

Abstract
Background and Purpose
Immunotherapeutic intervention is one of the most promising strategies for the prevention and treatment of Alzheimer's disease (AD). Although they showed great success in AD mouse models, the clinical trials of many immune approaches failed due to low efficacy and safety. Thus, an animal model which can show the potential side effects of vaccines or antibodies is urgently needed. In this study, we generated EAE/AD mice by crossing APP/PS1 mice with experimental autoimmune encephalomyelitis (EAE) mice. We then investigated the efficacy and safety of two vaccines: the immunogens of which were Aβ1‐42 aggregates (Aβ42 vaccine) and an oligomer‐specific conformational epitope (AOE1 vaccine), respectively.
Experimental Approach
EAE/AD mice were immunized with the Aβ42 vaccine or AOE1 vaccine five times at biweekly intervals. After the final immunization, cognitive function was evaluated by the Morris water maze, Y maze, and object recognition tests. Neuropathological changes in the mouse brains were analysed by immunohistochemistry and ELISA.
Key Results
In contrast to previous findings in conventional AD animal models, Aβ42 immunization promoted neuroinflammation, enhanced Aβ levels and plaque burden, and failed to restore cognitive deficits in EAE/AD mice. By contrast, AOE1 immunization dramatically attenuated neuroinflammation, reduced Aβ levels, and improved cognitive performance in EAE/AD mice.
Conclusion and Implications
These results suggest that the EAE/AD mouse model can exhibit the potential side effects of AD immune approaches that conventional AD animal models fail to display. Furthermore, strategies specifically targeting Aβ oligomers may be safe and show clinical benefit for AD treatment.
Abbreviations
- AD
Alzheimer's disease
- APP
amyloid precursor protein
- ARIAs
amyloid‐related imaging abnormalities
- Aβ
amyloid‐β
- CFA
complete Freund's adjuvant
- EAE
experimental autoimmune encephalomyelitis
- GFAP
glial fibrillary acidic protein
- Iba‐1
ionized calcium‐binding adapter molecule 1
- IFA
incomplete Freund's adjuvant
- MWM
Morris water maze
What is already known
Many AD immunotherapies failed in clinical trials although they exhibited success in animal models.
Animal models which show the potential side effects of AD vaccines are urgently needed.
What this study adds
The Aβ42 vaccine induced neuroinflammation and enhanced Aβ pathology in EAE/AD mice.
What is the clinical significance
The EAE/AD mouse model can express the potential side effects of AD immunotherapies.
Immunotherapies targeting Aβ oligomers may be safe and effective in the treatment of AD.
1. INTRODUCTION
The pathological hallmarks of Alzheimer's disease (AD) include extracellular senile plaques containing https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4865 and intracellular neurofibrillary tangles composed of hyperphosphorylated https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=9275 (Blennow, de Leon, & Zetterberg, 2006). The Aβ hypothesis proposes that Aβ is the major cause of AD and that Aβ oligomers, but not the monomers or fibrils, are the major inducers of neuropathology (Selkoe & Hardy, 2016). However, decreasing Aβ levels or neutralizing its toxicity failed to show clinical benefit in large clinical trials. Similarly, trials of active and passive immunotherapies targeting Aβ also failed to meet their primary endpoints (Panza, Lozupone, Logroscino, & Imbimbo, 2019).
Immunotherapeutic agents significantly differ with regard to their selective recognition to the domains and aggregated forms of Aβ (Long & Holtzman, 2019). The first therapeutic vaccine to be developed was AN‐1972, the immunogen of which was Aβ aggregates, but the Phase IIa clinical trial of this vaccine was halted when 6% of patients developed meningoencephalitis (Gilman et al., 2005). To avoid similar side effects, a number of second‐generation vaccines such as CAD106, ACC‐001, and AD02 were then developed. These used the Aβ N‐terminus as immunogen that only contain B‐cell epitopes (Pasquier et al., 2016; Schneeberger et al., 2015; Winblad et al., 2012). Nevertheless, these new generation vaccines still showed serious adverse effects including amyloid‐related imaging abnormalities (ARIAs; Pasquier et al., 2016; Vandenberghe et al., 2017). Similarly, with respect to passive anti‐Aβ immunotherapies, the clinical trials of many monoclonal antibody candidates failed due to low therapeutic efficacy or serious adverse events. A Phase III clinical trial of https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=6930 (an anti‐Aβ1‐5 antibody) was terminated because the primary clinical endpoints were not met and it was shown to increase the risk of vasogenic cerebral oedema (Salloway et al., 2014). A Phase II/III study of https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=6933 (recognizes the N‐terminal and central regions of Aβ) was prematurely discontinued due to lack of effect and an increase in the risk of ARIAs (Ostrowitzki et al., 2017). Moreover, ARIA‐vasogenic oedema occurred in 3–31% of https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=8325 (recognizes soluble Aβ aggregates and insoluble fibrils) recipients in a dose‐dependent manner in a Phase Ib study (Sevigny et al., 2016). It should be noted that all of the vaccines and antibodies tested in clinical trials had already shown marked benefit without apparent side effects in preclinical studies on animal models (Bohrmann et al., 2012; DeMattos et al., 2001; Schenk et al., 1999; Sevigny et al., 2016; Wiessner et al., 2011), demonstrating that the existing animal models fail to show all of the potential adverse effects of immunotherapies.
Therefore, a more sensitive animal model which demonstrates the complete side effect profiles of vaccines and antibody candidates during preclinical tests is urgently needed. It is conceivable that using a physiological molecule from the body as an immunogen would tend to induce autoimmune responses. Thus, non‐self‐molecules such as pathogenic factors themselves or conformational epitopes abstracted from self‐ or non‐self‐produced pathogenic factors may be less likely to induce autoimmune responses while incurring a specific immune response beneficial for disease treatment. Aβ oligomers, the most toxic form of Aβ aggregates, play a critical role in AD progression (Kayed & Lasagna‐Reeves, 2013; Viola & Klein, 2015). They contain unique structures which do not exist in Aβ monomers and can be recognized by Thioflavin T, Congo red, and oligomer‐specific antibodies (Kayed et al., 2003; Kayed & Glabe, 2006; Wang et al., 2009). However, the safety of vaccines containing spatial epitopes of Aβ oligomers remains unknown.
Experimental autoimmune encephalomyelitis (EAE) is the most commonly used animal model to study multiple sclerosis (Robinson, Harp, Noronha, & Miller, 2014). During the course of EAE, autoreactive T cells and monocytes in the blood and peripheral lymphoid organs penetrate the blood–brain barrier and target proteins expressed by neurons and glia, inducing inflammatory lesions, axonal damage, and chronic irreversible functional impairments within the CNS (De Feo et al., 2017). To evaluate the safety and therapeutic benefits of different vaccines for AD, we constructed a novel mouse model by crossing mice overexpressing amyloid precursor protein (APP) and https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2402 (PS1) with EAE mice. We then compared the therapeutic effects of two distinct vaccines on the cognitive function and neuropathology of these mice. The Aβ42 vaccine consists of pre‐aggregated full length Aβ42 and was the first clinically relevant active anti‐Aβ immunotherapy (AN1792) to be developed (Schenk et al., 1999). This Aβ42 vaccine significantly reduced Aβ burden and preserved cognitive function in APP transgenic mice, but a Phase IIa clinical trial was halted because 6% of the immunized patients developed meningoencephalitis (Gilman et al., 2005). Moreover, in C57BL/6 mice, active immunization with Aβ caused memory impairments and promoted immune cell infiltration into the CNS through https://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=316 2/4‐dependent activation of the innate immune system (Vollmar et al., 2010). Similarly, in AD mice overproducing TGF‐β1, Aβ immunization increased T cell recruitment to the CNS (Buckwalter et al., 2006). The AOE1 vaccine is a yeast cell‐based vaccine developed by our group, in which Aβ oligomer‐specific mimotopes are displayed on the yeast cell surface. This vaccine has been shown to effectively attenuate cognitive deficits and neuropathology in APP/PS1 mice (Wang et al., 2017).
2. METHODS
2.1. Vaccine preparation
To prepare the Aβ42 vaccine, Aβ42 peptide (human Aβ1‐42; Chinese Peptide Company, Hangzhou, China) was dissolved in PBS to a concentration of 1 mg·ml−1 and was incubated overnight at 37°C to allow aggregation.
The AOE1 vaccine was prepared according to a previously described method (Wang et al., 2017). Briefly, a DNA fragment encoding the oligomer‐specific mimotope peptide was inserted into a modified vector of pCTCON2, transfected into EBY100 (Saccharomyces cerevisiae), and expressed on the yeast cell surface. The surface display levels of the oligomer‐specific mimotope were measured by flow cytometry with Alexa Fluor 488‐conjugated anti‐c‐Myc antibody (1:100, Santa Cruz Biotechnology Cat# sc‐40, RRID:AB_627268) and imaged by confocal microscopy. Following this, the yeast cells were heat‐inactivated (56°C, 1 hr) and stored at −80°C until use.
2.2. Mice
All animal care and experimental procedures complied with and were approved by the Institutional Animal Care and Use Committee of Tsinghua University. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny, Browne, Cuthill, Emerson, & Altman, 2010) and with the recommendations made by the British Journal of Pharmacology. APP/PS1 mice (RRID:MGI:3665286) and EAE mice (RRID:IMSR_JAX:000457) were obtained from the Jackson Laboratory (Bar Harbor, ME, USA). APP/PS1 mice were bred with EAE mice to generate EAE/AD mice. Their WT littermates were used as controls. All mice were group‐housed (five mice per cage) in a colony room at 22 ± 2°C and 45 ± 10% humidity under a reverse 12 hr light/dark cycle and had ad libitum access to food and water.
2.3. Immunization protocols
For Aβ42 immunization, 6‐month‐old male EAE/AD mice were first injected subcutaneously in the scruff of the neck with 100 μg of Aβ42 (100 μl) emulsified in an equal volume of complete Freund's adjuvant (CFA) and then boosted with 100 μg of Aβ42 (100 μl) emulsified in an equal volume of incomplete Freund's adjuvant (IFA), four times at biweekly intervals (n = 8). In parallel, 6‐month‐old male EAE mice and APP/PS1 mice were immunized with Aβ42 according to the same protocol (n = 8). EAE/AD, EAE, and APP/PS1 control mice were injected with CFA/IFA alone (n = 8). For AOE1 immunization, 6‐month‐old male EAE/AD mice were immunized with 6 × 107 yeast cells of the AOE1 vaccine (n = 8) five times at biweekly intervals as described previously (Wang et al., 2017). Blood samples (300 μl ) were collected from the tail vein before the immunization protocol was initiated (pre‐bleed) and 10 days after each immunization. Behavioural studies were performed 14 days after the final immunization. The mice were then deeply anesthetized (avertin) and transcardially perfused with ice‐cold PBS containing heparin (10 U ml‐1), and killed by cervical dislocation. The neuropathological changes in their brains were analysed (see below).
2.4. Morris water maze test
The Morris water maze (MWM) consisted of a pool (110 cm in diameter) containing opaque water (22 ± 1°C) and a platform (10 cm in diameter) submerged 1 cm under the water (Zha et al., 2016). Hidden platform training was conducted twice per day for five consecutive days with an inter‐trial interval of 3–4 hr. Mice were allowed to swim for 60 s to locate the platform. Upon finding the platform, they were allowed to stay on it for 10 s. The trial ended when the mouse located the platform. Mice unable to locate the platform were guided to it. Then, 24 hr after the final acquisition trial, a probe trial in which the hidden platform was absent was conducted in order to assess the spatial memory of the mice. The performance of each mouse was monitored using a video camera (Sony, Tokyo, Japan) mounted over the maze and automatically recorded via a video tracking system.
2.5. Y maze test
The Y maze test was performed as described previously with minor modifications (Liu et al., 2018). The Y maze consisted of three arms (8 × 30 × 15 cm) separated by 120° angles. The Y maze test comprised two trials separated by an interval of 1 hr. In the first trial, which was 10 min long, the mouse was allowed to explore only two arms (the start and familiar arms) of the maze, and the third arm (the novel arm) was blocked. In the second trial, the mouse was put back in the same starting arm as in Trial 1 and had free access to all three arms for 5 min. The time spent in the novel arm was recorded by a video tracking system. The arms were cleaned with 70% ethanol between trials.
2.6. Object recognition test
The object recognition test was performed as previously described with some modifications (Lueptow, 2017; Zhang et al., 2018). Briefly, mice were individually habituated to the behavioural arena (50 cm × 50 cm × 25 cm white plastic box, empty) for 5 min 1 day before the test was initiated. For the training session (Trial 1), two identical objects were placed in the upper two corners of the box, and the mice were allowed to explore the box for 5 min. For the testing session (Trial 2), after a 24‐hr retention period, the object in the upper right corner was replaced with a novel object, and the mice were reintroduced to the box and allowed to explore for 5 min. The time spent exploring and sniffing each object was recorded. The discrimination index was then calculated by subtracting the time spent exploring the familiar object from the time spent exploring the novel object and dividing this by the total exploration time. To exclude olfactory cues, the box was cleaned with 70% alcohol before each test.
2.7. Immunohistochemistry and histology
The immuno‐related procedures used comply with the recommendations made by the British Journal of Pharmacology (Alexander et al., 2018). Mice were deeply anaesthetized with avertin (250 mg·kg−1), transcardially perfused with ice‐cold PBS containing heparin (10 U·ml−1), and then killed by cervical dislocation. Their brains were immediately removed and divided along the sagittal plane. The left brain hemisphere was fixed in 4% paraformaldehyde at 4°C overnight and processed into either frozen or paraffin‐embedded sections. For EAE/AD mice, coronal paraffin‐embedded serial sections of 5 μm thickness were cut using a Lecia CM1850 microtome (Leica Biosystems, Buffalo Grove, IL, USA). For EAE mice, coronal frozen sections of 20 μm thickness were produced. For immunostaining, floating sections were permeabilized and blocked with a 10% normal goat serum in 0.3% Triton X‐100 PBST for 1 hr at room temperature. Thereafter, the sections were incubated overnight with primary antibodies at 4°C. Following this, the sections were incubated with the appropriate HRP‐conjugated secondary antibodies for 1 hr at room temperature and visualized with diaminobenzidine. The following primary antibodies were used: anti‐Aβ1‐16 (clone 6E10, 1:100, BioLegend Cat# 803002, RRID:AB_2564654), anti‐ionized calcium‐binding adapter molecule 1 (Iba‐1) (1:100, GeneTex Cat# GTX100042, RRID:AB_1240434), anti‐glial fibrillary acidic protein (1:100, Cell Signaling Technology Cat# 3670, RRID:AB_561049), and anti‐CD68 (1:100, Abcam Cat# ab955, RRID:AB_307338). Haemosiderin deposits were detected via Prussian Blue staining. All images were acquired with an Olympus IX73 inverted microscope with a DP80 camera. Immunostained regions were quantified using ImageJ software (RRID:SCR_003070). Aβ deposits were presented as the number of plaques·mm−2 of each brain region or the per cent area occupied by 6E10‐immunoreactive deposits. For the other immunostaining experiments, the values obtained for the adjuvant‐treated control mice were averaged, and the values obtained for the other groups were calculated as a percentage of that mean.
2.8. Brain lysate preparation
The right brain hemisphere was homogenized in RIPA buffer containing a protease inhibitor cocktail (Sigma) consisting of 50‐mM Tris (pH 7.4), 150‐mM NaCl, 1% Triton X‐100, 1% sodium deoxycholate, and 0.1% SDS. The tissues were then centrifuged at 14,000× g for 30 min at 4°C, and the supernatant (RIPA soluble fraction) containing soluble Aβ was collected. The pellets were resuspended in guanidine buffer (5.0‐M guanidine‐HCl/50‐mM Tris–HCl, pH 8.0) and centrifuged at 14,000× g for 1 hr at 4°C to obtain the supernatant containing insoluble Aβ (guanidine soluble fraction).
2.9. Measurement of Aβ and proinflammatory cytokine levels
The Aβ levels in the RIPA soluble and RIPA insoluble (guanidine soluble) fractions of the brain lysates of mice were quantified by ELISA using Aβ40 and Aβ42 immunoassay kits (Immuno‐Biological Laboratories Co., Ltd.). The levels of soluble and insoluble Aβ were expressed as pg of Aβ per mg of brain tissue. With respect to proinflammatory cytokines, the levels of https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5074 and https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4974 in the brain lysates of mice were determined using ELISA kits (Neobioscience Technology, Beijing, China) according to the manufacturer's protocols.
2.10. Data and statistical analysis
The data and statistical analysis comply with the recommendations of the British Journal of Pharmacology on experimental design and analysis in pharmacology (Curtis et al., 2018). All mice were randomly assigned to different treatment groups, and the experimenters were blind to the treatment of the animals while performing the behavioural, immunohistochemical, and biochemical studies and analysing the data. Statistical analyses for group comparisons were undertaken only if group size n ≥ 5. Data from group sizes (n < 5) were not subjected to statistical analysis. The studies were designed to generate mouse groups of equal size with n ≥ 5. The group size was based on the expected difference from our pilot studies using similar protocols in mice without carrying out a formal power analysis. The group size was the number of independent values, and statistical analysis was performed using independent values corresponding to the data obtained from different mice. To allow for comparison between individual data sets, data from the immunohistochemistry experiments were normalized to the mean value of adjuvant‐treated control mice and expressed as percentage means ± SEM. Data were analysed with GraphPad Prism v.5 (RRID:SCR_002798). Statistical significance was assessed using the Student's t test or one‐ or two‐way ANOVA, as appropriate. The Bonferroni post hoc tests were used only if F was significant, and there was no variance inhomogeneity. The results were expressed as group means ± SEM, and P < .05 was considered statistically significant. All data were included in the statistical analysis as all fell within two SDs of the group mean.
2.11. Materials
The EBY100 yeast was a kind gift from Dr. Xiang‐mei Liu (State Key Laboratory of Microbial Technology, Shandong University, China). Sigma‐Aldrich supplied the CFA (Cat# F5881) and IFA (Cat# F5506); QS‐21 (Cat# 20‐2‐177) was supplied by Desert King International (San Diego, CA).
2.12. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2019/20 (Alexander, Fabbro et al., 2019a, 2019b).
3. RESULTS
3.1. Immunization with AOE1, but not Aβ42, restored cognitive deficits in the EAE/AD mouse
In previous studies, Aβ42 and AOE1 vaccination effectively induced the production of anti‐Aβ antibodies and improved cognitive performance in AD transgenic mice (Schenk et al., 1999; Wang et al., 2017). In this study, we compared the efficacy of Aβ42 and AOE1 vaccines in EAE/AD mice in parallel. Aβ42 vaccination induced robust humoral immune responses, and the anti‐Aβ antibody titre in vaccinated mice peaked after four immunizations, while the antibody titre induced by AOE1 immunization peaked after the fifth immunization (Figure 1a).
FIGURE 1.
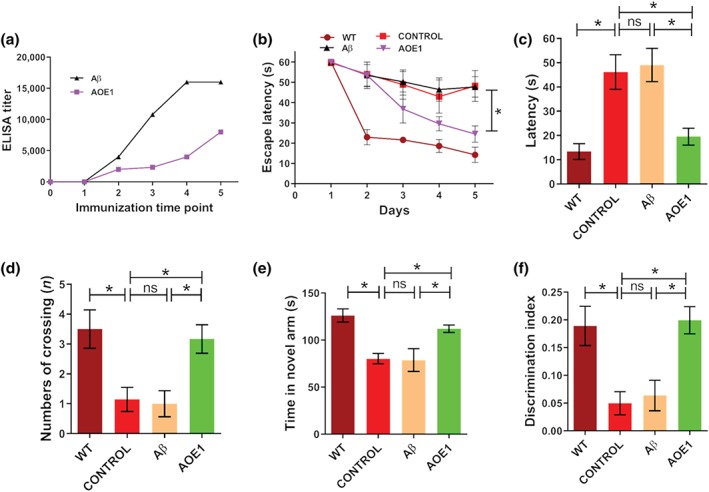
Immunization with AOE1, but not Aβ42, restored cognitive deficits in the EAE/AD mice. (a) Antibody titres in the serum of EAE/AD mice immunized with Aβ42 or AOE1. (b–d) To assess spatial learning and memory retention, EAE/AD mice immunized with Aβ42 or AOE1 were subjected to the Morris water maze test. The latency to find the hidden platform (b), the latency to find the position of the removed platform (c), and the number of platform crossings (d) were determined. (e) The short‐term working memory of EAE/AD mice immunized with Aβ42 or AOE1 was evaluated by the Y maze test, and the length of time spent in the novel arm was measured. (f) EAE/AD mice immunized with Aβ42 or AOE1 were subjected to the object recognition test, and the discrimination index was calculated. Data shown are means ± SEM, from n = 8 mice per group. *P < .05, significantly different as indicated; ns, not significantly different as indicated
To investigate the effects of Aβ42 and AOE1 vaccination on cognitive function in EAE/AD mice, we performed the MWM, Y maze, and object recognition tests. During the acquisition phase of the MWM test, AOE1 immunization significantly improved learning and memory retention in EAE/AD mice, and no difference in swimming speed was noted between the groups. In contrast, Aβ42‐immunized EAE/AD mice did not show any improvement in spatial memory, compared to the adjuvant‐treated control mice (Figure 1b). Similarly, in the probe trials, AOE1‐, but not Aβ42‐, immunized EAE/AD mice exhibited spatially oriented swimming behaviour, shorter escape latencies (Figure 1c), and a greater number of platform crossings (Figure 1d) than adjuvant‐treated EAE/AD mice. Consistent with these findings, immunization with AOE1, but not Aβ42, improved spatial working memory in the Y maze test. AOE1‐vaccinated EAE/AD mice spent more time in the novel arm, whereas adjuvant‐treated and Aβ42‐vaccinated EAE/AD mice exhibited no preference for the novel arm, showing impaired memory retention (Figure 1e). In the object recognition test, adjuvant‐treated and Aβ42‐vaccinated EAE/AD mice showed no preference for the novel object, while AOE1‐vaccinated mice showed a significant preference for the novel object (Figure 1f). These findings indicated that immunization with AOE1 ameliorated memory deficits in EAE/AD mice, whereas Aβ42 vaccination did not improve spatial memory or cognition in the EAE/AD mice.
To compare the effects of Aβ42 immunization on EAE/AD and conventional APP/PS1 mice, both mouse models were immunized with Aβ42, and the cognitive performance of the animals was assessed after the final immunization. We observed that EAE/AD and APP/PS1 mice exhibited comparable serum antibody titres against Aβ42. The serum antibody titres of both groups were higher than 1:16,000. In contrast, adjuvant‐treated control mice had negligible antibody titres against Aβ42. In the Y maze test, the adjuvant‐treated EAE/AD and APP/PS1 mice demonstrated no preference for the novel arm. EAE/AD mice vaccinated with Aβ42 showed spatial memory impairments (Figure S1). In contrast, Aβ42 immunization resulted in significant memory improvement in APP/PS1 mice (Figure S1).
3.2. Immunization with Aβ42, but not AOE1, enhanced Aβ pathology in the EAE/AD mouse
To assess the effects of Aβ42 and AOE1 immunization on Aβ pathology in the brains of EAE/AD mice, we performed 6E10‐immunostaining to detect Aβ plaques (Figure 2a). As expected, AOE1 vaccination significantly decreased the number of plaques and the per cent area occupied by plaques in the brains of EAE/AD mice. Surprisingly, Aβ42 immunization significantly increased plaque burden (Figure 2b,c). In clinical trials, QS‐21 was used as the adjuvant for AN1792 (Gilman et al., 2005). We found that when QS‐21 was used as the adjuvant for Aβ42 immunization in EAE/AD mice, this vaccine also significantly enhanced Aβ plaque burden (Figure S2A), which is consistent with the results obtained when CFA/IFA was used as the adjuvant.
FIGURE 2.
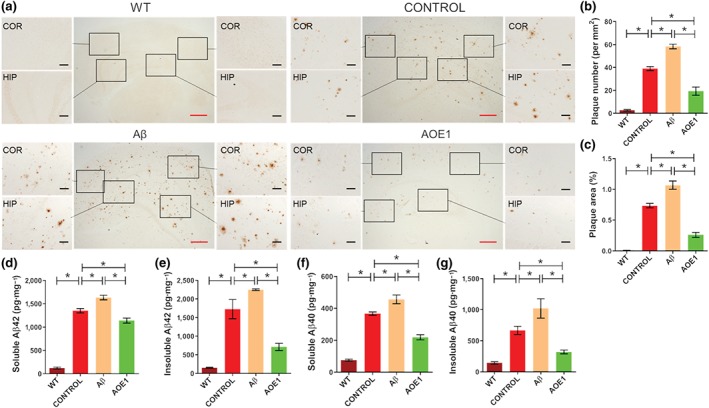
Immunization with Aβ42, but not AOE1, enhanced Aβ pathology in the EAE/AD mice. (a) 6E10 immunostaining for plaques in the brains of EAE/AD mice immunized with Aβ42 or AOE1. Scale bars: red, 500 μm; black, 100 μm. (b, c) Quantification of plaque number (b) and area (c) in the brain sections of EAE/AD mice immunized with Aβ42 or AOE1. (d–g) The levels of soluble Aβ42 (d), insoluble Aβ42 (e), soluble Aβ40 (f), and insoluble Aβ40 (g) in the brain lysates of EAE/AD mice immunized with Aβ42 or AOE1 were detected by ELISA. Data shown are means ± SEM, from n = 8 mice per group. *P < .05, significantly different as indicated
We then determined the levels of Aβ40 and Aβ42 in the brain lysates of vaccinated EAE/AD mice. AOE1 immunization significantly decreased the levels of soluble and insoluble Aβ40 and Aβ42 in EAE/AD mouse brains compared to adjuvant treatment. In contrast, Aβ42 immunization markedly increased Aβ levels (Figure 2d–g). These results indicated that Aβ42 vaccination did not attenuate Aβ burden but rather exacerbated Aβ pathology in the EAE/AD mouse model.
3.3. Immunization with Aβ42, but not AOE1, increased neuroinflammation in the EAE/AD mouse
Neuroinflammation is a critical pathological hallmark of AD. The sustained activation of microglia and astrocytes and continued release of proinflammatory cytokines exacerbate neuroinflammation and contribute to neurodegeneration (Heneka et al., 2015). To compare inflammation‐related pathology in the brains of EAE/AD mice immunized with Aβ42 and AOE1, we evaluated microgliosis and astrocytosis by Iba‐1 (Figure 3a) and GFAP immunostaining (Figure 4a) respectively. AOE1 immunization significantly decreased microgliosis (Figure 3b,c) and astrocytosis (Figure 4b,c) in the cortex and hippocampus of EAE/AD mice, compared with adjuvant treatment. In contrast, Aβ42 vaccination significantly increased the areas of Iba‐1 (Figure 3b,c) and GFAP immunostaining (Figure 4b,c), and thus exacerbated gliosis. Moreover, we evaluated microglial activation by CD68 immunostaining (Figure 3d). Consistent with the above results, AOE1 immunization significantly reduced microglial activation in the cortex and hippocampus of EAE/AD mice. In contrast, Aβ42 immunization increased microglial activation in these brain areas (Figure 3e,f).
FIGURE 3.
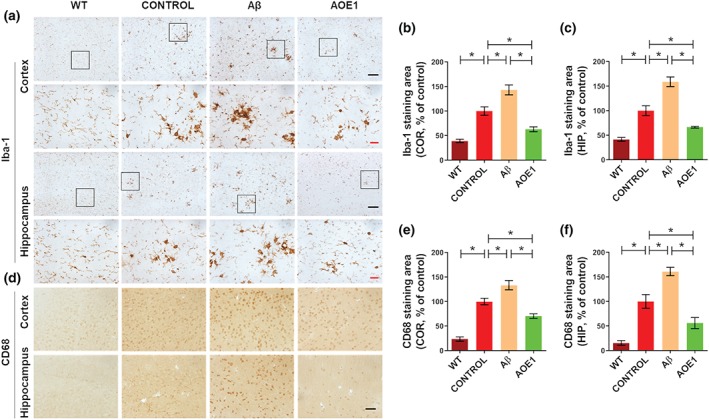
Immunization with Aβ42, but not AOE1, increased microgliosis in the EAE/AD mice. (a) Iba‐1 immunostaining for microglia in the brains of EAE/AD mice immunized with Aβ42 or AOE1. Scale bars: black, 100 μm; red, 20 μm. (b, c) Quantification of Iba‐1‐positive area in the cortex (b) and hippocampus (c). (d) CD68 immunostaining for microglia in the brains of EAE/AD mice immunized with Aβ42 or AOE1. Scale bar: 50 μm. (e, f) Quantification of CD68‐positive area in the cortex (e) and hippocampus (f). Data shown are means ± SEM, from n = 8 mice per group. *P < .05, significantly different as indicated
FIGURE 4.
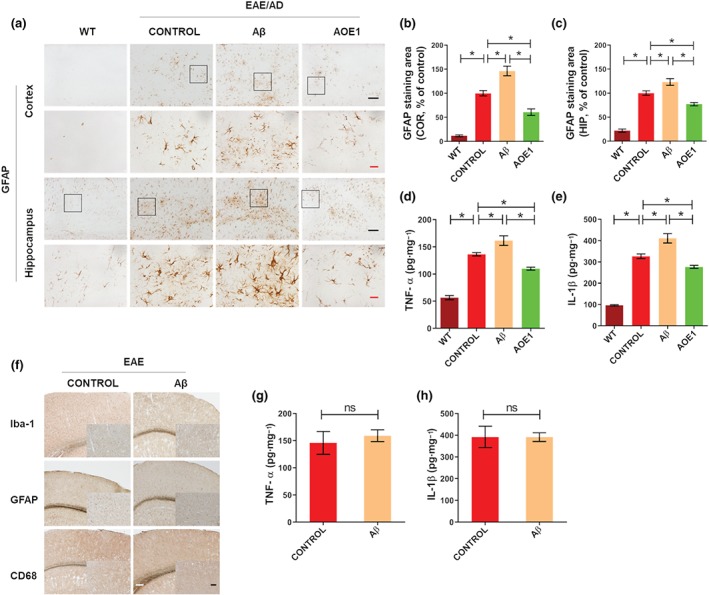
Immunization with Aβ42 increased astrocytosis and the levels of proinflammatory factors in the EAE/AD mice but did not exacerbate neuroinflammation in the EAE mice. (a) GFAP immunostaining for astrocytes in the brains of EAE/AD mice immunized with Aβ42 or AOE1. Scale bars: black, 100 μm; red, 20 μm. (b, c) Quantification of GFAP‐positive area in the cortex (b) and hippocampus (c). (d, e) The levels of TNF‐α (d) and IL‐1β (e) in the brain lysates of EAE/AD mice immunized with Aβ42 or AOE1 were detected by ELISA. (f) Iba‐1, GFAP, and CD68 immunostaining for gliosis in the brains of EAE mice immunized with Aβ42 or adjuvant. Scale bars: white, 200 μm; black, 100 μm. (g, h) The levels of TNF‐α (g) and IL‐1β (h) in the brain lysates of EAE mice immunized with Aβ42 or adjuvant were detected by ELISA. Data shown are means ± SEM, from n = 8 mice per group. *P < .05, significantly different as indicated; ns, not significantly different as indicated
To further compare the effects of Aβ42 and AOE1 immunization on proinflammatory cytokine levels, the levels of TNF‐α and IL‐1β in the brain lysates of vaccinated EAE/AD mice were determined by ELISA. Adjuvant‐treated EAE/AD mice exhibited higher levels of TNF‐α and IL‐1β compared to their WT littermates. AOE1 vaccination significantly reduced the levels of TNF‐α and IL‐1β by 19.4% (Figure 4d) and 15.3% (Figure 4e), respectively. However, immunization with Aβ42 significantly increased the levels of these proinflammatory cytokines compared to adjuvant treatment (Figure 4d,e). Correlating with this, using QS‐21 as the adjuvant for Aβ42 immunization in EAE/AD mice also promoted neuroinflammation, as demonstrated by significantly elevated gliosis (Figure S2A) and increased levels of proinflammatory cytokines (Figure S2B, C).
3.4. Aβ42 immunization exacerbated cerebral haemorrhage in the EAE/AD mouse
Aβ immunotherapy increases the risk of cerebral haemorrhage (Pfeifer et al., 2002; Wilcock et al., 2004), which may underlie the neuroinflammatory complications of Aβ immunization seen in clinical trials (Schenk, 2002). In this study, we assessed the effect of Aβ42 and AOE1 vaccination on microhaemorrhages in the brains of EAE/AD mice (Figure 5a). A greater number of microhaemorrhages were observed in the brains of EAE/AD mice than in the brains of the WT mice, and Aβ42 immunization significantly increased the number of cerebral haemorrhages in EAE/AD mice. Comparable results were obtained when QS‐21 was used as the adjuvant (Figure S2A). However, AOE1 immunization did not induce more haemorrhages than adjuvant treatment (Figure 5b,c).
FIGURE 5.
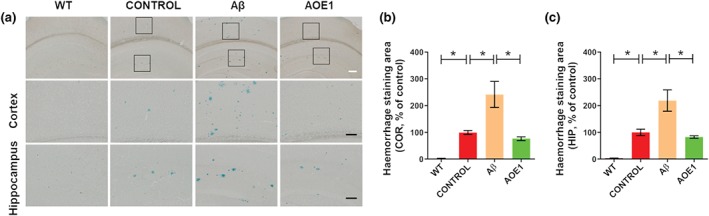
Aβ42 immunization exacerbated cerebral haemorrhage in the EAE/AD mouse model. (a) Hemosiderin staining for microhaemorrhages in the brains of EAE/AD mice immunized with Aβ42 or AOE1. Scale bars: white, 200 μm; black, 100 μm. (b, c) Quantification of hemosiderin‐positive area in the cortex (b) and hippocampus (c). Data shown are means ± SEM, from n = 8 mice per group. *P < .05, significantly different as indicated
3.5. Immunization with Aβ42 did not induce neuroinflammation in the EAE mouse
To investigate the effect of Aβ42 immunization on neuroinflammation in the EAE mouse, we immunized EAE mice with Aβ42 and assessed neuroinflammation in their brains by immunohistochemistry using anti‐Iba‐1, anti‐GFAP, and anti‐CD68 antibodies. Compared to adjuvant treatment, Aβ42 vaccination did not induce significant changes in microgliosis or astrocytosis in the brains of EAE mice (Figure 4f). Moreover, no difference in the levels of TNF‐α (Figure 4g) and IL‐1β (Figure 4h) was observed in the brain lysates of adjuvant‐treated and Aβ42‐vaccinated EAE mice. These findings suggest that EAE/AD mice, but not EAE mice, exhibit Aβ42 immunization‐induced neuropathology.
4. DISCUSSION
Aβ and tau are both physiological proteins which undergo aggregation to form neurotoxic oligomers, protofibrils, and fibrils at the disease state and promote the pathological progression of AD (Polanco et al., 2018; Selkoe & Hardy, 2016; Shafiei, Guerrero‐Munoz, & Castillo‐Carranza, 2017). Immunotherapeutic approaches aimed at directly targeting Aβ or tau with vaccines or antibodies are among the most promising strategies for AD treatment (Congdon & Sigurdsson, 2018; Panza et al., 2019). In the past decade, a tremendous amount of effort has been put into translating immunotherapies into cognitive benefits in human clinical trials (Graham, Bonito‐Oliva, & Sakmar, 2017; Long & Holtzman, 2019). However, a number of clinical trials failed because of serious side effects or inadequate therapeutic efficacy (Gilman et al., 2005; Landen et al., 2017; Pasquier et al., 2016; Salloway et al., 2014; Schneeberger et al., 2015; Vandenberghe et al., 2017).
Elan Pharmaceuticals conducted the first clinical trial of active anti‐Aβ immunotherapy in 2001 by administering pre‐aggregated Aβ42 (AN‐1792) with a Th1 cell‐activating QS‐21 adjuvant (Gilman et al., 2005). However, this Phase IIa trial was halted after 6% of the patients developed severe meningoencephalitis, a side effect that was not seen in the preclinical mouse studies. The most likely cause of this adverse effect is that AN‐1792 elicited a T cell‐mediated immunological response by promoting T lymphocyte infiltration into the brain and exacerbating neuroinflammation. Vaccines containing safer Aβ antigens targeting smaller epitopes in the N‐terminus of Aβ were later developed but still induced side effects and did not produce clinical benefits.
Consequently, passive immunization with monoclonal antibodies directed at Aβ was developed as an alternative therapeutic strategy. However, these antibodies, namely, bapineuzumab (Salloway et al., 2014), gantenerumab (Ostrowitzki et al., 2017), and aducanumab (Sevigny et al., 2016), still failed to demonstrate cognitive or clinical benefit in patients and caused adverse effects such as symptomatic ARIAs and cerebral microhaemorrhages. In preclinical studies in transgenic animals, some immunization strategies exhibited encouraging results and decreased neuropathology and improved cognition function with few side effects. However, in subsequent clinical trials, ARIAs and other side effects frequently occurred in AD patients treated with these same strategies (Graham et al., 2017; Panza et al., 2019). Thus, the development of an animal model which replicates the mechanism of treatment and demonstrates the detrimental effects that would be seen in patients would advance the selection and assessment of anti‐AD drugs.
In the present study, we generated a novel mouse model, EAE/AD, by crossing APP/PS1 mice with EAE mice. EAE mice, also named B10.RIII mice, develop chronic and relapsing EAE after immunization with myelin basic protein. Notably, our EAE/AD model exhibited considerable neuroinflammation following Aβ immunization, even without myelin basic protein induction. This suggests that Aβ, acting as a self‐protein, can induce autoimmune reactions in EAE/AD mice, which is consistent with previous reports (Marchese et al., 2014; Sardi et al., 2011; Wang et al., 2014). In contrast, AOE1 immunization did not promote neuroinflammation or detrimental immune responses. These results demonstrated that a specific conformational epitope of Aβ oligomers is much safer than Aβ itself when used as an immunogen and that the EAE/AD mouse model can distinguish the safety of these two types of vaccines.
In previous studies, vaccines containing Aβ42 significantly decreased soluble and insoluble Aβ levels and plaque burden, inhibited the production of proinflammatory factors, restored cognitive deficits, and did not show any noticeable side effects in conventional AD animal models (Schenk et al., 1999). However, when administered to EAE/AD mice, this vaccine induced neuroinflammation and enhanced Aβ levels and plaque burden. Autoimmune responses would occur when active vaccines or passive antibodies targeted Aβ on the surface of neurons or APP, a membrane protein with an extracellular Aβ domain. The binding of Aβ‐targeted antibodies to Aβ or APP on neurons would induce complement activation, thus damaging neurons and promoting microglia to release more inflammatory factors. The existing APP transgenic mouse is not sensitive enough to show these immune responses, but the EAE/AD mice generated in this study clearly exhibited these processes. In comparison, the vaccine containing a conformational epitope of Aβ exhibited beneficial effects in APP and EAE/AD mice without inducing side effects.
Microglia are the resident innate immune cells of the CNS and have an essential function in maintaining homeostasis by constantly surveying the environment and clearing apoptotic debris and pathogens (Salter & Stevens, 2017). During AD pathogenesis, Aβ aggregates induce a switch in microglial phenotype from a resting ramified state to an activated state. These cells then release inflammatory mediators such as cytokines, chemokines, and ROS (Hansen, Hanson, & Sheng, 2018; Sarlus & Heneka, 2017). Moreover, over‐activated microglia exhibit a reduced ability to phagocytose and clear Aβ, resulting in the formation of more Aβ aggregates and further inflammation (Heneka et al., 2013). Excessive inflammation impairs the function of neurons and microglia, which obstructs the clearance of Aβ and creates a vicious cycle (Kinney et al., 2018). Numerous reports have indicated that the antibodies induced by Aβ vaccines contribute to Aβ clearance by promoting microglial activation (Heneka et al., 2015; Xiang et al., 2016). Indeed, our previous reports also demonstrated that the AOE1 vaccine significantly decreased the levels of soluble and fibrillar Aβ and attenuated neuroinflammation in APP/PS1 mice (Wang et al., 2017). However, immunization with Aβ42 promoted inflammation and enhanced Aβ burden in the brains of EAE/AD mice, suggesting that the immune responses triggered by the vaccine induced further microglial activation, promoted neuroinflammation, and suppressed Aβ clearance. Over‐activated microglia lose their ability to phagocytose Aβ and release proinflammatory cytokines such as IL‐1β, which can aggravate plaque formation by modulating APP expression and proteolysis (Heneka et al., 2015). Additionally, even systemic inflammation can impair microglial Aβ clearance and increase amyloid burden in transgenic murine models of amyloidosis through activation of https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1770 inflammasomes (Lee et al., 2008; Tejera et al., 2019; Wendeln et al., 2018). This damaged brain environment then propagates neuronal damage, leading to further cognitive deficits, which is consistent with previous reports showing that neuroinflammation directly contributes to AD (Ransohoff, 2016).
Substantial evidence has demonstrated that Aβ oligomers, not monomers or fibrils, play a critical role in the progression of AD (Ferreira, Lourenco, Oliveira, & De Felice, 2015; Selkoe & Hardy, 2016; Viola & Klein, 2015). Theoretically, antibodies targeting Aβ would preferentially bind to monomers and insoluble fibrils as they are more abundant in the brain, so fewer antibodies would be available to neutralize the more neurotoxic oligomeric and protofibrillar forms of Aβ. This may underlie the failure of previous clinical trials of immunotherapies. Compared to Aβ vaccines, anti‐Aβ monoclonal antibodies target only a single epitope of the multi‐epitopic Aβ molecule, so toxic Aβ oligomers in which the specific epitope was hidden would evade antibody neutralization (Rosenblum, 2014). This suggests that active immunization may be more effective than passive immunization when the antibody titre induced by a vaccine is sufficiently high. During aggregation, Aβ forms oligomers which contain unique structures not present in Aβ monomers. Therefore, oligomer‐specific epitopes have become promising targets of immunotherapeutic interventions in AD (Guerrero‐Munoz, Castillo‐Carranza, & Kayed, 2014). Indeed, our previous reports and the results of the present study demonstrated that oligomer‐specific vaccines and antibodies significantly reduced brain pathology and prevented cognitive deficits in models of AD in mice (Wang et al., 2017; Zha et al., 2016; Zhao et al., 2014).
In summary, the EAE/AD mouse model displayed distinct outcomes when immunized with vaccines containing Aβ42 and oligomer‐specific epitopes. Therefore, this mouse model is suitable for the selection and evaluation of anti‐AD immunotherapeutic strategies. Our results further confirmed that neuroinflammation markedly exacerbates AD progression and demonstrated that interventions specifically targeting Aβ oligomers may be safe and effective in the treatment of AD.
AUTHOR CONTRIBUTIONS
X.‐L.Y. and R.‐T.L. designed the experiments. X.‐M.L. and P.‐X.X. performed the behavioural experiments. J.Z. and Y.Z. conducted the immunohistochemistry and biochemistry experiments. X.‐L.Y. and R.‐T.L. wrote the manuscript.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for https://bpspubs.onlinelibrary.wiley.com/doi/abs/10.1111/bph.14207, https://bpspubs.onlinelibrary.wiley.com/doi/abs/10.1111/bph.14208 and https://bpspubs.onlinelibrary.wiley.com/doi/abs/10.1111/bph.14206, and as recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1. Aβ42 immunization rescued cognitive deficits in APP/PS1 but not EAE/AD mouse model. The short‐term working memory of APP/PS1 and EAE/AD mice immunized with Aβ42 or adjuvant was evaluated by Y‐maze, the time spent in the novel arm (A) and the number of entries to the novel arm (B) were measured. n = 8 mice/group. Data represent means ± SEM. *P < 0.05
Figure S2. Aβ42 immunization in the presence of QS‐21 adjuvant enhanced neuropathology in EAE/AD mouse model. (A) 6E10 immunostaining for plaques, Iba‐1 and GFAP immunostaining for gliosis, and hemosiderin staining for microhemorrhages in the brains of EAE/AD mice immunized with Aβ42 plus QS‐21 or QS‐21 alone. Scale bars: black, 200 μm; white, 100 μm. (B‐C) The levels of TNF‐α (B) and IL‐1β (C) in the brain lysates of EAE/AD mice immunized with Aβ42 plus QS‐21 or QS‐21 alone were detected by ELISA. n = 8 mice/group. Data represent means ± SEM. *P < 0.05
ACKNOWLEDGEMENTS
This work was supported by grants from the National Natural Science Foundation of China (81971610) and the National Science and Technology Major Projects of New Drugs (2018ZX09733001‐001‐008).
Yu X‐L, Zhu J, Liu X‐m, Xu P‐x, Zhang Y, Liu R‐t. Vaccines targeting the primary amino acid sequence and conformational epitope of amyloid‐β had distinct effects on neuropathology and cognitive deficits in EAE/AD mice. Br J Pharmacol. 2020;177:2860–2871. 10.1111/bph.15015
REFERENCES
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Mathie, A. , Peters, J. A. , Veale, E. L. , … Collaborators, C. G. T. P. (2019a). The Concise Guide To PHARMACOLOGY 2019/20: Catalytic receptors. British Journal of Pharmacology, 176, S247–S296. 10.1111/bph.14751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Mathie, A. , Peters, J. A. , Veale, E. L. , … Collaborators, C. G. T. P. (2019b). The Concise Guide To PHARMACOLOGY 2019/20: Enzymes. British Journal of Pharmacology, 176, S297–S396. 10.1111/bph.14752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Roberts, R. E. , Broughton, B. R. S. , Sobey, C. G. , George, C. H. , Stanford, S. C. , … Ahluwalia, A. (2018). Goals and practicalities of immunoblotting and immunohistochemistry: A guide for submission to the British Journal of Pharmacology. British Journal of Pharmacology, 175, 407–411. 10.1111/bph.14112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blennow, K. , de Leon, M. J. , & Zetterberg, H. (2006). Alzheimer's disease. Lancet, 368, 387–403. 10.1016/S0140-6736(06)69113-7 [DOI] [PubMed] [Google Scholar]
- Bohrmann, B. , Baumann, K. , Benz, J. , Gerber, F. , Huber, W. , Knoflach, F. , … Loetscher, H. (2012). Gantenerumab: A novel human anti‐Aβ antibody demonstrates sustained cerebral amyloid‐β binding and elicits cell‐mediated removal of human amyloid‐β. Journal of Alzheimer's Disease: JAD, 28, 49–69. 10.3233/JAD-2011-110977 [DOI] [PubMed] [Google Scholar]
- Buckwalter, M. S. , Coleman, B. S. , Buttini, M. , Barbour, R. , Schenk, D. , Games, D. , … Wyss‐Coray, T. (2006). Increased T cell recruitment to the CNS after amyloid β 1‐42 immunization in Alzheimer's mice overproducing transforming growth factor‐β1. The Journal of Neuroscience, 26, 11437–11441. 10.1523/JNEUROSCI.2436-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Congdon, E. E. , & Sigurdsson, E. M. (2018). Tau‐targeting therapies for Alzheimer disease. Nature Reviews Neurology, 14, 399–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis, M. J. , Alexander, S. , Cirino, G. , Docherty, J. R. , George, C. H. , Giembycz, M. A. , … Ahluwalia, A. (2018). Experimental design and analysis and their reporting II: Updated and simplified guidance for authors and peer reviewers. British Journal of Pharmacology, 175, 987–993. 10.1111/bph.14153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Feo, D. , Merlini, A. , Brambilla, E. , Ottoboni, L. , Laterza, C. , Menon, R. , … Bacigaluppi, M. (2017). Neural precursor cell‐secreted TGF‐β2 redirects inflammatory monocyte‐derived cells in CNS autoimmunity. The Journal of Clinical Investigation, 127, 3937–3953. 10.1172/JCI92387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMattos, R. B. , Bales, K. R. , Cummins, D. J. , Dodart, J. C. , Paul, S. M. , & Holtzman, D. M. (2001). Peripheral anti‐Aβ antibody alters CNS and plasma Aβ clearance and decreases brain Aβ burden in a mouse model of Alzheimer's disease. Proceedings of the National Academy of Sciences of the United States of America, 98, 8850–8855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira, S. T. , Lourenco, M. V. , Oliveira, M. M. , & De Felice, F. G. (2015). Soluble amyloid‐β oligomers as synaptotoxins leading to cognitive impairment in Alzheimer's disease. Frontiers in Cellular Neuroscience, 9, 191 10.3389/fncel.2015.00191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilman, S. , Koller, M. , Black, R. S. , Jenkins, L. , Griffith, S. G. , Fox, N. C. , … AN1792(QS‐21)‐201 Study Team . (2005). Clinical effects of Aβ immunization (AN1792) in patients with AD in an interrupted trial. Neurology, 64, 1553–1562. 10.1212/01.WNL.0000159740.16984.3C [DOI] [PubMed] [Google Scholar]
- Graham, W. V. , Bonito‐Oliva, A. , & Sakmar, T. P. (2017). Update on Alzheimer's disease therapy and prevention strategies. Annual Review of Medicine, 68, 413–430. 10.1146/annurev-med-042915-103753 [DOI] [PubMed] [Google Scholar]
- Guerrero‐Munoz, M. J. , Castillo‐Carranza, D. L. , & Kayed, R. (2014). Therapeutic approaches against common structural features of toxic oligomers shared by multiple amyloidogenic proteins. Biochemical Pharmacology, 88, 468–478. 10.1016/j.bcp.2013.12.023 [DOI] [PubMed] [Google Scholar]
- Hansen, D. V. , Hanson, J. E. , & Sheng, M. (2018). Microglia in Alzheimer's disease. The Journal of Cell Biology, 217, 459–472. 10.1083/jcb.201709069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … NC‐IUPHAR . (2018). The IUPHAR/BPS Guide to pharmacology in 2018: Updates and expansion to encompass the new guide to immunopharmacology. Nucleic Acids Research, 46, D1091–D1106. 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka, M. T. , Carson, M. J. , El Khoury, J. , Landreth, G. E. , Brosseron, F. , Feinstein, D. L. , … Herrup, K. (2015). Neuroinflammation in Alzheimer's disease. The Lancet Neurology, 14, 388–405. 10.1016/S1474-4422(15)70016-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka, M. T. , Kummer, M. P. , Stutz, A. , Delekate, A. , Schwartz, S. , Vieira‐Saecker, A. , … Golenbock, D. T. (2013). NLRP3 is activated in Alzheimer's disease and contributes to pathology in APP/PS1 mice. Nature, 493, 674–678. 10.1038/nature11729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayed, R. , & Glabe, C. G. (2006). Conformation‐dependent anti‐amyloid oligomer antibodies. Methods in Enzymology, 413, 326–344. 10.1016/S0076-6879(06)13017-7 [DOI] [PubMed] [Google Scholar]
- Kayed, R. , Head, E. , Thompson, J. L. , McIntire, T. M. , Milton, S. C. , Cotman, C. W. , & Glabe, C. G. (2003). Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science, 300, 486–489. 10.1126/science.1079469 [DOI] [PubMed] [Google Scholar]
- Kayed, R. , & Lasagna‐Reeves, C. A. (2013). Molecular mechanisms of amyloid oligomers toxicity. Journal of Alzheimer's Disease: JAD, 33(Suppl 1), S67–S78. [DOI] [PubMed] [Google Scholar]
- Kilkenny, C. , Browne, W. , Cuthill, I. C. , Emerson, M. , & Altman, D. G. (2010). Animal research: Reporting in vivo experiments: The ARRIVE guidelines. British Journal of Pharmacology, 160, 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinney, J. W. , Bemiller, S. M. , Murtishaw, A. S. , Leisgang, A. M. , Salazar, A. M. , & Lamb, B. T. (2018). Inflammation as a central mechanism in Alzheimer's disease. Alzheimers Dement (N Y), 4, 575–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landen, J. W. , Cohen, S. , Billing, C. B. Jr. , Cronenberger, C. , Styren, S. , Burstein, A. H. , … Binneman, B. (2017). Multiple‐dose ponezumab for mild‐to‐moderate Alzheimer's disease: Safety and efficacy. Alzheimers Dement (N Y), 3, 339–347. 10.1016/j.trci.2017.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, J. W. , Lee, Y. K. , Yuk, D. Y. , Choi, D. Y. , Ban, S. B. , Oh, K. W. , & Hong, J. T. (2008). Neuro‐inflammation induced by lipopolysaccharide causes cognitive impairment through enhancement of β‐amyloid generation. Journal of Neuroinflammation, 5, 37 10.1186/1742-2094-5-37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, S. Y. , Lu, S. , Yu, X. L. , Yang, S. G. , Liu, W. , Liu, X. M. , … Liu, R. T. (2018). Fruitless Wolfberry‐sprout extract rescued cognitive deficits and attenuated neuropathology in Alzheimer's disease transgenic mice. Current Alzheimer Research, 15, 856–868. 10.2174/1567205015666180404160625 [DOI] [PubMed] [Google Scholar]
- Long, J. M. , & Holtzman, D. M. (2019). Alzheimer disease: An update on pathobiology and treatment strategies. Cell, 179, 312–339. 10.1016/j.cell.2019.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lueptow, L. M. (2017). Novel object recognition test for the investigation of learning and memory in mice. Journal of Visualized Experiments, e55718 10.3791/55718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchese, M. , Cowan, D. , Head, E. , Ma, D. , Karimi, K. , Ashthorpe, V. , … Sakic, B. (2014). Autoimmune manifestations in the 3xTg‐AD model of Alzheimer's disease. Journal of Alzheimer's Disease: JAD, 39, 191–210. 10.3233/JAD-131490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrowitzki, S. , Lasser, R. A. , Dorflinger, E. , Scheltens, P. , Barkhof, F. , Nikolcheva, T. , … Klein, G. (2017). A phase III randomized trial of gantenerumab in prodromal Alzheimer's disease. Alzheimer's Research & Therapy, 9, 95 10.1186/s13195-017-0318-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panza, F. , Lozupone, M. , Logroscino, G. , & Imbimbo, B. P. (2019). A critical appraisal of amyloid‐β‐targeting therapies for Alzheimer disease. Nature Reviews Neurology, 15, 73–88. 10.1038/s41582-018-0116-6 [DOI] [PubMed] [Google Scholar]
- Pasquier, F. , Sadowsky, C. , Holstein, A. , Leterme Gle, P. , Peng, Y. , Jackson, N. , … ACC‐001 (QS‐21) Study Team . (2016). Two phase 2 multiple ascending‐dose studies of Vanutide Cridificar (ACC‐001) and QS‐21 adjuvant in mild‐to‐moderate Alzheimer's disease. Journal of Alzheimer's Disease: JAD, 51, 1131–1143. 10.3233/JAD-150376 [DOI] [PubMed] [Google Scholar]
- Pfeifer, M. , Boncristiano, S. , Bondolfi, L. , Stalder, A. , Deller, T. , Staufenbiel, M. , … Jucker, M. (2002). Cerebral hemorrhage after passive anti‐Aβ immunotherapy. Science, 298, 1379 10.1126/science.1078259 [DOI] [PubMed] [Google Scholar]
- Polanco, J. C. , Li, C. , Bodea, L. G. , Martinez‐Marmol, R. , Meunier, F. A. , & Gotz, J. (2018). Amyloid‐β and tau complexity—Towards improved biomarkers and targeted therapies. Nature Reviews Neurology, 14, 22–39. 10.1038/nrneurol.2017.162 [DOI] [PubMed] [Google Scholar]
- Ransohoff, R. M. (2016). How neuroinflammation contributes to neurodegeneration. Science, 353, 777–783. 10.1126/science.aag2590 [DOI] [PubMed] [Google Scholar]
- Robinson, A. P. , Harp, C. T. , Noronha, A. , & Miller, S. D. (2014). The experimental autoimmune encephalomyelitis (EAE) model of MS: Utility for understanding disease pathophysiology and treatment. Handbook of Clinical Neurology, 122, 173–189. 10.1016/B978-0-444-52001-2.00008-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenblum, W. I. (2014). Why Alzheimer trials fail: removing soluble oligomeric β amyloid is essential, inconsistent, and difficult. Neurobiology of Aging, 35, 969–974. [DOI] [PubMed] [Google Scholar]
- Salloway, S. , Sperling, R. , Fox, N. C. , Blennow, K. , Klunk, W. , Raskind, M. , … Bapineuzumab 301 and 302 Clinical Trial Investigators . (2014). Two phase 3 trials of bapineuzumab in mild‐to‐moderate Alzheimer's disease. The New England Journal of Medicine, 370, 322–333. 10.1056/NEJMoa1304839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salter, M. W. , & Stevens, B. (2017). Microglia emerge as central players in brain disease. Nature Medicine, 23, 1018–1027. 10.1038/nm.4397 [DOI] [PubMed] [Google Scholar]
- Sardi, F. , Fassina, L. , Venturini, L. , Inguscio, M. , Guerriero, F. , Rolfo, E. , & Ricevuti, G. (2011). Alzheimer's disease, autoimmunity and inflammation. The good, the bad and the ugly. Autoimmunity Reviews, 11, 149–153. 10.1016/j.autrev.2011.09.005 [DOI] [PubMed] [Google Scholar]
- Sarlus, H. , & Heneka, M. T. (2017). Microglia in Alzheimer's disease. The Journal of Clinical Investigation, 127, 3240–3249. 10.1172/JCI90606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenk, D. (2002). Amyloid‐β immunotherapy for Alzheimer's disease: The end of the beginning. Nature Reviews. Neuroscience, 3, 824–828. 10.1038/nrn938 [DOI] [PubMed] [Google Scholar]
- Schenk, D. , Barbour, R. , Dunn, W. , Gordon, G. , Grajeda, H. , Guido, T. , … Seubert, P. (1999). Immunization with amyloid‐β attenuates Alzheimer‐disease‐like pathology in the PDAPP mouse. Nature, 400, 173–177. 10.1038/22124 [DOI] [PubMed] [Google Scholar]
- Schneeberger, A. , Hendrix, S. , Mandler, M. , Ellison, N. , Burger, V. , Brunner, M. , … Imarhiagbe, D. (2015). Results from a Phase II study to assess the clinical and immunological activity of AFFITOPE(R) AD02 in patients with early Alzheimer's disease. The Journal of Prevention of Alzheimer's Disease, 2, 103–114. 10.14283/jpad.2015.63 [DOI] [PubMed] [Google Scholar]
- Selkoe, D. J. , & Hardy, J. (2016). The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO Molecular Medicine, 8, 595–608. 10.15252/emmm.201606210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevigny, J. , Chiao, P. , Bussiere, T. , Weinreb, P. H. , Williams, L. , Maier, M. , … O'Gorman, J. (2016). The antibody aducanumab reduces Aβ plaques in Alzheimer's disease. Nature, 537, 50–56. 10.1038/nature19323 [DOI] [PubMed] [Google Scholar]
- Shafiei, S. S. , Guerrero‐Muñoz, M. J. , & Castillo‐Carranza, D. L. (2017). Tau oligomers: Cytotoxicity, propagation, and mitochondrial damage. Frontiers in Aging Neuroscience, 9, 83 10.3389/fnagi.2017.00083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tejera, D. , Mercan, D. , Sanchez‐Caro, J. M. , Hanan, M. , Greenberg, D. , Soreq, H. , … Heneka, M. T. (2019). Systemic inflammation impairs microglial Aβ clearance through NLRP3 inflammasome. The EMBO Journal, 38, e101064 10.15252/embj.2018101064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenberghe, R. , Riviere, M. E. , Caputo, A. , Sovago, J. , Maguire, R. P. , Farlow, M. , … Graf, A. (2017). Active Aβ immunotherapy CAD106 in Alzheimer's disease: A phase 2b study. Alzheimers Dement (N Y), 3, 10–22. 10.1016/j.trci.2016.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viola, K. L. , & Klein, W. L. (2015). Amyloid β oligomers in Alzheimer's disease pathogenesis, treatment, and diagnosis. Acta Neuropathologica, 129, 183–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vollmar, P. , Kullmann, J. S. , Thilo, B. , Claussen, M. C. , Rothhammer, V. , Jacobi, H. , … Hemmer, B. (2010). Active immunization with amyloid‐β 1‐42 impairs memory performance through TLR2/4‐dependent activation of the innate immune system. Journal of Immunology, 185, 6338–6347. 10.4049/jimmunol.1001765 [DOI] [PubMed] [Google Scholar]
- Wang, S. , Yu, Y. , Geng, S. , Wang, D. , Zhang, L. , Xie, X. , … Wang, B. (2014). A coimmunization vaccine of Aβ42 ameliorates cognitive deficits without brain inflammation in an Alzheimer's disease model. Alzheimer's Research & Therapy, 6, 26 10.1186/alzrt256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, S. W. , Liu, D. Q. , Zhang, L. X. , Ji, M. , Zhang, Y. X. , Dong, Q. X. , … Liu, R. T. (2017). A vaccine with Aβ oligomer‐specific mimotope attenuates cognitive deficits and brain pathologies in transgenic mice with Alzheimer's disease. Alzheimer's Research & Therapy, 9, 41 10.1186/s13195-017-0267-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, X. P. , Zhang, J. H. , Wang, Y. J. , Feng, Y. , Zhang, X. , Sun, X. X. , … Liu, R. T. (2009). Conformation‐dependent single‐chain variable fragment antibodies specifically recognize β‐amyloid oligomers. FEBS Letters, 583, 579–584. 10.1016/j.febslet.2008.12.064 [DOI] [PubMed] [Google Scholar]
- Wendeln, A. C. , Degenhardt, K. , Kaurani, L. , Gertig, M. , Ulas, T. , Jain, G. , … Neher, J. J. (2018). Innate immune memory in the brain shapes neurological disease hallmarks. Nature, 556, 332–338. 10.1038/s41586-018-0023-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiessner, C. , Wiederhold, K. H. , Tissot, A. C. , Frey, P. , Danner, S. , Jacobson, L. H. , … Staufenbiel, M. (2011). The second‐generation active Aβ immunotherapy CAD106 reduces amyloid accumulation in APP transgenic mice while minimizing potential side effects. The Journal of Neuroscience, 31, 9323–9331. 10.1523/JNEUROSCI.0293-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcock, D. M. , Rojiani, A. , Rosenthal, A. , Subbarao, S. , Freeman, M. J. , Gordon, M. N. , & Morgan, D. (2004). Passive immunotherapy against Aβ in aged APP‐transgenic mice reverses cognitive deficits and depletes parenchymal amyloid deposits in spite of increased vascular amyloid and microhemorrhage. Journal of Neuroinflammation, 1, 24 10.1186/1742-2094-1-24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winblad, B. , Andreasen, N. , Minthon, L. , Floesser, A. , Imbert, G. , Dumortier, T. , … Graf, A. (2012). Safety, tolerability, and antibody response of active Aβ immunotherapy with CAD106 in patients with Alzheimer's disease: Randomised, double‐blind, placebo‐controlled, first‐in‐human study. The Lancet Neurology, 11, 597–604. 10.1016/S1474-4422(12)70140-0 [DOI] [PubMed] [Google Scholar]
- Xiang, X. , Werner, G. , Bohrmann, B. , Liesz, A. , Mazaheri, F. , Capell, A. , … Haass, C. (2016). TREM2 deficiency reduces the efficacy of immunotherapeutic amyloid clearance. EMBO Molecular Medicine, 8, 992–1004. 10.15252/emmm.201606370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zha, J. , Liu, X. M. , Zhu, J. , Liu, S. Y. , Lu, S. , Xu, P. X. , … Liu, R. T. (2016). A scFv antibody targeting common oligomeric epitope has potential for treating several amyloidoses. Scientific Reports, 6, 36631 10.1038/srep36631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, L. F. , Yu, X. L. , Ji, M. , Liu, S. Y. , Wu, X. L. , Wang, Y. J. , & Liu, R. T. (2018). Resveratrol alleviates motor and cognitive deficits and neuropathology in the A53T α‐synuclein mouse model of Parkinson's disease. Food & Function, 9, 6414–6426. 10.1039/c8fo00964c [DOI] [PubMed] [Google Scholar]
- Zhao, M. , Wang, S. W. , Wang, Y. J. , Zhang, R. , Li, Y. N. , Su, Y. J. , … Liu, R. T. (2014). Pan‐amyloid oligomer specific scFv antibody attenuates memory deficits and brain amyloid burden in mice with Alzheimer's disease. Current Alzheimer Research, 11, 69–78. 10.2174/15672050113106660176 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Aβ42 immunization rescued cognitive deficits in APP/PS1 but not EAE/AD mouse model. The short‐term working memory of APP/PS1 and EAE/AD mice immunized with Aβ42 or adjuvant was evaluated by Y‐maze, the time spent in the novel arm (A) and the number of entries to the novel arm (B) were measured. n = 8 mice/group. Data represent means ± SEM. *P < 0.05
Figure S2. Aβ42 immunization in the presence of QS‐21 adjuvant enhanced neuropathology in EAE/AD mouse model. (A) 6E10 immunostaining for plaques, Iba‐1 and GFAP immunostaining for gliosis, and hemosiderin staining for microhemorrhages in the brains of EAE/AD mice immunized with Aβ42 plus QS‐21 or QS‐21 alone. Scale bars: black, 200 μm; white, 100 μm. (B‐C) The levels of TNF‐α (B) and IL‐1β (C) in the brain lysates of EAE/AD mice immunized with Aβ42 plus QS‐21 or QS‐21 alone were detected by ELISA. n = 8 mice/group. Data represent means ± SEM. *P < 0.05