

Abstract
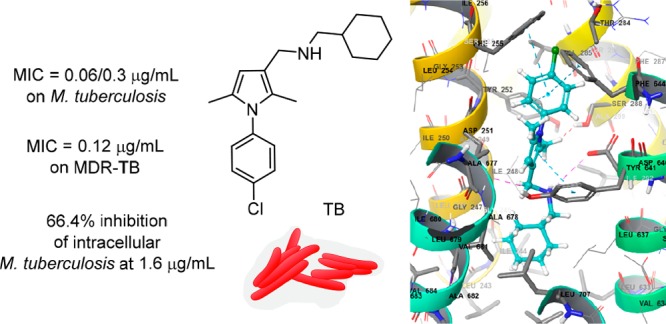
A series of N-phenyl-2,5-dimethylpyrrole derivatives, designed as hybrids of the antitubercular agents BM212 and SQ109, have been synthesized and evaluated against susceptible and drug-resistant mycobacteria strains. Compound 5d, bearing a cyclohexylmethylene side chain, showed high potency against M. tuberculosis including MDR-TB strains at submicromolar concentrations. The new compound shows bacteriostatic activity and low toxicity and proved to be effective against intracellular mycobacteria too, showing an activity profile similar to isoniazid.
Keywords: Tuberculosis, antimycobacterial drug, pyrroles, drug resistance, intracellular tuberculosis
The treatment of infectious diseases, especially those caused by bacterial pathogens such as Mycobacterium tuberculosis (MTB) responsible for many forms of tuberculosis (TB), is becoming a major challenge for the World Healthcare Systems.1 During the 20th century, there was a growing confidence that infectious diseases such as TB would have been quickly annihilated thanks to the discovery of efficient antitubercular drugs2 and the development of new therapeutic regimens which made tuberculosis a curable disease. However, the increasing ability of mycobacteria and other pathogens to adapt and develop resistance to almost all the known antibiotics undermined the initial optimism and forced us to move into the “postantibiotic era of infectious diseases”.3 As declared by the World Health Organization (WHO),4 MTB today represents a global health emergency5 due to the emergence of multi-, extensively-, and even totally-drug resistant tuberculosis (MDR-,6 XDR-,7 and TDR-TB,8 respectively) strains which make the disease difficult, if not impossible, to treat with currently available drugs.9 In addition, the ability of MTB to evade macrophage responses and to survive and replicate within those cells actually designed to kill invading microbes makes the disease even harder to be fully eradicated.10−12
The standard antitubercular therapy still relies on the use of drugs introduced in the clinic in the middle of the 20th century, with the exception of the fluoroquinolones. In the past decade, only two new drugs, bedaquiline,13 which nonetheless suffers from some major side effects,14 and pretomanid,15 have been approved for the treatment of TB, and only a limited number of drug candidates are currently undergoing clinical trials or preclinical development.16
For many years, drug discovery programs have relied heavily on target-based high-throughput screening (HTS) of large compound libraries. Nowadays, it is undeniable that such an approach suffers from limitations and that deficiencies in current compound collections have emerged, as shown by the continuing decline of success in drug discovery. With the aim to identify new anti-TB treatments, we recently adopted an alternative strategy which led to the discovery of the N-aryl-2,5-dimethylpyrroles 1 and 2 as hybrid derivatives of the antitubercular drugs BM212 and SQ10917 (Figure 1).
Figure 1.

Pyrrole hybrid derivatives 1 and 2 and the rational approach of the proposed work.
Compounds 1 and 2 showed good activity against a panel of mycobacteria and an improved selectivity index (SI) compared to the parent compounds. However, in our previous work, only a limited series of pyrrole analogues were designed and only a few structure-activity relationships (SARs) were evaluated. In addition, no studies on the activity of these compounds on intramacrophagic mycobacteria were carried out. Herein, we describe the design, synthesis, and biological evaluation of a series of N-aryl-2,5-dimethylpyrrole derivatives with improved potency against susceptible, drug-resistant, and intracellular mycobacteria. The chemical space around the 2,5-dimethylpyrrole nucleus has been explored through the replacement of the cyclohexyl and benzyl rings of 1–2 with different aromatic or aliphatic substituents. This chemical modification was carried out with the aim to confer on the new compounds a sufficient degree of lipophilicity to penetrate mycobacteria and macrophages and, in turn, inhibit intracellular mycobacteria themselves. The effect of electron-withdrawing and electron-donating substituents on the N-aryl ring on the antimycobacterial activity was also evaluated, as well as the role of the pyrrole nucleus. A series of analogues bearing pyrazole, triazole, and imidazopyridine rings in place of the 2,5-dimethylpyrrole core was also designed and synthesized (Figure 1).
A series of pyrrole derivatives 5a–q was first synthesized according to Scheme 1 (path a).18 The 2,5-hexandione was reacted with different anilines to afford N-arylpyrroles 3. The latter were formylated and then treated with appropriate amines in the presence of Na(AcO)3BH as reducing agent to give the desired products 5a–q in good overall yields. Compounds 5a–d were designed with the aim to evaluate the effect of different substituents on the C3 of the pyrrole ring other than cyclohexyl and benzyl moieties.
Scheme 1. Synthesis of Antimycobacterial Derivatives 5, 7, 9, and 11.

For compounds 5e–q, the side chain in C3 was left unchanged and the effects of substituents on the N-phenyl ring were investigated. The pyrrole ring of 1 and 2 was then replaced with different heterocyclic rings leading to derivatives 7, 9, and 11. Compounds 7a–c have a pyrazole ring and were designed taking into account our previous works on antitubercular pyrazolones (Scheme 1, path b).19 The reaction of phenylhydrazine and p-chlorophenylhydrazine with the 2,4-pentadione led to pyrazoles 6a–b, which were in turn formylated and treated with different amines under reductive amination conditions affording the desired pyrazoles 7a–c. The pyrrole nucleus of 1–2 was then replaced by a triazole ring in derivatives 9a–b (Scheme 1, path c). The p-chloroaniline was converted into the corresponding azide, which was then reacted with N-Boc-propargylamine via a Huisgen 1,3-dipolar cycloaddition reaction in the presence of CuSO4 and Na-ascorbate. The resulting triazole 8 was then deprotected and reacted with benzaldehyde or cyclohexanone in the presence of Na(AcO)3BH affording the desired triazole derivatives 9a–b. Finally, the rigidified bicyclic analogues 11a–b were designed and synthesized from 5-chloro-2-aminopyridine (Scheme 1, path d). This latter compound was reacted with ethyl bromopyruvate to afford the bicyclic ester 10, which was in turn converted into 11a–b via DIBAL-H reduction followed by reductive amination with the appropriate amine. The structures of compounds 5, 7, 9, and 11 are reported in Table 1.
Table 1. Structures of Antimycobacterial Derivatives 5, 7, 9, and 11.

| Cmpd | R | R1 |
|---|---|---|
| 5a | Cl | 2-norbornyl |
| 5b | Cl | α-methylbenzyl |
| 5c | Cl | phenylaminoethyl |
| 5d | Cl | cyclohexanemethyl |
| 5e | 3,5-di-Cl | cyclohexyl |
| 5f | 2,5-di-Me | cyclohexyl |
| 5g | 2-CF3 | cyclohexyl |
| 5h | 4-F | cyclohexyl |
| 5i | 4-iPr | cyclohexyl |
| 5j | 2-F | cyclohexyl |
| 5k | 4-CN | cyclohexyl |
| 5l | 4-OMe | cyclohexyl |
| 5m | 2,5-di-Me | benzyl |
| 5n | 3,5-di-Cl | benzyl |
| 5o | 4-iPr | benzyl |
| 5p | 4-F | benzyl |
| 5q | 2-CF3 | benzyl |
| 7a | H | cyclohexyl |
| 7b | 4-Cl | cyclohexyl |
| 7c | 4-Cl | benzyl |
| 9a | cyclohexyl | |
| 9b | benzyl | |
| 11a | cyclohexyl | |
| 11b | benzyl |
In order to quickly identify potential candidates for testing against drug-resistant and intracellular mycobacteria, compounds 5a–q were initially screened against M. bovis BCG by determining the minimum inhibitory concentrations (MIC). Results are reported in Table 2. As a general trend, all the compounds 5a–q showed good to excellent antimycobacterial activity with MIC values in the range of 0.125–2 μg/mL, thus underscoring the significance of the N-phenyl-2,5-dimethylpyrrole scaffold as a template for the development of antitubercular agents. SAR considerations showed that the presence of bulky, aliphatic and lipophilic substituents on the methyleneamine side chain on the C3 of the pyrrole nucleus is fundamental to improve the antimycobacterial activity. In fact, 5e–l showed a better biological profile than the corresponding 5m–q bearing a benzyl group in place of a cyclohexyl moiety. Also, the replacement of the cyclohexyl of 1 with the larger and more lipophilic 2-norbornyl and cyclohexanemethyl substituents led to 5a and 5d, respectively, which showed excellent and improved activity against M. bovis BCG (MIC = 0.25 μg/mL for 5a and 0.125 μg/mL for 5d). The introduction of a more hydrophilic phenylaminoethyl chain resulted in a loss of activity in 5c while 5b, bearing an α-methylbenzyl group, showed a similar biological profile to 2. When the chlorine atom on the N-aryl ring of 2 was replaced with various substituents (5m–q), a decrease in activity was observed. Only the derivative 5p bearing a fluorine in place of the chlorine showed a similar biological profile (MIC = 0.5 μg/mL) to 2. The series of compounds 5e–l, bearing a cyclohexyl group on C3 and different substituents on the N-phenyl ring, showed excellent activity instead with the only exceptions of derivatives 5g and 5k bearing the electron-withdrawing groups −CF3 and −CN, respectively. It is evident that the electronic density on the N-phenyl ring has a crucial role on the antimycobacterial activity of the pyrroles 5 as discussed later.
Table 2. Preliminary Screening and Antimycobacterial Activity of 5a–q against M. bovis BCG.
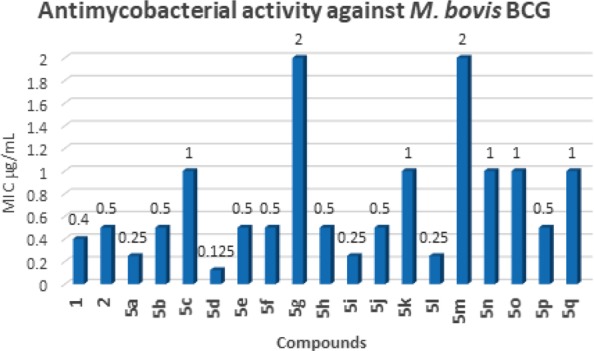
These data are in perfect agreement with previous pharmacophoric hypotheses.19−21 As further confirmation, the 5i and 5l, bearing the electron-donating substituents -iPr and -OMe, respectively, showed excellent MIC = 0.25 μg/mL.
Four compounds, namely 5a, 5d, 5i, and 5l, were then selected from the first screening and further assayed on M. tuberculosis H37Rv as well as on clinical MDR and TB sensitive strains. The results are reported in Table 3. The clinical MDR strain CCM11.1 is identified as lineage 2 Beijing strain resistant to isoniazid, rifampicin, ethambutol, and ethionamide, while the sensitive clinical strain CCM10.1 is identified as lineage 2 Indian strain with no known resistances. Both strains were obtained from clinical isolates. Compounds 5a, 5i, and 5l showed activity against MTB H37Rv similar to that observed against M. bovis BCG with MIC values ranging between 0.12 and 0.5 μg/mL. Derivative 5d showed an excellent profile against M. tuberculosis H37Rv with MIC = 0.06–0.3 μg/mL as well low cytotoxicity with an excellent Selectivity Index (SI). All four compounds also showed excellent activity against CCM11.1 and CCM10.1 TB strains. In particular, 5d proved to be a potent inhibitor of the MDR TB strain CCM11.1 with MIC of 0.12 μg/mL and the sensitive clinical strain CCM10.1 with MIC < 0.625 ng/mL.
Table 3. Biological Evaluation of Compounds against Bacterial and Intracellular Strains.
|
M. tuberculosis MIC (μg/mL) |
|||||
|---|---|---|---|---|---|
| CCM | CCM | ||||
| Cmpd | H37Rva | 11.1a | 10.1a | GIC50 | SIb |
| 5a | 0.5–0.25 | 1 | 0.0025 | 23.4 | 47–94 |
| 5d | 0.06a-0.3c | 0.12 | <0.000625 | 11.7 | 195–39 |
| 5i | 0.25–0.12 | 0.5 | 0.00125 | 5.8 | 23–48 |
| 5l | 0.25–0.12 | 0.5 | <0.000625 | 3.9 | 16–32 |
| INH | 0.24 | ||||
| Rif | >0.06 | ||||
M. tuberculosis clinical strains from Royal Free Hospital NHS Trust, London.
The selectivity index is calculated as the ratio of the GIC50 and the MIC observed against M. tuberculosis H37Rv.
MIC obtained from clinical strains from the Tuberculosis Research Laboratory, Sao Paulo.22
Finally, the nonpyrrole derivatives 7a–c, 9a–b, and 11a–b were tested against M. tuberculosis H37Rv strain. However, all these derivatives showed poor activity (MIC > 25 μg/mL) and were thus excluded from further studies. However, these data confirm the key role of the 2,5-dimethylpyrrole nucleus for antitubercular activity.
Encouraged by the biological profile displayed by 5d, additional assays were conducted to assess its toxicity and intracellular activity. The acute toxicity of 5din vivo in Galleria mellonella larvae was first investigated. At the administered dose of 12 mg/kg, the pyrrole 5d elicited no apparent toxic effects on the wax moth larvae and no signs of melanization were observed. This promising data confirmed the low toxicity of 5d and its suitability for further development.23 A time-kill assay was then performed to understand if the most active compound 5d was bactericidal or bacteriostatic at a determined concentration (Figure 2).24 The bactericidal activity is characterized by 3 Log10 CFU/mL or 99.9% reduction of the bacterial inoculum. Pyrrole 5d presents a bacteriostatic effect at 0.32 μg/mL while the reference antibiotic moxifloxacin (MOX) did not inhibit the MTB growth at 0.07 μg/mL but showed bactericidal effect at 0.35 μg/mL. The bactericidal and bacteriostatic effect is dependent on the concentration.
Figure 2.
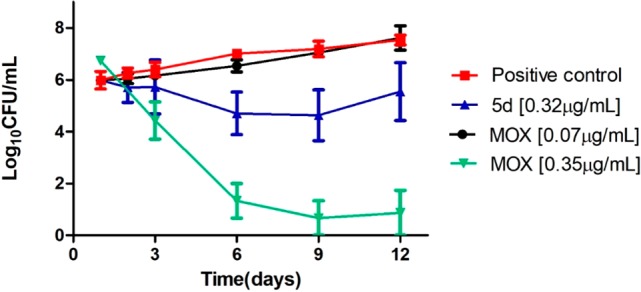
Time-kill of MTB H37Rv according to time (days) in the presence of 5d (0.32 μg/mL) or moxifloxacin (MOX – 0.07 μg/mL) or untreated (growth control).
Since the MTB is able to survive inside the macrophages, a set of experiments to evaluate the intramacrophagic activity of 5d was then planned. The inhibition of intracellular MTB by 5d was initially assayed at concentrations higher than or equal to MIC values and compared with those of isoniazid (INH) and moxifloxacin (MOX).25 The concentrations tested were chosen to not be toxic to macrophages within 72 h of the assay. The data are reported in Figure 3a and clearly show that the intramacrophage activity of 5d is comparable to that of INH and MOX. Even if this experiment is not able to confirm if the activity of 5d is directly based on the elimination of the bacillus once it penetrates the cellular barriers or if 5d is able to enhance the macrophage killing action, it clearly proves the ability of 5d to act on intramacrophagic mycobacteria at low concentrations.
Figure 3.
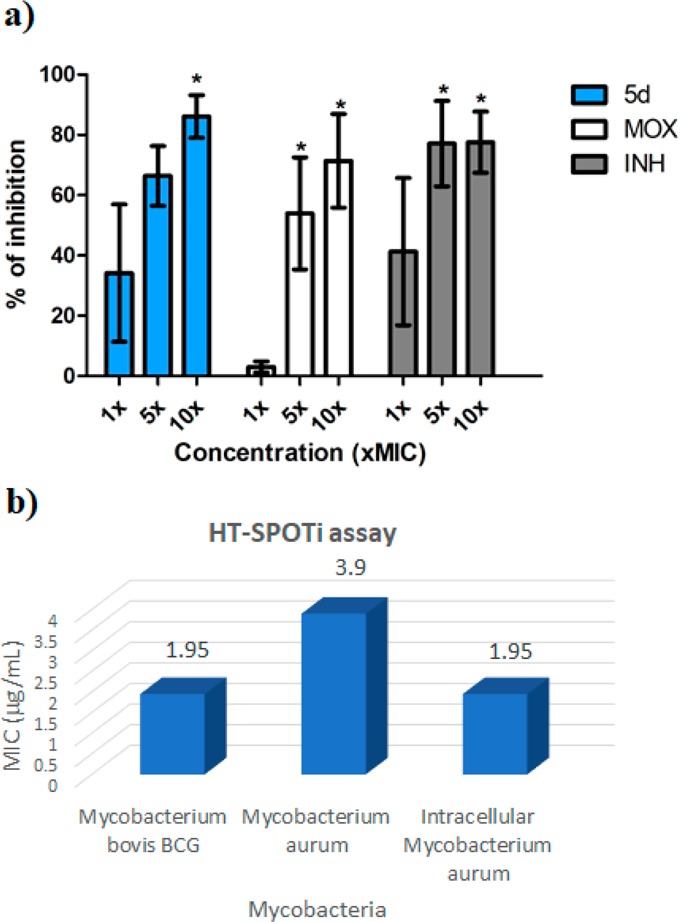
(a) Percent inhibition of intramacrophage MTB (Cell line J774A.1, murine origin) in the presence of 5d, moxifloxacin (MOX), or isoniazid (INH). Concentrations: 5d (1 x MIC = 0.32 μg/mL; 5 x MIC = 1.6 μg/mL, and 10 x MIC = 3.2 μg/mL); INH (1 x MIC = 0.06 μg/mL; 5 x MIC = 0.30 μg/mL, and 10 x MIC = 0.60 μg/mL); MOX (1 x MIC = 0.07 μg/mL; 5 x MIC = 0.35 μg/mL, and 10 x MIC = 0.70 μg/mL). The results are the mean and standard error of experiments performed in duplicate. * = Statistically different from the untreated control (P < 0.05) by GraphPad Prism (One-way ANOVA with Dunnett’s Multiple Comparison Post Test). (b) Intracellular activity of 5d via HT-SPOTi assay.
As further confirmation of the intramacrophagic activity and to estimate the intracellular MIC of compound 5d, an intracellular HT-SPOTi assay on Mycobacterium aurum was performed. The HT-SPOTi is an assay principally based on the growth of an organism on agar medium containing a range of different concentrations of drugs or inhibitors, and it can be employed to screen the antimicrobial potency with high reliability and reproducibility. The in vitro activity of 5d on M. bovis BCG and M. aurum using HT-SPOTi was first investigated. The pyrrole 5d showed good inhibition of both species at micromolar concentration (1.95 and 3.9 μg/mL, respectively). The higher activity observed is related to the assay conditions, which, however, are able to better mimic the conditions at which mycobacteria survive in vivo. The activity of 5d on intracellular M. aurum was then assayed, showing inhibition at a concentration (1.95 μg/mL) lower than that of free mycobacteria (Figure 3b). This data further corroborates the potentiality of 5d as promising antitubercular agent.
Finally, in silico studies were carried out in an attempt to explain the mode of action of 5. Both the parent compounds of hybrids 5, namely BM212 and SQ109, have been demonstrated to inhibit MTB by binding to the mycolic acid transporter MmpL3. Due to the structural similarity between the hybrid derivative 5 and BM212, we hypothesized a similar mode of action for both compounds and, with the aim to understand their interactions with MmpL3, a molecular docking study was carried out.
The crystal structure of MmpL3 from M. smegmatis is available in the RCSB Protein Data Bank (PDB, 6AJI, 6AJH, and 6AJJ)26 and was used to generate a homology model for the MTB MmpL3 transporter. Subsequently, the pyrrole derivative 5d was docked into the MTB MmpL3 homology model and compared to BM212 and SQ109 binding poses. The N1 piperazine nitrogen of BM212 shows a charge-reinforced hydrogen bond interaction with the carboxyl terminal group of Asp640, which is known to be involved in the electrochemical proton gradient for the MmpL3 transporter.27 Similarly, SQ109 binds MmpL3 in a such a way that its secondary amine groups form two charge-reinforced hydrogen bonds with the carboxyl moiety of Asp640. Similar interactions with Asp251 further stabilize the binding. Moreover, the adamantyl moiety forms hydrophobic interactions with Tyr252, Phe255, and Phe244, while the geranyl side chain interacts with Leu633, Ser295, Ala296, Ala632, and Thr314. Finally, most conformers of 5d displayed a binding mode similar to that of BM212 with the cyclohexyl ring accommodated within the region of space where the piperazine ring of BM212 was located. In detail, the cyclohexyl ring of 5d was embedded within the hydrophobic region constituted by Ile674, Phe644, Ile673, and Ala677. A charge-reinforced hydrogen bond between its secondary amine moiety and Asp640 is observed, as already found for BM212 and SQ109, thus suggesting this interaction of pivotal importance for binding ligands to MmpL3. In addition, the N1-chlorophenyl side chain makes hydrophobic contacts with Leu243, Ala685, Ile243, and Val684. The main structural difference between 5d and BM212 is represented by the presence of a methyl group on C5 in place of a 4-chlorophenyl group. This simple structural modification leads 5d to have a second binding mode in the MmpL3 binding pocket with a different orientation than BM212 as shown in Figure 4. In the inverted binding mode (Figure 4, left), the N1-chlorophenyl moiety of 5d replaces the piperazine of BM212 in the binding pocket. This orientation of 5d allows the pyrrole ring to form an extra π–π interaction with Tyr641, which is also reported to be involved in the electrochemical proton gradient with the MmpL3 transporter. Moreover, a π–π interaction between the 4-chlorobenzene ring of 5d and Tyr252 can also be observed in one of the MmpL3 subpockets. Tyr252 is a highly conserved residue among MmpL3 isophorms and is located with proximity to Gly253, which has been shown to mutate with exposure to SQ109 resulting in a decreased inhibition. Further reinforcement in the binding of aromatic portions of 5d to MmpL3 is favored by the π–π stacking interactions with Phe255 and Phe644, as well as by hydrophobic interactions with Val285 and Ala677. The secondary amine group of 5d forms a hydrogen bond with the carbonyl group of Asp640 and Asp251. Finally, a hydrophobic subpocket of the MmpL3 binding site is able to accommodate the cyclohexyl ring of 5d that interacts with Ile244, Leu703, Leu633, Ile248, and Gly247. The complexes formed by 5d with the MmpL3 model provide a compelling validation of the ability of the protein to accommodate the binding of dimethyl pyrrole structures. Despite the lack of a second phenyl ring, the pyrrole 5d is still capable of binding in the MmpL3 binding site. This would confirm that the presence of a second phenyl substituent plays a marginal role in the interactions of pyrrole derivatives with MmpL3. Docking simulations of 5d have shown that it may bind to MmpL3 through two binding modes, even though with different relevance in terms of both docking scores and relative abundance. Docking calculations of 5a, which differs from 5d for the presence of a norbornyl side chain, were also performed. The secondary amine provides stabilization through a hydrogen bond with Asp640 and a charge-reinforced hydrogen bond with Asp251, as found in 5d.
Figure 4.
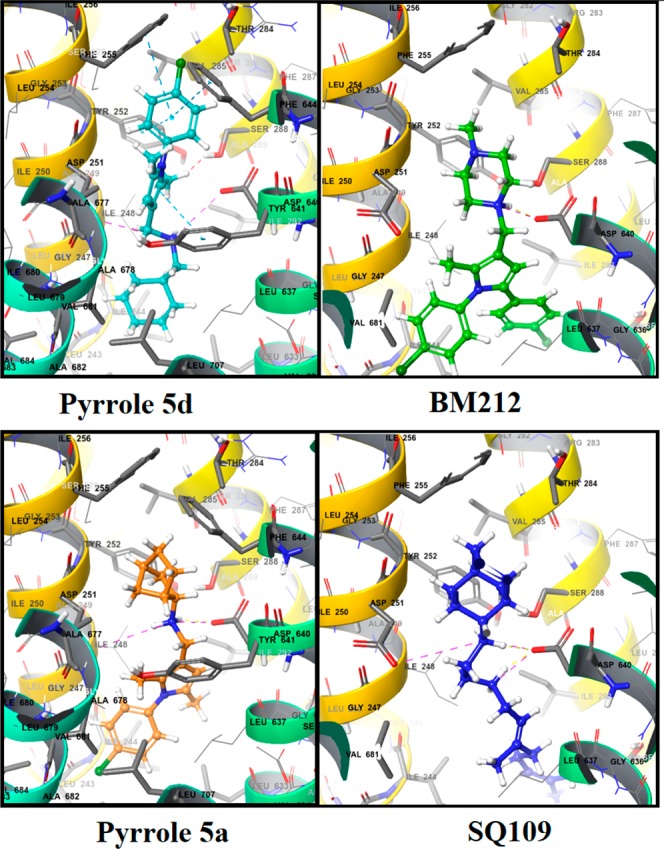
Binding of 5d (top left), 5a (bottom left), BM212 (top right), and SQ109 (bottom right) with the M. tuberculosis MmpL3 homology model.
However, the binding mode of 5a is inverted with respect to 5d, with the norbornyl ring placed in the position of the 4-chlorophenyl moiety. This allows 5d to form π–π stacking interactions with Phe255 and Tyr252 that are not observed with 5a and can explain the higher antitubercular activity of 5d in comparison with 5a and other compounds. The inverted binding mode for 5a could be explained by the bulkier and more rigid nature of the norbornyl ring that is sterically unfavored within the pocket defined by Leu243, Ile244, and Leu744. Docking of 5i and 5l showed a binding mode similar to that of 5a with interactions between the isopropyl and methoxy groups with the hydrophobic subpocket (Figure 5). This confirms that minimal structural modifications can change the binding mode of the different pyrrole analogues, possibly contributing to modulate their antitubercular activity.
Figure 5.
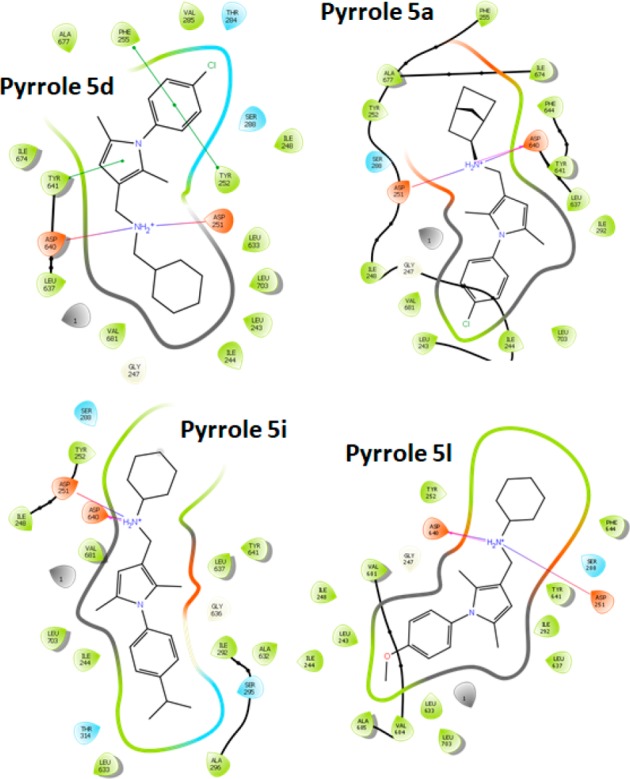
Binding interactions of pyrroles 5a, 5d, 5i, and 5l.
In conclusion, a new series of N-aryl-2,5-dimethylpyrrole derivatives has been synthesized and evaluated as antitubercular agents. Compound 5d, bearing a cyclohexylmethylene side chain, showed an improved activity profile when compared to the parent drug BM212 and low toxicity, both in vitro as well as in preliminary in vivo assay on Galleria mellonella models. Pyrrole 5d also showed bacteriostatic and intracellular activity against M. tuberculosis at concentrations similar to antibiotics moxifloxacin and isoniazid as well as inhibitory activity of the MDR strain CCM11.1. Finally, computational studies suggest that the mycolic acid transporter MmpL3 is a plausible target for 5d, which binds the protein similarly to BM212 and SQ109. Further studies to confirm this last hypothesis are in progress.
Acknowledgments
We gratefully acknowledge EPSRC (Global Challenges Competition King’s College London), Global Challenges Research Fund at Birkbeck, University of London and Royal Society (RG160870) for research funding and financial support. MT acknowledge King’s College London for a period of leave. DS acknowledges the South African National Research Foundation-SARChI for financial support. We gratefully acknowledge NC3Rs (National Centre for the Replacement, Refinement and Reduction of Animals in Research, NC/T001240/1) for financial support and Carolyn Lam and Simona di Blasio for their help and training on Galleria mellonella assays. FRP and CMR acknowledge Sao Paulo Research Foundation (FAPESP - grant 2018/00163-0) for financial support.
Glossary
Abbreviations
- TB
tuberculosis
- MTB
Mycobacterium tuberculosis
- MDR
multidrug resistant
- XDR
extensively drug-resistant
- TDR
totally drug-resistant
- HTS
high-throughput screening
- SARs
structure-activity relationships
- BCG
Bacillus Calmette–Guérin
- MIC
minimum inhibitory concentration
- PDB
Protein Data Bank.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.9b00515.
General procedures for the synthesis and biological evaluation of the compounds. Full characterization of compounds 5, 7, 9, and 11. (PDF)
Author Present Address
∇ (A.G.) Faculty of Infectious Diseases, London School of Hygiene & Tropical Medicine, London WC1E 7HT, United Kingdom.
The authors declare no competing financial interest.
Dedication
This manuscript is dedicated to the memory of Prof. Maurizio Botta, to whose research, teachings, vision and ability to always look beyond the obvious, we all owe much. Thank you, Prof!
Supplementary Material
References
- World Health Organization . Antimicrobial resistance: global report on surveillance, 2014. https://www.who.int/drugresistance/documents/surveillancereport/en (December 10, 2019). [Google Scholar]
- Fox W.; Ellard G. A.; Mitchison D. A. Studies on the treatment of tuberculosis undertaken by the British Medical Research Council Tuberculosis Units, 1946–1986, with relevant subsequent publications. Int. J. Tuberc Lung Dis. 1999, 3, 231–279. [PubMed] [Google Scholar]
- Howard S. J.; Catchpole M.; Watson J.; Davies S. C. Antibiotic resistance: global response needed. Lancet Infect. Dis. 2013, 13, 1001–1003. 10.1016/S1473-3099(13)70195-6. [DOI] [PubMed] [Google Scholar]
- World Health Organization . Tuberculosis. http://www.who.int/en/news-room/fact-sheets/detail/tuberculosis (December 2, 2019).
- Grange J. M.; Zumla A. J. The global emergency of tuberculosis: what is the cause?. J. R. Soc. Promot. Health 2002, 122, 78–81. 10.1177/146642400212200206. [DOI] [PubMed] [Google Scholar]
- Eker B.; Ortmann J.; Migliori G. B.; Sotgiu G.; Muetterlein R.; Centis R.; Hoffmann H.; Kirsten D.; Schaberg T.; Ruesch-Gerdes S.; Lange C. Multidrug- and extensively drug-resistant tuberculosis, Germany. Emerging Infect. Dis. 2008, 14, 1700–1706. 10.3201/eid1411.080729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migliori G. B.; Loddenkemper R.; Blasi F.; Raviglione M. C. 125 years after Robert Koch’s discovery of the tubercle bacillus: the new XDR-TB threat. Is ″science″ enough to tackle the epidemic?. Eur. Respir. J. 2007, 29, 423–427. 10.1183/09031936.00001307. [DOI] [PubMed] [Google Scholar]
- Velayati A. A.; Farnia P.; Masjedi M. R. The totally drug resistant tuberculosis (TDR-TB). Int. J. Clin. Exp. Med. 2013, 6, 307–309. [PMC free article] [PubMed] [Google Scholar]
- Caminero J. A.; Sotgiu G.; Zumla A.; Migliori G. B. Best drug treatment for multidrug-resistant and extensively drug-resistant tuberculosis. Lancet Infect. Dis. 2010, 10, 621–629. 10.1016/S1473-3099(10)70139-0. [DOI] [PubMed] [Google Scholar]
- Hartkoorn R. C.; Chandler B.; Owen A.; Ward S. A.; Bertel Squire S.; Back D. J.; Khoo S. H. Differential drug susceptibility of intracellular and extracellular tuberculosis, and the impact of P-glycoprotein. Tuberculosis 2007, 87, 248–255. 10.1016/j.tube.2006.12.001. [DOI] [PubMed] [Google Scholar]
- Chanwong S.; Maneekarn N.; Makonkawkeyoon L.; Makonkawkeyoon S. Intracellular growth and drug susceptibility of Mycobacterium tuberculosis in macrophages. Tuberculosis 2007, 87, 130–133. 10.1016/j.tube.2006.06.001. [DOI] [PubMed] [Google Scholar]
- Gupta A.; Kaul A.; Tsolaki A. G.; Kishore U.; Bhakta S. Mycobacterium tuberculosis: Immune evasion, dormancy and resuscitation. Immunobiology 2012, 217, 363–374. 10.1016/j.imbio.2011.07.008. [DOI] [PubMed] [Google Scholar]
- Diacon A. H.; Pym A.; Grobusch M.; Patientia R.; Rustomjee R.; Page-Shipp L.; Pistorius C.; Krause R.; Bogoshi M.; Churchyard G.; Venter A.; Allen J. The diarylquinoline TMC207 for multidrug-resistant tuberculosis. N. Engl. J. Med. 2009, 360, 2397–405. 10.1056/NEJMoa0808427. [DOI] [PubMed] [Google Scholar]
- Pontali E.; Sotgiu G.; D’Ambrosio L.; Centis R.; Migliori G. B. Bedaquiline and multidrug-resistant tuberculosis: a systematic and critical analysis of the evidence. Eur. Respir. J. 2016, 47, 394–402. 10.1183/13993003.01891-2015. [DOI] [PubMed] [Google Scholar]
- Lenaerts A. J.; Gruppo V.; Marietta K. S.; Johnson C. M.; Driscoll D. K.; Tompkins N. M.; Rose J. D.; Reynolds R. C.; Orme I. M. Preclinical testing of the nitroimidazopyran PA-824 for activity against Mycobacterium tuberculosis in a series of in vitro and in vivo models. Antimicrob. Agents Chemother. 2005, 49, 2294–301. 10.1128/AAC.49.6.2294-2301.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiberi S.; du Plessis N.; Walzl G.; Vjecha M. J.; Rao M.; Ntoumi F.; Mfinanga S.; Kapata N.; Mwaba P.; McHugh T. D.; Ippolito G.; Migliori G. B.; Maeurer M. J.; Zumla A. Tuberculosis: progress and advances in development of new drugs, treatment regimens, and host-directed therapies. Lancet Infect. Dis. 2018, 18, 183–198. 10.1016/S1473-3099(18)30110-5. [DOI] [PubMed] [Google Scholar]
- Bhakta S.; Scalacci N.; Maitra A.; Brown A. K.; Dasugari S.; Evangelopoulos D.; McHugh T.; Mortazavi P. N.; Twist A.; Petricci E.; Manetti F.; Castagnolo D. Design and synthesis of 1-((1,5-bis(4-chlorophenyl)-2-methyl-1H-pyrrol-3-yl)methyl)-4-methylpiperazine (BM212) and N-Adamantan-2-yl-N’-((E)-3,7-dimethyl-octa-2,6-dienyl)-ethane-1,2-diamine (SQ109) pyrrole hybrid derivatives: discovery of potent anti-tubercular agents effective against multi-drug resistant mycobacteria. J. Med. Chem. 2016, 59, 2780–2793. 10.1021/acs.jmedchem.6b00031. [DOI] [PubMed] [Google Scholar]
- Masci D.; Hind C.; Islam M. K.; Toscani A.; Clifford M.; Coluccia A.; Conforti I.; Touitou M.; Memdouh S.; Wei X.; La Regina G.; Silvestri R.; Sutton J. M.; Castagnolo D. Switching on the activity of 1,5-diaryl-pyrrole derivatives against drug-resistant ESKAPE bacteria: Structure-activity relationships and mode of action studies. Eur. J. Med. Chem. 2019, 178, 500–514. 10.1016/j.ejmech.2019.05.087. [DOI] [PubMed] [Google Scholar]
- Manetti F.; Magnani M.; Castagnolo D.; Passalacqua L.; Botta M.; Corelli F.; Saddi M.; Deidda D.; De Logu A. Ligand-Based Virtual Screening, Parallel Solution-Phase and Microwave-Assisted Synthesis as Tools to Identify and Synthesize New Inhibitors of Mycobacterium tuberculosis. ChemMedChem 2006, 1, 973–989. 10.1002/cmdc.200600026. [DOI] [PubMed] [Google Scholar]
- Manetti F.; Corelli F.; Biava M.; Fioravanti R.; Porretta G. C.; Botta M. Building a pharmacophore model for a novel class of antitubercular compounds. Farmaco 2000, 55, 484–491. 10.1016/S0014-827X(00)00072-0. [DOI] [PubMed] [Google Scholar]
- Biava M.; Porretta G. C.; Poce G.; De Logu A.; Saddi M.; Meleddu R.; Manetti F.; De Rossi E.; Botta M. 1,5-Diphenylpyrrole derivatives as antimycobacterial agents. Probing the influence on antimycobacterial activity of lipophilic substituents at the phenyl rings. J. Med. Chem. 2008, 51, 3644–3648. 10.1021/jm701560p. [DOI] [PubMed] [Google Scholar]
- The different activity observed is imputable to the different strains and different methods used in London and Sao Paulo.
- Details of the assay are reported in the Supporting Information. The preliminary observations of toxicity in vivo will be followed by further testing at higher doses and will be later corroborated by in vivo experimentation in small mammals. These studies are in progress and will be published later.
- de Steenwinkel J. E. M.; de Knegt G. J.; Kate M. T.; Belkum A. Van; Verbrugh H. A.; Kremer K.; Van Soolingen D.; Bakker-Woudenberg I. A. J. M. Time–Kill Kinetics Of Anti-Tuberculosis Drugs, And Emergence Of Resistance, In Relation To Metabolic Activity Of Mycobacterium tuberculosis. J. Antimicrob. Chemother. 2010, 65, 2582–2589. 10.1093/jac/dkq374. [DOI] [PubMed] [Google Scholar]
- Dos Santos Fernandes G. F.; De Souza P. C.; Moreno-Viguri E.; Santivañez-Veliz M.; Paucar R.; Pérez-Silanes S.; Chegaev K.; Guglielmo S.; Lazzarato L.; Fruttero R.; Man Chin C.; Da Silva P. B.; Chorilli M.; Solcia M. C.; Ribeiro C. M.; Silva C. S. P.; Marino L. B.; Bosquesi P. L.; Hunt D. M.; De Carvalho L. P. S.; De Souza Costa C. A.; Cho S. H.; Wang Y.; Franzblau S. G.; Pavan F. R.; Dos Santos J. L. Design, Synthesis, And Characterization Of N-Oxide-Containing Heterocycles With In Vivo Sterilizing Antitubercular Activity. J. Med. Chem. 2017, 60, 8647–8660. 10.1021/acs.jmedchem.7b01332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B.; Li J.; Yang X.; Wu L.; Zhang J.; Yang Y.; Zhao Y.; Zhang L.; Yang X.; Yang X.; Cheng X.; Liu Z.; Jiang B.; Jiang H.; Guddat L. W.; Yang H.; Rao Z. Crystal Structures of Membrane Transporter MmpL3, an Anti-Tb Drug Target. Cell 2019, 176, 636–648. 10.1016/j.cell.2019.01.003. [DOI] [PubMed] [Google Scholar]
- Li W.; Upadhyay A.; Fontes F. L.; North E. J.; Wang Y.; Crans D. C.; Grzegorzewicz A. E.; Jones V.; Franzblau S. G.; Lee R. E.; Crick D. C.; Jackson M. Novel insights into the mechanism of inhibition of MmpL3, a target of multiple pharmacophores in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2014, 58, 6413–6423. 10.1128/AAC.03229-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.