

Abstract

Due to the biological liability of diketo acid (DKA) chain, we transferred this element of our previously reported anti-HIV-1 pyrrolyl derivatives to a non-DKA scaffold, obtaining a series of pyrrolyl–pyrazole carboxylic acids as new RNase H inhibitors. Among the newly synthesized derivatives, oxyphenylpyrrolyl–pyrazoles demonstrated inhibitory activities within the low micromolar/submicromolar range with compound 11b being the most potent. Interestingly, all tested compounds showed up to 2 orders of magnitude of selectivity for RNase H vs integrase. Docking studies within the RNase H catalytic site, coupled with site-directed mutagenesis, showed the key structural features that could confer the ability to establish specific interactions within RNase H. Furthermore, they proved the ability of our compounds to interact with amino acids highly conserved among HIV-1 subspecies isolated among patients carrying drug-resistant variants. In the end, the newly discovered pyrazole carboxylic acid derivatives feature promising serum stability with respect to their corresponding DKAs.
Keywords: AIDS, HIV, RNase H inhibitors, Diketo acid, Pyrazole
Introduction
The human immunodeficiency virus type 1 (HIV-1) is the etiological agent of one of the most threatening human diseases, referred to as acquired immune deficiency syndrome (AIDS). Currently, there are close to 40 million people living with HIV/AIDS worldwide, about 1 million AIDS-related deaths, and about 1.8 million new infections were estimated in 2017.1 Even though the large therapeutic armory (ART, antiretroviral therapy) allowed reaching remarkable milestones, life-long treatments present multiple drawbacks.2,3 Thus, there is still the pressing demand for a constant effort toward the identification of new targets and the development of new effective anti-HIV agents.4 Along this line, the development of compounds able to inhibit crucial but unexplored viral processes is an attractive solution to raise the genetic barrier to resistance.
The conversion of the viral RNA into DNA is a critical step in the HIV-1 lifecycle initiated by the reverse transcriptase (RT), a well-established drug target. RT possesses two functional domains: an RNA- and DNA-dependent DNA polymerase domain (RDDP and DDDP, respectively) and a ribonuclease H domain (RNase H). All current anti-HIV drugs acting on RT are targeting the polymerase domain. The RNase H domain catalyzes the selective hydrolysis of the RNA strand of the RNA:DNA heteroduplex, an essential step for viral replication.5−7 Indeed, several lines of evidence suggest that selectively inhibiting RNase H confers an antiviral phenotype, defining RNase H as an attractive target for rational anti-HIV-1 drug design.8,9 The discovery of RNase H inhibitors (RHIs) is still in its infancy and none of them has reached the clinical pipeline.8
Actually, many RHIs have been reported in the literature as effective inhibitors in enzyme assays;5 nevertheless, they showed high toxicity or they did not exhibit selective inhibition over the human RNase H,8 RT-associated RDDP, and/or integrase (IN) activities.9−12 In this field of research, our group has previously reported DKA derivatives that prove to be dual inhibitors of IN and RH13,14 or IN strand transfer inhibitors (INSTIs) endowed with marginal RH inhibition activities.15,16 Among the dual inhibitors, notable examples are the phenylpyrrolyl derivative 1,17 the pyrrolyl diketo hexenoic ester 2, which was the first DKA derivative reported as RHI to have an antiviral effect,18 and its chlorine analogue 3, which was proven to inhibit viral replication by selectively blocking RNase H function (Figure 1).19
Figure 1.

Inhibitors of HIV-1 RNase H function of RT and/or IN enzyme.
Despite the relevance of the DKA branch in the inhibitory activity, it is widely reported that this group is biologically liable due to high metabolic turnover and because it shows poor cell membrane penetration. Furthermore, it has been proven to have a time-dependent decrease in the activity in solutions at room temperature, even during short periods of time.20 Likewise, we observed a time-dependent decay of the inhibition of viral replication using the RHI 3 as an anti-HIV agent in cell-based assays.19
For this reason, a variety of compounds were developed by mimicking the DKA element into scaffolds characterized by improved druglike qualities, thus overcoming the biological liability of the DKA moiety. A success case in the HIV field is the discovery of Raltegravir (4), a hydroxypirimidinone derivative, the first in class INSTI approved in 2007.21 Another example reported in the literature are potential INSTIs designed by the conversion of the 1,3-diketo unit into heteroaromatic motifs, obtaining azole carboxylic acid derivatives.22 In the end, in the field of RHIs, naphthyridinones, like compound 5, resulted in good anti-RH agents8 but were inactive in multicycle viral replication assays due to high toxicity (Figure 1).
A further drawback reported for DKAs is their low selectivity vs enzyme targets. In fact, in general, they inhibit both IN and RH enzymes. Our works widely demonstrated that the quinolonyl and pyrrolyl DKA derivatives13−17,19 were dual RH/IN inhibitors, or RH agents endowed with very poor selectivity, 2–3 fold at the best.
So the aim of this work is the design of new RHIs starting from the pyrrolyl DKAs 1–3 to obtain a new class of compounds endowed with improved stability and selectivity/specificity against HIV RH vs IN enzyme.
Design of New RHIs
In the present study we synthesized and biologically evaluated new compounds designed starting from our previously reported DKA inhibitors. In particular, starting from the structure of 1, we decided to mimic the α–γ-diketo unit of the DKAs with a pyrazole ring (Figure 2), to obtain potentially more stable pyrrolyl–pyrazole carboxylic acid derivatives (Chart 1).
Figure 2.

Design of pyrrolyl–pyrazole carboxylic acids.
Chart 1. Structures of the Newly Designed Pyrazole Derivatives 6a–h, 7a–h, 8a–c, 9a–c, 10a–i, and 11a–i.

This modification was suggested also by results reported by Zeng et al.,22 who described pyrazole carboxylate derivatives designed as DKA isosteres. These compounds proved to be very weak or null INSTIs and therefore their antiviral activity was supposed to be related to a mechanism of action different from the IN inhibition.
Thus, we designed the compounds reported in Chart 1, by keeping fixed the pyrrolyl–pyrazole carboxylic moiety, we conceived a new series of derivatives (6 and 7) characterized by one or more of the following modifications: (i) the removal of the substituent in 1- and/or 4-position of the pyrrole core; (ii) the introduction of various alkyl or arylalkyl substituents replacing the p-fluorobenzyl ring in position 1 of the pyrrole nucleus; (iii) the replacement of a naphth-2-yl group replacing the phenyl ring in position 4 of the pyrrolyl skeleton. Noteworthy, within this series, derivatives 6a and 7a, ester and acid, respectively, represent the non-DKA analogues of 1. Furthermore, a series of oxyphenylpyrrolyl–pyrazole derivatives (10 and 11) was designed mainly by introducing various substituents endowed with different degrees of freedom or steric hindrance on the oxyphenyl portion linked in 4-position to the pyrrole core and/or by replacing the p-fluorobenzyl ring with a methylnaphth-1-yl group on the pyrrolyl nitrogen. Notably, in order to define more extensive structure–activity relationships (SARs), we decided to include a small series of non-pyrrolyl derivatives (8 and 9) characterized by the replacement of the pyrrole core linked in 3-position to the pyrazole ring with a methyl,23 phenyl,24 or biphenyl substituent. The newly synthesized compounds have been evaluated in vitro for their ability to inhibit the specific enzymatic activity of recombinant RNase H and IN, for their cytotoxicity and antiviral activity against HIV-1 in human cells. In addition, a rationalization of the interaction with the biological target has been proposed on the basis of docking studies using the crystal structure of RNase H and validated by site-directed mutagenesis on the residues indicated as crucial for the binding. Finally, mass spectrometry (MS) experiments allowed probing the human serum stability of the most active pyrrolyl–pyrazole compounds in comparison with their diketobutanoic congeners.
Results and Discussion
The synthesis of derivatives 6a–h and 7a–h is outlined in Scheme 1.
Scheme 1. Synthetic Route to 6a–h and 7a–h Derivatives.

Reagents and conditions: (i) TosMIC, NaH, 2:1 Et2O dry/DMSO dry, N2, room temp, 1 h, 100% yield; (ii) proper arylalkyl halide, DMF, K2CO3, 80 °C, overnight, 55–70% yield; (iii) diethyl oxalate, EtONa, THF dry, N2, room temperature, 2 h, 97–100% yield; (iv) MeOH, NH2NH2 dihydrochloride, Et3N, CH3COOH gl, room temperature, 2 h, 60–100% yield; (v) THF, NaOH 20%, 90 or 100 °C, 6–12 h, 65–98% yield.
Scheme 2 describes the synthetic route to obtain derivatives 8c and 9c.
Scheme 2. Synthetic Route to 8c and 9c Derivatives.

Reagents and conditions: (i) MeOH, NH2NH2 dihydrochloride, Et3N, CH3COOH gl, room temperature, 2 h, 100% yield; (ii) THF, NaOH 20%, 90 °C, 12 h, 78% yield.
Synthesis of oxyphenylpyrrolyl–pyrazole derivatives 10a–h and 11a–h were performed as reported in Scheme 3. For a detailed description of all synthetic procedures and characterization of final compounds and intermediates, see the Supporting Information.
Scheme 3. Synthetic Route to 10a–i and 11a–i Derivatives.

Reagents and conditions: (i) acetone, NaOH 1 N, 80 °C, 5 h, 41–63% yield; (ii) TosMIC, NaH, 2:1 Et2O dry/DMSO dry, N2, room temp, 1 h, 87–100% yield; (iii) proper arylalkyl halide, DMF, K2CO3, 80 °C, 2–24 h, 15–59% yield; (iv) diethyl oxalate, EtONa, THF dry, N2, room temp, 2 h, 55–100% yield; (v) MeOH, NH2NH2 dihydrochloride, Et3N, CH3COOH gl, room temp, 2 h, 20–65% yield; (vi) THF, NaOH 20%, 90 °C, 12 h, 62–100% yield.
In Vitro Screening for RNase H Inhibitory Activity
All the newly synthesized compounds 6a–h, 7a–h, 8a–c, 79a–c, 10a–i, and 11a–i were tested in vitro against recombinant HIV-1 RT-associated RNase H activity (Table 1). In general, compounds belonging to series 6 and 7 demonstrated less interesting results, with 6 compounds (out of 16) active in the micromolar range. Differently, derivatives of series 8 and 9 proved to be inactive, suggesting that the pyrrole core is essential for efficient inhibition of RNase H.
Table 1. RNase H Inhibition of the Newly Synthesized Compounds 6a–h, 7a–h, 8a–c, 9a–c, 10a–i, and 11a–i.
| cpd | IC50 (μM)a | cpd | IC50 (μM)a | cpd | IC50 (μM)a |
|---|---|---|---|---|---|
| 6a | 14.3 ± 2.35 | 7g | 63 ± 7.0 | 10g | 6.8 ± 0.4 |
| 6b | >100 | 7h | 13.7 ± 1.3 | 10h | 7.9 ± 0.7 |
| 6c | >100 | 8a | >100 | 10i | 25.8 ± 3.7 |
| 6d | >100 | 8b | >100 | 11a | 3.21 ± 0.94 |
| 6e | >100 | 8c | >100 | 11b | 0.27 ± 0.05 |
| 6f | 9.4 ± 0.2 | 9a | >100 | 11c | 2.38 ± 0.59 |
| 6g | 97 ± 5.0 | 9b | >100 | 11d | 4.50 ± 0.60 |
| 6h | 9.4 ± 0.7 | 9c | >100 | 11e | 1.73 ± 0.31 |
| 7a | 75 ± 4.5 | 10a | 5.4 ± 1.2 | 11f | 70 ± 8 |
| 7b | >100 | 10b | 17.8 ± 1.2 | 11g | 23 ± 3 |
| 7c | >100 | 10c | 8.5 ± 1.6 | 11h | 5.8 ± 1.4 |
| 7d | >100 | 10d | 5.50 ± 0.93 | 11i | 9.2 ± 0.4 |
| 7e | >100 | 10e | 4.15 ± 0.55 | 1 | 10 ± 1.2 |
| 7f | 25.3 ± 5.3 | 10f | 8.1 ± 0.4 |
Inhibitory concentration 50% (μM) determined from dose–response curves: experiments performed against HIV-1 RT-associated RNase H activity.
Bn, benzyl.
Differently, derivatives of series 10 and 11 proved to be the best RHIs among the newly synthesized derivatives, showing 17 compounds (out of 18) to be active in the low micromolar/submicromolar range (11b). In particular, these latter derivatives are characterized by the introduction of various substituents endowed with different degrees of freedom or steric hindrance on the oxyphenyl portion and/or by the replacement of the p-fluorobenzyl ring with a methylnaphth-1-yl group on the pyrrolyl nitrogen.
The sole exception to this trend was observed for the acid 11f (IC50 = 70 μM), where the presence of a N-Ph-4-carboxyacetamide substituent along with the acid function had a detrimental effect with respect to the ester analogue 10f (IC50 = 8.1 μM). Interestingly, the presence of a methylnaphth-1-yl in position 1 of the pyrrole nucleus confers similar potency to esters and their acidic counterparts (couples 10c–11c, 10e–11e, 10h–11h with IC50 values ranging from 1.7 to 8.5 μM). Conversely, the compounds characterized by the presence of the p-fluorobenzyl ring in position 1 of the pyrrole core, showed different inhibitory potency within the ester/acid couple (except for derivatives 10a,d, and their acids 11a,d). In particular, the presence of a phenylbutyl (10b, IC50 = 17.8 μM; 11b, IC50 = 0.27 μM) or phenylpentyl (10i, IC50 = 25.8 μM; 11i, IC50 = 9.2 μM) substituent on the oxyphenyl portion led to an increase in potency for acid derivatives while the presence of a N-Ph-4-carboxyacetamide (10f, IC50 = 8.1 μM; 11f, IC50 = 70 μM) or a N-phenylacetamide (10g, IC50 = 6.8 μM and 11g, IC50 = 23 μM) substituent on the oxyphenyl portion caused an increase in inhibitory activity for the ester counterparts.
To determine the effect of compounds on viral replication four acid–ester couples of compounds (6a,h–7a,h; 10a,b–11a,b) were chosen to test the antiviral activity in HeLa-CD4-LTR-β-gal cells. This was initially assessed using 3 (IC50 value against RNase H of 7.3 μM)18 and 4 as reference compounds (Table S1 in the Supporting Information). Notably, only the ester derivatives gave measurable EC50 values that ranged from 4 to 41 μM. Although compound 6a showed the lowest EC50 of the series; nevertheless, 6h and 10b were still active but not cytotoxic up to high concentration (200 μM), showing SIs > 10.
Because of the structural similarity between RNase H and IN, the potential anti-IN activity in vitro has been assayed for compounds 6a,h, 10a,b, and 11a. Using an HTRF-based solution assay in the presence of recombinant WT IN and LEDGF, only compound 6a was slightly active against IN catalytic activity (IC50 value of 75 μM, Table S1) showing a 5-fold selectivity for RNase H inhibition over IN. Compounds 6h, 10a, and 10b showed no activity resulting in selectivity indexes of >10.6, >18.5, and >5.6 for RNase H inhibition compared to IN, respectively.
Docking Studies
To rationalize the activity of the newly synthesized compounds, molecular docking studies were undertaken. For the docking calculations AutoDock Vina25 was employed, whose accuracy in correctly predicting binding poses and energies was already demonstrated in our previous works.26 The high-resolution crystal structure of HIV-1 RT in complex with a small molecule RHI (PDB code 3QIP)27 was chosen as macromolecule for docking calculation. Compounds 6a, 6h, 10a, and 10b were selected for in silico experiments, given their promising anti-RNase H activity in vitro. The docked pose for 6a (Figure 3a) indicates that the pyrazole nitrogens and the ester moiety chelate the two magnesium ions in a position that is consistent with other cocrystallized RNase H ligands.27,28 This pose allows for an H-bonding interaction between the ester group and H539. Of note, the p-fluorobenzyl group is pointing toward the positively charged K540, probably engaging in a cation−π interaction. Moreover, the adjacent W535 also establishes a T-shaped charge-transfer interaction with the same p-fluorobenzyl moiety. Also, the phenyl moiety is lodged in a cleft lined by residues S499, Y501, Q475, E478, N474, and Q475. Consistently with previous mutagenesis studies with ligands featuring comparable binding modes (compound 1),18 a parallel-displaced π interaction takes place between Y501 and the phenyl moiety. The latter would also engage in a cation−π interaction with R448. In agreement with RNase H inhibitory activity, it could be postulated that two aromatic rings, one in the cleft comprising S499, Y501, Q475, E478, N474, and Q475, and the other in the gorge with W535 and K540, represent critical structural requirements for RNase H inhibition in our compounds. This might account for the lack of activity shown by compounds devoid of one or both aromatic rings.
Figure 3.
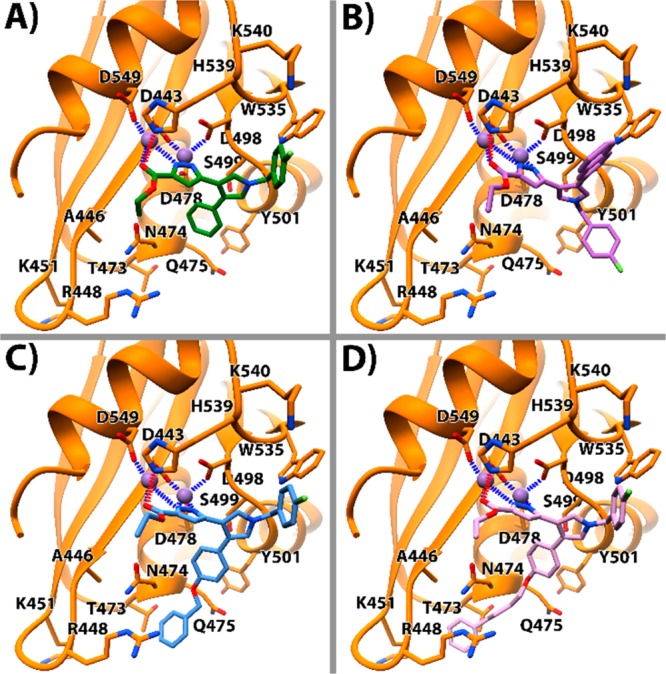
Binding poses of 6a (a), 6h (b), 10a (c), and 10b (d) in the HIV RNase H binding site (PDB 3QIP). Important residues are labeled. 6a is represented as green sticks, 6h as dark purple sticks, 10a as cyan sticks, 10b as pink sticks. The magnesium ions are represented in purple and their coordination with the nearby atoms is represented in blue. The H-bonds are represented as a dashed red line. The protein is represented as orange ribbons and sticks.
As inferred in our previous studies,18 the ester group featured in the compounds of the series 6 and 10, in place of the carboxyl acid in the series 7 and 11, contributes to the charge neutrality of the binding site, in some cases favorably influencing the stability of the ligand/protein complex. This trend is verified for 6a/7a, 6f/7f, 6h/7h, 10f/11f, and 10g/11g ester/acid couples. Regarding compound 6h, the binding mode of the inhibitor is similar to that observed for compounds 6a (Figure 3b). One key difference is to be found in the positions of naphthyl and p-fluorobenzyl groups, which appeared switched compared to the corresponding rings in 6a. The bulky naphthyl group can be better accommodated in the cleft harboring the RNase H residues W535 and K540. The enhancement of the interaction with these latter residues could explain the higher activity of compounds carrying this substituent. In the same vein, compound 10a shares a similar binding mode compared to that of 6a, with the flexible pendant benzyloxy portion lodged closer to the RNase H residue R448, thereby enhancing the strength of the cation−π interactions (Figure 3c). Finally, thanks to its long and flexible phenylbutoxy chain, compound 10b delves into an additional pocket, adjacent to the main binding site common with 6a and 10a. Thus, 10b establishes additional contacts with RNase H residues N474, Q475, T473, A446, and R448. In addition to the aforementioned cation−π interaction with R448, also K451 might favorably interact with phenylbutoxy moiety of 10b.
Site-Directed Mutagenesis Experiments
To experimentally verify the binding model suggested by computational studies, seven-point mutants of HIV-1 RNase H were generated by introducing an alanine substitution at residues R448, K451, N474, Q475, Y501, W535A, and K540. Compounds 6a, 6h, 10a, and 10b were tested for their ability to inhibit these mutated RT variants. In parallel, we used RHI 3(12) as control (Figure 4). In agreement with computational calculations, mutations N474A, Q475A, Y501A, and W535A induced drastic reductions of inhibitory potency (generally more than 5 folds) of all compounds (Figure 4 and Table S2 in the Supporting Information).
Figure 4.
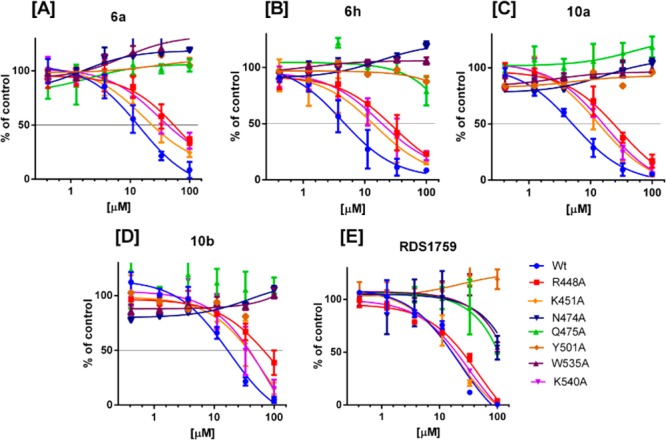
Inhibition of HIV-1 RT-associated RNase H activity of mutated HIV-1 RTs by non-DKA pyrrolyl derivatives: [A], 6a; [B], 6h; [C], 10a; [D], 10b, [E], 3.
Notably, all these amino acids are highly conserved among HIV-1 subspecies, even in drug-resistant variants isolated from patients.10 In the case of substitutions K540A and R448A, the loss of potency was less prominent but still significant for all the non-DKA pyrrolyl derivatives, while it was not significant for control 3 that lacks the aromatic substituent in position 4 of the pyrrole ring. These data are consistent with the structural requirements postulated in computational studies suggesting the need for a second aromatic ring in the gorge formed by the RNase H residues W535 and K540. In particular, the interaction with the aforementioned residues should compensate for the absence of DKA moiety (3), which should per se provide higher inhibitory potency than the pyrrole group. In this respect, it is worth noting that compound 6h, carrying the naphthyl group in the 4-position, is the one that exhibited the most significant variation in potency of inhibition against K540A (p-value <0.0002), as predicted. In the case of the K451A substitution, only moderate, but still significant, increases in IC50 values were observed for 6h and 10a and no effect was observed for 6a, while compound 10b, with the long and flexible phenylbutoxy chain, exhibited the most significant variation in potency (p-value <0.0008) as predicted. The presence of K451A substitution had no effect on the potency of inhibition of compound 3, as expected.
In Vitro Serum Stability
To probe whether our strategy of replacing the biologically labile diketobutanoic scaffold with the pyrrolyl–pyrazole carboxylic moiety resulted in more biologically stable compounds, liquid chromatography–mass spectrometry experiments were performed. In particular, compound 1, its pyrazole derivative 6a, and the corresponding carboxylic acid derivative 7a were assessed for their serum stability by incubating them in 90% human serum at 37 °C according to a previously described protocol (see Supporting Information for detailed experimental methods).29 As shown in Figure 5, 1 starts to be significantly degraded after 3 h while its carboxylic analogue is instantly degraded upon serum exposure. Interestingly, we were able to identify a common degradation product for 1 and its carboxylic analogue, which should be ascribed to the retro-Claisen adduct (see Supporting Information Table S3 and Figure S1). Similar results were already obtained by other authors for structural congeners of 1.20 Conversely, in our assay both 6a and 7a remained unaltered for the entire course of the experiment, confirming that the employed lead-optimization strategy was successful to yield more biologically stable RHIs.
Figure 5.

Stability of 1, its carboxylic derivative (1 COOH), 6a, and 7a at different intervals of incubation with 90% human serum. Relative concentrations of compounds were determined by integration of the A220 peaks from analytical HPLC.
Conclusions
In this work, we used the pyrazole ring as a non-DKA scaffold replacing the DKA chain to overcome its biological limitations. We reached the first goal of this work, obtaining non-DKA pyrrolyl derivatives with selective RNase H activity. Among the newly synthesized derivatives, the oxyphenylpyrrolyl–pyrazole subseries exhibited the best inhibitory potencies, being active within the low micromolar/submicromolar range. The most potent compound was 11b that inhibited RNase H with an IC50 = 0.27 μM. The selected compounds that have been tested in cell-based assays against HIV, showed low cytotoxicity, with moderate SIs.
Interestingly, all tested compounds showed up to 2 orders of magnitude of selectivity for RNase H vs IN. Docking studies, coupled with site-directed mutagenesis, allowed us to identify the structural requirements that could confer the possibility to establish specific interactions within the RNase H domain, retaining good selectivity against IN. Indeed, the presence of two aromatic rings on the pyrrole nucleus of non-DKA derivatives represents a critical requirement for RNase H inhibition. It is also worth noting that our non-DKA derivatives showed the ability to interact with residues highly conserved among HIV-1 subspecies isolated among patients carrying drug-resistant variants. In the end, the newly discovered pyrazole carboxylic acid derivatives, feature promising serum stability with respect to their corresponding DKAs, thereby confirming the achievement of the second goal of our design strategy.
Acknowledgments
We thank UB’L3 facility for providing access to the BSL3 facility of Bordeaux University.
Glossary
ABBREVIATIONS
- HIV-1
human immunodeficiency virus type 1
- AIDS
acquired immune deficiency syndrome
- ART
antiretroviral therapy
- RT
reverse transcriptase
- PI
protease inhibitor
- IN
integrase
- INSTI
integrase strand transfer inhibitor
- ART
antiretroviral therapy
- RDDP
RNA-dependent DNA polymerase
- DDDP
DNA-dependent DNA polymerase
- RNase H
ribonuclease H
- RHI
RNase H inhibitor
- DKA
diketo acid
- SAR
structure–activity relationship
- TosMIC
toluene-4-sulfonylmethylisocyanide
- HPLC
high-performance liquid chromatography
- GP
general procedure
- IR
infrared
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.9b00617.
Chemistry experimental section, including general chemistry and instrumentation, general and specific experimental procedures, and characterization of compounds 6a–h, 7a–h, 8a–c, 9a–c, 10a–i, and 11a–i, 12, 13f–h, 14b,d–h, 15b,d,f,g,i, 16ab,d,fg,i, 17a–i, 18a–i; molecular modeling methods section; biology experimental section (including tables of cytotoxicity, antiviral, and anti-IN activities, non-DKA pyrrolyl derivatives, and ESI-MS data); MS experimental section (PDF)
Author Contributions
^ The manuscript was written through the contributions of all authors. All authors have given approval to the final version of the manuscript. A.M., A.C., and V.N.M. contributed equally.
This work was supported by the “Sapienza” University of Rome to R.C. (Ateneo 2017). The following reagent was obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH: HeLa-CD4-LTR-β-gal from Dr. Michael Emerman.
The authors declare no competing financial interest.
Supplementary Material
References
- WHO . HIV/AIDS data and statistics. https://www.who.int/hiv/data/en (accessed August 06, 2019).
- Deeks S. G. The end of AIDS: HIV infection as a chronic disease. Lancet 2013, 382 (9903), 1525–1533. 10.1016/S0140-6736(13)61809-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margot N. A.; Wong P.; Kulkarni R.; White K.; Porter D.; Abram M. E.; Callebaut C.; Miller M. D. Commonly transmitted HIV-1 drug resistance mutations in reverse-transcriptase and protease in antiretroviral treatment-naive patients and response to regimens containing tenofovir disoproxil fumarate or tenofovir alafenamide. J. Infect. Dis. 2017, 215 (6), 920–927. 10.1093/infdis/jix015. [DOI] [PubMed] [Google Scholar]
- Guasch L.; Zakharov A. V.; Tarasova O. A.; Poroikov V. V.; Liao C.; Nicklaus M. C. Novel HIV-1 Integrase Inhibitor Development by Virtual Screening Based on QSAR Models. Curr. Top. Med. Chem. 2016, 16 (4), 441–448. 10.2174/1568026615666150813150433. [DOI] [PubMed] [Google Scholar]
- Wang X.; Gao P.; Menendez-Arias L.; Liu X.; Zhan P. Update on recent developments in small molecular HIV-1 RNase H inhibitors (2013–2016): opportunities and challenges. Curr. Med. Chem. 2018, 25 (14), 1682–1702. 10.2174/0929867324666170113110839. [DOI] [PubMed] [Google Scholar]
- Boyer P. L.; Smith S. J.; Zhao X. Z.; Das K.; Gruber K.; Arnold E.; Burke T. R. J.; Hughes S. H. Developing and Evaluating Inhibitors against the RNase H Active Site of HIV-1 Reverse Transcriptase. J. Virol. 2018, 92 (13), e02203–e02217. 10.1128/JVI.02203-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schatz O.; Cromme F.; Naas T.; Lindemann D.; Gruninger-Leitch F.; Mous J.; Le Grice S. F. J.. Inactivation of the RNase H domain of HIV-1 reverse transcriptase blocks viral infectivity. In Oncogenesis and AIDS; Papas T. S., Eds.; Portfolio Publishing Company: Houston, 1990; pp 293–404. [Google Scholar]
- Corona A.; Masaoka T.; Tocco G.; Tramontano E.; Le Grice S. F. Active site and allosteric inhibitors of the ribonuclease H activity of HIV reverse transcriptase. Future Med. Chem. 2013, 5 (18), 2127–2139. 10.4155/fmc.13.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corona A.; Esposito F.; Tramontano E. Can the ever-promising target HIV reverse transcriptase-associated RNase H become a success story for drug development?. Future Virol. 2014, 9, 445–448. 10.2217/fvl.14.24. [DOI] [Google Scholar]
- Tang J.; Do H. T.; Huber A. D.; Casey M. C.; Kirby K. A.; Wilson D. J.; Kankanala J.; Parniak M. A.; Sarafianos S. G.; Wang Z. Pharmacophore-based design of novel 3-hydroxypyrimidine-2,4-dione subtypes as inhibitors of HIV reverse transcriptase-associated RNase H: tolerance of a nonflexible linker. Eur. J. Med. Chem. 2019, 166, 390–399. 10.1016/j.ejmech.2019.01.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tramontano E.; Di Santo R. HIV-1 RT-associated RNase H function inhibitors: recent advances in drug development. Curr. Med. Chem. 2010, 17 (26), 2837–2853. 10.2174/092986710792065045. [DOI] [PubMed] [Google Scholar]
- Di Santo R. Diketo acids derivatives as dual inhibitors of human immunodeficiency virus type. Curr. Med. Chem. 2011, 18, 3335–3342. 10.2174/092986711796504619. [DOI] [PubMed] [Google Scholar]
- Costi R.; Metifiot M.; Esposito F.; Cuzzucoli Crucitti G.; Pescatori L.; Messore A.; Scipione L.; Tortorella S.; Zinzula L.; Novellino E.; Pommier Y.; Tramontano E.; Marchand C.; Di Santo R. 6-(1-Benzyl-1H-pyrrol-2-yl)-2,4-dioxo-5-hexenoic acids as dual inhibitors of recombinant HIV-1 integrase and ribonuclease H, synthesized by a parallel synthesis approach. J. Med. Chem. 2013, 56, 8588–8598. 10.1021/jm401040b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corona A.; Di Leva F. S.; Rigogliuso G.; Pescatori L.; Madia V. N.; Subra F.; Delelis O.; Esposito F.; Cadeddu M.; Costi R.; Cosconati S.; Novellino E.; Di Santo R.; Tramontano E. New insights into the interaction between pyrrolyl diketoacids and HIV-1 integrase active site and comparison with RNase H. Antiviral Res. 2016, 134, 236–243. 10.1016/j.antiviral.2016.09.008. [DOI] [PubMed] [Google Scholar]
- Pescatori L.; Métifiot M.; Chung S.; Masoaka T.; Cuzzucoli Crucitti G.; Messore A.; Pupo G.; Madia V. N.; Saccoliti F.; Scipione L.; Tortorella S.; Di Leva F. S.; Cosconati S.; Marinelli L.; Novellino E.; Le Grice S. F.; Pommier Y.; Marchand C.; Costi R.; Di Santo R. N-Substituted quinolinonyl diketo acid derivatives as HIV integrase strand transfer inhibitors and their activity against RNase H function of reverse transcriptase. J. Med. Chem. 2015, 58 (11), 4610–4623. 10.1021/acs.jmedchem.5b00159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costi R.; Métifiot M.; Chung S.; Cuzzucoli Crucitti G.; Maddali K.; Pescatori L.; Messore A.; Madia V. N.; Pupo G.; Scipione L.; Tortorella S.; Di Leva F. S.; Cosconati S.; Marinelli L.; Novellino E.; Le Grice S. F.; Corona A.; Pommier Y.; Marchand C.; Di Santo R. Basic quinolinonyl diketo acid derivatives as inhibitors of HIV integrase and their activity against RNase H function of reverse transcriptase. J. Med. Chem. 2014, 57, 3223–3234. 10.1021/jm5001503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuzzucoli Crucitti G.; Metifiot M.; Pescatori L.; Messore A.; Madia V. N.; Pupo G.; Saccoliti F.; Scipione L.; Tortorella S.; Esposito F.; Corona A.; Cadeddu M.; Marchand C.; Pommier Y.; Tramontano E.; Costi R.; Di Santo R. Structure-activity relationship of pyrrolyl diketo acid derivative as dual inhibitors of HIV-1 integrase and reverse transcriptase ribonuclease H domain. J. Med. Chem. 2015, 58 (4), 1915–1928. 10.1021/jm501799k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tramontano E.; Esposito F.; Badas R.; Di Santo R.; Costi R.; La Colla P. 6-[1-(4-Fluorophenyl)methyl-1H-pyrrol-2-yl)]-2,4-dioxo-5-hexenoic acid ethyl ester a novel diketo acid derivative which selectively inhibits the HIV-1 viral replication in cell culture and the ribonuclease H activity in vitro. Antiviral Res. 2005, 65, 117–124. 10.1016/j.antiviral.2004.11.002. [DOI] [PubMed] [Google Scholar]
- Corona A.; Di Leva F. S.; Thierry S.; Pescatori L.; Cuzzucoli Crucitti G.; Subra F.; Delelis O.; Esposito F.; Rigogliuso G.; Costi R.; Cosconati S.; Novellino E.; Di Santo R.; Tramontano E. Identification of highly conserved residues involved in inhibition of HIV-1 RNase H function by diketo acid derivatives. Antimicrob. Agents Chemother. 2014, 58 (10), 6101–6110. 10.1128/AAC.03605-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieger I. V.; Freundlich J. S.; Gawandi V. B.; Roberts J. P.; Gawandi V. B.; Sun Q.; Owen J. L.; Fraile M. T.; Huss S. I.; Lavandera J. L.; Ioerger T. R.; Sacchettini J. C. Structure-guided discovery of phenyl-diketo acids as potent inhibitors of M. tuberculosis malate synthase. Chem. Biol. 2012, 19 (12), 1556–1567. 10.1016/j.chembiol.2012.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summa V.; Petrocchi A.; Bonelli F.; Crescenzi B.; Donghi M.; Ferrara M.; Fiore F.; Gardelli C.; Gonzalez Paz O.; Hazuda D. J.; Jones P.; Kinzel O.; Laufer R.; Monteagudo E.; Muraglia E.; Nizi E.; Orvieto F.; Pace P.; Pescatore G.; Scarpelli R.; Stillmock K.; Witmer M. V.; Rowley M. Discovery of raltegravir, a potent, selective orally bioavailable HIV-integrase inhibitor for the treatment of HIV-AIDS infection. J. Med. Chem. 2008, 51 (18), 5843–5855. 10.1021/jm800245z. [DOI] [PubMed] [Google Scholar]
- Zeng L. F.; Zhang H. S.; Wang Y. H.; Sanchez T.; Zheng Y. T.; Neamati N.; Long Y. Q. Efficient synthesis and utilization of phenyl-substituted heteroaromatic carboxylic acids as aryl diketo acid isosteres in the design of novel HIV-1 integrase inhibitors. Bioorg. Med. Chem. Lett. 2008, 18 (16), 4521–4524. 10.1016/j.bmcl.2008.07.047. [DOI] [PubMed] [Google Scholar]
- Zhang D.; Xu G. F.; Fan Z. J.; Wang D. Q.; Yang X. L.; Yuan D. K. Synthesis and anti-TMV activity of novel N-(3-alkyl-1H-pyrazol-4-yl)-3-alkyl-4-substituted-1H-pyrazole-5-carboxamides. Chin. Chem. Lett. 2012, 23 (6), 669–672. 10.1016/j.cclet.2012.04.010. [DOI] [Google Scholar]
- Gharbaoui T.; Skinner P. J.; Shin Y. J.; Averbuj C.; Jung J. K.; Johnson B. R.; Duong T.; Decaire M.; Uy J.; Cherrier M. C.; Webb P. J.; Tamura S. Y.; Zou N.; Rodriguez N.; Boatman P. D.; Sage C. R.; Lindstrom A.; Xu J.; Schrader T. O.; Smith B. M.; Chen R.; Richman J. G.; Connolly D. T.; Colletti S. L.; Tata J. R.; Semple G. Agonist lead identification for the high affinity niacin receptor GPR109a. Bioorg. Med. Chem. Lett. 2007, 17 (17), 4914–4919. 10.1016/j.bmcl.2007.06.028. [DOI] [PubMed] [Google Scholar]
- Trott O.; Olson A. J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31 (2), 455–461. 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amendola G.; Di Maio D.; La Pietra V.; Cosconati S. Best matching protein conformations and docking programs for a virtual screening campaign against SMO receptor. Mol. Inf. 2016, 35, 340–349. 10.1002/minf.201501021. [DOI] [PubMed] [Google Scholar]
- Lansdon E. B.; Liu Q.; Leavitt S. A.; Balakrishnan M.; Perry J. K.; Lancaster-Moyer C.; Kutty N.; Liu X.; Squires N. H.; Watkins W. J.; Kirschberg T. A. Structural and binding analysis of pyrimidinol carboxylic acid and N-hydroxy quinazolinedione HIV-1 RNase H inhibitors. Antimicrob. Agents Chemother. 2011, 55 (6), 2905–2915. 10.1128/AAC.01594-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kankanala J.; Kirby K. A.; Huber A. D.; Casey M. C.; Wilson D. J.; Sarafianos S. G.; Wang Z. Design, synthesis and biological evaluations of N-hydroxy thienopyrimidine-2,4-diones as inhibitors of HIV reverse transcriptase-associated RNase H. Eur. J. Med. Chem. 2017, 141, 149–161. 10.1016/j.ejmech.2017.09.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercurio M. E.; Tomassi S.; Gaglione M.; Russo R.; Chambery A.; Lama S.; Stiuso P.; Cosconati S.; Novellino E.; Di Maro S.; Messere A. Switchable protecting strategy for solid phase synthesis of DNA and RNA interacting nucleopeptides. J. Org. Chem. 2016, 81, 11612–11625. 10.1021/acs.joc.6b01829. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.