

Abstract
We report here the synthesis and human carbonic anhydrases (CA, EC 4.2.1.1) inhibitory properties of a series of 4′-substituted 1,1′-biphenyl-4-sulfonamides incorporating a 2″- or 3″-amino- or carboxyphenyl unit. Most compounds showed significant variations in their inhibition profiles against CA II and IX when compared to previously reported analogs 12–18 bearing a 4″-amino or a 4″-carboxy group. In particular, compounds 1–11 showed considerable improvement of the CA II inhibitory efficacy with KI values in the subnanomolar range (KIs spanning between 0.57 and 31.0 nM), a drop of activity against CA IX (KIs in the range 92.0 to 555.7 nM) and were as potent as 12–18 toward CA I (KIs in the range 5.9–217.7 nM). Docking and molecular dynamics were used to gain insights on the inhibition profiles. The reported inhibition data show that 1–11 have potential as novel agents to treat ocular pathologies, such as glaucoma, because of the potent and selective targeting of CA II, which is the isoform most implicated in this disease.
Keywords: Carbonic anhydrase, inhibitor, biphenyl, subnanomolar, glaucoma
Carbonic anhydrase (CA, EC 4.2.1.1) zinc(II) metalloenzymes catalyze the reversible conversion of carbon dioxide and water into monohydrogen carbonate.1 CAs fall in at least seven genetically distinct families that include the human CA (hCA) isoforms, all belonging to the α-class.2,3 CAs are associated with many key physiological processes and pathological conditions.4 For this reason, hCAs have been an ever growing focus of attention as a therapeutic strategy for the design of new agents to treat a variety of diseases.5−8 Most CA inhibitors reported so far are characterized by the presence of a primary sulfonamide group that in its anion form coordinates the zinc(II) atom of the catalytic site. The isoforms CA I and CA II are widely spread across the human body. In particular, CA I is responsible for retinal and cerebral edema; CA II inhibition is involved in the treatment of glaucoma8 and epilepsy,9 as well as in preventing acute mountain sickness.10 Following recognition that hCA inhibitors (hCAIs) may have therapeutic potential, many efforts have been spent to synthesize novel inhibitors of selected hCA isoforms in the past decade.11 The design of new CAI was mainly accomplished by the growing knowledge of the topography of the CA active site. Protein Data Bank has reported more than one hundred structures cocrystallized with the hCA II as well as other isoforms, such as hCA I, IV, IX, XII, XIII, and XIV, among others.12
In our previous work,13 we identified 1,1′-biphenyl sulfonamide derivatives as an interesting class of isoform-selective CAIs targeting CA XIV, an isoform involved in several serious ocular and renal diseases for which only few isoform-selective inhibitors are reported in the literature. Herein, we report that moving the amino or carboxy groups from 4″ to 3″ or 2″ position led to an unpredicted selective inhibition of the hCAII isoform. These new compounds have potential as novel therapeutic agents to treat ocular pathologies, such as glaucoma.
No cure for glaucoma exists. Current available intraocular pressure lowering treatments are based on eye drops, prostaglandin analogs, autonomic nervous system agents, and CAIs.14 First generation CAIs are still used as systemic drugs to treat glaucoma. Second generation inhibitors show fewer side effects compared to the first CAIs and are used topically as eye drops. However, these treatments are also associated with side effects, such as blurred vision, eye itching and irritation, spots on the cornea, and systemic adverse reactions. New improved agents to treat glaucoma are pursued, either by the synthesis of new scaffolds or hybrid inhibitors, or by the identification of new therapeutic targets.15,16
1,1′-Biphenyl-4-sulfonamides 1–5 were prepared as depicted in Scheme 1. Treatment of 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzenesulfonamide (28)17 with an appropriate aryl bromide 19–21 in the presence of cesium carbonate and Pd(II) acetate in aqueous 1-methyl-2-pyrrolidinone at 110 °C under MW irradiation at 100 W for 15 min gave 22–24. Heating of 20, 22, or 23 with sodium borohydride in aqueous tetrahydrofuran (THF) at reflux temperature for 2 h gave the corresponding alcohols 27, 25, or 26. Compound 27 was reduced to a methylene 21 in the presence of triethylsilane and trifluoromethanesulfonic acid in chloroform. Nitro derivatives 22–26 were reduced to amines 1–3 with tin(II) chloride dihydrate in boiling ethyl acetate or to 4 and 5 with hydrogen (2 atm) in the presence of Pd/C in methanol and THF.
Scheme 1. Synthesis of Compounds 1–5.

Reagents and reaction conditions: (a) CsCO3, Pd(II) acetate, NMP/H2O, 110 °C, MW, 100 W, 15 min, closed vessel, 36–78%; (b) SnCl2·2H2O, AcOEt, reflux, 3 h, 22–64%; (c) NaBH4, THF/H2O, reflux, 2 h, 31–89%; (d) H2, 2 atm, Pd/C, MeOH/THF, 25 °C 4 h, 20–43%; (e) triethylsilane, trifluoromethanesulfonic acid, CHCl3, 25 °C, overnight, 65%.
1,1′-Biphenyl-4-sulfonamides 6–11 were obtained as shown in Scheme 2. Methyl 2- and 3-(4-bromobenzoyl)benzoates 33 and 34 were synthesized starting from the corresponding 2- and 3-tolyl ketones 29 and 30, which were oxidized with potassium permanganate in aqueous tert-butanol at 80 °C for 3 h to obtain the carboxylic acids 31 and 32. Subsequent esterification with methanol at reflux in the presence of sulfuric acid for 12 h provided 33 and 34. Sodium borohydride reduction of 33 and 34 furnished the lactone 35, due to the concomitant intramolecular cyclization of the intermediate hydroxy-carboxylic acid, and the alcohol 36, respectively. Alcohol 36 was converted to 39 by reaction with triethylsilane and trifluoromethanesulfonic acid in chloroform at 25 °C for 12 h. Reaction of 35 with chlorotrimethylsilane and iodomethane in acetonitrile at 80 °C overnight gave acid 37. The latter was esterified to 38 in methanol in the presence of sulfuric acid. 1,1′-Biphenyl-4-sulfonamides 6–11 were obtained by treatment of 28 with an appropriate aryl bromide 35, 33, 38, 34, 39, or 36, respectively, potassium phosphate tribasic, and 1,1′-bis(diphenylphosphino)-ferrocene]dichloropalladium(II) complex with dichloromethane (1:1) (Pd(dppf)Cl2·CH2Cl2) in N,N-dimethylformamide (DMF) at 60 °C for 2 h under Ar stream to afford 6 and 40–44. Lithium hydroxide hydrolysis of esters 40–44 in aqueous THF at 25 °C for 12 h furnished the desired carboxylic acids 7–11.
Scheme 2. Synthesis of Compounds 6–11.

Reagents and reaction conditions: (a) t-ButOH, H2O, 50 °C, KMnO4, 80 °C, 5 h, 73%; (b) MeOH, H2SO4, reflux, 12 h, 63–81%; (c) NaBH4, THF/H2O, reflux, 2 h, 73%; (d) NaBH4, EtOH, reflux, 2 h, 38%; (e) TMCS, NaI, CH3CN, Ar stream, reflux, overnight, 67%; (f) 28, K3PO4, Pd(dppf)Cl2·CH2Cl2, DMF, 60 °C, 2 h, Ar stream, 30–69%; (g) triethylsilane, trifluoromethanesulfonic acid, CHCl3, 25 °C, overnight, 68%; (h) LiOH·H2O, THF/H2O, 25 °C, 12 h, 77–95%.
Inhibition data of 4,4′-biphenylsulfonamides 1–11 against human isoforms CA I, II, and IX were measured by a stopped flow CO2 hydrase assay;18 the KI values are depicted in Table 1. Acetazolamide (AAZ), a clinically used sulfonamide inhibitor, was used as reference drug. Inhibition profiles are displayed in comparison with those of the previously reported 4,4′-biphenylsulfonamides 12–18.13 The following structure–activity relationships (SAR) can be gathered from the inhibition data reported in Table 1.
Table 1. Inhibition of hCA I, II, and IX (KI values nM) Isoforms by 4,4′-Biphenylsulfonamides 1–18 and AAZ as Reference Inhibitor by a Stopped-Flow CO2 Hydrase Assay18.


Mean data from three different assays. SD: standard deviations ranged from ±5% to ±10% of the indicated KI values.
SI: hCA II selectivity index obtained as ratio between KIs of the indicated hCA and hCA II KIs.
From ref (13).
The cytosolic isoform hCA I was quite effectively inhibited by the 4,4′-biphenylsulfonamides 1–11 here reported, with KI values in a low to medium nanomolar range (5.9–217.7 nM). The hCA I inhibition potency tightly depended on the nature and position of the substituent at the terminal phenyl ring as well as on the type of linker X. As inhibitors of hCA I, methylene compounds 3 and 8, alcohols 4 and 11, and ketone 9 reported single-digit KI values ranging from 5.9 to 8.4 nM, whereas ketone 2 and lactone 6 showed higher two-digits KI values of 87.3 and 80.1 nM. The 2″-aminophenyl ketone 1 exhibited the weakest CA I inhibition among the newly investigated compounds 1–11 (KI of 217.7 nM). Similarly to 1, the previously reported derivatives 12–16, with a 4″-amino or a 4″-carboxylic acid substituent at the terminal aromatic ring, inhibited the hCA I with KIs in the range of 62.0–589.0 nM. As inhibitors of CA I, 3, 4, 8, 9, and 11 were equipotent to the previously reported ester (17) and nitro (18) compounds.13
Switching the amino and carboxy substituents on the outer phenyl ring from 4″- to 2″- or 3″-positions had a positive impact on the hCA II inhibition potency of the new compounds 1–11 compared to 12–18, with the exception of derivatives 1 and 2. In fact, most new derivatives yielded subnanomolar KI values spanning between 0.57 and 0.97 nM; meanwhile, 12–18 yielded KI values from 4.8 to 814 nM. The 2″-aminophenyl ketone 1 was a weak hCA II inhibitor (KI of 31.0 nM), although it was more potent than the corresponding 4″-amino analog 12 (KI of 86.2 nM). The improvement of inhibition reached out to 3 orders of magnitude for the couple 11 (KI of 0.83 nM)/16 (KI of 814 nM).
A dramatic drop of activity against hCA IX arose with the newly reported compounds 1–11 with respect to 12–16. In fact, the KI values of 1–11 span in a medium nanomolar range from 92.0 to 555.7 nM and were comparable to 17 and 18 (KI of 710 and 678 nM, respectively). In contrast, the KIs of 12–16 against the CA IX were in the range between 6.0 and 31.4 nM. As an inhibitor of the hCA IX, the 4″-carboxyphenyl derivative 16 (KI of 450 nM) was 4-fold less potent than the corresponding 3″-analog 11 (KI of 88.4 nM). Most new compounds exhibited significant CA II/CAI selectivity indexes (SI) ranging between 6.4 and 114.3. Alongside, 2–10 also showed exceptional CA II/IX SIs, which had not stood out for 4″-substituted parent compounds 12–16. Previously reported 17 and 18 yielded negligible CA II/I SIs, but their CA II/XI SIs were at the same level of 2–10.
CA II is thoroughly implicated in aqueous humor production and represents an important antiglaucoma drug target (together with CAs IV and XII). As CA I is off target in the treatment of such an ailment, compounds showing subnanomolar inhibition of the CA II with high II/I selectivity might be of interest for developing new antiglaucoma agents. Besides, selectivity for CA II over CA IX might be beneficial because CA IX is not a target for glaucoma treatment, and although mainly overexpressed in cancer, it is found in some healthy tissues such as duodenum, jejunem, and ileum.
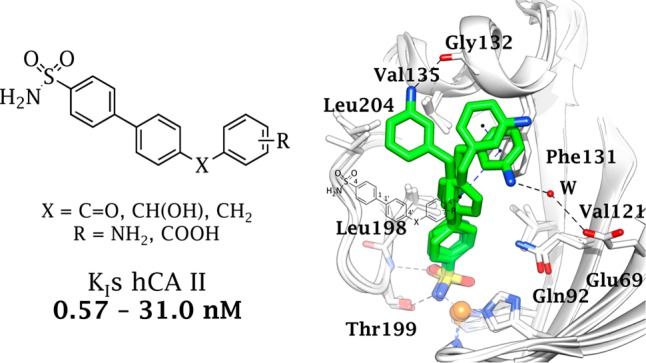
In silico studies were performed to point out the ligand/target interactions driving the outstanding hCA II inhibition efficacy of this set of biphenyl compounds. The subnamolar inhibitors 3 and 10 were chosen as representative within the series and docked into hCA II active site. Poses showing the best score values and favorable binding interactions were subjected to a cycle of 100 ns molecular dynamics (MD). The protein root-mean-square deviation (RMSD) evolution as a function of time indicated that the protein experienced only small conformational variations. In contrast, RMSD plotted for inhibitors 3 and 10 over the 100 ns approximately spanned in the 1–2 Å range (Figure S1, Supporting Information).
This was found to be related to the movements of their molecular X–R tails within the hCA II cavity, as it is reflected in the root-mean-square fluctuation (RMSF) plotted for each heavy atom of 3 and 10 in Figure S2, Supporting Information. In detail, binding orientations extrapolated from the dynamic simulations are depicted in Figure 1A (3) and 1B (10). The benzenesulfonamides 3 and 10 form the typical coordination bond to hCAs2 between the zinc ion and the SO2NH– group. The sulfonamido group is also involved in two H-bonds with Thr199, while the aromatic rings form π-alkyl contacts with Val121, Val135, and Leu198.
Figure 1.

Binding poses extrapolated from the 100 ns MD performed on the adduct of (A) 3 and (B) 10 with hCA II. (C) Best docked binding orientations of 15 with hCA II. H-bonds and π–π interactions are represented as black and blue dashed lines, respectively.
The low to subnanomolar hCA II inhibition efficiency of the biphenylsulfonamides 1–18 (except 16), in which another substituted phenyl ring is linked to the main scaffold by a CH2, CHOH or C=O linker, is apparently related to the strong interactions between the ligands tail and Phe131. The absence of the aromatic residue Phe131 in hCA I and IX, replaced by a Leu and Val, respectively, may impair key contacts of the ligand/target adduct. In fact, Phe131 is involved in π–π or hydrophobic interactions with at least one of the two outer aromatic rings of 3 and 10 for more than 80% of the MD time. The rotation of the X–R bond during the MD simulation time favors the formation of various interactions involving the 3″-NH2 (3) or 3″-COOH (10) groups and the active site residues. The amino group of 3 was found to be in H-bond distance with Gly132 carbonyl group or water-bridged with Glu69 (Figure 1A), while the carboxylic group of 10 was found in H-bond distance with Arg58 or Lys133 or, in addition, water-bridged with Gln136 (Figure 1B).
In an attempt to understand the remarkable increase of the inhibitory potency of 1–11, up to an order of magnitude compared to 12–16, compound 15, the 4″-substituted analog of 10, was docked into hCA II active site. As depicted in Figure 1C, the all-para-substitutions at the three phenyl rings force compounds 12–18 to assume more outward orientations compared to corresponding ortho- and meta-substituents, thus narrowing the overall interactions in the binding pocket. On the contrary, the ortho- and meta-substituents, due to their orientation, can form more effective interactions with the target.
In conclusion, we have synthesized a series of 4,4′-biphenylsulfonamides with a new 2″- and 3″-amino- or carboxyphenyl unit. Most compounds showed significant variations in their inhibition profiles against hCAs II and IX upon swapping the substituent on the terminal phenyl ring from para to ortho or meta position. In fact, compounds 1–11 report an enhancement of hCA II inhibitory efficacy and a drop of activity against hCA IX maintaining instead a comparable one with analogs 12–16 toward hCA I.
Docking and molecular dynamic simulations were used to gain insights on the potent hCA II inhibitory action of biphenylsulfonamides 1–16 and on the more efficient inhibition induced by 2″- and 3″-derivatives 1–11 than the 4″-analogs 12–16 previously reported. Inhibition data depicted in Table 1 show potential for the development of agents for the treatment of ocular pathologies, such as glaucoma, because of the potent and selective targeting of CA II, which is the isoform most implicated in this disease.
Glossary
ABBREVIATIONS
- CAI(s)
carbonic anhydrase inhibitor(s)
- AAZ
acetazolamide
- (h)CA
(human) carbonic anhydrase
- KI
inhibition constant
- THF
tetrahydrofuran
- DMF
N,N-dimethylformamide
- SAR
structure–activity relationships
- MD
molecular dynamics
- RMSD
root-mean-square deviation
- RMSF
root-mean-square fluctuation
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.9b00437.
Synthetic procedures, compounds characterization, in vitro kinetic procedure, in silico supplemental graphs, and methods (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
This work was supported by Italian Ministry of Education, Universities and Research, Dipartimenti di Eccellenza, grant n. L. 232/2016 (to G.L.R., M.P., M.N., and R.S.).
The authors declare no competing financial interest.
Supplementary Material
References
- Supuran C. T. Carbonic anhydrases. Bioorg. Med. Chem. 2013, 21, 1377–1378. 10.1016/j.bmc.2013.02.026. [DOI] [PubMed] [Google Scholar]
- Alterio V.; Di Fiore A.; D’Ambrosio K.; Supuran C. T.; De Simone G. Multiple binding modes of inhibitors to carbonic anhydrases: how to design specific drugs targeting 15 different isoforms?. Chem. Rev. 2012, 112, 4421–4468. 10.1021/cr200176r. [DOI] [PubMed] [Google Scholar]
- De Simone G.; Di Fiore A.; Capasso C.; Supuran C. T. The zinc coordination pattern in the η-carbonic anhydrase from Plasmodium falciparum is different from all other carbonic anhydrase genetic families. Bioorg. Med. Chem. Lett. 2015, 25, 1385–1389. 10.1016/j.bmcl.2015.02.046. [DOI] [PubMed] [Google Scholar]
- Nocentini A.; Supuran C. T. Advances in the structural annotation of human carbonic anhydrases and impact on future drug discovery. Expert Opin. Drug Discovery 2019, 14, 1175–1197. 10.1080/17460441.2019.1651289. [DOI] [PubMed] [Google Scholar]
- Neri D.; Supuran C. T. Interfering with pH regulation in tumours as a therapeutic strategy. Nat. Rev. Drug Discovery 2011, 10, 767–777. 10.1038/nrd3554. [DOI] [PubMed] [Google Scholar]
- Supuran C. T.; Scozzafava A. Carbonic anhydrases as targets for medicinal chemistry. Bioorg. Med. Chem. 2007, 15, 4336–4350. 10.1016/j.bmc.2007.04.020. [DOI] [PubMed] [Google Scholar]
- Supuran C. T. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat. Rev. Drug Discovery 2008, 7, 168–181. 10.1038/nrd2467. [DOI] [PubMed] [Google Scholar]
- Mincione F.; Scozzafava A.; Supuran C. T. The development of topically acting carbonic anhydrase inhibitors as antiglaucoma agents. Curr. Pharm. Des. 2008, 14, 649–654. 10.2174/138161208783877866. [DOI] [PubMed] [Google Scholar]
- Hen N.; Bialer M.; Yagen B.; Maresca A.; Aggarwal M.; Robbins A. H.; McKenna R.; Scozzafava A.; Supuran C. T. Anticonvulsant 4-aminobenzenesulfonamide derivatives with branched-alkylamide moieties: X-ray crystallography and inhibition studies of human carbonic anhydrase isoforms I, II, VII, and XIV. J. Med. Chem. 2011, 54, 3977–3981. 10.1021/jm200209n. [DOI] [PubMed] [Google Scholar]
- Swenson E. R.; Teppema L. J. Prevention of acute mountain sickness by acetazolamide: as yet an unfinished story. J. Appl. Physiol. 2007, 102, 1305–1307. 10.1152/japplphysiol.01407.2006. [DOI] [PubMed] [Google Scholar]
- Supuran C. T. Structure-based drug discovery of carbonic anhydrase inhibitors. J. Enzyme Inhib. Med. Chem. 2012, 27, 759–772. 10.3109/14756366.2012.672983. [DOI] [PubMed] [Google Scholar]
- Alterio V.; Di Fiore A.; D’Ambrosio K.; Supuran C. T.; De Simone G. Multiple binding modes of inhibitors to carbonic anhydrases: how to design specific drugs targeting 15 different isoforms?. Chem. Rev. 2012, 112, 4421–4468. 10.1021/cr200176r. [DOI] [PubMed] [Google Scholar]
- La Regina G.; Coluccia A.; Famiglini V.; Pelliccia S.; Monti L.; Vullo D.; Nuti E.; Alterio V.; De Simone G.; Monti S. M.; Pan P.; Parkkila S.; Supuran C. T.; Rossello A. Silvestri. Discovery of 1,1’-Biphenyl-4-sulfonamides as a New Class of Potent and Selective Carbonic Anhydrase XIV Inhibitors. J. Med. Chem. 2015, 58, 8564–8572. 10.1021/acs.jmedchem.5b01144. [DOI] [PubMed] [Google Scholar]
- Weinreb R. N.; Aung T.; Medeiros F. A. The pathophysiology and treatment of glaucoma. JAMA 2014, 311, 1901–1911. 10.1001/jama.2014.3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scozzafava A.; Supuran C. T. Glaucoma and the applications of carbonic anhydrase inhibitors. Subcell. Biochem. 2014, 75, 349–359. 10.1007/978-94-007-7359-2_17. [DOI] [PubMed] [Google Scholar]
- Kolko M. Present and new treatment strategies in the management of glaucoma. Open Ophthalmol. J. 2015, 9, 89–100. 10.2174/1874364101509010089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollbuck B.; Denholm A.; Eder J.; Hersperger R.; Janser P.; Revesz L.; Schlapbach A.; Waelchli R.. Preparation of aminopyrimidines as IKK inhibitors for treating autoimmune disease and inflammations. PCT Int. Appl. WO 2004089913 A1, October 21, 2004.
- Khalifah R. G. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop-flow kinetic studies on the native human isoenzymes B and C. J. Biol. Chem. 1971, 246, 2561–2573. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.