

Abstract
The renin–angiotensin system (RAS) now underlies the successful treatment of almost 50% of the patients in cardiovascular medicine, with serious possibilities of extension to diabetes, Alzheimer's disease and cancer. This clinical transformation started just over 50 years ago, with the unexpected identification of a bradykinin‐potentiating peptide from snake venom, as a potent inhibitor of ACE which led to the development of the first synthetic inhibitor, captopril, followed by the angiotensin receptor blockers. This article analyses the transformation of the RAS into its different stages, from academic experiments to clinical use and back to the laboratory, identifying the critical events involved, both clinical and scientific. The analysis also assesses the contributions of chance, coincidence, and conviction that were crucial in this transformation. Although questions remain, the transformation of the RAS over the past five decades provides a success story for medicine, for pharmacology, and, most significantly, for patients.
Abbreviations
- Ang1‐7
angiotensin 1‐7
- ARB
angiotensin receptor blocker
- BPF
bradykinin‐potentiating factor
- ESRD
end‐stage renal disease
- NEP
neutral endopeptidase, neprilysin
- RAS
renin–angiotensin system
1. INTRODUCTION
The renin–angiotensin system (RAS) had its beginnings at the end of the 19th century (Tigerstedt & Bergman, 1898), and, over the following six decades, all of its components were identified and functionally characterised (see Fasciolo, 1990; Page & Bumpus, 1961). https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2413, released from the kidney, cleaved a decapeptide from a plasma protein, renin substrate (angiotensinogen). This decapeptide (https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=583) was biologically inactive but was hydrolysed to the active octapeptide, https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2504, by another plasma protein, https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1613. Angiotensin II was a potent vasoconstrictor and stimulator of https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2872 secretion, and these activities were lost after any further cleavage to smaller peptides (Figure 1). Thus, in 1965, an authoritative Pharmacological Review of the RAS concluded “The renin‐angiotensin system has emerged as a truly hormonal controlling system intimately concerned with electrolyte balance …” and “It seems unlikely that the system usually operates as a direct pressor system” (Peart, 1965).
FIGURE 1.
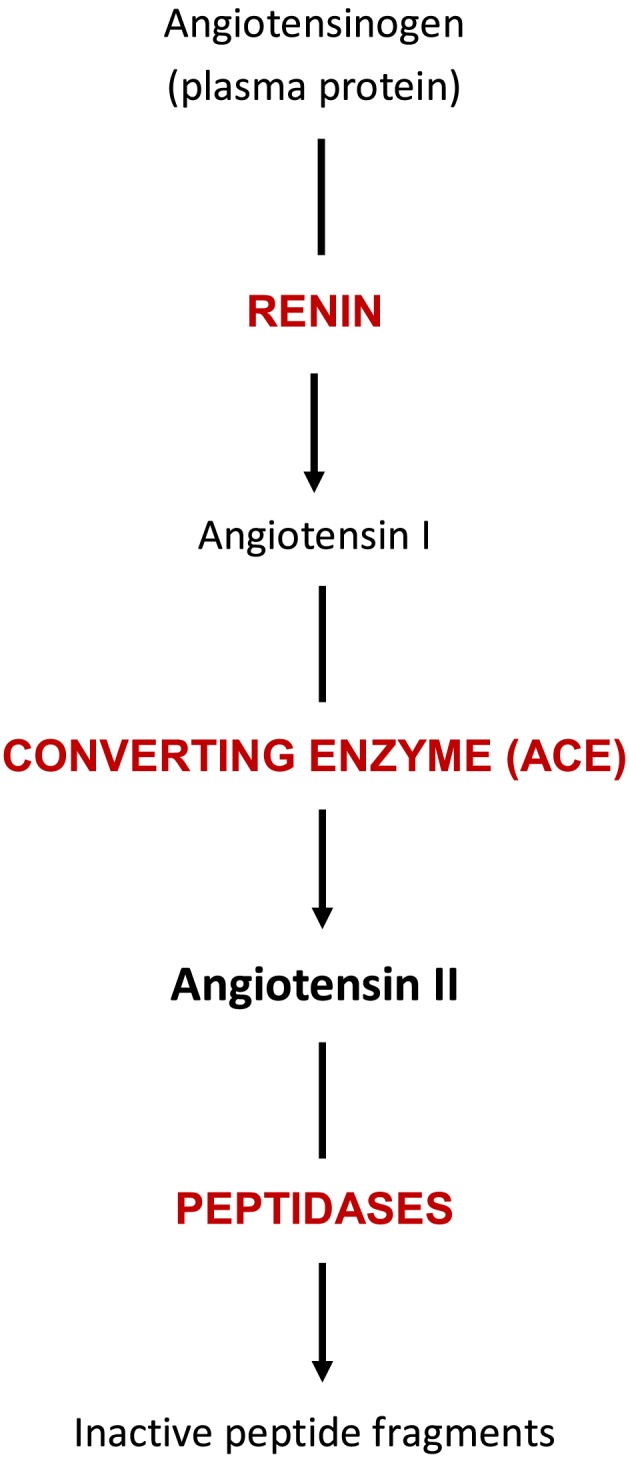
The RAS in 1965. Angiotensinogen, or renin substrate, is a plasma protein synthesised, like other α‐globulins, by the liver. Renin is synthesised in the juxtaglomerular cells of the kidney and secreted into the renal blood vessels and then into the systemic circulation. There renin cleaves a decapeptide, angiotensin I, from renin substrate. This peptide has little or no biological activity and is a substrate for several plasma peptidases, one of which, the dipeptidyl carboxypeptidase ACE, forms the octapeptide, angiotensin II. This peptide was the only fragment of angiotensin I with biological activity as a vasopressor agent and a stimulator of aldosterone release. Subsequent cleavage of angiotensin II into smaller fragments by other plasma peptidases yields biologically inactive peptides
However, over the next six decades, the RAS, in all its aspects was transformed well beyond any predictions, possibilities or fanciful speculations. Now, in 2020, the RAS underlies the clinical treatment of at least 50% of the patient population in cardiovascular medicine (including hypertension!) and has brought undoubted benefit to many millions of patients. This clinical transformation has been accompanied by an equal transformation of the scientific basis of the RAS. Particularly relevant, in the present context, is that in 1965, Peart could adequately cover the topic of the ACE in one short paragraph of 42 words. In 2013, the Pharmacological Review on ACE had eight authors and 36 pages of printed text (Bernstein et al., 2013).
The present article seeks to analyse this transformation into its different stages, to elucidate how and why those stages developed and to identify events critical to that development. Because the present state of the RAS represents a success story for pharmacology, such an analysis could provide insights into this success, insights that could help generate other pharmacological successes. Particularly, the analysis could identify the contribution of non‐scientific factors such as chance, coincidence and conviction, to the scientific advances in the RAS and its application to medicine. However, what is clear is that this remarkable development of the RAS had, as its starting point, the discovery of the ACE inhibitors.
2. EARLY WORK IN ACE INHIBITION
That discovery actually started in the same year as Peart's review, but in a very different system, that of a relatively newly discovered endogenous peptide, https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=649. In 1965, a young Brazilian pharmacologist, Sergio Ferreira, published a paper in the British Journal of Pharmacology based on his PhD thesis, describing the properties of an extract of snake venom, a partly purified mixture of several short peptides (5–11 residues) and called bradykinin‐potentiating factor (BPF). This BPF had no direct myotropic activity, but it did potentiate the actions of the nonapeptide bradykinin, most likely due to inhibition of the metabolism of bradykinin to inactive fragments (Ferreira, 1965). Importantly, although there were chemicals, such as 2,3‐dimercaptopropanol, which also potentiated bradykinin by blocking its metabolism, the BPF was the first naturally occurring bradykininase inhibitor.
In the same year, Ferreira joined John Vane's group at the Pharmacology Department in the Royal College of Surgeons in London, as a post‐doctoral fellow. This was not his original plan, which had been to join Paton at the Oxford Department of Pharmacology, for post‐doctoral study. However, his wife had secured a research post in London, and as Paton and Vane had been colleagues at the Department at the Royal College of Surgeons, it was arranged that Ferreira should transfer his fellowship to London. Vane was just starting his study of the fate of endogenous vasoactive substances in the circulation, later summarised in his Gaddum Lecture (Vane, 1969), using a newly developed bioassay, the blood‐bathed organ technique (Vane, 1964), to measure levels of vasoactive hormones in vivo in experimental animals. As Ferreira's experience had been with bradykinin, he was allocated this peptide for investigation. His first paper on the fate of bradykinin in the circulation (Ferreira & Vane, 1967) showed that there was extensive (80%) inactivation of this peptide on a single passage through the pulmonary circulation. This inactivation was selective in that https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2086, a peptide closely related to bradykinin, did not undergo such a high level of inactivation and, furthermore, the inactivation of bradykinin was blocked by the BPF extract, administered intravenously. While Ferreira was carrying out his studies on bradykinin, another post‐doctoral fellow, Kevin Ng, started to assess the fate of another endogenous vasoactive plasma peptide, angiotensin II, in vivo using the same blood‐bathed organ technique and including studies of its biologically inactive precursor, the decapeptide, angiotensin I. From these experiments (Ng & Vane, 1967, 1968), two interesting results emerged. One was that passage through the pulmonary circulation did not inactivate angiotensin II but did convert angiotensin I to angiotensin II. Furthermore, this conversion in the pulmonary vasculature was more extensive and more rapid than that after incubation with blood, which was, then, the sole location of ACE activity (Helmer, 1955; Skeggs, Kahn, & Shumway, 1956). These findings implied that there was a peptidase activity associated with the pulmonary vascular bed that was highly active and selective towards the angiotensin peptides. Because the earlier studies with bradykinin had also implied a highly active but selective peptidase activity in the pulmonary circulation, Ng and Vane suggested that these two activities might be properties of the same enzyme (Ng & Vane, 1968). Another further relevant coincidence was that a cell‐free extract of dog lung with ACE activity and very little angiotensinase activity had been prepared, in Vane's group. Thus, in 1968, it was immediately possible to test, in vitro, the lung ACE for bradykininase activity and then to find out if Ferreira's BPF inhibited both the ACE and bradykininase activities of the lung preparation. The results of these highly speculative experiments showed clearly that both ACE and bradykininase activities were present in the same subcellular fraction of dog lung and, critically, that both activities were inhibited by the BPF (Bakhle, 1968).
As the BPF was a mixture of peptides, the next stage was for Ferreira, in a successful collaboration with Lewis Greene, to isolate six separate peptides from BPF, one pentapeptide and the others, nonapeptides and decapeptides. The profile of activity of these purified peptides as bradykinin potentiators (Ferreira, Bartelt, & Greene, 1970) was the same as their profile as ACE inhibitors (Ferreira, Greene, et al., 1970), further supporting the possibility that ACE was also the potent lung bradykininase.
These early years of ACE inhibition were crucially influenced by chance events and coincidences. If Ferreira had gone to Oxford as he originally intended, it is unlikely that he would have carried out the experiments that Vane was planning, and Vane would probably not have used bradykinin as a substrate to study but would have concentrated on the angiotensin peptides. It is even more unlikely that the BPF would have been tested for activity against ACE. So Ferreira's “second choice” turned out to be the critical choice in the discovery of the ACE inhibitors and the subsequent transformation of the RAS.
Further, the work with angiotensin I, carried out by Kevin Ng, was made possible by a ready supply of synthetic angiotensin I provided by Wilkinson, a chemist at the Wellcome Laboratories in Kent, who used the solid phase synthesis, then newly developed by Merrifield. The first experiments showing inhibition of ACE by the BPF were more than highly speculative; they were entirely dependent on temporal and spatial coincidences. The results of Ferreira and of Ng had to be fresh in the minds of Vane's group, and the lung enzyme and BPF, along with a ready supply of angiotensin I all had to be available at the same time. In these circumstances, it was easier to do the experiment first and to assess its results, if necessary, later. A more extensive scientific analysis of the reasons for testing BPF against ACE would only have discouraged the experiments.
3. DEVELOPMENT AT E.R. SQUIBB
However, real pharmacological progress with the BPF peptides as ACE inhibitors required much greater technical expertise and effort than either Ferreira, now back in Brazil, or Vane in London could provide. So Ferreira and Vane took the project to E.R. Squibb, who were then a relatively small pharmaceutical company in New Jersey. They were able to persuade Squibb that a synthetic, low MW analogue of the BPF nonapeptide (the most potent peptide isolated) would provide a compound with potential use as an anti‐hypertensive drug, based on the inhibition of the formation of the known vasopressor angiotensin II. Although the majority of the clinical opinions obtained, at the time, did not support that possibility {Smith & Vane, 2003). Squibb assigned two researchers to this project, a biochemist, David Cushman, and a peptide chemist, Miguel Ondetti. Over the next 6 years, Cushman devised the first spectrophotometric assay for ACE activity using purified ACE from rabbit lung along with a synthetic substrate (Cushman & Cheung, 1971), an assay that has been used with variations ever since. Ondetti succeeded in sequencing and then synthesising the most potent nonapeptide from BPF, known as SQ 20881 or teprotide (Ondetti et al., 1971), which allowed it to be used in a few patients to demonstrate its efficacy as an anti‐hypertensive agent (Case et al., 1976; Gavras et al., 1974). However, Cushman and Ondetti did not succeed in their primary aim, to design a potent, low MW, orally active analogue of the nonapeptide, and so Squibb decided to close the project. According to Cushman (personal communication), it was while he was clearing out his ACE‐related reprints that he came across a paper by Byers and Wolfenden (1973) on carboxypeptidase inhibitors, which inspired him to design and Ondetti to synthesise and to test another dozen low MW compounds. Among these few compounds was SQ 14225 (Ondetti, Rubin, & Cushman, 1977) which, as https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5158, was approved by the FDA in 1981 for hypertension. Many other ACE inhibitors (https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=6322, https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=6360, https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=6339 and https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=6367) from other companies followed soon after.
At Squibb, success of the search for low MW, orally available, ACE inhibitors depended crucially on the actions and interactions of the people most directly involved, particularly John Vane, Arnold Welch and David Cushman. Vane and Welch had met some years earlier at the Department of Pharmacology at Yale Medical School, when Welch was Chairman of the Department and Vane an Assistant Professor. In 1967, Welch moved to E.R. Squibb as Director of the Squibb Institute for Medical Research and soon afterwards Vane joined as Senior Consultant in Pharmacology. When Ferreira and Vane brought their results and ideas about BPF and ACE inhibition to Squibb, Welch was convinced of the viability of the project to use the BPF peptides as lead compounds in the design of synthetic non‐peptide inhibitors. However, there was little evidence of the clinical utility of ACE inhibitors and most clinical opinion was negative. Welch's decision to initiate the search was strongly influenced by his good relationship with Vane and by his trust in Vane's scientific judgement. The personality of the biochemist selected by Squibb to lead the project, Dave Cushman, was also crucial to its eventual success, as shown by his persistence with the project when it had been officially closed. Cushman was also convinced, beyond reason for many, that a low MW, non‐peptide ACE inhibitor was just waiting to be found. In 1973, Vane gave up his consultancy at Squibb on his appointment as Research Director at the Wellcome Laboratories at Beckenham. This meant that the final design, synthesis and testing of captopril and its immediate analogues took place without his active support and encouragement.
4. CLINICAL OUTCOMES OF ACE INHIBITION
The clinical success of captopril in https://www.guidetopharmacology.org/GRAC/DiseaseDisplayForward?diseaseId=458—it was both effective and had relatively few side‐effects—had a number of consequences, not all immediately predictable. First and most predictable from the high prevalence of primary hypertension worldwide was that many millions of patients have received significant clinical benefit. The same features provided, very rapidly, a wide clinical experience with ACE inhibitors which gave clinicians the confidence to use these agents “off‐label” in a variety of cardiovascular pathologies. In this way, ACE inhibitors were soon used, again with success, to treat heart failure (Turini, Brunner, Gribic, Waeber, & Gavras, 1979; Gavras & Brunner, 2001; Sayer & Bhat, 2014) and in the aftermath of myocardial infarction (AIRE study 1993; Borghi et al., 2018). Another clinical area in which ACE inhibitors have provided real benefit is renal dysfunction. Initially, these inhibitors were used to treat the hypertension that often accompanies end‐stage renal disease (ESRD), the stage immediately before dialysis and transplantation. In this condition, two results quickly emerged. As expected, ACE inhibitors were effective anti‐hypertensive agents, but they also appeared to delay the progressive loss of renal function, i.e. they appeared to be reno‐protective. This protection did not correlate with the level of hypertension and patients with ESRD but without hypertension also showed improved renal function. Further studies showed that ACE inhibitors were reno‐protective before the end stage was reached and benefit could be obtained in patients with macroalbuminuria and then in patients with microalbuminuria, independent of any anti‐hypertensive action (Persson, Lindhardt, Rossing, & Parving, 2016). Such early prevention of renal damage and dysfunction is of particular relevance in diabetic nephropathy (Ma, Kam, Yan, & Lam, 2010; Ruggenenti, 2017). There is now a strong suggestion (Persson et al., 2016) that ACE inhibitors should be used prophylactically to prevent renal dysfunction in patients with either Type 1 or Type 2 diabetes, with or without hypertension. The case for such prophylactic use is strengthened by the benefit provided by ACE inhibitors in another microvascular complication of diabetes, retinopathy (Wang et al., 2015). Prophylactic use of ACE inhibitors is already known to be effective in cardiovascular disease, as a component of the polypill, a fixed dose combination of aspirin, a statin and an anti‐hypertensive agent. Most recently, three reports have shown polypill treatment, for 12 months or 5 years, to reduce the incidence and risk of cardiovascular events in patient populations lacking access to adequate medical care (Muñoz et al., 2019; Roshandel et al., 2019; Selak et al., 2019).
A further possible target for ACE inhibitors is the microvasculature of tumours, suggesting that these inhibitors might be effective adjunct therapy for cancer (George, Thomas, & Hannan, 2010; Radin, Krebs, Maqsudlu, & Patel, 2018; Xu et al., 2017). However, the growing interest and data accumulation in another clinical area, dementia, may reflect non‐vascular effects of the RAS (Farag et al., 2017; Santos et al., 2018; Wharton et al., 2018).
5. PHARMACOLOGICAL OUTCOMES OF ACE INHIBITION
The first pharmacological consequence of the rapidly established benefit of ACE inhibitors in primary hypertension was the powerful impetus it provided to the search for angiotensin receptor antagonists. Although earlier work (Brunner, Gavras, Ribeiro, & Posternak, 1976; Streeten, Anderson, Freiberg, & Dalakos, 1975) had shown that https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=598 (Sar1‐Ala8‐angiotensin II) exerted anti‐hypertensive effects, this compound had two major disadvantages, it was a peptide and thus could not be administered orally and, pharmacologically, it was actually a very weak partial agonist, capable of raising BP before blocking angiotensin receptors. Furthermore, at the time, there was not enough evidence that angiotensin receptor antagonists (later called angiotensin receptor blockers, ARBs) would be clinically useful, and thus, there was little support for further research. However, once the clinical benefits of ACE inhibition had been demonstrated, the search for ARBs was re‐invigorated, successfully, in many pharmaceutical companies (see Timmermans et al., 1993).
The first ARB in clinical use was https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=590, approved by the FDA in 1995, followed very soon by many others including https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=587, https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=3937 and https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=592. They are now used almost as widely as the ACE inhibitors and their main clinical advantage is the absence of the dry cough associated with inhibition of ACE. In parallel with this pharmaceutical development, two receptors for angiotensin II, https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=34 and https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=35, with significantly different structures, ligands and actions were identified and the receptors cloned (Karnik et al., 2015). The ARBs in clinical use are antagonists at the AT1 receptor, with minimal effects at the AT2 receptor. This selectivity, together with the almost opposite effects of AT2 receptor agonists on the vasculature, has raised the possibility that some of the benefit of the ARBs could derive from increased activation of AT2 receptors and not only from the blockade of AT1 receptors (Carey, 2017; Wang et al., 2017).
6. PHYSIOLOGICAL OUTCOMES OF ACE INHIBITION
The clinical success of the ARBs, added to that of the ACE inhibitors, has undoubtedly driven and sustained the further expansion of the physiology of the RAS (Figure 2). The major advance has been the uncovering of the parallel, “alternative,” or “depressor” RAS, with a different converting enzyme, https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1614 (Kuba, Imai, Ohto‐Nakanishi, & Penninger, 2010), a new endogenous peptide agonist, the heptapeptide https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=582, that acts on a new receptor, https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=150, for which a selective synthetic agonist and antagonist have been identified (Karnik, Singh, Tirupula, & Unal, 2017). The effects of Ang1‐7 and other agonists at Mas1 receptors are mostly opposed to the effects of angiotensin II at AT1 receptors, further supporting the concept of a RAS maintained in balance, by the opposing actions of endogenous angiotensin peptides (Santos et al., 2018). Recent work on the alternative, parallel RAS has identified another bioactive heptapeptide, https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=6065 (Ala1‐Ang1‐7), as an agonist at another orphan receptor, https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=152 (Oliveira et al., 2019; Santos et al., 2018; Villela, Passos‐Silva, & Santos, 2014). Alamandine is generated from Ang1‐7 by a decarboxylase (Figure 2), not a peptidase, raising the possibility of other bioactive Ala1‐peptides derived from angiotensin I and II (Santos et al., 2019). Although the clinical relevance of these newest components of the RAS is not yet established (Tetzner et al., 2018), the prospect, of another RAS‐based intervention in cardiovascular disease will continue to drive research in this area.
FIGURE 2.
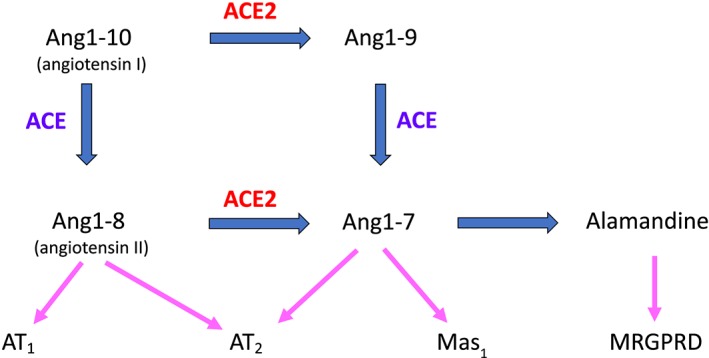
Development of the RAS since 1965. The first advance was the identification of the angiotensin AT1 and AT2 receptors, with the same endogenous agonist, angiotensin II (Ang1‐8) but with significantly different, almost antagonistic, outcomes. The clinically used ARBs are selective antagonists of the AT1 receptor. The alternative RAS comprises a carboxypeptidase (ACE2) removing one peptide residue at a time, for which angiotensin II is a substrate; both properties are marked differences from ACE. The product of ACE2 activity on angiotensin II is Ang1‐7 which is an agonist at both AT2 receptors and its own receptor, Mas1 (where angiotensin II is not an agonist). The most recent component identified is alamandine, another heptapeptide derived from Ang1‐7 by decarboxylation of the N‐terminal Asp. Alamandine has its own receptor, an orphan GPCR known as MRGPRD, at which neither angiotensin II nor Ang1‐7 are agonists
The scientific expansion of the RAS has also been anatomical, as the components of the RAS are now found in many organs, apart from the kidney, including the heart (Zhuo et al., 1998) and, particularly, the CNS (Ganten et al., 1971; McKinley et al., 2003; Santos et al., 2018). Consequently, circulating levels of angiotensin peptides and/or of renin may not reflect the activity of the RAS in any given tissue which may control its own levels of angiotensin peptides, distinct from that of any other tissue or the circulating levels. For the CNS, the blood–brain barrier ensures that the RAS located here is separated from the peripheral RAS and all the necessary substrates, enzymes, receptors and ligands are present in the CNS, along with the required control processes of stimulation or inhibition. The location of the RAS in the CNS has generated a new range of physiological and/or pharmacological target areas, including anxiety, learning, memory, physiological responses to stress and ischaemic stroke, apart from the centrally regulated effects on the peripheral cardiovascular system (Gebre, Altaye, Atey, Tuem, & Berhe, 2018; Gironacci, Vicario, Cerezo, & Silva, 2018; Santos et al., 2018).
7. CHARACTERISTIC ASPECTS OF THE TRANSFORMATION OF THE RAS
The advances noted above form only a part of the expansion of the RAS over the past 50 years, as almost every scientific aspect of this system has experienced a significant and sustained increase in knowledge. The new, transformed RAS has, however, to be seen in the context of the marked increase in pharmacological knowledge over the last five decades. For instance, 5‐HT, the PGs, the endogenous opioids, the endocannabinoid systems and NO have all developed into major signalling systems since 1968. Against such a background of continuous pharmacological discovery, the expansion and transformation of the RAS would seem to be just one example among many.
There are, nonetheless, some aspects peculiar to the RAS that deserve comment. For example, the most striking characteristic of the expansion of RAS‐based clinical therapies is that it has been clinically led, through clinicians using the ACE inhibitors off‐label, at first, and subsequently validating positive results with fully randomised clinical trials (see Sadat‐Ebrahimi et al., 2018). Two factors underlie this empirical, rather than the more usual, predicted, development of drug use. One is the relatively low level of side‐effects of the ACE inhibitors. The most common side‐effect is a dry cough which is perhaps less acceptable to companions, colleagues and family than to the patient, and the other, angioedema, is relatively very rare. Such low levels of possible harm would make off‐label use by clinicians easier to propose and execute, both ethically and practically.
The other factor is the lack of a clear physiological or pharmacological explanation for the effects of the ACE inhibitors. The immediate consequence of ACE inhibition has always been clear, a decreased amount of angiotensin II and consequent block of the biological activity of the RAS. The BPF nonapeptide (SQ 20881, teprotide) and captopril lowered BP in models and in patients with high‐renin hypertension (Gavras et al., 1974; Laffan et al., 1978), providing a clear proof of concept. However, there is no clear evidence that, in primary hypertension, the activity of the RAS is increased, relative to that in normotensive subjects. Equally, there was no clear reason to include an animal model of primary hypertension—the SHR strain of Wistar–Kyoto rats—in the definitive report on SQ 14225 (captopril), which recorded the anti‐hypertensive effects of ACE inhibition in the SHR, but without further comment (Laffan et al., 1978). A subsequent paper from the Squibb team (Antonaccio et al., 1979) showed chronic treatment of the SHR strain with SQ 14225 to be effective, and here, the authors considered and rejected several possible mechanisms. However, neither paper gave any reason for including a model of primary hypertension in studies of an agent particularly designed for high‐renin hypertension. Nevertheless, these experimental results provided support for testing captopril in patients with primary hypertension, tests which showed that patients responding to ACE inhibitors exhibited low, normal, or high levels of renin activity in plasma, with no correlation of response with levels of renin activity (Gavras, Faxon, Berkoben, Brunner, & Ryan, 1978).
This divergence from a simple relationship of beneficial effect with the hypertensive activity of the RAS was much clearer in renal disease where the benefits of ACE inhibitors did not correlate with the severity of hypertension (Manley, 2000). Laragh, an early and vigorous supporter of the relevance of the RAS to primary hypertension, proposed that renin alone is not the critical factor and that renin levels needed to be combined with sodium excretion in each patient to explain the response to RAS blockade (Laragh, 1981). However, this proposition has not been converted into a reliable predictor of sensitivity to RAS blockade, capable of identifying those clinical conditions that would respond to ACE inhibitors (or ARBs). In the absence of easily assessable markers of sensitivity to ACE inhibition, the empirical approach of careful clinical observation followed by off‐label use (“try this for a week and see if it helps”) is probably the best method of assessing the possible clinical value of ACE inhibition.
The expansion of the science of the RAS has, however, been much less empirical, using all the powerful new techniques, assays and concepts of the last five decades, including molecular biology, the application of MS and NMR to biology and all the “‘omics” available, to provide more knowledge and understanding of biological events. These advances have uncovered many actions of angiotensin II apart from vasoconstriction and aldosterone secretion, actions such as its proliferative effects on vascular smooth muscle and the generation of ROS characteristic of inflammatory responses. Added to these, the emergence of the alternative RAS (ACE2, Ang1‐7, alamandine and their bioactivity; Santos et al., 2018, 2019) provides many new and different possible explanations of the clinical effects of ACE inhibition. Nevertheless, even this scientific expansion has been made possible by the clinical successes. The RAS as the basis of important medical advances is much more “grant‐worthy” than the RAS as just another physiological system. “Translation” works both ways.
8. CONTRIBUTION OF BRADYKININ TO THE EFFECTS OF ACE INHIBITORS
Another question relevant to the action of the ACE inhibitors that has remained unanswered since the early work with the BPF is the contribution of bradykinin potentiation to the therapeutic benefits of ACE inhibition (Regoli & Gobeil, 2017; Taddei & Bortolotto, 2016). All the advances in science have confirmed the early suggestion that the ACE protein was also an important bradykininase and that inhibition of angiotensin II conversion is accompanied by an increased survival of bradykinin (Bernstein et al., 2013). The dry cough that is a side‐effect of ACE inhibition was attributed to the effects of raised levels of bradykinin, as was the rare case of angioedema. The latter effect had a higher incidence when ACE inhibition was combined with inhibition of https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1611), another enzyme inactivating bradykinin, and this was a major reason for the rejection by the FDA of omapatrilat (Campbell, 2018). The low levels of angioedema in the successful clinical trials of LCZ696 (https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=7857/valsartan), in which the NEP inhibitor is combined with an ARB, would support this assumption (Hubers & Brown, 2016). There is, therefore, little doubt that ACE inhibitors can and do increase bradykinin levels in vivo. Does such an increase make a clinical difference to the effects of ACE inhibitors?
A significant contribution by bradykinin to clinical outcomes is likely because of the indirect effects of this peptide. Bradykinin is known to stimulate the production of two major endogenous vasodilators, https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1915 and https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2509 (de Nucci, Warner, & Vane, 1988; Hannan, Davis, & Widdop, 2003), to stimulate the release of https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2392 (Pretorius, Rosenbaum, Vaughan, & Brown, 2003), to suppress apoptosis in endothelial cells (Kono et al., 2002) and to improve survival of endothelial progenitor cells (Sheng et al., 2013), all effects likely to provide benefit in cardiovascular disease. Comparison of the effects of ACE inhibitors with those of ARBs or of the https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4812 (Pantzaris, Karanikolas, Tsiotsios, & Velissaris, 2017), does show some differences, but they are not marked and not simple to evaluate. There are, for instance, differences between ACE inhibitors in the treatment of heart failure and in the aftermath of myocardial infarction (Borghi et al., 2018; Di Nicolantonio, Hu, Lavie, O'Keefe, & Bangalore, 2014) which could reflect differences in potency as bradykininase inhibitors. Also, and unexpectedly, bradykinin levels in vivo are raised during treatment with the ARB losartan (Campbell, Krum, & Esler, 2005).
As few studies have actually assayed bradykinin levels in patients during treatment with ACE inhibitors, the possible relevance of bradykinin to the action of ACE inhibitors will remain without a satisfactory answer. Overall, there has been less scientific progress with the https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2865–bradykinin system, relative to that of the RAS, even though these two peptide systems have been exposed to the same advances in scientific concepts and methods over the past 50 years. This difference may reflect the lack of a clinical use for a bradykininase inhibitor and/or a receptor antagonist, to provide the “justification” necessary to drive the research.
These unanswered questions do not, however, detract from the considerable benefits to patients and real scientific advances that have emerged in the last five decades of the development of the RAS. Such benefits and advances constitute a pharmacological success story and illustrate the power of the scientific method—experiments, analysis, and deduction—using all the new techniques and methods that have become available, to explore and develop the potential of new discoveries. However, this success story also demonstrates the crucial contributions made by non‐scientific factors, particularly chance, coincidence and conviction, to the new discoveries and to their successful development.
9. FINAL COMMENTS
The pharmacological success story of the development of the RAS over the past five decades exemplifies very clearly the two characteristic aims of Pharmacology, analysis of biological events into pathways, mechanisms and molecules and synthesis, reconstituting those molecules and mechanisms into clinically useful medicines. A comparison of the RAS with its companion plasma protein system, kallikrein–bradykinin, suggests that, although the logical application of scientific techniques and concepts to identify new compounds, new receptors, new enzymes and new physiological pathways has been essential and very effective in the development of the full potential of a “discovery,” the potential for translation into clinical medicine may be an intrinsic property of that biological system. Excellent science is necessary but not sufficient for a therapeutic advance. As for the initiating event, the “discovery” itself, that still depends, in Pasteur's words, on Chance and a prepared mind. This certainly was true for the transformation of the RAS over the last 50 years, as the initiating event was the discovery of ACE inhibition by the BPF peptides. The ACE inhibitors have been the key, not to Pandora's Box, but to a treasure chest of patient benefit and scientific advance.
NOMENCLATURE OF TARGETS AND LIGANDS
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org/, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2019/20 (Alexander, Christopoulos, et al., 2019; Alexander, Fabbro, et al., 2019).
CONFLICT OF INTEREST
The author declares no conflicts of interest.
ACKNOWLEDGEMENTS
It is a pleasure to acknowledge, gratefully, my colleagues who have been exposed to several versions of this article and have given me their time, their corrections, and their comments. However, I have to take sole responsibility for this final version.
Bakhle YS. How ACE inhibitors transformed the renin–angiotensin system. Br J Pharmacol. 2020;177:2657–2665. 10.1111/bph.15045
REFERENCES
- AIRE (Acute Infarction Ramipril Efficacy) study investigators . (1993). Effect of ramipril on mortality and morbidity of survivors of acute myocardial infarction with clinical evidence of heart failure. Lancet, 342, 821–828. [PubMed] [Google Scholar]
- Alexander, S. P. H. , Christopoulos, A. , Davenport, A. P. , Kelly, E. , Mathie Alistair, P. , … CGTP Collaborators . (2019). The concise guide to pharmacology 2019/20: G protein‐coupled receptors. British Journal of Pharmacology, 176, S21–S141. 10.1111/bph.14748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Mathie, A. , Peters, J. A. , Veale, E. L. , … CGTP Collaborators . (2019). The concise guide to pharmacology 2019/20: Enzymes. British Journal of Pharmacology, 176, S297–S396. 10.1111/bph.14752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonaccio, M. J. , Rubin, B. , Horovitz, Z. P. , Laffan, R. J. , Goldberg, M. E. , High, J. P. , … Zaidi, I. (1979). Effects of chronic treatment with captopril (SQ 14,225), an orally active inhibitor of angiotensin I‐converting enzyme, in spontaneously hypertensive rats. Japanese Journal of Pharmacology, 29, 285–294. 10.1254/jjp.29.285 [DOI] [PubMed] [Google Scholar]
- Bakhle, Y. S. (1968). Conversion of angiotensin I to angiotensin II by cell‐free extracts of dog lung. Nature, 220, 919–921. 10.1038/220919a0 [DOI] [PubMed] [Google Scholar]
- Bernstein, K. E. , Ong, F. S. , Blackwell, W. L. , Shah, K. H. , Giani, J. F. , Gonzalez‐Villalobos, R. A. , … Touyz, R. M. (2013). A modern understanding of the traditional and non‐traditional biological functions of angiotensin‐converting enzyme. Pharmacological Reviews, 65, 1–46. 10.1124/pr.112.006809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borghi, C. , Omboni, S. , Novo, S. , Vinereanu, D. , Ambrosio, G. , & Ambrosioni, E. (2018). Efficacy and safety of zofenopril versus ramipril in the treatment of myocardial infarction and heart failure: A review of the published and unpublished data of the randomized double‐blind SMILE‐4 study. Advances in Therapy, 35, 604–618. 10.1007/s12325-018-0697-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunner, H. R. , Gavras, H. , Ribeiro, A. B. , & Posternak, L. (1976). Angiotensin II blockade in normal man and patients with essential hypertension. Blood pressure effects depending on renin and sodium balance. Progress in Biochemical Pharmacology, 12, 145–162. [PubMed] [Google Scholar]
- Byers, L. D. , & Wolfenden, R. (1973). Binding of the biproduct analog benzylsuccinic acid by carboxypeptidase A. Biochemistry, 12, 2070–2078. 10.1021/bi00735a008 [DOI] [PubMed] [Google Scholar]
- Campbell, D. J. (2018). Neprilysin inhibitors and bradykinin. Front Med (Lausanne), 5, 257 10.3389/fmed.2018.00257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell, D. J. , Krum, H. , & Esler, M. D. (2005). Losartan increases bradykinin levels in hypertensive humans. Circulation, 111, 315–320. 10.1161/01.CIR.0000153269.07762.3B [DOI] [PubMed] [Google Scholar]
- Carey, R. M. (2017). AT2 receptors: Potential therapeutic targets for hypertension. American Journal of Hypertension, 30, 339–347. 10.1093/ajh/hpw121 [DOI] [PubMed] [Google Scholar]
- Case, D. B. , Wallace, J. M. , Keim, H. J. , Weber, M. A. , Drayer, J. I. M. , White, R. P. , … Laragh, J. H. (1976). Estimating renin participating in hypertension: Superiority of converting enzyme inhibitor over saralasin. The American Journal of Medicine, 61, 790–796. [DOI] [PubMed] [Google Scholar]
- Cushman, D. W. , & Cheung, H. S. (1971). Spectrophotometric assay and properties of the angiotensin‐converting enzyme of rabbit lung. Biochemical Pharmacology, 20, 1637–1648. 10.1016/0006-2952(71)90292-9 [DOI] [PubMed] [Google Scholar]
- de Nucci, G. , Warner, T. , & Vane, J. R. (1988). Effect of captopril on the bradykinin‐induced release of prostacyclin from guinea‐pig lungs and bovine aortic endothelial cells. British Journal of Pharmacology, 95, 783–788. 10.1111/j.1476-5381.1988.tb11705.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Nicolantonio, J. J. , Hu, T. , Lavie, C. J. , O'Keefe, J. H. , & Bangalore, S. (2014). Perindopril vs enalapril in patients with systolic heart failure: Systematic review and meta‐analysis. The Ochsner Journal, 14, 350–358. [PMC free article] [PubMed] [Google Scholar]
- Farag, E. , Sessler, D. I. , Ebrahim, Z. , Kurz, A. , Morgan, J. , Ahuja, S. , … John Doyle, D. (2017). The renin angiotensin system and the brain: New developments. Journal of Clinical Neuroscience, 46, 1–8. 10.1016/j.jocn.2017.08.055 [DOI] [PubMed] [Google Scholar]
- Fasciolo, J. C. (1990). The experimental observation that led to the discovery of angiotensin. 1939 Buenos Aires, Argentina. Hypertension, 16, 194–198. 10.1161/01.HYP.16.2.194 [DOI] [PubMed] [Google Scholar]
- Ferreira, S. H. (1965). A bradykinin‐potentiating factor (BPF) present in the venom of Bothrops jararaca . British Journal of Pharmacology and Chemotherapy, 24, 163–169. 10.1111/j.1476-5381.1965.tb02091.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira, S. H. , Bartelt, D. C. , & Greene, L. J. (1970). Isolation of bradykinin potentiating peptides from Bothrops jararaca venom. Biochemistry, 9, 2583–2593. 10.1021/bi00815a005 [DOI] [PubMed] [Google Scholar]
- Ferreira, S. H. , Greene, L. H. , Alabaster, V. A. , Bakhle, Y. S. , & Vane, J. R. (1970). Activity of various fractions of bradykinin potentiating factor against angiotensin I converting enzyme. Nature, 225, 379–380. 10.1038/225379a0 [DOI] [PubMed] [Google Scholar]
- Ferreira, S. H. , & Vane, J. R. (1967). The disappearance of bradykinin and eledoisin in the circulation and vascular beds of the cat. British Journal of Pharmacology and Chemotherapy, 30, 417–424. 10.1111/j.1476-5381.1967.tb02148.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganten, D. , Minnich, J. L. , Granger, P. , Hayduk, K. , Brecht, H. M. , Barbeau, A. , … Genest, J. (1971). Angiotensin‐forming enzyme in brain tissue. Science, 173, 64–65. 10.1126/science.173.3991.64 [DOI] [PubMed] [Google Scholar]
- Gavras, H. , & Brunner, H. R. (2001). Role of angiotensin and its inhibition in hypertension, ischemic heart disease, and heart failure. Hypertension, 37, 342–345. 10.1161/01.HYP.37.2.342 [DOI] [PubMed] [Google Scholar]
- Gavras, H. , Brunner, H. R. , Laragh, J. H. , Sealey, J. E. , Gavras, I. , & Vukovich, R. A. (1974). An angiotensin converting‐enzyme inhibitor to identify and treat vasoconstrictor and volume factors in hypertensive patients. The New England Journal of Medicine, 291, 817–821. 10.1056/NEJM197410172911603 [DOI] [PubMed] [Google Scholar]
- Gavras, H. , Faxon, D. P. , Berkoben, J. , Brunner, H. R. , & Ryan, T. J. (1978). Angiotensin converting enzyme inhibition in patients with congestive heart failure. Circulation, 58, 770–776. 10.1161/01.CIR.58.5.770 [DOI] [PubMed] [Google Scholar]
- Gebre, A. K. , Altaye, B. M. , Atey, T. M. , Tuem, K. B. , & Berhe, D. F. (2018). Targeting renin‐angiotensin system against Alzheimer's disease. Frontiers in Pharmacology, 9, 440 10.3389/fphar.2018.00440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- George, A. J. , Thomas, W. G. , & Hannan, R. D. (2010). The renin angiotensin system and cancer: Old dog, new tricks. Nature Reviews. Cancer, 10, 745–759. 10.1038/nrc2945 [DOI] [PubMed] [Google Scholar]
- Gironacci, M. M. , Vicario, A. , Cerezo, G. , & Silva, M. G. (2018). The depressor axis of the renin‐angiotensin system and brain disorders: A translational approach. Clinical Science (London, England), 132, 1021–1038. 10.1042/CS20180189 [DOI] [PubMed] [Google Scholar]
- Hannan, R. E. , Davis, E. A. , & Widdop, R. E. (2003). Functional role of angiotensin II AT2 receptor in modulation of AT1 receptor‐mediated contraction in rat uterine artery: Involvement of bradykinin and nitric oxide. British Journal of Pharmacology, 140, 987–995. 10.1038/sj.bjp.0705484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … NC‐IUPHAR (2018). The IUPHAR/BPS guide to pharmacology in 2018: Updates and expansion to encompass the new guide to immunopharmacology. Nucleic Acids Research, 46, D1091–D1106. 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmer, O. M. (1955). A factor in plasma that enhances contraction produced by angiotonin on rabbit aortic strips. Federation Proceedings, 14(1), 728.13262134 [Google Scholar]
- Hubers, S. A. , & Brown, N. J. (2016). Combined angiotensin receptor antagonism and neprilysin inhibition. Circulation, 133, 1115–1124. 10.1161/CIRCULATIONAHA.115.018622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karnik, S. S. , Singh, K. D. , Tirupula, K. , & Unal, H. (2017). Significance of angiotensin 1–7 coupling with MAS1 receptor and other GPCRs to the renin‐angiotensin system: IUPHAR Review 22. British Journal of Pharmacology, 174, 737–753. 10.1111/bph.13742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karnik, S. S. , Unal, H. , Kemp, J. R. , Tirupula, K. C. , Eguchi, S. , Vanderheyden, P. M. , & Thomas, W. G. (2015). International Union of Basic and Clinical Pharmacology. XCIX. Angiotensin receptors: Interpreters of pathophysiological angiotensinergic stimuli [corrected]. Pharmacological Reviews, 67, 754–819. 10.1124/pr.114.010454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kono, Y. , Sawada, S. , Kawahara, T. , Tsuda, Y. , Higaki, T. , Yamasaki, S. , … Nakagawa, M. (2002). Bradykinin inhibits serum‐depletion‐induced apoptosis of human vascular endothelial cells by inducing nitric oxide via calcium ion kinetics. Journal of Cardiovascular Pharmacology, 39, 251–261. 10.1097/00005344-200202000-00012 [DOI] [PubMed] [Google Scholar]
- Kuba, K. , Imai, Y. , Ohto‐Nakanishi, T. , & Penninger, J. M. (2010). Trilogy of ACE2: A peptidase in the renin‐angiotensin system, a SARS receptor, and a partner for amino acid transporters. Pharmacology & Therapeutics, 128, 119–128. 10.1016/j.pharmthera.2010.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laffan, R. J. , Goldberg, M. E. , High, J. P. , Schaeffer, T. R. , Waugh, M. H. , & Rubin, B. (1978). Antihypertensive activity in rats for SQ 14,225, an orally active inhibitor of angiotensin I‐converting enzyme. The Journal of Pharmacology and Experimental Therapeutics, 204, 281–288. [PubMed] [Google Scholar]
- Laragh, J. H. (1981). Anti‐renin system therapy. A new horizon for understanding and treating hypertension. American Heart Journal, 101, 364–368. 10.1016/0002-8703(81)90214-3 [DOI] [PubMed] [Google Scholar]
- Ma, T. K. , Kam, K. K. , Yan, B. P. , & Lam, Y. Y. (2010). Renin‐angiotensin‐aldosterone system blockade for cardiovascular diseases: Current status. British Journal of Pharmacology, 160, 1273–1292. 10.1111/j.1476-5381.2010.00750.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manley, H. J. (2000). Role of angiotensin‐converting‐enzyme inhibition in patients with renal disease. American Journal of Health‐System Pharmacy, 57(Suppl 1), S12–S18. [DOI] [PubMed] [Google Scholar]
- McKinley, M. J. , Albiston, A. L. , Allen, A. M. , Mathai, M. L. , May, C. N. , McAllen, R. M. , … Chai, S. Y. (2003). The brain renin‐angiotensin system: Location and physiological roles. The International Journal of Biochemistry & Cell Biology, 35, 901–918. 10.1016/s1357-2725(02)00306-0 [DOI] [PubMed] [Google Scholar]
- Muñoz, D. , Uzoije, P. , Reynolds, C. , Miller, R. , Walkley, D. , Pappalardo, S. , … Wang, T. J. (2019). Polypill for cardiovascular disease prevention in an underserved population. The New England Journal of Medicine, 381, 1114–1123. 10.1056/NEJMoa1815359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng, K. K. , & Vane, J. R. (1967). Conversion of angiotensin I to angiotensin II. Nature, 216(5117), 762–766. [DOI] [PubMed] [Google Scholar]
- Ng, K. K. , & Vane, J. R. (1968). Fate of angiotensin I in the circulation. Nature, 218(5137), 144–150. 10.1038/218144a0 [DOI] [PubMed] [Google Scholar]
- Oliveira, A. C. , Melo, M. B. , Motta‐Santos, D. , Peluso, A. A. , Souza‐Neto, F. , da Silva, R. F. , … Santos, R. A. S. (2019). Genetic deletion of the alamandine receptor MRGD leads to dilated cardiomyopathy in mice. American Journal of Physiology. Heart and Circulatory Physiology1, 316, H123–H133. 10.1152/ajpheart.00075.2018 [DOI] [PubMed] [Google Scholar]
- Ondetti, M. A. , Rubin, B. , & Cushman, D. W. (1977). Design of specific inhibitors of angiotensin‐converting enzyme: New class of orally active antihypertensive agents. Science, 196, 441–444. 10.1126/science.191908 [DOI] [PubMed] [Google Scholar]
- Ondetti, M. A. , Williams, N. J. , Sabo, E. F. , Pluscec, J. , Weaver, E. R. , & Kocy, O. (1971). Angiotensin‐converting enzyme inhibitors from the venom of Bothrops jararaca: Isolation, elucidation of structure and synthesis. Biochemistry, 10, 4033–4039. 10.1021/bi00798a004 [DOI] [PubMed] [Google Scholar]
- Page, I. H. , & Bumpus, F. M. (1961). Angiotensin. Physiological Reviews, 41, 331–390. 10.1152/physrev.1961.41.2.331 [DOI] [PubMed] [Google Scholar]
- Pantzaris, N. D. , Karanikolas, E. , Tsiotsios, K. , & Velissaris, D. (2017). Renin inhibition with Aliskiren: A decade of clinical experience. Journal of Clinical Medicine, 6pii: E61. 10.3390/jcm6060061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peart, W. S. (1965). The renin‐angiotensin system. Pharmacological Reviews, 17, 143–182. [PubMed] [Google Scholar]
- Persson, F. , Lindhardt, M. , Rossing, P. , & Parving, H. H. (2016). Prevention of microalbuminuria using early intervention with renin‐angiotensin system inhibitors in patients with type 2 diabetes: A systematic review. Journal of the Renin‐Angiotensin‐Aldosterone System, 17pii: 1470320316652047. 10.1177/1470320316652047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pretorius, M. , Rosenbaum, D. , Vaughan, D. E. , & Brown, N. J. (2003). Angiotensin‐converting enzyme inhibition increases human vascular tissue‐type plasminogen activator release through endogenous bradykinin. Circulation, 107, 579–585. 10.1161/01.CIR.0000046268.59922.A4 [DOI] [PubMed] [Google Scholar]
- Radin, D. P. , Krebs, A. , Maqsudlu, A. , & Patel, P. (2018). Our ACE in the HOLE: Justifying the use of angiotensin‐converting enzyme inhibitors as adjuvants to standard chemotherapy. Anticancer Research, 38, 45–49. 10.21873/anticanres.12190 [DOI] [PubMed] [Google Scholar]
- Regoli, D. , & Gobeil, F. (2017). Kallikrein‐kinin system as the dominant mechanism to counteract hyperactive renin‐angiotensin system. Canadian Journal of Physiology and Pharmacology, 95, 1117–1124. 10.1139/cjpp-2016-0619 [DOI] [PubMed] [Google Scholar]
- Roshandel, G. , Khoshnia, M. , Poustchi, H. , Hemming, K. , Kamangar, F. , Gharavi, A. , … Malekzadeh, R. (2019). Effectiveness of polypill for primary and secondary prevention of cardiovascular diseases (PolyIran): A pragmatic, cluster‐randomised trial. Lancet, 394, 672–683. 10.1016/S0140-6736(19)31791-X [DOI] [PubMed] [Google Scholar]
- Ruggenenti, P. (2017). Dual renin‐angiotensin system blockade for nephroprotection. Néphrologie & Thérapeutique, 13(Suppl 1), S43–S45. 10.1016/j.nephro.2017.02.006 [DOI] [PubMed] [Google Scholar]
- Sadat‐Ebrahimi, S. R. , Parnianfard, N. , Vahed, N. , Babaei, H. , Ghojazadeh, M. , Tang, S. , & Azarpazhooh, A. (2018). An evidence‐based systematic review of the off‐label uses of lisinopril. British Journal of Clinical Pharmacology, 84, 2502–2521. 10.1111/bcp.13705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos, R. A. S. , Oudit, G. Y. , Verano‐Braga, T. , Canta, G. , Steckelings, U. M. , & Bader, M. (2019). The renin‐angiotensin system: Going beyond the classical paradigms. American Journal of Physiology. Heart and Circulatory Physiology, 316, H958–H970. 10.1152/ajpheart.00723.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos, R. A. S. , Sampaio, W. O. , Alzamora, A. C. , Motta‐Santos, D. , Alenina, N. , Bader, M. , & Campagnole‐Santos, M. J. (2018). The ACE2/angiotensin‐(1–7)/MAS axis of the renin‐angiotensin system: Focus on angiotensin‐(1–7). Physiological Reviews, 98, 505–553. 10.1152/physrev.00023.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayer, G. , & Bhat, G. (2014). The renin‐angiotensin‐aldosterone system and heart failure. Cardiology Clinics, 32(1), 21–32. 10.1016/j.ccl.2013.09.002 [DOI] [PubMed] [Google Scholar]
- Selak, V. , Webster, R. , Stepien, S. , Bullen, C. , Patel, A. , Thom, S. , … Rodgers, A. (2019). Reaching cardiovascular prevention guideline targets with a polypill‐based approach: A meta‐analysis of randomised clinical trials. Heart, 105, 42–48. 10.1136/heartjnl-2018-313108 [DOI] [PubMed] [Google Scholar]
- Sheng, Z. , Yao, Y. , Li, Y. , Yan, F. , Huang, J. , & Ma, G. (2013). Bradykinin preconditioning improves therapeutic potential of human endothelial progenitor cells in infarcted myocardium. PLoS ONE, 8, e81505 10.1371/journal.pone.0081505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skeggs, L. T. Jr. , Kahn, J. R. , & Shumway, N. P. (1956). The preparation and function of the hypertensin‐converting enzyme. The Journal of Experimental Medicine, 103, 295–299. 10.1084/jem.103.3.295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, C. G. , & Vane, J. R. (2003). The discovery of captopril. The FASEB Journal, 17, 788–789. 10.1096/fj.03-0093life [DOI] [PubMed] [Google Scholar]
- Streeten, D. H. , Anderson, G. H. , Freiberg, J. M. , & Dalakos, T. G. (1975). Use of an angiotensin II antagonist (saralasin) in the recognition of “angiotensinogenic” hypertension. The New England Journal of Medicine, 292, 657–662. 10.1056/NEJM197503272921301 [DOI] [PubMed] [Google Scholar]
- Taddei, S. , & Bortolotto, L. (2016). Unraveling the pivotal role of bradykinin in ACE inhibitor activity. American Journal of Cardiovascular Drugs, 16, 309–321. 10.1007/s40256-016-0173-4 [DOI] [PubMed] [Google Scholar]
- Tetzner, A. , Naughton, M. , Gebolys, K. , Eichhorst, J. , Sala, E. , Villacañas, Ó. , & Walther, T. (2018). Decarboxylation of Ang‐(1–7) to Ala1‐Ang‐(1–7) leads to significant changes in pharmacodynamics. European Journal of Pharmacology, 833, 116–123. 10.1016/j.ejphar.2018.05.031 [DOI] [PubMed] [Google Scholar]
- Tigerstedt, R. , & Bergman, P. G. (1898). Niere und kreislauf. Scand Arch Physiol, 8, 223–271. 10.1111/j.1748-1716.1898.tb00272.x [DOI] [Google Scholar]
- Timmermans, P. B. , Wong, P. C. , Chiu, A. T. , Herblin, W. F. , Benfield, P. , Carini, D. J. , … Smith, R. D. (1993). Angiotensin II receptors and angiotensin II receptor antagonists. Pharmacological Reviews, 45, 205–251. [PubMed] [Google Scholar]
- Turini, G. A. , Brunner, H. R. , Gribic, M. , Waeber, B. , & Gavras, H. (1979). Improvement of chronic congestive heart‐failure by oral captopril. Lancet, 1, 1213–1215. 10.1016/s0140-6736(79)91897-x [DOI] [PubMed] [Google Scholar]
- Vane, J. R. (1964). The use of isolated organs for detecting active substances in the circulating blood. British Journal of Pharmacology and Chemotherapy, 23, 360–373. 10.1111/j.1476-5381.1964.tb01592.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vane, J. R. (1969). The release and fate of vaso‐active hormones in the circulation. British Journal of Pharmacology, 35, 209–242. 10.1111/j.1476-5381.1969.tb07982.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villela, D. C. , Passos‐Silva, D. G. , & Santos, R. A. (2014). Alamandine: A new member of the angiotensin family. Current Opinion in Nephrology and Hypertension, 23, 130–134. 10.1097/01.mnh.0000441052.44406.92 [DOI] [PubMed] [Google Scholar]
- Wang, B. , Wang, F. , Zhang, Y. , Zhao, S. H. , Zhao, W. J. , Yan, S. L. , & Wang, Y. G. (2015). Effects of RAS inhibitors on diabetic retinopathy: A systematic review and meta‐analysis. The Lancet Diabetes and Endocrinology, 3, 263–274. 10.1016/S2213-8587(14)70256-6 [DOI] [PubMed] [Google Scholar]
- Wang, Y. , Del Borgo, M. , Lee, H. W. , Baraldi, D. , Hirmiz, B. , Gaspari, T. A. , … Widdop, R. E. (2017). Anti‐fibrotic potential of AT2 receptor agonists. Frontiers in Pharmacology, 8, 564 10.3389/fphar.2017.00564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wharton, W. , Goldstein, F. C. , Tansey, M. G. , Brown, A. L. , Tharwani, S. D. , Verble, D. D. , … Kehoe, P. G. (2018). Rationale and design of the mechanistic potential of antihypertensives in preclinical Alzheimer's (HEART) trial. Journal of Alzheimer's Disease, 61, 815–824. 10.3233/JAD-161198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, J. , Fan, J. , Wu, F. , Huang, Q. , Guo, M. , Lv, Z. , … Jin, Y. (2017). The ACE2/angiotensin‐(1–7)/Mas receptor axis: Pleiotropic roles in cancer. Frontiers in Physiology, 8, 276 10.3389/fphys.2017.00276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuo, J. , Moeller, I. , Jenkins, T. , Chai, S. Y. , Allen, A. M. , Ohishi, M. , & Mendelsohn, F. A. (1998). Mapping tissue angiotensin‐converting enzyme and angiotensin AT1, AT2 and AT4 receptors. Journal of Hypertension, 16, 2027–2037. 10.1097/00004872-199816121-00026 [DOI] [PubMed] [Google Scholar]