

Abstract
Background and Purpose
Antibodies targeting cell surface receptors are considered to enable highly selective therapeutic interventions for immune disorders and cancer. Their biological profiles are found, generally, to represent the net effects of antibody–target interactions. The former therapeutic anti‐integrin αLβ2 antibody efalizumab seems to defeat this paradigm by eliciting, via mechanisms currently unknown, much broader effects than would be predicted based on its target specificity.
Experimental Approach
To elucidate the mechanisms behind these broad effects, we investigated in primary human lymphocytes in vitro the effects of anti‐αLβ2 antibodies on the expression of αLβ2 as well as unrelated α4 integrins, in comparison to Fab fragments and small‐molecule inhibitors.
Key Results
We demonstrate that anti‐αLβ2 mAbs directly induce the internalization of α4 integrins. The endocytotic phenomenon is a direct consequence of their antibody nature. It is inhibited when monovalent Fab fragments or small‐molecule inhibitors are used. It is independent of crosslinking via anti‐Fc mAbs and of αLβ2 activation. The cross‐modulatory effect is unidirectional and not observed in a similar fashion with the α4 integrin antibody natalizumab.
Conclusion and Implications
The present study identifies endocytotic cross‐modulation as a hitherto unknown non‐canonical functionality of anti‐αLβ2 antibodies. This cross‐modulation has the potential to fundamentally alter an antibody's benefit risk profile, as evident with efalizumab. The newly described phenomenon may be of relevance to other therapeutic antibodies targeting cluster‐forming receptors. Thus, pharmacologists should be cognizant of this action when investigating such antibodies.
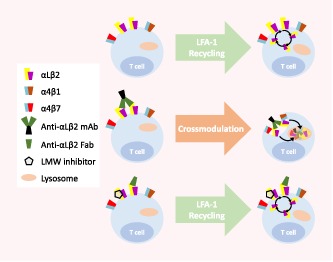
Abbreviations
- APC
allophycocyanin
- CD
cluster of differentiation
- Cy7
cyanine‐7
- Fab
antigen‐binding fragment
- Fc
fragment crystallizable
- FITC
fluorescein isothiocyanate
- ICAM‐1
intercellular adhesion molecule‐1
- LFA‐1
leukocyte‐function associated antigen‐1
- mAb
monoclonal antibody
- PBMC
peripheral blood mononuclear cell
- PE
phycoerythrin
- PerCP
peridinin‐chlorophyll‐protein
- PMA
phorbol 12‐myristate 13‐acetate
What is already known
Efalizumab unexpectedly reduces the expression of major immune receptors on patients circulating T cells.
The mechanism/s are unknown; altered T‐cell trafficking remains a potential explanation.
What this study adds
This study clarifies the mechanism by which anti‐αLβ2 mAbs, including efalizumab, directly down‐regulate α4 integrins.
The study describes endocytotic cross‐modulation as a novel non‐canonical antibody functionality.
What is the clinical significance
Endocytotic cross‐modulation has the potential to fundamentally alter the effect profiles of therapeutic antibodies.
Pharmacologists should be aware of this when developing therapeutic antibodies targeting cluster‐forming receptors.
1. INTRODUCTION
Antibodies targeting cell surface receptors are generally considered to enable highly selective therapeutic interventions. Their biological profiles are expected and generally found to represent the net effect of antibody binding to the target via their antigen‐binding fragment (Fab) regions, resulting in target inhibition or activation. Additional functionalities of therapeutic antibodies may derive from their interaction with the immune system through their fragment crystallizable (Fc) portion. The interaction of the Fc part with complement triggers complement‐dependent cytotoxicity. Further, by binding of the Fc region to Fc receptors, antibodies can induce antibody‐dependent cell‐mediated cytotoxicity, phagocytosis and Fc receptor‐mediated trogocytosis. The recruitment of these effectors is dependent on the antibody's isotype and its ability to interact with complement or effector cells (Chames, Van Regenmortel, Weiss, & Baty, 2009; Smith, 2015; Taylor et al., 2015).
Against this background, an antibody whose effect profile does not seem to match generally accepted concepts of canonical antibody functions caught our attention. http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=6593, a recombinant humanized monoclonal antibody (mAb), which had been approved in 2003 for the treatment of patients with moderate‐to‐severe chronic plaque psoriasis and was withdrawn from markets in 2009 for a serious side effect not anticipated based on this antibody's target leukocyte‐function associated antigen‐1 (https://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=760#2582, LFA‐1 and CD11a/CD18; Lebwohl et al., 2003; Seminara & Gelfand, 2010).
αLβ2 is a α/β heterodimeric immune receptor belonging to the integrin family and expressed on leukocytes. The major ligand of αLβ2 is intercellular adhesion molecule‐1 (http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=6757), up‐regulated on endothelial cells and leukocytes (Tan, 2012). αLβ2 orchestrates leukocyte migration and extravasation to sites of inflammation as well as trafficking. Moreover, αLβ2 is an important component of the T‐cell immune synapse, a large‐scale molecular assembly of defined immune receptors, controlling T‐cell activation and differentiation. Given these central roles within the immune response, the function of αLβ2 is tightly regulated by bidirectional signalling processes. Signals from inside the cell convert αLβ2 from an inactive into an active ligand‐binding state, while upon ligand‐binding αLβ2 is capable to convey signals back into cells (Verma & Kelleher, 2017; Walling & Kim, 2018). Moreover, endocytotic pathways contribute to the control of αLβ2 function by modulating αLβ2 surface expression (Fabbri et al., 2005; Walling & Kim, 2018).
Unexpectedly, efalizumab was clinically found to down‐regulate the surface expression of αLβ2 as well as a broad spectrum of other major immune receptors, leading to a state of profound T‐cell hyporesponsiveness. In patients on continuous long‐term efalizumab therapy (>3 years), this state was associated with the occurrence of a life‐threatening opportunistic viral infection afflicting the CNS, termed progressive multifocal leukoencephalopathy (Seminara & Gelfand, 2010). Efalizumab was withdrawn from markets, in consequence. Of note, αLβ2 genetic deficiency states in mice and men are found to mount adequate antiviral immune responses (Etzioni, 2010; Schmits et al., 1996).
Strikingly, the immune receptors down‐regulated by efalizumab included other integrin family members, specifically the integrins http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2443 (very late antigen‐4, CD49d/CD29) and http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2770 (Guttman‐Yassky et al., 2008; Vugmeyster et al., 2004). Like αLβ2, α4 integrins are expressed on lymphocytes and involved in lymphocyte migration and trafficking (Bertoni, Alabiso, Galetto, & Baldanzi, 2018; Chigaev & Sklar, 2012). The integrin α4β1, specifically, is recognized to play pivotal roles in CNS immune surveillance (Lodygin et al., 2019; Rothhammer et al., 2014; Wilson, Weninger, & Hunter, 2010). Both α4β7 and α4β1 are targets of http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=6591, a humanized mAb directed to the α4 integrin chain and approved for the treatment of multiple sclerosis and Crohn's disease (Rudick, Polman, Clifford, Miller, & Steinman, 2013). https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=6591 also had to be withdrawn from markets due to increased progressive multifocal leukoencephalopathy risk. However, natalizumab returned to markets in 2006 under a restricted distribution and monitoring programme (Rudick et al., 2013).
While it is established that efalizumab‐engaged αLβ2 is internalized and preferentially targeted to lysosomes for degradation (Coffey et al., 2004; Mancuso, Welzenbach, Steinberger, Krahenbuhl, & Weitz‐Schmidt, 2016), the mechanisms by which this antibody affects the surface expression of other immune receptors has remained elusive, to date. Specifically, it has remained unclear whether the broad downmodulation of α4 integrins, together with other immune receptors, observed in efalizumab‐treated patients is caused by a direct effect on these receptors' surface expression or, for an alternative explanation, by altered trafficking of lymphocyte subsets with differential immune receptor expression patterns.
In order to elucidate the mechanisms behind efalizumab's broad immunomodulatory actions, we characterized the effects of various anti‐αLβ2 mAbs, Fab fragments and small‐molecule αLβ2 inhibitors on the surface expression of integrins αLβ2, α4β1 and α4β7 in primary human lymphocytes in vitro. To the best of our knowledge, we demonstrate here for the first time at single cell level that anti‐αLβ2 antibodies, but not the respective Fab fragments or small‐molecule αLβ2 inhibitors induce the internalization of α4 integrins. This internalization is independent of crosslinking via anti‐Fc mAbs and αLβ2 activation, excluding earlier described phenomena such as trogocytosis. In consequence, the results of the present study identify and characterize a hitherto unknown non‐canonical functionality of anti‐αLβ2 antibodies which may be of relevance to other antibodies targeting cluster‐forming receptors.
2. METHODS
2.1. Antibodies and reagents
Anti‐CD11a clone R7.1 (Bio X Cell Cat# BE0192, RRID:AB_10948991). Anti‐CD11a clone TS1/22 (Thermo Fisher Scientific Cat# MA11A10, RRID:AB_223513). Anti‐CD11a FITC clone R7.1 (Thermo Fisher Scientific Cat# BMS102FI, RRID:AB_10598522). Anti‐CD11a PerCP clone TS2/4 (BioLegend Cat# 350608, RRID:AB_10662249). Anti‐CD11a/CD18‐AlexaFluor488 clone m24 (BioLegend Cat# 363404, RRID:AB_2565289). Anti‐CD18‐PE clone MEM148 (Sigma‐Aldrich Cat# SAB4700402, RRID:AB_10897633). Anti‐CD2‐APC clone RPA‐2.10 (BioLegend Cat# 300213, RRID:AB_10900259). Anti‐CD2‐PE clone RPA‐2.10 (BioLegend Cat# 300208, RRID:AB_314032). Anti‐CD29‐APC clone TS2/16 (BioLegend Cat# 303007, RRID:AB_314323). Anti‐CD29‐PE clone HUTS‐21 (BD Biosciences Cat# 556049, RRID:AB_396320). Anti‐CD3 clone OKT3 (BioLegend Cat# 317325, RRID:AB_11147370). Anti‐CD49d clone 9F10 (BioLegend Cat# 304310, RRID:AB_2130039). Anti‐CD49d‐PE/Cy7 clone 9F10 (BioLegend Cat# 304313, RRID:AB_10642817). Anti‐CD69‐PE clone FN50 (BioLegend Cat# 310905, RRID:AB_314840). Anti‐human IgG Fc clone HP6017 (BioLegend Cat# 409302, RRID:AB_10900247). Anti‐mouse IgG Fc clone Poly4053 (BioLegend Cat# 405301, RRID:AB_315005). Anti‐β7‐APC/Fire clone FIB504 (BioLegend Cat# 321224, RRID:AB_2715981). hIgG1 (Sigma‐Aldrich Cat# I5154, RRID:AB_1163610). hIgG4 clone QA16A15 (BioLegend Cat# 403701). mIgG1 clone MOPC‐21 (BioLegend Cat# 400140, RRID:AB_493443). Efalizumab (=Raptiva®; Merck‐Serono PubChem SID: http://2.208.78.58) was kindly provided by Peter Steinberger. Natalizumab (=Tysabri®; Biogen, PubChem SID: http://2.245.199.90) was kindly provided by Raija Lindberg Gasser. Vedolizumab (=Entyvio®; Takeda Oncology, PubChem SID: 96024773) was kindly provided by Petr Hruz. BIRT‐377 (Tocris Cat# 4776, PubChem CID: 9803375). Firategrast (MedChemTronica Cat# 402567‐16‐2, PubChem CID: http://0.151.155.65). LFA878 (Novartis Pharma AG, PubChem CID: http://0.6.218.146) was kindly provided by Berndt Oberhauser. Lifitegrast (=Xiidra®; Shire Plc, PubChem CID: 11965427). RO0505376 (Hoffmann‐La Roche, PubChem CID: 9806910). XVA143 (=RO0281607‐000; Hoffmann‐La Roche, PubChem CID: http://0.150.164.205) was kindly provided by Paul Gillespie. DAPI (=4′,6‐diamidino‐2‐phenylindole; BioLegend Cat# 422801). Concanamycin A (abcam Cat# 80890‐47‐7, PubChem CID: http://0.98.61.7). Dynasore (abcam Cat# 304448–55‐3, PubChem CID: 135533054). Fixation Buffer (BioLegend Cat# 420801). Intracellular Staining Permeabilization Wash Buffer (10X; BioLegend Cat# 421002). Ionomycin (Sigma‐Aldrich Cat# I3909). PMA (=phorbol 12‐myristate 13‐acetate; Sigma‐Aldrich Cat# P1585). R7.1‐F(ab) and R7.1 F(ab′)2 were prepared following manufacturer's instructions, Pierce™ Mouse IgG1 Fab and F(ab′)2 Preparation Kit (Thermo Fisher Scientific Cat# 44980).
2.2. Preparation of mAbs and compounds
Test compounds were dissolved at 10 mM in DMSO (stock), serially diluted in DMSO to avoid precipitation, followed by dilution in medium or assay buffer. The final DMSO concentration during the experiment was 0.1%. Efalizumab was kept at −80°C in RPMI 1640 at a concentration of 10 mg·ml−1. Natalizumab was kept at 4°C in its original formulation at a concentration of 1.9 mg·ml−1. https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=7437 was kept at 4°C at a concentration of 1 mg·ml−1. Serial dilutions of the antibodies were prepared in RPMI 1640.
2.3. Isolation and culture of peripheral blood mononuclear cells (PBMCs) and HL‐60 cells
Blood samples were collected at the Blutspendezentrum SRK beider Basel. Peripheral blood mononuclear cells were isolated from heparin samples through centrifugation in Ficoll® Paque Plus, GE Healthcare (Chicago, IL), following manufacturer's instructions. Peripheral blood mononuclear cells and HL‐60 (ATCC Cat# CCL‐240, RRID:CVCL_0002) were cultured in RPMI 1640 containing 10% (v/v) heat inactivated FBS, 1% minimum essential medium non‐essential amino acids, amphotericin B 1 μg·ml−1, and gentamycin 10 μg·ml−1. All cell culture media and reagents were purchased from Gibco Life Technologies (Paisely, UK).
2.4. Surface expression analysis
Peripheral blood mononuclear cells were treated for 24 hr at 37°C with anti‐αL (efalizumab, R7.1, and TS1/22), anti‐α4 (natalizumab), anti‐α4β7 (vedolizumab) mAbs, and respective isotype controls (hIgG1, mIgG1, and hIgG4) at 10 μg·ml−1. Small‐molecule inhibitors targeting αL (LFA878 and BIRT377), αLβ2 (XVA143 and lifitegrast) and α4β1/α4β7 (firategrast and RO0505376) were used at 10 μM. The concentration used of each integrin targeting intervention was well above reported IC50 and/or reported saturation levels respectively. Moreover, therapeutic antibodies were used at therapeutically relevant concentrations (Table S1). DMSO served as solvent control. Single‐cell suspensions were stained with anti‐CD2‐PE (RPA‐2.10), anti‐CD11a‐PerCP (TS2/4; recognizes the αLβ2 complex, does not compete with efalizumab for binding; Mancuso et al., 2016), anti‐CD49d‐PE/Cy7 (9F10; does not compete with natalizumab for binding; Schneider‐Hohendorf, Philipp, Husstedt, Wiendl, & Schwab, 2014), anti‐CD29‐APC (TS2/16) and anti‐β7‐APC/Fire (FIB504) at 4°C for 30 min in Stain Buffer, BD Biosciences (Franklin Lakes, NJ). CD2, αLβ2, α4, β1, and β7 expression was thereafter analysed by flow cytometry using a Cytoflex cytometer, Beckman Coulter (Indianapolis, IN). For the flow cytometry gating strategy, singlets were first identified by a forward scatter area and forward scatter height gate and then lymphocytes by a forward scatter area and side scatter area gate. Lymphocyte CD2+ cells were identified by a FL‐2 channel and a side scatter area gate. Samples were analysed using FlowJo software, Tree Star (Ashland, OR, RRID:SCR_008520).
2.5. ImageStream analysis
Peripheral blood mononuclear cells were treated at 37°C with anti‐α4 natalizumab, anti‐αL efalizumab and respective isotype controls (hIgG4 and hIgG1) at 10 μg·ml−1. Cells were fixed and permeabilized using fixation buffer and intracellular staining permeabilization wash buffer, respectively, Biolegend (San Diego, CA). Cells were stained at RT with anti‐CD2‐APC (RPA‐2.10), anti‐α4 (CD49d)‐PE/Cy7 (9F10) and DAPI after 24 hr of incubation for extracellular and intracellular staining of α4 integrin, and after 14 hr of incubation for intracellular staining of the α4 integrin. A pre‐saturation step of extracellular α4 with unlabelled anti‐CD49d (9F10) was conducted at 4°C on samples incubated for 14 hr before proceeding with the staining. The samples were acquired using ImageStream Amnis®, Merck Millipore (Burlington, MA), at 60× magnification which provides a numerical aperture of 0.9, and a pixel dimension of 0.3 μm × 0.3 μm, scale bars in the image is 7 μm. Typical files contained imagery for 20,000 cells. Cell imagery was analysed using the software IDEAS, Version 6.0, Merck Millipore (Burlington, MA). Cells in best focus were selected using the feature Brightfield Gradient RMS, a measurement of image contrast that excludes out‐of‐focus events. Doublets, aggregates, dead cells and debris were excluded by using side scatter. A normalized frequency of cells versus intensity of fluorescence of α4 integrin histogram, and a frequency of cells versus intensity of fluorescence of α4 integrin histogram, were used for extracellular and intracellular staining, and intracellular staining respectively. Intensity and all analyses were restricted to single cells as described previously (George et al., 2006; Mancuso et al., 2016).
2.6. Effect of concanamycin A on mAb R7.1‐ or natalizumab‐induced downmodulation of αLβ2 and α4
Peripheral blood mononuclear cells were treated for 24 hr at 37°C with anti‐αL mAb (mAb R7.1, efalizumab, and efalizumab crosslinked), anti‐α4 mAb (natalizumab), and respective isotype controls (mIgG1, hIgG1, hIgG1 crosslinked, and hIgG4) at 10 μg·ml−1. Treatments were performed in the presence of concanamycin A (1 μM) or DMSO controls. Cells were fixed and permeabilized using fixation buffer and intracellular staining permeabilization buffer respectively. Cells were stained at RT with anti‐CD2‐PE (RPA‐2.10), anti‐CD11a PerCP (TS2/4) and anti‐α4 (CD49d)‐PE/Cy7. Surface expression of αLβ2 and α4 on CD2+ T cells was measured by flow cytometry as described above.
2.7. Integrin conformation analysis
Peripheral blood mononuclear cells were treated for 40 min at 37°C with anti‐αL (efalizumab, R7.1, and TS1/22), anti‐α4 (natalizumab), anti‐α4β7 (vedolizumab) mAbs and respective isotype controls (hIgG1, mIgG1, and hIgG4) at 10 μg·ml−1 at 37°C. Small‐molecule inhibitors targeting αL (LFA878 and BIRT377), αLβ2 (XVA143 and https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=7533) and α4β1/α4β7 (firategrast and RO0505376) integrins were used at 10 μM. DMSO served as solvent control. Manganese (2 mM) and ionomycin/PMA (1 μg·ml−1/0.05 μg·ml−1) were used as positive controls. Exposure of activation epitopes on the β2 chain of αLβ2 and the β1 chain of α4β1 on CD2+ T cells was measured by flow cytometry as described above. Anti‐CD2‐APC (RPA‐2.10), anti‐CD11a/CD18‐AlexaFluor488 (m24), and anti‐CD18‐PE (MEM148) were used for the detection of CD2, m24 epitope and MEM148 epitope respectively. Anti‐CD2‐APC (RPA‐2.10) and anti‐CD29‐PE (HUTS‐21) were used for the detection of CD2 and HUTS‐21 epitope respectively.
2.8. T‐cell activation analysis
Peripheral blood mononuclear cells were treated for 24 hr at 37°C with efalizumab, natalizumab and respective isotype controls (hIgG1 and hIgG4) at 10 μg·ml−1. Soluble anti‐CD3 mAb (OKT3; 500 ng·ml−1) was used as positive control for the induction of CD69 surface expression. Surface expression of CD69 on CD2+ T cells was measured by flow cytometry as described above. Anti‐CD2‐APC (RPA‐2.10) and anti‐CD69‐PE (FN50) were used for the detection of CD2 and CD69 respectively.
2.9. Preparation and analysis of mAb R7.1 Fab and F(ab′)2 fragments
Fab and F(ab′)2 fragments of mAb R7.1 were generated using Pierce™ Mouse IgG1 Fab and F(ab′)2 Preparation Kit following manufacturer's instructions. The fragments were analysed by reducing SDS‐PAGE. NuPAGE™ 4–12% Bis‐Tris Protein Gel wells were loaded with samples containing 2.5 μg of protein. The Coomassie Blue staining method was used for the detection of proteins and PageRuler™ Prestained Protein Ladder; 10 to 180 kDa was used as protein MW The binding of mAb R7.1 and fragments to HL‐60 cells was assessed by flow cytometry as described above. Briefly, HL‐60 cells were treated for 30 min at 4°C with anti‐αL R7.1 IgG1, R7.1 F(ab′)2, R7.1 Fab, or mIgG1 at 10 μg·ml−1 and then stained for 30 min at 4°C with anti‐CD11a FITC (R7.1) in Stain Buffer.
2.10. Effect of mAb R7.1 and fragments on αLβ2 and α4 integrin surface expression
Peripheral blood mononuclear cells were treated for 24 hr at 37°C with anti‐αL R7.1 IgG1, R7.1 F(ab′)2, R7.1 Fab, and mIgG1 at 10 μg·ml−1. Surface expression of αLβ2, α4β1, and α4β7 on CD2+ T cells was measured by flow cytometry as described above. Anti‐CD2‐PE (RPA‐2.10), anti‐CD11a‐PerCP (TS2/4), anti‐CD49d‐PE/Cy7 (9F10), anti‐CD29‐APC (TS2/16), and anti‐β7‐APC/Fire (FIB504) were used for the detection of CD2, αLβ2, α4, β1 and β7 respectively.
2.11. Effect of dynasore on R7.1 or natalizumab‐induced downmodulation of αLβ2 and α4
Peripheral blood mononuclear cells were treated for 24 hr at 37°C with anti‐αL R7.1, anti‐α4 natalizumab, and respective isotype controls (mIgG1 and hIgG4) at 10 μg·ml−1. Treatments were performed in the presence of dynasore (40 μM) or DMSO controls. Surface expression of αLβ2 and α4 on CD2+ T cells was measured by flow cytometry as described above. Anti‐CD2‐PE (RPA‐2.10), anti‐CD11a PerCP (TS2/4) and anti‐CD49d‐PE/Cy7 (9F10) were used for the detection of CD2, αLβ2, and α4 respectively.
2.12. Statistical analysis
The data and statistical analysis comply with the recommendations of the British Journal of Pharmacology on experimental design and analysis in pharmacology (Curtis et al., 2015). Specifically, the number of independent experiments (n) is provided in the figure legends for all experiments. The statistical analysis was performed using these independent values. Randomization was not required in the current study because equal aliquots of the same PBMC preparations were used to compare different conditions. Blinding was not used for this study as each experiment was controlled by detailed protocols and generated objective readout parameters not considered to be affected by subjective bias. In order to control for potential intra‐experimental bias, analyses were routinely not performed until the experimental data set was complete. At least five independent experiments were performed for each data set with the exception of the exploratory experiment described in Figure 3c,d (n = 4). Data are expressed as means ± SEM. Statistical significance of differences was determined by one‐way ANOVA or two‐way ANOVA. Differences were considered significant for P < .05. Statistical analysis was performed using Prism 8.0, GraphPad (San Diego, CA, RRID:SCR_002798).
Figure 3.
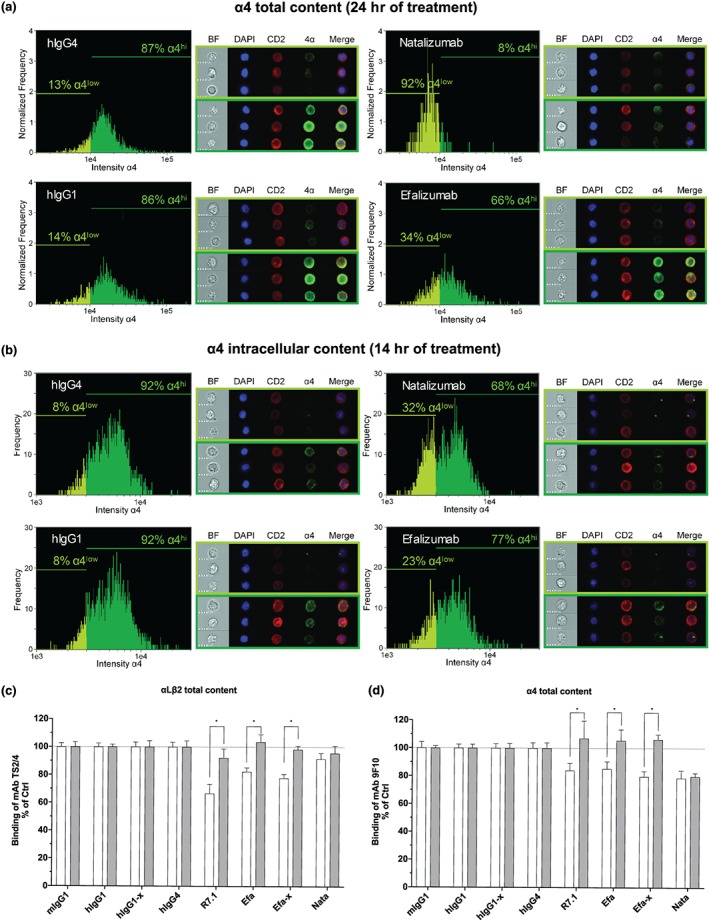
Expression and localization of α4 chain in CD2+ T cells by ImageStream flow‐based imaging (a,b) and effect of concanamycin A on mAb R7.1 or natalizumab‐induced downmodulation of αLβ2 and α4 chain (c,d). Total content (extracellular and intracellular) (a) and intracellular content (b) of CD2+ T‐cell α4 chain after 24 and 14 hr of treatment (hIgG4: 10 μg·ml−1, hIgG1: 10 μg·ml−1, natalizumab: 10 μg·ml−1, and efalizumab:10 μg·ml−1) respectively. ImageStream cell images acquired in flow are shown on the right of each histogram by multispectral imaging of brightfield (BF), DNA staining (DAPI), CD2 staining (CD2), α4 integrin staining (α4), and overlay of DAPI, CD2 and α4 (merge) respectively. Scale bars represent 7 μm. Representative images of cells with low expression of α4 (defined as “α4low” in the histogram) and high expression of α4 (defined as “α4hi” in the histogram) are shown in the light green frame and dark green frame respectively. The intensity levels of α4 chain expression is shown in the histogram plots. Total content of αLβ2 (c) and α4 integrin (d) on CD2+ T cells after 24 hr of treatment with anti‐αL R7.1, efa (efalizumab, 10 μg·ml−1), efa‐x (efalizumab‐crosslinked, 10 μg·ml−1 of efalizumab plus 10 μg·ml−1 of anti‐Fc), nata (natalizumab, 10 μg·ml−1), and respective isotype controls (mIgG1, hIgG1, hIgG1‐x, and hIgG4). Treatments were performed in the presence of concanamycin A (1 μM, grey bars) or DMSO controls (white bars). Each bar represents the mean value ± SEM of four independent experiments using blood samples from different donors. Statistical significance was determined by using two‐way ANOVA, $ P < .05, versus incubation without concanamycin A
2.13. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2019/20 (Alexander et al., 2019).
3. RESULTS
3.1. Anti‐αLβ2 mAbs downmodulate α4 integrins in vitro
Anti‐αLβ2 antibodies were clinically found to not only reduce the surface expression of αLβ2 yet also of structurally diverse α4 integrins. The mechanisms of this cross‐modulation have remained speculative, to date. In order to further elucidate the mechanisms by which efalizumab affects integrins other than αLβ2, we assessed the effect of humanized or murine αLβ2‐specific mAbs efalizumab, TS1/22 and R7.1 on the surface expression of αLβ2, α4β1 and α4β7 in vitro utilizing primary CD2+ T cells. These mAbs bind to the http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2451&familyId=760&familyType=CATALYTICRECEPTOR inserted (I) domain of αLβ2 either close to (efalizumab and TS1/22) or distant from the ligand‐binding site (R7.1) and block αLβ2 function (Figure 1 and Table 1; Li et al., 2009; Lu, Shimaoka, Salas, & Springer, 2004; Weitz‐Schmidt, Schurpf, & Springer, 2011). Efalizumab, TS1/22 and R7.1 downmodulated αLβ2 by 21%, 31.5% and 39.9%, respectively, when incubated with CD2+ cells for 24 hr. The 24 hr time point was selected because a time course experiment (including 48 hr of incubation) indicated that maximum effects of anti‐αLβ2 mAbs on αLβ2 downmodulation in CD2+ were already achieved at this time point (Figure S1). Concomitantly, the surface expression of α4 integrins was reduced by more than 50% as compared to respective IgG controls (Figure 2a,b). These results clarify that similar clinical observations in psoriasis patients treated with efalizumab for 14 days (Guttman‐Yassky et al., 2008; Vugmeyster et al., 2004) are attributable to direct cellular effects of the antibody rather than to redistribution phenomena of T‐cell subsets.
Figure 1.
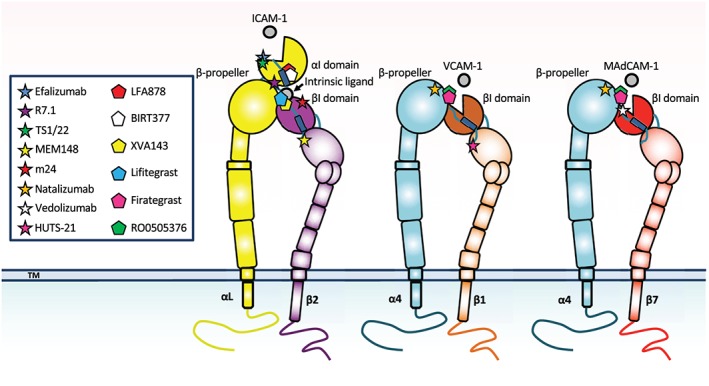
Schematic figure of integrins αLβ2, α4β1, and α4β7 with different mAb and small‐molecule inhibitor binding sites shown. The natural integrin ligands (ICAM‐1, VCAM‐1 and MAdCAM‐1, respectively) are displayed, as well. The integrin α/β heterodimers are shown in their active, extended conformation with an open headpiece allowing ligand binding. The binding sites of mAbs and compounds used in the study are indicated. Anti‐β2 chain mAb MEM148, anti‐β2 chain mAb m24 and the anti‐β1 chain mAb HUTS‐21 recognize activation epitopes only exposed in activated integrins
Table 1.
Integrin pharmacology and targeted integrin subunit(s)
| mAb | Isotype | Target |
|---|---|---|
| Efalizumab (Raptiva®) | hIgG1 | αL |
| R7.1 | mIgG1 | αL |
| TS1/22 | mIgG1 | αL |
| Natalizumab (Tysabri®) | hIgG4 | α4 |
| Vedolizumab (Entyvio®) | hIgG1 | α4β7 |
| Compound | Class | Target |
|---|---|---|
| LFA878 | Statin derivative | αL |
| BIRT377 | Hydantoin derivative | αL |
| XVA143 | Peptidomimetic | αLβ2 |
| Lifitegrast (Xiidra®) | Peptidomimetic | αLβ2 |
| Firategrast | Peptidomimetic | α4β1/α4β7 |
| RO0505376 | Peptidomimetic | α4β1/α4β7 |
Abbreviations: h, human; m, mouse.
Figure 2.
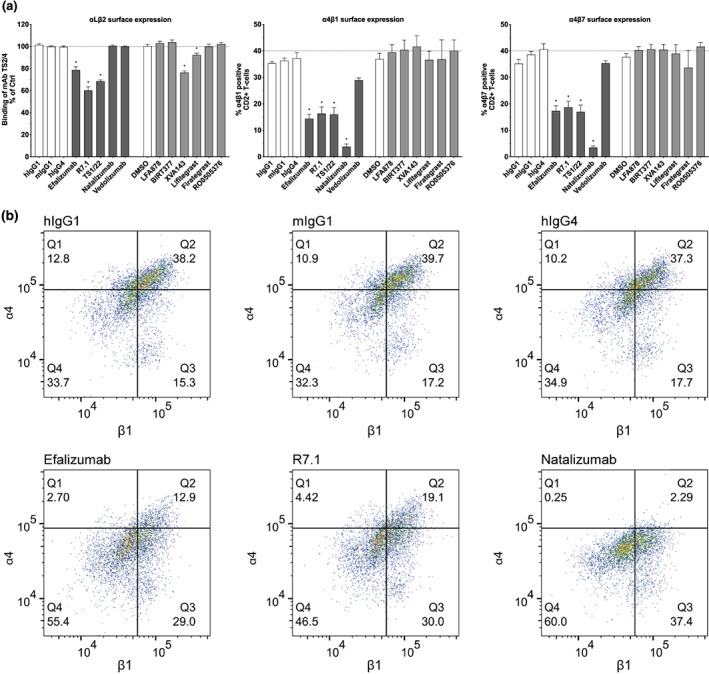
Effect of mAbs and compounds on integrin αLβ2, α4β1, and α4β7 surface expression. (a) Surface expression of αLβ2, α4β1, and α4β7 on CD2+ T cells after 24 hr of treatment with anti‐αL (efalizumab, R7.1, and TS1/22), anti‐α4 (natalizumab), anti‐α4β7 (vedolizumab) mAbs (all 10 μg·ml−1), small‐molecule inhibitors targeting αL (LFA878 and BIRT377), αLβ2 (XVA143 and lifitegrast), and α4β1/α4β7 (firategrast and RO0505376; all 10 μM) and respective controls. Each bar represents the mean value ± SEM of five independent experiments using blood samples from different donors. Statistical significance was determined by using one‐way ANOVA, *P < .05, versus control. (b) Flow cytometry dot plots of α4β1 CD2+ T cells. Representative flow cytometry dot plots of CD2+ T cells treated for 24 hr with efalizumab, R7.1, natalizumab and respective isotype controls are shown. Dots in the upper right quadrant indicate α4β1 positive CD2+ T cells. The samples were acquired using a Cytoflex cytometer and analysed using FlowJo software
Vice versa, we assessed the effect of the anti‐α4 mAb natalizumab on the expression of α4 integrins and αLβ2. Natalizumab binds to the β‐propeller of the α4 subunit shared by α4β1 and α4β7, close to the natural ligand‐binding pocket formed at the interface between the α4 β‐propeller and β I domain (Yu, Schurpf, & Springer, 2013; Figure 1 and Table 1). Consistent with previous studies (Benkert et al., 2012; Defer et al., 2012; Jilek et al., 2014), we found natalizumab to reduce in vitro the surface expression of its target integrins α4β1 and α4β7 by 89.8% and 91.4%, respectively (Figure 2a,b). However, despite this pronounced effect on α4 integrins, natalizumab did not downmodulate αLβ2. This finding suggests that the decrease of α4 integrins induced by anti‐αLβ2 mAbs is a unidirectional phenomenon. To further substantiate this conclusion, we investigated the effect of vedolizumab on α4 integrin and αLβ2 expression. In contrast to natalizumab, vedolizumab selectively binds to α4β7 and is used to treat Crohn's disease and ulcerative colitis (Feagan et al., 2013; Sandborn et al., 2013; Soler et al., 2009). The selectivity of vedolizumab can be derived from its binding site located on the β7 βI domain at the interface with the α4 β‐propeller domain (Figure 1 and Table 1; Yu et al., 2012). To our surprise, we found vedolizumab to neither reduce the expression of α4β7 nor to affect the expression of integrins α4β1 and αLβ2 (Figure 2a). Of note, a previous in vitro study focusing on memory T cells, a subpopulation of CD2+ T cells, reported reduced α4β7 surface expression in the presence of vedolizumab (Wyant, Yang, & Fedyk, 2013). In this study, however, the fate of fluorescently labelled vedolizumab was used to deduce the fate of α4β7, whereas we used a non‐competing anti‐β7 antibody to assess the cell surface expression of α4β7 itself. These differences in experimental design could explain why we were able to document the unaltered expression of α4β7 in the presence of vedolizumab, in contrast to the earlier study (Wyant et al., 2013).
3.2. Small‐molecule αLβ2 inhibitors do not affect α4 integrins
Next, we asked the question whether downmodulation of α4 integrins may be observed also in the presence of small‐molecule αLβ2 inhibitors. Two different classes of small‐molecule αLβ2 inhibitors were investigated. http://birt377 and LFA878 belong to the class of α I allosteric inhibitors which bind to an allosteric pocket of the αL I domain, thereby stabilizing αLβ2 in its inactive conformation (Figure 1 and Table 1; Kelly et al., 1999; Mancuso et al., 2016; Weitz‐Schmidt et al., 2001). XVA143 and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=7533 are ligand mimetic inhibitors, derived from amino acids of the αLβ2 ligand ICAM‐1 critical for binding to αLβ2 (Figure 1 and Table 1; Gadek et al., 2002; Semba & Gadek, 2016; Shimaoka, Salas, Yang, Weitz‐Schmidt, & Springer, 2003). Whether these ligand mimetics directly compete with ICAM‐1 binding to the αL I domain of αLβ2 or indirectly by preventing the interaction of an intrinsic ligand to the http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2456&familyId=760&familyType=CATALYTICRECEPTOR I domain is under debate (Semba & Gadek, 2016; Shimaoka et al., 2003). Of note, lifitegrast is approved as a topical treatment of dry eye disease (Semba & Gadek, 2016). We found that BIRT377 and LFA878 did not reduce αLβ2 expression and did not downmodulate α4 integrins (Figure 2a). Likewise, XVA143 and lifitegrast did not affect α4 integrin expression, although, as previously shown for XVA143, they downmodulated αLβ2 by 23.7% and 7.8%, respectively, as compared to solvent control (Figure 2a). These results demonstrate that small‐molecule inhibitors of αLβ2 with different modes of action do not cross‐modulate α4β1 and α4β7, in contrast to mAbs inhibiting αLβ2. We also investigated the effect of the dually acting α4β1/α4β7 ligand mimetic inhibitors firategrast and RO0505376 on α4 and αLβ2 expression (Miller et al., 2012; Yu et al., 2012). This class of inhibitors has been shown to bind across the integrin α4 β propeller–βI domain interface, which forms the ligand‐binding site (Figure 1 and Table 1; Miller et al., 2012; Yu et al., 2012). These compounds neither internalized α4β1 and α4β7 (in contrast to natalizumab) nor did they affect αLβ2 expression (Figure 2). Taken together these data indicate that small‐molecule and antibody‐based αLβ2 or α4 integrin inhibition result in different effect profiles regarding target internalization and selectivity.
3.3. Antibody‐engaged αLβ2 reduces extracellular as well as intracellular α4 integrin
Previous studies established that endocytosed integrins are predominantly recycled back to the membrane rather than being targeted to lysosomal compartments for degradation (Moreno‐Layseca, Icha, Hamidi, & Ivaska, 2019). In contrast, efalizumab‐ and natalizumab‐engaged integrins have been described to be preferentially routed towards the lysosomal pathway (Benkert et al., 2012; Coffey et al., 2004; Mancuso et al., 2016). We therefore extended our investigation to also analyse the intracellular fate of non‐occupied α4 integrins cross‐modulated by efalizumab, with natalizumab included as a positive control. Using ImageStream analysis, we demonstrated reduced overall extracellular/intracellular staining (=total content) of α4 integrins after 24 hr of anti‐αLβ2 antibody exposure, as compared to IgG controls (Figure 3a). As expected, anti‐α4 natalizumab triggered the same, even to more pronounced degrees (Figure 3a). These results establish that antibody‐induced internalization of α4 integrins does not lead to an accumulation of α4 within the cells, irrespective of whether this internalization is induced by efalizumab (cross‐modulation) or by natalizumab (target internalization). Apparently, in both instances, α4 integrins follow the degradative pathway and are removed from the normal integrin recycling route. To substantiate this interpretation, we determined the subcellular localization of internalized α4 integrin in cells treated with the antibodies for 14 hr using ImageStream analysis. This shorter exposure time was chosen because previous studies indicated that lysosomal localization of integrins is best detectable within the first 14 hr rather than at later time points of more advanced degradation (Coffey et al., 2004; Lobert et al., 2010). We once again found intracellular α4 staining to be decreased with both antibodies efalizumab and natalizumab, consistent with the results described above (Figure 3b). Moreover, the α4 staining observed was mostly limited to single dots, substantiating lysosomal localization (Figure 3b). To further ascertain the involvement of lysosomal pathways, we next performed experiments in the presence of the antibiotic concanamycin A. Concanamycin A specifically inhibits vacuolar‐type ATPases, thereby retarding lysosomal functions (Huss & Wieczorek, 2009). In agreement with above described results, we found that anti‐αLβ2 mAb‐induced downmodulation of αLβ2 and α4 integrin was prevented to similar degrees in the presence of concanamycin A (Figure 3bc,d). This finding confirms that the reductions of both αLβ2 and α4 integrin expression induced by anti‐αLβ2 mAbs are indeed due to lysosomal protein degradation. Other pathways, for example, pathways involving regulation at transcriptional levels, are considered less likely to play important roles. Numerous studies have shown that regulation of integrin function occurs mainly at conformational and endocytotic levels rather than transcriptional levels (Walling & Kim, 2018).
Interestingly, other than the fates of αLβ2 bound and α4 integrin cross‐modulated by anti‐αLβ2 mAb, the fate of α4 integrin bound by natalizumab is not affected by concanamycin A treatment under the experimental conditions applied (Figure 3c,d). These data suggest that the α4 cross‐modulation mediated by anti‐αL antibodies and the α4 downmodulation mediated by anti‐α4 antibody binding represent different phenomena. This clear differential between the two phenomena had been apparent already from our observation that anti‐α4 natalizumab does not cross‐modulate αLβ2, while anti‐LFA‐1 efalizumab conversely cross‐modulates α4 integrins. Hypothetically, natalizumab‐occupied α4 integrin may follow degradative pathways not sensitive to concanamycin A in CD2+ lymphocytes. Alternatively, concanamycin A may not be adequately potent to prevent lysosomal degradation of natalizumab/α4 complexes. Further investigations outside the scope of the current study will be required to characterize the pathway involved in the α4 downmodulation by anti‐α4 antibodies.
3.4. Antibody‐engaged αLβ2 downmodulates α4 integrins in the absence of activation
There is strong evidence that both inactive and active forms of integrins can be internalized (Arjonen, Alanko, Veltel, & Ivaska, 2012; Nader, Ezratty, & Gundersen, 2016). Efalizumab‐bound αLβ2 is known to adopt an inactive conformation (Li et al., 2009; Mancuso et al., 2016). We assessed the activation status of both mAb‐engaged αLβ2 and anti‐αLβ2 cross‐modulated α4β1 by probing the induction of well‐characterized activation epitopes located on the β2 chain (m24 and MEM148) and the http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2455&familyId=760&familyType=CATALYTICRECEPTOR chain (HUTS‐21) respectively (Figure 1; Chigaev et al., 2009; Njus et al., 2009; Schurpf & Springer, 2011). This activation epitope analysis revealed that none of the anti‐αLβ2 mAbs used in this study activated αLβ2 or cross‐activated α4β1 (Figure 4a–c). Similarly, natalizumab and vedolizumab did not affect the conformational status of their target integrins (Figure 4a–c).
Figure 4.
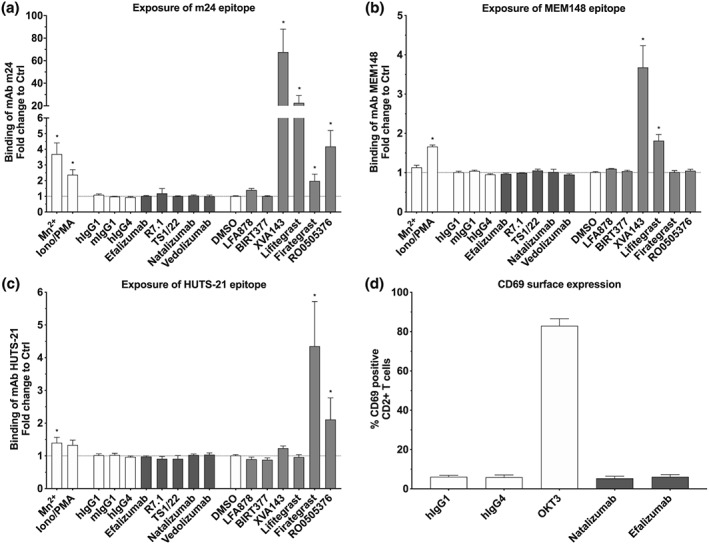
Effect of αLβ2 inhibitors on the conformational state of αLβ2, α4β1, and CD69 expression. Exposure of (a) m24 epitope, (b) MEM148 epitope, and (c) HUTS‐21 on CD2+ T cells after 40 min of treatment with anti‐αL (efalizumab, R7.1, and TS1/22), anti‐α4 (natalizumab), anti‐α4β7 (vedolizumab) mAbs (all 10 μg·ml−1), small‐molecule inhibitors targeting αL (LFA878 and BIRT377), αLβ2 (XVA143 and lifitegrast) and α4 (firategrast and RO0505376; all 10 μM), and respective controls. (d) Surface expression of CD69 on CD2+ T cells after 24 hr of treatment with efalizumab (10 μg·ml−1), natalizumab (10 μg·ml−1), and respective isotype controls (hIgG1 and hIgG4). Anti‐CD3 (OKT3) was used as positive control for the induction of CD69 surface expression. Each bar represents the mean value ± SEM of five independent experiments using blood samples from different donors. Statistical significance was determined by using one‐way ANOVA, *P < .05, versus control
As paradoxical induction of β chain activation epitopes is a common characteristic of ligand mimetic integrin inhibitors but not of α I allosteric αLβ2 inhibitors, we included both classes of inhibitors as controls (Figure 1 and Table 1; Ahrens & Peter, 2008; Shimaoka et al., 2003). As expected, the ligand mimetic αLβ2 inhibitors XVA143 and lifitegrast induced β2 chain activation epitopes m24 and MEM148, whereas the β1 chain activation epitope HUTS‐21 was not affected (Figure 4a–c). Vice versa, the ligand mimetic α4 integrin inhibitors firategrast and RO0505376 triggered the β1 HUTS‐21, as expected (Figure 4c). Interestingly, however, both compounds also induced the exposure of the m24 activation epitope of the β2 I domain of αLβ2 but not of the β2 MEM148 activation epitope (Figure 4a,b), located more distally from the integrin headpiece (Figure 1). The m24 exposure induced by α4 ligand mimetics is considerably weaker than the induction observed with αLβ2 ligand mimetics (Figure 4a,b). It is likely explained by a direct interaction of the compounds with the αLβ2 β2 I domain, reflecting the high homology between the I domains of integrin α and β subunits. Intriguingly, these data also show that the degree of ligand mimetic‐induced αLβ2 activation correlates with the magnitude of ligand mimetic‐induced αLβ2 internalization (Figures 2a and 4a,b), suggesting a clear differentiation between the phenomena of activation‐dependent αLβ2 internalization triggered by αLβ2 ligand mimetics and activation‐independent αLβ2 internalization triggered by anti‐αLβ2 antibodies. The α I allosteric inhibitors BIRT377 and LFA878 did not cause β chain activation epitope exposure, confirming earlier studies (Mancuso et al., 2016; Shimaoka et al., 2003), similar to anti‐αLβ2 mAbs (Figure 4a–c). Other than anti‐αLβ2 antibodies, however, they neither triggered αLβ2 internalization or α4 cross‐modulation, establishing the categorical differentiation of these two types of actions (Figure 2a). Further control reagents such as http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2341/ionomycin (activation of αLβ2 via inside‐out signalling) and Mn2+ (artificial extracellular activation) resulted in expected activation epitope induction profiles (Chigaev et al., 2009; Mancuso et al., 2016; Schurpf & Springer, 2011).
We next addressed the question whether the internalization of antibody‐engaged αLβ2 per se may activate CD2+ T cells, thereby evoking the downmodulation of α4 integrins. We assessed the effect of efalizumab on CD69 expression, a T‐cell activation marker (Gonzalez‐Amaro, Cortes, Sanchez‐Madrid, & Martin, 2013). We found that efalizumab did not up‐regulate CD69, in contrast to cells activated with the control anti‐http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2742&familyId=852&familyType=OTHER mAb http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=6889 (Figure 4d). In addition, natalizumab did not induce CD69 expression (Figure 4d). These results together indicate that mAb‐induced internalization of αLβ2 and α4 integrins is neither associated with an active conformation of these integrins nor with states of T‐cell activation, as characterized by CD69 expression.
3.5. Anti‐αLβ2 mAb bivalency is crucial for α4 integrin cross‐modulation
We hypothesized that mAb‐induced clustering of αLβ2 may solely drive the simultaneous internalization of αLβ2 and α4 integrins. To further test this hypothesis, F(ab′)2 and Fab fragments of the mAb R7.1 were prepared, purified, and shown to bind to αLβ2 (Figure 5a,b). R7.1 (Fab′)2 fragments still reduced αLβ2 and α4 surface expression yet to lesser extents than full‐size R7.1 IgG (Figure 5c,d).
Figure 5.
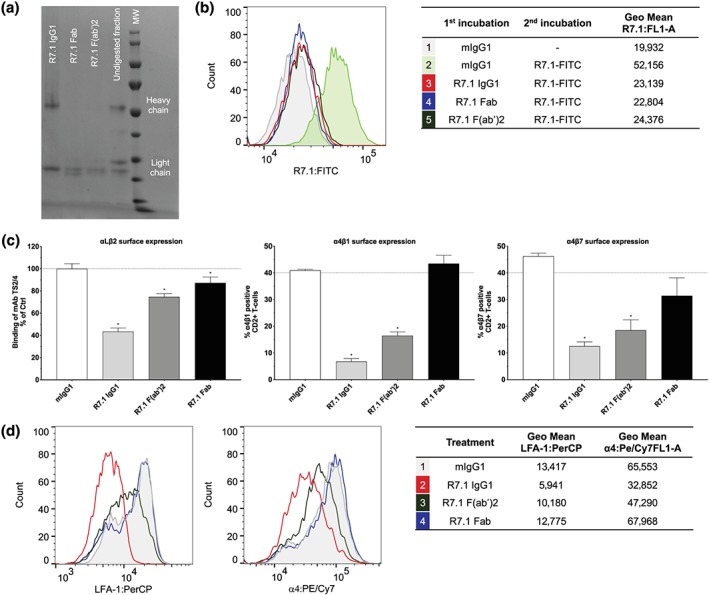
Effect of R7.1 IgG1, R7.1 F (ab′)2, and R7.1 Fab on αLβ2, α4β1, and α4β7 surface expression. (a) Analysis of R7.1 IgG1, R7.1 Fab, and R7.1 F(ab′)2 by reducing SDS‐PAGE. (b) Binding of mAb and fragments (all 10 μg·ml−1) to αLβ2 on HL‐60 cells. (c) Surface expression of αLβ2 and α4 integrin on CD2+ T cells after 24 hr of treatment with anti‐αL R7.1 IgG1, R7.1 F (ab′)2, R7.1 Fab and mIgG1 (all 10 μg·ml−1). Each bar represents the mean value ± SEM of five independent experiments using blood samples from different donors. Statistical significance was determined by using one‐way ANOVA, *P < .05, versus control. (d) Representative histogram plots of αLβ2 and α4 expression on CD2+ T cells. The samples were acquired using the Cytoflex cytometer and analysed applying the FlowJo software
This partial loss of down‐modulatory capacity with murine R7.1 (Fab′)2 fragments was unexpected because human Fc receptors reportedly have poor cross‐reactivities with murine Fc (Bruhns, 2012). We therefore decided to more directly assess the potential impact of Fc receptor‐mediated crosslinking in our assay system by using anti‐murine Fc antibodies (mimicking Fc receptor‐mediated crosslinking). Not surprisingly, this incremental crosslinking resulted in more prominent internalization of αLβ2 as compared to non‐crosslinking conditions (Table 2). Intriguingly, however, it did not impact the cross‐modulatory effects of anti‐αLβ2 mAbs on α4 integrin surface expression (Table 2).
Table 2.
Fold increase of anti‐αLβ2 mAb induced αLβ2, α4β1 and α4β7 internalization upon anti‐Fc crosslinking
| mAb | αLβ2 | α4β1 | α4β7 |
|---|---|---|---|
| Efalizumab | 2.60 ± 0.23* | 1.02 ± 0.02 (ns) | 1.04 ± 0.03 (ns) |
| R7.1 | 1.35 ± 0.05 (ns) | 1.00 ± 0.05 (ns) | 0.97 ± 0.07 (ns) |
| TS1/22 | 1.55 ± 0.02* | 1.03 ± 006 (ns) | 1.02 ± 0.10 (ns) |
Note. Mean value ± SEM of six independent experiments using blood samples from different donors are shown. Statistical significance was determined by using one‐way ANOVA.
Abbreviation: ns, not significant.
P < .05.
versus control.
Monovalent R7.1 Fab fragments minimally reduced αLβ2 expression and completely lost the capacity to decrease α4 integrin expression (Figure 5c,d).
Taken together, these data show that anti‐αLβ2 mAb bivalency is crucial for both αLβ2 downmodulation and α4 integrin cross‐modulation, while further crosslinking via the Fc portion of the antibodies only impacts αLβ2 downmodulation yet not α4 integrin cross‐modulation.
3.6. Dynamin‐dependent pathways contribute to mAb‐induced αLβ2 and α4 integrin internalization
Previous studies established that integrins are internalized at the plasma membrane via several types of endocytosis, including clathrin‐dependent and clathrin‐independent routes (Bridgewater, Norman, & Caswell, 2012; Moreno‐Layseca et al., 2019). To study which endocytotic mechanism may be involved in anti‐αLβ2‐triggered αLβ2 and α4 integrin downmodulation, we tested the effect of dynasore on αLβ2 and α4 surface expression in mAb R7.1‐treated CD2+ T cells. Dynasore is an inhibitor of dynamin, a GTPase acting at fission sites for clathrin‐, caveolin‐ and http://il-2 receptor‐mediated endocytosis (Blouin & Lamaze, 2013; Ferguson & De Camilli, 2012). We found that dynasore partially prevented the internalization of αLβ2 and α4 integrins induced by mAb R7.1 (Figure 6a,b). The effect size of dynasore on the internalization of these integrins was similar to the magnitude of effect elicited in cells co‐transfected with αLβ2 and a dominant negative mutant of dynamin (Fabbri et al., 2005). Similarly, natalizumab‐induced α4 integrin downmodulation was inhibited by dynasore; however, this effect did not reach significance. Further, in control experiments, dynasore inhibited anti‐CD3 mAb OKT3‐induced CD3 internalization, whose dynamin‐dependent endocytosis has been documented before (Figure S2; Compeer et al., 2018; Yudushkin & Vale, 2010).
Figure 6.
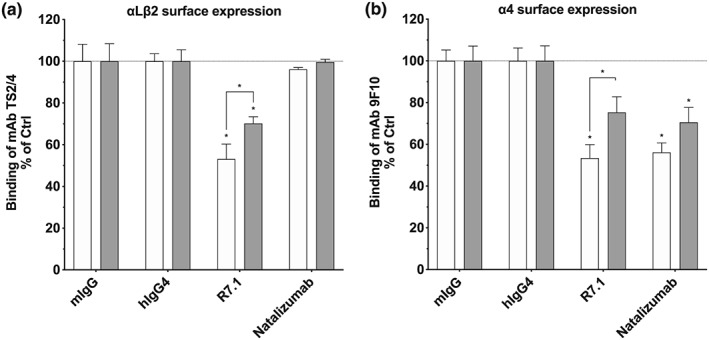
Effect of dynasore on R7.1 or natalizumab‐induced downmodulation of αLβ2 and α4. Surface expression of (a) αLβ2 and (b) α4 integrins on CD2+ T cells after 24 hr of treatment with anti‐αL R7.1 (10 μg·ml−1), anti‐α4 natalizumab (10 μg·ml−1) and respective isotype controls (mIgG1 and hIgG4). Treatments were performed in presence of dynasore (grey bars) or DMSO controls (white bars). Each bar represents the mean value ± SEM of six independent experiments using blood samples from different donors. Statistical significance was determined by using one‐way ANOVA, *P < .05, versus control and two‐way ANOVA, $ P < .05 versus incubation without dynasore
Taken together, these results provide evidence that both anti‐αLβ2 mAb‐induced αLβ2 internalization and endocytotic α4 integrin cross‐modulation involves dynamin‐dependent endocytosis.
4. DISCUSSION
This study sheds new light on the unexpected clinical observation of reduced α4β1 and α4β7 expression in peripheral blood lymphocytes of patients treated with anti‐αLβ2 efalizumab (Guttman‐Yassky et al., 2008; Vugmeyster et al., 2004) and the mechanisms thereof. Our investigation establishes that this alteration of α4 expression involves a hitherto unknown direct downstream effect of efalizumab binding to αLβ2, involving dynamin‐dependent endocytosis and concanamycin A sensitive lysosomal degradation. The phenomenon is referred to as endocytotic cross‐modulation. The present study further elucidates the specific requirements and mechanisms of anti‐αLβ2 antibody‐mediated α4 integrin cross‐modulation. We found that antibody bivalency was a strict requirement for integrin cross‐modulation; that is, monovalent Fab fragments lost the capacity to downmodulate α4 integrins. This suggests that αLβ2 clustering is critical to integrin cross‐modulation. This notion is further supported by the fact that monovalent small‐molecule inhibitors of αLβ2 without the capacity to cluster αLβ2 did not affect α4 integrin expression.
The aberrancy of antibody‐induced targeting of αLβ2 to lysosomal pathways, which have been proposed as pathways by which efalizumab is cleared in vivo (Coffey et al., 2004), is a further prerequisite for the α4 endocytic cross‐modulation described here. We could positively affirm this by showing that ligand mimetic αLβ2 inhibitors, such as XVA143, do not affect α4 integrins although they also reduce αLβ2 surface expression in vitro to degrees observed with anti‐αLβ2 mAbs by a different mechanism. The ligand mimetic class of αLβ2 inhibitors is thought to alter the kinetics of αLβ2 recycling by stabilizing a primed, semi‐active conformer of αLβ2 rather than directing αLβ2 to the lysosome as observed with inhibitory anti‐αLβ2 mAbs (Arjonen et al., 2012; Mancuso et al., 2016).
Notably, anti‐αLβ2 induced cross‐modulation of α4 integrins neither required integrin activation nor cellular activation. It occurred in antibody‐treated resting cells. Thus, the effect is fundamentally different from the integrin crosstalk between αLβ2 and α4β1 reported previously (Gronholm et al., 2016; Porter & Hogg, 1997; Uotila et al., 2014). This crosstalk requires activated αLβ2 and is associated with signalling cascades shutting‐off α4β1 function. Interestingly, however, both integrin crosstalk and endocytotic cross‐modulation have in common that they are unidirectional, at least when studied in in vitro systems (Gronholm et al., 2016; Porter & Hogg, 1997).
We were interested to further understand the mechanisms leading to the downmodulation of α4 integrins by mAb‐engaged αLβ2. It is tempting to speculate that clustering of inactive αLβ2 induced by antibodies evokes endocytotic signals, which aberrantly internalize αLβ2 together with co‐localized and/or co‐recruited receptors such as α4 integrins as additional cargo. Indeed, we were able to provide evidence for the dynamin dependency of the cross‐modulatory phenomenon observed. Moreover, a potential linkage between αLβ2 and α4 integrin endocytosis is biologically plausible because these integrins need to act in concert in order to control leukocyte migration and immune synapse function (Bertoni et al., 2018; Chigaev & Sklar, 2012; Mittelbrunn et al., 2004; Walling & Kim, 2018). The known hierarchy of integrin usage in these processes (αLβ2 is the dominant integrin; Bertoni et al., 2018; Porter & Hogg, 1997) may also explain the directionality of these processes; that is, mAb‐engaged αLβ2 downmodulates α4 integrins but not vice versa.
A relevant question is whether integrin cross‐modulation as observed may represent Fc receptor‐mediated trogocytosis. This Fc‐mediated functionality of mAbs involves a rapid intercellular transfer of membrane fragments associated with antibody–antigen immune complexes during intercellular contact (Taylor et al., 2015). Of note, earlier studies have established already that anti‐αLβ2 antibodies, in contrast to, for example, anti‐T‐cell receptor, anti‐CD3ε and http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2863&familyId=852&familyType=OTHER antibodies, do not elicit and in fact suppress trogocytosis (Hudrisier, Aucher, Puaux, Bordier, & Joly, 2007; Miyake et al., 2017). Thus, having used anti‐αLβ2 antibodies at saturation levels and having established, further, that integrin cross‐modulation does not require αLβ2 activation (which would be a prerequisite for its binding to ICAM‐1), we can exclude αLβ2‐mediated trogocytotic processes as a relevant mechanism for antibody‐induced reduction of integrin cell surface expression and cross‐modulation. This conclusion is affirmed further by the observation that murine anti‐αLβ2 antibodies such as R7.1 or TS1/22 (having Fc parts not or only poorly recognized by human Fc receptor; Bruhns, 2012) elicit αLβ2 internalization and α4 cross‐modulation to degrees similar to human anti‐αLβ2 efalizumab. Taken together, these results exclude trogocytosis as the mechanism behind the endocytotic cross‐modulation described in this study.
This in vitro study investigates, at the single cell level, the non‐canonical phenomenon of α4 integrin cross‐modulation by anti‐αLβ2 antibodies. The study utilized primary cells from healthy donors and employed integrin targeting actions at therapeutically relevant concentrations. Therefore, its findings are expected to be relevant for the in vivo situation and to shed further light on the hitherto unexplained downmodulation of α4 integrins observed in patients treated with the anti‐αLβ2 antibody efalizumab (Guttman‐Yassky et al., 2008; Vugmeyster et al., 2004). This study does not speak, however, to integrin‐mediated leukocyte trafficking, compartmental redistribution and chronic adaptive changes only observable in vivo, assessment of which will require dedicated in vivo studies. Further, the current in vitro study deliberately focused on α4 integrins for the important functional redundancies these integrins can provide if αLβ2 is inhibited. The current study does not investigate the mechanisms responsible for the downmodulation of other major immune receptors observed in efalizumab‐treated patients in vivo (Guttman‐Yassky et al., 2008; Vugmeyster et al., 2004). It is intuitive to speculate that the downmodulation of these other receptors under anti‐αLβ2 efalizumab therapy in vivo may follow similar endocytotic cross‐modulatory mechanisms as established for α4 integrins here. The availability of αLβ2 targeting action of diverse downstream effect profiles may help to further elucidate these still incompletely understood effects of anti‐αLβ2 antibodies in future in in vivo studies.
In conclusion, the findings of this study identify and characterize, for the first time to our knowledge, at the single cell level a hitherto unknown non‐canonical function of anti‐αLβ2 antibodies entirely attributable to their antibody nature. This phenomenon is of utmost therapeutic relevance because, in the case of anti‐αLβ2 efalizumab, it abrogates key functional redundancies provided by α4 integrins under αLβ2 blockade, specifically in terms of CNS immune surveillance. Given this level of potential clinical impact, the phenomenon described here should also be considered with other antibodies targeting cluster‐forming receptors, specifically receptors assembled within the immune synapse.
As a direct learning, we submit that internalizing antibodies targeting cluster‐forming receptors, such as the anti‐αLβ2 antibodies assessed here, should be considered to become tested for in vitro phenomena of non‐canonical endocytotic receptor cross‐modulation because such cross‐modulation has the potential to alter an antibody's benefit/risk profile, most fundamentally.
AUTHOR CONTRIBUTIONS
All authors contributed to the planning and analysis of the study as well as the writing of the manuscript. R.V.M. and J.C. conducted the experiments.
CONFLICT OF INTEREST
G.W.S. and R.V.M. are co‐inventors of a patent covering small‐molecule LFA‐1 inhibitors. G.W.S. and A.G.S. are shareholders of AlloCyte Pharmaceuticals AG. LFA‐1 inhibitors patented and pursued by AlloCyte Pharmaceuticals AG are not assessed in the current study.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for Design &Analysis, and as recommended by funding agencies, publishers and other organisation engaged with supporting research.
Supporting information
Table S1: Properties of small molecules and antibodies used in the present study.
Figure S1: Effect of efalizumab on integrin αLβ2 surface expression. Surface expression of αLβ 2 on CD2+ T cells after 24 hours and 48 hours of treatment with anti‐ αLβ 2 efalizumab (10 μg mL−1), and respective isotype control (hIgG1). Each bar represents a single determination using blood from one donor.
FIGURE S2: Effect of dynasore on mAb R7.1‐induced αLβ2 and α4 downmodulation, natalizumab induced α4 downmodulation and mAb OKT3‐induced downmodulation of CD3. Surface expression of (A) αLβ2 and (B) α4 integrin on CD2+ T cells after 24 hr of treatment with anti‐αL R7.1 (10 μg mL1), anti‐ α4 natalizumab (10 μg mL−1), and respective isotype controls (mIgG1, hIgG4). Treatments were performed in the presence of dynasore (40 μM, grey bars) or DMSO (white bars). Each bar represents the mean value ± SEM of 6 independent experiments using blood samples from different donors. Statistical significance was determined by using one‐way ANOVA, ***P < 0.001, ****P < 0.0001 vs control, and two‐way ANOVA, $$P < 0.01 vs incubation without dynasore. (C) Surface expression of CD3 on CD2+ T cells after 24 hours of treatment with anti‐CD3 OKT3 (1 ng mL−1, 10 ng mL−1, 100 ng mL1) in the presence of dynasore (40 μM, grey bars) or DMSO (white bars). Each bar represents a single determination, using blood from one donor.
ACKNOWLEDGEMENTS
The authors thank Peter Steinberger, Medical University of Vienna, Austria, for providing efalizumab, Berndt Oberhauser, Novartis Pharma AG Basel, for providing compound LFA878, Paul Gillespie, Hoffmann‐La Roche Inc. Nutley, for supplying compound RO0281607‐000 (also termed XVA143) and compound RO0505376, Raija Lindberg Gasser, University Hospital Basel, Switzerland, for providing natalizumab, and Petr Hruz, University Hospital Basel, for vedolizumab. This research was supported by Innosuisse (Grant 19080.29).
Mancuso RV, Casper J, Schmidt AG, Krähenbühl S, Weitz‐Schmidt G. Anti‐αLβ2 antibodies reveal novel endocytotic cross‐modulatory functionality. Br J Pharmacol. 2020;177:2696–2711. 10.1111/bph.14996
REFERENCES
- Ahrens, I. , & Peter, K. (2008). Therapeutic integrin inhibition: Allosteric and activation‐specific inhibition strategies may surpass the initial ligand‐mimetic strategies. Thrombosis and Haemostasis, 99, 803–804. 10.1160/TH08-03-0194 [DOI] [PubMed] [Google Scholar]
- Alexander, S. P. , Kelly, E. , Mathie, A. , Peters, J. A. , Veale, E. L. , Amstrong, J. F. , … Collaborators, GTP (2019). The Concise Guide to PHARMACOLOGY 2019/20: Introduction and Other Protein Targets. British Journal of Pharmacology, 176, S1–S20. 10.1111/bph.14747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arjonen, A. , Alanko, J. , Veltel, S. , & Ivaska, J. (2012). Distinct recycling of active and inactive β1 integrins. Traffic, 13, 610–625. 10.1111/j.1600-0854.2012.01327.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benkert, T. F. , Dietz, L. , Hartmann, E. M. , Leich, E. , Rosenwald, A. , Serfling, E. , … Berberich‐Siebelt, F. (2012). Natalizumab exerts direct signaling capacity and supports a pro‐inflammatory phenotype in some patients with multiple sclerosis. PLoS ONE, 7, e52208 10.1371/journal.pone.0052208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertoni, A. , Alabiso, O. , Galetto, A. S. , & Baldanzi, G. (2018). Integrins in T cell physiology. International Journal of Molecular Sciences, 19, 485. 10.3390/ijms19020485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blouin, C. M. , & Lamaze, C. (2013). Interferon gamma receptor: The beginning of the journey. Frontiers in Immunology, 4, 267 10.3389/fimmu.2013.00267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridgewater, R. E. , Norman, J. C. , & Caswell, P. T. (2012). Integrin trafficking at a glance. Journal of Cell Science, 125, 3695–3701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruhns, P. (2012). Properties of mouse and human IgG receptors and their contribution to disease models. Blood, 119, 5640–5649. 10.1182/blood-2012-01-380121 [DOI] [PubMed] [Google Scholar]
- Chames, P. , Van Regenmortel, M. , Weiss, E. , & Baty, D. (2009). Therapeutic antibodies: Successes, limitations and hopes for the future. British Journal of Pharmacology, 157, 220–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chigaev, A. , & Sklar, L. A. (2012). Aspects of VLA‐4 and LFA‐1 regulation that may contribute to rolling and firm adhesion. Frontiers in Immunology, 3, 242 10.3389/fimmu.2012.00242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chigaev, A. , Waller, A. , Amit, O. , Halip, L. , Bologa, C. G. , & Sklar, L. A. (2009). Real‐time analysis of conformation‐sensitive antibody binding provides new insights into integrin conformational regulation. The Journal of Biological Chemistry, 284, 14337–14346. 10.1074/jbc.M901178200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffey, G. P. , Stefanich, E. , Palmieri, S. , Eckert, R. , Padilla‐Eagar, J. , Fielder, P. J. , & Pippig, S. (2004). In vitro internalization, intracellular transport, and clearance of an anti‐CD11a antibody (Raptiva) by human T‐cells. The Journal of Pharmacology and Experimental Therapeutics, 310, 896–904. 10.1124/jpet.104.067611 [DOI] [PubMed] [Google Scholar]
- Compeer, E. B. , Kraus, F. , Ecker, M. , Redpath, G. , Amiezer, M. , Rother, N. , … Rossy, J. (2018). A mobile endocytic network connects clathrin‐independent receptor endocytosis to recycling and promotes T cell activation. Nature Communications, 9, 1597 10.1038/s41467-018-04088-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis, M. J. , Bond, R. A. , Spina, D. , Ahluwalia, A. , Alexander, S. P. , Giembycz, M. A. , … McGrath, J. (2015). Experimental design and analysis and their reporting: New guidance for publication in BJP. British Journal of Pharmacology, 172, 3461–3471. 10.1111/bph.12856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Defer, G. , Mariotte, D. , Derache, N. , Toutirais, O. , Legros, H. , Cauquelin, B. , & le Mauff, B. (2012). CD49d expression as a promising biomarker to monitor natalizumab efficacy. Journal of the Neurological Sciences, 314, 138–142. 10.1016/j.jns.2011.10.005 [DOI] [PubMed] [Google Scholar]
- Etzioni, A. (2010). Defects in the leukocyte adhesion cascade. Clinical Reviews in Allergy & Immunology, 38, 54–60. 10.1007/s12016-009-8132-3 [DOI] [PubMed] [Google Scholar]
- Fabbri, M. , Di Meglio, S. , Gagliani, M. C. , Consonni, E. , Molteni, R. , Bender, J. R. , … Pardi, R. (2005). Dynamic partitioning into lipid rafts controls the endo‐exocytic cycle of the αL/β2 integrin, LFA‐1, during leukocyte chemotaxis. Molecular Biology of the Cell, 16, 5793–5803. 10.1091/mbc.e05-05-0413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feagan, B. G. , Rutgeerts, P. , Sands, B. E. , Hanauer, S. , Colombel, J. F. , Sandborn, W. J. , … GEMINI 1 Study Group (2013). Vedolizumab as induction and maintenance therapy for ulcerative colitis. The New England Journal of Medicine, 369, 699–710. 10.1056/NEJMoa1215734 [DOI] [PubMed] [Google Scholar]
- Ferguson, S. M. , & De Camilli, P. (2012). Dynamin, a membrane‐remodelling GTPase. Nature Reviews Molecular Cell Biology, 13, 75–88. 10.1038/nrm3266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadek, T. R. , Burdick, D. J. , McDowell, R. S. , Stanley, M. S. , Marsters, J. C. , Paris, K. J. Jr. , … Lee, W. P. (2002). Generation of an LFA‐1 antagonist by the transfer of the ICAM‐1 immunoregulatory epitope to a small molecule. Science, 295, 1086–1089. 10.1126/science.295.5557.1086 [DOI] [PubMed] [Google Scholar]
- George, T. C. , Fanning, S. L. , Fitzgerald‐Bocarsly, P. , Medeiros, R. B. , Highfill, S. , Shimizu, Y. , … Lynch, D. H. (2006). Quantitative measurement of nuclear translocation events using similarity analysis of multispectral cellular images obtained in flow. Journal of Immunological Methods, 311, 117–129. 10.1016/j.jim.2006.01.018 [DOI] [PubMed] [Google Scholar]
- Gonzalez‐Amaro, R. , Cortes, J. R. , Sanchez‐Madrid, F. , & Martin, P. (2013). Is CD69 an effective brake to control inflammatory diseases? Trends in Molecular Medicine, 19, 625–632. 10.1016/j.molmed.2013.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gronholm, M. , Jahan, F. , Bryushkova, E. A. , Madhavan, S. , Aglialoro, F. , Soto Hinojosa, L. , … Gahmberg, C. G. (2016). LFA‐1 integrin antibodies inhibit leukocyte α4β1‐mediated adhesion by intracellular signaling. Blood, 128, 1270–1281. 10.1182/blood-2016-03-705160 [DOI] [PubMed] [Google Scholar]
- Guttman‐Yassky, E. , Vugmeyster, Y. , Lowes, M. A. , Chamian, F. , Kikuchi, T. , Kagen, M. , … Krueger, J. G. (2008). Blockade of CD11a by efalizumab in psoriasis patients induces a unique state of T‐cell hyporesponsiveness. The Journal of Investigative Dermatology, 128, 1182–1191. 10.1038/jid.2008.4 [DOI] [PubMed] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … NC‐IUPHAR (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: Updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Research, 46, D1091–D1106. 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudrisier, D. , Aucher, A. , Puaux, A. L. , Bordier, C. , & Joly, E. (2007). Capture of target cell membrane components via trogocytosis is triggered by a selected set of surface molecules on T or B cells. Journal of Immunology, 178, 3637–3647. [DOI] [PubMed] [Google Scholar]
- Huss, M. , & Wieczorek, H. (2009). Inhibitors of V‐ATPases: old and new players. The Journal of Experimental Biology, 212, 341–346. 10.1242/jeb.024067 [DOI] [PubMed] [Google Scholar]
- Jilek, S. , Mathias, A. , Canales, M. , Lysandropoulos, A. , Pantaleo, G. , Schluep, M. , & du Pasquier, R. A. (2014). Natalizumab treatment alters the expression of T‐cell trafficking marker LFA‐1 α‐chain (CD11a) in MS patients. Multiple Sclerosis, 20, 837–842. 10.1177/1352458513513208 [DOI] [PubMed] [Google Scholar]
- Kelly, T. A. , Jeanfavre, D. D. , McNeil, D. W. , Woska, J. R. Jr. , Reilly, P. L. , Mainolfi, E. A. , … Rothlein, R. (1999). Cutting edge: A small molecule antagonist of LFA‐1‐mediated cell adhesion. Journal of Immunology, 163, 5173–5177. [PubMed] [Google Scholar]
- Lebwohl, M. , Tyring, S. K. , Hamilton, T. K. , Toth, D. , Glazer, S. , Tawfik, N. H. , … Efalizumab Study Group (2003). A novel targeted T‐cell modulator, efalizumab, for plaque psoriasis. The New England Journal of Medicine, 349, 2004–2013. 10.1056/NEJMoa030002 [DOI] [PubMed] [Google Scholar]
- Li, S. , Wang, H. , Peng, B. , Zhang, M. , Zhang, D. , Hou, S. , … Ding, J. (2009). Efalizumab binding to the LFA‐1 αL I domain blocks ICAM‐1 binding via steric hindrance. Proceedings of the National Academy of Sciences of the United States of America, 106, 4349–4354. 10.1073/pnas.0810844106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobert, V. H. , Brech, A. , Pedersen, N. M. , Wesche, J. , Oppelt, A. , Malerod, L. , & Stenmark, H. (2010). Ubiquitination of α5β1 integrin controls fibroblast migration through lysosomal degradation of fibronectin‐integrin complexes. Developmental Cell, 19, 148–159. 10.1016/j.devcel.2010.06.010 [DOI] [PubMed] [Google Scholar]
- Lodygin, D. , Hermann, M. , Schweingruber, N. , Flugel‐Koch, C. , Watanabe, T. , Schlosser, C. , … Reichardt, H. M. (2019). Publisher correction: β‐Synuclein‐reactive T cells induce autoimmune CNS grey matter degeneration. Nature, 567: E15, 503–508. [DOI] [PubMed] [Google Scholar]
- Lu, C. , Shimaoka, M. , Salas, A. , & Springer, T. A. (2004). The binding sites for competitive antagonistic, allosteric antagonistic, and agonistic antibodies to the I domain of integrin LFA‐1. Journal of Immunology, 173, 3972–3978. [DOI] [PubMed] [Google Scholar]
- Mancuso, R. V. , Welzenbach, K. , Steinberger, P. , Krahenbuhl, S. , & Weitz‐Schmidt, G. (2016). Downstream effect profiles discern different mechanisms of integrin αLβ2 inhibition. Biochemical Pharmacology, 119, 42–55. 10.1016/j.bcp.2016.09.002 [DOI] [PubMed] [Google Scholar]
- Miller, D. H. , Weber, T. , Grove, R. , Wardell, C. , Horrigan, J. , Graff, O. , … Montalban, X. (2012). Firategrast for relapsing remitting multiple sclerosis: A phase 2, randomised, double‐blind, placebo‐controlled trial. Lancet Neurology, 11, 131–139. 10.1016/S1474-4422(11)70299-X [DOI] [PubMed] [Google Scholar]
- Mittelbrunn, M. , Molina, A. , Escribese, M. M. , Yanez‐Mo, M. , Escudero, E. , Ursa, A. , … Sanchez‐Madrid, F. (2004). VLA‐4 integrin concentrates at the peripheral supramolecular activation complex of the immune synapse and drives T helper 1 responses. Proceedings of the National Academy of Sciences of the United States of America, 101, 11058–11063. 10.1073/pnas.0307927101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyake, K. , Shiozawa, N. , Nagao, T. , Yoshikawa, S. , Yamanishi, Y. , & Karasuyama, H. (2017). Trogocytosis of peptide‐MHC class II complexes from dendritic cells confers antigen‐presenting ability on basophils. Proceedings of the National Academy of Sciences of the United States of America, 114, 1111–1116. 10.1073/pnas.1615973114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno‐Layseca, P. , Icha, J. , Hamidi, H. , & Ivaska, J. (2019). Integrin trafficking in cells and tissues. Nature Cell Biology, 21, 122–132. 10.1038/s41556-018-0223-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nader, G. P. , Ezratty, E. J. , & Gundersen, G. G. (2016). FAK, talin and PIPKIgamma regulate endocytosed integrin activation to polarize focal adhesion assembly. Nature Cell Biology, 18, 491–503. 10.1038/ncb3333 [DOI] [PubMed] [Google Scholar]
- Njus, B. H. , Chigaev, A. , Waller, A. , Wlodek, D. , Ostopovici‐Halip, L. , Ursu, O. , … Sklar, L. A. (2009). Conformational mAb as a tool for integrin ligand discovery. Assay and Drug Development Technologies, 7, 507–515. 10.1089/adt.2009.0203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter, J. C. , & Hogg, N. (1997). Integrin cross talk: Activation of lymphocyte function‐associated antigen‐1 on human T cells alters α4β1‐ and α5β1‐mediated function. The Journal of Cell Biology, 138, 1437–1447. 10.1083/jcb.138.6.1437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothhammer, V. , Muschaweckh, A. , Gasteiger, G. , Petermann, F. , Heink, S. , Busch, D. H. , … Korn, T. (2014). α4‐integrins control viral meningoencephalitis through differential recruitment of T helper cell subsets. Acta Neuropathologica Communications, 2, 27 10.1186/2051-5960-2-27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudick, R. , Polman, C. , Clifford, D. , Miller, D. , & Steinman, L. (2013). Natalizumab: Bench to bedside and beyond. JAMA Neurology, 70, 172–182. [DOI] [PubMed] [Google Scholar]
- Sandborn, W. J. , Feagan, B. G. , Rutgeerts, P. , Hanauer, S. , Colombel, J. F. , Sands, B. E. , … GEMINI 2 Study Group (2013). Vedolizumab as induction and maintenance therapy for Crohn's disease. The New England Journal of Medicine, 369, 711–721. 10.1056/NEJMoa1215739 [DOI] [PubMed] [Google Scholar]
- Schmits, R. , Kundig, T. M. , Baker, D. M. , Shumaker, G. , Simard, J. J. , Duncan, G. , … Ohashi, P. S. (1996). LFA‐1‐deficient mice show normal CTL responses to virus but fail to reject immunogenic tumor. The Journal of Experimental Medicine, 183, 1415–1426. 10.1084/jem.183.4.1415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider‐Hohendorf, T. , Philipp, K. , Husstedt, I. W. , Wiendl, H. , & Schwab, N. (2014). Specific loss of cellular L‐selectin on CD4+ T cells is associated with progressive multifocal leukoencephalopathy development during HIV infection. Aids, 28, 793–795. [DOI] [PubMed] [Google Scholar]
- Schurpf, T. , & Springer, T. A. (2011). Regulation of integrin affinity on cell surfaces. The EMBO Journal, 30, 4712–4727. 10.1038/emboj.2011.333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semba, C. P. , & Gadek, T. R. (2016). Development of lifitegrast: a novel T‐cell inhibitor for the treatment of dry eye disease. Clinical Ophthalmology, 10, 1083–1094. 10.2147/OPTH.S110557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seminara, N. M. , & Gelfand, J. M. (2010). Assessing long‐term drug safety: Lessons (re)learned from raptiva. Seminars in Cutaneous Medicine and Surgery, 29, 16–19. 10.1016/j.sder.2010.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimaoka, M. , Salas, A. , Yang, W. , Weitz‐Schmidt, G. , & Springer, T. A. (2003). Small molecule integrin antagonists that bind to the β2 subunit I‐like domain and activate signals in one direction and block them in the other. Immunity, 19, 391–402. 10.1016/s1074-7613(03)00238-3 [DOI] [PubMed] [Google Scholar]
- Smith, A. J. (2015). New horizons in therapeutic antibody discovery: Opportunities and challenges versus small‐molecule therapeutics. Journal of Biomolecular Screening, 20, 437–453. 10.1177/1087057114562544 [DOI] [PubMed] [Google Scholar]
- Soler, D. , Chapman, T. , Yang, L. L. , Wyant, T. , Egan, R. , & Fedyk, E. R. (2009). The binding specificity and selective antagonism of vedolizumab, an anti‐α4β7 integrin therapeutic antibody in development for inflammatory bowel diseases. The Journal of Pharmacology and Experimental Therapeutics, 330, 864–875. 10.1124/jpet.109.153973 [DOI] [PubMed] [Google Scholar]
- Tan, S. M. (2012). The leucocyte β2 (CD18) integrins: The structure, functional regulation and signalling properties. Bioscience Reports, 32, 241–269. [DOI] [PubMed] [Google Scholar]
- Taylor, A. , Foo, S. S. , Bruzzone, R. , Dinh, L. V. , King, N. J. , & Mahalingam, S. (2015). Fc receptors in antibody‐dependent enhancement of viral infections. Immunological Reviews, 268, 340–364. 10.1111/imr.12367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uotila, L. M. , Jahan, F. , Soto Hinojosa, L. , Melandri, E. , Gronholm, M. , & Gahmberg, C. G. (2014). Specific phosphorylations transmit signals from leukocyte β2 to β1 integrins and regulate adhesion. The Journal of Biological Chemistry, 289, 32230–32242. 10.1074/jbc.M114.588111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma, N. K. , & Kelleher, D. (2017). Not just an adhesion molecule: LFA‐1 contact tunes the T lymphocyte program. Journal of Immunology, 199, 1213–1221. [DOI] [PubMed] [Google Scholar]
- Vugmeyster, Y. , Kikuchi, T. , Lowes, M. A. , Chamian, F. , Kagen, M. , Gilleaudeau, P. , … Krueger, J. G. (2004). Efalizumab (anti‐CD11a)‐induced increase in peripheral blood leukocytes in psoriasis patients is preferentially mediated by altered trafficking of memory CD8+ T cells into lesional skin. Clinical Immunology, 113, 38–46. 10.1016/j.clim.2004.06.001 [DOI] [PubMed] [Google Scholar]
- Walling, B. L. , & Kim, M. (2018). LFA‐1 in T cell migration and differentiation. Frontiers in Immunology, 9, 952 10.3389/fimmu.2018.00952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weitz‐Schmidt, G. , Schurpf, T. , & Springer, T. A. (2011). The C‐terminal αI domain linker as a critical structural element in the conformational activation of αI integrins. The Journal of Biological Chemistry, 286, 42115–42122. 10.1074/jbc.M111.282830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weitz‐Schmidt, G. , Welzenbach, K. , Brinkmann, V. , Kamata, T. , Kallen, J. , Bruns, C. , … Hommel, U. (2001). Statins selectively inhibit leukocyte function antigen‐1 by binding to a novel regulatory integrin site. Nature Medicine, 7, 687–692. 10.1038/89058 [DOI] [PubMed] [Google Scholar]
- Wilson, E. H. , Weninger, W. , & Hunter, C. A. (2010). Trafficking of immune cells in the central nervous system. The Journal of Clinical Investigation, 120, 1368–1379. 10.1172/JCI41911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyant, T. , Yang, L. , & Fedyk, E. (2013). In vitro assessment of the effects of vedolizumab binding on peripheral blood lymphocytes. MAbs, 5, 842–850. 10.4161/mabs.26392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, Y. , Schurpf, T. , & Springer, T. A. (2013). How natalizumab binds and antagonizes α4 integrins. The Journal of Biological Chemistry, 288, 32314–32325. 10.1074/jbc.M113.501668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, Y. , Zhu, J. , Mi, L. Z. , Walz, T. , Sun, H. , Chen, J. , & Springer, T. A. (2012). Structural specializations of α4β7, an integrin that mediates rolling adhesion. The Journal of Cell Biology, 196, 131–146. 10.1083/jcb.201110023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yudushkin, I. A. , & Vale, R. D. (2010). Imaging T‐cell receptor activation reveals accumulation of tyrosine‐phosphorylated CD3ζ in the endosomal compartment. Proceedings of the National Academy of Sciences of the United States of America, 107, 22128–22133. 10.1073/pnas.1016388108 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: Properties of small molecules and antibodies used in the present study.
Figure S1: Effect of efalizumab on integrin αLβ2 surface expression. Surface expression of αLβ 2 on CD2+ T cells after 24 hours and 48 hours of treatment with anti‐ αLβ 2 efalizumab (10 μg mL−1), and respective isotype control (hIgG1). Each bar represents a single determination using blood from one donor.
FIGURE S2: Effect of dynasore on mAb R7.1‐induced αLβ2 and α4 downmodulation, natalizumab induced α4 downmodulation and mAb OKT3‐induced downmodulation of CD3. Surface expression of (A) αLβ2 and (B) α4 integrin on CD2+ T cells after 24 hr of treatment with anti‐αL R7.1 (10 μg mL1), anti‐ α4 natalizumab (10 μg mL−1), and respective isotype controls (mIgG1, hIgG4). Treatments were performed in the presence of dynasore (40 μM, grey bars) or DMSO (white bars). Each bar represents the mean value ± SEM of 6 independent experiments using blood samples from different donors. Statistical significance was determined by using one‐way ANOVA, ***P < 0.001, ****P < 0.0001 vs control, and two‐way ANOVA, $$P < 0.01 vs incubation without dynasore. (C) Surface expression of CD3 on CD2+ T cells after 24 hours of treatment with anti‐CD3 OKT3 (1 ng mL−1, 10 ng mL−1, 100 ng mL1) in the presence of dynasore (40 μM, grey bars) or DMSO (white bars). Each bar represents a single determination, using blood from one donor.