

Abstract
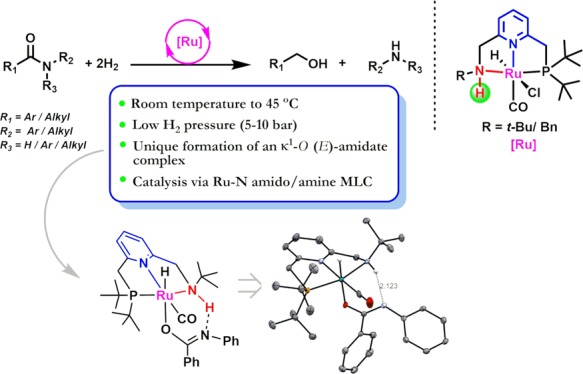
We report a room-temperature protocol for the hydrogenation of various amides to produce amines and alcohols. Compared with most previous reports for this transformation, which use high temperatures (typically, 100–200 °C) and H2 pressures (10–100 bar), this system proceeds under extremely mild conditions (RT, 5–10 bar of H2). The hydrogenation is catalyzed by well-defined ruthenium-PNNH pincer complexes (0.5 mol %) with potential dual modes of metal–ligand cooperation. An unusual Ru-amidate complex was formed and crystallographically characterized. Mechanistic investigations indicate that the room-temperature hydrogenation proceeds predominantly via the Ru–N amido/amine metal–ligand cooperation.
Keywords: amide, hydrogenation, metal−ligand cooperation, ruthenium-pincer, room-temperature, ruthenium amidate complex
Reduction of the amide group has useful applications in diverse areas of chemical transformations including synthesis of amines, alcohols, peptide chemistry, total synthesis, CO2 recycling, and others.1,2 The traditional methods to reduce amides are noncatalytic and use stoichiometric amounts of hydride reducing agents, thus producing a stoichiometric amount of waste.3 In this context, development of efficient catalytic hydrogenation systems for their reduction is desirable.4 While hydrogenations of amides have been reported, they are more challenging than that of most other carboxylic derivatives because of the low electrophilic character of the amide carbonyl group.5
Amide hydrogenation can proceed either via C–N bond cleavage or by C–O bond cleavage (Scheme 1).5a,5b We are particularly interested in the C–N bond cleavage pathway, which is challenging because of the water losing tendency of the hemiaminal intermediate to form imine. The first example of amide hydrogenation with C–N bond cleavage appeared in 20106a followed by reports of several other research groups.6 Hydrogenation and dehydrogenative formation of the amide bond has also been instrumental in the recent development of promising liquid organic hydrogen carrier (LOHC) systems based on alcohols and amines.7 However, the reported hydrogenation reactions require harsh reaction conditions (typically 100–150 °C and/or 10–100 bar of H2 pressure). In this regard, amide hydrogenation protocols to form amines and alcohols under mild conditions are clearly required. Development of such protocols would also facilitate the development of low-temperature amide-based LOHC systems as well as low-temperature CO2 recycling to methanol systems.8
Scheme 1. Two Possible Pathways of Amide Hydrogenation.

Saito and co-workers have reported an example of Ru-catalyzed dimethylformamide hydrogenation at 60 °C under high H2 pressure of 80 bar,6k but higher temperature (120 °C) was required at lower H2 pressures (20 bar) to obtain similar yields (60%). Langer and co-workers reported examples of amide hydrogenation at 70 °C with an iron complex using high catalyst loading (10 mol %) at 50 bar H2;6g however, this interesting system exhibited limited scope and required 100 °C for completion at lower catalyst loadings (2 mol %). Recently, Beller and co-workers reported a Mo-pincer catalyzed amide hydrogenation at 80 °C, but this system employed high H2 pressure (50 bar) and the substrate scope was synthetically limited to N-methylphenylformamides.6l Bergens reported asymmetric hydrogenolysis of some specific functionalized α-phenoxy amides at RT and low pressure, but the system requires high catalyst loading (10 mol %) and more than stoichiometric amounts of strong base (120–250 mol % of t-BuOK/i-PrONa) for effective catalysis.6p
As a part of our ongoing research endeavor, we seek to develop metal complexes to facilitate (de)hydrogenation reactions under mild conditions. To this extent, we have reported a new class of pincer complexes (based on PNNH ligands) where dual modes of metal ligand cooperation (MLC) are possible (via ligand dearomatization and metal amido bond formation) (Figure 1).7e,9 Here, we report a system for room-temperature amide hydrogenation to amines and alcohols catalyzed by the Ru-PNNH complexes at low catalyst loading (0.5 mol %) under low H2 pressures (5–10 bar).
Figure 1.

Amide hydrogenation reported here as compared with previous reports.
We started our investigation with benzanilide as the model substrate (Table 1). When benzanilide in THF was subjected to hydrogenation (10 bar of H2) at 45 °C with complex Ru-PNNtBuH (Ru-1; 0.5 mol %) as the precatalyst in the presence of catalytic t-BuOK (2 mol %), 67% of amide conversion was observed by GC after 20 h (entry 1). We observed the formation of benzyl alcohol and aniline as the reaction products by GC-MS, revealing the C–N bond cleavage to be the favorable pathway under the reaction conditions. Changing the N-substitution of the catalyst from t-Bu to benzyl group (Ru-2), the catalytic activity increased even further, and complete conversion of the amide to the amine and alcohol was observed after 20 h (entry 2). However, changing the P substitution of the PNNH ligand from t-Bu to Ph (Ru-3) resulted in a significant decrease in the catalytic activity (entry 3). The traditional Ru-PNNEt catalyst Ru-4, lacking a N–H bond, was unable to efficiently catalyze the hydrogenation at 45 °C (entry 4). With Ru-2, the hydrogenation can proceed to completion within 20 h even at the lower temperature of 35 °C (entry 5). Furthermore, full conversion of benzanilide to benzyl alcohol and aniline can be obtained even at RT (19–24 °C), although a longer reaction time (68 h) is required (entries 6–7). Toluene is not as good a solvent as THF for this hydrogenation, possibly because of the limited solubility of benzanilide in toluene (entry 8).
Table 1. Optimization of Catalytic Hydrogenation of Benzanilidea.

| entry | cat. | T (°C) | t (h) | solvent | conv. (%)b | 2a (%)b | 3a (%)b |
|---|---|---|---|---|---|---|---|
| 1 | Ru-1 | 45 | 20 | THF | 67 | 63 | 62 |
| 2 | Ru-2 | 45 | 20 | THF | 99 | 94 | 91 |
| 3 | Ru-3 | 45 | 20 | THF | 20 | 14 | 12 |
| 4 | Ru-4 | 45 | 20 | THF | 3 | 3 | 2 |
| 5 | Ru-2 | 35 | 20 | THF | 99 | 95 | 91 |
| 6 | Ru-2 | RTc | 20 | THF | 52 | 49 | 47 |
| 7 | Ru-2 | RT | 68 | THF | 97 | 91 | 93 |
| 8 | Ru-2 | RT | 68 | toluene | 79 | 73 | 75 |
Reaction conditions: benzanilide (0.5 mmol), cat. (0.5 mol %), t-BuOK (2 mol %), THF (3 mL), H2 (10 bar at RT), T (as specified, bath temperature), t (as specified).
Conversions and yields are determined by GC with mesitylene as an internal standard.
RT = room temperature (19–24 °C)
Subsequently, we explored the substrate scope of this catalytic hydrogenation of amides under mild conditions (Table 2). Several substituted benzanilides were quantitatively converted to the corresponding alcohol and aniline at RT (under 10 bar H2) with catalyst Ru-2 (entries 1–6). Formamides, such as N-(4-chlorophenyl)formamide, N-(4-methylphenyl)formamide can also be hydrogenated at room temperature to the corresponding amine and methanol (entry 7–8). Other amides, such as N-acetylmorpholine; N,N-diphenylbenzamide; N-acetylaniline; N-hexylbenzamide; N-phenyloctanamide; N-benzylformamide; and N-heptyloctanamide were hydrogenated at either RT or at a slightly elevated temperature of 35–45 °C (entries 9–15), which is still significantly lower than the hydrogenation temperature reported previously with other catalysts.
Table 2. Hydrogenation of Various Amides at or Near RT§.


Reaction conditions: amide (0.5 mmol), Ru-2 (0.5 mol %), t-BuOK (2 mol %), THF (3 mL), H2 (10 bar at RT), T (as specified), t (68 h).
Conversions and yields were determined by GC with mesitylene as an internal standard. Products were characterized by GC-MS.
Yield determined by 1H NMR.
Yields of methanol and ethanol are lower than that of the corresponding amine due to volatility.
With catalyst Ru-1 (0.5 mol %), H2 (5 bar), t (20 h).
Next, we carried out mechanistic investigations to further understand the high catalytic activity of the Ru-PNNH complexes compared to the traditional PNNEt complex Ru-4. Upon addition of 2 equiv of t-BuOK to a solution of complex Ru-1 (dissolved in THF), an intensely purple-colored solution of the anionic ruthenium enamido complex Ru-1A was formed.9d Upon addition of two equiv of benzanilide to this solution, the intense purple color disappeared to produce a light-green solution, and the 31P NMR spectrum exhibited only a single peak at 108.5 ppm (Figure S13). This is due to the formation of the ruthenium amidate complex Ru-1B, which was characterized by 1H, 31P, and 13C NMR spectroscopy (Scheme 2a, Figures S8–S11) and a single-crystal X-ray diffraction study (see SI). As exhibited by the crystal structure, the amidate ligand displays κ1-O binding to the Ru metal center, which is uncommon for late transition metal complexes and, to the best of our knowledge, has not been observed previously in ruthenium amidate complexes.10 The κ1-O geometry is also characterized by an elongated C–O bond length (1.279 Å) and a shortened C–N bond length (1.307 Å) of the amidate ligand as compared to free benzanilide (1.227 and 1.363 Å, respectively, for C–O and C–N bond).11 Furthermore, the coordinated (E) isomer of κ1-ORu-1B was observed in the structure, which is rare in any late transition metal κ1-O amidate complex due to steric reasons.10 Interestingly, the N-H proton of the PNNH ligand and the N atom of the amidate ligand are aligned in close spatial proximity (2.123 Å) with the possibility of a hydrogen bonding interaction, thus providing additional stability to Ru-1B. Indeed, in the 1H NMR spectrum of Ru-1B, the chemical shift of the N-H proton is strongly downfield shifted (∼10.5 ppm), indicating hydrogen bonding in solution (Figure S8). The hydrogen bonding is crucial to the formation of the κ1-O (E)-amidate complex as the analogous amidate complex of Ru-4 lacking an N–H bond, was not observed under similar conditions (Scheme 2b). Notably, a somewhat similar amidate complex has been observed by Bernskoetter, Hazari, and co-workers in a Fe-PNP system, although with κ1-N coordination.6i
Scheme 2. Mechanistic Reactions.
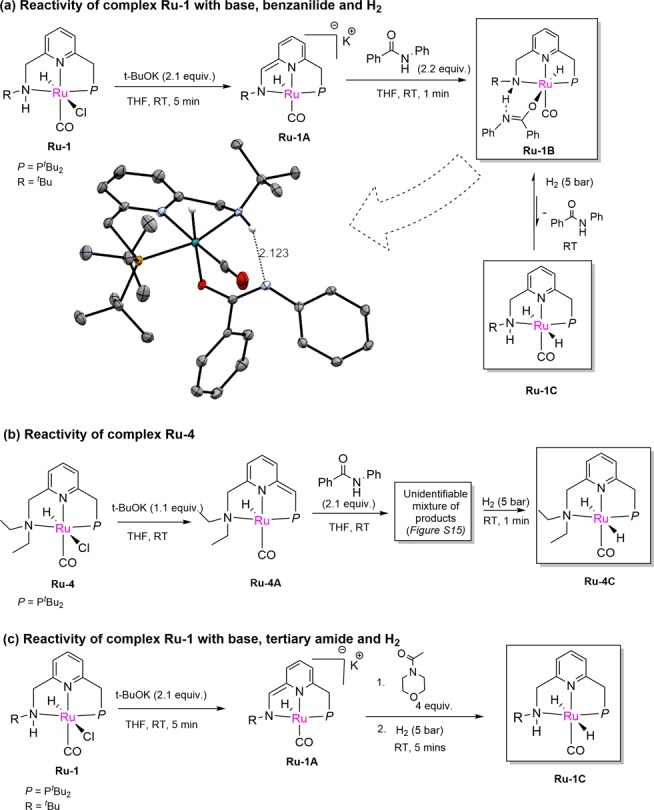
Formation of the amidate complex, while interesting, is off-cycle and may also somewhat decrease the catalytic efficiency in the hydrogenation of the secondary amides. Ru-1B undergoes rapid dissociation and reassociation of the amide in solution, as verified by an amide exchange experiment (Figure S20). Under 5 bar of H2 pressure, complex Ru-1B is in equilibrium with the dihydride complex Ru-1C (Scheme 2a, Scheme S1). This equilibrium favors the amidate complex Ru-1B (Figure S13, panel 4). The equilibrium can also be indirectly observed in the presence of D2, in which case rapid suppression of the N-H proton and one of the P–CH2 protons of Ru-1B was observed due to H/D scrambling (Figure S18–S19). To probe the resting state of the catalyst during the reaction, we conducted a control catalytic hydrogenation experiment in an NMR tube with Ru-1B as catalyst using 5 equiv of benzanilide substrate under 5 bar H2. Monitoring the reaction progress at 45 °C, only the amidate complex Ru-1B was observed in the solution while the dihydride complex was not observed (Figure S14). Thus, complex Ru-1B acts as the catalytic resting state during the reaction.
Returning to the reactivity of the enamido complex Ru-1A, upon addition of a substrate lacking a N–H proton, such as N-acetylmorpholine, the solution of complex Ru-1A remained purple even after the addition of 4 equiv of amide. No formation of any new complex was observed by NMR spectroscopy. After the reaction was pressurized with 5 bar of H2, immediate formation of the dihydride complex was observed at RT (Scheme 2c; Figure S16). The dihydride peaks remained the major peak in both 31P and 1H NMR spectra until completion of the hydrogenation, indicating that the reaction of the amide with the dihydride complex is the rate-determining step in the catalytic cycle (vide infra, Scheme 3).
Scheme 3. Plausible Mechanistic Cycle.

On the basis of these insights and prior investigations reported, we propose a catalytic cycle as depicted in Scheme 3.6a,6k,9a,12 For amides bearing an N-H proton, the catalytic resting state is the amidate complex Ru-XB. In the presence of H2, Ru-XB forms the dihydride Ru-XC, which through hydride attack on the amide carbonyl group and proton abstraction of the N-H group of the PNNH ligand forms the hemiaminal, along with the intermediate complex Ru-XD. This step can also occur via proton abstraction from the CH2 group of the P-side arm instead of the N–H group (to generate Ru-XD′). However, this pathway does not significantly contribute to the overall hydrogenation at lower temperatures (RT to 45 °C) (compare the hydrogenation activity of Ru-1/Ru-2 with that of Ru-4). It is likely that the acidic nature of the N-H proton of PNNH ligand assists in lowering the energy requirement of this rate-determining step, thus allowing the hydrogenation to proceed even at RT via the amido pathway. This can also explain the higher catalytic activity of Ru-2 compared with Ru-1, where the Bn substitution of the N donor atom makes the ligand N-H of Ru-2 less basic as compared with Ru-1 with t-Bu group in the N atom. Alternatively, the lower steric bulk of the benzyl group can also contribute to the observed increased hydrogenation rate of Ru-2 compared to Ru-1. Subsequently, the hemiaminal quickly binds to the ruthenium center to form the hemiaminoxy complex Ru-XE, which, through a proton abstraction from the ligand, releases an amine molecule with concomitant formation of the aldehyde complex Ru-XF (or Ru-XF′). Dissociation of the weakly coordinating aldehyde from Ru center, followed by the H2 splitting across the Ru–N bond generates the dihydride complex Ru-XC. Outer-sphere hydride transfer from Ru-XC to the aldehyde then forms the alkoxy complex Ru-XG, which releases an alcohol molecule, generating the amido complex Ru-XD or the dearomatized complex Ru-XD′. Ru-XD (or Ru-XD′) then binds another amide molecule to regenerate the amidate complex Ru-XB. In case of a tertiary amide, the hydrogenation proceeds through a similar pathway but without the involvement of Ru-amidate complex.
In conclusion, selective hydrogenation of amides to alcohols and amines is reported at RT under mild H2 pressure. The ruthenium-PNNH complexes display excellent catalytic activity in this hydrogenation. The high activity of Ru-PNNH complexes is due to the MLC pathway via Ru–N amido/amine pathway. Using Ru-2 as the catalyst, a series of amides were hydrogenated at, or near, RT under 5–10 bar of H2 pressure. We believe that this protocol can facilitate the development of CO2 recycling (via hydrogenation) as well as LOHC systems that may operate at considerably lower temperatures (<50 °C) compared with the existing ones. Both avenues are currently being explored in our group.
Acknowledgments
This research was supported by the European Research Council (ERC AdG 692775). D.M. holds the Israel Matz Professorial Chair of Organic Chemistry. S.K. acknowledges the Sustainability and Energy Research Initiative (SAERI) of the Weizmann Institute of Science for a research fellowship. M.R. acknowledges the Zuckerman STEM Leadership Program for a research fellowship.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscatal.0c01406.
Author Present Address
# A.K.: School of Chemistry, University of St. Andrews, North Haugh, St. Andrews, KY169ST, Scotland, U.K.
The authors declare no competing financial interest.
Supplementary Material
References
- Hudlicky M.Reductions in Organic Chemistry; Ellis Horwood Ltd.: Chichester, U.K., 1984; pp 164–172. [Google Scholar]
- Andersson P. G.; Munslow I. J.. Modern Reduction Methods; Wiley: New York, 2008; p 423. [Google Scholar]
- Seyden-Penne J.Reductions by the Alumino- and Borohydrides in Organic Synthesis, 2nd ed.; Wiley: New York, 1997; pp 1–13. [Google Scholar]
- a Gunanathan C.; Milstein D. Bond Activation and Catalysis by Ruthenium Pincer Complexes. Chem. Rev. 2014, 114, 12024–12087. 10.1021/cr5002782. [DOI] [PubMed] [Google Scholar]; b Filonenko G. A.; van Putten R.; Hensen E. J. M.; Pidko E. A. Catalytic (de)hydrogenation promoted by non-precious metals – Co, Fe and Mn: recent advances in an emerging field. Chem. Soc. Rev. 2018, 47, 1459–1483. 10.1039/C7CS00334J. [DOI] [PubMed] [Google Scholar]
- a Smith A. M.; Whyman R. Review of Methods for the Catalytic Hydrogenation of Carboxamides. Chem. Rev. 2014, 114, 5477–5510. 10.1021/cr400609m. [DOI] [PubMed] [Google Scholar]; b Pritchard J.; Filonenko G. A.; van Putten R.; Hensen E. J. M.; Pidko E. A. Heterogeneous and homogeneous catalysis for the hydrogenation of carboxylic acid derivatives: history, advances and future directions. Chem. Soc. Rev. 2015, 44, 3808–3833. 10.1039/C5CS00038F. [DOI] [PubMed] [Google Scholar]; c Chardon A.; Morisset E.; Rouden J.; Blanchet J. Recent Advances in Amide Reductions. Synthesis 2018, 50, 984–997. 10.1055/s-0036-1589144. [DOI] [Google Scholar]; d Zhou Y.; Khan R.; Fan B.; Xu L. Ruthenium-Catalyzed Selective Reduction of Carboxylic Esters and Carboxamides. Synthesis 2019, 51, 2491–2505. 10.1055/s-0037-1611524. [DOI] [Google Scholar]
- a Balaraman E.; Gnanaprakasam B.; Shimon L. J. W.; Milstein D. Direct Hydrogenation of Amides to Alcohols and Amines under Mild Conditions. J. Am. Chem. Soc. 2010, 132, 16756–16758. 10.1021/ja1080019. [DOI] [PubMed] [Google Scholar]; b John J. M.; Bergens S. H. A Highly Active Catalyst for the Hydrogenation of Amides to Alcohols and Amines. Angew. Chem., Int. Ed. 2011, 50, 10377–10380. 10.1002/anie.201103137. [DOI] [PubMed] [Google Scholar]; c Ito M.; Ootsuka T.; Watari R.; Shiibashi A.; Himizu A.; Ikariya T. Catalytic Hydrogenation of Carboxamides and Esters by Well-Defined Cp*Ru Complexes Bearing a Protic Amine Ligand. J. Am. Chem. Soc. 2011, 133, 4240–4242. 10.1021/ja1117254. [DOI] [PubMed] [Google Scholar]; d Miura T.; Held I. E.; Oishi S.; Naruto M.; Saito S. Catalytic hydrogenation of unactivated amides enabled by hydrogenation of catalyst precursor. Tetrahedron Lett. 2013, 54, 2674–2678. 10.1016/j.tetlet.2013.03.047. [DOI] [Google Scholar]; e Cabrero-Antonino J. R.; Alberico E.; Drexler H.-J.; Baumann W.; Junge K.; Junge H.; Beller M. Efficient Base-Free Hydrogenation of Amides to Alcohols and Amines Catalyzed by Well-Defined Pincer Imidazolyl–Ruthenium Complexes. ACS Catal. 2016, 6, 47–54. 10.1021/acscatal.5b01955. [DOI] [Google Scholar]; f Garg J. A.; Chakraborty S.; Ben-David Y.; Milstein D. Unprecedented iron-catalyzed selective hydrogenation of activated amides to amines and alcohols. Chem. Commun. 2016, 52, 5285–5288. 10.1039/C6CC01505K. [DOI] [PubMed] [Google Scholar]; g Schneck F.; Assmann M.; Balmer M.; Harms K.; Langer R. Selective Hydrogenation of Amides to Amines and Alcohols Catalyzed by Improved Iron Pincer Complexes. Organometallics 2016, 35, 1931–1943. 10.1021/acs.organomet.6b00251. [DOI] [Google Scholar]; h Shi L.; Tan X.; Long J.; Xiong X.; Yang S.; Xue P.; Lv H.; Zhang X. Direct Catalytic Hydrogenation of Simple Amides: A Highly Efficient Approach from Amides to Amines and Alcohols. Chem. - Eur. J. 2017, 23, 546–548. 10.1002/chem.201604904. [DOI] [PubMed] [Google Scholar]; i Jayarathne U.; Zhang Y.; Hazari N.; Bernskoetter W. H. Selective Iron-Catalyzed Deaminative Hydrogenation of Amides. Organometallics 2017, 36, 409–416. 10.1021/acs.organomet.6b00816. [DOI] [Google Scholar]; j Papa V.; Cabrero-Antonino J. R.; Alberico E.; Spanneberg A.; Junge K.; Junge H.; Beller M. Efficient and selective hydrogenation of amides to alcohols and amines using a well-defined manganese–PNN pincer complex. Chem. Sci. 2017, 8, 3576–3585. 10.1039/C7SC00138J. [DOI] [PMC free article] [PubMed] [Google Scholar]; k Miura T.; Naruto M.; Toda K.; Shimomura T.; Saito S. Multifaceted catalytic hydrogenation of amides via diverse activation of a sterically confined bipyridine–ruthenium framework. Sci. Rep. 2017, 7, 1586. 10.1038/s41598-017-01645-z. [DOI] [PMC free article] [PubMed] [Google Scholar]; l Leischner T.; Artús Suarez L.; Spannenberg A.; Junge K.; Nova A.; Beller M. Highly selective hydrogenation of amides catalysed by a molybdenum pincer complex: scope and mechanism. Chem. Sci. 2019, 10, 10566–10576. 10.1039/C9SC03453F. [DOI] [PMC free article] [PubMed] [Google Scholar]; m Kar S.; Goeppert A.; Kothandaraman J.; Prakash G. K. S. Manganese-Catalyzed Sequential Hydrogenation of CO2 to Methanol via Formamide. ACS Catal. 2017, 7, 6347–6351. 10.1021/acscatal.7b02066. [DOI] [Google Scholar]; n Kar S.; Sen R.; Kothandaraman J.; Goeppert A.; Chowdhury R.; Munoz S. B.; Haiges R.; Prakash G. K. S. Mechanistic Insights into Ruthenium-Pincer-Catalyzed Amine-Assisted Homogeneous Hydrogenation of CO2 to Methanol. J. Am. Chem. Soc. 2019, 141, 3160–3170. 10.1021/jacs.8b12763. [DOI] [PubMed] [Google Scholar]; o Kar S.; Goeppert A.; Prakash G. K. S. Catalytic Homogeneous Hydrogenation of CO to Methanol via Formamide. J. Am. Chem. Soc. 2019, 141, 12518–12521. 10.1021/jacs.9b06586. [DOI] [PubMed] [Google Scholar]; p Rasu L.; John J. M.; Stephenson E.; Endean R.; Kalapugama S.; Clément R.; Bergens S. H. Highly Enantioselective Hydrogenation of Amides via Dynamic Kinetic Resolution Under Low Pressure and Room Temperature. J. Am. Chem. Soc. 2017, 139, 3065–3071. 10.1021/jacs.6b12254. [DOI] [PubMed] [Google Scholar]
- a Hu P.; Fogler E.; Diskin-Posner Y.; Iron M. A.; Milstein D. A novel liquid organic hydrogen carrier system based on catalytic peptide formation and hydrogenation. Nat. Commun. 2015, 6, 6859. 10.1038/ncomms7859. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Xie Y.; Hu P.; Ben-David Y.; Milstein D. A Reversible Liquid Organic Hydrogen Carrier System Based on Methanol-Ethylenediamine and Ethylene Urea. Angew. Chem., Int. Ed. 2019, 58, 5105–5109. 10.1002/anie.201901695. [DOI] [PubMed] [Google Scholar]; c Hu P.; Ben-David Y.; Milstein D. Rechargeable Hydrogen Storage System Based on the Dehydrogenative Coupling of Ethylenediamine with Ethanol. Angew. Chem., Int. Ed. 2016, 55, 1061–1064. 10.1002/anie.201505704. [DOI] [PubMed] [Google Scholar]; d Kothandaraman J.; Kar S.; Sen R.; Goeppert A.; Olah G. A.; Prakash G. K. S. Efficient Reversible Hydrogen Carrier System Based on Amine Reforming of Methanol. J. Am. Chem. Soc. 2017, 139, 2549–2552. 10.1021/jacs.6b11637. [DOI] [PubMed] [Google Scholar]; e Kumar A.; Janes T.; Espinosa-Jalapa N. A.; Milstein D. Selective Hydrogenation of Cyclic Imides to Diols and Amines and Its Application in the Development of a Liquid Organic Hydrogen Carrier. J. Am. Chem. Soc. 2018, 140, 7453–7457. 10.1021/jacs.8b04581. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Suárez A., Hydrogenation of carbonyl compounds of relevance to hydrogen storage in alcohols. In Phys. Sci. Rev., 2018; Vol. 3;. 10.1515/psr-2017-0028 [DOI] [Google Scholar]; g Shao Z.; Li Y.; Liu C.; Ai W.; Luo S.-P.; Liu Q. Reversible interconversion between methanol-diamine and diamide for hydrogen storage based on manganese catalyzed (de)hydrogenation. Nat. Commun. 2020, 11, 591. 10.1038/s41467-020-14380-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Rezayee N. M.; Huff C. A.; Sanford M. S. Tandem Amine and Ruthenium-Catalyzed Hydrogenation of CO2 to Methanol. J. Am. Chem. Soc. 2015, 137, 1028–1031. 10.1021/ja511329m. [DOI] [PubMed] [Google Scholar]; b Kothandaraman J.; Goeppert A.; Czaun M.; Olah G. A.; Prakash G. K. S. Conversion of CO2 from Air into Methanol Using a Polyamine and a Homogeneous Ruthenium Catalyst. J. Am. Chem. Soc. 2016, 138, 778–781. 10.1021/jacs.5b12354. [DOI] [PubMed] [Google Scholar]; c Kar S.; Goeppert A.; Prakash G. K. S. Integrated CO2 Capture and Conversion to Formate and Methanol: Connecting Two Threads. Acc. Chem. Res. 2019, 52, 2892–2903. 10.1021/acs.accounts.9b00324. [DOI] [PubMed] [Google Scholar]
- a Kumar A.; Janes T.; Espinosa-Jalapa N. A.; Milstein D. Manganese Catalyzed Hydrogenation of Organic Carbonates to Methanol and Alcohols. Angew. Chem., Int. Ed. 2018, 57, 12076–12080. 10.1002/anie.201806289. [DOI] [PubMed] [Google Scholar]; b Kumar A.; Espinosa-Jalapa N. A.; Leitus G.; Diskin-Posner Y.; Avram L.; Milstein D. Direct Synthesis of Amides by Dehydrogenative Coupling of Amines with either Alcohols or Esters: Manganese Pincer Complex as Catalyst. Angew. Chem., Int. Ed. 2017, 56, 14992–14996. 10.1002/anie.201709180. [DOI] [PubMed] [Google Scholar]; c Kumar A.; Daw P.; Espinosa-Jalapa N. A.; Leitus G.; Shimon L. J. W.; Ben-David Y.; Milstein D. CO2 activation by manganese pincer complexes through different modes of metal–ligand cooperation. Dalton Transactions 2019, 48, 14580–14584. 10.1039/C9DT03088C. [DOI] [PubMed] [Google Scholar]; d Fogler E.; Garg J. A.; Hu P.; Leitus G.; Shimon L. J. W.; Milstein D. System with Potential Dual Modes of Metal–Ligand Cooperation: Highly Catalytically Active Pyridine-Based PNNH–Ru Pincer Complexes. Chem. - Eur. J. 2014, 20, 15727–15731. 10.1002/chem.201405295. [DOI] [PubMed] [Google Scholar]
- Drover M. W.; Love J. A.; Schafer L. L. 1,3-N,O-Complexes of late transition metals. Ligands with flexible bonding modes and reaction profiles. Chem. Soc. Rev. 2017, 46, 2913–2940. 10.1039/C6CS00715E. [DOI] [PubMed] [Google Scholar]
- Bowes K. F.; Glidewell C.; Low J. N.; Skakle J. M. S.; Wardell J. L. A triclinic polymorph of benzanilide: disordered molecules form hydrogen-bonded chains. Acta Crystallogr., Sect. C: Cryst. Struct. Commun. 2003, 59, o1–o3. 10.1107/S0108270102019996. [DOI] [PubMed] [Google Scholar]
- a Das U. K.; Kumar A.; Ben-David Y.; Iron M. A.; Milstein D. Manganese Catalyzed Hydrogenation of Carbamates and Urea Derivatives. J. Am. Chem. Soc. 2019, 141, 12962–12966. 10.1021/jacs.9b05591. [DOI] [PubMed] [Google Scholar]; b Hou C.; Jiang J.; Li Y.; Zhao C.; Ke Z. When Bifunctional Catalyst Encounters Dual MLC Modes: DFT Study on the Mechanistic Preference in Ru-PNNH Pincer Complex Catalyzed Dehydrogenative Coupling Reaction. ACS Catal. 2017, 7, 786–795. 10.1021/acscatal.6b02505. [DOI] [Google Scholar]; c Artús Suàrez L.; Culakova Z.; Balcells D.; Bernskoetter W. H.; Eisenstein O.; Goldberg K. I.; Hazari N.; Tilset M.; Nova A. The Key Role of the Hemiaminal Intermediate in the Iron-Catalyzed Deaminative Hydrogenation of Amides. ACS Catal. 2018, 8, 8751–8762. 10.1021/acscatal.8b02184. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.