

Abstract
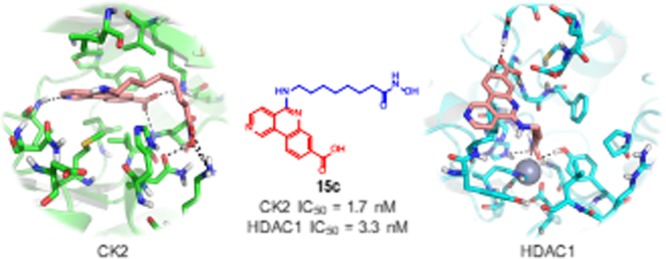
Four potent CK2 inhibitors derived from CX-4945 are described. They also provided nanomolar activity against HDAC1, therefore having promising utility as dual-target agents for cancer. The linker length between the hydroxamic acid and the CX-4945 scaffold plays an important role in dictating balanced activity against the targeted enzymes. The seven-carbon linker (compound 15c) was optimal for inhibition of both CK2 and HDAC1. Remarkably, 15c showed 3.0 and 3.5 times higher inhibitory activity than the reference compounds CX-4945 (against CK2) and SAHA (against HDAC1), respectively. Compound 15c exhibited micromolar activity in cell-based cytotoxic assays against multiple cell lines.
Keywords: Multitarget drugs, CK2, HDAC1, dual inhibitors, docking, molecular dynamics
Cancer is a multifactorial complex disease that is caused by multiple dysfunctions in genes or pathways.1 Combination therapies directed to two or more molecular targets have been widely used in the treatment of this disease.2 Nevertheless, combination therapy has serious disadvantages such as patient incompliance, difficulty to predict side effects and to optimize the dose ratio, as well as unwanted drug–drug interactions and unpredictable pharmacokinetics.2 Multitarget single agents are expected to improve the efficacy of the treatments, by exploiting synergistic interactions, avoiding problems of drug–drug interactions, decreasing drug resistance, and making the pharmacokinetic studies easier to perform.3
Histone deacetylases (HDACs) are a family of epigenetic enzymes that control the transcription and regulation of genes as well as cell proliferation, differentiation, migration, death, and angiogenesis.4−6 Also, overexpression of HDACs has been found in many human cancers.7 Therefore, inhibiting HDACs have been recognized as a promising approach for treating cancer. To date, four HDAC inhibitors (HDACi) were approved by the FDA,6 vorinostat (SAHA), romidepsin (FK-228), belinostat, (PXD101) and panobinostat (LBH589), and another one was approved by the Chinese FDA,8 chidamide (tucidinostat, HBI-8000). On the other hand, almost 18 HDACi are in clinical trials.6
Protein kinase 2 (CK2) is an ubiquitously expressed and constitutively active serine/threonine kinase that phosphorylates an impressive array of substrates including HDACs.9,10 Overexpression of CK2 is involved in several human cancers and has also been linked to poor prognosis and disease progression.11 Several CK2 inhibitors have been discovered in the past, but among them only two inhibitors, CX-4945 (NCT03904862) and CIGB-300 (NCT01639625), have recently entered into Phase II clinical trials as potential anticancer drugs.12
While many HDACi are in preclinical and clinical studies, they have been found to induce drug resistance13 and their results in the treatment of solid cancers have been disappointing.4−6 Interestingly, a SAR study confirms that the cap group in HDACi is flexible and tolerates modification. Thus, the zinc-binding group present in most of them can be easily linked to other anticancer scaffolds providing numerous multitargeting agents with higher potency than the parent compounds.4−6,14,15 Some examples are CUDC-101,16 CUDC-907,17 and 4SC-20218 (Figure 1) which have already entered clinical trials.14 These results strongly support that a single compound that simultaneously inhibits HDAC1 and other oncological targets could be beneficial in cancer resistance over single-acting agents, through the interference with multiple pathways.
Figure 1.

HDACi based dual inhibitors in clinical trials.
Protein kinase 2 (CK2) regulates the dimerization of HDAC1 and HDAC2 during mitosis.19 Under hypoxic conditions, CK2 phosphorylates HDAC leading to HDAC activation. Activated HDAC contributes to tumor growth and vasculogenesis by Hippel-Lindau protein (pVHL) downregulation and, hence, to the increased expression and stabilization of hypoxia-inducible factor HIF-1α protein.20 Since both CK2 and HDAC are involved in the related cancer-relevant biological pathways,21−24 we anticipated that simultaneously inhibiting these two targets by a multitarget single molecule should improve efficacy compared to single-target agents.
In previous publications,25,26 we developed a series of dual CK2/HDAC1 inhibitors using TBB and DMAT as scaffolds to promote CK2 inhibition and a hydroxamate zinc binding group (ZBG) to interact with the zinc present in the active site of HDAC1 and simulating the structure of SAHA, a potent inhibitor of HDAC1. The synthesized dual-acting agents exhibited promising inhibitory activities with the best compound showing IC50 of 5 μM for both enzymes and micromolar activity in cell-based assays (Figure 2).
Figure 2.

Design strategy for dual CK2/HDAC1 inhibitors.
Encouraged by these results, we have designed a new series of CK2/HDAC1 dual inhibitors where the TBB scaffold has been substituted by CX-4945, a 500-fold more potent CK2 inhibitor than TBB.12,27 Herein we describe the computational design and synthesis of these compounds, together with some promising preliminary assays of their biological activity.
Results and Discussion
Computational Docking and Molecular Dynamics
The binding to the target proteins was analyzed by means of docking and molecular dynamics (MD). The binding mode to CK2 of 15a–d was similar to that of CX-4945 in the reference crystal structure28 (PDB code 3PE1) (Figure S1, Supporting Information). The carboxylic acid moiety interacts with the side chain of Lys68 and the backbone NH of Asp175, whereas the pyridine nitrogen interacts with the backbone NH of Val116 in the hinge region. The linkers oriented the hydroxamic moieties toward the entrance of the nucleotide-binding site establishing different hydrogen bonds with the amino acid side chains depending on the length of the linker (Figure S2, Supporting Information). The stability of the binding mode of compound 15c (Figure 3) was assessed by a MD simulation that proved that, despite the high mobility of the linker, the initial binding mode and the interactions of the CX-4945 moiety were maintained, whereas the original hydrogen bonds established by the hydroxamic acid varied but always maintained an interaction with the entrance of the binding site (Figure S3, Supporting Information). These results demonstrate that the incorporation of an alkyl chain with a hydroxamic acid to the CX-4945 core does not impair the proper orientation of the molecules in the ATP binding site, as well as the establishment of the essential interactions that guarantee the high potency found for the reference compound CX-4945.
Figure 3.
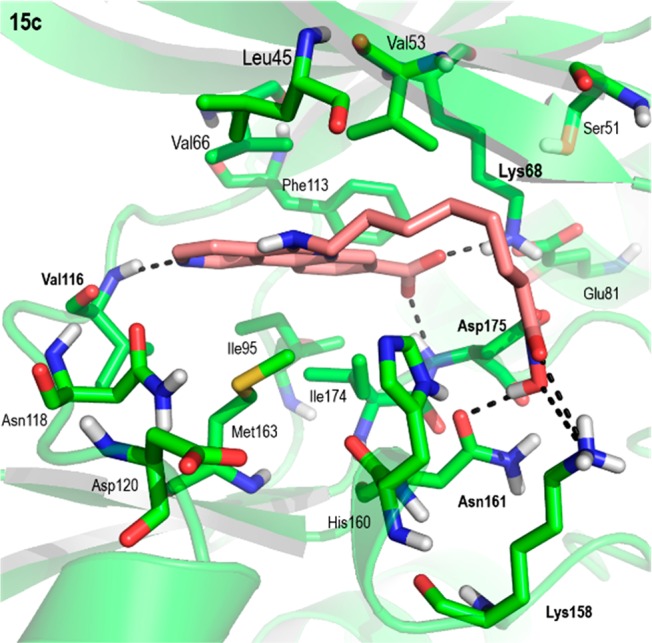
PyMOL stick and cartoon representation of the best docking pose of compound 15c to CK2. For the sake of clarity, only polar hydrogens are shown, and hydrogen bonds have been highlighted with dashed lines.
The binding of compounds 15a–d to HDAC1 was similar to that of SAHA (Vorinostat) to different HDACs in the reference crystal structures.29−31 (Figure S4, Supporting Information). All compounds established with HDAC1 a bidentate chelation to the catalytic Zn2+ ion stabilized by hydrogen bonds with the side chains of His140 and Tyr303 and oriented the CX-4945 moiety to establish different interactions with amino acids in the surface of the protein that also varied depending on the length of the linker (Figure S5, Supporting Information). The stability of the binding mode of compound 15c to HDAC1 (Figure 4) was also assessed by means of MD simulations. During the simulation the CX-4945 moiety explored the surface of the protein establishing different interactions through the carboxylic acid moiety but mainly establishing π-stacking interactions with the side chain of Phe205 (Figure S6, Supporting Information). The movement brought about by the linker and the CX-4945 moiety destabilized some of the initial hydrogen bonding interactions between the hydroxamate moiety and the side chains of His140 and Tyr303; however, a new and stable hydrogen bond is established with the side chain of His141. These results suggested that the interaction between the CX-4945 cap and residue Phe205 and an optimal linker length to establish bidentate Zn2+ coordination are important for the HDAC1 inhibitory potency.
Figure 4.
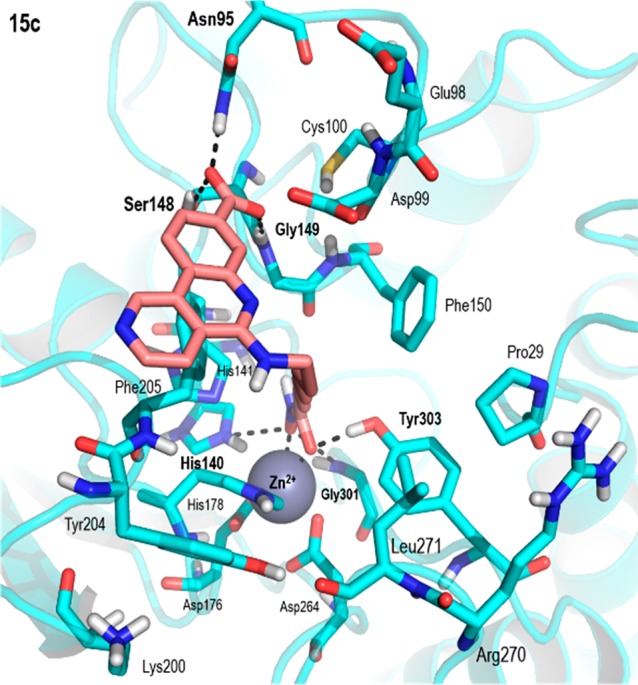
PyMOL stick and cartoon representation of the best docking pose of compound 15c to HDAC1. For the sake of clarity, only polar hydrogens are shown, and hydrogen bonds have been highlighted with dashed lines.
These results predict an overall good affinity of 15a–d for CK2 and HDAC1, so they were forwarded for synthesis and enzymatic studies.
Chemistry
A convergent strategy was developed for the synthesis of this new generation of dual inhibitors. First, chloroquinoline 8 which possesses a chlorine atom that allows the substitution with different nucleophiles was synthesized. Then, different Ω-amino benzyloxy-protected compounds 12a–d were selected and synthesized in order to assess the linker that best fits in the catalytic site of HDAC1 (Scheme 1).
Scheme 1. Convergent Strategy for Dual Inhibitor Synthesis.

Compound 8 was efficiently synthesized adapting the procedure described in the literature32 (Scheme 2). Compounds 3 and 6 were synthesized from commercially available carboxylic acids 1 and 2 as described in Scheme 2. Then, lactam 7 was obtained through a one-pot Suzuki coupling/intramolecular amide cyclization between the boronic ester 6 and the ethyl ester 3. Treatment of lactam 7 with phosphorus oxychloride afforded chloroquinoline 8 which was ready for substitution with the different amines 12a–d.
Scheme 2. Synthesis of Compound 8.

Reagents and conditions: (a) MeOH, H2SO4, reflux, 12 h; (b) KOAc, Pd(dppf)Cl2, bis(pinacolate)diboron, DCM, dioxane, 80 °C, overnight; (c) H2, Pd/C 10%, EtOAc, RT, 12 h; (d) MeOH, H2SO4, reflux, 12 h; (e) NaOAc, Pd(dppf)Cl2 DCM, dioxane, 125 °C, 12 h; (f) neat POCl3, reflux, 2 h.
Amines 12a–b were prepared as depicted in Scheme 3. Nucleophilic substitution of the commercially available Ω-bromo carboxylic acids 9a–d gave the corresponding azido carboxylic acids 10a–d. Amide coupling of 10a–d with the O-benzyl-hydroxylamine, followed by selective catalytic hydrogenation of the formed azides 11a–d, gave the desired amines 12a–d. The use of ethyl acetate as solvent is the key condition to achieve selective reduction of the azide, without deprotecting the benzyl group present in the molecule. When more polar solvents such as methanol were used, both reductions occurred.
Scheme 3. Synthesis of Ω-Amine (Benzyloxy)amino Compounds 12a–d as HDAC1 Scaffold.

Reagents and conditions: (a) NaN3, DMF, 77 °C, 48 h; (b) HCl·H2N-OBn, HBTU, DIPEA, DMF, RT, 12 h; (c) H2, Pd/C, EtOAc, RT, 2 h.
Finally, as depicted in Scheme 4, chloroquinoline 8 and amines 12a–d were reacted to yield methyl benzo[c][2,6]naphthyridine-8-carboxylates 13a–d. After the O-debenzylation by catalytic hydrogenation at high pressure in methanol, these compounds were further converted to the corresponding carboxylic acids 15a–d that were purified by HPLC and isolated as trifluoroacetic salts with the necessary purity (>95%) for biological assays.
Scheme 4. Synthesis of Dual Inhibitors 15a–d Isolated as TFA Salts.

Reagents and conditions: (a) DMF, K2CO3; 135 °C, 55 min, MW irradiation; (b) for 14a–c: H2, Pd/C 10%, MeOH, 4 bar, RT, 12 h; for 14d: H2 atmosphere and MeOH:THF 1:1 as solvent, 12 h; (c) LiOH, RT, 96 h.
Enzymatic Inhibitory Evaluation
The inhibitory activities of the synthesized compounds were determined against CK2 and HDAC1, and the IC50 values are collected in Table 1. The activity toward human recombinant HDAC1 was tested using a fluorometric method.25 CK2 inhibition was measured by a luminometric assay using ADP-Glo kinase assay (Promega).33
Table 1. Inhibitory Activity (IC50, nM) on HDAC1, HDAC6, and CK2.
The four compounds presented an inhibitory activity on CK2 in the low nanomolar range. This result demonstrates that the introduction of a carbon chain containing the hydroxamic group in the CX-4945 scaffold does not interfere in the interaction of these compounds with the active site of CK2 and supports our hypothesis that CX-4945 is an appropriate scaffold for the design of dual CK2/HDAC1 inhibitors.
The best result was found for 15c, with a value of IC50 = 1.7 nM, similar to that obtained for the control compound (CX-4945). What is more interesting, 15c is also a potent inhibitor of HDAC1, with a value of IC50 = 3.3 nM, which is 1.7-fold lower than the value obtained for TSA in our hands. In the design of dual inhibitors, it is especially important to achieve a good balance in the activity against the two target enzymes. This is the case of 15c, a promising compound with a remarkable potency for both enzymes.
Interestingly, this compound showed potent activity in HDAC6 (see Table 1). SAHA is also highly potent against both HDAC1 (IC50: 34 nM) and HDAC6 (IC50: 33 nM) while it is remarkably less potent in other isoforms such as HDAC4 (IC50: >1000 nM) and HDAC8 (IC50: 776 nM).34
When the chain connecting the two selected pharmacophores was shortened (compound 15b) or elongated (compound 15d), the inhibitory potency in CK2 was maintained, while some activity in HDAC1 was lost. These results demonstrated that the best length for a dual interaction is provided by a seven-carbon chain. Our efforts in the design of the next series of inhibitors will concentrate in the modification of the nature of the connecting chain, keeping the same length, and the modification of the ZBG.
Cytotoxic Activity in Cells
The cytotoxicity of the four compounds 15a–d in comparison with reference compounds SAHA and CX-4945 under identical conditions was investigated by using the resazurin assay.35 In vitro cytotoxicity for CX-4945 was reported already against a panel of cancer cell lines, including LNCaP, PC3, MCF-7, and A549, and in vivo efficacy was tested in PC3 xenografts.32 Similarly, the HDAC inhibitor SAHA showed low micromolar cytotoxicity against LNCaP, PC3, MCF-7, and A549 cell lines.36−38 To directly compare the potency of our compounds with CX-4945 and SAHA, we selected these four human cancer cell lines, namely lung (A549), breast (MCF-7), and prostate (PC3 and LNCaP) cell lines (Table 2).
Table 2. IC50 Values of Compounds 15a–d, SAHA, and CX-4945 against Four Human Tumor Cell Lines.
| IC50 values (μM) |
||||
|---|---|---|---|---|
| Cpd | LNCaP | PC3 | MCF-7 | A549 |
| 15a | >100 | >100 | >100 | >100 |
| 15b | >100 | >100 | >100 | >100 |
| 15c | 16.31 ± 0.7 | 40.42 ± 4.4 | 52.48 ± 10.0 | 104.73 ± 7.9 |
| 15d | 66.70 ± 5.0 | >100 | >100 | >100 |
| SAHA | 0.69 ± 0.1 | 2.03 ± 0.1 | 1.26 ± 0.1 | 3.14 ± 0.2 |
| CX-4945 | 6.52 ± 0.6 | 10.87 ± 0.6 | 9.41 ± 0.3 | 11.60 ± 1.8 |
Interestingly, only 15c showed activity in the cell experiments, which is in accordance with the docking and enzymatic assay results. The best activity was observed in LNCaP, an androgen-sensitive human prostate adenocarcinoma cell line, with an IC50 of 16.31 μM. The activity in this cell line was lower than the activities obtained for the parent compounds. Considering the results of inhibitory activity of 15c in isolated enzymes, we postulate that the lower activity exerted by 15c in cells could be due to a low permeability, which we will try to overcome in the next series of inhibitors.
In order to evaluate the effect on the cytotoxicity of the TFA salts,39,40 we carried out a cell assay on LNCaP cell line using CX-4945 as a TFA salt. At the concentrations used in the cytotoxicity study we observed slight differences compared to the free compound. (CX-4945 6.52 μM; CX-4945 as a TFA salt 3.70 μM).
In conclusion, we found four remarkably potent CK2 inhibitors, showing that the introduction of chains of different lengths provided with hydroxamate groups does not affect the interaction of the CX-4945 ring with the active site of CK2.
The presence of the hydroxamate allows the interaction with HDAC1, providing only one promising dual CK2/HDAC1 inhibitory agent 15c, with nanomolar activity in both enzymes. The modeling work has provided plausible binding modes that can account for these results.
Preliminary assays in human cell lines confirm the interest of this compound as a cytotoxic agent. Future studies are necessary to improve the pharmacokinetic profile of this new hit compound and examine its activity in other human cell lines.
Acknowledgments
This work was supported by MICIU/FEDER, UE grant number RTI2018-093539-B-I00. This project received funding from the European Union’s Horizon 2020 research and innovation programme under the Marie-Sklodowska-Curie grant agreement number DUALITY 746225.
Glossary
Abbreviations
- HDAC
histone deacetylase
- CK2
protein kinase type 2
- SAHA
suberoylanilide hydroxamic acid (Vorinostat)
- TBB
4,5,6,7-tetrabromo-2H-benzotriazole
- DMAT
2-dimethylamino-4,5,6,7-tetrabromo-1H-benzimidazole
- dppf
1,1′-bis(diphenylphosphino)ferrocene
- DMF
dimethylformamide
- MW
microwave
- RT
room temperature
- THF
tetrahydrofuran
- ZBG
zinc binding group
- TFA
trifluoroacetic acid
- TSA
trichostatin A
- HBTU
O-(benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate
- DIPEA
N,N-diisopropylethylamine
- DCM
dichloromethane
- THF
tetrahydrofuran
- PDB
protein data bank
- ATP
adenosine 5′-triphosphate
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.9b00561.
Synthetic procedures, spectroscopic and analytical data for all compounds, in vitro enzymatic assay, resazurin assay, and computational methods (PDF)
Author Contributions
‡ L.R. and I.O. contributed equally to this work. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Wu S.; Zhu W.; Thompson P.; Hannun Y. A. Evaluating intrinsic and non-intrinsic cancer risk factors. Nat. Commun. 2018, 9 (1), 3490. 10.1038/s41467-018-05467-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez J. S.; Banerji U. Combine and conquer: challenges for targeted therapy combinations in early phase trials. Nat. Rev. Clin. Oncol. 2017, 14 (1), 57–66. 10.1038/nrclinonc.2016.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J.; Jiang X.; He S.; Jiang H.; Feng F.; Liu W.; Qu W.; Sun H. Rational design of multitarget-directed ligands: strategies and emerging paradigms. J. Med. Chem. 2019, 62 (20), 8881–8914. 10.1021/acs.jmedchem.9b00017. [DOI] [PubMed] [Google Scholar]
- Stazi G.; Fioravanti R.; Mai A.; Mattevi A.; Valente S. Histone deacetylases as an epigenetic pillar for the development of hybrid inhibitors in cancer. Curr. Opin. Chem. Biol. 2019, 50, 89–100. 10.1016/j.cbpa.2019.03.002. [DOI] [PubMed] [Google Scholar]
- Luan Y.; Li J.; Bernatchez J. A.; Li R. Kinase and histone deacetylase hybrid inhibitors for cancer therapy. J. Med. Chem. 2019, 62 (7), 3171–3183. 10.1021/acs.jmedchem.8b00189. [DOI] [PubMed] [Google Scholar]
- Suraweera A.; O’Byrne K. J.; Richard D. J. Combination therapy with histone deacetylase inhibitors (HDACi) for the treatment of cancer: achieving the full therapeutic potential of HDACi. Front. Oncol. 2018, 8, 92. 10.3389/fonc.2018.00092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.; Seto E.. HDACs and HDAC inhibitors in cancer development and therapy. Cold Spring Harbor Perspect. Med. 2016, 6 ( (10), ), a026831. 10.1101/cshperspect.a026831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- https://en.wikipedia.org/wiki/Chidamide (accessed 2020-03-17).
- Perea S. E.; Baladrón I.; Valenzuela C.; Perera Y. CIGB-300: A peptide-based drug that impairs the Protein Kinase CK2-mediated phosphorylation. Semin. Oncol. 2018, 45 (1–2), 58–67. 10.1053/j.seminoncol.2018.04.006. [DOI] [PubMed] [Google Scholar]
- Masłyk M.; Janeczko M.; Martyna A.; Kubiński K. CX-4945: the protein kinase CK2 inhibitor and anti-cancer drug shows anti-fungal activity. Mol. Cell. Biochem. 2017, 435 (1–2), 193–196. 10.1007/s11010-017-3068-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trembley J. H.; Wang G.; Unger G.; Slaton J.; Ahmed K. Protein kinase CK2 in health and disease: CK2: a key player in cancer biology. Cell. Mol. Life Sci. 2009, 66 (11–12), 1858–67. 10.1007/s00018-009-9154-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao Y.; Chen T.; Yang H.; Chen Y.; Lin H.; Qu W.; Feng F.; Liu W.; Guo Q.; Liu Z.; Sun H. Small molecule modulators targeting protein kinase CK1 and CK2. Eur. J. Med. Chem. 2019, 181, 111581. 10.1016/j.ejmech.2019.111581. [DOI] [PubMed] [Google Scholar]
- Ni X.; Li L.; Pan G. HDAC inhibitor-induced drug resistance involving ATP-binding cassette transporters (Review). Oncol. Lett. 2015, 9 (2), 515–521. 10.3892/ol.2014.2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schobert R.; Biersack B. Multimodal HDAC inhibitors with improved anticancer activity. Curr. Cancer Drug Targets 2017, 18 (1), 39–56. 10.2174/1568009617666170206102613. [DOI] [PubMed] [Google Scholar]
- Hesham H. M.; Lasheen D. S.; Abouzid K. A. M. Chimeric HDAC inhibitors: Comprehensive review on the HDAC-based strategies developed to combat cancer. Med. Res. Rev. 2018, 38 (6), 2058–2109. 10.1002/med.21505. [DOI] [PubMed] [Google Scholar]
- https://clinicaltrials.gov/ct2/results?cond=&term=CUDC-101&cntry=&state=&city=&dist= (accessed 2020-03-17).
- https://clinicaltrials.gov/ct2/show/NCT02674750 (accessed 2020-03-17).
- https://clinicaltrials.gov/ct2/results?cond=&term=4SC-202&cntry=&state=&city=&dist= (accessed 2020-03-17).
- Khan D. H.; He S.; Yu J.; Winter S.; Cao W.; Seiser C.; Davie J. R. Protein kinase CK2 regulates the dimerization of histone deacetylase 1 (HDAC1) and HDAC2 during mitosis. J. Biol. Chem. 2013, 288 (23), 16518–28. 10.1074/jbc.M112.440446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pluemsampant S.; Safronova O. S.; Nakahama K.; Morita I. Protein kinase CK2 is a key activator of histone deacetylase in hypoxia-associated tumors. Int. J. Cancer 2008, 122 (2), 333–41. 10.1002/ijc.23094. [DOI] [PubMed] [Google Scholar]
- Patil H.; Wilks C.; Gonzalez R. W.; Dhanireddy S.; Conrad-Webb H.; Bergel M. Mitotic activation of a novel histone deacetylase 3-linker histone H1.3 protein complex by protein kinase CK2. J. Biol. Chem. 2016, 291 (7), 3158–72. 10.1074/jbc.M115.643874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H. S.; Chang Y. G.; Bae H. J.; Eun J. W.; Shen Q.; Park S. J.; Shin W. C.; Lee E. K.; Park S.; Ahn Y. M.; Park W. S.; Lee J. Y.; Nam S. W. Oncogenic potential of CK2α and its regulatory role in EGF-induced HDAC2 expression in human liver cancer. FEBS J. 2014, 281 (3), 851–61. 10.1111/febs.12652. [DOI] [PubMed] [Google Scholar]
- Chen M. C.; Chen C. H.; Chuang H. C.; Kulp S. K.; Teng C. M.; Chen C. S. Novel mechanism by which histone deacetylase inhibitors facilitate topoisomerase IIα degradation in hepatocellular carcinoma cells. Hepatology 2011, 53 (1), 148–59. 10.1002/hep.23964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H.; Song C.; Ding Y.; Pan X.; Ge Z.; Tan B. H.; Gowda C.; Sachdev M.; Muthusami S.; Ouyang H.; Lai L.; Francis O. L.; Morris C. L.; Abdel-Azim H.; Dorsam G.; Xiang M.; Payne K. J.; Dovat S. Transcriptional regulation of JARID1B/KDM5B histone demethylase by ikaros, histone deacetylase 1 (HDAC1), and casein kinase 2 (CK2) in B-cell acute lymphoblastic leukemia. J. Biol. Chem. 2016, 291 (8), 4004–18. 10.1074/jbc.M115.679332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purwin M.; Hernández-Toribio J.; Coderch C.; Panchuk R.; Skorokhyd N.; Filipiak K.; de Pascual-Teresa B.; Ramos A. Design and synthesis of novel dual-target agents for HDAC1 and CK2 inhibition. RSC Adv. 2016, 6 (71), 66595–66608. 10.1039/C6RA09717K. [DOI] [Google Scholar]
- Martínez R.; Geronimo B. D.; Pastor M.; Zapico J. M.; Coderch C.; Panchuk R.; Skorokhyd N.; Maslyk M.; Ramos A.; de Pascual-Teresa B. Multitarget Anticancer Agents Based on Histone Deacetylase and Protein Kinase CK2 inhibitors. Molecules 2020, 25 (7), 1497. 10.3390/molecules25071497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiqui-Jain A.; Drygin D.; Streiner N.; Chua P.; Pierre F.; O’Brien S. E.; Bliesath J.; Omori M.; Huser N.; Ho C.; Proffitt C.; Schwaebe M. K.; Ryckman D. M.; Rice W. G.; Anderes K. CX-4945, an orally bioavailable selective inhibitor of protein kinase CK2, inhibits prosurvival and angiogenic signaling and exhibits antitumor efficacy. Cancer Res. 2010, 70 (24), 10288–98. 10.1158/0008-5472.CAN-10-1893. [DOI] [PubMed] [Google Scholar]
- Battistutta R.; Cozza G.; Pierre F.; Papinutto E.; Lolli G.; Sarno S.; O’Brien S. E.; Siddiqui-Jain A.; Haddach M.; Anderes K.; Ryckman D. M.; Meggio F.; Pinna L. A. Unprecedented selectivity and structural determinants of a new class of protein kinase CK2 inhibitors in clinical trials for the treatment of cancer. Biochemistry 2011, 50 (39), 8478–88. 10.1021/bi2008382. [DOI] [PubMed] [Google Scholar]
- Schuetz A.; Min J.; Allali-Hassani A.; Schapira M.; Shuen M.; Loppnau P.; Mazitschek R.; Kwiatkowski N. P.; Lewis T. A.; Maglathin R. L.; McLean T. H.; Bochkarev A.; Plotnikov A. N.; Vedadi M.; Arrowsmith C. H. Human HDAC7 harbors a class IIa histone deacetylase-specific zinc binding motif and cryptic deacetylase activity. J. Biol. Chem. 2008, 283 (17), 11355–63. 10.1074/jbc.M707362200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somoza J. R.; Skene R. J.; Katz B. A.; Mol C.; Ho J. D.; Jennings A. J.; Luong C.; Arvai A.; Buggy J. J.; Chi E.; Tang J.; Sang B. C.; Verner E.; Wynands R.; Leahy E. M.; Dougan D. R.; Snell G.; Navre M.; Knuth M. W.; Swanson R. V.; McRee D. E.; Tari L. W. Structural snapshots of human HDAC8 provide insights into the class I histone deacetylases. Structure 2004, 12 (7), 1325–34. 10.1016/j.str.2004.04.012. [DOI] [PubMed] [Google Scholar]
- Lauffer B. E.; Mintzer R.; Fong R.; Mukund S.; Tam C.; Zilberleyb I.; Flicke B.; Ritscher A.; Fedorowicz G.; Vallero R.; Ortwine D. F.; Gunzner J.; Modrusan Z.; Neumann L.; Koth C. M.; Lupardus P. J.; Kaminker J. S.; Heise C. E.; Steiner P. Histone deacetylase (HDAC) inhibitor kinetic rate constants correlate with cellular histone acetylation but not transcription and cell viability. J. Biol. Chem. 2013, 288 (37), 26926–43. 10.1074/jbc.M113.490706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierre F.; Chua P. C.; O’Brien S. E.; Siddiqui-Jain A.; Bourbon P.; Haddach M.; Michaux J.; Nagasawa J.; Schwaebe M. K.; Stefan E.; Vialettes A.; Whitten J. P.; Chen T. K.; Darjania L.; Stansfield R.; Anderes K.; Bliesath J.; Drygin D.; Ho C.; Omori M.; Proffitt C.; Streiner N.; Trent K.; Rice W. G.; Ryckman D. M. Discovery and SAR of 5-(3-chlorophenylamino)benzo[c][2,6]naphthyridine-8-carboxylic acid (CX-4945), the first clinical stage inhibitor of protein kinase CK2 for the treatment of cancer. J. Med. Chem. 2011, 54 (2), 635–54. 10.1021/jm101251q. [DOI] [PubMed] [Google Scholar]
- Liu S.; Hsieh D.; Yang Y. L.; Xu Z.; Peto C.; Jablons D. M.; You L. Coumestrol from the national cancer Institute’s natural product library is a novel inhibitor of protein kinase CK2. BMC Pharmacol. Toxicol. 2013, 14, 36. 10.1186/2050-6511-14-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu C. C.; Pan S. L.; Chao S. W.; Liu S. P.; Hsu J. L.; Yang Y. C.; Li T. K.; Huang W. J.; Guh J. H. A novel small molecule hybrid of vorinostat and DACA displays anticancer activity against human hormone-refractory metastatic prostate cancer through dual inhibition of histone deacetylase and topoisomerase I. Biochem. Pharmacol. 2014, 90 (3), 320–30. 10.1016/j.bcp.2014.06.001. [DOI] [PubMed] [Google Scholar]
- Nikolić S.; Rangasamy L.; Gligorijević N.; Arand̵elović S.; Radulović S.; Gasser G.; Grgurić-Šipka S. Synthesis, characterization and biological evaluation of novel Ru(II)-arene complexes containing intercalating ligands. J. Inorg. Biochem. 2016, 160, 156–65. 10.1016/j.jinorgbio.2016.01.005. [DOI] [PubMed] [Google Scholar]
- Butler L. M.; Agus D. B.; Scher H. I.; Higgins B.; Rose A.; Cordon-Cardo C.; Thaler H. T.; Rifkind R. A.; Marks P. A.; Richon V. M. Suberoylanilide hydroxamic acid, an inhibitor of histone deacetylase, suppresses the growth of prostate cancer cells in vitro and in vivo. Cancer Res. 2000, 60 (18), 5165–70. [PubMed] [Google Scholar]
- Munster P. N.; Troso-Sandoval T.; Rosen N.; Rifkind R.; Marks P. A.; Richon V. M. The histone deacetylase inhibitor suberoylanilide hydroxamic acid induces differentiation of human breast cancer cells. Cancer Res. 2001, 61 (23), 8492–7. [PubMed] [Google Scholar]
- Leoni F.; Zaliani A.; Bertolini G.; Porro G.; Pagani P.; Pozzi P.; Donà G.; Fossati G.; Sozzani S.; Azam T.; Bufler P.; Fantuzzi G.; Goncharov I.; Kim S. H.; Pomerantz B. J.; Reznikov L. L.; Siegmund B.; Dinarello C. A.; Mascagni P. The antitumor histone deacetylase inhibitor suberoylanilide hydroxamic acid exhibits antiinflammatory properties via suppression of cytokines. Proc. Natl. Acad. Sci. U. S. A. 2002, 99 (5), 2995–3000. 10.1073/pnas.052702999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikora K.; Jaśkiewicz M.; Neubauer D.; Bauer M.; Bartoszewska S.; Barańska-Rybak W.; Kamysz W. Counter-ion effect on antistaphylococcal activity and cytotoxicity of selected antimicrobial peptides. Amino Acids 2018, 50 (5), 609–619. 10.1007/s00726-017-2536-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tipps M. E.; Iyer S. V.; John Mihic S. Trifluoroacetate is an allosteric modulator with selective actions at the glycine receptor. Neuropharmacology 2012, 63 (3), 368–73. 10.1016/j.neuropharm.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.