

Abstract

Tankyrases (TNKSs) have recently gained great consideration as potential targets in Wnt/β-catenin pathway-dependent solid tumors. Previously, we reported the 2-mercaptoquinazolin-4-one MC2050 as a micromolar PARP1 inhibitor. Here we show how the resolution of the X-ray structure of PARP1 in complex with MC2050, combined with the computational investigation of the structural differences between TNKSs and PARP1/2 active sites, provided the rationale for a structure-based drug design campaign that with a limited synthetic effort led to the discovery of the bis-quinazolinone 5 as a picomolar and selective TNKS2 inhibitor, endowed with antiproliferative effects in a colorectal cancer cell line (DLD-1) where the Wnt pathway is constitutively activated.
Keywords: Tankyrases, adenosine subpocket ligands, selective inhibition, quinazolinones, antitumor
ADP-ribosylation consists in the catalytic transfer of the ADP-ribose moiety from the nicotinamide adenine dinucleotide (NAD+) cofactor to acceptor proteins and is mediated by the 17-membered family of ADP-ribosyltransferases diphtheria toxin-like (ARTDs), formerly known as poly(ADP-ribose) polymerases (PARPs).1,2 Although all family members catalyze the initial attachment of ADP-ribose monomers on different amino acid side chains, only a subset of them can further elongate and/or branch the ADP-ribose tail by polymerization (PARP1/2 and PARP5a/b).2 Among these, tankyrase 1 (TNKS1, PARP-5a) and tankyrase 2 (TNKS2, PARP5b) are peculiar since, in addition to the conserved C-terminal catalytic polyADP-ribosylating domain, they encompass two distinct protein–protein interaction domains: a sterile α-motif and a five ankyrin repeat cluster, mediating self-multimerization and target protein recognition, respectively.3 Tankyrases (TNKSs) are ubiquitously expressed and have a tissue-specific cytoplasmic sublocalization according to their cellular functions. Although TNKS1 was initially identified as a negative regulator of telomeric repeat binding factor-1, a component of telomere-end architecture, the most investigated function of TNKSs is the regulatory effect on the Wnt/β-catenin signaling pathway.4 Indeed, ADP-ribosylation triggers proteasomal degradation of axin proteins, the process-limiting and core component of β-catenin destruction complex, allowing β-catenin to evade degradation and to prime the downstream transcription of Wnt pathway genes.3,4 In addition to this cancer related mechanism, TNKSs play roles in membrane translocation of the GLUT4 transporter, in the assembly of microtubules at spindle poles, and viral replication.3,5 The chemical modulation of ADP-ribosylation started with the development of inhibitors for PARP1/2, two enzymes engaged in DNA break repair.6 Recently, some of these compounds moved forward to clinical applications as chemosensitizers in cytotoxic therapeutic blueprints or as single agents in BRCA-defected tumors.7−9 The sustained interest in PARPs as anticancer targets together with the therapeutic potential of the above-mentioned TNKS functions increased the appeal of TNKS1/2 in medicinal chemistry.3,10 Indeed, TNKS inhibitors especially as Wnt/β-catenin pathway modulators have solid perspectives in oncology, particularly in colorectal cancer (CRC) and nonsmall cell lung cancer (NSCLC).3−5,10,11 In addition, tackling TNKS-mediated ADP-ribosylation may provide a valuable alternative in viral infections and fibrotic diseases treatments.3,5,10,11 Until now, most TNKS inhibition approaches have focused on occupying the NAD+ binding site, formally divided into nicotinamide (NAM) and adenosine (AD) subsites, which is outlined by the donor D-loop, the glycine-rich G-loop and the Phe-containing F-loop.7,12 Leveraging interactions in the NAM subpocket represented a validated strategy to inhibit ADP-ribosylation, and different chemotypes were found active in this sense.6,7,11 Unfortunately, the high grade of sequence homology and the spatial organization in this region renders NAM-mimicking compounds promiscuous PARP inhibitors rather than selective chemical tools.12 Recently, a fruitful approach to elicit selectivity, especially in TNKS inhibition, consisted in targeting the AD subsite or both NAM and AD subsites simultaneously.13,14 This strategy benefited from the isoform-specific chemical environment within the AD pocket. Moreover, spanning the length of the whole NAD+ binding cleft may provide further interaction sites between active site and inhibitors. The D-loop in TNKSs is in fact shorter than the homologue in PARP1/2, contains bulkier hydrophobic amino acids, and offers a reduced space to host ligands in this region.15
Previously, we reported the 2-mercaptoquinazolin-4-one derivative MC2050 as a (sub)micromolar PARP1 inhibitor selective over PARP2, and correlated this activity with the cytoprotective effect in neuroblastoma SH-SY5Y cells and with the modulation of viral gene expression in EBV-infected Burkitt’s lymphoma cells.16,17 Here, our aim has been to develop a series of MC2050 analogues able to potently inhibit TNKSs while sparing PARP1/2 (Figure 1).
Figure 1.
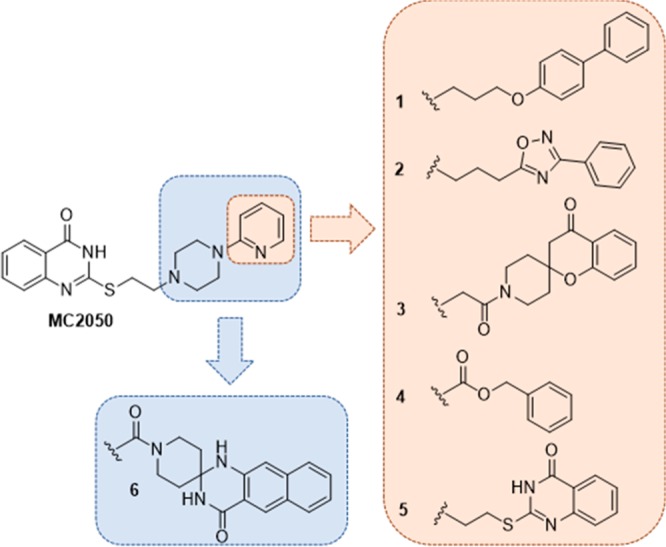
Analogues of MC2050 investigated in the study.
We started by shedding light on the MC2050 binding in the PARP1 catalytic domain by X-ray crystallography. These insights, combined with structural differences between TNKSs and PARP1/2, set the stage for a structure-based drug design campaign. We kept intact the NAM-mimicking 2-mercaptoquinazolin-4-one scaffold and synthesized a set of derivatives protruding out of the NAM-binding site to elicit further and specific interactions with the TNKS AD subpocket. Biological evaluation on a panel of different PARP enzymes led to the identification of a couple of selective (sub)nanomolar TNKS inhibitors endowed with antiproliferative effect in DLD-1 human colon carcinoma cells.
The structure of human PARP1 (hPARP1c) in complex with MC2050 was solved at 2.0 Å resolution and refined to R/Rfree values of 22.6/26.5% (Table S1). A significant anomalous signal was detected during processing, and the anomalous electron density revealed six Ni2+ ions from the crystallization solution, two inside each protein molecule in the asymmetric unit and two between their interfaces (Figure 2A). A nickel dependency of PARP1 has not been described and a potential physiological relevance of the ion sites remains to be clarified. Inspection of the active site revealed well-defined electron density for the inhibitor MC2050 (Figure 2B). The ligand is mainly bound through the quinazolinone group, which mimics the nicotinamide moiety of the NAD+ substrate. N3 and the keto group of the 4-quinazolinone nucleus form H-bonds to Gly863 and Ser904, respectively (Figure 2C, D), while His862 and Tyr907 render the pocket suitable to harbor the phenyl ring of this NAM-mimetic moiety. Besides the H-bond connecting N1 of the pyridine group to Asp770, the piperazine and pyridine moieties lack H-bond interactions, but appear also tightly bound, as indicated by the absence of fragmented or smeared density that would indicate flexibility or second conformations. Rigid binding is achieved by hydrophobic interactions to the side chains of Glu763, Asp766, Asn767, Leu769, Asp770, Asn868, Arg878, and Ile879, which create a binding cleft stabilized through a H-bond network. Thus, MC2050 is dominantly bound via π-stacking and other hydrophobic interactions, as observed for almost all structurally characterized PARP inhibitors.12,14,15
Figure 2.
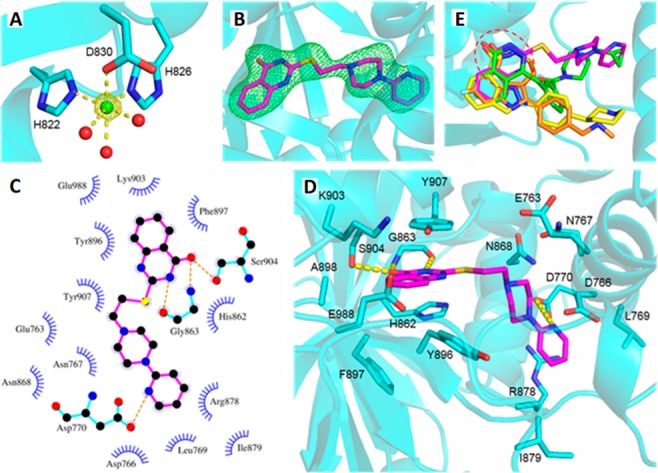
(A) Octahedral binding of Ni2+ (green sphere) in PARP1, with 2.2 ± 0.1 Å coordination distances. Red spheres represent water molecules. The anomalous density is contoured at 5σ. (B) Active site of the hPARP1c/MC2050 complex. The inhibitor (magenta) is overlaid with Fo – Fc omit density contoured at 3σ. (C and D) Binding mode of MC2050 in the active site of hPARP1c. (E) Structural overlay of PARP complexes with the inhibitors olaparib (green, 5DS3), rucaparib (orange, 4RV6), niraparib (yellow, 4R6E), and MC2050 (magenta). The NAM-binding site is indicated by the red dashed circle.
However, a comparison of the binding site interactions of MC2050 and of olaparib, rucaparib, and niraparib visualizes differences for our parent compound (Figure 2E, PDB: 5DS3,184RV6, and 4R6E(19)). All these chemotypes share the same interaction pattern within the PARP1 NAM-binding site, establishing H-bonds to Gly863 and Ser904. Although the cyclopropane of olaparib lies in the AD subsite, like for the pyridine tail of MC2050, the connecting piperazinyl and fluorophenyl portions fill a different active site area (next to Tyr896) than the MC2050 linker (next to Asn767). Deviating even more from the binding pose of MC2050 and extending toward Arg878, the distal [(methylamino)methyl]phenyl of rucaparib and 3-phenylpiperidine of niraparib occupy a different pocket comprising Gln759, Val762, Glu763, and Tyr889.
Based on the experimental data on MC2050 interactions with hPARP1c, we evaluated potential interactors of the binding site for designing selective TNKS inhibitors.
We superposed the crystal structures of the human TNKS catalytic domains (residues 1105–1315 of TNKS1, PDB: 4MSG and residues 956–1161 of TNKS2, PDB: 3U9Y(14)) with the hPARP1c/MC2050 crystal structure (Figure 3). It revealed a high degree of similarity at the 2-mercaptoquinazolin-4-one binding site but also substantial differences in key residues in the AD subsite, which accommodates the MC2050 pyridine moiety. In particular, Asp770 and Arg878 in the PARP1 AD cavity are replaced by Phe and His (Phe1188 and His1201 in TNKS1 and Phe1035 and His1048 in TNKS2) suggesting that (1) removing the pyridin-2-yl ring could induce the loss of interaction with Asp770 and reduce inhibition of PARPs; (2) in order to interact with TNKSs, MC2050 should avoid the steric clash with Phe1188/Phe1035 and adopt a different conformation of the ethylene linker and piperazine ring, pointing toward His1201/His1048; (3) increasing the length of the linker between the piperazine and a proper not-basic (hetero)aromatic ring could allow the aromatic tail of potential MC2050 analogues to establish a favorable stacking interaction with His1201/His1048 in TNKSs, and at the same time could lead to a clash with the bulkier side-chain of Arg878 in PARP1.
Figure 3.
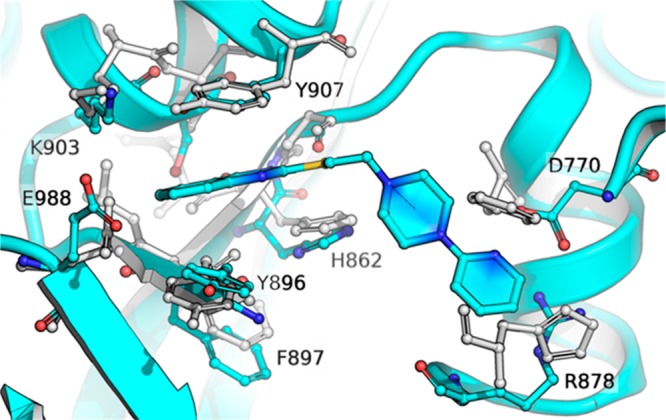
Structural comparison between the crystal structures of the human TNKS2 ARTD catalytic domain (light gray) and MC2050 bound to the active site of PARP1 (cyan). The structural superposition underscores differences in key residues in the AD-binding site, in particular Asp770 and Arg878 in PARP1 (cyan) are replaced by Phe1188/Phe1035 and His1201/His1048 in TNKS1/2 enzymes (residue numbering refers to PDB codes 4MSG(20) and 3U9Y,14 respectively).
Given the interaction details of hPARP1c/MC2050, we decided to leverage the PARP1/TNKSs structural differences from our in-silico comparison to design a small library of analogues of our lead (Figure 1). To explore the molecular space in the AD cavity of the ARTD catalytic domain, we maintained the 2-mercaptoquinazolin-4-one as a NAM-mimetic scaffold since it evokes favorable H-bonds with Gly863 and Ser904 in PARP1, corresponding to Gly1185-Ser1221 and Gly1032-Ser1068 in TNKS1/2, respectively. Additionally, the π-staking between the phenyl ring of the 4-quinazolinone nucleus and Tyr907 and His862 of PARP1 might be preserved in TNKS1/2 (with Tyr1224-His1184 and Tyr1071-His1031). Since the thioethylene-piperazinyl moiety adapts into a molecular gorge covering the space between Glu763/Asn868 and Leu769/Arg878 in the hPARP1c/MC2050 structure, we reasoned that this portion would span this length also in TNKS1/2. Our crystallographic investigation also showed that the MC2050 pyridin-2-yl group plays a relevant role in PARP1 binding by H-bonding the Asp770 side chain. This evidence and our computational modeling studies suggested to replace this aromatic ring with a flexible spacer (i.e., 2–4 methylene units or similar) in order to attain a dual effect for TNKSs selectivity: removing an important structural element for PARP1 interaction and averting a steric clash with Phe from TNKS1/2. Moreover, this linker by projecting from the NAM-binding site, where the 2-mercaptoquinazolin-4-one is harbored, could orientate proper aromatic moieties into the AD pocket. Indeed, using a computational scaffold hopping approach with a library of aromatic rings, we selected a small series of best hits (compounds 1–5 in Figure 1) to be connected through the spacer to the thioethylene-piperazinyl moiety of MC2050 and predicted to exploit the structural differences in the AD pockets of PARP1 and TNKS1/2. With the only exception of compound 6, designed by an alternative strategy relying on the fusion of both pyridine and piperazine moieties into a rigid spiro tetracyclic system, the piperazine ring as connecting moiety between the NAM- and AD-mimetic portions was kept in all compounds because it offers the proper geometry to connect these two portions and at the same time may act as inner solubility group.
The general synthetic route for the preparation of final compounds 1–6 is illustrated in Scheme 1. The commercially available 4-phenylphenol and N′-hydroxybenzimidamide were first converted into the corresponding alkyl bromides (7 and 9) by alkylation with 1,3-dibromopropane and cyclization with 4-bromobutanoyl chloride, respectively, and then treated with commercial 2-(piperazin-1-yl)ethan-1-ol affording the related intermediates 8 and 10. These alcohols then underwent Mitsunobu reaction with commercial 2-mercaptoquinazolin-4-one in the presence of diisopropyl azodicarboxylate and trimethyl phosphine under microwave irradiation to provide final derivatives 1 and 2 (Scheme 1A and 1B). The condensation between the commercially available 2-hydroxyacetophenone and tert-butyloxycarbonylpiperidin-4-one followed by N-Boc deprotection provided the spiro-4-oxocromane intermediate 11, which was then acylated with bromoacetyl chloride to give the intermediate 12 that was finally converted into 3 by alkylation with 2-(piperazin-1-yl)ethan-1-ol followed by reaction of the intermediate 13 with 2-mercaptoquinazolin-4-one under the same Mitsunobu conditions as for 1 and 2 (Scheme 1C). Final compounds 4 and 5 were both prepared starting from the commercially available 2-(piperazin-1-yl)ethanol (Scheme 1D). In the former case the alcohol was first acylated with benzyloxycarbonyl chloride and then converted into the corresponding bromide (15) that was finally alkylated with 2-mercaptoquinazolin-4-one to provide 4. The symmetric compound 5 was instead obtained by Mitsunobu reaction of two equivalents of 2-mercaptoquinazolin-4-one with the diol 16 previously prepared by alkylation of 2-(piperazin-1-yl)ethanol with 2-bromoethanol. The final compound 6 was synthesized by coupling between the spiropiperidine intermediate 18 and the quinazolinone-containing propionic acid derivative 20 in the presence of the 1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide/hydroxybenzotriazole system (Scheme 1E). The intermediate 18 was prepared by conversion of the commercial 3-amino-2-naphthoic acid into the corresponding amide 17 followed by condensation of this intermediate with the piperid-4-one under acidic conditions. The 3-((4-oxo-3,4-dihydroquinazolin-2-yl)thio)propanoic acid 20 was obtained by alkylation of 2-mercaptoquinazolin-4-one with commercially available ethyl 3-bromopropionate followed by hydrolysis of the ethyl ester 19 under basic conditions.
Scheme 1. Synthetic Routes to Compounds 1–6.

Reagents and conditions: (a) 1,3-dibromopropane, K2CO3, dry CH3CN, reflux; (b) 2-(piperazin-1-yl)ethan-1-ol, K2CO3, NaI, dry DMF, 60 °C; (c) 2-mercapto-4(3H)-quinazolinone, DIAD, 1 M trimethylphosphine in toluene, dry DMF, microwave, 40 °C; (d) (i) 4-bromobutanoyl chloride, TEA, dry DCM, rt, (ii) toluene, reflux; (e) tert-butyl 4-oxopiperidine-1-carboxylate, pyrrolidine, dry MeOH, reflux; (f) 4 M HCl in dioxane, dry THF; (g) bromoacetyl chloride, TEA, dry DCM, rt; (h) benzyloxycarbonyl chloride, TEA, dry THF, rt; (i) CBr4, triphenylphosphine, dry THF, rt; (j) 2-mercapto-4(3H)-quinazolinone, K2CO3, NaI, dry DMF, rt; (k) 2-bromoethan-1-ol, K2CO3, dry CH3CN, reflux; (l) NH3 aq., EDCI, HOBt, 4-methylmorpholine, dry THF, rt; (m) piperidin-4-one hydrochloride, concentrated H2SO4, CH3COOH, rt; (n) EDCI, HOBt, TEA, dry DMF, rt; (o) 2 M potassium hydroxide, EtOH, rt.
The capability of 1–6 to inhibit poly(ADP)ribosylating activity was assessed in vitro against PARP1/2 and TNKS1/2, and it was compared with the activity of the parent PARP1 inhibitor MC2050,16 of the unselective PARP inhibitor PJ34,21 and of the selective TNKS inhibitor IWR-114 (Table 1). All synthesized compounds substantially lost PARP1 inhibitory potency and selectivity over PARP2 in comparison with MC2050 and, with the sole exception of 6 (substantially inactive against all enzymes), displayed a submicromolar activity against TNKS1 (IC50 ranging from 673 to 6.1 nM) and TNKS2 (IC50 ranging from 588 to 0.3 nM). Significant differences were observed among compounds 1 and 2, that are characterized by a not-condensed bicyclic system linked to the piperazine ring through a polymethylene spacer. Indeed, the presence of a 4-(4′-biphenyl) moiety (1) induced a loss of activity in comparison with MC2050 not only against PARP1 but also versus TNKS1/2, whereas the introduction of a 3-phenyl-1,2,4-oxadiazol-5-yl moiety (2) produced a significant increase of inhibitory potency against TNKSs (IC50 = 37.4 nM on TNKS1 and IC50 = 11.7 nM on TNKS2) joined to selectivity over PARP1 (IC50 = 1480 nM) and, to a lesser extent, PARP2 (IC50 = 370 nM). The introduction on the spacer between the piperazine and the aromatic ring pointing toward the AD subpocket of a carbonyl function (carboxamide or carbamate) joined to either a bulky spiro tricyclic system (3) or a simple benzene ring (4) caused an almost complete loss of selectivity for TNKSs over PARP1/2 leading to the unselective (sub)micromolar PARP inhibitors 3 and 4. Interestingly, when the pyridine ring of MC2050 was substituted with a 2-mercaptoquinazolin-4-one linked to the central piperazine through an ethylene spacer providing the symmetrical compound 5, a massive increase in activity on TNKSs and selectivity over PARP1/2 was re-established. In particular, endowed with subnanomolar inhibitory potency against TNKS2 (IC50 = 0.3 nM) and with an important selectivity not only over PARP1/2 (more than 100-fold) but also over the closely related TNKS1 (more than 20-fold), 5 displayed a significant increase of TNKS2 inhibitory potency and selectivity not only in comparison with the pan-PARP inhibitor PJ34 (TNKS2 IC50 = 52.7 nM) but also with the TNKS selective inhibitor IWR-1 (TNKS2 IC50 = 29.2 nM). Overall these results confirmed that leveraging favorable interactions within the AD subpocket, as previously reported,13 may be advantageous also to gain isoform selectivity within the TNKS subfamily of PARPs.
Table 1. Inhibitory Activity of Derivatives 1–6 on PARP1/2 and TNKS1/2.
| IC50 (nM)a |
||||
|---|---|---|---|---|
| Compd | PARP-1 | PARP-2 | TNKS1 | TNKS2 |
| MC2050 | 9.3 ± 0.09 | 549 ± 12 | 218 ± 8 | 193 ± 7 |
| 1 | 2040 ± 61 | 340 ± 11 | 673 ± 28 | 588 ± 19 |
| 2 | 1480 ± 58 | 370 ± 8 | 37.4 ± 0.4 | 11.7 ± 0.3 |
| 3 | 1280 ± 49 | 347 ± 13 | 552 ± 18 | 103 ± 4 |
| 4 | 356 ± 14 | 963 ± 39 | 398 ± 14 | 318 ± 13 |
| 5 | 89.6 ± 2.2 | 37.8 ± 1.1 | 6.1 ± 0.07 | 0.3 ± 0.005 |
| 6 | 26% inhib. (@200 μM) | 132000 ± 987 | NIb (@200 μM) | NI (@200 μM) |
| PJ34 | 9.1 ± 0.09 | 14.7 ± 0.4 | 39.2 ± 0.9 | 52.7 ± 1.2 |
| IWR-1 | NI (@200 μM) | NI (@200 μM) | 303 ± 9 | 29.2 ± 0.8 |
Values are means ± SD determined from at least two independent experiments, each one done in triplicate.
NI, no inhibition.
A retrospective structural investigation of the interaction mode of 5 into the AD binding cleft of PARP1 and TNKS2 using computational docking suggested a possible explanation for the observed selectivity (Figure S1). In the energetically minimized geometry of 5, the second quinazolinone ring is placed in a perfect stacking interaction with His1048, and at the same time is predicted to be stabilized by favorable interactions with the residues of the TNKS2 D-loop (Phe1035, Ala1038, Lys1042, Asp1045). Conversely, the same geometry would sterically clash with Arg878 of PARP1.
Since endowed with (sub)nanomolar inhibitory potency against TNKSs with a significant selectivity over PARP1/2, compounds 2 and 5 were selected to be further evaluated in a colony forming assay on DLD-1 human CRC cells, a cell line characterized by a truncated form of the APC protein, one of the components of the β-catenin destruction complex, that shows a constitutive activation of the canonical Wnt signaling pathway and is frequently used to validate TNKS inhibitors in cells.4,13,22 The selectedcompounds were testedat 1 and 10 μM (2) or at 0.1 and 1 μM (5) depending on the relative potency in enzyme assays (see Table 1). As shown in Figure 4, compounds 2 and 5 were able to inhibit formation of DLD-1 cell colonies in a dose-dependent manner, with compound 5 showing a strong antiproliferative effect at 1 μM, better than that of the reference TNKS inhibitor IWR-1 at the same concentration.
Figure 4.
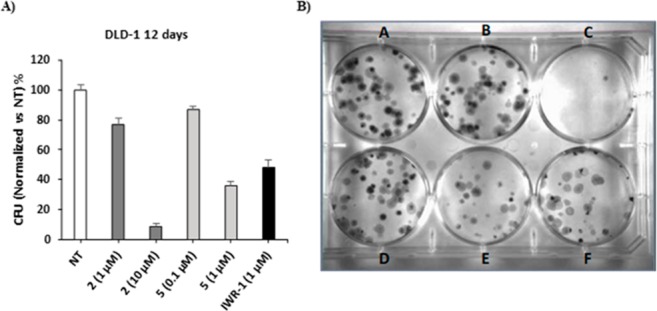
(A) Bar diagram of antiproliferative effects on DLD-1 CRC cells of 2 and 5. (B) Colony forming assay on DLD-1 cancer cells. (A) untreated cells; (B) compound 2 at 1 μM; (C) compound 2 at 10 μM; (D) compound 5 at 0.1 μM; (E) compound 5 at 1 μM; (F) IWR-1 at 1 μM.
The development of PARP inhibitors historically focused on the NAM subpocket of the NAD+ binding cavity but mostly afforded polypharmacological agents rather than selective molecular tools. The establishment of accessory interplays with the regulatory α-helical subdomain next to the cofactor-binding site in PARP1/2 (and lacking in TNKSs) led to development of selective inhibitors, and some of them are now on the market.8 Similarly, exploring the AD subpocket as well as exploiting the unique structural features of the D-loop, such as the hydrophobic nature of the sequence and the narrower space, resulted in a more selective inhibition of TNKSs. Moreover, the simultaneous targeting of different portions of the cofactor donor site, i.e. by dual binders, might not only lead to selectivity among PARPs, but might also prevent inhibition of other NAD+ dependent enzymes, possibly reducing unpredictable off-target effects.7,11−15
Here we took advantage of the combination of crystallographic and computational techniques to generate a small collection of quinazolinone-based compounds able to span the length of the NAD+ binding groove. The inspection of the X-ray structure of the PARP1 inhibitor MC2050 in complex with hPARP1c revealed that its quinazolinone ring is embedded in the NAM subsite of the cofactor-binding pocket engaging π–π stacking interactions and H-bonds. Superimposition with TNKSs catalytic domains uncovered that the quinazolinone H-bonding could be preserved and paved the way for the rational design of a focused library of compounds in which the 2-mercaptoquinazolin-4-one scaffold was functionalized with different aromatic systems installed through a proper spacer on the piperazine moiety of the parent compound MC2050. Biological tests highlighted 2 and 5 as (sub)nanomolar TNKS2 inhibitors with a significant selectivity over PARP1/2, inverting the selectivity profile of MC2050. 5 showed a dramatic increase of activity on TNKSs (up to 600-fold). Moreover, it exhibited a 2 orders of magnitude higher potency on TNKS2 with respect to PARP1 and PARP2 and a selectivity of about 20-fold on the closely related TNKS1. Compounds 2 and 5 also showed dose-dependent antiproliferative effects in colony forming assays, comparable to the TNKSs inhibitor IWR-1, confirming the efficaciousness of TNKS inhibition in affecting cancer cell growth.
In conclusion, herein we reported a structure-based computationally driven approach, that with a limited, but extremely focused, synthetic effort led to the discovery of the bis-quinazolinone-based NAM-AD dual-binder 5 as a picomolar and selective TNKS2 inhibitor, that has been selected as lead compound for further optimization studies.
Acknowledgments
This work was supported by funds from PRIN 2015 (prot.20152TE5PK) (A.M.), AIRC IG 2016 (n. 19162) (A.M.), and AIRC MFAG2017 (n. 20447) (A.P. and C.E.). Thanks are due to Dr. Sébastien Moniot for helpful discussion. We thank Swiss Light Source (SLS; Villigen, Switzerland) for beamtime and support. The authors dedicate the work to the memory of Prof. Maurizio Botta, a great friend and continuous source of inspiration for med-chem studies.
Glossary
Abbreviations
- AD
adenosine
- ARTDs
ADP-ribosyltransferases diphtheria toxin-like
- CRC
colorectal cancer
- NAD+
nicotinamide adenine dinucleotide
- NAM
nicotinamide
- NSCLC
nonsmall cell lung cancer
- PARPs
poly(ADP-ribose) polymerases
- TNKSs
tankyrases
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.9b00654.
Experimental procedures for the synthesis and physicochemical characterization of all new compounds (1–20); materials and methods for enzyme inhibition assays, crystallographic (PDB: 6XVW) and in silico studies, and cell-based assays (PDF)
Author Contributions
S.T., A.P., C.S. A.M., and D.R. manuscript preparation and data interpretation. S.T., E.N., N.M., A.R., and D.R. compound synthesis and enzymatic tests. J.P., M.P., and C.S. crystallographic studies. A.P. and C.E. computational studies. D.P. and R.P. cell-based assays. A.P., C.S. A.M., and D.R. conceived the study and followed the whole project. All authors have approved the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Hottiger M. O.; Hassa P. O.; Lüscher B.; Schüler H.; Koch-Nolte F. Toward a unified nomenclature for mammalian ADP-ribosyltransferases. Trends Biochem. Sci. 2010, 35, 208–219. 10.1016/j.tibs.2009.12.003. [DOI] [PubMed] [Google Scholar]
- Langelier M. F.; Eisemann T.; Riccio A. A.; Pascal J. M. PARPfamily enzymes: regulation and catalysis of the poly(ADP-ribose) posttranslational modification. Curr. Opin. Struct. Biol. 2018, 53, 187–198. 10.1016/j.sbi.2018.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riffell J. L.; Lord C. J.; Ashworth A. Tankyrase-targeted therapeutics: expanding opportunities in PARP family. Nat. Rev. Drug Discovery 2012, 11, 923–936. 10.1038/nrd3868. [DOI] [PubMed] [Google Scholar]
- Mariotti L.; Pollock K.; Guettler S. Regulation of Wnt/β-catenin signalling by tankyrase-dependent poly(ADP-ribosyl)ation and scaffolding. Br. J. Pharmacol. 2017, 174, 4611–4636. 10.1111/bph.14038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M. K. Novel insight into the function of tankyrase. Oncol. Lett. 2018, 16, 6895–6902. 10.3892/ol.2018.9551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferraris D. V. Evolution of poly(ADP-ribose) polymerase-1 (PARP-1) inhibitors. From concept to clinic. J. Med. Chem. 2010, 53, 4561–4584. 10.1021/jm100012m. [DOI] [PubMed] [Google Scholar]
- Ekblad T.; Camaioni E.; Schüler H.; Macchiarulo A. PARP inhibitors: polypharmacology versus selective inhibition. FEBS J. 2013, 280, 3563–3575. 10.1111/febs.12298. [DOI] [PubMed] [Google Scholar]
- Kim G.; Ison G.; McKee A. E.; Zhang H.; Tang S.; Gwise T.; Sridhara R.; Lee E.; Tzou A.; Philip R.; Chiu H. J.; Ricks T. K.; Palmby T.; Russell A. M.; Ladouceur G.; Pfuma E.; Li H.; Zhao L.; Liu Q.; Venugopal R.; Ibrahim A.; Pazdur R. FDA approvalsummary: Olaparib monotherapy in patients with deleterious germlineBRCA-mutated advanced ovarian cancer treated with three or morelines of chemotherapy. Clin. Cancer Res. 2015, 21, 4257–4261. 10.1158/1078-0432.CCR-15-0887. [DOI] [PubMed] [Google Scholar]
- Curtin N. J.; Szabo C. Therapeutic applications of PARPinhibitors: anticancer therapy and beyond. Mol. Aspects Med. 2013, 34, 1217–1256. 10.1016/j.mam.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehtiö L.; Chi N. W.; Krauss S. Tankyrases as Drug Targets. FEBS J. 2013, 280, 3576–3593. 10.1111/febs.12320. [DOI] [PubMed] [Google Scholar]
- Kamal A.; Riyaz Sd.; Srivastava A. K.; Rahim A. Tankyraseinhibitors as therapeutic targets for cancer. Curr. Top. Med. Chem. 2014, 14, 1967–1976. 10.2174/1568026614666140929115831. [DOI] [PubMed] [Google Scholar]
- Thorsell A. G.; Ekblad T.; Karlberg T.; Löw M.; Pinto A. F.; Trésaugues L.; Moche M.; Cohen M. S.; Schüler H. Structural Basis for Potency and Promiscuity in Poly(ADP-ribose) Polymerase (PARP) and Tankyrase Inhibitors. J. Med. Chem. 2017, 60, 1262–1271. 10.1021/acs.jmedchem.6b00990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathubhai A.; Haikarainen T.; Koivunen J.; Murthy S.; Koumanov F.; Lloyd M. D.; Holman G. D.; Pihlajaniemi T.; Tosh D.; Lehtiö L.; Threadgill M. D. Highly potent and isoform selective dual site binding Tankyrase/Wnt signaling inhibitors that increase cellular glucose uptake and have antiproliferative activity. J. Med. Chem. 2017, 60, 814–820. 10.1021/acs.jmedchem.6b01574. [DOI] [PubMed] [Google Scholar]
- Narwal M.; Venkannagari H.; Lehtiö L. Structural basis of selective inhibition of human Tankyrases. J. Med. Chem. 2012, 55, 1360–1367. 10.1021/jm201510p. [DOI] [PubMed] [Google Scholar]
- Wahlberg E.; Karlberg T.; Kouznetsova E.; Markova N.; Macchiarulo A.; Thorsell A. G.; Pol E.; Frostell A.; Ekblad T.; Öncü D.; Kull B.; Robertson G. M.; Pellicciari R.; Schüler H.; Weigelt J. Family wide chemical profiling and structural analysis of PARP and tankyrase inhibitors. Nat. Biotechnol. 2012, 30, 283–288. 10.1038/nbt.2121. [DOI] [PubMed] [Google Scholar]
- Mosca L.; Rotili D.; Tempera I.; Masci A.; Fontana M.; Chiaraluce R.; Mastromarino P.; d’Erme M.; Mai A. Biological effects of MC2050, a quinazoline-based PARP-1 inhibitor in human neuroblastoma and EBV-positive Burkitt’s lymphoma cells. ChemMedChem 2011, 6, 606–611. 10.1002/cmdc.201000536. [DOI] [PubMed] [Google Scholar]
- Martire S.; Fuso A.; Rotili D.; Tempera I.; Giordano C.; De Zottis I.; Muzi A.; Vernole P.; Graziani G.; Lococo E.; Faraldi M.; Maras B.; Scarpa S.; Mosca L.; d’Erme M. PARP-1 modulates amyloid beta peptide-induced neuronal damage. PLoS One 2013, 8, e72169 10.1371/journal.pone.0072169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawicki-McKenna J. M.; Langelier M. F.; DeNizio J. E.; Riccio A. A.; Cao C. D.; Karch K. R.; McCauley M.; Steffen J. D.; Black B. E.; Pascal J. M. PARP-1 activation requires local unfolding of an autoinhibitory domain. Mol. Cell 2015, 60, 755–768. 10.1016/j.molcel.2015.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorsell A. G.; Ekblad T.; Karlberg T.; Löw M.; Pinto A. F.; Trésaugues L.; Moche M.; Cohen M. S.; Schüler H. Structural Basis for Potency and Promiscuity in Poly(ADP-ribose) Polymerase (PARP) and Tankyrase Inhibitors. J. Med. Chem. 2017, 60, 1262–1271. 10.1021/acs.jmedchem.6b00990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua Z.; Bregman H.; Buchanan J. L.; Chakka N.; Guzman-Perez A.; Gunaydin H.; Huang X.; Gu Y.; Berry V.; Liu J.; Teffera Y.; Huang L.; Egge B.; Emkey R.; Mullady E. L.; Schneider S.; Andrews P. S.; Acquaviva L.; Dovey J.; Mishra A.; Newcomb J.; Saffran D.; Serafino R.; Strathdee C. A.; Turci S. M.; Stanton M.; Wilson C.; Dimauro E. F. Development of noveldual binders as potent, selective, and orally bioavailable tankyrase inhibitors. J. Med. Chem. 2013, 56, 10003–10015. 10.1021/jm401317z. [DOI] [PubMed] [Google Scholar]
- Kirby C. A.; Cheung A.; Fazal A.; Shultz M. D.; Stams T. Structure of human tankyrase 1 in complex with small-molecule inhibitors PJ34 and XAV939. Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun. 2012, 68, 115–118. 10.1107/S1744309111051219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizutani A.; Yoshiroda Y.; Muramatsu Y.; Yoshida H.; Chikada T.; Tsumura T.; Okue M.; Shirai F.; Fukami T.; Yoshida M.; Seimiya H. RK-287107, a potent and specific tankyrase inhibitor, blocks colorectal cancer cell growth in a preclinical model. Cancer Res. 2018, 109, 4003–4014. 10.1111/cas.13805. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.