

Abstract

A series of 3-methyl-2-phenyl-1H-indoles was prepared and investigated for antiproliferative activity on three human tumor cell lines, HeLa, A2780, and MSTO-211H, and some structure–activity relationships were drawn up. The GI50 values of the most potent compounds (32 and 33) were lower than 5 μM in all tested cell lines. For the most biologically relevant derivatives, the effect on human DNA topoisomerase II relaxation activity was investigated, which highlighted the good correlation between the antiproliferative effect and topoisomerase II inhibition. The most potent derivative, 32, was shown to induce the apoptosis pathway. The obtained results highlight 3-methyl-2-phenyl-1H-indole as a promising scaffold for further optimization of compounds with potent antiproliferative and antitopoisomerase II activities.
Keywords: 3-Methyl-2-phenyl-1H-indole, antiproliferative activity, topoisomerase II, apoptosis
Cancer is one of the leading causes of mortality worldwide, and its incidence is rapidly growing.1,2 The use of conventional chemotherapeutic agents, such as alkylating cytostatics, nucleoside analogues, anthracyclines, and compounds that stabilize microtubules is often limited because of their severe side effects. Drugs such as tyrosine kinase inhibitors and monoclonal antibodies that target specific signaling oncoproteins have been developed to increase specificity and thus to reduce side effects, but these can also have limitations, such as development of resistance.3 Despite the considerable progress made with new targeted therapies, for many patients the therapeutic options remain limited. Thus, new chemotherapeutics with improved pharmaco-toxicological profiles are urgently needed.4
One of the important targets for anticancer drug discovery is human DNA topoisomerase II (topo II).3,5−9 Topoisomerases are enzymes that catalyze changes in DNA topology, and they are crucial for processes such as DNA replication and transcription and chromosomal segregation. They introduce transient breaks in the DNA molecule by cleaving one (type I topoisomerases) or both (type II topoisomerases) strands.10,11 Topo II is a type II topoisomerase, and two isoforms, α and β, are expressed in mammalian cells. Topo IIβ is expressed constitutively throughout the cell cycle, whereas topo IIα is predominantly in highly proliferating cells.
Based on their mechanism of action, compounds targeting topo II can be divided into two large groups: poisons12 and catalytic inhibitors.6,13−16 Topo II poisons stabilize the covalent DNA–topo II complex, leading to DNA damage and apoptotic cell death and are used in the clinic for the treatment of breast, lung, prostate, and hematological cancers and sarcomas.17 They included etoposide, doxorubicin, and mitoxantrone. A common side effect of DNA poisons is the risk of secondary malignancies and cardiotoxicity, which are mostly connected to the effects of these drugs on topo IIβ.18 On the other hand, catalytic inhibitors affect one of the steps in the catalytic cycle of the enzyme. In particular, they can block binding of the DNA molecule to the enzyme, compete for binding with ATP, inhibit cleavage of the DNA molecule, or prevent hydrolysis of ATP,12 and interestingly, they do not increase DNA cleavage, which could indicate an improved safety profile.5
We report here the design and synthesis of a series of 3-methyl-2-phenyl-1H-indole analogues with antiproliferative and topo II inhibitory effects (Figure 1A). The compounds were designed based on the 3-methyl-2-phenyl-1H-indole hit I (Figure 1A), which was identified through screening of an in-house library of compounds against three selected human tumor cell lines: cervix adenocarcinoma (HeLa), ovarian carcinoma (A2780), and biphasic mesothelioma (MSTO-211H). Compound I showed good antiproliferative activity, with growth inhibition values (GI50) in the micromolar range. Based on compound I, a series of analogues was designed and synthesized, through varying the substituents on R1–R4 (Figure 1A). The antiproliferative activities of compounds were evaluated in vitro against HeLa, A2780, and MSTO-211H cells. Because of structural similarities of our compounds with some naturally occurring flavonoids that inhibit topo II, such as quercetin and luteolin (Figure 1B),19−21 the topo II inhibitory effects of the most potent analogues were investigated. For compound 32, cytofluorimetric analysis was performed to investigate the mechanism of cell death.
Figure 1.

Design of the 3-methyl-2-phenyl-1H-indoles as antiproliferative compounds, based on screening hit I (A); structures of flavonoids quercetin and luteolin with topo II inhibitory activities (B).
The designed compounds were prepared according to synthetic pathways presented in Schemes 1 and 2. Full synthetic procedures and analytical data are available in the Supporting Information. The synthesis of the central indole scaffold started with benzyl protection of hydroxyl group(s) of propiophenones 1 and 2, to obtain ethers 3 and 4 (Scheme 1). These ethers were α-brominated with bromine under acidic conditions, to yield 5 and 6, which were then reacted with 4-benzyloxyaniline in the Bischler–Möhlau indole synthesis, to give indoles 8–9. Indole 7 was obtained by Fischer indole synthesis from ketone 3 and phenylhydrazine under acidic conditions. Removal of the benzyl protecting groups of 7–9 to obtain phenols 10–12 was achieved by hydrogenation, using palladium on charcoal as catalyst. In addition, esters 13 and 14 were prepared from phenol 10: for 13, with 4-methylthiazole-5-carboxylic acid using 1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide (EDC) as coupling reagent, and for 14, from 1H-imidazole-4-carboxylic acid using 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethylaminium tetrafluoroborate (TBTU). Moreover, the benzyl protected indoles 7–9 were reacted with different haloalkanes using NaH as the base in N,N-dimethylformamide (DMF), to give N-alkylated indoles 15–25, which were deprotected by catalytic hydrogenation to obtain the final compounds 26, 27, and 29–37 (Scheme 2). Carboxylic acid derivative 28 was obtained by alkaline hydrolysis of ester 27. In addition, methylation of 37 with methyl iodide in the presence of cesium carbonate gave ether 38.
Scheme 1. Synthesis of Compounds 10–14.

Reagents and conditions: (a) benzyl bromide, K2CO3, acetone, reflux, 18 h; (b) for 7: phenylhydrazine, H2SO4, acetone, reflux, 24 h; (c) Br2, glacial acetic acid, 0 °C, 1 h; (d) for 8, 9: 4-benzyloxyaniline·HCl, Et3N, DMF, 120 °C, 2 h, then 150 °C, 3 h; (e) H2, 10% Pd/C, EtOH, r.t., 18 h; (f) for 13: 4-methylthiazole-5-carboxylic acid, EDC, 4-dimethylaminopyridine, CH2Cl2, r.t., 15 h; for 14: 1H-imidazole-4-carboxylic acid, TBTU, NMM, DMF, r.t., 15 h.
Scheme 2. Synthesis of Compounds 26–38.

Reagents and conditions: (a) for 15, 18, 23: 1-bromopropane; for 16: ethyl bromoacetate; for 17, 21, 25: (4-bromobutoxy)benzene; for 20, 24: (3-bromopropoxy)benzene; for 19: (2-bromoethoxy)benzene; for 22: methyl iodide; NaH, DMF, r.t., 3 h; (b) H2, 10% Pd/C, THF/EtOH, r.t., 18 h; (c) 1 M NaOH; (d) methyl iodide, Cs2CO3, acetone, 60 °C, 48 h.
The antiproliferative effect exerted by the new compounds 10–14 and 26–38 and by the well-known inhibitors of topo II m-amsacrine (m-AMSA) and quercetin was assayed on three human tumor cell lines: HeLa, A2780, and MSTO-211H. The results are expressed as GI50 values, i.e. the concentration (μM) of compound able to inhibit the growth in 50% of cells with respect to a control culture. The obtained data are shown in Table 1.
Table 1. Chemical Structures and Cell Growth Inhibition in the Presence of Compounds 10–14 and 26–38 and of m-AMSA and Quercetin as Reference.
![]()

Values are the mean ± SD of at least three independent experiments.
Compound I.
Reference compounds.
Most of the tested compounds had significant antiproliferative effect on the selected cell lines, showing GI50 values ranging from 2.0 to 18 μM. In general, the analogues 10–14, with the unsubstituted R3 (R3 = H), showed a rather low antiproliferative effect. Nevertheless, their GI50 values allowed us to draw some preliminary structure–activity relationships. In particular, it can be seen that 10 (unsubstituted at R1 and R2; hydroxyl group at R4) did not have any effect at the tested concentrations. The introduction of a hydroxyl group at R2 (11) induced a small increase in antiproliferative activity against HeLa and A2780 cells, while the insertion of a hydroxyl group at both R1 and R2 (12) resulted in a GI50 value of 2.2 μM on A2780 cells. Finally, the data obtained for compounds 13 and 14 suggest that large arylcarboxy substituents at R4 do not improve the cell effect.
As regards compounds 26–38, characterized by alkyl substituents at R3, they appeared generally more potent than 10–14. This can be seen by comparing the cell growth inhibition data of 10 (GI50 > 20 μM, on all tested cell lines) with those of 26, 27, and 29. Interestingly, 28 with a carboxymethylene substituent at R3 was not active (GI50 > 20 μM), which indicated that introduction of groups with low pKa is detrimental for cellular activity. Similarly as for 10, 26, 27, and 29, also the activity of 12 (R1, R2, and R4 = OH) on HeLa and MSTO-211H cells increased when alkyl substituents were introduced at R3 (12, GI50 > 20 μM; 34–37, GI50 from 3.4 to 18 μM). On the other hand, the activity of 12 on A2780 cells (GI50 = 2.2 μM) was higher compared to that of 34 (methyl group at R3; GI50 = 14 μM), 35 (propyl group at R3; GI50 = 8.4 μM), 36 (phenoxypropyl group at R3; GI50 = 7.4 μM), and 37 (phenoxybutyl group at R3; GI50 = 4.3 μM).
The compounds with a hydroxyl group at R2 (11, 12, 30–37) were generally more potent than those with unsubstituted R2 (10, 13, 26–29), as demonstrated by the comparison between 26 (unsubstituted R2) and 30 (hydroxyl group at R2). A similar behavior can also be observed for 29 (unsubstituted R2) with respect to 33, even if the differences in GI50 values appeared notably reduced.
The presence of an OH group at R1 had unfavorable effects on the antiproliferative activity as demonstrated by the higher potencies of 30 (R1 = H) versus 35 (R1 = OH), 32 (R1 = H) versus 36 (R1 = OH), and 33 (R1 = H) versus 37 (R1 = OH). The good antiproliferative profile of 30 (GI50 = 7.9 μM, 2.3 μM and 2.9 μM for HeLa, A2780 and MSTO-211H, respectively) was further improved by the addition of a phenoxy group to the propyl side chain of R3, to obtain the 3-phenoxypropyl derivative 32. Promising activity was maintained even when the length of the alkyl linker was increased from propylene to butylene, as for 33. The GI50 values of 32 and 33 were in the low micromolar range for all cell lines tested, and these values were the lowest among all of the studied analogues, thus highlighting 32 and 33 as the most promising compounds of the series. The comparison between the antiproliferative activities of 32 (3-phenoxypropyl substituent at R3) and 31 (3-phenoxyethyl substituent at R3) showed a significant decrease in activity for the latter in all tested cell lines. This result indicates that the appropriate length of the side chain at R3 is crucial for the biological effects.
Finally, alkylation of the hydroxyl groups of 37 to obtain the trimethoxy derivative 38 resulted in a significant decrease in the antiproliferative effect, which indicates the possibility of hydrogen bonding between the OH groups and the biological target.
Based on the structural similarities of the prepared compounds with some flavonoid inhibitors of topo II (Figure 1B) and with the aim to investigate the molecular mechanism responsible for the antiproliferative activity, we evaluated the effect of the most interesting analogues on the relaxation of supercoiled plasmid DNA catalyzed by topo II.22,23
In the series of compounds with unsubstituted R3 (10–14), the activities of the most potent derivatives, 11 and 12, were analyzed and m-AMSA (Figure 2) and quercetin (Figure 1SI) were used as references.
Figure 2.

Effect of compounds 11 and 12 on relaxation of supercoiled pBR322 DNA by human recombinant topoisomerase II. Supercoiled DNA (DNA) was incubated with the enzyme in the absence (topo II) and in the presence of 11 and 12 at 100 μM (A) or 25 μM and 50 μM (B). Twenty μM m-AMSA was used as reference.
Both 11 and 12 are able to inhibit topo II relaxation activity at 100 μM concentration (Figure 2A). In detail, compound 12 showed an electrophoretic pattern with almost solely supercoiled DNA, which suggests that it completely inhibited topo II activity. For compound 11, as well as the presence of supercoiled DNA, some topoisomers were formed, thus suggesting a slightly lower inhibitory activity. To further confirm these effects, topo II was incubated in the presence of 11 and 12 at 25 μM and 50 μM (Figure 2B). At the lower concentration, only 12 inhibited the activity of topo II, while 11 had no measurable effect. Interestingly, these results correlate with the antiproliferative effects on A2780 cells, where 11 showed a GI50 value (GI50 = 11 μM) about 5 times higher with respect to 12 (GI50 = 2.2 μM) (Table 1).
As regards the compounds having alkyl substituents at the R3 position (26–38), the effects on topo II activity of 100 μM 26–29 (unsubstituted at R1 and R2) are shown in Figure 3. For 26–28, the electrophoretic patterns appeared comparable to that of topo II alone, with a series of topoisomers as a result of the enzyme relaxation activity. On the other hand, as well as a weak formation of topoisomers, in the presence of 29, a strong band corresponding to supercoiled DNA also appears, indicative of an inhibitory effect, and in agreement with the antiproliferative data shown in Table 1.
Figure 3.

Effect of compounds 26–29 on relaxation of supercoiled pBR322 DNA by human recombinant topoisomerase II. Supercoiled DNA (DNA) was incubated with topo II in the absence (topo II) and in the presence of 26–29 at 100 μM.
Compounds 30–33 (hydroxyl substituent at R2) were characterized by low micromolar GI50 values (Table 1), and actually they showed significant topo II inhibitory activity at 100 μM concentration (Figure 4A). In particular, in these experimental conditions, for 30, 32, and 33 a marked band of supercoiled DNA was observed. For 31, a slightly lower inhibitory activity was detected, in agreement with its lower antiproliferative effect with respect to 30, 32, and 33 (Table 1). In addition, incubation of the enzyme with 25 μM and 50 μM concentrations (Figure 4B) highlighted 32 as the most effective and 31 as the least effective of this group of compounds. Indeed, in the presence of 32 a complete block of DNA relaxation can be observed at 50 μM, while no effect was obtained in the presence of 31 at the same concentration.
Figure 4.

Effect of compounds 30–33 on the relaxation of supercoiled pBR322 DNA by human recombinant topoisomerase II. Supercoiled DNA (DNA) was incubated with topo II in the absence (topo II) and in the presence of 30–33 at 100 μM (A), or at 25 μM and 50 μM (B).
Finally, topo II inhibition by 34–38, which showed intermediate antiproliferative profiles, was also investigated (Figure 5). Incubation of topo II in the presence of 100 μM 34–38 indicates compounds 34 and 38 as the less active derivatives of this series, in agreement with their relatively low antiproliferative effect, while a more pronounced inhibitory activity is exerted by 35–37, according with their more significant inhibition of cell proliferation (Figure 5; Table 1).
Figure 5.

Effect of compounds 34–38 on the relaxation of supercoiled pBR322 DNA by human recombinant topoisomerase II. Supercoiled DNA (DNA) was incubated with topo II in the absence (topo II) and in the presence of 34–38 at 100 μM.
The interesting biological profile of 32 prompted us to investigate more in depth its effects on cells, and in particular the mechanism involved in cell death. For this purpose, the most sensitive A2780 cells were treated with increasing concentrations of 32, incubated with the fluorescent probe JC-1, and then analyzed by flow cytometry, as previously described.24 The green monomeric fluorescent dye JC-1 accumulates in the mitochondrial matrix driven by the membrane potential, negative inside. This uptake leads to an increase in JC-1 concentration in mitochondria that promotes the formation of aggregates, which are characterized by high red fluorescence. The activation of the apoptosis pathway can result in collapse of mitochondrial transmembrane potential and release of mitochondrial proapoptotic factors in the cytoplasm. This collapse allows the leakage of JC-1, with a consequent decrease in red fluorescence. Figure 6 shows that A2780 cells incubated in the presence of 32 underwent a concentration-dependent decrease of mitochondrial transmembrane potential, with about 40% of cells showing depolarized mitochondria at 50 μM concentration.
Figure 6.

Mitochondrial membrane potential assessed by flow cytometry in A2780 cells after JC-1 staining. Cells were incubated for 28 h in the absence (control) and in the presence of compound 32 at the indicated concentrations. The percentages of cells with polarized (above) and depolarized (below) mitochondrial membranes are shown.
The ability of 32 to induce apoptosis was further confirmed by the analysis of hypodiploid cells stained with propidium iodide. Indeed, an important hallmark of apoptosis is the activation of endonucleases that cleave DNA at internucleosomal sites, thus creating a large number of small fragments of nucleic acid. Then, on DNA content histograms, apoptotic cells show DNA content as sub-G0 or hypodiploid peak.25,26
Figure 7 shows the results obtained in A2780 cells incubated with 10 μM and 50 μM 32, stained with propidium iodide and analyzed for DNA/cell content. Compound 32 induced a significant increase in the sub-G0 phase, thus indicating apoptotic events in about 30% of cells treated with the higher concentration (Figure 7).
Figure 7.
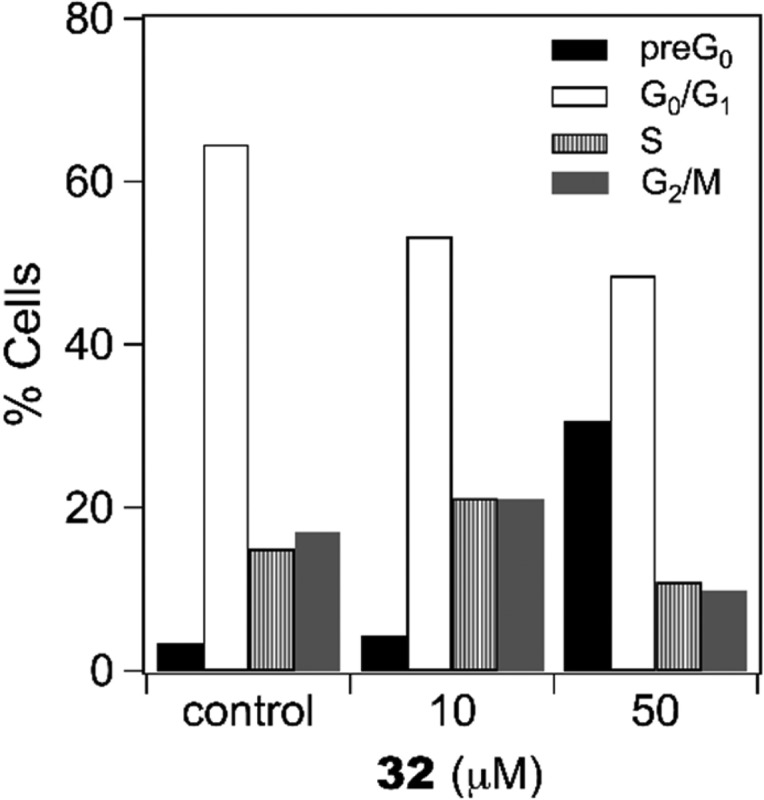
Flow cytometry analysis of A2780 cells treated for 28 h with compound 32 at the indicated concentrations and stained with propidium iodide.
A series of 3-methyl-2-phenyl-1H-indoles (10–14 and 26–38) was prepared and evaluated for the antiproliferative effect on three human tumor cell lines, HeLa, A2780, and MSTO-211H. The role of different substituents on the two aromatic moieties of these compounds was discussed. In particular, compounds with an unsubstituted R1 were generally more potent than those with a hydroxyl or a methoxy group at R1. On the other hand, at R2 and R4, hydroxyl groups were generally preferred. The most active compounds contained alkyl or phenoxyalkyl substituents at R3, with the size of these substituents important for the activity. The most interesting compound was 32, showing GI50 values of 4.4 μM, 2.2 μM, and 2.4 μM on HeLa, A2780, and MSTO-211H cell lines, respectively. Notably, for six compounds (12, 29–31, 33, 37), GI50 values lower than 5 μM against at least one cell line were obtained.
For selected compounds (11, 12, 26–38), the effect on the relaxation of supercoiled plasmid DNA catalyzed by topo II was also measured. The obtained results highlighted a good correlation between the inhibition of enzyme and the antiproliferative effect. For the most promising compound 32, cytofluorimetric analysis showed the ability to induce a concentration-dependent collapse of mitochondrial transmembrane potential. Moreover, a significant increase in the sub-G0 phase was measured in cells incubated with 32, confirming the occurrence of apoptosis. Overall, these data highlight 3-methyl-2-phenyl-1H-indole as a promising scaffold for the design of compounds with significant antiproliferative and antitopo II activities and provide new ideas for further optimization.
Acknowledgments
This work was supported by the Slovenian Research Agency (Grant No. P1-0208) and by Italian MIUR (Ministero dell’Istruzione, dell’Università e della Ricerca). The authors thank Žiga Hodnik for the help with chemical synthesis and Dušan Žigon (Mass Spectrometry Center, Jožef Stefan Institute, Ljubljana, Slovenia) for recording mass spectra.
Glossary
Abbreviations
- DMF
N,N-dimethylformamide
- DIAD
diisopropylazodicarboxylate
- DMSO
dimethyl sulfoxide
- EDC
1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide
- EDTA
ethylenediaminetetraacetic acid
- m-AMSA
meta-amsacrine
- NMM
N-methylmorpholine
- TBTU
O-(benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium tetrafluoroborate
- THF
tetrahydrofuran
- topo II
human DNA topoisomerase II
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.9b00557.
General information—chemistry; synthetic procedures and analytical data; 1H and 13C spectra of representative compounds; materials and methods—biological studies; Figure 1 SI: Effect of quercetin on the relaxation of supercoiled pBR322 DNA by human recombinant topoisomerase II (topo II); references (PDF)
Author Contributions
The manuscript was written using contributions from all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- World Cancer Report 2014; Stewart B.W., Wild C.P., Eds.; WHO, 2014.
- Bray F.; Ferlay J.; Soerjomataram I.; Siegel R. L.; Torre L. A.; Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. Ca-Cancer J. Clin. 2018, 68 (6), 394–424. 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- Dang C. V.; Reddy E. P.; Shokat K. M.; Soucek L. Drugging the ’undruggable’ cancer targets. Nat. Rev. Cancer 2017, 17, 502–508. 10.1038/nrc.2017.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoelder S.; Clarke P. A.; Workman P. Discovery of small molecule cancer drugs: successes, challenges and opportunities. Mol. Oncol. 2012, 6 (2), 155–176. 10.1016/j.molonc.2012.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitiss J. L. Targeting DNA topoisomerase II in cancer chemotherapy. Nat. Rev. Cancer 2009, 9 (5), 338–350. 10.1038/nrc2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pommier Y. Drugging topoisomerases: lessons and challenges. ACS Chem. Biol. 2013, 8 (1), 82–95. 10.1021/cb300648v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang X. X.; Wu Q.; Luan S. X.; Yin Z. Q.; He C. L.; Yin L. Z.; Zou Y. F.; Yuan Z. X.; Li L. X.; Song X.; He M.; Lv C.; Zhang W. A comprehensive review of topoisomerase inhibitors as anticancer agents in the past decade. Eur. J. Med. Chem. 2019, 171, 129–168. 10.1016/j.ejmech.2019.03.034. [DOI] [PubMed] [Google Scholar]
- Delgado J. L.; Hsieh C. M.; Chan N. L.; Hiasa H. Topoisomerases as anticancer targets. Biochem. J. 2018, 475 (2), 373–398. 10.1042/BCJ20160583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hevener K. E.; Verstak T. A.; Lutat K. E.; Riggsbee D. L.; Mooney J. W. Recent developments in topoisomerase-targeted cancer chemotherapy. Acta Pharm. Sin. B 2018, 8 (6), 844–861. 10.1016/j.apsb.2018.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Champoux J. J. DNA topoisomerases: structure, function, and mechanism. Annu. Rev. Biochem. 2001, 70, 369–413. 10.1146/annurev.biochem.70.1.369. [DOI] [PubMed] [Google Scholar]
- Parker M. W.; Botchan M. R.; Berger J. M. Mechanisms and regulation of DNA replication initiation in eukaryotes. Crit. Rev. Biochem. Mol. Biol. 2017, 52 (2), 107–144. 10.1080/10409238.2016.1274717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pogorelčnik B.; Perdih A.; Solmajer T. Recent developments of DNA poisons - human DNA topoisomerase II alpha inhibitors - as anticancer agents. Curr. Pharm. Des. 2013, 19 (13), 2474–2488. 10.2174/1381612811319130016. [DOI] [PubMed] [Google Scholar]
- Khadka D. B.; Cho W. J. Topoisomerase inhibitors as anticancer agents: a patent update. Expert Opin. Ther. Pat. 2013, 23 (8), 1033–1056. 10.1517/13543776.2013.790958. [DOI] [PubMed] [Google Scholar]
- Chene P.; Rudloff J.; Schoepfer J.; Furet P.; Meier P.; Qian Z.; Schlaeppi J. M.; Schmitz R.; Radimerski T. Catalytic inhibition of topoisomerase II by a novel rationally designed ATP-competitive purine analogue. BMC Chem. Biol. 2009, 9, 1. 10.1186/1472-6769-9-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pogorelčnik B.; Brvar M.; Zegura B.; Filipič M.; Solmajer T.; Perdih A. Discovery of mono- and disubstituted 1H-pyrazolo[3,4]pyrimidines and 9H-purines as catalytic inhibitors of human DNA topoisomerase IIalpha. ChemMedChem 2015, 10 (2), 345–359. 10.1002/cmdc.201402459. [DOI] [PubMed] [Google Scholar]
- Janežič M.; Pogorelčnik B.; Brvar M.; Solmajer T.; Perdih A. 3-substituted-1H-indazoles as catalytic inhibitors of the human DNA topoisomerase II alpha. ChemistrySelect 2017, 2 (1), 480–488. 10.1002/slct.201601554. [DOI] [Google Scholar]
- Jensen L. H.; Thougaard A. V.; Grauslund M.; Sokilde B.; Carstensen E. V.; Dvinge H. K.; Scudiero D. A.; Jensen P. B.; Shoemaker R. H.; Sehested M. Substituted purine analogues define a novel structural class of catalytic topoisomerase II inhibitors. Cancer Res. 2005, 65 (16), 7470–7477. 10.1158/0008-5472.CAN-05-0707. [DOI] [PubMed] [Google Scholar]
- Pogorelčnik B.; Brvar M.; Zajc I.; Filipič M.; Solmajer T.; Perdih A. Monocyclic 4-amino-6-(phenylamino)-1,3,5-triazines as inhibitors of human DNA topoisomerase II alpha. Bioorg. Med. Chem. Lett. 2014, 24 (24), 5762–5768. 10.1016/j.bmcl.2014.10.042. [DOI] [PubMed] [Google Scholar]
- Cantero G.; Campanella C.; Mateos S.; Cortes F. Topoisomerase II inhibition and high yield of endoreduplication induced by the flavonoids luteolin and quercetin. Mutagenesis 2006, 21 (5), 321–325. 10.1093/mutage/gel033. [DOI] [PubMed] [Google Scholar]
- Mittra B.; Saha D.; Chowdhury A. R.; Pal C.; Mandal S.; Mukhopadhyay S.; Bandyopadhyay S.; Majumder H. K. Luteolin, an abundant dietary component is a potent anti-leishmanial agent that acts by inducing topoisomerase II-mediated kinetoplast DNA cleavage leading to apoptosis. Mol. Med. 2000, 6 (6), 527–541. 10.1007/BF03401792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constantinou A.; Mehta R.; Runyan C.; Rao K.; Vaughan A.; Moon R. Flavonoids as DNA topoisomerase antagonists and poisons - structure-activity-relationships. J. Nat. Prod. 1995, 58 (2), 217–225. 10.1021/np50116a009. [DOI] [PubMed] [Google Scholar]
- Nitiss J. L. DNA topoisomerase II and its growing repertoire of biological functions. Nat. Rev. Cancer 2009, 9 (5), 327–337. 10.1038/nrc2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pommier Y.; Sung Y. L.; Huang S. Y. N.; Nitiss J. L. Roles of eukaryotic topoisomerases in transcription, replication and genomic stability. Nat. Rev. Mol. Cell Biol. 2016, 17 (11), 703–721. 10.1038/nrm.2016.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalla Via L.; Garcia-Argaez A. N.; Adami A.; Grancara S.; Martinis P.; Toninello A.; Dell’Amico D. B.; Labella L.; Samaritani S. Synthesis, antiproliferative and mitochondrial impairment activities of bis-alkyl-amino transplatinum complexes. Bioorg. Med. Chem. 2013, 21 (22), 6965–6972. 10.1016/j.bmc.2013.09.025. [DOI] [PubMed] [Google Scholar]
- Ormerod M. G. The study of apoptotic cells by flow cytometry. Leukemia 1998, 12 (7), 1013–1025. 10.1038/sj.leu.2401061. [DOI] [PubMed] [Google Scholar]
- Wlodkowic D.; Skommer J.; Darzynkiewicz Z. Cytometry of apoptosis. Historical perspective and new advances. Exp. Oncol. 2012, 34 (3), 255–262. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.