

Abstract
A strategy for top-down analysis of branched proteins has been reported earlier, which relies on electron transfer dissociation assisted by collisional activation, and software designed for graphic interpretation of tandem mass spectra and adapted for branched proteins. In the present study, the strategy is applied to identify unknown and novel products of reactions in which rationally mutated proteoforms of Rub1 are used to probe the selectivity of E1 and E2 enzymes normally active in ubiquitination. To test and demonstrate this application, components and attachment sites of three branched dimers are deduced and the mutations are confirmed.
Keywords: branched proteins, graphical software, Rub1, rubylation, top-down mass spectrometry
1 |. INTRODUCTION
Post-translational modification of proteins by covalent attachment of ubiquitin (Ub) and its homologous proteins Rub1 (NEDD8 in mammals) and SUMO determines protein function and fate in eukaryotic cells. Attachment at the sidechain of lysine residues is usually catalyzed by sequential actions of three enzymes, among which the activating enzyme (E1) binds and activates the modifying protein which is then recognized by and transferred to the conjugating enzyme (E2) to form a transient thioester, while the ligase (E3) binds both the substrate protein and the activated E2 carrying the modifying protein.1 Some E2 enzymes have been shown to form covalent ubiquitin dimers (branched proteins) in the absence of E3 by conjugating activated ubiquitin to another ubiquitin bound as substrate to the same E2. It is of both theoretical and therapeutic interest to determine the basis for selective recognition and activation of ubiquitin by its cognate E1 and E2 enzymes, and an approach under development in our laboratories involves challenging ubiquitin’s E1 and E2 with Rub1 and variations of Rub1, which have been mutated to imitate potentially the binding surface and structure of ubiquitin. Despite their strong sequence and structure similarity,2 ubiquitin and Rub1 have their own dedicated activation/conjugation enzymes and signal for distinct cellular outcomes. We aim at understanding at the amino-acid residue levels the features of these two proteins that allow enzymes and other receptors to distinguish ubiquitin from Rub1 and define ubiquitin and Rub1 as separate cellular signals. The composition and structures of any dimeric conjugation products of ubiquitin and/or Rub1 can reveal the mechanisms and preferences for enzymatic recognition, activation, and ligation. For this work to proceed rapidly and reliably, a method is needed to characterize the components and lysine attachment sites of the branched protein products and confirm the presence of expected mutations.
Most of what is known about the cellular occurrence of proteins modified by ubiquitin (or Rub1) has been acquired by analysis of the tryptic products of ubiquitinated proteins. Ubiquitin is cleaved at R74 to leave GG tags on the modified lysines in peptides from the substrate proteins.3–6 This bottom-up proteomic strategy has the drawback that the GG tag does not allow the distinction of ubiquitination from rubylation (because both ubiquitin and Rub1 have the same C-terminal LRGG sequence) and does not provide information on the length and connectivity of polymeric modifiers.7–9 It also requires that the GG-tagged peptide be located within a complex mixture of tryptic peptide products.
Top-down mass spectrometry is an attractive alternative technique, providing molecular mass and structural information on intact proteins.10–12 It has been successfully demonstrated for analyses of a series of intact polyubiquitins7–9 and a set of ubiquitinated polypeptide substrates.13 Top-down analysis of these branched proteins is facilitated by the high resolution, high mass accuracy, and extensive activation provided by contemporary tandem instrumentation. However, these large complex spectra are not easily interpreted manually. We have developed a quasi-interpretative approach supported by a novel use of graphic interpretation programs14,15 to recognize branched proteins, distinguish anchor proteins and modifying proteins, locate mutations and modifications, and assign lysine attachment sites.7,8,13 This strategy is modified (Scheme 1) and further tested in the present study, whose objective is to characterize primary structures and branching sites of novel Rub1-containing branched proteins formed by conjugation catalyzed by with ubiquitin cognate E1 and E2 enzymes in which Rub1 is the target protein or both the target and the modifier.
SCHEME 1.
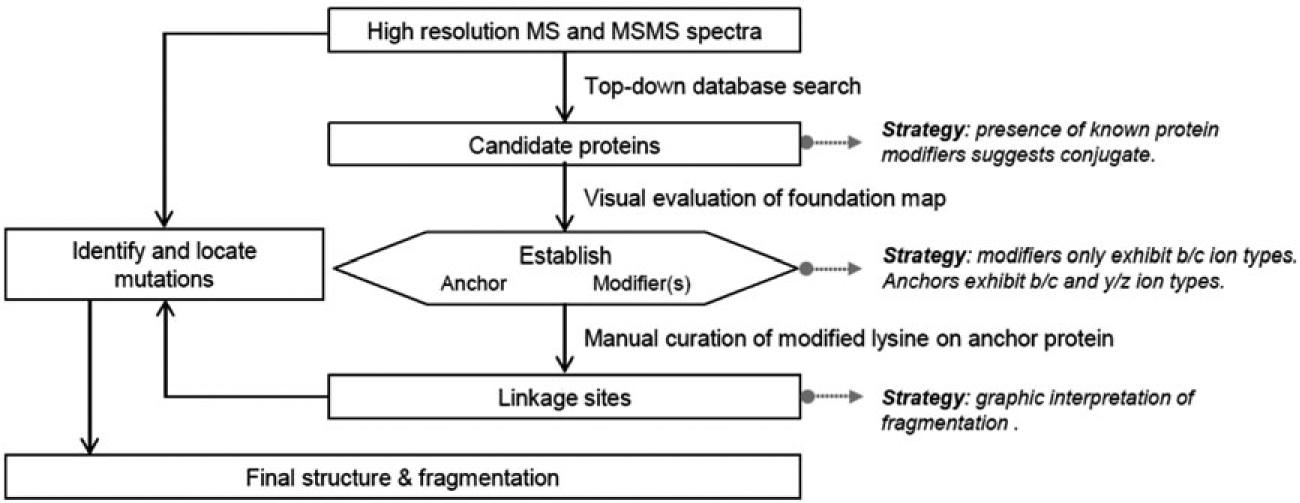
Strategy used here for the identification and characterization of novel synthetic branched proteins. Modified from reference 13
2 |. EXPERIMENTAL
2.1 |. Expression and purification of ubiquitin (UbΔGG)
Escherichia coli BL21(DE3) cells carrying a chloramphenicol-marked helper plasmid pJY2 were used to express untagged Ub variant (UbΔGG) containing residues 1 to 74 and lacking the C-terminal GG motif. This modification prevented ubiquitin from forming homo-conjugates or ubiquitination of Rub1 and allowed control of the composition of the conjugated product, in this case being rubylated ubiquitin, ie, Rub1 attached to a lysine in ubiquitin. Harvested cells were lysed using sonication, and ubiquitin was purified using perchloric acid precipitation followed by fractionation on a cation-exchange column (GE Healthcare Life Sciences) as detailed elsewhere.16 The purity of each UbΔGG fraction was determined using sodium dodecyl sulfate-polyacrylamide-gel electrophoresis (SDS-PAGE) and mass spectrometry.
2.2 |. Expression and purification of Rub1 variants
In this study, Rub1 (Resembles Ubiquitin Protein 1, from Saccharomyces cerevisiae) contained the following mutations: E63K and T72R (Rub1E63K,T72R), K4F and T72R (Rub1K4F,T72R), or T72R (Rub1T72R). Rub1 was expressed in E. coli BL21(DE3) cells as a fusion with an intein tag containing a chitin-binding domain. The cells were lysed using sonication and then centrifuged at 6000 rpm. The supernatant was fractionated on a chitin-bead column as detailed elsewhere.2 The purity of Rub1 fractions was determined using SDS-PAGE and mass spectrometry.
2.3 |. Enzymatic assembly of Rub1E63K,T72R-UbΔGG heterodimer
Rub1E63K,T72R and UbΔGG (10 mg each) were incubated with 20-μM ubiquitin-conjugating E2 enzyme (UBE2K, aka E2–25 K), 500-nM ubiquitin-activating E1 enzyme (UBE1, aka UBA1), 10 mM creatine phosphate, 5 mM MgCl2, 5 mM ATP, and creatine phosphokinase in 50 mM Tris-HCl buffer (pH 8) at room temperature for approximately 16 hours. The solution was centrifuged at 13 000 rpm in order to remove the precipitated enzymes (E1 and E2) and further fractionated using FPLC (GE Healthcare Life Sciences) as detailed elsewhere.2
2.4 |. Enzymatic assembly of Rub1K4F,T72R-Rub1K4F, T72R homodimer
Rub1K4F,T72R (20 mg) was incubated with 20-μM ubiquitin-conjugating E2 enzyme UBE2K, 500-nM UBE1, 10 mM creatine phosphate, 5 mM MgCl2, creatine phosphokinase in 50 mM Tris-HCl buffer (pH 8), and 5 mM ATP for approximately 16 hours. The solution was centrifuged at 13 000 rpm in order to remove the precipitated enzymes (E1 and E2) and then fractionated using FPLC (GE Healthcare Life Sciences) as detailed elsewhere.2 The reaction was monitored by SDS PAGE (Figure 1A).
FIGURE 1.
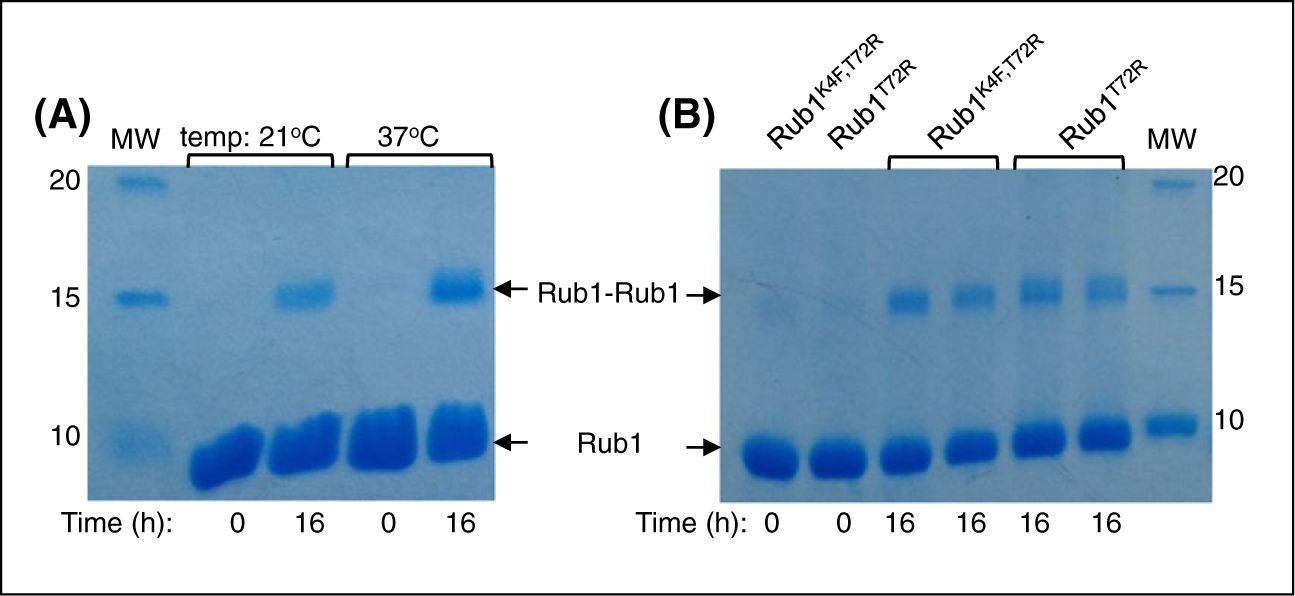
Assembly of Rub1-Rub1 homodimers monitored by Coomassie stained SDS-PAGE. A, Formation of Rub1K4F,T72R-Rub1K4F,T72R homodimer catalyzed by ubiquitin E1 and UBE2K (as E2). B, Formation of Rub1T72R-Rub1T72R homodimers catalyzed by ubiquitin E1 and UBE2S (as E2). The products are shown in two lanes: one collected after the reaction and the other after centrifugation to remove precipitated E1 and E2
2.5 |. Enzymatic assembly of Rub1T72R-Rub1T72R homodimer
Rub1T72R (10 mg) was incubated with 20-μM ubiquitin-conjugating E2 enzyme UBE2S (UBE2S-UBD, Addgene #66713), 500-nM UBE1, 10 mM creatine phosphate, 5 mM MgCl2, 5 mM ATP, and creatine phosphokinase in 50-mM Tris-HCl buffer (pH 8) for approximately 16 hours. The solution was centrifuged at 13 000 rpm in order to remove the precipitated enzymes (E1 and E2). The reaction was monitored by SDS PAGE (Figure 1B) The supernatant was then injected slowly onto a 5-mL cation-exchange column at 1 mL/min using FPLC (GE Healthcare Life Sciences). The Rub1-Rub1 containing species were eluted with cation buffer B (50 mM ammonium acetate containing 1 M NaCl, pH 4.5), and the purity was checked using SDS-PAGE.
2.6 |. Liquid chromatography-tandem mass spectrometry
Intact rubylated proteins (Rub1E63K,T72R-UbΔGG, Rub1K4F,T72R-Rub1K4F,T72R and Rub1T72R-Rub1T72R) were analyzed using an Ultimate 3000 ultrahigh-performance liquid chromatographic system coupled online to an orbitrap Fusion Lumos mass spectrometer equipped with a nanoelectrospray ionization source (Thermo Fisher). Optimally, mixtures containing 100 ng of each branched protein construct were loaded onto a PepSwift Monolithic trap (200 μm × 5 mm). Reverse-phase LC separation was performed using a ProSwift RP-4H column (100 μm × 25 cm, Thermo Fisher). The flow-rate was 1.5 μL/min. The gradient elution consisted of 5% to 55% mobile phase B (75% acetonitrile and 25% water in 0.1% formic acid) in 25 minutes. Mobile phase A consisted of 97.5% water and 2.5% acetonitrile in 0.1% formic acid. The auto-sampler and column oven temperatures were set to 4°C and 35°C, respectively. The mass spectrometer was operated in data-dependent mode using Xcalibur software (Thermo Fischer). The measurements were performed in intact protein mode, with nitrogen pressure of 3 mTorr in the ion-routing multipole. The potential for in-source fragmentation was maintained at 15 V. A resolving power of 120 000 was used to acquire both the precursor and fragment ions. The automatic gain control (AGC) was set at 106 during both precursor and fragment ion acquisitions.
Ion activation was provided by electron transfer dissociation supported by high-energy collision-induced dissociations (EThcD) with a 4-ms electron transfer dissociation reaction time and supplemental activation with 10% normalized high-energy collisions. The reagent anion AGC target was set to 106, with 200-ms injection time.7,8,13 All MS/MS spectra were acquired across a mass range of 150 to 2000 m/z. A tandem spectrum was constructed for Rub1E63K,T72R-UbΔGG, using Xcalibur to combine tandem spectra from precursor ions with charge states +19, +18, and +17. Tandem spectra from precursors with charge states +18 and +17 were combined for Rub1K4F, T72R-Rub1K4F,T72R. The most abundant precursor charge states (+16 and + 17) of the scarce Rub1T72R-Rub1T72R dimer were targeted for fragmentation and combined. Precursor ion isolation was performed by the quadruple module using a window of 1.6 m/z.
2.7 |. Data analysis
Raw data was uploaded into the ProSight PD node 1.1 in Proteome Discoverer 2.2 and deconvoluted using Xtract in the Xcalibur suite. Each file was searched against a database containing 7965 yeast-protein entries from UniProt and the predicted amino acid sequences of Rub1E63K,T72, Rub1T72R, and UbΔGG. For top-down data analysis, the yeast database was curated using ProSight PC 4.0 considering N-terminal methionine cleavage and annotated post-translational modifications. The searches were performed in Absolute Mass Search using a 9000-Da precursor mass tolerance and 10-ppm fragment mass tolerance. The large precursor mass tolerance was used in order to accommodate possible additions of small protein modifiers, while high mass accuracy (10 ppm) was required for analysis of the fragment ions and subsequent identification of the conjugated protein. Analyses of the three combined tandem spectra were conducted using ProSight Lite13 software with mass tolerances of 10 ppm. Sequences and linkage sites were interrogated using manual interpretation assisted by fragmentation graphics from ProSight Lite. In all fragmentation maps presented here, bond cleavages that form b and y ions are indicated in blue, while cleavages that form c and z ions are marked in red.7,8,13
3 |. RESULTS AND DISCUSSION
A strategy is shown (Scheme 1) for interpreting the tandem mass spectra to characterize dimeric products expected in the constrained syntheses studied here. This strategy is modified from the more general approach published earlier by this group.13 Following the strategy, the presence of a branched protein structure is first confirmed by molecular masses and by the automated identification of fragment ions formed from ubiquitin, Rub1, and potentially mutated homologs.13 Next, the strategy identifies which protein is the modifier and which protein is the anchor (substrate). A useful guide is the mechanistic requirement that the modifier protein be covalently bonded via an isopeptide bond between its C-terminus and a lysine sidechain of the anchor protein.1 When fragment ions are mapped onto the individual sequences, as in the Foundation maps presented in Figure 2, primarily b/c ion series will be identified on the modifying protein. Series of b/c and y/z ions will all be assigned to the sequence of the anchor or substrate protein. In the third step (Scheme 1), the fragment ions in the MS/MS spectrum are mapped onto dimer structures in which attachment is tested at each lysine residue in the anchor protein. The correct linkage site is assigned based on quasi-manual interpretation (see below) and consideration of the number of peptide bond cleavages characterized as a per cent of the total available. The optimal fragmentation map establishes the site of attachment. The mutations, predicted in the present case by the molecular biology, are confirmed (or not) by molecular masses, and the mutations are located by dense fragmentation or localized within each branch by incomplete fragmentation (Figure 3).
FIGURE 2.

Graphic fragmentation maps interpreting the tandem mass spectrum of a heterodimer formed between UbΔGG and Rub1E63K,T72R. Bottom: Foundation map of UbΔGG. Top: Foundation map of Rub1E63K,T72R
FIGURE 3.

Fragment ion map and structure assigned to the branched product of the conjugation of Rub1E63K,T72R and UbΔGG. Cleavage is assigned at 94% of the amide bonds
3.1 |. Conjugation of UbΔGG with Rub1E63K,T72R
These two proteins were incubated (see Experimental) with UBE1 (E1) and UBE2K (E2) enzymes. The E63K and T72R mutations in Rub1 were designed to mimic the respective ubiquitin residues. In particular, the T72R substitution was introduced to facilitate Rub1 binding to and activation by ubiquitin’s E1.17 The C-terminal truncation prevented UbΔGG from being activated by E1, thus rendering UbΔGG a potential anchor but not modifier protein in this reaction. The molecular mass of the branched product indicates that it contains one molecule from each mutated protein present in the incubation. Foundation maps are shown in Figure 2 in which all fragment ions in the spectrum are mapped against the sequence of each protein, assuming the predicted mutations are present. Although a significant number of peptide bonds is found to be cleaved in each protein, the difference in the fragmentation patterns readily distinguishes UbΔGG (bottom) as the anchor protein and Rub1E63K,T72R (top) as the modifier protein. Specifically, all fragment ions mapped on the Rub1E63K,T72R sequence (top) are b/c ions, and one compelling interpretation is that no y/z ions are assigned because the C-terminus is modified. In Figure 2 (bottom), both b/c and y/z ion series are identified on the sequence of UbΔGG. These two series do not overlap and are separated by a gap at K48. This fragmentation pattern indicates13 that UbΔGG is the anchor protein and that K48 is the site of Rub1 attachment on UbΔGG.
In Figure 3, fragment ions from the MS/MS spectrum are mapped over the sequence of the branched protein with Rub1E63K,T72R attached at K48 in UbΔGG. Now, both b/c and y/z series are detected in both the modifier and the anchor chains. The molecular mass confirms the presence of mutations E63K and T72R in Rub1E63K,T72R and the missing terminal GG in ubiquitin. Extensive fragmentation confirms truncation of ubiquitin, locates the mutation at E63K, and localizes the mutation T72R to residue 72. The deconvoluted product ion spectrum from precursor ions with a single charge state (+19) ions is shown in Figure 4, with interpretation consistent with Figure 3.
FIGURE 4.
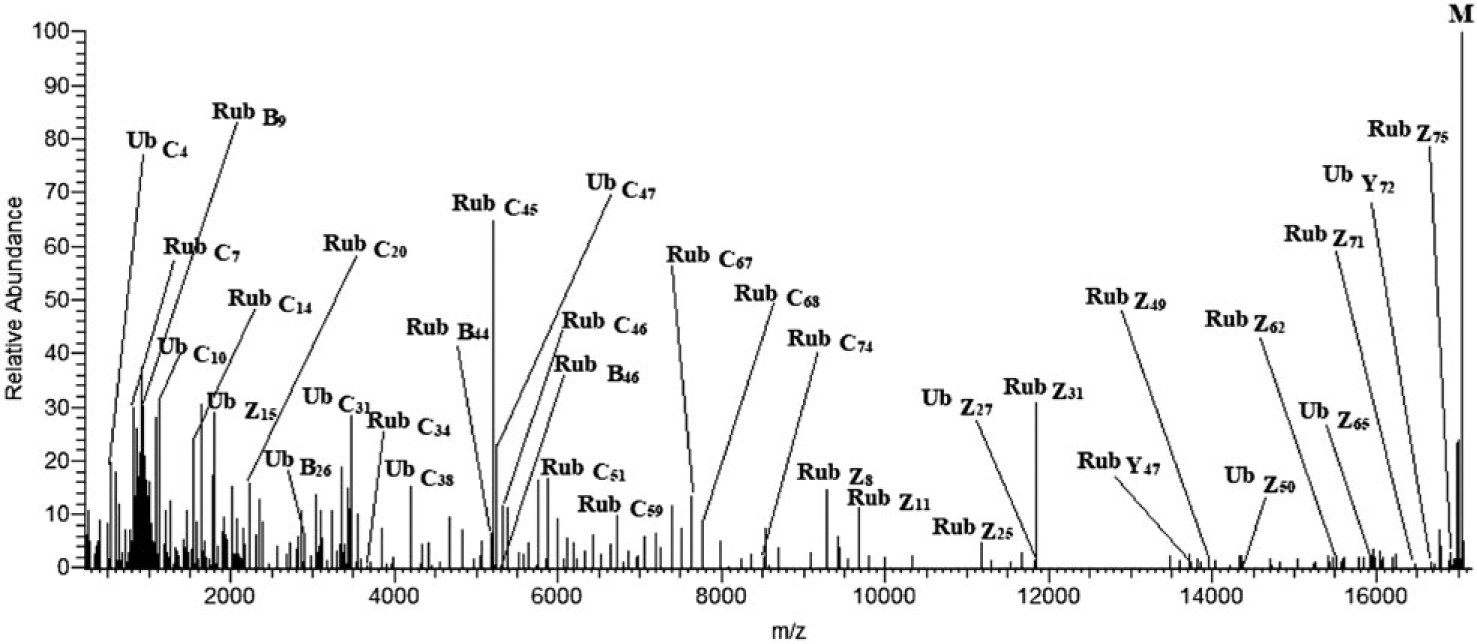
Deconvoluted product ion spectrum of the +19 ions of branched protein Rub1E63K,T72R-UbΔGG
This product can be compared with the product formed by conjugation of Ub-D77 (ubiquitin having an additional aspartate (D77) at its carboxyl terminus) and Rub1T72R, with the same E1 and E2 enzymes, which we reported earlier.13 In that case, Rub1T72R was attached to ubiquitin at the same site, K48. In the present work, the replacement of an acidic residue with a basic residue at position 63 in Rub1T72R does not modify the course of the conjugation reaction, to the extent that this is reflected in the attachment site for rubylation of ubiquitin. (The modifications at the structurally flexible carboxyl terminus of ubiquitin in these two examples prevent it from being activated by its E1 enzyme but do not impact the ubiquitin fold.16,18)
3.2 |. Conjugation of Rub1K4F,T72R with Rub1K4F,T72R
This homodimer was produced by incubating the Rub1K4F,T72R monomer with ubiquitin enzymes UBE1 (E1) and UBE2K (E2). As in the previous example, these mutations in Rub1 were designed to mimic the respective residues in ubiquitin. The foundation map for this homodimer is presented in Figure 5, which indicates a gap at residue K48 between the b/c fragmentation series and the y/z series. K48 is assigned as the site of attachment. The fragmentation map of the complete homodimer is shown in Figure 6, where b/c and y/z ions are now seen to be formed throughout the length of both the anchor and modifier proteins and additional peptide bond cleavages are detected. The presence of the expected mutations is confirmed by the molecular mass. The position of K4F is well defined in each branch by dense fragmentation, and T72R is localized within two and three residues in the anchor and modifier chains, respectively.
FIGURE 5.

Foundation map of the tandem mass spectrum of a homodimer of Rub1K4F, T72R
FIGURE 6.

Fragment ion map and structure assigned to the product of the incubation of Rub1K4F,T72R with UBE1 and UBE2K. Cleavage is assigned at 82% of the amide bonds
These results show that the K4F mutation in Rub1T72R does not prevent its activation by ubiquitin E1. Furthermore, ubiquitin E2 (UBE2K) recognizes and treats this Rub1 variant as both the modifier protein and the anchor (substrate), as if it were ubiquitin, and, moreover, it forms the same linkage (via K48) as in the case of ubiquitin homodimer. Until now, we (and others) have been able to use Ub machinery to conjugate Rub1 as the modifier; however, attempts to trick E2 to use it as the anchor/substrate have failed.2 This is the first time a Rub1 proteoform is being recognized by E2 in both capacities necessary for forming a homodimer.
3.3 |. Conjugation of Rub1T72R with Rub1T72R
A homodimer was found to be formed in small amounts when the protein was incubated with UBE1 (E1) and a different ubiquitin-conjugating E2 enzyme, UBE2S. The foundation map (Figure 7) shows a gap at [52–57] between the b/c series and a sparse y/z series. This sequence contains one lysine residue, K53. Based on the canonical role of lysine as the attachment site in conjugation by this enzyme family,1 Figure 8 presents the fragmentation map of a branched dimer linked at K53.
FIGURE 7.

Foundation map of the tandem mass spectrum of a homodimer of Rub1T72R
FIGURE 8.

Fragment ion map and structure assigned to the product of the conjugation of Rub1 T72R. Cleavage is assigned at 50% of the amide bonds
As in the previous example, our results demonstrate that ubiquitin E2 (UBE2S) recognizes Rub1T72R as both the modifier protein and the anchor (substrate), as if it were ubiquitin. However, the resulting conjugation site is different from that expected for the ubiquitin homodimer. The UBE2S enzyme predominantly makes ubiquitin chains linked via K11, with only a small percentage of K63 linkages and possibly K48 linkages.19 Rub1 lacks lysine at position 63, and conjugation at neither K11 nor K48 was detected in the Rub1T72R homodimer formed by this enzyme. Instead, the dimer appears to be linked through K53. Ubiquitin does not have a lysine at position 53; thus, this linkage site appears to be unique for Rub1 conjugation by UBE2S.
4 |. CONCLUSIONS
Rub1 (NEDD8) is the closest kin of ubiquitin among all ubiquitin-like proteins. With ca. 60% sequence identity and above 70% homology, Rub1 and ubiquitin are structurally superimposable and share the same surface hydrophobic residues responsible for ubiquitin recognition by the majority of ubiquitin receptors.2 Furthermore, five out of the seven ubiquitin lysines are preserved in Rub1. And yet the two proteins act as molecular signals in separate cellular pathways and have their own dedicated activating enzymes, ligases, and conjugation targets. Furthermore, in contrast to ubiquitin, Rub1-Rub1 conjugates have not been reported yet. In this study, we were able to rubylate ubiquitin using ubiquitin’s E1 and E2 enzymes and succeeded, for the first time, to form, isolate, and analyze Rub1 homodimers. Because the enzymes used for this were not cognate to Rub1, it was necessary to characterize these conjugates in terms of amino acid sequences and the linkage sites. We found that UBE2K (E2–25 K) retains its linkage specificity for K48 when forming Rub1-Ub and Rub1-Rub1 conjugates. UBE2S, however, does not retain its preference for K11-linkage and instead links Rub1 to Rub1 through a different lysine (K53) not present in ubiquitin. Structural mechanisms responsible for this, and functional implications of these findings will be addressed in future studies.
Top-down protein analysis is used successfully here to provide rapid and reliable characterization of three novel branched proteins. A bottom-up study based on tryptic or Arg-C digestion could not have distinguished between Ub-Rub1 and Rub1-Rub1 conjugates. An interpretation strategy has been developed for this analysis, supported by graphic interpretation software, which defines the constituents and their sites of attachments and confirms and locates mutations in each branch. While cleavage at every bond remains the ideal in top-down protein analysis, we argue that the canonical understanding that only lysine residues can serve as branching points in our system allows these sites to be recognized by bracketing. A similar systematic interpretation has been applied successfully to a variety of polyubiquitins,7,8 and, we expect that it will be reliably applicable to branched proteins formed by conjugation of other ubiquitin-like modifiers, eg, SUMO, ISG15, FAT10.
ACKNOWLEDGEMENTS
The work was supported by grants GM021248 and OD019938 from the US National Institutes of Health and MCB1818280 from the National Science Foundation.
Funding information
National Science Foundation, Grant/Award Number: MCB1818280; US National Institutes of Health, Grant/Award Numbers: GM021248 and OD019938
REFERENCES
- 1.Pickart CM. Mechanisms underlying ubiquitination. Annu Rev Biochem. 2001;70(1):503–533. [DOI] [PubMed] [Google Scholar]
- 2.Singh RK, Zerath S, Kleifeld O, Scheffner M, Glickman MH, Fushman D. Recognition and cleavage of related to ubiquitin 1 (Rub1) and Rub1-ubiquitin chains by components of the ubiquitin-proteasome system. Mol Cell Proteomics. 2012;11(12):1595–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Peng J, Schwartz D, Elias JE, et al. A proteomics approach to understanding protein ubiquitination. Nat Biotechnol. 2003;21(8):921–926. [DOI] [PubMed] [Google Scholar]
- 4.Kim W, Bennett EJ, Huttlin EL, et al. Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol Cell. 2011;44(2):325–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Udeshi ND, Mertins P, Svinkina T, Carr SA. Large-scale identification of ubiquitination sites by mass spectrometry. Nat Protoc. 2013;8(10):1950–1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burke MC, Wang Y, Lee AE, et al. Unexpected trypsin cleavage at ubiquitinated lysines. Anal Chem. 2015;87(16):8144–8148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee AE, Geis-Asteggiante L, Dixon EK, et al. Preparing to read the ubiquitin code: characterization of ubiquitin trimers by top-down mass spectrometry. J Mass Spectrom. 2016;51(4):315–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee AE, Geis-Asteggiante L, Dixon EK, et al. Preparing to read the ubiquitin code: top-down analysis of unanchored ubiquitin tetramers. J Mass Spectrom. 2016;51(8):629–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cannon JR, Martinez-fonts KL, Rogotham SA, Mataouschek AT, Brodbelt JS. Top-down 193 nm ultraviolet photodissociation mass spectrometry for simultaneous determination of polyubiquitin chain length and topology. AnalChem. 2015;87:1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Greer SM, Brodbelt JS. Top-down characterization of heavily modified histones using 193 nm ultraviolet photodissociation mass spectrometry. J Proteome Res. 2018;17(3):1138–1145. [DOI] [PubMed] [Google Scholar]
- 11.Catherman AD, Skinner OS, Kelleher NL. Top down proteomics: facts and perspectives. Biochem Biophys Res Commun. 2014;445(4):683–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ansong C, Wu S, Meng D, et al. Top-down proteomics reveals a unique protein S-thiolation switch in Salmonella typhimurium in response to infection-like conditions. Proc Natl Acad Sci. 2013;110(25):10153–10158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen D, Gomes F, Abeykoon D, et al. Top-down analysis of branched proteins using mass spectrometry. Anal Chem. 2018;90(6):4032–4038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.DeHart CF, Fellers RT, Fornelli L, Kelleher NL, Thomas PM. Bioinformatics analysis of top-down mass spectrometry data with ProSight Lite In: Wu CH, Arighi CN, Ross KE, eds. Protein Bioinformatics: From Protein Modifications and Networks to Proteomics. New York, NY: Springer New York; 2017:381–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cai W, Guner H, Gregorich ZR, et al. MASH suite pro: a comprehensive software tool for top-down proteomics. Mol Cell Proteomics. 2016;15(2):703–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pickart CM, Raasi S. Controlled synthesis of polyubiquitin chains. Methods Enzymol. 2005;399:21. [DOI] [PubMed] [Google Scholar]
- 17.Whitby FG, Xia G, Pickart CM, Hill CP. Crystal structure of the human ubiquitin-like protein NEDD8 and interactions with ubiquitin pathway enzymes. J Biol Chem. 1998;273(52):34983–34991. [DOI] [PubMed] [Google Scholar]
- 18.Castaneda CA, Chaturvedi A, Camara CM, Curtis JE, Krueger S, Fushman D. Linkage-specific conformational ensembles of non-canonical polyubiquitin chains. Phys Chem Chem Phys. 2016;18(8):5771–5788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bremm A, Freund SM, Komander D. Lys11-linked ubiquitin chains adopt compact conformations and are preferentially hydrolyzed by the deubiquitinase Cezanne. Nat Struct Mol Biol. 2010;17(8):939–947. [DOI] [PMC free article] [PubMed] [Google Scholar]