to be the length and architecture of a loop close to the active site. This loop forms the substrate access channel and seems to be highly flexible in firmicute ChdCs.2,3
Prior to this study, only crystal structures of firmicute ChdCs were solved. Apo structures are available from Geobacillus stearothermophilus (GsChdC, PDB code 1T0T) and LmChdC (4WWS);11 holo structures from GsChdC in complex with Mn-coproheme (5T2K)12 and LmChdC in complex with iron coproheme (6FXJ) and the three-propionate intermediate (6FXQ)9 (Figure S1). As one tyrosine is essential for both decarboxylation reactions, a single active site was identified for the oxidative decarboxylation. Therefore, the substrate needs to reorient in the active site after the first decarboxylation. This was proposed on the basis of the MMD-LmChdC crystal structure (6FXQ) and verified by extensive UV–vis and resonance Raman spectroscopic analyses of LmChdC mutants, probing the environment of all four propionate groups in the resting state13,14 and during turnover.9 The mechanism of hydrogen peroxide activation and Compound I formation in ChdCs from Firmicutes remains unknown, as a potential distal base for deprotonation of hydrogen peroxide is lacking.
In this study we present the first actinobacterial crystal structures of ChdC from Corynebacterium diphtheriae in complex with coproheme and with MMD, which prove the proposed substrate reorientation for actinobacterial representatives. Analogously to the catalytic tyrosine in Firmicutes, Y135 is shown to be essential for catalysis.8,9,12 Most interestingly, we demonstrate—on the basis of crystal structures and UV–vis stopped-flow kinetics data—that actinobacterial ChdCs use a conserved distal histidine residue (H118 in CdChdC) as base for deprotonation and heterolytic cleavage of hydrogen peroxide. For the first time, an apparent bimolecular reaction rate for Compound I formation with the physiological oxidant hydrogen peroxide is presented. This in-depth study of wild-type CdChdC and the variants Y135A, H118A, and H118A/Y135A allows the identification of H118 as a second catalytic residue and verifies Y135 as the redox-active site. Finally we propose a molecular mechanism for the entire catalytic cycle of heme b formation mediated by this actinobacterial ChdC.
2. Experimental Procedures
2.1. Expression and Purification of Wild-Type CdChdC and Variants
CdChdC gene (DIP1394) was synthesized and subcloned into a pD441-NH vector (ATUM, Newark, California) containing an N-terminal His-tag. The synthesized vector was complemented with a HRV 3C protease cleavage site between the tag and the gene of interest by overlap extension PCR. Recombinant protein production was performed in E. coli Tuner (DE3) (Merck/Novagen, Darmstadt, Germany) cells in LB-medium containing 100 μg mL–1 kanamycin. A 500 mL portion of the medium was inoculated with 1 mL of overnight culture and cultivated in shaking flasks at 37 °C and 180 rpm for 3 h. The temperature was lowered to 16 °C prior to induction with 0.5 mM isopropyl-β-d-thiogalactopyranoside (IPTG) and kept for further cultivation overnight. Cells were harvested by centrifugation (4 °C, 2700g, 30 min) and resuspended in 50 mL of lysis buffer (50 mM phosphate buffer pH 7.4, with 500 mM NaCl, 5% glycerol, and 0.5% Triton X-100) and lysed by three 1 min cycles of pulsed ultrasonication (1 s of sonication with 1 s between pulses, 90% power) on ice. The resulting lysate was centrifuged for 30 min at 38720g and 4 °C, and the supernatant was filtered (0.45 μm pore sized filter). For purification, the filtrate was loaded on a His-trap affinity column (5 mL, GE Healthcare) pre-equilibrated with binding buffer (50 mM phosphate buffer, pH 7.4, with 500 mM NaCl) on an ÄKTA system. The column was further washed with binding buffer and equilibrated with cleavage buffer (50 mM Tris-HCl with 150 mM NaCl and 1 mM EDTA). The protein was eluted with cleavage buffer after on-column cleavage from the His-tag with a likewise His-tagged HRV 3C PreScission Protease overnight at 4 °C. The eluate was concentrated using a centrifugal filter unit (Amicon Ultra-15, Merck Millipore Ltd., Tullagreen, Carrigtwohill Co. Cork, Ireland, 50 kDa cutoff) by centrifuging at 4500 g for 10–20 min in order to attain a volume of 1–3 mL for further purification via size exclusion chromatography (SEC) using a HiLoad 16/600 Superdex 200 pg, GE Healthcare column equilibrated with 100 mM phosphate buffer pH 7.4, with 100 mM NaCl. The collected fractions were pooled and again concentrated via a centrifugal filter unit to a concentration of 750–1000 μM. Finally, the solution was stored at −80 °C in 20 μL aliquots. CdChdC was produced in its apo form and supplemented with coproheme directly before analysis in order to avoid unintended decarboxylation activity during storage.
For crystallization and circular dichroism experiments, the enzyme was reconstituted with iron coproporphyrin III chloride (coproheme; Frontier Scientific, Logan, UT, USA) prior to SEC purification, and the SEC-purified protein was directly used for experiments. Mutants of CdChdC were produced by site-directed mutagenesis using the previously described vector as a template and expressed and purified equally (Table 1).
Table 1. List of Primers for Cloning of CdChdC Wild-Type via Gibson Assembly into Vector pD441-NH and Variants Y135A, H118A, and H118A/Y135A (Site-Directed Mutagenesis).
| primer for Gibson assembly | ||
|---|---|---|
| wild-type (fragment 1) | forward | aattgtccttttaacagcgatcgcgtatttcgcctcgctcag |
| wild-type (fragment 2) | reverse | ctgagcgaggcgaaatacgcgatcgctgttaaaaggacaatt |
| backbone | forward | ggactggaagttctgttccaggggcccatggcagagaaattgaatttcgaag |
| backbone | reverse | gggcccctggaacagaacttccagtccatggtggtggtggtgatgtttcatt |
| primer for site-directed mutagenesis | ||
|---|---|---|
| H118A | forward | ctgagttcaaccgcagcgctctgccgtcgttcatta |
| H118A | reverse | taatgaacgacggcagagcgctgcggttgaactcag |
| Y135A | forward | cccgggcgattggattacggttgcaccgttcgtgcgc |
| Y135A | reverse | gcgcacgaacggtgcaaccgtaatccaatcgcccggg |
2.2. Coproheme Decarboxylase Activity
H2O2-induced conversion of supplied coproheme to MMD and heme b was investigated by titration of 1000 μL of the enzyme solution in 50 mM phosphate buffer, pH 7, with around 15 μM apo-enzyme and 10 μM coproheme in a Cary 60 spectrophotometer (Agilent Technologies) with a resolution of 1.5 nm. Subequimolar amounts of H2O2 were added, and spectra were taken after each titration step. Samples from this solution (10 μL) were drawn and analyzed using a Dionex Ultimate 3000 system directly linked to a QTOF mass spectrometer (maXis 4G ETD, Bruker) equipped with the standard ESI source in the positive ion mode. MS scans were recorded within a range from m/z 400 to 3800, and the instrument was tuned to detect both the rather small free heme derivatives and intact proteins in a single run. For separation of the analytes a Thermo ProSwift RP-4H analytical separation column (250 × 0.200 mm) was used.
A gradient from 99% solvent A and 1% solvent B (solvent A, 0.05% trifluoroacetic acid (TFA); solvent B, 80.00% acetyl cyanide (ACN) and 20% solvent A) to 65% B in 11 min was applied, followed by a 2 min gradient from 65% B to 95% B, at a flow rate of 8 μL min–1 and at 65 °C. A blank run (5.0 μL of H2O) was performed after each sample to minimize carryover effects. Relative amounts of formed MMD and heme b as well as oxidized coproheme and heme b upon addition of hydrogen peroxide were determined.
2.3. Steady-State Ligand Binding
The steady-state
kinetics of cyanide binding were determined with a Cary 60 spectrophotometer
(Agilent Technologies). Typically, 10 μM holo enzyme was titrated
with NaCN using a DOSY titration device (DOSTAL) to a 15–20-fold
excess of ligand. The absorbance change at 412/413 nm was plotted
against ligand concentration and fitted in Sigma Plot with a single
rectangular hyperbola with three parameters (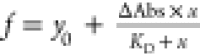 ) in which y0 is the absorbance at zero
ligand concentration, ΔAbs the absorbance
difference at maximum saturation, and KD the dissociation constant.
) in which y0 is the absorbance at zero
ligand concentration, ΔAbs the absorbance
difference at maximum saturation, and KD the dissociation constant.
2.4. Stopped-Flow Spectroscopy
Pre-steady-state spectroscopic changes upon addition of the oxidant H2O2 or the ligand NaCN were monitored using a stopped-flow apparatus equipped with a photodiode array detector (SX-18MV, Applied Photophysics). The path length and volume of the optical quartz cell were 10 mm and 20 μL, respectively. The first spectrum was taken 1 ms after mixing. All measurements were performed in 100 mM phosphate buffer, pH 7, at 25 °C. Apo CdChdC (4 μM) was reconstituted with 2 μM coproheme directly before the measurements. Oxidant/ligand concentrations were varied from 0.1 to 2 mM.
Rate constants for hydrogen peroxide mediated Compound I formation
as well as cyanide binding were determined using a stopped-flow apparatus
equipped with a monochromator (Pi-star, Applied Photophysics). Time
traces were taken at 390/412 nm to follow the formation of Compound
I/binding of cyanide. Typically, the enzyme concentration was around
2 μM and oxidant/ligand concentrations were varied from 0.1
to 1.2 mM. For the single-wavelength measurements, a minimum of three
runs were performed for each ligand or substrate concentration. kobs values for each substrate/ligand concentration
were determined by fitting the respective time traces with a single-exponential
fit (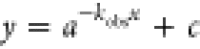 ), in which a is the absorbance
at the beginning of the reaction/ligand binding, kobs is the absorbance change per second, and c is the absorbance at the end of the reaction/ligand binding. kobs values were fitted using the Pro-Data viewer
software and plotted against the respective oxidant/substrate concentration.
), in which a is the absorbance
at the beginning of the reaction/ligand binding, kobs is the absorbance change per second, and c is the absorbance at the end of the reaction/ligand binding. kobs values were fitted using the Pro-Data viewer
software and plotted against the respective oxidant/substrate concentration.
2.5. Crystallization and Structure Determination
CdChdC reconstituted with coproheme was purified by size exclusion chromatography and was stored at −80 °C for crystallization. Crystallization experiments were performed using SWISSCI 96-well 3-drop MRC crystallization plates (Molecular Dimensions, Newmarket, U.K.), adopting the vapor diffusion method. Crystallization drops were set using a Mosquito LCP (TTP Labtech, Melbourn Science Park, Melbourn, U.K.). Protein stocks were diluted in 50 mM phosphate buffer, pH 7, with or without 10 mM NaCN to a concentration of about 6.8 μg μL–1 as a stock for setting up crystallization plates. Seed solutions were prepared by harvesting a single drop with crystallized protein and diluting it in 50 μL of 50 mM phosphate buffer, pH 7. Ten glass beads with a diameter of 1.0 mm were added to the solution, which was vortexed for 1 min and kept on ice for 1 min; this was repeated three times. Dilutions 1:1000 in 50 mM phosphate buffer, pH 7, were used for seeding in the crystallization plates. The reservoir was filled with 40 μL of the crystallization solution. Single drops were set up with ratios of 150:200:100, 200:200:100, and 250:200:100 protein (nL):crystallization (nL):seed (nL). Crystallization plates were sealed and stored at 22 °C. Wild-type CdChdC crystallized in 16–27% PEG3500 and 0.10–0.25% MgCl2. For cryoprotection, the crystallization conditions were supplemented with 25% glycerol. All crystals were harvested using cryoloops and flash-vitrified in liquid nitrogen. Data sets were collected at beamline ID-29 of the European Synchrotron Radiation Facility (Grenoble, France). Processed data sets from the EDNA15 pipeline were used for structure refinement. The phase problem was solved by molecular replacement using Phaser-MR16 taking PDB structure 3DTZ of putative chlorite dismutase TA0507 from Thermoplasma acidophilum and further by the achieved CdChdC structure described in this paper. The models were further improved by model building using maximum likelihood refinement phenix.refine and manual model building using COOT.17,18 The phenix.french_wilson script converted intensities into amplitudes using the French and Wilson algorithm. Restraints for coproheme, MMD, and heme b (ligand ID: RM9, VOV, and FEC) were generated using eLBOW using an sdf file as input and applying the final-geometry option. Final stages of refinement included translation liberation screw (TLS) parameters with the number of TLS groups determined using phenix.tls, isotropic B-factor model, automated addition of hydrogens and water molecules, optimization of X-ray/ADP weight, and optimization of X-ray/stereochemistry weight. Figures were prepared with PYMOL (http://www.pymol.org).
2.6. Molecular Dynamics Simulations
Molecular dynamics simulations of CdChdC with coproheme and MMD as redox-active substrates, respectively, were performed using the GROMOS1119 software for biomolecular simulation. We investigated hydrogen-bonding interactions of the redox-active substrate within the active sites and analyzed the dynamics of H118. Interactions were described by the 54a8 force field parameter set.20 Coproheme and MMD were parametrized in analogy to heme b in the ferric form.21 Water was modeled explicitly and described by means of the simple point charge (SPC) water model.22 The first 10 amino acids of the N-termini of each CdChdC monomer were truncated. The initial binding poses were obtained from the respective crystal structures. MMD was additionally simulated in the coproheme position.
The CdChdC pentamers with the respective substrates were solvated in pre-equilibrated periodic rectangular computational boxes of SPC water with box sizes of approximately 10.2 nm × 10.4 nm × 11.0 nm. The box was cosolvated with 25 mM sodium chloride, and in order to neutralize the systems, additional sodium atoms were added (45 in the case of coproheme, 40 in the case of MMD). The simulation of one CdChdC comprises five porphyrin substrate ligands, which is comparable to five separate subunit simulations. Equations of motion were integrated using the leapfrog algorithm,23 and bond lengths were constrained using the SHAKE algorithm,24 which allowed for integration time steps of 2 fs. Nonbonded interactions were treated using a twin-range cutoff (short range 0.8 nm, long range 1.4 nm). For long-range interactions, a reaction field contribution with a relative dielectric permittivity of 61, as appropriate for the SPC water model, was added.25 Initial velocities were sampled from a Maxwell–Boltzmann distribution at 60 K. The equilibration of the systems was performed by starting with a 100 ps simulation at 60 K, keeping the solutes positionally restrained by a harmonic potential with a force constant of 2.5 × 104 kJ mol–1. In the subsequent equilibration steps the system was heated by 60 K in each step and the force constant was decreased by a factor of 10. The final equilibration step was conducted at 298 K for 150 ps, where position constraints were replaced by rototranslational restraints. After equilibration, production simulations were performed for 20 ns each. The temperature was maintained at 298 K by using a weak-coupling thermostat26 of solute and solvent to two separate heat baths with a coupling time of 0.1 ps, the pressure kept constant at 1 atm with a coupling time of 0.5 ps, and an isothermal compressibility of 4.575 × 10–4 kJ nm3. Trajectories were written every 0.2 ps. Analyses of the coordinate trajectories were performed using gromos++ programs.27
2.7. Assessment of Secondary Structure Integrity of CdChdC Wild-Type and Variants
In order to assess the structural integrity of the created single mutants and double mutant, electronic circular dichroism (ECD) spectra in the far (180–260 nm)- and near-UV (250–500 nm) regions were taken (Chirascan, Applied Photophysics, Leatherhead, U.K.). Conditions were as follows: spectral bandwidth, 1 nm; scan speed, 10 s nm–1; path length, 1 mm; temperature, 20 °C.
Samples were prepared with 5 μg μL–1 (far-UV) or 10 μg μL–1 (near-UV) apo or coproheme bound protein in 10 mM phosphate buffer, pH 7. In order to ensure fully coproheme bound spectra, the protein was supplemented with an excess of coproheme prior to an SEC purification as described in section 2.1 before CD spectral analysis.
2.8. High-Pressure Liquid Size Exclusion Chromatography with Multiangle Light Scattering (HPLC-SEC-MALS)
The homogeneity and oligomeric state of the purified proteins were determined by HPLC and size exclusion chromatography (SEC) coupled to multiangle light scattering (MALS). A LC20 prominence high-pressure liquid chromatography (HPLC) system with the refractive index detector RIF-10A, the photodiode array detector SPD-M20A (Shimadzu), and MALS Heleos Dawn8+ with QELY detector (Wyatt Technology) was used for analysis. Superdex 200 10/300 GL (GE Healthcare) was equilibrated with running buffer (phosphate buffered saline (PBS) with 200 mM NaCl (pH 7.4)). Experiments were performed at a flow rate of 0.75 mL min–1 and 25 °C, and the resulting data were analyzed using the ASTRA 6 software (Wyatt Technology). Proper performance of the molar mass calculation was verified by analysis of a bovine serum albumin sample. A 80 μg portion of the protein (in 5–100 μL) was loaded per run. For runs with more than one protein, 80 μg per protein was loaded. All samples were prepared in running buffer. The samples were further centrifuged (17000g, 10 min) and filtered through an Ultrafree-MC filter with a pore size of 0.1 μM (Merck Millipore) before loading to the column.
3. Results
3.1. General Assessment of Protein Quality and Structural Integrity of Produced Wild-Type CdChdC and Variants
Wild-type CdChdC and the variants Y135A, H118A, and Y135A/H118A were obtained in highly pure form in good yield (5–10 mg L–1E. coli culture). All four recombinant homopentameric proteins were monodisperse and eluted as single peaks in HPLC-SEC-MALS. They exhibited a wild-type-like overall secondary structure composition, as demonstrated by electronic circular dichroism (ECD) spectroscopy (Figure S2).
3.2. Structure of CdChdC
Crystal structures of CdChdC in a complex with coproheme as well as with MMD were solved at a resolution of 1.8 Å (see full statistics in Table 2). To obtain the coproheme bound structure, the crystallization solution was supplemented with cyanide in order to inhibit residual activity. In the absence of cyanide, small amounts of hydrogen peroxide present in aqueous solutions were sufficient to enzymatically decarboxylate the propionate at position 2 (p2) during the crystallization process, yielding MMD. Structures are deposited in the PDB (coproheme-ChdC, 6XUC; MMD-ChdC, 6XUB).
Table 2. Data Collection and Refinement Statisticsa.
| coproheme-CdChdC | MMD-CdChdC | |
|---|---|---|
| wavelength (Å) | 1.0723 | 0.9762 |
| resolution range (Å) | 47.38–1.87 (1.94–1.87) | 48.17–1.78 (1.84–1.78) |
| space group | P1211 | P1211 |
| unit cell | ||
| a (pm) | 61.02 | 60.97 |
| b (pm) | 123.16 | 123.38 |
| c (pm) | 77.89 | 78.01 |
| α (deg) | 90 | 90 |
| β (deg) | 98.4990 | 98.71 |
| γ (deg) | 90 | 90 |
| total no. of rflns | 306975 (26356) | 327938 (30459) |
| no. of unique rflns | 92358 (8365) | 107260 (10074) |
| multiplicity | 3.3 (3.1) | 3.1 (2.9) |
| completeness (%) | 98.43 (89.58) | 97.87 (92.72) |
| mean I/σ(I) | 7.67 (0.91) | 7.47 (0.76) |
| Wilson B factor | 30.67 | 28.78 |
| Rmeas (%) | 11.29 (128.0) | 10.36 (156.3) |
| CC1/2 | 99.6 (32.7) | 99.6 (33.1) |
| no. of rflns used in refinement | 92282 (8365) | 106574 (10073) |
| no. of rflns used for Rfree | 4520 (451) | 5271 (482) |
| Rwork | 0.1647 (0.2956) | 0.1751 (0.3554) |
| Rfree | 0.2230 (0.3316) | 0.2266 (0.3777) |
| no. of non-H atoms | 10228 | 10311 |
| macromolecules | 9415 | 9344 |
| ligands | 245 | 230 |
| solvents | 568 | 737 |
| no. of protein residues | 1147 | 1142 |
| RMS(bonds) | 0.012 | 0.012 |
| RMS (angles) | 1.17 | 1.12 |
| Ramachandran favored (%) | 98.15 | 97.97 |
| Ramachandran allowed (%) | 1.67 | 1.94 |
| Ramachandran outliers (%) | 0.18 | 0.09 |
| rotamer outliers (%) | 0.21 | 0.11 |
| clashscore | 2.83 | 4.68 |
| av B factor | 39.44 | 40.23 |
| macromolecules | 39.51 | 39.78 |
| ligands | 42.25 | 62.72 |
| solvent | 37.07 | 38.98 |
| no. of TLS groups | 39 | 40 |
Statistics for the highest-resolution shell are shown in parentheses.
In the homopentameric structure the subunits are organized in a ringlike shape (Figure 1). An individual subunit comprises one C-terminal and one N-terminal ferredoxin-like domain, the latter containing the redox-active substrate (Figure 1). Direct comparison of the structure of CdChdC with that of ChdC from Listeria monocytogenes (LmChdC) (6FXJ) reveals a flexible loop (residues 112–125 CdChdC numbering) which is positioned close to the active site of CdChdC (Figure 2), while in LmChdC it points away from the substrate and does not contribute to the architecture of the active site.9 Most interesting about this loop in the actinobacterial structure is the conserved (in clade 2) H118, which is located about 5 Å above the heme iron. H118 further takes part in stabilization of coproheme by establishing an H-bond to propionate at position 7 (p7) (Figure 3). With regard to the higher catalytic efficiency of CdChdC in comparison to other ChdCs,3 this histidine is of particular interest since it could act as a distal base and contribute to deprotonation of hydrogen peroxide. It has to be mentioned that in LmChdC the flexible loop contains a histidine (H117), which is positioned at a distance of approximately 17 Å from the coproheme iron, according to structural data (6FXJ).9 The loop position in 6FXJ is stabilized by crystal-packing contacts in two out of five subunits and most probably is a crystallographic artifact and is highly flexible in solution. The role of H117 in LmChdC and a possible movement of the loop has not yet been elucidated.
Figure 1.

Overall structure of coproheme decarboxylase from Corynebacterium diphtheriae (CdChdC). (left) Crystal structure of pentameric CdChdC. The five subunits, each containing one coproheme, are organized in a ringlike shape. (right) One monomer is comprised of one C- and one N-terminal domain, the latter containing the substrate binding site.
Figure 2.

Differences in structure of loop 112–115 (CdChdC numbering) between Actinobacterial and Firmicutes ChdCs: (left) CdChdC; (right) ChdC from Listeria monocytogenes (LmChdC). (center) Overlay of the flexible loop of CdChdC (orange) and LmChdC (gray) depicts the difference in length and orientation. H118 in CdChdC and H117 in LmChdC are shown in stick representation.
Figure 3.

Noncovalent interactions of coproheme and monovinyl monopropionate deteroheme (MMD) in CdChdC. (left) Carbon atoms of amino acid residues involved in interactions with coproheme (crystal structure of coproheme-CdChdC) are shown as green sticks. Water molecules are shown as red spheres. The color code of carbon atoms of propionates p2, p4, p6, and p7 in coproheme is depicted in the middle subpanel and corresponds to that of Table 3. (right) Carbon atoms of amino acid residues involved in interactions with MMD (crystal structure of MMD-CdChdC) are shown as blue sticks. The color code of vinyl and propionates v2, p4, p6, and p7 in coproheme is depicted in the middle subpanel and corresponds to that of Table 3 (p2, v2, yellow; p4, orange; p6, cyan; p7: magenta).
The position and orientation of coproheme in the active site is stabilized by several H-bond interactions between the propionate groups and active site residues (Figure 3). p2 forms two hydrogen bonds to R139 via one water molecule. It further interacts with R208 and T205 (mediated by water). p4 interacts with R139 as well, and in addition p4 forms H-bonds with W143 and with E113 bridged by a water molecule. A complex hydrogen-bonding network is formed between p6 and the peptide backbone (residues 112–114). One hydrogen bond is established between p6 and N115. There are several hydrogen bonds to p7 (H118, peptide backbone 69–70).
Electron densities clearly show that MMD is positioned in a different orientation in comparison to coproheme (Figure 4). This can be seen in all five subunits of the pentameric structure. While p2 of coproheme faces the catalytic tyrosine, MMD is rotated by 90°, thereby allowing p4 to occupy this position. In MMD-ChdC only minor changes in the hydrogen-bonding network can be seen for p4 and p6 (corresponding to p2 and p4 in the coproheme structure; Figure 3). A water-mediated hydrogen bond between p6 and p7 in the coproheme structure is not present in the MMD-bound structure. The vinyl group at position 2 (v2) of MMD is in no proximity to any ligands to form noncovalent interactions.
Figure 4.

Orientation of coproheme and MMD in CdChdC. The crystal structure of CdChdC in a complex with carbon atoms of coproheme in green (6XUC) and carbon atoms of MMD in cyan (6XUB) clearly show a reorientation of the three-propionate reaction intermediate (MMD) after conversion of propionate at position 2 (p2) to vinyl (v2). 2|Fo| – |Fc| electron density maps are shown at σ = 1.5 of chain A of the respective structures.
3.3. Molecular Dynamics Simulations
Molecular dynamics simulations were performed in order to elucidate hydrogen-bonding interactions between the propionates and the residues in the active site. CdChdC was simulated in a complex with coproheme and MMD in the native, catalytically active poses (according to the crystal structures) or with MMD in the coproheme pose after cleavage of p2 and prior to the 90° rotation.
H-bonds involving propionates of the substrate were analyzed considering hydrogen bonds to be present if the distance between a hydrogen atom connected to a donor atom is within 0.25 nm from an acceptor and the donor–hydrogen–acceptor angle is larger than 135°. Two types of hydrogen bonds were distinguished: (i) direct hydrogen bonds (solute–solute bonds) between propionates of the substrates and amino acid residues and (ii) bridging hydrogen bonds (solute–solvent–solute bridges), where a water molecule facilitates the hydrogen bonding. In Table 3 the mean number of H-bonds over a 20 ns simulation time averaged over the five monomers per propionate and residue are given. H-bonds were considered for residues with the occurrence of more than 0.01 H-bond per residue. In addition, the sums of direct H-bonds and bridging H-bonds per propionate, per substrate, and the total sums are included in Table 3. The total H-bond occurrence is highest for the coproheme substrate, mainly because of its four propionate groups and thus more hydrogen bond acceptors. After cleavage of the p2 carboxylic group, the MMD in the coproheme pose forms similar hydrogen bonds. The maximum difference is observed with p4, which interacts more strongly in the simulations of the three-propionate intermediate. The H-bond occurrence of MMD after rotation (pose that corresponds to the crystal structure) is higher than that in the initial pose. The residues with the most interactions are R139 and W143 in both poses, but the interactions are facilitated through p4 and p6, respectively. The H-bonding interactions suggest that the MMD pose after rotation is slightly more favorable.
Table 3. Hydrogen-Bonding Interactions between Substrate-Propionates and Amino Acid Residues in the Active Site of CdChdC for the Three Simulated Systems: i.e., Coproheme in the Resting State, MMD (Monovinyl Monopropionyl Deuteroheme) in Coproheme Pose, and MMD in Catalytically Active Pose (90° Rotation)a.

Direct and bridging hydrogen bonds are given separately.
The flexibility of H118 was evaluated by analyzing the distance of the δ nitrogens to the iron centers of the substrates. The distance–time series for the five subunits of the three different systems coproheme (native pose) and MMD (native and coproheme poses) were generated (Figure 5). The normalized densities of the distances from the δ nitrogen of H118 to the iron in nanometers are plotted for the five individual subunits of the three systems (Figure 5). Pronounced peaks are observed at a distance of 0.43 nm, especially for coproheme in its native position and MMD in the coproheme pose. This indicates a relatively low flexibility of the histidine residue. In the MMD simulation (native pose) just one subunit shows this behavior. In the coproheme simulation, two subunits show a slightly higher flexibility. The H118 residues in the MMD simulation are fluctuating with maximum distances of up to 1.6 nm; the normalized densities are much broader and spread toward higher distances (Figure 5).
Figure 5.

Flexibility of H118 in coproheme CdChdC and MMD CdChdC. Distance analysis of the three simulated systems: i.e., coproheme in the resting state, MMD in the coproheme pose, and MMD in the catalytically active pose (90° rotation). The normalized densities for the distances (in nanometers) of the δ nitrogen of H118 to the iron centers of the substrates of the individual monomers are plotted.
The differences in flexibility of H118 can also be deduced from an H-bond analysis. It interacts quite strongly with coproheme and MMD in the coproheme pose, forming an average of one hydrogen bond with p7 over the complete trajectory when direct and bridging H-bonds are added up. With MMD in its catalytically active pose, p7 adapts a completely different position due to the 90° rotation and interacts with fewer than 0.1 H-bonds on average.
3.4. Coproheme Decarboxylase Activity
Next we investigated the influence of Y135 and H118 on the decarboxylase activity of CdChdC by exchanging the respective amino acids with alanine. The coproheme complexes of wild-type CdChdC and the variants Y135A and H118A were titrated with hydrogen peroxide in order to follow the stepwise decarboxylation from coproheme via MMD to heme b by UV–vis spectroscopy and mass spectrometry (Figures 6A,B).
Figure 6.

Activity of wild-type coproheme decarboxylase (CdChdC) and the variants Y135A and H118A followed by UV–vis spectroscopy and mass spectrometry. (A) Spectral transition upon mixing of 10 μM wild-type (wt), Y135A, and H118A CdChdC with hydrogen peroxide up to 30 μM. Spectra of the coproheme bound proteins are shown in light green. (B) Mass spectrometric analysis of the same reaction mix from (A) showing the ratios of heme species present in the reaction mix. (C) Time traces of 2 μM wt, H118A, and Y135A CdChdC with 50 μM H2O2 followed at 412 nm.
High-spin wild-type coproheme CdChdC shows a Soret maximum at 392 nm (ε392 = 68000 M–1 cm–1) and a shoulder at around 387 nm as well as Q and charge transfer bands (CT) at 497 and 635 nm, respectively. Upon addition of H2O2, coproheme CdChdC is converted to heme b CdChdC with a Soret maximum at 404 nm, Q bands at 498 and 531 nm, and a CT band at 640 nm.
The inactive coproheme complex of Y135A exhibits wild-type-like spectral properties, including the extinction coefficient of the Soret maximum. Upon addition of H2O2, a decrease of absorbance in the Soret maximum is observed without a concomitant red shift. In addition, a prominent peak at 580 nm appears.
The spectrum of H118A differs from that of the wild-type enzyme. Its Soret maximum is red-shifted to 398 nm and is sharper (ε398 = 120000 M–1 cm–1), whereas Q and CT bands are slightly blue shifted to 494 and 612 nm, respectively. The resulting spectrum after addition of a 3-fold excess of hydrogen peroxide resembles that of the wild-type enzyme with a Soret maximum at 404 nm, Q bands at 498 and 531 nm (shoulder), a weak band at 587 nm, and the CT band at 640 nm.
Insets to Figure 6A show the decrease in absorbance at the Soret maximum of coproheme and the increase of the red-shifted Soret maximum of heme b upon addition of H2O2 to the wild-type enzyme and the mutants. Wild-type CdChdC reaches a plateau after addition of a 2-fold excess of hydrogen peroxide, whereas in Y135A only a decrease in absorbance was observed due to coproheme bleaching by the oxidant. In the variant H118A the absorbance at 404 nm increases until addition of a 1.5-fold excess of hydrogen peroxide but finally decreases at higher hydrogen peroxide to protein ratios, indicating bleaching of the redox-active substrate.
The ratios of heme species present in the respective reaction mix determined by mass spectrometry show that, in wild-type CdChdC and the H118A variant, addition of equimolar amounts of H2O2 leads primarily to formation of MMD (662.2 Da), while only minor amounts of the final product heme b (616.2 Da) are formed (Figure 6B). Further addition of the oxidant leads to conversion to heme b, which is predominant in the wild-type enzyme (∼75%) but <50% in H118A. In fact, in the H118A variant oxidized heme b (632.2 Da) is detected after addition of a 3-fold excess of hydrogen peroxide, while minor amounts of the substrate coproheme (708.2 Da) are still present. No oxidized three-propionate intermediate was detected in any analysis.
Exchange of the catalytic Y135 by alanine leads to complete loss of decarboxylation activity. Upon addition of hydrogen peroxide, only oxidation of coproheme (724.2 Da) was detectable by mass spectrometry. The chemical and electronic structures of the oxidized coproheme species in Y135A are unknown and need to be investigated in future studies.
Kinetic studies monitoring the change in absorbance at 412 nm as a readout for heme b formation allowed direct comparison of the enzymatic activity of wild-type CdChdC and the variants H118A and Y135A (Figure 6C). With wild-type CdChdC the reaction is complete within a few seconds (3–5 s), whereas with H118A it takes approximately 10 times longer (>40 s). No change in absorbance is observed with Y135A in the same experimental setup.
3.5. Histidine 118 Promotes Heterolytic Cleavage of Hydrogen Peroxide
It has been postulated that a catalytically active Compound I* (oxoiron(IV) Y•), which attacks p2 in coproheme ChdC and p4 in MMD ChdC, is rapidly formed by electron transfer from the catalytic tyrosine to Compound I (oxoiron(IV) porphyryl radical).8,9 As a consequence, ChdC Compound I can only be trapped kinetically in the absence of the redox-active tyrosine (Y135 in CdChdC). In order to assess whether H118 is involved in the catalysis of Compound I formation in actinobacterial CdChdC, we have studied the wild-type protein and the variants Y135A and H118A/Y135A by stopped-flow spectroscopy (Figure 7).
Figure 7.

Kinetics of Compound I formation of wild-type CdChdC and variants. Spectral transition upon mixing of 2 μM wild-type (wt) CdChdC (A), Y135A (B), or H118A/Y135A (C) with 200 μM H2O2 in 50 mM phosphate buffer, pH 7.0. Initial spectra depicting the coproheme ferric resting state are shown in light green, and spectra of prominent intermediates are highlighted. In addition, representative monophasic time traces at 390 nm and single-exponential fits are shown together with linear plots of calculated kobs values versus hydrogen peroxide concentration.
Despite the fact that upon reaction of wild-type CdChdC with hydrogen peroxide multiple spectral species are observed, it is possible to trace the formation of Compound I by a single-exponential fit of the initial rapid decrease of absorbance of the Soret maximum (kapp = (4.90 ± 1.25) × 104 M–1 s–1). The high y intercept is caused by further competing reactions after Compound I formation. A red shift is already observed after 0.07 s (Figure 7A, dark green trace, partially Compound I) due to the decarboxylation reaction following Compound I formation. The resulting intermediate spectrum has its Soret maximum at 414 nm and bands at 532 and 551 nm (Figure 7A, cyan trace). After 23.63 s a heme b like spectrum (407, 538, and 581 nm) has evolved.
Upon mixing of the variant Y135A with hydrogen peroxide, formation of Compound I is observed with hypochromicity at the Soret maximum at 392 nm and prominent bands at 536, 583, and 645 nm (Figure 7B). During spectral conversion, clear isosbestic points at 345, 435, and 550 nm are observed. The time traces at 390 nm were monophasic, and kobs values were calculated from single-exponential fits. From the plot of kobs values versus H2O2 concentration an apparent bimolecular rate constant (kapp) was calculated to be (1.45 ± 0.21) × 104 M–1 s–1 (Table 4).
Table 4. Apparent Bimolecular Rate Constants for Hydrogen Peroxide-Mediated Compound I Formation of Wild-Type CdChdC and the Variants Y135A and H118A/Y135A at pH 7 and 25 °C.
| kapp (M–1 s–1) | |
|---|---|
| CdChdC wild-type | (4.90 ± 1.25) × 104 |
| CdChdC Y135A | (1.45 ± 0.21) × 104 |
| CdChdC H118A/Y135A | (6.30 ± 0.02) × 102 |
In order to evaluate the role of H118 in Compound I formation, we have designed the double variant H118A/Y135A (Figure 7C). Upon its reaction with H2O2 a less pronounced Compound I spectrum is observed in comparison to CdChdC Y135A with a red-shifted Soret maximum at 397 nm and bands at 541, 573, and 628 nm. During spectral conversion isosbestic points at 345, 420, 462, and 539 nm are observed. The initial time traces monitored at 390 nm are monophasic and were fitted by single-exponential means, allowing calculation of kapp = 6.3 × 102 M–1 s–1, which is about 2 orders of magnitude slower in comparison to Compound I formation in wild-type CdChdC and Y135A.
3.6. Cyanide Binding
To assess the accessibility
of the coproheme iron as well as to mirror Compound I formation, cyanide
binding was studied kinetically by conventional stopped-flow spectroscopy
as well as thermodynamically by spectrophotometric titration. Figure 8A shows the spectral
conversion of five-coordinated high-spin (5cHS) wild-type coproheme CdChdC to the six-coordinated low-spin (6cLS) cyanide coproheme
complex, which includes a shift of the Soret maximum from 392 to 410
nm and of the Q band from 498 to 534 nm. Similar spectral transitions
can be seen in the mutants. The absorbance changes at 412 nm were
fitted by single-exponential calculations, and kobs values were plotted against cyanide concentrations in order
to determine kon from the slope of the
plot and koff from the y intercept. Calculated kon and koff values are within (1.3–5.1) ×
103 and 0.049–0.129 M–1 s–1 for all studied proteins (Table 5). The calculated KD values for wild-type CdChdC (20.7 μM),
Y135A (52.5 μM), H118A (27.5 μM), and the double variant
H118A/Y135A (99.2 μM) are similar to those calculated from the
hyperbolic fits (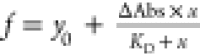 ) of the respective titration
experiments:
i.e., wild-type CdChdC (29.1 μM), Y135A (39.8
μM), H118A (36.4 μM), and the double variant H118A/Y135A
(100.4 μM) (Figure 8B and Table 5).
) of the respective titration
experiments:
i.e., wild-type CdChdC (29.1 μM), Y135A (39.8
μM), H118A (36.4 μM), and the double variant H118A/Y135A
(100.4 μM) (Figure 8B and Table 5).
Figure 8.

Kinetics and thermodynamics of cyanide binding to wild-type CdChdC and variants. (A) Kinetics of spectral transition of reaction between wild-type CdChdC (2 μM) and sodium cyanide (200 μM) in 50 mM phosphate buffer pH 7. The first spectrum (1 ms) is highlighted in orange, the 6cLS spectrum after 1.679 s in black, and intermediate spectra in gray. Typical time traces at 412 nm as well as linear plots of calculated kobs values versus cyanide concentrations are shown for wild-type CdChdC (orange), Y135A (blue), H118A (red), and H118A/Y135A (purple), respectively. (B) Titration of cyanide to wild-type CdChdC (orange), Y135A (blue), H118A (red), and H118A/Y135A (purple), respectively, followed at 412 nm.
Table 5. Kinetics and Thermodynamics of Cyanide Complex Formation of Wild-Type CdChdC and the Variants H118A, Y135A, and H118A/Y135Aa.
| kon (103 M–1 s–1) | koff (s–1) | KD (μM) (=koff/kon) | KD (μM) | |
|---|---|---|---|---|
| CdChdC wild-type | 5.1 | 0.105 | 20.7 | 29.1 |
| CdChdC Y135A | 1.7 | 0.087 | 52.5 | 39.8 |
| CdChdC H118A | 1.8 | 0.049 | 27.5 | 36.4 |
| CdChdC H118A/Y135A | 1.3 | 0.129 | 99.2 | 100.4 |
The kinetic rate constants kon and koff are derived from conventional stopped-flow experiments with KD = koff/kon (pH 7.0, 25 °C). The KD values derived from titration experiments (end-point titration) are given.
4. Discussion
Recent kinetic data have demonstrated that actinobacterial (clade 2) CdChdC is a better catalyst for conversion of coproheme to heme b in comparison to ChdCs from Firmicutes (clade 1, Figure S1).3 The experimental data presented in this work provide the explanation for its higher catalytic power. This allows us to present a mechanistic model of the entire reaction cycle of actinobacterial coproheme decarboxylases.
Preceding studies on ChdCs from Firmicutes provided the first results on the biochemical mechanism of coproheme decarboxylation.8,9,12 They demonstrated that the reaction strictly depends on (i) hydrogen peroxide, (ii) a redox-active ferric substrate (coproheme or MMD), and (iii) the presence of a catalytic redox-active (conserved) tyrosine residue. Apo-ChdC has been shown to rapidly bind the substrate coproheme, whose redox-active nature is required for the reaction.28 It has been proposed that H2O2 oxidizes coproheme/MMD to a Compound I like species similar to Compound I formation in heme peroxidases and catalases. However, so far it has been impossible to trap this redox intermediate using the physiological oxidant H2O2.9 The proposed Compound I like intermediate was suggested to be immediately converted to Compound I* by internal quenching of the porphyryl radical and formation of a distinct oxidized catalytic tyrosine (Y135 in CdChdC).8,9 The neutral tyrosyl radical abstracts a hydrogen atom from the β carbon of p2, followed by migration of the unpaired propionyl electron to the substrate and reduction of the oxoiron(IV) species to the ferric state. Concomitantly the substituent is stabilized by elimination of carbon dioxide, thereby forming the vinyl substituent at position 2 (v2).9 The resulting MMD is proposed to rotate by 90° in order to form the catalytically active MMD-ChdC with p4 in close vicinity to catalytic tyrosine. After formation of MMD-Compound I and MMD-Compound I* mediated by another hydrogen peroxide molecule, p4 is attacked by the tyrosyl radical and the second decarboxylation reaction follows the sequence as described above.
So far, spectral observations of Compound I in firmicute ChdCs have only been reported with the inactive LmChdC variant Y147A in reaction with chlorite.9 Rate constants were determined to be in the range of 104 M–1 s–1. It has to be noted that, in contrast to the natural substrate hydrogen peroxide, Compound I formation with chlorite at pH 7.0 does not require deprotonation of the oxidant (pKa of chlorite is 1.97). In Firmicutes a potential distal base for deprotonation of H2O2, which is required for efficient formation of Compound 0 (Fe(III)-–OOH) has not yet been identified. In CdChdC Y135A we were able to detect an explicit, fully established electronic absorbance Compound I spectrum in reaction with the natural substrate H2O2. We could determine apparent rate constants for the initial reaction step for the inactive variant Y135A ((1.45 ± 2.10) × 104 M–1 s–1) as well as the wild-type ((4.9 ± 1.25) × 104 M–1 s–1; Figure 7B). The higher catalytic efficiency with a protonated oxidant indicates the presence of a distal base in this actinobacterial enzyme.
Thus, the distal histidine (H118), discovered in the crystal structure of CdChdC (Figure 3), is positioned at an optimal distance from the coproheme iron (∼5 Å) and may act as the Lewis base for Compound 0 formation. This histidine is situated on the flexible loop previously shown to define the substrate channel in ChdCs (Figure 2).3 In the well-studied firmicute LmChdC the loop points away from the coproheme cavity and the position of H118 in CdChdC remains vacant. Importantly, a previously reported I-TASSER model of CdChdC predicted an alternate loop conformation which rendered identification of H118 as a distal base almost impossible (Figure S3). However, the different orientation of the loop does not influence the accessibility of the porphyrin iron, as shown by the similar kinetics of cyanide binding (Table 5 and Figure 8).11,28
Compound I formation of an inactive Y135A variant lacking H118 is 2 orders of magnitude slower in comparison to wild-type CdChdC. The decreased kapp of Compound I formation of H118A/Y135A is comparable to coproheme conversion rates in LmChdC (1.8 × 102 M–1 s–1)7 and Staphylococcus aureus ChdC (SaChdC), where a kobs value of 0.3 min–1 was determined with 60 μM hydrogen peroxide, which corresponds to approximately 0.8 × 102 M–1 s–1.6 This suggests that in firmicute ChdCs uncatalyzed Compound I formation represents the rate-limiting step in the overall coproheme decarboxylation cycle.
Despite the presence of the distal catalytic histidine, Compound I formation in CdChdC is about 3 orders of magnitude slower in comparison to conventional heme peroxidases that typically use a distal histidine–arginine pair for efficient catalysis of heterolytic cleavage of hydrogen peroxide. For example, in horseradish peroxidase (HRP) Compound I formation occurs at a rate of 1.7 × 107 M–1 s–1 29 and follows the classical Poulos–Kraut mechanism.30,31 In HRP the distal arginine residue promotes polarization of the oxygen–oxygen bond, thereby facilitating heterolytic cleavage.31 A comparable positively charged interaction partner for H118 is missing in CdChdC, which may well explain the slower rate.
The significantly higher reactivity of CdChdC toward H2O2 in comparison to LmChdC described above was evidenced by reaction with very small amounts of hydrogen peroxide present in aerated aqueous buffer solutions that led to formation of MMD even in the absence of intentionally added H2O2. This allowed us to obtain the structure of pure MMD-CdChdC (PDB code: 6XUB) in addition to the crystal structure of coproheme-CdChdC (PDB code: 6XUC). The position and number of the respective propionate (and vinyl) groups are clearly visible in four out of five subunits in both structures. This strongly suggests reorientation of MMD after conversion of p2 to v2 (Figures 2, 3, and 9). The two independent, well distinguishable structures confirm what has been recently indicated in a single data set collected from LmChdC, in which partial conversion of coproheme to MMD was proposed and considered in structure refinement, finally leading to better statistics.9 The favorability of the rotated and catalytically active position of MMD (which brings p4 in close vicinity to the catalytic tyrosine) is supported by MD simulations and quantification of hydrogen bonds established between coproheme CdChdC and CdChdC in complex with MMD in its initial and rotated poses (Table 3). The overall amount of noncovalent interactions between MMD and nearby amino acids is slightly higher in the catalytically active pose.
Figure 9.

Complete catalytic cycle of coproheme decarboxylation in actinobacterial CdChdC. The reaction cycle starts with oxidation of the ferric resting state of coproheme CdChdC by hydrogen peroxide to Compound I (oxoiron(IV) porphyryl radical) and water. Histidine 118 acts as a distal base and promotes heterolytic cleavage of H2O2. Compound I is rapidly converted to Compound I* (oxoiron(IV) Y135•) by internal electron transfer. The neutral tyrosine radical performs a nucleophilic attack on the β carbon of propionate at position 2 (p2), thereby initiating its decarboxylation and formation of vinyl (v2). The resulting monovinyl monopropionate deuteroheme (MMD) undergoes a rotation of approximately 90°, thereby moving p4 in the position close to Y135. The second half of the reaction cycle starts with oxidation of the ferric resting state of MMD CdChdC by hydrogen peroxide to MMD-Compound I which is rapidly converted to MMD-Compound I* that attacks p4, thereby initiating its decarboxylation and formation of vinyl (v4). Finally, heme b is released and delivered to proteins.
Not only time-resolved spectroscopic and mass spectrometric data of CdChdC but also data obtained from representatives of Firmicutes suggest that the first half-reaction (conversion of p2 to v2) is faster in comparison to the second half-reaction (conversion of p4 to v4). The crystal structures and MD simulations both show significant differences in noncovalent interactions and flexibility of H118 between coproheme-CdChdC and MMD-CdChdC (Figures 3 and 4). In MMD-CdChdC p7 adapts a completely different position and loses interaction with H118. While the distance of H118 to the coproheme iron is relatively constant in all five subunits (0.4–0.6 nm), the residue is highly flexible, moving back and forth from the heme iron within 1.2 nm in the majority of the subunits in the MMD complex (Figure 5). A less stable conformation and higher flexibility of the catalytic H118 are likely to impair Compound I formation and decelerate the formation of heme b from MMD. Moreover, diminished interaction with p7 should result in decreased basicity of H118 in MMD-CdChdC in comparison to coproheme-CdChdC and consequently in a decreased rate of Compound I formation. This could be the reason we were not able to monitor a spectrum representative for MMD-Compound I (Figure 7A).
Summing up, our comprehensive biochemical and biophysical study on CdChdC allows us to propose the molecular mechanism of actinobacterial ChdCs, as illustrated in Figure 9. Due to the presence of H118 formation of Compound I and subsequently of Compound I* of clade 2, ChdCs are significantly faster in comparison to firmicute ChdCs. As a consequence, actinobacterial ChdCs exhibit a higher catalytic efficiency in heme b formation and are less prone to oxidative damage of the redox-active substrate. Coproheme-Compound I formation is faster in comparison to MMD-Compound I formation due to changes in noncovalent interactions of H118 after rotation of MMD. Clade 1 and 2 representatives seem to behave similarly with respect to the radical mechanism of decarboxylation of p2 by coproheme-Compound I* (oxoiron(IV) Y135•) and p4 by MMD-Compound I* (oxoiron(IV) Y135•) (Figure 9). Rotation of MMD by 90° after formation of the vinyl group at position 2, as proposed for firmicute ChdCs,8,9 is supported by the first high-resolution crystal structure of an actinobacterial MMD-ChdC. However, the mechanism of movement of MMD, e.g. whether it partially exits and re-enters the active site during rotation, is still unknown.
Acknowledgments
This project was supported by EQ-BOKU VIBT GmbH and the BOKU Core Facility Biomolecular & Cellular Analysis. We thank Daniel Maresch for technical support in performing mass spectrometry analysis. This project was supported by the Austrian Science Fund, FWF project P29099, and FWF doctoral program BioToP (W1224). Data were collected at beamline ID29 of the European Synchrotron Radiation Facility (proposal MX1998).
Glossary
Abbreviations
- 5cHS
five-coordinated high spin
- 6cLS
six-coordinated low spin
- Cd
Corynebacterium diphtheriae
- ChdC
coproheme decarboxylase
- CT
charge transfer
- ECD
electronic circular dichroism
- IPTG
isopropyl-β-d-thiogalactopyranoside
- Lm
Listeria monocytogenes
- MALS
multiangle light scattering
- MD
molecular dynamics
- MMD
monovinyl monopropionyl deuteroheme
- p2
propionate at position 2
- p4
propionate at position 4
- p6
propionate at position 6
- p7
propionate at position 7
- Sa
Staphylococcus aureus
- SEC
size exclusion chromatography
- SPC
simple point charge
- v2
vinyl at position 2
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscatal.0c00411.
Structural data and phylogeny of ChdCs, far- and near-UV circular dichroism spectra and HPLC-SEC-MALS data of recombinant wild-type (wt) CdChdC and variants, and comparison of CdChdC structures derived from X-ray crystallography and homology modeling (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Dailey H. A.; Dailey T. A.; Gerdes S.; Jahn D.; Jahn M.; O’Brian M. R.; Warren M. J. Prokaryotic Heme Biosynthesis: Multiple Pathways to a Common Essential Product. Microbiol. Mol. Biol. Rev. 2017, 81, e00048-16 10.1128/MMBR.00048-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dailey H. A.; Gerdes S. HemQ: An Iron-Coproporphyrin Oxidative Decarboxylase for Protoheme Synthesis in Firmicutes and Actinobacteria. Arch. Biochem. Biophys. 2015, 574, 27–35. 10.1016/j.abb.2015.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfanzagl V.; Holcik L.; Maresch D.; Gorgone G.; Michlits H.; Furtmüller P. G.; Hofbauer S. Coproheme Decarboxylases - Phylogenetic Prediction Versus Biochemical Experiments. Arch. Biochem. Biophys. 2018, 640, 27–36. 10.1016/j.abb.2018.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dailey H. A.; Gerdes S.; Dailey T. A.; Burch J. S.; Phillips J. D. Noncanonical Coproporphyrin-Dependent Bacterial Heme Biosynthesis Pathway that Does Not Use Protoporphyrin. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 2210–2215. 10.1073/pnas.1416285112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobo S. A.; Scott A.; Videira M. A.; Winpenny D.; Gardner M.; Palmer M. J.; Schroeder S.; Lawrence A. D.; Parkinson T.; Warren M. J.; Saraiva L. M. Staphylococcus aureus Haem Biosynthesis: Characterisation of the Enzymes Involved in Final Steps of the Pathway. Mol. Microbiol. 2015, 97, 472–487. 10.1111/mmi.13041. [DOI] [PubMed] [Google Scholar]
- Celis A. I.; Streit B. R.; Moraski G. C.; Kant R.; Lash T. D.; Lukat-Rodgers G. S.; Rodgers K. R.; DuBois J. L. Unusual Peroxide-Dependent, Heme-Transforming Reaction Catalyzed by HemQ. Biochemistry 2015, 54, 4022–32. 10.1021/acs.biochem.5b00492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofbauer S.; Mlynek G.; Milazzo L.; Pühringer D.; Maresch D.; Schaffner I.; Furtmüller P. G.; Smulevich G.; Djinovic-Carugo K.; Obinger C. Hydrogen Peroxide-Mediated Conversion of Coproheme to Heme b by HemQ - Lessons from the First Crystal Structure and Kinetic Studies. FEBS J. 2016, 283, 4386–4401. 10.1111/febs.13930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streit B. R.; Celis A. I.; Moraski G. C.; Shisler K. A.; Shepard E. M.; Rodgers K. R.; Lukat-Rodgers G. S.; DuBois J. L. Decarboxylation Involving a Ferryl, Propionate, and a Tyrosyl Group in a Radical Relay Yields Heme b.. J. Biol. Chem. 2018, 293, 3989–3999. 10.1074/jbc.RA117.000830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milazzo L.; Gabler T.; Pühringer D.; Jandova Z.; Maresch D.; Michlits H.; Pfanzagl V.; Djinovic-Carugo K.; Oostenbrink C.; Furtmüller P. G.; Obinger C.; Smulevich G.; Hofbauer S. Redox Cofactor Rotates during Its Stepwise Decarboxylation: Molecular Mechanism of Conversion of Coproheme to Heme b.. ACS Catal. 2019, 9, 6766–6782. 10.1021/acscatal.9b00963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dailey T. A.; Boynton T. O.; Albetel A. N.; Gerdes S.; Johnson M. K.; Dailey H. A. Discovery and Characterization of HemQ: an Essential Heme Biosynthetic Pathway Component. J. Biol. Chem. 2010, 285, 25978–25986. 10.1074/jbc.M110.142604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofbauer S.; Hagmüller A.; Schaffner I.; Mlynek G.; Krutzler M.; Stadlmayr G.; Pirker K. F.; Obinger C.; Daims H.; Djinović-Carugo K.; Furtmüller P. G. Structure and Heme-Binding Properties of HemQ (Chlorite Dismutase-like Protein) from Listeria monocytogenes. Arch. Biochem. Biophys. 2015, 574, 36–48. 10.1016/j.abb.2015.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celis A. I.; Gauss G. H.; Streit B. R.; Shisler K.; Moraski G. C.; Rodgers K. R.; Lukat-Rodgers G. S.; Peters J. W.; DuBois J. L. Structure-Based Mechanism for Oxidative Decarboxylation Reactions Mediated by Amino Acids and Heme Propionates in Coproheme Decarboxylase (HemQ). J. Am. Chem. Soc. 2017, 139, 1900–1911. 10.1021/jacs.6b11324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milazzo L.; Hofbauer S.; Howes B. D.; Gabler T.; Furtmüller P. G.; Obinger C.; Smulevich G. Insights into the Active Site of Coproheme Decarboxylase from Listeria monocytogenes. Biochemistry 2018, 57, 2044–2057. 10.1021/acs.biochem.8b00186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milazzo L.; Gabler T.; Pfanzagl V.; Michlits H.; Furtmüller P. G.; Obinger C.; Hofbauer S.; Smulevich G. The Hydrogen Bonding Network of Coproheme in Coproheme Decarboxylase from Listeria monocytogenes: Effect on Structure and Catalysis. J. Inorg. Biochem. 2019, 195, 61–70. 10.1016/j.jinorgbio.2019.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Incardona M. F.; Bourenkov G. P.; Levik K.; Pieritz R. A.; Popov A. N.; Svensson O. EDNA: a Framework for Plugin-Based Applications Applied to X-Ray Experiment Online Data Analysis. J. Synchrotron Radiat. 2009, 16, 872–879. 10.1107/S0909049509036681. [DOI] [PubMed] [Google Scholar]
- McCoy A. J.; Grosse-Kunstleve R. W.; Adams P. D.; Winn M. D.; Storoni L. C.; Read R. J. Phaser Crystallographic Software. J. Appl. Crystallogr. 2007, 40, 658–674. 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams P. D.; Afonine P. V.; Bunkóczi G.; Chen V. B.; Davis I. W.; Echols N.; Headd J. J.; Hung L. W.; Kapral G. J.; Grosse-Kunstleve R. W.; McCoy A. J.; Moriarty N. W.; Oeffner R.; Read R. J.; Richardson D. C.; Richardson J. S.; Terwilliger T. C.; Zwart P. H. PHENIX: a Comprehensive Python-Based System for Macromolecular Structure Solution. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 213–221. 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Lohkamp B.; Scott W. G.; Cowtan K. Features and Development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 486–501. 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid N.; Christ C.; Christen M.; Eichenberger A.; van Gunsteren W. Architecture, Implementation and Parallelisation of the GROMOS Software for Biomolecular Simulation. Comput. Phys. Commun. 2012, 183, 890–903. 10.1016/j.cpc.2011.12.014. [DOI] [Google Scholar]
- Reif M. M.; Hünenberger P. H.; Oostenbrink C. New Interaction Parameters for Charged Amino Acid Side Chains in the GROMOS Force Field. J. Chem. Theory Comput. 2012, 8, 3705–3723. 10.1021/ct300156h. [DOI] [PubMed] [Google Scholar]
- Zou C.; Larisika M.; Nagy G.; Srajer J.; Oostenbrink C.; Chen X.; Knoll W.; Liedberg B.; Nowak C. Two-Dimensional Heterospectral Correlation Analysis of the Redox-Induced Conformational Transition in Cytochrome c Using Surface-Enhanced Raman and Infrared Absorption Spectroscopies on a Two-Layer Gold Surface. J. Phys. Chem. B 2013, 117, 9606–9614. 10.1021/jp404573q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berendsen H. J. C.; Postma J. P. M.; van Gunsteren W. F.; Hermans J.. Interaction Models for Water in Relation to Protein Hydration; Reidel: Dordrecht, The Netherlands, 1981; pp 331–342. [Google Scholar]
- Hockney R. W. The Potential Calculation and Some Applications. Methods Comput. Phys. 1970, 9, 136–211. [Google Scholar]
- Ryckaert J. P.; Ciccotti G.; Berendsen H. J. C. Numerical Integration of Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of n-Alkanes. J. Comput. Phys. 1977, 23, 327–341. 10.1016/0021-9991(77)90098-5. [DOI] [Google Scholar]
- Heinz T. N.; van Gunsteren W. F.; Hünenberger P. H. Comparison of Four Methods to Compute the Dielectric Permittivity of Liquids from Molecular Dynamics Simulations. J. Chem. Phys. 2001, 115, 1125–1136. 10.1063/1.1379764. [DOI] [Google Scholar]
- Berendsen H. J. C.; Postma J. P. M.; van Gunsteren W. F.; DiNola A.; Haak J. R. Molecular-Dynamics with Coupling to an External Bath. J. Chem. Phys. 1984, 81, 3684–3690. 10.1063/1.448118. [DOI] [Google Scholar]
- Eichenberger A. P.; Allison J. R.; Dolenc J.; Geerke D. P.; Horta B. A.; Meier K.; Oostenbrink C.; Schmid N.; Steiner D.; Wang D.; van Gunsteren W. F. GROMOS++ Software for the Analysis of Biomolecular Simulation Trajectories. J. Chem. Theory Comput. 2011, 7, 3379–3390. 10.1021/ct2003622. [DOI] [PubMed] [Google Scholar]
- Hofbauer S.; Dalla Sega M.; Scheiblbrandner S.; Jandova Z.; Schaffner I.; Mlynek G.; Djinovic-Carugo K.; Battistuzzi G.; Furtmüller P. G.; Oostenbrink C.; Obinger C. Chemistry and Molecular Dynamics Simulations of Heme b-HemQ and Coproheme-HemQ. Biochemistry 2016, 55, 5398–5412. 10.1021/acs.biochem.6b00701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunford H. B.; Hewson W. D. Effect of Mixed Solvents on the Formation of Horseradish Peroxidase Compound I. The Importance of Diffusion-Controlled Reactions. Biochemistry 1977, 16, 2949–2957. 10.1021/bi00632a023. [DOI] [PubMed] [Google Scholar]
- Poulos T. L.; Freer S. T.; Alden R. A.; Edwards S. L.; Skogland U.; Takio K.; Eriksson B.; Xuong N.; Yonetani T.; Kraut J. The Crystal Structure of Cytochrome c Peroxidase. J. Biol. Chem. 1980, 255, 575–580. [PubMed] [Google Scholar]
- Poulos T. L.; Kraut J. The Stereochemistry of Peroxidase Catalysis. J. Biol. Chem. 1980, 255, 8199–8205. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.